 Hiral Aghara
Hiral Aghara Prashsti Chadha
Prashsti Chadha Palash Mandal
Palash Mandal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 September 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1205821
This article is part of the Research Topic Immune Response Changes in Alcohol and non-Alcohol Associated Tissue Injury View all 11 articles
Alcoholic liver disease (ALD) poses a significant threat to human health, with excessive alcohol intake disrupting the immunotolerant environment of the liver and initiating a cascade of pathological events. This progressive disease unfolds through fat deposition, proinflammatory cytokine upregulation, activation of hepatic stellate cells, and eventual development of end-stage liver disease, known as hepatocellular carcinoma (HCC). ALD is intricately intertwined with stress mechanisms such as oxidative stress mediated by reactive oxygen species, endoplasmic reticulum stress, and alcohol-induced gut dysbiosis, culminating in increased inflammation. While the initial stages of ALD can be reversible with diligent care and abstinence, further progression necessitates alternative treatment approaches. Herbal medicines have shown promise, albeit limited by their poor water solubility and subsequent lack of extensive exploration. Consequently, researchers have embarked on a quest to overcome these challenges by delving into the potential of nanoparticle-mediated therapy. Nanoparticle-based treatments are being explored for liver diseases that share similar mechanisms with alcoholic liver disease. It underscores the potential of these innovative approaches to counteract the complex pathogenesis of ALD, providing new avenues for therapeutic intervention. Nevertheless, further investigations are imperative to fully unravel the therapeutic potential and unlock the promise of nanoparticle-mediated therapy specifically tailored for ALD treatment.
The liver, as one of the largest immune organs in the human body, plays a crucial role in maintaining overall immunity by creating an immunotolerogenic environment (1–3). Comprised mainly of hepatocytes, which make up 70% of its composition, the liver also contains various non-parenchymal cells such as hepatic sinusoidal endothelial cells, hepatic stellate cells (HSC), Kupffer cells (liver macrophages), and pit cells (liver-specific natural killer cells) (4). The parenchymal cells of the liver produce innate immunity proteins, bactericidal proteins, and opsonins, contributing significantly to the body’s innate immune response (5, 6). Additionally, the liver has unique characteristics where liver allografts are more readily accepted due to heightened innate immunity and suppressed adaptive immunity compared to other organs (5, 7). However, if the liver’s immunotolerant environment is disrupted by factors such as viral infections, an unhealthy diet, or certain drugs, it can lead to liver disease.
Excessive and prolonged alcohol intake is a major cause of liver disease, specifically known as alcoholic liver disease (ALD). ALD, along with non-alcoholic liver disease (NAFLD), which is now referred to as metabolism-associated liver disease, pose significant health problems worldwide. Alcohol abuse is considered the leading risk factor for disease and disability globally, contributing to a mortality rate of 4.8% worldwide, with approximately 10% of these mortalities occurring in India (8). Furthermore, alcohol’s negative impact extends to socioeconomic activities. To provide context, a standard beer or wine cooler typically contains 5% alcohol, with 355mL of the drink containing 14 grams of pure alcohol. Malt liquor contains around 7% alcohol, with 355mL of the drink containing 19.60 grams of pure alcohol. Furthermore, 740mL of 12-14% wine contains approximately 70-80 grams of pure alcohol. The dietary guidelines in the United States suggest that consuming one drink per day for women and two drinks per day for men is considered moderate and does not typically lead to liver disease (9).
The pattern of ALD progression is illustrated in Figure 1 (10). If an individual consumes more than 40g of pure alcohol per day for an extended period, they may develop alcoholic fatty liver disease (AFL). In AFL condition, excess alcohol would interrupt fat oxidation by inhibiting β oxidation of fatty acid and 5ʹ-AMP-activated protein kinase (AMPK). If alcohol consumption is halted at this stage, AFL can be reversed to a healthy state. However, if an individual continues to consume high amounts of alcohol, inflammation will occur along with fat deposition, leading to the stage called alcoholic steatohepatitis (ASH). At this stage gut dysbiosis also takes place and it interacts with liver residential macrophages which further activate quiescent hepatic stellate cells. It will produce collagen and extra cellular matrix protein. Some growth factors like TGF-β and platelet derived growth factor- β are most known for HSC proliferation. Due to quiescent Hepatic Stellate Cells (HSCs) activation, the disease progresses to liver fibrosis and at this stage retinol storage decreases and extracellular matrix increases. Activated quiescent HSCs produce more collagen which would further promote capillarization of hepatic sinusoidal endothelial cells and interrupt nutrient transport. Further, the disease progresses to alcoholic cirrhosis and hepatocellular carcinoma, which denote end-stage liver disease (11, 12).
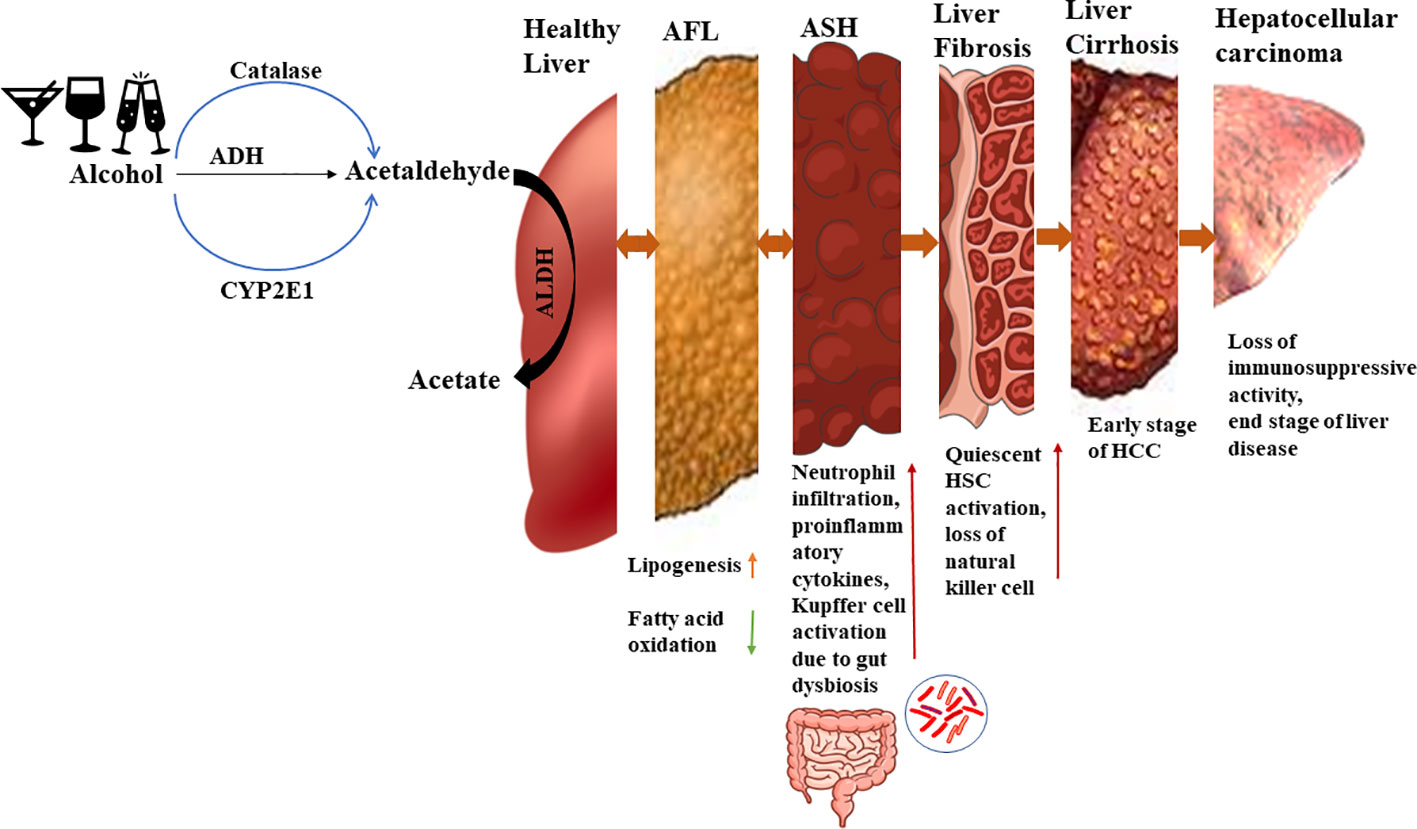
Figure 1 Alcohol metabolism and ALD progression. In a healthy state, ethanol is converted to acetaldehyde by alcohol dehydrogenase and then to acetate by aldehyde dehydrogenase. But in presence of excess ethanol, the MEOS pathway is activated leading to increased acetaldehyde levels. During the initial stage of alcoholic liver disease (ALD), there is an increase in lipid generation and reduction in fatty acid oxidation, resulting in lipid accumulation in the liver. Approximately 10-35% of individuals progress to alcoholic steatohepatitis, characterized by sterile inflammation and elevated cytokines and chemokines. Kupffer cells are activated by gut endotoxins during this stage. Continued heavy alcohol consumption leads to liver fibrosis, where around 40-50% of individuals experiencing activation of quiescent hepatic stellate cells due to Kupffer cell activation. Natural killer cell activity declines, further promoting hepatic stellate cell activation which accelerates fibrosis. Subsequently, 8-20% of individuals develop liver cirrhosis, an early stage of end-stage liver disease, and approximately 3-10% progress to hepatocellular carcinoma, an advanced stage of liver disease.
Alcohol metabolism occurs via two main pathways, the alcohol dehydrogenase (ADH) pathway and the non-ADH pathway which is also recognized as the microsomal ethanol oxidizing system (MEOS) (13). The ADH pathway is a primary pathway for ethanol metabolism in liver cells and occurs in cytosol due to presence of necessary enzymes. The MEOS pathway involves enzymes like catalase and CYP2E1 which are present in the peroxisomes and microsomes of cells. Pathways involving all the above enzymes are oxidative pathways. The non-oxidative metabolism of ethanol is not prominent but leads to the formation of fatty acid ethyl esters (FAEEs) or phosphatidic acid (PA) (14). The literature reports that if the alcohol concentration in blood is less than 10M/L, then alcohol metabolism will take place via the ADH pathway (15). As the blood alcohol concentration elevates, the other two pathway will also participate in ethanol metabolism.
Alcohol catabolism occurs, and initially acetaldehyde is formed and further converted into acetate via acetaldehyde dehydrogenase (ALDH). Since acetaldehyde is a toxic compound and acts as a mutagen, it is rapidly catabolized to acetate (16). However, as the alcohol concentration increases, more acetaldehyde is formed, posing a risk to cells. Acetaldehyde can bind to proteins and DNA, leading to structural and functional changes, the generation of neoantigens, and activation of the immune system (17). The progression of ethanol metabolism is accompanied by an increase in reactive oxygen species (ROS). These ROS contribute to the formation of lipid peroxides, which can further deform the structure of DNA and proteins through binding (18). Excessive lipid peroxidation results in the formation of electrophile products like Malondialdehyde (MDA) and 4 Hydroxynonenal (4-HNE), which bind with essential proteins, disrupting cellular homeostasis (18, 19). Figure 1 shows alcohol metabolism and ALD progression.
With an increase in alcohol intake, the cellular antioxidant defense mechanism weakens. Antioxidants such as glutathione and superoxide dismutase (SOD) are naturally present in cells, and their levels decrease as alcohol consumption rises. This results in an inverse relationship between the concentration of free radicals and antioxidants within the cells. Furthermore, excessive alcohol intake can also have an impact on vitamin levels. The elevated alcohol concentration triggers various stress mechanisms within the body.
A proper balance between antioxidants and reactive oxygen species (ROS) is generally maintained in every cell (20). ROS, being highly unstable free radicals, are generated by cellular mechanisms and play essential roles in cellular communication and other processes. Various species of ROS, including H2O2, O2-, and `OH are produced due to increased NOX4 activity (21). While ROS have several detrimental effects, they are generally important for transmitting messages and eliciting responses. There are two types of antioxidant mechanisms that remove excess ROS (22, 23) and both are equally vital for cellular function. Maintaining homeostasis between these two mechanisms is crucial. Imbalances in these mechanisms can lead to disease conditions (24, 25) which may arise from factors such as lifestyle choices, drug use, chemotherapy, and other epigenetic factors. Within cells, two types of antioxidant mechanisms exist. The first is an endogenous or enzymatic antioxidant mechanism generated within cells, while the second is an exogenous or non-enzymatic antioxidant mechanism supplemented by food products (26). Elevated ROS levels can damage cells and activate apoptosis-related mechanisms. ROS also play significant roles in various cell signaling pathways, such as NFκB, MAPK, ion channeling, and the ubiquitin proteasome response (27). These pathways can further trigger Endoplasmic reticulum (ER) response and other pathways, which will be discussed later. Figure 2 illustrates the damage caused by ROS to cells (28–32).
