 Jay V. Patankar
Jay V. Patankar Marvin Bubeck
Marvin Bubeck Miguel Gonzalez Acera
Miguel Gonzalez Acera Christoph Becker
Christoph Becker- 1Department of Medicine 1, University of Erlangen-Nuremberg, Erlangen, Germany
- 2Deutsches Zentrum Immuntherapie (DZI), Erlangen, Germany
A delicate balance between programmed cell death and proliferation of intestinal epithelial cells (IEC) exists in the gut to maintain homeostasis. Homeostatic cell death programs such as anoikis and apoptosis ensure the replacement of dead epithelia without overt immune activation. In infectious and chronic inflammatory diseases of the gut, this balance is invariably disturbed by increased levels of pathologic cell death. Pathological forms of cell death such as necroptosis trigger immune activation barrier dysfunction, and perpetuation of inflammation. A leaky and inflamed gut can thus become a cause of persistent low-grade inflammation and cell death in other organs of the gastrointestinal (GI) tract, such as the liver and the pancreas. In this review, we focus on the advances in the molecular and cellular understanding of programmed necrosis (necroptosis) in tissues of the GI tract. In this review, we will first introduce the reader to the basic molecular aspects of the necroptosis machinery and discuss the pathways leading to necroptosis in the GI system. We then highlight the clinical significance of the preclinical findings and finally evaluate the different therapeutic approaches that attempt to target necroptosis against various GI diseases. Finally, we review the recent advances in understanding the biological functions of the molecules involved in necroptosis and the potential side effects that may occur due to their systemic inhibition. This review is intended to introduce the reader to the core concepts of pathological necroptotic cell death, the signaling pathways involved, its immuno-pathological implications, and its relevance to GI diseases. Further advances in our ability to control the extent of pathological necroptosis will provide better therapeutic opportunities against currently intractable GI and other diseases.
1 Introduction
The GI tract is lined by a single layer of columnar epithelial cells, the enterocytes, which protect the host by forming a tight barrier to the external environment. The intestinal epithelial cells (IEC) form one of the largest barrier surfaces with the absorptive surface of the adult human small intestine alone having an estimated surface area of 250m2, which is in constant contact with a myriad of environmental and microbial components (1, 2). This barrier is crucial in mediating a healthy host-commensal symbiosis and therefore an uncontrolled breach of this barrier can quickly become a cause of microbial dysbiosis (1, 2). Dysbiosis can allow opportunistic pathogens access to the portal circulation increasing the likelihood of further inflammation in organs such as the liver and the pancreas. It has long been speculated that a defective gut barrier in humans may be a precursor to liver diseases such as steatohepatitis and cholangitis. The hypothesis that a leaky gut may be a harbinger of long-term inflammatory changes in distant tissues has been demonstrated using preclinical models and has shown positive association in patients (3–7).
Like other contact surfaces of the body such as the skin, the IECs are constantly shedding and sloughing into the intestinal lumen, a process regulated by physiological cell death programs such as apoptosis and anoikis. These coordinated cell death programs ensure that the tight junctions of neighboring non-dying cells seal the gap as the dying IEC exits the epithelial lining. To counteract this tight epithelial barrier, opportunistic pathogens have evolved mechanisms to induce cell death of IEC in order to breach the barrier to gain systemic access. For example, effector proteins such as Cif released by the enteropathogenic E. coli induce IEC apoptosis and the enterotoxin released by C. perfringens binds to the tight junction protein claudin-4, loosening the barrier and eventually resulting in IEC death (8, 9). The damaged IECs must be replaced to restore homeostasis, and one of the protective host mechanisms that accomplishes this is the secretion of TNF by macrophages and other myeloid cells that are activated by invading bacteria. However, TNF is a double-edged sword, inducing cell shedding and apoptosis of compromised IECs, but at sufficient concentrations can also trigger cell death of neighboring, healthy IECs. In addition to these effector mechanisms used by the pathogenic bacteria, viruses also employ strategies that inhibit and exploit various components of the programmed cell death machinery. For example, the virulence factor vIRA of mouse cytomegalovirus can target the RHIM domain-mediated interactions between key programmed cell death components RIP1, RIP3, and TRIF, whereas the K13 inhibitor produced by the Kaposi’s sarcoma-associated herpesvirus prevents the activation of caspase-8 (10). Such strategies employed by pathogens induce host proinflammatory responses that eliminate infected cells via natural killer and cytotoxic lymphocytes. However, immune over-activation or failure of immune suppression after pathogen clearance can cause disproportionate cytokine production, wreaking havoc by inducing pathological levels of IEC death, barrier disruption and immune activation (11). Once pathogens gain systemic access, the next target organ via the portal circulation is the liver, which like the gut is a tolerogenic, but an immune privileged organ (12). Pathogen access is mitigated mainly via the liver resident macrophages namely the Kupffer cells, although cholangiocytes remain susceptible to recurrent cytokine insults leading to pathological cell death (13–15). This establishes a vicious cycle of inflammation - cell death - barrier damage - further inflammation, and once such a self-perpetuating vicious cycle is established, it leads to the chronicity of intestinal and hepatic inflammation such as that observed in patients with inflammatory bowel disease (IBD) and primary sclerosing cholangitis (PSC).
Therefore, several therapeutic strategies have been explored that attempt to block the onset of necrosis in the gut. The desired outcome of a successful therapy is to achieve a long-term restoration of the IEC barrier (2). An example of a treatment that targets IEC death pathways and restores the barrier is that of the TNF inhibitors such as infliximab, adalimumab, and etanercept, however not all patients respond to the treatment and some side effects have also been reported (16–19). Therefore, efforts to understand the molecular and cellular complexity of pathologic cell death in the intestine have been a focus of research. Research has been spearheaded by efforts to draw a detailed picture of the intracellular signaling pathways and molecular nodes that regulate different forms of cell death, followed by research in the design and testing of pharmacological agents that effectively inhibit specific components of the programmed cell death machinery. In the following sections, we will discuss the details of the intracellular machinery involved in programmed necrosis, highlight the findings from the preclinical studies, and review the results of clinical trials and the associated challenges in developing interventions that target necroptosis.
2 Molecular machinery of necroptosis
2.1 Brief background on necroptosis in the GI tract
Since this review focuses on necroptosis, we refer the reader to other excellent reviews detailing the molecular machinery involved in other programmed death modes in cells of the GI tract (20–24). Necroptosis or programmed necrosis is an alternative form of coordinated cell suicide that culminates in lytic necrosis of the cell. The formal adoption of the term necroptosis is attributable to Degterev et al. in their 2005 report on this non-apoptotic form of cell death (25). Under circumstances where the normal execution of apoptosis cannot occur, necroptosis is triggered and, by extension, is the default mode of caspase-independent cell death that occurs in several cell types. Table 1 summarizes the evidence for necroptosis susceptibility in various tissue parenchymal cells of the GI tract. It is evident that several resident cell types that make up the majority of GI tissues are susceptible to necroptosis with the exception of hepatocytes, which is still under debated (26–35). Interestingly, there are no reports to date demonstrating whether other resident GI cell types including intestinal fibroblasts, smooth muscle cells, and enteric neurons and glia are susceptible to necroptosis. It is important to note that in the context of GI inflammatory diseases such as IBD, the repertoire of some of these cellular subsets is increased, and in some cases may end up completely replacing the parenchymal cells (36, 37). This is exemplified in intestinal fibrosis and during the inflammation-induced thickening of the intestinal muscle wall, hinting at a potential resistance of these cells to undergo necroptosis in a context-dependent manner that has not yet been formally tested. These cell type-specific differences in necroptosis susceptibility emerge due to the unique expression patterns of specific molecular components involved in the necroptosis pathway. In a later part of this section, we will revisit some of the recent findings on the relative expression abundances of the key components of necroptosis in gut IEC and liver hepatocytes cells and discuss how some GI cells are spared from necroptosis even when the necessary conditions are present.