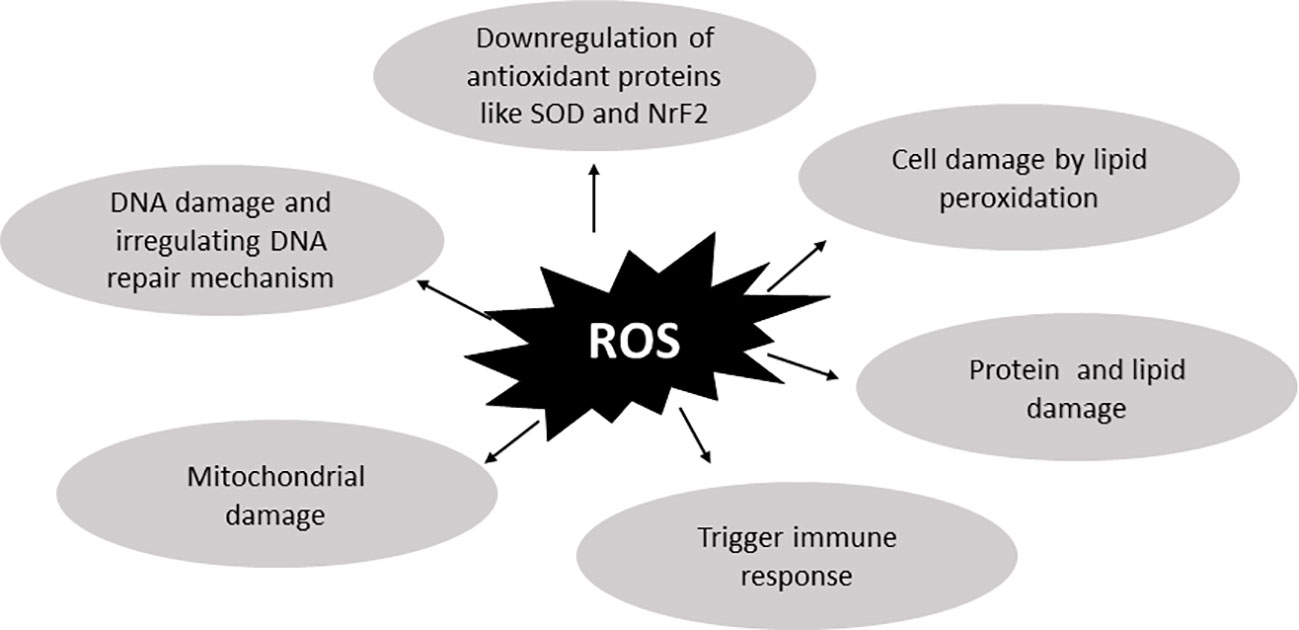
Figure 2 ROS-related damage to cells. Reactive oxygen species are important for cell-to-cell crosstalk but in excessive amounts they lead to diseased conditions. By binding protein, lipid, and DNA, they damage the cellular mechanism and also dysregulate antioxidant activities. They also dysregulate the DNA repair mechanism and cause more DNA damage. Further increased ROS concentrations damage cells by generating lipid peroxidation which further damages cell membrane and causes cell death.
Ethanol and acetaldehyde both are toxic to cells. The presence of these toxins results in mitochondrial damage further affecting various mechanisms which damage the cells (32). There are numerous factors involved in ROS generation. Therefore, it is important to understand these mechanisms and the harm caused by ROS generation in ALD.
At the early stage of ALD, fat generation occurs and the oxidation rate of fatty acids is slower (33). Even at the first stage, there are many elements that help ROS generation. Some are mentioned below:
● NAD+: NADH ratio: As the conversion of ethanol to acetaldehyde and acetate occurs, it also generates NADH, which dysregulates the cellular redox potential (10). Numerous metabolic processes depend on NAD+ since it is essential for maintaining the cellular levels of sirtuins (34), which play a crucial role in the antioxidant system and DNA repair in cells (35, 36). Additionally, sirtuins are important for the brain in maintaining circadian rhythm (37). A reduced NAD+:NADH ratio impacts histone deacetylase activity (38). The increased NADH also affects the antioxidant defense mechanism by reducing its power (39).
● Sterol regulatory binding protein (SREBP1C) and other early growth responses (EGR1) are activated by the CYP2E1 cycle, which in turn activates the lipogenic gene (33, 40). According to (41–44), CYP2E1 is crucial in the detoxification of xenobiotics and other hazardous compounds. The detoxification of bioactive compounds generates toxic compounds or carcinogenic products which further contribute to disease progression (45, 46). In liver cells, ROS generation is significantly affected by the activity of CYP2E1 (47, 48).
● According to Vasiliou et al., (49), the microsomal ethanol-oxidizing system (MEOS) pathway plays a significant role in ethanol sensitivity. An increased acetaldehyde concentration further reduces the ability of ALDH2 to convert acetaldehyde to acetate (13, 50, 51). Acetaldehyde is a carcinogenic substance (52) that further alters many biomolecule components and produces ROS (16, 53). It also leads to the formation of DNA adducts, causing mutations, and induces changes in protein structure, resulting in the formation of neoantigens. These neoantigens are further detected by T cell immune cells and form inflammatory responses (17). In addition, acetaldehyde also interacts with other mechanisms (54–56).
● Excessive alcohol consumption damages the mitochondria, which leads to ROS production. An elevated amount of NADH can hinder ATP production and negatively impact the electron transport chain. Excessive alcohol consumption also decreases the function of mitochondrial respiratory complexes, specifically complex I, III, IV, and V, which are involved in converting ADP to ATP. This metabolic state promotes higher H2O2 production. Ethanol further amplifies the imbalance of oxidative phosphorylation and ROS generation (25, 57–59). According to (60), increased NADH levels contribute to more ROS formation, which can interact with cellular components such as lipids, proteins, nucleic acids, and especially mitochondrial DNA (mtDNA), enhancing oxidative stress and leading to apoptosis. ROS production affects mtDNA and compromises cellular energy metabolism, resulting in the production of even more ROS. In summary, higher concentrations of ROS damage mitochondria, leading to further elevated concentrations of ROS.
● Overall, in all the other mechanisms, there is one more mechanism which has been studied and is still being explored regarding ALD progression. It also damages mitochondria, produces more ROS, and contributes to disease progression. Necroptosis is a cell death mechanism which is activated due to RIPK1 and RIPK3 (receptor interacting protein kinase 1 and 3) (61, 62). As mitochondrial ROS generation increases, it also triggers inflammation. Damage associated molecular patterns (DAMP), interferon, death ligand-like tumor necrosis factor, Fas ligand, toll-like receptors, and Tumor necrosis factor-related apoptosis-inducing ligand can activate RIPK1 and RIPK3. As alcohol accumulation increases, it activates RIPK1 and RIPK3. Individually RIPK1 can cause inflammatory activity by neutrophil infiltration and producing proinflammatory cytokines and chemokines. Furthermore, RIPK3 is also elevated due to overexpression of cytochrome p450E1. As RIPK3 phosphorylates and it interacts with mixed lineage kinase domain-like protein (MLKL) and leads to membrane disruption and necroptosis (63, 64). By that mechanism is also increases oxidative stress by generating more ROS and inflammatory response (65). Further molecular mechanisms still need to be clarified to prevent the progression of the disease.
ROS not only affects main pathways but also hinders non-relevant pathways, such as Brahma-related gene 1 (BRG1) expression (31). BRG1 is responsible for the activation of various antioxidant mechanisms which are mutated due to excess ROS (66). Even the overexpression of BRG1 further correlates with SREBP1c and contributes to lipid metabolism (67–69). Research is still underway to explore the connection between BRG1 and other inflammatory genes and pathways, as there are conflicting findings in some studies regarding the formation and role of BRG1 (70). The relationship between BRG1 and other pathways needs to be explored in regard to ALD.
ROS and acetaldehyde have the ability to disrupt the structure of proteins. In the presence of intracellular stress factors such as the accumulation of misfolded proteins in the endoplasmic reticulum (ER), a chaperone-mediated stress response is activated to aid in the recovery and restoration of ER function. However, in cases where recovery is not feasible and the stress becomes overwhelming, the unfolded protein response (UPR) directs the cell towards either autophagy, which helps maintain cellular homeostasis, or apoptosis, leading to the clearance of the affected cell. Autophagy pathways, including PERK/ATF4, IRE1α, ATF6, and Ca+, can be activated and play a dual role. They can inhibit apoptosis by dampening the activity of apoptosis-associated caspases, thereby promoting cell recovery. Alternatively, they can also induce apoptosis, leading to the removal of accumulated proteins, lipid droplets, and damaged cellular components through autophagy (71). However, excessive alcohol accumulation can inhibit autophagy, which contributes to disease progression by generating endoplasmic reticulum stress (72).
Cell damage can be caused not only by oxidative stress but also by ER stress, which acts as a secondary hit model for disease progression (73). The ER-associated degradation (ERAD) reaction is activated in response to unfolded or damaged proteins, aiming to maintain homeostasis. Under adverse conditions, ERAD supports the preservation of cellular equilibrium. Oxidative stress increases the production of ROS, which, in turn, triggers the activation of ER-associated genes due to stress metabolites, protein adducts, and protein misfolding. Additionally, other cellular responses, such as increased synthesis of CYP2E1 and a decreased liver S-adenosylmethionine (SAM)/S-adenosylhomocysteine (SAH) ratio, contribute to the development of ER stress (74, 75). SAM acts as a methyl donor and aids in the formation of glutathione, an important antioxidant. SAH, on the other hand, acts as a potential inhibitor of methylation. Furthermore, decreased NAD+ levels during alcohol metabolism directly increase the concentration of N-nicotinamide methyltransferase (NNMT), which plays a crucial role in maintaining the balance between NAD+ metabolism and the methionine cycle. Increased NNMT levels further elevate SAH. The decreased SAM : SAH ratio leads to an increase in CYP2E1, lipid peroxidation, the upregulation of ER stress marker genes, and prevention of DNA methylation (76–79). In ER stress, the activation of the PERK-ATF4 pathway plays a critical role in hepatic NNMT activation. Some researchers have reported that NNMT inhibition protects against alcohol-induced fatty liver disease and the generation of hepatic de novo lipogenesis (80, 81). There is a connection between oxidative stress and ER stress which still needs to be explored.
Various types of proteins are targeted in ALD. Excessive alcohol intake and its byproducts can cause denaturation or deformation of many proteins. As hepatocytes are rich in ER, they play a crucial role in maintaining protein homeostasis within cells. In adverse conditions, the unfolded protein response (UPR) becomes more prominent and helps maintain cellular homeostasis. However, prolonged activation of UPR can lead to inflammation and interaction with other pathways (82). The UPR consists of three main branches that increase the folding capacity of the ER, promoting protein folding and reducing protein translation by affecting mechanisms involved in protein synthesis and polypeptide formation (75, 83). This process increases ER-associated degradation (ERAD) and reduces protein translation by affecting mechanisms involved in protein synthesis and polypeptide formation (75). Within the ER, transmembrane sensors such as BiP, GRP78, and intraluminal chaperones are inactivated when they detect calcium depletion. The UPR is activated only after three transmembrane proteins, namely, inositol-requiring protein 1 (IRE1), dsRNA-activated protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6), dissociate from BiP/GRP78, which serves as an inhibitory binding chaperone.
In healthy states, due to IRE1 activation, XBP1 is spliced (sXBP1) and plays a vital role in activating other ERAD pathway genes, which further halt protein translation. Furthermore, PERK initiates phosphorylation of elF2-α subunit and activates ATF4, and it helps in activating ERAD, amino acid metabolism, antioxidant stress response, CHOP, and GADD34 activation (84). ATF6 is activated in the Golgi and it functions as a transcription factor that promotes the expression of XBP1, CHOP, and ER chaperons. Among all of them, CHOP is crucial for ER associated apoptosis. In normal conditions, all the above-mentioned factors play key roles in maintaining homeostasis in cells; but in adverse conditions, their overexpression induces inflammatory response by activation of NFκB and IL-1β (74, 82). Additionally, the JNK pathway is activated by the ER stress-induced response, which is critical for the progression of this disease (84). ER stress-induced apoptosis is caused due to the upregulation of proapoptotic proteins like CHOP, cell cycle arrest, and DNA damage (gene/protein) such as GADD34/135. Caspase 12 and 4, JNK, and IFN3 signaling components are increased as a result of ER stress and cause cell death (85).
The lipid peroxidation products MDA, MAA, 4-HNE, and acrolein are highly active and can impair structure and activity, ultimately leading to the generation of ER stress. Smathers et al., (86), concluded in their study that 4-HNE contributes to modifying Hsp70, Hsp90, and protein disulfide isomerase (PDI) in an animal model. All these targeted proteins are important for ATPase activity, the rearrangement of misfolded protein, and dysfunctional restoration of mismatched impaired protein disulphide bonds. These are the mechanisms by which 4-HNE contributes to ER stress and hepatic lipid accumulation. Acrolein is also one of the lipid peroxidation byproducts, which is not well explored in alcoholic liver disease. Chen et al., (87), showed in their work that alcohol consumption leads to hepatic acrolein accumulation and a decrease in glutathione-s-transferase-Pi (GSTP) that helps to metabolize acrolein. Acrolein induces proapoptotic signals and increases in ER stress. Furthermore, adducts formed due to acetaldehyde bind to advanced glycation end products and increase ROS generation, hepatocyte ballooning, apoptosis, and steatosis by hepatic fatty degeneration and gut leakiness (88). Acetaldehyde also impairs mitochondrial glutathione and sensitize cells to TNF (89).
In addition to the aforementioned mechanisms, it has been demonstrated that ER stress contributes to both inflammation and lipogenesis. The presence of essential enzymes on ER would further renders the crucial site for lipid metabolism (90). ER stress genes, predominantly the PERK/ATF4 pathway, can activate SREBP1c, which in turn elevates the concentrations of FAS and ACC (91, 92). According to (93)‘s research, ER stress causes elevated levels of in lipogenic enzyme due to the cleavage of SREBP1C by site 1 protease (S1P) and site 2 protease (S2P). Many researchers describes ER stress-mediated disease progression as the second stage of alcohol-related liver disease as a result of upregulated ER stress inflammatory and steatosis pathways.
Prolonged ER stress promotes cell apoptosis by activating NFκB and inhibiting the translation of IκB, leading to macrophage activation generating an inflammatory response. Along with these other mechanisms, ER stress also influences the activation of lipogenic factors such SREBP1C (84, 94). Currently, it can be said that oxidative stress generated due to ethanol metabolism is an important factor for ALD progression. Researchers have also carried out experiments with zebrafish to prove this theory (95). As fat accumulation and inflammation increase in the liver, they can also disrupt the gut environment and contribute to disease progression.
Collectively, the bacteria, fungi, viruses, archaea, and eukarya colonizing the GI tract are termed ‘gut microbiota’ (96). The gut microbiota are vital components of our daily lives, actively contributing to vitamin synthesis, the creation of both essential and non-essential amino acids, and the production of short-chain fatty acids. They form a mutualistic relationship with the host and protect against environmental factors and some antigens by generating immunogenic responses (97). The gut contains more than 1012 to 1014 microbial cells, which is nearly equivalent to the total number of the cells in entire body (98). Scientists became interested in the mutualistic link between the host and gut microbiota because changes in gut bacteria are critical in the development of disease and also affect the metabolism of many vitamins, amino acids, enzymes, and short chain fatty acids. From infancy until adulthood, several bacterial alterations take place to maintain homeostasis.
Alcohol does not only affect liver; it also affects other organs like the brain (99), gut intestinal track (100), kidney (101), breast (102), and heart (103). Due to excessive alcohol consumption, gut microflora also gets disturbed (104). According to (105), when an excessive amount of alcohol is administered, the established bacterial community will seek more alcohol because it will assist to maintain its dominance in the gut ecosystem. Additionally, they looked at the higher Actinobacterial concentration in the gut of the alcohol group. Firmicutes and Bacteroidetes made up the majority of the gut microbiota, while Proteobacteria, Verrucomicrobia, Actinobacteria, and Fusobacteria were also present but in the minority (106).
Firmicutes, a class of bacteria with a predominance of gram-positive bacteria, and Bacteroidetes, a class of bacteria with a predominance of gram-negative bacteria, are more prevalent in the gut microbiota in normal conditions, making up approximately 90% of it (96, 106). But, comparing Firmicutes and Bacteroidetes, the concentration of Firmicutes is higher. Other bacteria classes include Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia (106–108). Firmicutes are composed of many bacteria like Lactobacillus and other opportunistic pathogenic bacteria like Clostridium which are relatively low. The Firmicutes and Bacteroidetes (F/B) ratio is important to maintain intestinal homeostasis. As the F/B ratio is increased, depending on lifestyle, food intake, and other environmental factors, gut homeostasis is affected. Due to a heavy alcohol intake, the F/B ratio gets disturbed and increases and, because of this, gut dysbiosis is generated (109). Additionally (110), mentioned that due to a high fat diet, Bacteroides and Actinobacteria are majorly benefited while the opposite goes for Firmicutes and Proteobacteria. This explains that diet and lifestyle affect gut microbial composition. Similarly, alcohol changes gut microbiota by enhancing the F/B ratio (111). showed that due to excessive alcohol consumption, Proteobacteria and many species of Bacteroidetes benefit but most abundant phyla of Firmicutes are inhibited in the presence of alcohol. Mainly, Lactobacillus and Bifidobacterium along with Faecalibacterium prausnitzii and Akkermansia muciniphila are inhibited (112). Such a shift from good bacteria to harmful bacteria results in gut dysbiosis and affect bacterial products. It further triggers other immune regulators and helps in disease progression.
The gut microbiota is composed of a mixture of bacteria, fungi, viruses, and archaea. While the bacterial component of the microbiota is well understood, other microbial groups have not been extensively studied. Nevertheless, they also play a significant role in maintaining eubiosis and promoting gut homeostasis (113). Ethanol consumption leads to fungal dysbiosis, which in turn contributes to the generation of immune responses (114). Common fungal species found in the gut include Candida spp., Saccharomyces cerevisiae, Penicillium commune, and Aspergillus versicolor (115). These species are present in higher concentrations, while others such as Galactomyces, Debaryomyces, Cladosporium, and Trichosporon are present in lower abundance. With increased alcohol consumption, the concentration of Candida albicans, a pathogenic fungus known to induce inflammatory responses, also increases (114). Conversely, the concentrations of Galactomyces, Debaryomyces, and Saccharomyces cerevisiae decrease (116, 117). Patients with alcoholic hepatitis have been found to exhibit reduced fungal diversity due to the overgrowth of Candida albicans (118). In patients with alcoholic hepatitis, the presence of anti-Saccharomyces cerevisiae antibodies indicates a systemic immune response against fungal products (116).
Gut viruses are different in every person and because of that are recognized as virus fingerprints. Some scientists describe them as ‘dark matter’ in the intestine. The human gut contains 10 times more phages than symbiotic bacteria. According to GVD, 97.7% of the viral population are phages, 2.1% are eukaryotic viruses, and the remaining are archaeal viruses (119). In normal conditions, the majority of phages come from the Caudovirales order. In chronic ethanol feeding, gut virome abundance is also disrupted and the concentration of Lactobacillus phages and Propionibacterium phages decrease while Streptococcus and Lactococcus phages are increased and help in the progression of disease severity (120, 121).
The shift from good to bad bacteria disrupts many intestinal mechanisms such as the production of many short chain fatty acids (SCFA) like Butyrate, change in the mucus layer integrity, endotoxemia, increased bacterial acetaldehyde dehydrogenase activity, and intestinal impermeability development. Due to all these conditions, gut leakiness can occur. In every step, molecular mechanisms are involved which are directly or indirectly related to the gut dysbiosis generated due to heavy alcohol intake (122, 123).
● Gut microbes help to ferment indigested dietary products into SCFAs. Due to alcohol consumption, this process is negatively impacted in the conversion of Butyrate co-A to Butyrate. It occurs due to change in 2 butyryl-CoA: acetate CoA transferase (BUT) and the butyrate kinase (BUK) gene (124). The impact of ethanol on gut tight junctions and gut metabolites is still unknown. Researchers have shown interest in decoding these particular mechanisms in order to discover how ethanol changes intestinal metabolite composition and how those metabolites protect against ethanol driven injury. Several researchers have explored the positive impact of exogenously administered SCFAs, namely butyrate, propionate, and acetate. These compounds appear to enhance AMPK activity and reduce metabolic stress. Even the tight junctions of Caco2 monolayer cells are restored after treatment with SCFAs (125–128). SCFAs are further helpful in generating immune signal cascades coupled with G protein (129). CPT1A gene expression is important for fatty acid oxidation. Due to a heavy alcohol intake, CPT1A expression decreases. But when the butyric treatment is given, it acts as an inhibitor to histone deacetylase (HDAC) and increases CPT1A expression (130). In this way, alcohol consumption changes butyric concentrations, which is one of the essential SCFAs.
● Muc2 overexpression is observed in alcoholics, which further correlates with antibiotic resistance and gut leakiness (permeability). Due to gut dysbiosis, Muc2 expression is increased which further acts as a glycan source for the bacteria and the overgrowth of undesirable bacteria is observed. In many muc2 deficient mice, alcoholic steatohepatitis is delayed (131–135). But there are still some contradictory studies that deny these observations. Therefore, further studies still need to be performed in order to understand the mechanism.
● Gut leakiness is generated due to ethanol which affects the tight junctions of intestinal cells, which leads to alterations of gut permeability. Increased acetaldehyde concentrations brought on by bacterial alcohol dehydrogenase activity causes leaky gut (136). Tight junctions and adherent junction protein’s integrity mainly depend on protein phosphorylation and dephosphorylation. Many proteins are involved in tight junction integrity. Acetaldehyde is one of the major effective molecules that rearranges the components of tight junction proteins. Due to increase in acetaldehyde, tyrosine phosphorylation of ZO-1, E cadherin also increases. This phosphorylation activity can be attenuated via protein tyrosine phosphatase activity which is hindered because of the increased concentration of acetaldehyde (137, 138). Further endotoxins trigger the dysregulation of tight junction proteins by activating NFκB, TNF-α mediated damage, and the downregulation of ZO-1 (139, 140).
● Zinc is a trace element that helps to maintain cell homeostasis, detoxification, antioxidant defense, and in gene regulation. Zinc acts as a nutraceutical substance to maintain the barrier function of the epithelium (141). Zinc deficiency is another factor for gut permeability by disassembling tight junction proteins (142), and, due to zinc deficiency, antioxidant mechanisms also get disrupted and inflammatory response gets activated (143–146). It is also associated with hepatic nuclear factor 4α (HNF4α) activity and causes damage to lipid metabolism by dysregulating PPARα (147–149). Zinc helps to maintain the intestinal barrier integrity and regeneration of impaired epithelium. It helps in invading molecular ions and pathogens by occluding proteolysis and occluding transcription. By barrier disfunction, neutrophil infiltration will be carried out and mucosal inflammation will be carried away (150, 151).
● Reg3 gene encodes a number of regenerating islet-derived genes which show antimicrobial and bactericidal activity (109, 152, 153). This family of genes plays an important role in maintaining gut homeostasis and acts as an antimicrobial defense mechanism (154, 155). Chronic ethanol feeding reduces the expression of Reg3 in the intestine (156–158). This compromised mechanism further contributes to disease progression.
There are still different mechanisms which need to be uncovered and found to understand the mechanisms behind disease progression so that some therapeutic targets can be generated which can help in disease amelioration. For instance, Reg3 protein genes have antimicrobial and bactericidal activity and are connected to the MYD88 pathway but the cell signaling behind these still needs to be explored (159). Further, gut dysbiosis due to ethanol dysregulates indole 2 acetic acid (IAA), type 3 innate lymphoid cells, and aryl hydrocarbon receptors (AHR), and causes an increase in inflammation (156).