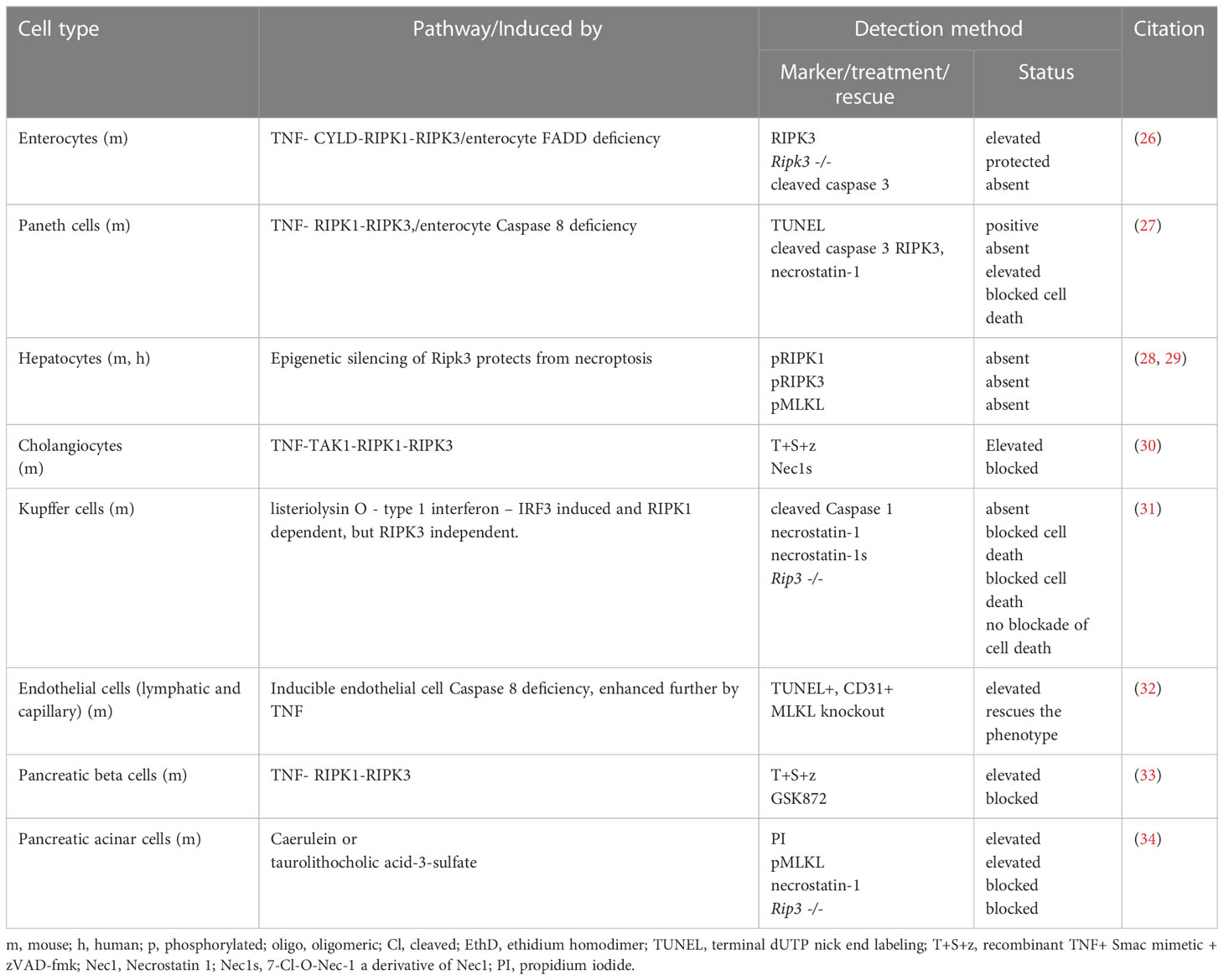
Table 1 Various GI cell types in which necroptosis has been investigated.
2.2 Initiation of the molecular cascades leading to necroptosis
Analogous to extrinsic and intrinsic apoptosis, necroptosis has both extracellular and intracellular activators. Before delving into the details, the readers is referred to Box 1 for a brief overview of the most important molecular players in necroptosis. Several signals are known to induce necroptosis in susceptible cells including extracellular TNF, TRAIL, FASL, LPS, dsRNA, and intracellular dsDNA, which can be recognized by cells expressing the specific receptors, namely TNFR1, DR4/DR5, FAS, various TLRs, and ZBP1 respectively. Upon ligand binding, the cytosolic domains of the TNFR, FAS, and DR4/DR5, and TLR receptors assemble into unique signalosome complexes. Under conditions of cellular stress, and when the magnitude of the trigger is sufficient to suppress the downstream NF-κB mediated survival queues, CASPASE-8 inhibits RIPK1 and CYLD and activates the effector caspases CASPASE 3/7, leading to apoptotic death. Recent studies have also linked environmental stresses such as heat shock and osmotic shock as inducers of necroptosis (38). In particular, heat shock protein 90, which is activated in hyperthermic conditions, has been implicated in a non-canonical activation of RIPK3 and MLKL during necroptosis in the colorectal cancer cell line HT-29 (39, 40). Furthermore, hyperosmotic stress triggers necroptosis by increasing intracellular pH (41). It is important to note that in all of these triggering conditions, a contextual prerequisite for necroptosis to occur is a defective initiation of apoptotic cascades. This may occur when initiator caspases such as CASPASE-8 are blocked biologically (inherited mutations, virally encoded inhibitors, host factors c-FLIP, or reduced expression) or pharmacologically (10, 42, 43). Furthermore, biological CASPASE-8 blockade was recently shown to occur via phosphorylation of pro-Caspase 8 at Thr265 by p90 ribosomal S6 kinase downstream of the TNFR-PDK1 (44). In such cellular contexts, the formation of a multi-protein signaling scaffold called the “necrosome” occurs, leading to necroptosis. The necrosome is essentially a multi-protein signaling complex containing activated RIPK1, RIPK3, and MLKL, with activated RIPK1recruiting RIPK3 via the homotypic interaction motif (RHIM domain) (45). The importance of the RHIM domain in mediating the interaction between RIPK1 and RIPK3 is underscored by the finding that mutation of the RHIM domain of RIPK3 attenuates necroptosis (46). Lack of CASPASE-8 activity and deubiquitination of RIPK1 by CYLD are also critical for the stability of the complex (47, 48). Another important requirement for the formation of the necrosome is the inhibition of the A20 deubiquitinase, which blocks the progression of necroptosis by modifying the ubiquitination on RIPK3 (48, 49).
Box 1: Critical nodes regulating necroptosis:
◼ FADD: Fas-associated protein with death domain
◼ CYLD: Deubiquitinase responsible for deubiquitinating RIPK1 and TRAF2 promoting necroptosis via stabilizing RIPK1 and RIPK3 interaction and blocking TRAF2 mediated inhibition of MLKL
◼ A20: Deubiquitinase and ubiquitin ligase responsible for blocking RIPK3 – RIPK1 interaction, as ubiquitinated RIPK3 is required for necroptosis progression
◼ TAK1: Transforming growth factor-β (TGF-β)-activated kinase 1 is the MAP kinase kinase kinase family member 7 a crucial signaling kinase which dictates mode of programed cell death downstream of TLR and TNFR.
◼ CASPASE 8: Cysteine-aspartate protease (CASPASE) is an initiator caspase downstream of TNF family receptors, death receptors, and FAS receptor. Initiates signaling via FADD and RIPK1 that culminates on the activation of effector caspases such as Caspase 3 and 7.
◼ CASPASE 10: An initiator caspase that once activated, cleaves and activates effector caspases. Pro-caspase 10 is activated in a FADD-dependent manner at the TRAIL and CD95 death-inducing signaling complexes, but cannot functionally replace Caspase 8.
◼ RIPK1: Receptor-interacting serine/threonine-protein kinase 1 acts downstream of TNF family receptors and death receptors and regulates phosphorylation and formation of signaling scaffolds that control apoptosis and necroptosis. More recently, RIPK1 was shown to also regulate inflammasome function.
◼ RIPK3: Receptor-interacting serine/threonine-protein kinase 1 is activated via phosphorylation by RIPK1 and in the absence of CASPASE 8 activity and/or activation of ZBP1, forms the signaling scaffold ‘necrosome’ that phosphorylates and activates MLKL.
◼ ZBP1: Z-DNA binding protein 1 is an interferon induced sensor of intracellular DNA and triggers the recruitment and activation of RIPK3 upon binding DNA of intracellular pathogens.
◼ MLKL: Mixed lineage kinase domain-like protein, is phosphorylated and activated by RIPK3. Some reports have indicated that further MLKL activation may require actions of other kinases including TAM and CAMK2. Upon full activation, MLKL undergoes oligomerization and inserts into the plasma membrane via its N-terminal helix bundle that binds to phosphatidyl inositol phosphates.
2.3 Formation of the core necrosome
Phosphorylation of RIPK1 at multiple sites in its intermediate domain is one of the most upstream events in necrosome formation and is mediated by the kinase activity of TAK1, NIK, and the autophosphorylation of mouse and human RIPK1 at Ser166 (50, 51). On the other hand, several kinases including MK2, IKKα/β, TBK1/IKKϵ, phosphorylate and inhibit mouse and human RIPK1 activity via the addition of inhibitory phosphates at Ser320, Ser321, Ser336, and Ser25 (52–56). Interestingly, another study suggested that TAK1-dependent phosphorylation of RIPK1 at Ser321 is essential for its association with FADD and the execution of RIPK1-dependent apoptosis (57). The authors used genetic and pharmacological ablation of TAK1 to demonstrate its critical involvement in phosphorylation of Ser321, challenging the inhibitory role of Ser321 on RIPK1 activity (57). However, a recent report revealed that the non-receptor tyrosine kinases JAK1 and SRC phosphorylate RIPK1 at Tyr384 (human) and Tyr383 (mouse), which are inhibitory phosphates critical in suppressing RIPK1 activation (58). The authors further showed that mice carrying mutations of these Tyr residues that prevent their phosphorylation develop spontaneous systemic inflammation, reinforcing the critical role of the complex post-translational modifications of RIPK1 that regulate this key component of the cell death machinery (58). Recently, we have shown that the activation of the EP4 receptor via endogenous or pharmacological agonists leads to a suppression of RIPK1 phosphorylation at Ser166. We have also implicated TAK1 downstream of the EP4 receptor in this suppressed activation and blocking of necrosome formation under conditions of CASPASE 8 inhibition in IECs (59). These studies highlight the critical role of the complex post-translational modifications of RIPK1 that regulate this key component of the cell death machinery.
Following RIPK3 recruitment, activated RIPK1 in the necrosome phosphorylates and activates RIPK3 at Ser227 (human) and Ser232/Thr231 (mouse) (60, 61). Once activated, RIPK3 undergoes autophosphorylation at Tyr224/Ser227 (human), Tyr231/Ser232 (mouse), and promotes the assembly of heterodimeric oligomers with RIPK1 and with itself via the RHIM domain, forming filamentous fibril-like structures (46, 62). These fibrils can bind thioflavin T and Congo red, and display circular dichroism, and bear structural features of β-amyloids (62). Structurally, activated RIPK1 and RIPK3 form heterotypic alternating stacks that form β-sheets, two of which bind together to form the polymeric fibrils that form the core of the necrosome (63). In addition, the reader is encouraged to note the complexity of the post-translational regulation of RIPK3 as well as that for RIPK1. For example, in certain contexts, RIPK1 is also able to negatively regulate RIPK3 oligomerization and necroptosis, as cells lacking RIPK1 undergo spontaneous RIPK3-mediated death, whereas cells expressing a catalytically inactive form of RIPK1 are protected from cell death (64). Interestingly, RIPK3 activity can be restricted by ubiquitination of the beyond-the-RHIM (BTR) domain, and its TSC1/mTOR - TRIM11-mediated ubiquitination and degradation in IECs alleviates necroptosis and hence colitis (65, 66).
Box 2: Molecular identification of necroptotic cells: (See limitations in text section 2.4 below)
Immunohistochemically:
◼ Co-immunolabelling – Cleaved Caspase 3/7, terminal deoxyuridine nick end labelling (TUNEL), and pMLKL. Here cells that are TUNEL(+), pMLKL(+), and Cleaved Caspase 3/7 (-) are considered necroptotic.
Molecular Biological:
◼ Western blotting – pRIPK3 (+), pMLKL (+), Cleaved Caspase 3/7 (-)
◼ Immuno co-precipitation – Necrosome members RIP1/pRIP3/pMLKL.
◼ Native PAGE – oligomerization of MLKL using anti-MLKL and/or anti-pMLKL antibodies.
Flow cytometric:
◼ Non-fixed cells – Cell permeable effector caspase (Caspase 3/7) activity probes (-), cell-impermeant DNA binding dyes such as propidium iodide (PI) (+), pMLKL (+). Cells that fall into the Caspase3/7 probe (-), PI (+), and pMLKL(+) gate are necroptotic.
◼ Fixed cells – Surface staining for pMLKL(+) before fixation followed by intracellular staining for TUNEL and cleaved Caspase 3/7.
2.4 Execution of necroptosis
Once assembled, the RIPK1-RIPK3 core necrosome recruits the ultimate executioner of necroptosis, namely the mixed lineage kinase domain like pseudokinase (MLKL). Canonical activation of MLKL occurs when activated RIPK3 with its kinase domain interacts with the pseudokinase domain of MLKL and phosphorylates it at Thr357 and Ser358 (20). After the critical phosphates are added to MLKL, it forms homotetramers, and prior to its release from the necrosome, two MLKL tetramers assemble to form homo-octamers, a process that does not depend on intramolecular disulfide bonds (21, 22). The oligomerization process of MLKL is a hotly debated topic, and although earlier work by Liu et al. using mouse and human cell lines provided evidence that MLKL tetramerization is a conserved event, newer evidence suggests otherwise (67). For instance, Petrie et al. discovered that RIPK3 phosphorylation of MLKL causes toggling of the pseudokinase domain activation loop, which promotes the necroptosis-inducing tetramerization of only human, but not mouse MLKL, suggesting species-specific differences in MLKL tetramerization (68). The molecular chronology of these events is still being investigated as exemplified by a recent study implicating a role of the TAM kinase family in promoting MLKL-mediated necroptosis via additional phosphorylation of MLKL at Tyr376 (69). The authors showed that inhibition or the absence of the TAM kinases prevents the progression of MLKL-mediated necroptosis and that TAM kinase activity is essential for the oligomerization step following necrosome release (69). However, further investigation is needed to determine the exact mechanism, the cells affected, and the critical dependence of necroptosis execution on the TAM kinase family. Moreover, octameric MLKL then forms Congo red dye-stainable amyloid fibrils of 5nm diameter, which are associated with necroptosis execution. However, it is not clear whether necroptosis execution critically depends on these MLKL amyloid-like polymers (23). One of the proposed mechanisms for oligomeric MLKL to execute cell death is its insertion into the membrane via its N-terminal helix bundle domain, which binds to the polar head groups of phosphatidylinositol phosphate in the membrane, creating a cationic large membrane pore (24, 25, 31). However, the exact mechanism of MLKL membrane insertion is currently under intense investigation and it is so far unclear how this process is exactly carried out. The membrane pore created by MLKL can cause the leakage of intracellular molecules of 10-kDa, leading to disruption of membrane integrity, osmotic imbalance, cell swelling, and ultimately lytic cell death (20). Thus, necroptotic cells are characterized by distinct changes in the cell morphology, cytosolic and nuclear swelling, and an inability to expel or prevent the influx of membrane-impermeable DNA binding dyes such as propidium iodide. However, these features alone are not sufficient to definitely diagnose necroptosis, and we therefore refer the reader to Box 2 for an overview of the methods and tools that enable the proper detection of necroptosis. The readers should note that the methods for necroptosis detection, especially in vivo, have several limitations. The closest proxy for determining necroptotic cell death is the simultaneous labelling of phosphorylated MLKL and cleaved executioner caspases. It is still unclear whether necroptotic cells can be reliably labelled for double-stranded DNA breaks (DSBs) using the terminal deoxynucleotidyl transferase dUTP nick-end labeling reaction. Some data indicate that late necroptotic cells acquire DSBs by an unknown mechanism. However, the lack of caspase activation indicates that cells at early stages of necroptosis would not have detectable DSBs. Another common way that necroptosis is inferred is by showing that the cell death in question is blocked by necrosulfonamide. This method suffers from a major drawback, as necrosulfonamide also inhibits gasdermin D oligomerization and insertion in the plasma membrane and as such a non-canonical gasdermin D cleavage remains a possibility. Besides these, in long-term live cell imaging experiments, incubation of cells with caspase activity probes is associated with reduced caspase availability on cognate targets and may lead to experimental artefacts. Similarly, long-term incubation with PI is also associated with undesirable adverse effects on cell viability and must be considered.