As gut dysbiosis takes place, endotoxin and other material from the gut first enter the liver via the portal vein. In normal conditions, liver sinusoidal endothelial cells (LSEC) help to maintain homeostasis by removing toxicants, viruses, waste products, and lipopolysaccharide (LPS). They also play a key role in maintaining quiescent hepatic stellate cell conditions. But in adverse conditions, they convert to their proinflammatory phenotype and further also help in activating quiescent hepatic stellate cells. LSEC produce proinflammatory cytokines like TNF-α, MCP-1, and macrophage inflammatory protein -1 (MIP-1). They also generate immune response and activate Kupffer cells and HSC.
Hepatic stellate cell activation is a significant feature in the progression of alcoholic liver disease. These cells, known for their role in lipid storage, particularly retinoic acid (Vitamin A), are impacted by increased alcohol intake. The activity of CYP2E1 interferes with retinyl ester formation and disrupts the balance of the extracellular matrix (160, 161). Furthermore, bacterial lipopolysaccharides (LPS) and endotoxins not only activate Kupffer cells but also stimulate hepatic stellate cells (162, 163). This activation triggers the MyD88-dependent pathway, leading to the production of inflammatory cytokines and transforming growth factor β (164). Activated Kupffer cells produce interferon regulatory factors, which in turn activate quiescent hepatic stellate cells, transforming them into myofibroblasts (165). Pit cells, also referred to as natural killer (NK) cells, normally exhibit antifibrotic and anti-inflammatory activities. However, in diseased conditions, their functionality is compromised, leading to an inability to carry out their intended functions (114). Additionally, their cytotoxicity towards activated hepatic stellate cells is reduced due to changes in their surface ligands and the accumulation of higher levels of TGF-β (166).
The interaction between the gut and liver plays an essential role. Liver residential macrophages- Kupffer cells play a significant role. There are two types of Kupffer cells, M1 and M2, and they switch from M2 to M1 as per immunogenic conditions. M1 is activated because of bacterial products like lipopolysaccharides (LPS) and produces proinflammatory molecules. The moment when M1 is activated, M2 macrophages, which are mainly known for anti-inflammatory activity, are suppressed (167, 168). As mentioned earlier, ethanol and its byproducts create gut dysbiosis, causing endotoxemia. As the release of LPS endotoxins increase, they can enter the blood circulation system and hence can travel to many organs. It can also cross the blood–brain barrier and liver parenchymal layer, causing maximum damage. Endotoxins travel through the portal vein and enter the liver. Due to LPS, liver Kupffer cells are activated via pathogen-associated molecular patterns (PAMPs); Tall like receptor (TLR4). LPS-binding protein (LPB) helps LPS to translocate and bind to CD14. It further facilitates binding to the TLR4/MD2 complex. These endotoxins or LPS bind to TLR4 in order to support MD2 and CD4+. This complex further targets two major mechanisms. The reason behind the triggering of specific mechanisms is still a gray area. Majorly, the MyD88 and TRIF pathways are activated. Both the pathways produce different types of inflammatory molecules and further cause more liver injury (169–173). Kupffer cell activation mechanisms are well known and have been extensively studied, but the activation resulting from fungal expression is not as well investigated. The C-type lectin-like receptor (CLEC7A) on Kupffer cells has the ability to bind to 1-3β glucan, which is present on the fungal cell wall. Upon activation, Kupffer cells produce IL-1β (174).
Alcohol damages not only the liver but also affects other organs and disrupts their physiological mechanisms. If the right abstinence conditions, diet, and healthy lifestyle are adopted, ALD can be treated in its early stages and reversed. There are some targeted drugs that are being observed in clinical trials. S-adenosyl methionine and granulocyte colony stimulation factor drugs produced average results in clinical phase 2 trials (175). Furthermore, there are other pharmaceutical agents are being studied clinically and, from them, metadoxine completed a phase IV trial and produced effective results for severe alcoholic hepatitis (176). There are several other steroid-like corticosteroid-based treatments available, but if positive results are not observed then they are discontinued. Other anti-inflammatory treatments, such as pentoxifylline (15), antioxidant treatments, like SOD, and natural antioxidant treatments, like silymarin (33), are also available. Research is ongoing for probiotics (177–179) and synbiotics (180) as well. VSL#3 is in clinical trials (Clinical.Trail.gov identifier no. NCT05007470). In the end stage of liver disease, liver transplantation is the only option and even after liver transplantation, proper lifestyle and diet changes should be maintained.
Though there are various treatments available, none are FDA approved and have some drawbacks. To minimalize the drawbacks, nanoparticles are used. There are various type of nanoparticle and nano-formulations are used to check against different types of liver disease, targeting the same mechanism involved in ALD. The use of nanoparticles for ALD should be studied more. Some of the research done on various liver diseases and nano-formulations are mentioned in Table 1. There are several bare nanoparticles used to treat liver disease mentioned in Table 2.
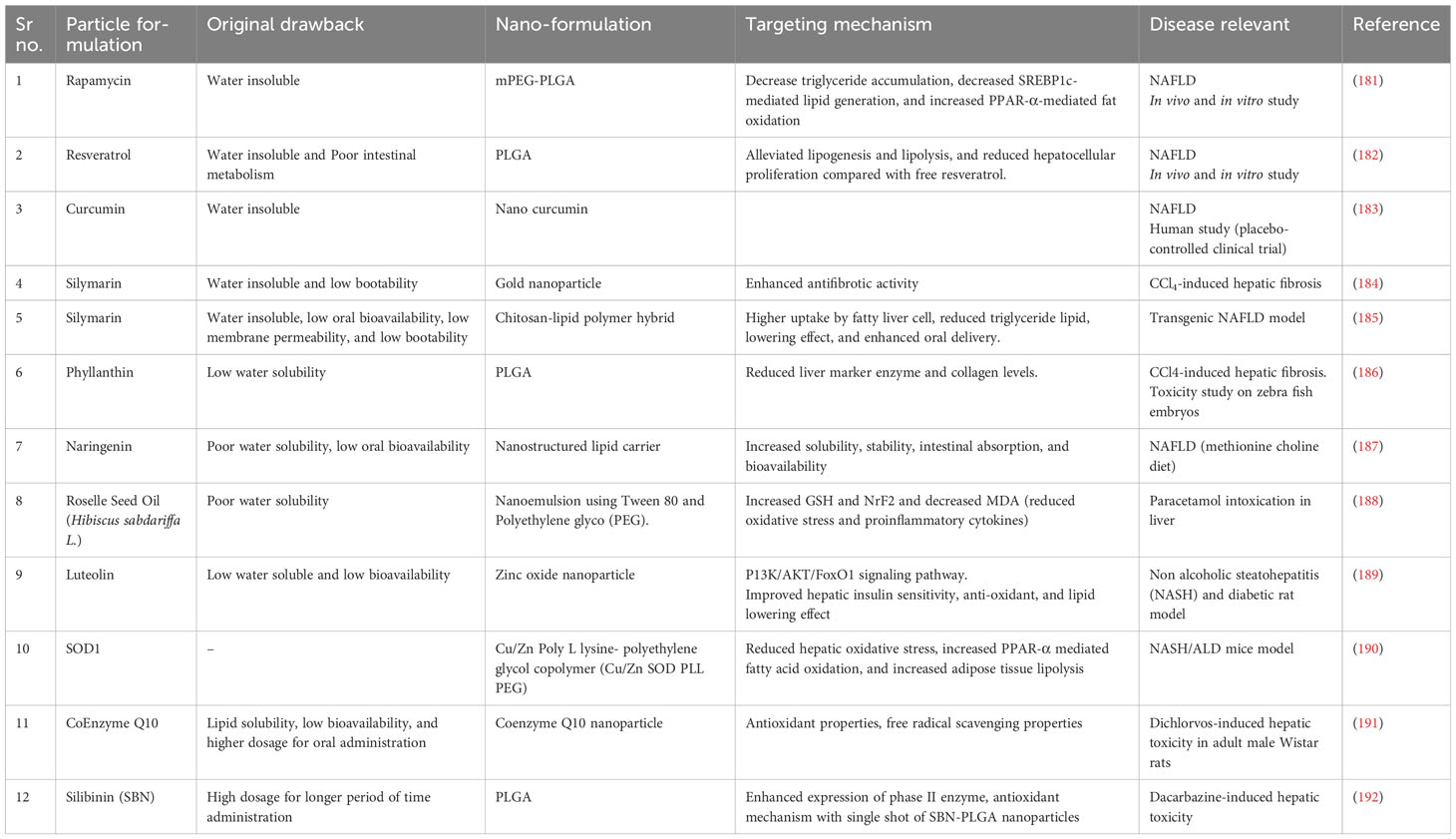
Table 1 Nano-formulations and their role in liver disease.
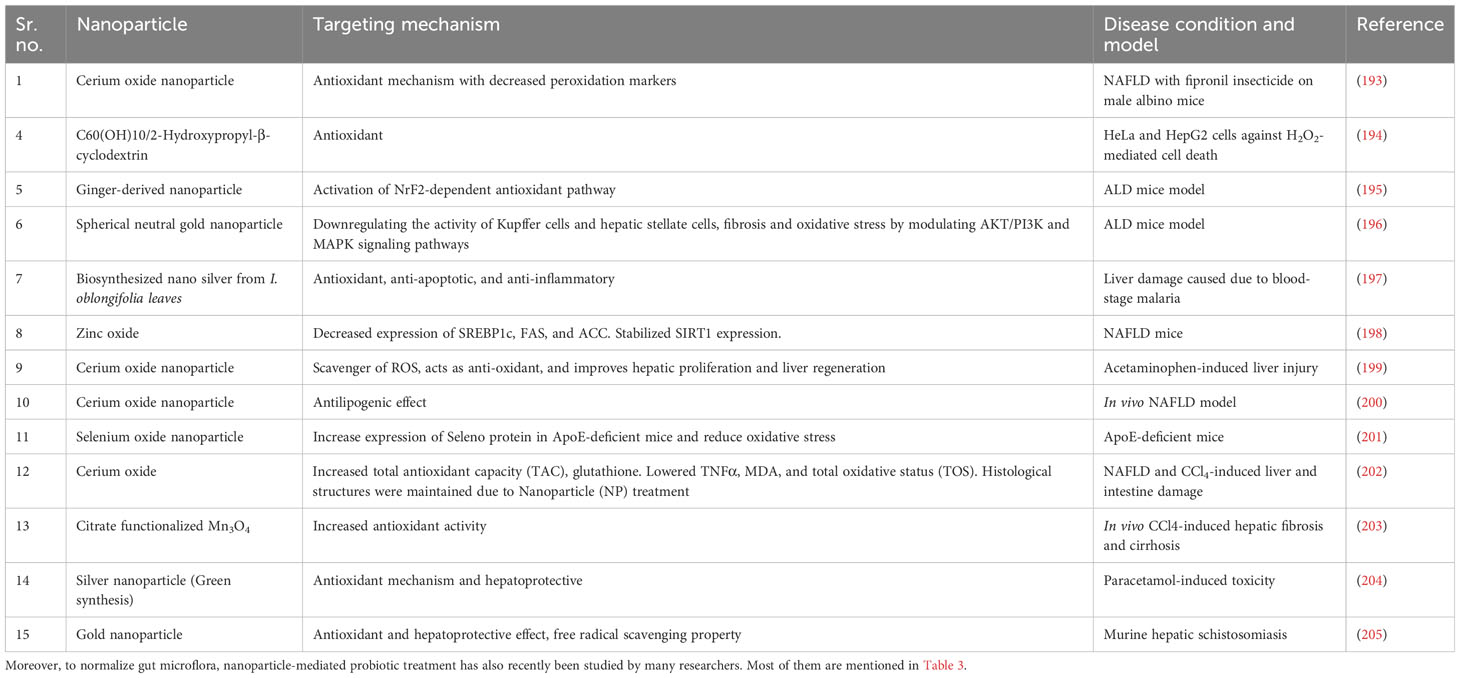
Table 2 Ameliorating effect of nanoparticles on liver disease.