2.5 Transcriptomics reveal cell type susceptibility to necroptosis
Several components of the necroptosis machinery are expressed in the cells of the GI tract. With the sweeping advances in the ease of performing of bulk and single cell transcriptomic analyses, it is now possible to assess the relative expression of the various molecular mediators of cell death in a cell type and organ specific manner. In Figure 1A we have summarized the expression levels of the key regulators of necroptosis in the outstanding study by Lu et al. comparing the bulk transcriptomes of freshly isolated mouse intestinal crypts and intestinal epithelial organoids (70). It is clear that some of the components such as Zbp1, which is expressed in the crypts, are barely detectable in the organoid cultures, suggesting a non-epithelial origin of expression in fresh crypts. The levels of Cyld and Tnfaip3 (gene encoding A20) were also lower in the organoids compared to the crypts (Figure 1A) (70). The only gene that tends to show slightly higher levels in the organoids is Ripk3, whereas the levels of Tlr4 are barely detectable in either organoids or crypts (Figure 1A) (70). Interestingly, the single cell transcriptomic survey of the mouse small intestine by Haber et al. also confirms the overall relative abundance of the molecules associated with the cell death machinery, with the highest expression detected for Casp8 and Tnfrsf1a (Figure 1B) (71). More importantly, the study allows the relative abundance of these transcripts to be examined in a cell type-specific manner. For example, as shown in Figure 1B, the expression of Casp8 and Tnfrsf1a, is the highest in mature enterocytes, whereas that of Ripk3 is the highest in early enterocyte progenitors (71). Only a small percentage of gut epithelial cell subsets express Zbp1, and among these, goblet cells are the highest expressers (Figure 1B) (71). On the other hand, the study by Nault et al. has revealed the single cell transcriptomes of mouse liver, allowing the exploration of the relative abundance of cell death associated molecules in different liver cell subsets (72, 73). As shown in Figure 1C, both centrolobular and periportal hepatocytes barely express any Ripk3 (72, 73). This is consistent with reports showing that methylation and silencing of the Ripk3 locus in hepatocytes renders them resistant to necroptosis even under conditions of immune and pathogen related stress (28, 29). Interestingly, hepatocytes express Map3k7 (TAK1) and Ripk1 (Figure 1C) (72, 73). These studies have shown how single cell transcriptomics can help to identify the susceptibility of specific GI cell types to necroptosis. Therefore, a pre-screening based on single cell transcriptomics followed by a systematic analysis using multiple molecular and cellular approaches will allow the identification of subtype-specific susceptibility to necroptosis in the future. Nevertheless, the readers are advised to interpret the results of the transcriptomic studies with caution given that transcript to protein expression correlations are especially poor in intestinal tissues (74).
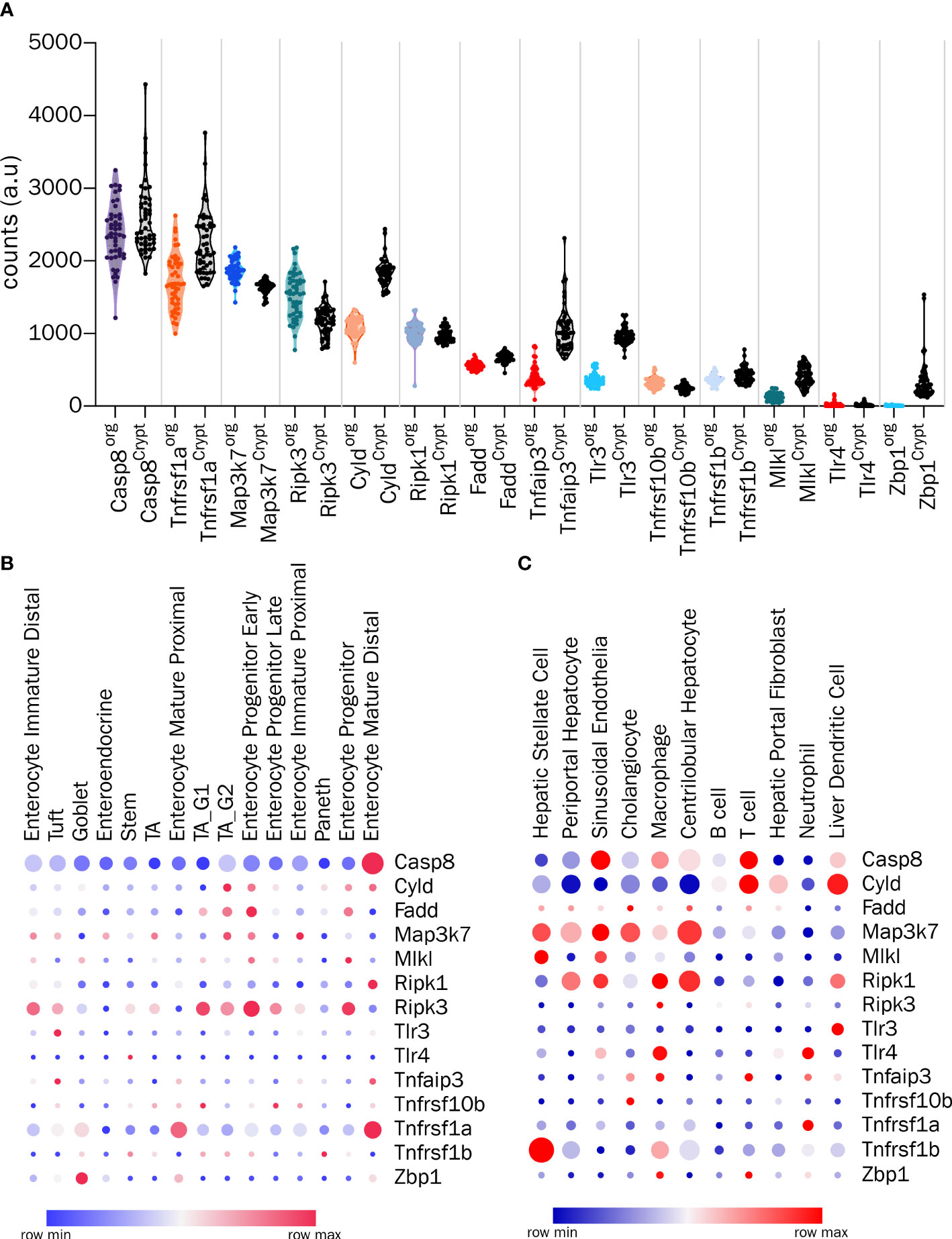
Figure 1 Transcriptomic analysis of the components involved in cell death in GI cells (A) Analysis of bulk transcriptomes from crypts and organoids for the indicated genes from GSE169368. (B) Expression profiles of indicated genes from single-cell RNA-Seq of intestinal epithelial cells (GSE92332) and (C) Expression profiles of indicated genes from single nucleus RNA-Seq of hepatic cells (GSE184506). In (B, C), circle color indicates the scaled mean expression, and the size indicates percent of cells expressing the transcript.
3 Necroptosis in preclinical models of GI diseases
3.1 Intestinal models with an involvement of necroptosis
Several studies have highlighted the delicate balance of different cell death cascades in IECs by conditional ablation of these molecules in mouse IECs, which, when disrupted leads to overt intestinal inflammation and potentially life-threatening systemic sepsis (26, 27, 75–78). These studies collectively highlighted the essential pro-survival functions of NF-κB in the IECs, provided the first insights into RIPK1 and FADD as critical regulators of IEC apoptosis and RIPK3-driven necroptosis, and demonstrated the importance of the initiator caspase, caspase-8 in regulating apoptotic cascades in IECs, the failure of which triggered RIPK3-dependent IEC necroptosis. These studies also laid the foundation for the detection, analysis and study of apoptotic and necroptotic cell death in IECs. Inspired by these studies, several more recent studies have established the roles of other key regulators of IEC necroptosis, further utilizing villin promoter-driven Cre recombinase expression in mouse IECs as preclinical models. In their 2018 study, Kattah et al. demonstrated the critical role of A20 and ABIN3 in IECs downstream of the TNFR in suppressing the activation of caspase-8 and RIPK1. Deletion of ABIN3 and A20 in IECs led to spontaneous cell death of IEC organoids, demonstrating that this is an effect intrinsic to IECs. The authors were able to completely rescue the cell death phenotype by inhibiting RIPK1 or deleting RIPK3 in conjunction with caspase inhibition (79). These findings were later extended further to show the ability of ABIN3 to coordinate with A20 to regulate the K63 deubiquitination and subsequent activation of RIPK3 (80).
In an interesting study, using the inducible version of the Cre recombinase (CreERT2) downstream of the villin promoter, Wang et al. showed how an IEC-specific deletion of the histone methyltransferase Setdb1 leads to a reactivation of endogenous genomic transposable elements (81). Reactivation of these dormant retrovirus-like elements triggered genomic instability in IEC stem cells and a ZBP1-mediated licensing of RIPK3 to induce necroptosis and intestinal inflammation. Pharmacological inhibition of RIPK3 was sufficient to cure the inflammatory phenotype of mice with the Setdb1-deficient IECs (81). Similar to viral dsDNA, intestinal ischemia-reperfusion injury is also associated with massive cell death via necroptosis and the release of mitochondrial DNA (mtDNA) into the tissue (82). The potential of mtDNA to induce necroptosis was directly addressed in a study by Zhang et al. The authors showed that a single intraperitoneal injection of mtDNA was sufficient to induce phosphorylation of both RIPK3 and MLKL, which could be inhibited by the administration of necrostatin-1 or by knocking out the gene Sting, a canonical DNA-sensor (83). Since the progression of necroptosis is critically dependent on the insertion of MLKL into the plasma membrane via its binding to membrane phospholipids, it is conceivable that altering the membrane lipid composition would have an impact on necroptosis. This speculation was supported by a recent study that examined the effect of IEC-specific deletion of the rate-limiting enzyme, cytidine triphosphate: phosphocholine cytidylyltransferase-α (CTα), which controls the synthesis of phosphatidylcholine (84). Inducible and conditional deletion of the CTα encoding gene Pcyt1a rendered mice susceptible to spontaneous colitis, increased number of TUNEL-positive cells in the colon, and increased RIPK3 mRNA and protein levels (84). Electron microscopy revealed characteristic features of necroptosis in both colonocytes and goblet cells (84).
The cognate physiological role of MLKL has been the subject of intense investigation, and studies have shown that MLKL may be involved in the regulation of autophagy upon its translocation to intracellular membranes (85). There is also evidence that lipidation of MLKL by very long chain fatty acids is critical for its function, although the mechanism is unknown (86). In an attempt to investigate the functions of the autophagy-associated protein ATG16L1 involved in LC3 lipidation, Matsuzawa-Ishimoto et al. uncovered an unexpected link of this autophagy regulator with IEC necroptosis. Using organoids generated from Atg16l1 sufficient and deficient IECs, the authors showed that ATG16L1 inhibits TNF-driven necroptosis by regulating mitochondrial homeostasis (87).
Interestingly, a non-mouse model has recently been reported by Liu et al. showing that necroptosis is active in an LPS-TLR4-induced inflammation model in piglets (88). The authors used the detection of phosphorylated MLKL and the mitochondrial and nuclear alarmins PGAM5 and HMGB1 as readouts after a sub-lethal dose of LPS; however, it should be noted that this is not an IEC-specific model. Another study by Wen et al. used a non-mouse model of intestinal ischemia-reperfusion injury to demonstrate the involvement of necroptosis, as both histological injury and alarmin release were attenuated by the administration of necrostatin-1. In addition, the authors were able to show lower levels of RIPK1-associated MLKL by immunoprecipitation, suggesting the involvement of necroptosis in this model. However, just as with the piglet model, it should be noted that this is not an IEC specific model (82). In addition, studies relying solely on necrostatin-1 should be interpreted with caution since it also blocks and inhibits indoleamine-2,3-dioxygenase, an enzyme that modulates inflammation (89).
The most commonly used mouse model to understand human intestinal inflammation is the dextran sulfate sodium-induced colitis model. Interestingly, a study by Zhang et al. using mice that lack MLKL showed that the IEC loss seen in the DSS model appears to have some contribution from necroptosis, as MLKL ablation was protective against DSS in wild type mice (90). Another recent study supports the involvement of necroptosis in DSS-induced colitis, since the authors showed that GAB1 is involved in RIPK3 dephosphorylation and inhibition of IEC necroptosis using a DSS colitis model (91). However, Alvarez-Diaz et al. challenged these results in their report showing that germline deficiency of MLKL or RIPK3 exacerbates the severity of DSS-induced colitis (92). Therefore, the DSS colitis model is not an exclusive necroptosis inducing model per se. However, in combination with the genetic ablation of key necroptosis blockers such as Casp8, this model shows a high involvement of necroptosis in the IECs (59). Some studies have investigated the potential involvement of necroptosis using the pharmacological inhibitor of MLKL, necrosulfonamide (NSA), in the 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced mouse model of colitis (93). However, these studies must be interpreted in light of the promiscuity of NSA, which also blocks gasdermin D-mediated pyroptosis direct inhibition of gasdermin D or via a proximal modulation of caspase 1 activity (94, 95).
Future studies systematically analyzing the contribution of necroptosis in commonly used models of intestinal inflammation, including DSS colitis, TNBS colitis, oxazolone-induced colitis, and adoptive T-cell transfer-induced colitis, are warranted.
3.2 Hepatic models with involvement of necroptosis
An initial report on the potential involvement of necroptosis in the liver came from a mouse model of vaccinia virus infection (96). The authors showed that in several organs including the liver, vaccinia virus infection induces a phosphorylation-dependent RIPK1-RIPK3 complex and that the formation of the complex is necessary for necroptosis of the infected cells and thus infection control (96). They found that RIPK3-deficient animals had impaired infection control and increased inflammation. However, the study did not identify the cell types in the liver that underwent necroptosis. Later, Upton et al. revealed that viral factors can also inhibit RIPK3-dependent necroptosis in multiple tissues, including the liver, in a murine cytomegalovirus infection model (97). However, in contrast to the strong evidence for necroptosis in IECs in intestinal models, the role of necroptosis in hepatocytes has been a matter of debate. The first studies suggesting the involvement of necroptosis in hepatocytes used the acetaminophen-induced liver injury model (98, 99). Using RIPK3 silencing or genetic ablation in the acetaminophen-induced liver injury model and primary hepatocytes, the authors concluded that suppression of RIPK3 ameliorated liver injury, implicating hepatocyte necroptosis (98). After this report, other studies also showed an improvement in acetaminophen-induced liver injury upon RIPK1 inhibition with necrostatin-1 (99). Using another infectious hepatitis model of the hepatocyte-infecting bacterium Listeria monocytogenes, Koschel et al. uncovered an important role for the deubiquitinating enzyme, OTU domain aldehyde binding-1 (OTUB1) in the regulation of liver necroptosis (100). Under normal circumstances, an infection of this nature results in profound hepatocyte apoptosis, however, mice with a conditional ablation of OTUB1 in liver parenchymal cells showed a shift from apoptotic to necroptotic cell death. The authors inferred necroptosis from reduced levels of activated caspases 3, 7, and 8. Furthermore, the lethal infection phenotype that was observed in mice lacking OTUB1 in the parenchymal cells was rescued by the administration of necrostatin-1s and by co-ablation of MLKL (100). Thus far multiple studies had indicated the existence of necroptosis in the liver, albeit most suffered from an interpretation bias in which off-target pharmacological effects such as necrostatin mediated inhibition of RIPK1-driven apoptosis or necrosulfonamide-mediated inhibition of pyroptosis were not considered. Another confounding factor was with the interpretation of hepatocytes undergoing necroptosis, although germline knockouts of MLKL and not hepatocyte specific knockouts were used. Therefore, the question of whether hepatocytes are the targets of necroptotic cell death in hepatic pathology remained unanswered. Adding to this conundrum, a recent study by Preston et al. showed that both mouse and human RIPK3 promoter are epigenetically silenced in hepatocytes, thereby sparing hepatocytes from MLKL-mediated necroptosis (29). The authors used multiple models of infectious hepatitis and showed that the silencing of the RIPK3 promoter in hepatocytes was not relieved under any conditions of immunological stress tested in their study, deepening the mystery of hepatocyte necroptosis further (29). This hypothesis has been supported by studies investigating the association between transplant-related ischaemia and reperfusion-induced liver injury and necroptosis. In rats, alanine and aspartate aminotransferase levels were elevated after liver transplantation, clearly indicating hepatocyte damage. Interestingly, however, an evaluation of potential necroptosis by immunohistochemistry revealed increased levels of phosphorylated MLKL protein only in periportal cholangiocytes, but not in hepatocytes, suggesting that hepatocytes were spared from necroptosis, but not from other forms of cell death (101). The findings of this study are consistent with another study where the authors reported a dominant role of effector caspases in suppressing RIPK1-dependent necroptosis in this model (102).Yet, further reports have emerged which implicate ischemia-reperfusion-related liver injury in the context of aging livers to be dependent on hepatocyte necroptosis and ER stress (103, 104). A separate study confirmed this showing elevated levels of phosphorylated RIPK1, RIPK3, and MLKL in aged livers compared to young livers. The levels of total MLKL protein and its tetramer and octomerization as determined by native PAGE were also significantly increased (104). However, the authors could not conclusively show that this was a hepatocyte-specific increase in necroptosis.