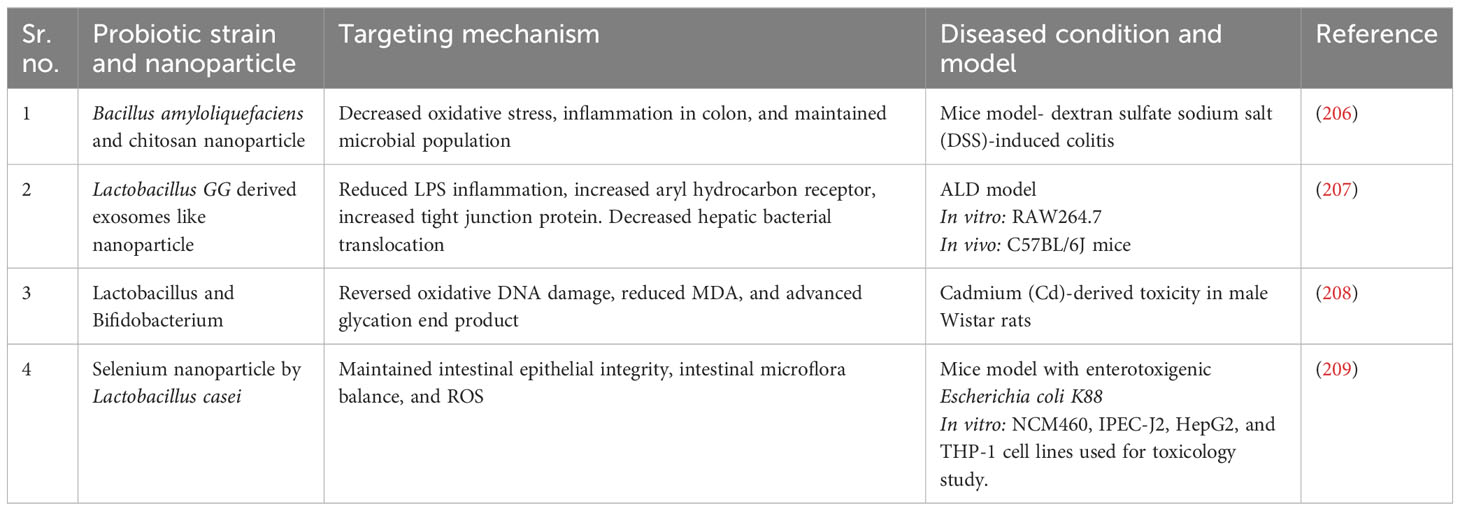
Table 3 Probiotic nanoparticles for intestinal gut dysbiosis.
Excessive alcohol consumption is a lifestyle-related issue that poses significant harm to various organs, with the liver being one of the most affected. It disrupts the balance of reactive oxygen species (ROS) and antioxidant mechanisms, leading to an imbalance in liver homeostasis. Alcohol also contributes to gut dysbiosis, further exacerbating the progression of the disease towards irreversible stages. While several drugs have been developed for the treatment of alcoholic liver disease (ALD), none have received FDA approval as targeted therapies. There are numerous underlying mechanisms involved in ALD, many of which remain unexplored. Recently, researchers have shown interest in nanomedicine as a potential targeted treatment approach. Nanoparticles are being investigated for various chronic liver conditions due to their ability to minimize side effects, deliver drugs specifically to the intended site, and enhance the bioavailability of natural compounds. Some nanoparticles have shown promising results by targeting the same mechanisms involved in ALD. Although finding a targeted drug for ALD is crucial, it is equally important to uncover other mechanisms implicated in the progression of the disease.
HA and DZ: conceptualization. PC and DZ: formal analysis and investigation. HA: writing—original draft preparation. PC and DZ: writing—review and editing.PM: supervision. All authors contributed to the article and approved the submitted version.
The author would like to acknowledge Govt. of Gujarat, India, for providing fellowship (SHODH) and Charotar University of Science and Technology for academic support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13:267–76. doi: 10.1038/cmi.2016.3
2. Qin L, Crawford JM. Anatomy and cellular functions of the liver. In: Zakim and boyer’s hepatology Elsevier, Philadelphia (2018).
3. Roth GS, Decaens T. Liver immunotolerance and hepatocellular carcinoma: Patho-physiological mechanisms and therapeutic perspectives. Eur J Cancer (2017) 87:101–12. doi: 10.1016/j.ejca.2017.10.010
4. Mandal P, Nagy LE. Chapter 10 - stellate cells in alcoholic hepatitis. In: Stellate cells in health and disease. Academic Press, United States (2015).
6. Zhou Z, Xu MJ, Gao B. Hepatocytes: A key cell type for innate immunity. Cell Mol Immunol (2016) 13:301–15. doi: 10.1038/cmi.2015.97
7. Wang QX, Ma X. Liver: a unique immune organ. Zhonghua Gan Zang Bing Za Zhi (2021) 29(6):497–9. doi: 10.3760/cma.j.cn501113-20210408-00174
8. Niu X, Zhu L, Xu Y, Zhang M, Hao Y, Ma L, et al. Global prevalence, incidence, and outcomes of alcohol related liver diseases: a systematic review and meta-analysis. BMC Public Health (2023) 23(1):1–21. doi: 10.1186/s12889-023-15749-x
9. Toshikuni N, Tsutsumi M, Arisawa T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J Gastroenterol (2014) 20:8393. doi: 10.3748/wjg.v20.i26.8393
10. Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. In: Nature reviews disease primers, vol. 4. Nature Publishing Group, UK (2018).
11. Gao B, Zakhari S. Epidemiology and pathogenesis of alcoholic liver disease. In: Zakim and boyer’s hepatology. Elsevier, Philadelphia (2018).
12. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: A translational success story. Gut (2015) 64(5):830–41. doi: 10.1136/gutjnl-2014-306842
13. Seitz HK, Neuman MG. The history of alcoholic liver disease: From an unrecognized disease to one of the most frequent diseases in hepatology. J Clin Med (2021) 10(4):858. doi: 10.3390/jcm10040858
14. Zakhari S. Overview: How is alcohol metabolized by the body? Alcohol Res Health (2006) 29:245. doi: 10.1159/000095013
15. Kong LZ, Chandimali N, Han YH, Lee DH, Kim JS, Kim SU, et al. Pathogenesis, early diagnosis, and therapeutic management of alcoholic liver disease. Int J Mol Sci (2019) 20:2712. doi: 10.3390/ijms20112712
16. Setshedi M, Wands JR, de la Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longevity (2010) 3:178–85. doi: 10.4161/oxim.3.3.12288
17. Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer (2007) 7:599–612. doi: 10.1038/nrc2191
18. Galicia-Moreno M, Gutiérrez-Reyes G. The role of oxidative stress in the development of alcoholic liver disease. Rev Gastroenterologia Mexico (2014) 79:135–44. doi: 10.1016/j.rgmxen.2014.06.007
19. Tan HK, Yates E, Lilly K, Dhanda AD. Oxidative stress in alcohol-related liver disease. World J Hepatol (2020) 12:332. doi: 10.4254/wjh.v12.i7.332
20. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J (2012) 5. doi: 10.1097/WOX.0b013e3182439613
21. Yang YM, Cho YE, Hwang S. Crosstalk between oxidative stress and inflammatory liver injury in the pathogenesis of alcoholic liver disease. Int J Mol Sci (2022) 23:774. doi: 10.3390/ijms23020774
22. Shields HJ, Traa A, van Raamsdonk JM. Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.628157
23. Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol (2009) 83:519–48. doi: 10.1007/s00204-009-0432-0
24. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E, et al. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol (2020) 11. doi: 10.3389/fphys.2020.00694
25. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
26. Moussa Z MA, Judeh Z, A. Ahmed S. Nonenzymatic exogenous and endogenous antioxidants. In: Free radical medicine and biology. IntechOpen (crosscheck) (2020).
27. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longevity (2016) 2016:4350965. doi: 10.1155/2016/4350965
28. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol (2014) 20:8082. doi: 10.3748/wjg.v20.i25.8082
29. Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J Gastroenterol (2014) 20(47):17756. doi: 10.3748/wjg.v20.i47.17756
30. di Ciaula A, Bonfrate L, Krawczyk M, Frühbeck G, Portincasa P. Synergistic and detrimental effects of alcohol intake on progression of liver steatosis. Int J Mol Sci (2022) 23:2636. doi: 10.3390/ijms23052636
31. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol (2019) 25:101084. doi: 10.1016/j.redox.2018.101084
32. Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis (2009) 29:141–54. doi: 10.1055/s-0029-1214370
33. Osna NA, Donohue TM, Kharbanda KK. Alcoholic liver disease: pathogenesis and current management. Alcohol research : Curr Rev (2017) 38:147.
34. Amjad S, Nisar S, Bhat AA, Shah AR, Frenneaux MP, Fakhro K, et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol Metab (2021) 49:101195. doi: 10.1016/j.molmet.2021.101195
35. Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology (2017) 18:447–76. doi: 10.1007/s10522-017-9685-9
36. Satoh A, Stein L, Imai S. The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb Exp Pharmacol (2011) 206:125–62. doi: 10.1007/978-3-642-21631-2_7
37. Xu J, Jackson CW, Khoury N, Escobar I, Perez-Pinzon MA. Brain SIRT1 mediates metabolic homeostasis and neuroprotection. Front Endocrinol (2018) 9. doi: 10.3389/fendo.2018.00702
38. D’Addario C, Maccarrone M. Alcohol and epigenetic modulations. In: Molecular aspects of alcohol and nutrition: A volume in the molecular nutrition series. Academic Press, United States (2016).
39. Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduction Targeted Ther (2020) 5:227. doi: 10.1038/s41392-020-00311-7
40. Thomes PG, Osna NA, Davis JS, Donohue TM. Cellular steatosis in ethanol oxidizing-HepG2 cells is partially controlled by the transcription factor, early growth response-1. Int J Biochem Cell Biol (2013) 45(2):454–63. doi: 10.1016/j.biocel.2012.10.002
41. Zhang Y, Yan T, Wang T, Liu X, Hamada K, Sun D, et al. Crosstalk between CYP2E1 and PPARα substrates and agonists modulate adipose browning and obesity. Acta Pharm Sin B (2022) 12:2224–38. doi: 10.1016/j.apsb.2022.02.004
42. Zeng T, Zhang CL, Zhao N, Guan MJ, Xiao M, Yang R, et al. Impairment of Akt activity by CYP2E1 mediated oxidative stress is involved in chronic ethanol-induced fatty liver. Redox Biol (2018) 14:295–304. doi: 10.1016/j.redox.2017.09.018
43. Hastings RC. Goodman and gilman’s the pharmacological basis of therapeutics. JAMA: J Am Med Assoc (1996) 276(12):1361–73. doi: 10.1001/jama.1996.03540120077042
44. Garciá-Suástegui WA, Ramos-Chávez LA, Rubio-Osornio M, Calvillo-Velasco M, Atzin-Méndez JA, Guevara J, et al. The role of CYP2E1 in the drug metabolism or bioactivation in the brain. Oxid Med Cell Longevity (2017) 2017. doi: 10.1155/2017/4680732
45. Massart J, Begriche K, Fromenty B. Cytochrome P450 2E1 should not be neglected for acetaminophen-induced liver injury in metabolic diseases with altered insulin levels or glucose homeostasis. Clinics Res Hepatol Gastroenterol (2021) 45:101470. doi: 10.1016/j.clinre.2020.05.018
46. Harjumäki R, Pridgeon CS, Ingelman-Sundberg M. Cyp2e1 in alcoholic and non-alcoholic liver injury. Roles of ros, reactive intermediates and lipid overload. Int J Mol Sci (2021) 22:8221. doi: 10.3390/ijms22158221
47. Stadtman ER. Oxidation of proteins by mixed-function oxidation systems: implication in protein turnover, ageing and neutrophil function. Trends Biochem Sci (1986) 11(1):11–2. doi: 10.1016/0968-0004(86)90221-5
48. di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longevity (2016) 2016. doi: 10.1155/2016/1245049
49. Vasiliou V, Ziegler TL, Bludeau P, Petersen DR, Gonzalez FJ, Deitrich RA. CYP2E1 and catalase influence ethanol sensitivity in the central nervous system. Pharmacogenet Genomics (2006) 16(1):51–8. doi: 10.7150/ijbs.42300
50. Wang W, Wang C, Xu H, Gao Y. Aldehyde dehydrogenase, liver disease and cancer. Int J Biol Sci (2020) 16:921. doi: 10.14218/JCTH.2020.00104
51. Wang Q, Chang B, Li X, Zou Z. Role of aldh2 in hepatic disorders: Gene polymorphism and disease pathogenesis. J Clin Trans Hepatol (2021) 9:90. doi: 10.1021/tx5001046
52. Lopachin RM, Gavin T. Molecular mechanisms of aldehyde toxicity: A chemical perspective. Chem Res Toxicol (2014) 27:1081–91. doi: 10.1016/j.jhep.2018.10.008
53. Ganne-Carrié N, Nahon P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J Hepatol (2019) 70:284–93. doi: 10.1016/j.livres.2018.11.002
54. Ohashi K, Pimienta M, Seki E. Alcoholic liver disease: A current molecular and clinical perspective. Liver Res (2018) 2:161–72. doi: 10.1038/nature25154
55. Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature (2018) 553(7687):171–7. doi: 10.3390/ijms22115717
56. Hyun J, Han J, Lee C, Yoon M, Jung Y. Pathophysiological aspects of alcohol metabolism in the liver. Int J Mol Sci (2021) 22:5717. doi: 10.1152/ajpgi.00270.2015
57. Sun Q, Zhong W, Zhang W, Zhou Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am J Physiol Gastrointest Liver Physiol (2016) 310(3):G205–14. doi: 10.1053/gast.2002.33613
58. Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology (2002) 122(7):2049–1063. doi: 10.1016/j.mam.2004.02.005
59. Cadenas E. Mitochondrial free radical production and cell signaling. Mol Aspects Med (2004) 25:17–26. doi: 10.3748/wjg.v20.i9.2136
60. Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol (2014) 20(9):2136–42. doi: 10.1038/nri2215
61. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol (2008) 8:279–89. doi: 10.1038/nature14191
62. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517:311–20. doi: 10.7150/ijbs.67533
63. Chen H, McKeen T, Chao X, Chen A, Deng F, Jaeschke H, et al. The role of MLKL in hepatic ischemia-reperfusion injury of alcoholic steatotic livers. Int J Biol Sci (2022) 18(3):1096. doi: 10.1016/j.tox.2021.152923
64. Shao Y, Wang X, Zhou Y, Jiang Y, Wu R, Lu C. Pterostilbene attenuates RIPK3-dependent hepatocyte necroptosis in alcoholic liver disease via SIRT2-mediated NFATc4 deacetylation. Toxicology (2021) 461:152923. doi: 10.1111/cpr.13193
65. Zhou Y, Wu R, Wang X, Bao X, Lu C. Roles of necroptosis in alcoholic liver disease and hepatic pathogenesis. Cell Proliferation (2022) 55:e13193. doi: 10.1155/2020/6095673
66. You S, Zhang Y, Xu J, Qian H, Wu S, Wu B, et al. The role of BRG1 in antioxidant and redox signaling. Oxid Med Cell Longevity (2020) 2020. doi: 10.1016/j.bbadis.2018.05.022
67. Li N, Li M, Hong W, Shao J, Xu H, Shimano H, et al. Brg1 regulates pro-lipogenic transcription by modulating SREBP activity in hepatocytes. Biochim Biophys Acta Mol Basis Dis (2018) 1864(9):2881–9. doi: 10.3389/fcell.2021.622866
68. Kong M, Zhu Y, Shao J, Fan Z, Xu Y. The chromatin remodeling protein BRG1 regulates SREBP maturation by activating SCAP transcription in hepatocytes. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2020.00259
69. Fan Z, Kong M, Li M, Hong W, Fan X, Xu Y. Brahma related gene 1 (Brg1) regulates cellular cholesterol synthesis by acting as a co-factor for SREBP2. Front Cell Dev Biol (2020) 8. doi: 10.3389/fcell.2019.00245
70. Li Z, Chen B, Dong W, Kong M, Shao Y, Fan Z, et al. The chromatin remodeler brg1 integrates ROS production and endothelial-mesenchymal transition to promote liver fibrosis in mice. Front Cell Dev Biol (2019) 7. doi: 10.1002/jcp.25785
71. Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol (2017) 232:2977–84. doi: 10.1258/ebm.2011.010360
72. Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (2011) 236:546–56. doi: 10.3350/cmh.2020.0173
73. Kim YS, Kim SG. Endoplasmic reticulum stress and autophagy dysregulation in alcoholic and non-alcoholic liver diseases. Clin Mol Hepatol (2020) 26:715. doi: 10.1016/j.livres.2019.01.002
74. Liu X, Green RM. Endoplasmic reticulum stress and liver diseases. Liver Res (2019) 3(1):55–64. doi: 10.1002/hep.24279
75. Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology (2011) 53(5):1752–63. doi: 10.1038/s41598-018-26882-8
76. Komatsu M, Kanda T, Urai H, Kurokochi A, Kitahama R, Shigaki S, et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD + metabolism. Sci Rep (2018) 8(1):8637. doi: 10.1159/000475432
77. Li Q, He M, Mao L, Wang X, Jiang YL, Li M, et al. Nicotinamide n-methyltransferase suppression participates in nickel-induced histone H3 Lysine9 dimethylation in BEAS-2B cells. Cell Physiol Biochem (2017) 41(5):2016–26. doi: 10.1111/j.1530-0277.2011.01547.x
78. Medici V, Virata MC, Peerson JM, Stabler SP, French SW, Gregory JF, et al. S-adenosyl-L-methionine treatment for alcoholic liver disease: A double-blinded, randomized, placebo-controlled trial. Alcohol Clin Exp Res (2011) 35(11):1960–5. doi: 10.1093/ajcn/86.1.14
79. Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N, et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am J Clin Nutr (2007) 86:14–24. doi: 10.1016/j.jhep.2020.04.038
80. Song Q, Chen Y, Wang J, Hao L, Huang C, Griffiths A, et al. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J Hepatol (2020) 73(4):783–93. doi: 10.1016/j.drudis.2021.05.011
81. Gao Y, Martin NI, van Haren MJ. Nicotinamide N-methyl transferase (NNMT): An emerging therapeutic target. Drug Discovery Today (2021) 26:2699–706. doi: 10.3389/fgene.2014.00242
82. Guo B, Li Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front Genet (2014) 5. doi: 10.1098/rsif.2019.0288
83. Stroberg W, Eilertsen J, Schnell S. Information processing by endoplasmic reticulum stress sensors. J R Soc Interface (2019) 16(158):20190288. doi: 10.1016/j.jhep.2010.11.005
84. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol (2011) 54:795–809. doi: 10.1053/j.gastro.2016.02.035
85. Nagy LE, Ding WX, Cresci G, Saikia P, Shah VH. Linking pathogenic mechanisms of alcoholic liver disease with clinical phenotypes. Gastroenterology (2016) 150(8):1756–68. doi: 10.1038/s12276-021-00561-7
86. Smathers RL, Galligan JJ, Stewart BJ, Petersen DR. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. In: Chemico-biological interactions. Elsevier, Philadelphia (2011).
87. Chen WY, Zhang J, Ghare S, Barve S, McClain C, Joshi-Barve S. Acrolein is a pathogenic mediator of alcoholic liver disease and the scavenger hydralazine is protective in mice. CMGH (2016) 2(5):685–700. doi: 10.1194/jlr.R067595
88. Rungratanawanich W, Qu Y, Wang X, Essa MM, Song BJ. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp Mol Med (2021) 53:168–88. doi: 10.3390/biom5021099
89. Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology (2003) 124(3):708–24. doi: 10.3389/fgene.2014.00112
90. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res (2016) 57:1329–38. doi: 10.1007/s11010-013-1694-7
91. Ji C. Advances and new concepts in alcohol-induced organelle stress, unfolded protein responses and organ damage. Biomolecules (2015) 5:1099–121. doi: 10.1155/2014/513787
92. Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet (2014) 5. doi: 10.1242/dmm.012195
93. Fang DL, Wan Y, Shen W, Cao J, Sun ZX, Yu HH, et al. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol Cell Biochem (2013) 381(1–2):127–37. doi: 10.1042/BCJ20160510
94. Ji C. New insights into the pathogenesis of alcohol-induced ER stress and liver diseases. Int J Hepatol (2014) 2014. doi: 10.1155/2014/513787
95. Tsedensodnom O, Vacaru AM, Howarth DL, Yin C, Sadler KC. Ethanol metabolism and oxidative stress are required for unfolded protein response activation and steatosis in zebrafish with alcoholic liver disease. DMM Dis Models Mech (2013) 6(5):1213–26. doi: 10.3390/microorganisms9061302
96. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J (2017) 474:1823–36. doi: 10.3389/fphar.2018.00081
97. Cresci GAM, Izzo K. Gut microbiome. In: Adult short bowel syndrome: nutritional, medical, and surgical management. Functional Food Science Journal, UK (2018).
98. Cani PD, de Hase EM, van Hul M. Gut microbiota and host metabolism: From proof of concept to therapeutic intervention. Microorganisms (2021) 9:1302. doi: 10.3390/microorganisms9061302
99. Ballerini P, Diwan V, Rossi B, Tarzami ST, Obad A, Peeran A, et al. Alcohol-mediated organ damages: Heart and brain. Front Pharmacol (2018) 9:81. doi: 10.20944/preprints201802.0120.v1
100. Seitz HK, Scherübl H. Alcohol use and gastrointestinal diseases. Visceral Med (2020) 36:157–9. doi: 10.1159/000507643
101. Fan ZA, Yun J, Yu SD, Yang Q, Song L. Alcohol consumption can be a “Double-edged sword” for chronic kidney disease patients. Medical science monitor : international medical journal of experimental and clinical research (2019) 25:7059–72. doi: 10.3389/fcimb.2022.840164
102. Dguzeh U, Haddad NC, Smith KTS, Johnson JO, Doye AA, Gwathmey JK, et al. Alcoholism: A multi-systemic cellular insult to organs. Int J Environ Res Public Health (2018) 15:1083. doi: 10.1038/s41398-022-01920-2
103. O’Keefe JH, Bhatti SK, Bajwa A, DiNicolantonio JJ, Lavie CJ. Alcohol and cardiovascular health: The dose makes the poison.or the remedy. Mayo Clinic Proc (2014) 89:382–93. doi: 10.1016/j.mayocp.2013.11.005
104. Day AW, Kumamoto CA. Gut microbiome dysbiosis in alcoholism: consequences for health and recovery. Front Cell Infection Microbiol (2022) 12. doi: 10.3389/fcimb.2022.840164