Other studies have also implicated hepatocyte necroptosis in different mouse models of liver disease. In one such report the authors used ethanol-induced liver and hepatocyte injury. In this model, the authors reported a direct RIPK3-FADD interaction downstream of TNFR and an obligate involvement of TNF in driving the liver phenotype. Although, similar to the drawbacks of the previous studies, even this study used germline RIPK3 knockout mice, which afforded protection from the alcohol-induced liver pathology. However, the authors failed to rescue the liver phenotype by pharmacological RIPK1 inhibition, raising speculations of a RIPK1-independent activation of RIPK3 in hepatocytes (105). Another study implicated hepatocyte necroptosis in a rat model of ischemic acute kidney injury, which causes liver damage. The authors inferred hepatocyte necroptosis based on electron microscopic evaluation, and immunohistochemistry for RIPK3, identifying dying hepatocytes with features of necroptosis (106). However, a careful dissection of the specific cell types undergoing necroptosis using genetic models was not conducted.
In an interesting study using parenchymal-specific Cre driver line to ablate Casp8, Gautheron et al. performed a direct evaluation of necroptosis in liver parenchymal cells using a mouse model of methionine-choline dietary deficiency (MCD) of non-alcoholic steatohepatitis (NASH) (107). The authors showed that the Casp8 deficiency in the parenchymal cells exacerbated MCD diet-induced liver injury and upregulated RIPK3 protein levels. Under these circumstances, when the authors co-ablated RIPK3, the liver phenotypes were rescued. Interestingly, the authors still observed a high level of CASPASE3 cleavage and activation in the liver demonstrating that MCD- triggered cell death in the liver could be dominated by RIPK1-depedent or classical apoptosis apoptosis in the absence of Caspase-8, via intrinsic pathways of effector caspase activation (107). In addition, the study failed to conclusively distinguish hepatocyte versus cholangiocyte necroptosis.
One of the direct evidences of necroptosis in hepatocytes was presented by Zhang et al. while uncovering an interesting role of O-linked β-N-acetylglucosamine (O-GlcNAc) modification in hepatocytes. Zhang et al. reported that the liver-specific conditional deletion of the O-GlcNAc transferase (OGT) gene causes hepatocyte necroptosis. The authors showed that primary hepatocytes with conditional ablation of OGT exhibited elevated levels of phosphorylated MLKL and cell death, which could be suppressed by treating the hepatocytes with GSK-872, a RIPK3 inhibitor, but not with z-VAD, a pan-caspase inhibitor suggesting a role for necroptosis in this mouse model (108). This study provided compelling ex vivo evidence that hepatocyte necroptosis can occur, however, potential off-target effects of GSK-872 were not considered and rescue using silencing of MLKL was not conducted.
In addition to these models, two other models of immune-mediated liver injury, namely concanavalin A (Con A) and α-galactosylceramide (α-GalCer)-induced injury have been widely used to investigate the mode of cell death in the liver (109). The ConA model is initiated by direct activation of CD4+ T- cells and NKT cells, which then target the hepatocytes in a manner that is independent of antigen presentation and TCR activation. On the other hand, the α-GalCer model is a milder, non-lethal model, that is predominantly an NKT cell-driven injury model in which T-cell involvement can be excluded (109). Using the Con A model and applying it to germline RIPK3-deficient mice, Deutsche et al. showed a mild degree of protection from liver injury, but not from lethal inflammation (110). Another study investigating the potential role of necroptosis in the Con A model also used RIPK3 knockout mice and found no significant differences in the extent of liver damage between the groups (111). Interestingly, when Gunther et al. applied the Con A model to mice deficient for the MLKL, they observed significant protection against liver injury (112). However, germline deficiency of RIPK3 gene could not protect against the Con A-induced liver injury. No detectable levels of RIPK3 were found in hepatocytes under any of the experimental conditions, raising the speculation of a RIPK3-independent mode of MLKL activation in hepatocytes (112). Similar to the caveats of the previous studies, no conclusive interpretation of hepatocyte necroptosis could be drawn based on these studies that have heavily relied on pharmacological inhibition and germline ablation.
Recently however, Hamon et al. reported that the hepatocyte cell death observed in the Con A model is non-necroptotic in nature, as mice with conditional ablation of MLKL in hepatic parenchymal cells were not protected from Con A-mediated liver injury and inflammation in their studies (28). In addition, some supporting evidence has emerged from the α-GalCer-induced liver injury model in the RIPK3 and MLKL deficient mice also with contrasting findings (113, 114). Therefore, the most plausible hypothesis at present is that necroptosis does occur in the liver during various pathophysiological conditions. However targeted approaches indicate that hepatocytes could be spared from canonical necroptosis owing to their failure to express RIPK3. Future investigations using targeted approaches will be necessary to prove the existence of potential novel mechanisms of necroptosis in hepatocytes. In summary, while new findings indicate that immune-mediated necroptosis mainly occurs in non-parenchymal cells of the liver, the precise role of necroptosis in diseases of the liver remains under debate.