105. Segovia-Rodríguez L, Echeverry-Alzate V, Rincón-Pérez I, Calleja-Conde J, Bühler KM, Giné E, et al. Gut microbiota and voluntary alcohol consumption. Transl Psychiatry (2022) 12(1):146. doi: 10.1038/s41398-022-01920-2
106. Kim JW, Lee SE, Kim NY, Jung JH, Jang JH, Park JW, et al. Role of microbiota-derived metabolites in alcoholic and non-alcoholic fatty liver diseases. International Journal of molecular sciences (2021) 23(1):426. doi: 10.3390/ijms23010426
107. Mohajeri MH, Fata G, Steinert RE, Weber P. Relationship between the gut microbiome and brain function. Nutrition reviews (2018) 76(7):481–96. doi: 10.1093/nutrit/nuy009
108. Liu J, Yang D, Wang X, Tetteh Asare P, Zhang Q, Na L, et al. Gut microbiota targeted approach in the management of chronic liver diseases. Frontiers in cellular and infection microbiology (2022) 12:774335. doi: 10.1016/j.bbr.2019.112196
109. Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology (Baltimore, Md.) (2010) 53(1):96–105. doi: 10.3390/microorganisms3040759
110. Bibbò S, Ianiro G, Giorgio V, Scaldaferri F, Masucci L, Gasbarrini A, et al. The role of diet on gut microbiota composition. Eur Rev Med Pharmacol Sci (2016) 20(22). doi: 10.3350/cmh.2017.0067
111. Qamar N, Castano D, Patt C, Chu T, Cottrell J, Chang SL. Meta-analysis of alcohol induced gut dysbiosis and the resulting behavioral impact. Behav Brain Res (2019) 376:112196. doi: 10.1080/19490976.2021.1984122
112. Fukui H. Gut microbiota and host reaction in liver diseases. Microorganisms (2015) 3(4):759–91. doi: 10.3390/microorganisms3040759
113. Cassard AM, Ciocan D. Microbiota, a key player in alcoholic liver disease. Clin Mol Hepatol (2018) 24:100–7. doi: 10.1002/hep.30832
114. Gao W, Zhu Y, Ye J, Chu H. Gut non-bacterial microbiota contributing to alcohol-associated liver disease. Gut Microbes (2021) 13:1984122. doi: 10.1016/j.livres.2019.09.001
115. Thomas H. Gut microbiota: Intestinal fungi fuel the inflammatory fire in alcoholic liver disease. Nat Rev Gastroenterol Hepatol (2017) 14:385–385. doi: 10.3389/fphys.2021.699253
116. Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M, et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology (2020) 71(2):522–38. doi: 10.1016/j.jhep.2021.08.003
117. Li F, McClain CJ, Feng W. Microbiome dysbiosis and alcoholic liver disease. Liver Res (2019) 3:218–26. doi: 10.1002/hep4.1947
118. Hartmann P, Lang S, Zeng S, Duan Y, Zhang X, Wang Y, et al. Dynamic changes of the fungal microbiome in alcohol use disorder. Front Physiol (2021) 12. doi: 10.3390/microorganisms11051181
119. Hsu CL, Duan Y, Fouts DE, Schnabl B. Intestinal virome and therapeutic potential of bacteriophages in liver disease. J Hepatol (2021) 75:1465–75. doi: 10.1016/j.cofs.2020.10.001
120. Hsu CL, Zhang X, Jiang L, Lang S, Hartmann P, Pride D, et al. Intestinal virome in patients with alcohol use disorder and after abstinence. Hepatol Commun (2022) 6(8):2058–69. doi: 10.3390/ijms20184568
121. Chen L, Hou X, Chu H. The novel role of phage particles in chronic liver diseases. Microorganisms (2023) 11:1181. doi: 10.1080/19490976.2021.1946367
122. Lee E, Lee JE. Impact of drinking alcohol on gut microbiota: recent perspectives on ethanol and alcoholic beverage. Curr Opin Food Sci (2021) 37:91–7. doi: 10.1016/j.cofs.2020.10.001
123. Meroni M, Longo M, Dongiovanni P. Alcohol or gut microbiota: who is the guilty? Int J Mol Sci (2019) 20:4568. doi: 10.1016/j.alcohol.2016.10.021
124. Singhal R, Donde H, Ghare S, Stocke K, Zhang J, Vadhanam M, et al. Decrease in acetyl-CoA pathway utilizing butyrate-producing bacteria is a key pathogenic feature of alcohol-induced functional gut microbial dysbiosis and development of liver disease in mice. Gut Microbes (2021) 13(1):1946367. doi: 10.1155/2018/9671919
125. Elamin EE, Masclee AA, Dekker J, Pieters HJ, Jonkers DM. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J Nutr (2013) 143(12):1872–81. doi: 10.1007/s00394-017-1445-8
126. Cresci GA, Glueck B, Nagy LE. Butyrate protects intestinal immune response during chronic-binge ethanol exposure. Alcohol (2017) 59:72. doi: 10.1016/j.alcohol.2016.10.021
127. Glueck B, Han Y, Cresci GAM. Tributyrin supplementation protects immune responses and vasculature and reduces oxidative stress in the proximal colon of mice exposed to chronic-binge ethanol feeding. Journal of immunology research (2018) 2018:9671919. doi: 10.1155/2018/9671919
128. Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, et al. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr (2018) 57:1–24. doi: 10.1002/hep.26321
129. Lavelle EC, Raghavan S, Patrick McEntee C, Hermoso MA, Parada Venegas D, de la Fuente MK, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol (2019) 1:277. doi: 10.3389/fimmu.2019.00277
130. Donde H, Ghare S, Joshi-Barve S, Zhang JW, Vadhanam MV, Gobejishvili L, et al. Tributyrin inhibits ethanol-induced epigenetic repression of CPT-1A and attenuates hepatic steatosis and injury. Cell Mol Gastroenterol Hepatol (2020) 9(4):569–85. doi: 10.1073/pnas.1006451107
131. Hartmann P, Chen P, Wang HJ, Wang L, Mccole DF, Brandl K, et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology (Baltimore, Md.) (2013) 58(1):108–19. doi: 10.1152/ajpgi.00245.2020
132. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science (2005) 307:1915–20. doi: 10.1016/j.livres.2017.12.004
133. Johansson MEV, Holmén Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U.S.A. (2011) 108(SUPPL. 1):4659–65. doi: 10.3748/wjg.v20.i44.16639
134. Khoshbin K, Camilleri M. Effects of dietary components on intestinal permeability in health and disease. Am J Physiol - Gastrointestinal Liver Physiol (2020) 319:G589–608. doi: 10.1002/hep.23009
135. Zhou Z, Zhong W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res (2017) 1(4):197–207. doi: 10.1111/j.1749-6632.2009.04054.x
136. Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M. Gut microbiota in alcoholic liver disease: Pathogenetic role and therapeutic perspectives. World J Gastroenterol (2014) 20(44):16639–48. doi: 10.3748/wjg.v20.i44.16639
137. Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology (Baltimore, Md.) (2009) 50(2):638–44. doi: 10.1155/2018/2645465
138. Rao R, Rao RK. Occludin phosphorylation in regulation of epithelial tight junctions hhs public access. Ann N Y Acad Sci (2009) 1165:62–8. doi: 10.1093/jn/138.9.1664
139. Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol Gastrointest Liver Physiol (2004) 286(3 49-3):G367–376. doi: 10.1152/ajpgi.00350.2009
140. Lee B, Moon KM, Kim CY. Tight junction in the intestinal epithelium: Its association with diseases and regulation by phytochemicals. J Immunol Res (2018) 2018. doi: 10.1016/j.mam.2005.07.002
141. Finamore A, Massimi M, Devirgiliis LC, Mengheri E. Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J Nutr (2008) 138(9):1664–70. doi: 10.1007/s10620-006-9462-0
142. Zhong W, McClain CJ, Cave M, Kang YJ, Zhou Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am J Physiol Gastrointest Liver Physiol (2010) 298(5):G625–33. doi: 10.1007/s11894-999-0021-7
143. Kang YJ, Zhou Z. Zinc prevention and treatment of alcoholic liver disease. Mol Aspects Med (2005) 26:391–404. doi: 10.1007/s00204-005-0009-5
144. Stamoulis I, Kouraklis G, Theocharis S. Zinc and the liver: An active interaction. Digestive Dis Sci (2007) 52:1595–612. doi: 10.1016/j.jhep.2018.05.021
145. Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep (1999) 1:398–403. doi: 10.1016/B978-0-12-814468-8.00008-9
146. Stefanidou M, Maravelias C, Dona A, Spiliopoulou C. Zinc: A multipurpose trace element. Arch Toxicol (2006) 80:1–9. doi: 10.1371/journal.pone.0076522
147. Shao T, Zhao C, Li F, Gu Z, Liu L, Zhang L, et al. Intestinal HIF-1α deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J Hepatol (2018) 69(4):886–95. doi: 10.4172/2161-069X.1000548
148. Zhong W, Zhou Z. Sealing the leaky gut represents a beneficial mechanism of zinc intervention for alcoholic liver disease. Dietary Interventions Gastrointestinal Diseases: Foods Nutrients Dietary Suppl (2019),91–106. doi: 10.1007/s10620-012-2328-8
149. Zhong W, Zhao Y, Sun X, Song Z, McClain CJ, Zhou Z. Dietary zinc deficiency exaggerates ethanol-induced liver injury in mice: involvement of intrahepatic and extrahepatic factors. PloS One (2013) 8(10):e76522. doi: 10.1136/gut.2008.168443
150. Usama U, Jaffar Khan M, Fatima S, Fatima S. Role of zinc in shaping the gut microbiome; proposed mechanisms and evidence from the literature. J Gastrointest Dig Syst (2018) 08(01). doi: 10.1128/IAI.06165-11
151. Wang X, Valenzano MC, Mercado JM, Zurbach EP, Mullin JM. Zinc supplementation modifies tight junctions and alters barrier function of CACO-2 human intestinal epithelial layers. Dig Dis Sci (2013) 58(1):77–87. doi: 10.1007/s10620-012-2328-8
152. Dessein R, Gironella M, Vignal C, Peyrin-Biroulet L, Sokol H, Secher T, et al. Toll-like receptor 2 is critical for induction of Reg3b expression and intestinal clearance of Yersinia pseudotuberculosis. Gut (2009) 58(6):771–6. doi: 10.1016/j.immuni.2014.12.028
153. van Ampting MTJ, Loonen LMP, Schonewille AJ, Konings I, Vink C, Iovanna J, et al. Intestinally secreted c-type lectin Reg3b attenuates salmonellosis but not listeriosis in mice. Infect Immun (2012) 80(3):1115–1120. doi: 10.1136/gutjnl-2018-317232
154. Vassallo G, Mirijello A, Ferrulli A, Antonelli M, Landolfi R, Gasbarrini A, et al. alcohol and gut microbiota-the possible role of gut microbiota modulation in the treatment of alcoholic liver disease. International journal of molecular sciences (2019) 20(18):4568. doi: 10.1016/j.chom.2016.01.003
155. Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity (2015) 42:28–39. doi: 10.1097/SHK.0b013e3182749f96
156. Hendrikx T, Duan Y, Wang Y, Oh JH, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut (2019) 68(8):1504–15. doi: 10.1084/jem.20070563
157. Wang L, Fouts DE, Stärkel P, Hartmann P, Chen P, Llorente C, et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe (2016) 19(2):227–39. doi: 10.1371/journal.pone.0261675
158. Rendon JL, Li X, Akhtar S, Choudhry MA. IL-22 modulates gut epithelial and immune barrier functions following acute alcohol exposure and burn injury $watermark-text $watermark-text $watermark-text. Nutrients (2012) 60153(1):11–8. doi: 10.3390/nu4050356
159. Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIIIγ and protect mice against intestinal Listeria monocytogenes infection. J Exp Med (2007) 204(8):1891–900. doi: 10.1111/jgh.12019
160. Ferdouse A, Agrawal RR, Gao MA, Jiang H, Blaner WS, Clugston RD. Alcohol induced hepatic retinoid depletion is associated with the induction of multiple retinoid catabolizing cytochrome P450 enzymes. PloS One (2022) 17(1 January):e0261675. doi: 10.1038/s41419-021-04377-1
161. Clugston RD, Blaner WS. The adverse effects of alcohol on vitamin A metabolism. Nutrients (2012) 4:356–71. doi: 10.3748/wjg.v22.i4.1348
162. Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol (Australia) (2013) 28:38–42. doi: 10.1111/jgh.12019
163. De Smet V, Eysackers N, Merens V, Kazemzadeh Dastjerd M, Halder G, Verhulst S, et al. Initiation of hepatic stellate cell activation extends into chronic liver disease. Cell Death Dis (2021) 12(12):1110. doi: 10.1053/j.gastro.2007.09.034
164. Seo W, Jeong W. Hepatic non-parenchymal cells: Master regulators of alcoholic liver disease? World J Gastroenterol (2016) 22:1348. doi: 10.1016/j.jhep.2013.12.025
165. Hegedús G. Pathology of alcoholic liver disease. Orvosi hetilap (2000) 141:331–6. doi: 10.3389/fimmu.2021.803037
166. Jeong W, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-γ Contributes to alcohol acceleration of liver fibrosis. Gastroenterology (2008) 134(1):248–58. doi: 10.1007/s00018-020-03656-y
167. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
168. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol (2021) 12. doi: 10.1111/acer.12704
169. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci (2021) 78:538. doi: 10.3389/fimmu.2016.00538
170. Slevin E, Baiocchi L, Wu N, Ekser B, Sato K, Lin E, et al. Kupffer cells: inflammation pathways and cell-cell interactions in alcohol-associated liver disease. Am J Pathol (2020) 190. doi: 10.20517/2394-5079.2019.29
171. Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcoholism: Clin Exp Res (2015) 39:913842. doi: 10.3389/fmed.2022.913842
172. Zeng T, Zhang CL, Xiao M, Yang R, Xie KQ. Critical roles of kupffer cells in the pathogenesis of alcoholic liver disease: From basic science to clinical trials. Front Immunol (2016) 7. doi: 10.21037/tgh.2020.01.04
173. Méndez-Sánchez N, Valencia-Rodriguez A, Vera-Barajas A, Abenavoli L, Scarpellini E, Ponciano-Rodriguez G. The mechanism of dysbiosis in alcoholic liver disease leading to liver cancer. Hepatoma Res (2020) 2020:305–13. doi: 10.1016/j.jhep.2018.10.026
174. Jung JH, Kim SE, Suk KT, Kim DJ. Gut microbiota-modulating agents in alcoholic liver disease: Links between host metabolism and gut microbiota. Front Med Front Media S.A (2022) 9. doi: 10.1155/2021/6636152
175. Asgharpour A, Dinani A, Friedman SL. Basic science to clinical trials in non-alcoholic fatty liver disease and alcohol-related liver disease: Collaboration with industry. Trans Gastroenterol Hepatol (2021) 6:2866–75. doi: 10.1016/j.ajpath.2011.08.039
176. Singal AK, Shah VH. Current trials and novel therapeutic targets for alcoholic hepatitis. J Hepatol (2019) 70:6224. doi: 10.3748/wjg.v26.i40.6224
177. Patel F, Parwani K, Patel D, Mandal P. Metformin and probiotics interplay in amelioration of ethanol-induced oxidative stress and inflammatory response in an in vitro and in vivo model of hepatic injury. Mediators Inflamm (2021) 2021:3285. doi: 10.3390/biomedicines10123285
178. Wang Y, Kirpich I, Liu Y, Ma Z, Barve S, McClain CJ, et al. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity and ameliorates alcohol-induced liver injury. Am J Pathol (2011) 179(6):407. doi: 10.3389/fchem.2020.00407
179. Jiang XW, Li YT, Ye JZ, Lv LX, Yang LY, Bian XY, et al. New strain of Pediococcus pentosaceus alleviates ethanol-induced liver injury by modulating the gut microbiota and short-chain fatty acid metabolism. World J Gastroenterol (2020) 26(40):181457. doi: 10.1098/rsos.181457
180. Patel D, Desai C, Singh D, Soppina V, Parwani K, Patel F, et al. Synbiotic intervention ameliorates oxidative stress and gut permeability in an in vitro and in vivo model of ethanol-induced intestinal dysbiosis. Biomedicines (2022) 10(12):3285. doi: 10.3390/biomedicines10123285
181. Zhao R, Zhu M, Zhou S, Feng W, Chen H. Rapamycin-loaded mPEG-PLGA nanoparticles ameliorate hepatic steatosis and liver injury in non-alcoholic fatty liver disease. Front Chem (2020) 8. doi: 10.3390/biomedicines9121767
182. Wan S, Zhang L, Quan Y, Wei K. Resveratrol-loaded PLGA nanoparticles: Enhanced stability, solubility and bioactivity of resveratrol for non-alcoholic fatty liver disease therapy. R Soc Open Sci (2018) 5(11):1–12. doi: 10.1186/s12951-018-0391-9
183. Ali Jazayeri-Tehrani S, Mahdi Rezayat S, Mansouri S, Qorbani M, Moayed Alavian S, Daneshi-Maskooni M, et al. Nano-curcumin improves glucose indices, lipids, inflammation, and Nesfatin in overweight and obese patients with non-alcoholic fatty liver disease (NAFLD): a double-blind randomized placebo-controlled clinical trial. Nutrition & Metabolism (2019) 16:8. doi: 10.1186/s12986-019-0331-1
184. Abdullah AS, Sayed IET, El-Torgoman AMA, Alghamdi NA, Ullah S, Wageh S, et al. Preparation and characterization of silymarin-conjugated gold nanoparticles with enhanced anti-fibrotic therapeutic effects against hepatic fibrosis in rats: Role of micrornas as molecular targets. Biomedicines (2021) 9(12):1767. doi: 10.1016/j.nano.2020.102310
185. Liang J, Liu Y, Liu J, Li Z, Fan Q, Jiang Z, et al. Chitosan-functionalized lipid-polymer hybrid nanoparticles for oral delivery of silymarin and enhanced lipid-lowering effect in NAFLD. J Nanobiotechnol (2018) 16(1):1–17. doi: 10.25135/rnp.133.19.03.1220
186. Krithika R, Vhora I, Verma RJ. Preparation, toxicity analysis and in vivo protective effect of phyllanthin-loaded PLGA nanoparticles against CCl4-induced hepatic fibrosis. J Drug Delivery Sci Technol (2019) 51:364–71. doi: 10.1177/03946320221137435
187. Hu R, Liu S, Anwaier G, Wang Q, Shen W, Shen Q, et al. Formulation and intestinal absorption of naringenin loaded nanostructured lipid carrier and its inhibitory effects on nonalcoholic fatty liver disease. Nanomedicine (2021) 32:102310. doi: 10.1152/ajpgi.00217.2019
188. Hussein ME, el Senousy AS, Abd-Elsalam WH, Ahmed KA, El-Askary HI, Mouneir SM, et al. Roselle seed oil and its nano-formulation alleviated oxidative stress, activated nrf2 and downregulated m-RNA expression genes of pro-inflammatory cytokines in paracetamol-intoxicated rat model. Records Natural Products (2020) 14(1):167–77. doi: 10.1002/tox.22505
189. Ahmed ES, Mohamed HE, Farrag MA. Luteolin loaded on zinc oxide nanoparticles ameliorates non-alcoholic fatty liver disease associated with insulin resistance in diabetic rats via regulation of PI3K/AKT/FoxO1 pathway. Int J Immunopathol Pharmacol (2022) 36:3946320221137430. doi: 10.3389/fbioe.2020.00495
190. Gopal T, Kumar N, Perriotte-Olson C, Casey CA, Donohue TM, Harris EN, et al. Nanoformulated SOD1 ameliorates the combined NASH and alcoholassociated liver disease partly via regulating CYP2E1 expression in adipose tissue and liver. Am J Physiol Gastrointest Liver Physiol (2020) 318(3):G428–38. doi: 10.1038/s41598-020-79479-5
191. Eftekhari A, Ahmadian E, Azami A, Johari-Ahar M, Eghbal MA. Protective effects of coenzyme Q10 nanoparticles on dichlorvos-induced hepatotoxicity and mitochondrial/lysosomal injury. Environ Toxicol (2018) 33(2):2959–65. doi: 10.1016/j.xphs.2016.04.033
192. Durymanov M, Permyakova A, Reineke J. Pre-treatment with PLGA/silibinin nanoparticles mitigates dacarbazine-induced hepatotoxicity. Front Bioeng Biotechnol (2020) 8. doi: 10.3402/jev.v4.28713
193. Wasef L, Nassar AMK, El-Sayed YS, Samak D, Noreldin A, Elshony N, et al. The potential ameliorative impacts of cerium oxide nanoparticles against fipronil-induced hepatic steatosis. Sci Rep (2021) 11(1):1–14. doi: 10.1016/j.ijpharm.2018.06.008
194. Iohara D, Umezaki Y, Anraku M, Uekama K, Hirayama F. In vitro and in vivo evaluation of hydrophilic C60(OH)10/2-hydroxypropyl-β-cyclodextrin nanoparticles as an antioxidant. J Pharm Sci (2016) 105(9):6254–9. doi: 10.1016/j.sjbs.2021.06.089
195. Zhuang X, Deng Zb, Mu J, Zhang L, Yan J, Miller D, et al. Ginger-derived nanoparticles protect against alcohol-induced liver damage. J Extracell Vesicles (2015) 4(1):28713. doi: 10.3402/jev.v4.28713
196. de Carvalho TG, Garcia VB, de Araújo AA, da Silva Gasparotto LH, Silva H, Guerra GCB, et al. Spherical neutral gold nanoparticles improve anti-inflammatory response, oxidative stress and fibrosis in alcohol-methamphetamine-induced liver injury in rats. Int J Pharm (2018) 548(1):1–14. doi: 10.1186/s12951-019-0544-5
197. Dkhil MA, Abdel-Gaber R, Alojayri G, Thagfan FA, Al-Shaebi EM, Al-Quraishy S. Biosynthesized nanosilver as anti-oxidant, anti-apoptotic and anti-inflammatory agent against Plasmodium chabaudi infection in the mouse liver. Saudi J Biol Sci (2021) 28(11):12848. doi: 10.1038/s41598-019-49262-2
198. Dogra S, Kar AK, Girdhar K, Daniel PV, Chatterjee S, Choubey A, et al. Zinc oxide nanoparticles attenuate hepatic steatosis development in high-fat-diet fed mice through activated AMPK signaling axis. Nanomedicine (2019) 17:210–22. doi: 10.1039/c9mt00215d
199. Córdoba-Jover B, Arce-Cerezo A, Ribera J, Pauta M, Oró D, Casals G, et al. Cerium oxide nanoparticles improve liver regeneration after acetaminophen-induced liver injury and partial hepatectomy in rats. J Nanobiotechnol (2019) 17(1):1–12. doi: 10.1016/j.metop.2021.100151
200. Carvajal S, Perramón M, Oró D, Casals E, Fernández-Varo G, Casals G, et al. Cerium oxide nanoparticles display antilipogenic effect in rats with non-alcoholic fatty liver disease. Sci Rep (2019) 9(1):12848. doi: 10.4155/fsoa-2016-0029
201. Guo L, Xiao J, Liu H, Liu H. Selenium nanoparticles alleviate hyperlipidemia and vascular injury in ApoE-deficient mice by regulating cholesterol metabolism and reducing oxidative stress. Metallomics (2020) 12(2):204–17. doi: 10.1016/j.onano.2016.03.001
202. Abbasi E, Vafaei SA, Naseri N, Darini A, Azandaryani MT, Ara FK, et al. Protective effects of cerium oxide nanoparticles in non-alcoholic fatty liver disease (NAFLD) and carbon tetrachloride-induced liver damage in rats: Study on intestine and liver. Metabol Open (2021) 12:100151. doi: 10.1016/j.metop.2021.100151
203. Adhikari A, Polley N, Darbar S, Bagchi D, Pal SK. Citrate functionalized Mn3O4 in nanotherapy of hepatic fibrosis by oral administration. Future Sci OA. (2016) 2(4):FSO146. doi: 10.1038/s41598-022-08915-5
204. Vasquez RD, Apostol JG, de Leon JD, Mariano JD, Mirhan CMC, Pangan SS, et al. Polysaccharide-mediated green synthesis of silver nanoparticles from Sargassum siliquosum J.G. Agardh: Assessment of toxicity and hepatoprotective activity. OpenNano (2016) 1:16–24. doi: 10.1002/hep4.1679
205. Dkhil MA, Bauomy AA, Diab MSM, Al-Quraishy S. Antioxidant and hepatoprotective role of gold nanoparticles against murine hepatic schistosomiasis. Int J Nanomed (2015) 10:7467–75. doi: 10.1016/j.jksus.2020.08.011
206. Alkushi AG, Abdelfattah-Hassan A, Eldoumani H, Elazab ST, Mohamed SAM, Metwally AS, et al. Probiotics-loaded nanoparticles attenuated colon inflammation, oxidative stress, and apoptosis in colitis. Sci Rep (2022) 12(1):5116. doi: 10.3389/fmicb.2018.01129
207. Gu Z, Li F, Liu Y, Jiang M, Zhang L, He L, et al. Exosome-like nanoparticles from lactobacillus rhamnosus GG protect against alcohol-associated liver disease through intestinal aryl hydrocarbon receptor in mice. Hepatol Commun (2021) 5(5):846–64. doi: 10.1097/01.fpc.0000182777.95555.56
208. Mohmmad Monadi Al-Enazi A, Virk P, Hindi A, Awad MA, Elobeid M, Qindeel R. Protective effect of probiotic bacteria and its nanoformulation against cadmium-induced oxidative stress in male Wistar rat. J King Saud Univ Sci (2020) 32(7):3045–51. doi: 10.1016/j.jksus.2020.08.011
209. Xu C, Guo Y, Qiao L, Ma L, Cheng Y, Roman A. Biogenic synthesis of novel functionalized selenium nanoparticles by Lactobacillus casei ATCC 393 and its protective effects on intestinal barrier dysfunction caused by Enterotoxigenic Escherichia coli K88. Front Microbiol (2018) 9(JUN). doi: 10.1016/j.jcmgh.2016.05.010
Keywords: alcoholic liver disease, metabolism, gut-liver, stress mechanism, nanoparticle mediated treatment
Citation: Aghara H, Chadha P, Zala D and Mandal P (2023) Stress mechanism involved in the progression of alcoholic liver disease and the therapeutic efficacy of nanoparticles. Front. Immunol. 14:1205821. doi: 10.3389/fimmu.2023.1205821
Received: 14 April 2023; Accepted: 07 September 2023;
Published: 29 September 2023.
Edited by:
Murali Ganesan, University of Nebraska Medical Center, United StatesReviewed by:
Pradeep Kumar Shukla, University of Tennessee Health Science Center (UTHSC), United StatesCopyright © 2023 Aghara, Chadha, Zala and Mandal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Palash Mandal, cGFsYXNobWFuZGFsLmJpb0BjaGFydXNhdC5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.