3.3 Pancreatic models with involvement of necroptosis
Cell death modes in pancreatic pathology have been intensively investigated, particularly in the context of pancreatic islet β-cell death. Transplantation of donor islets into recipient animals is often associated with increased host-versus-graft immunity and associated death of cells in the transplanted tissue. Zhao et al. used a model of CD4+ T-cell mediated autoimmune diabetes after transplantation of donor islets with altered expression of the proteins involved in the cell death machinery to investigate the role of different modes of cell death (115). Here, the authors assessed the autoimmune attack on islets upon adoptive transfer of FACS sorted CD4+ BDC2.5highCD25- diabetogenic T-cells into recipient mice. The authors showed that islets from RIPK3-deficient recipient mice were still equally susceptible to the autoimmune T-cell- mediated destruction (115). Moreover, recipient mice with a concomitant inhibition of both RIPK3 and BLC2 to block necroptotic and apoptotic cell death were still unable to protect the islets from the autoimmune attack (115). These results seem to be in line with a report that described rare MLKL mutations in individuals, which were associated with an increased risk of maturity-onset diabetes of the young potentially acting as an epistatic modifier of existing disease mutations in the PDX1 gene (116). The authors showed that the mutant form of MLKL was unable to induce necroptosis; however, the patients still developed diabetes and beta cell loss potentially via other modes of cell death, arguing against a role for necroptosis in β-cell loss (116). The induction of pancreatic β-cell death and the consequent diabetes in mice and rats is achieved by five consecutive intraperitoneal doses of streptozotocin (STZ). Between two and five days after STZ administration, mice begin to show reduced circulating insulin levels and increased glucose levels due to β-cell death (117, 118). A few studies have investigated the involvement of necroptosis in this model. For example, Contreras et al. showed that isolated pancreatic islets from the RIPK3 knockout mice were protected from TNF- and z-VAD- induced cell death compared to control islets (33). The authors also observed a significant reduction in the STZ induced hyperglycemia and glucose intolerance in mice with a germline deficiency of RIPK3. The authors concluded that in this model, the TNF-induced RIPK1-RIPK3 pathway is operational and STZ- induces necroptosis of β-cells (33). Taken together, the majority of studies indicate that islet cells of the pancreas are resistant to necroptotic cell death under conditions of autoimmune attack, however, a detailed molecular and biochemical characterization of the mode of cell death is still missing. Therefore, RIPK3-dependent necroptosis as a contributing factor to autoimmune islet cell death can be excluded for the time being. Unlike the autoimmune destruction of islets in diabetes, pancreatitis is frequently associated with a loss of acinar cells, a feature recapitulated in the mouse model of cerulein-induced acute pancreatitis. Using RIPK3 knockout mice, Zhang et al. and He et al. independently showed a crucial role of necroptosis in driving acinar cell loss, and tissue damage in the cerulein-induced pancreatitis model (119, 120). This was followed by the report by Louhimo et al. who showed a direct induction of MLKL phosphorylation in pancreatic acinar cells at supramaximal concentrations of cerulein or submicellar concentrations of the bile acid taurolithocholic acid-3-sulfate (34). The authors also reported an improvement in pancreatitis upon RIPK3 ablation or pharmacological inhibition of RIPK1 via necrostatin-1 (34). Another study by Zhao et al. employed the cerulein-induced acute pancreatitis model, in RIPK3 knockout mice as well as in mice carrying a mutation in the kinase domain of RIPK3, rendering it catalytically inactive. The authors showed a significant reduction in the number of dead pancreatic cells, again implying that the kinase function of RIPK3 was critical in mediating acinar cell loss in this pancreatitis model (121).
In contrast to these studies that clearly linked RIPK3-mediated necroptosis with cerulein-induced pancreatitis, a report by Newton et al. found no contribution of necroptosis to pancreatitis severity in histological and serological endpoints (122). The authors showed that in mice bearing a mutation in the RIPK1 kinase domain a germline deficiency of the RIPK1 kinase domain or of RIPK3 were not rescued from cerulein-induced pancreatitis. In addition, a recent report by Boonchan et al. provided more evidence against the involvement of necroptosis in acinar cell loss in acute pancreatitis (123). The authors used knockouts for both RIPK3 and MLKL and showed that RIPK3 deficiency resulted in a more severe pancreatic inflammation and disease, and MLKL deficiency rendered mice even more susceptible to tissue damage caused in the cerulein-induced acute pancreatitis model. Their findings showed that the loss of the key executioner of necroptosis shifted the balance of cell death towards apoptosis via a downregulation of Bclxl and Cflar, detectable as an increase in the number of TUNEL positive apoptotic cells (123). However, the authors did not investigate whether pharmacological pan-caspase blockade could mitigate the increase in disease severity ascribed to increased apoptosis. Several other studies have used the cerulein-induced acute pancreatitis mouse model to investigate necroptotic cell death in the pancreas (124, 125). However, given the conflicting results of some of the systematic investigations, we recommend that this model be used with caution when interpreting or implicating necroptosis as a driving factor until conclusive evidence on the role of necroptosis in the pancreas becomes available.
In summary, there is biochemical and genetic evidence showing that pancreatic acinar cells express the key components of the necroptotic machinery, but there are significant discrepancies regarding the evidence for necroptotic cell death in pancreatic pathology. The discrepancies may emerge from the different treatment times and concentrations used in these studies to induce acute pancreatitis. Although blocking RIPK3 seems to lower pancreatitis severity, the contribution of necroptosis-independent functions of RIPK3 needs to be investigated. Further research using targeted conditional and inducible knockout mice for various components of the necroptosis machinery is needed to fully address the role of necroptosis in pancreatitis.
4 Therapeutic discovery: necroptosis interventions
4.1 Translation of RIK1 Inhibitors, from bench to bedside
As our understanding of the molecular cascade and signaling involved in necroptosis has improved, the prevention of necroptotic cell death has become a therapeutically viable opportunity. This is particularly true in case of diseases where overt and excessive cell death occurs triggering further inflammation and tissue damage. Therefore, small molecule inhibitors to block necroptosis and enhance cytoprotection are being investigated for potential applications in organ preservation for transplantation, autoimmune diseases, and viral infectious diseases. As therapeutic targeting of necroptosis has evolved, three clear targets have emerged: 1. RIPK1, 2. RIPK3 and 3. MLKL. A kinase inhibitor screen identified several type I inhibitors that competitively inhibit the ATP binding site of the RIPK1 (126). Interestingly, several of these inhibitors are FDA- approved kinase inhibitors, such as sunitinib, that block a wide variety of kinases other than RIPK1 and are therefore not selective/specific (Table 2). Other compounds, such as the FDA-approved drug pazopanib, as well as compounds from the GSK kinase inhibitor library, have been identified as type II inhibitors that bind to RIPK1 in an inactive kinase conformation (DFG-out), sterically blocking the ATP binding site (127, 128). Unfortunately, further development of these type II inhibitors was stalled due to higher molecular weights, lower solubility in aqueous solvents, and several off-target kinase inhibitory effects (128). Finally, the necrostatin class of inhibitors has also proven to be a useful platform for pharmacological development of RIPK1 inhibition. Necrostatins bind to RIPK1 in its inactive conformation via the nitrogen atom in the indole ring of necrostatin, forming an H-bond with a hydroxyl group in the activation loop of RIPK1 (127, 129, 130). Several derivatives of necrostatin-1 have been developed, and protect preclinical mouse models of colitis, pancreatitis and steatohepatitis. However, like necrostatin-1, they suffer from poor pharmacokinetics, lack of efficacy, and off target effects due to the inhibition of other enzymes, particularly indoleamine-2,3-dioxygenase, which have critical functions in immune cells (131). Recently, a randomized, placebo controlled clinical trial of a new first-in-class RIPK1 kinase inhibitor, GSK2982772, in active ulcerative colitis was completed. The compound has better pharmacokinetic properties, higher potency and selectivity, and was well tolerated with no safety concerns (128, 132). Interestingly, at the 43-day trial mark, the GSK2982772 group had significantly lower fecal calprotectin levels and a modest reduction in mean circulating CRP levels compared to the placebo group. However, these differences disappeared at later time points and no demonstrable improvements in efficacy were observed in terms of improved Ulcerative Colitis endoscopic index of severity (132). Considering the short period of time during which improved levels of fecal calprotectin and CRP were observed, together with the recent observation of a stage-dependent effect of RIPK1 inhibition on inflammation, it would be necessary to identify the optimal therapeutic window of inhibition. Another recent discovery has raised the possibility of a co-regulatory core molecular platform called the PANoptosome which controls multiple inflammatory modes of cell death that has been termed “PANoptosis”. The core feature of panoptosis is the activation of RIPK1 by extracellular pathogens or danger signals, or of ZBP-1 via intracellular pathogens or danger signals. The potential therapeutic value of targeting PANoptosis in inflammatory diseases has also been recently assessed against severe COVID-19 given that SARS-CoV-2 increases RIPK1 activation and ZBP-1 expression to promote viral spread (133–135). The authors showed that human lung organoids infected with the SARS-CoV-2 virus had a lower viral load and reduced inflammation upon treatment with Nec-1s (134).
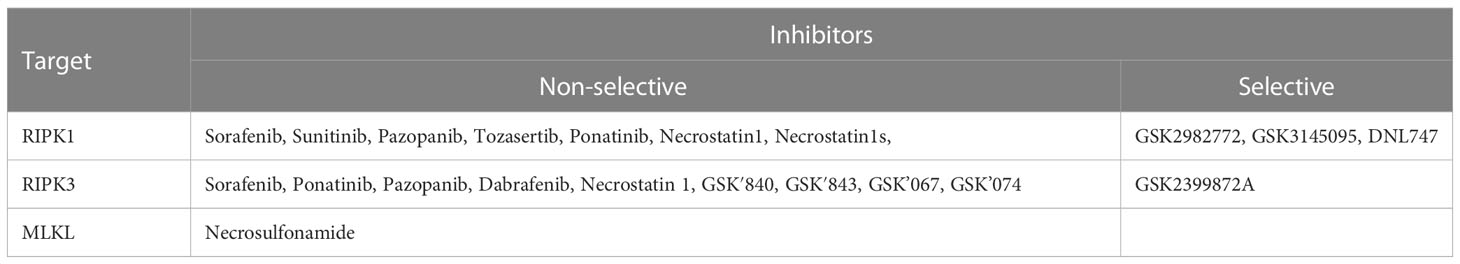
Table 2 Therapeutic and experimental inhibitors against necroptosis.
4.2 Potential of RIPK3 inhibitors
Few type I inhibitors of RIPK3 have entered therapeutic development, mainly due to the identification of toxic kinase-dead variants in which the aspartate in the conserved DFG motif is replaced by asparagine (136). This substitution resulted in uncontrollable RIPK1- and caspase-8- dependent apoptosis and midgestational death, raising safety concerns of overt apoptosis induction upon the inhibition of RIPK3 kinase functions (131, 136). Interestingly, however, the FDA-approved ATP-competitive BRAF inhibitor dabrafenib also inhibits human and mouse RIPK3 without cross-inhibition of RIPK1 and without adverse induction of RIPK1-dependent apoptosis and protects mice from acetaminophen-induced liver injury (137, 138). However, other type I inhibitors of RIPK3, such as GSK’872 and GSK’843, do not spare RIPK1-FADD-CASP8 complex formation and apoptosis induction, and therefore therapeutic exploration has been inconsequential (131). Interestingly, a recent study reported the effects of GSK’872 treatment in mice expressing human ACE2 in the nasal and lung epithelium infected with the SARS-CoV-2 viruses to inhibit the ZBP-1 branch of PANoptosis in a mouse model of COVID-19 (135). Treatment with GSK’872 significantly reduced MLKL phosphorylation, however, chemokine production remained unaffected. On the contrary, mice with a genetic ablation of RIPK3 displayed a significant reduction in chemokine levels upon SARS-CoV-2 infection compared against infected wildtype controls, implying that inhibition of the kinase functions of RIPK3 alone may not be sufficient to control inflammation (135). Several type II kinase inhibitors such as the FDA approved drugs ponatinib and sorafenib, which target RIPK1, also block the kinase activity of RIPK3 and can protect mice from TNF-induced systemic inflammatory response syndrome (SIRS) and renal ischemia-reperfusion injury (139). Attempts to develop newer RIPK3 inhibitors have met with some success. For example, Xia et al. recently identified Zharp-99, an unclassified small molecule kinase inhibitor of RIPK3, which blocked necroptosis in human and mouse cells without inducing RIPK1-dependent apoptosis and also blocked TNF-induced SIRS in mice (140). Interestingly, a recent study showed that blocking the oligomerization of RIPK3 can also be used as a new strategy to inhibit necroptosis without increasing apoptosis via the pharmacological antagonist of SRC, named 1-(tert-butyl)-3-(4-chlorophenyl)-1H-pyrazolo[3,4-d] pyrimidin-4-amine (also known as PP2). The authors found that PP2 specifically disrupted RIPK3 oligomerization in cell lines, without altering its autophosphorylation or its association with RIPK1 and MLKL (141). However, the in vivo safety, efficacy, and specificity of these newly developed compounds that block RIPK3 function remain a topic for further investigation.
4.3 MLKL as a potential future therapeutic target
Pharmacological inhibition of the final executioner of necroptosis, MLKL, is also promising but is still in its infancy due to the complications of targeting a pseudokinase where screening of well-established kinase libraries is not feasible. Mice carrying a germline mutation of MLKL are viable and show no apparent adverse phenotype at steady state (142). In addition, mutations in the brace region of MLKL, which generate a lethal constitutively active MLKL variant, are only toxic if homozygous, demonstrating that normal MLKL copies can induce clearance of toxic versions of MLKL (143). By screening a compound library of over 200,000 compounds in an anti- necroptosis cell screen, Sun et al. identified a small molecule called (E)-N-(4-(N-(3-methoxypyrazin-2-yl) sulfamoyl)phenyl)-3-(5-nitrothiophene-2-yl)acrylamide, which they named necrosulfonamide as a molecule that targets the Cys86 residue of MLKL (60). The authors showed that the targeting mechanism is specific to human MLKL, as the Cys86 residue is not conserved in mouse cells and therefore necrosulfonamide is unable to inhibit necroptosis in mouse cells (60). Recent evidence has challenged the specificity of necrosulfonamide by showing that in addition to MLKL-mediated necroptosis, necrosulfonamide also inhibits GSDMD-induced pyroptosis (144). Following this report, several studies showed that necrosulfonamide prevented disease outcomes in mouse models of diseases such as acute brain injury, colitis, acute liver failure, and myocardial dysfunction due to its pyroptosis inhibitory effects (144–147). Recently, a new class of xanthine and uracil derivatives has been reported to stabilize the inactive state of MLKL, thereby inhibiting MLKL oligomerization and membrane translocation, but their precise mechanism of action, in vivo safety, and off-target effects remain to be elucidated (148, 149). To the best of our knowledge, no clinical phase trials have been conducted for MLKL inhibitors so far. Future strategies of computational repurposing of approved drugs with the potential to cross-inhibit MLKL-mediated necroptosis, may achieve faster translational results (150).
5 Concluding remarks
A large body of evidence points to the role of necroptosis of various GI cells in the context of infectious and inflammatory diseases. The modeling of necroptosis in preclinical disease is driven by our growing understanding of the underlying molecular machinery and is fueled by conditional genetic and pharmacological screens. However, to date, the collective beneficial evidence from preclinical studies outweighs the translational efforts in the development of safe, efficacious, potent, and selective inhibitors, which target this cell death mechanism in human diseases. Future development of newer interventions, therapeutic strategies, and repurposing of existing drugs against necroptosis is needed.
Author contributions
JP and CB: conceptualization and composition. MG: analysis. MB: editing and proofreading. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TRR241 375876048 (A03, B05), SFB1181 (C05), and individual grants with project numbers 418055832 and 510624836. The project was also supported by the Interdisciplinary Centre for Clinical Research (IZKF: A76, A93, and ELAN P120).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol (2017) 4(1):33–46. doi: 10.1016/j.jcmgh.2017.03.007
2. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol (2017) 14(1):9–21. doi: 10.1038/nrgastro.2016.169
3. Ray K. NAFLD. leaky guts: intestinal permeability and NASH. Nat Rev Gastroenterol Hepatol (2015) 12(3):123. doi: 10.1038/nrgastro.2015.15
4. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut (2009) 58(8):1091–103. doi: 10.1136/gut.2008.165886
5. You Q, Shen Y, Wu Y, Li Y, Liu C, Huang F, et al. Neutrophil extracellular traps caused by gut leakage trigger the autoimmune response in nonobese diabetic mice. Front Immunol (2021) 12:711423. doi: 10.3389/fimmu.2021.711423
6. Singla S, Sahu C, Jena G. Association of type 1 diabetes with ulcerative colitis in BALB/c mice: investigations on sex-specific differences. J Biochem Mol Toxicol (2022) 36(4):e22980. doi: 10.1002/jbt.22980
7. Kwon J, Lee C, Heo S, Kim B, Hyun CK. DSS-induced colitis is associated with adipose tissue dysfunction and disrupted hepatic lipid metabolism leading to hepatosteatosis and dyslipidemia in mice. Sci Rep (2021) 11(1):5283. doi: 10.1038/s41598-021-84761-1
8. Samba-Louaka A, Nougayrede JP, Watrin C, Oswald E, Taieb F. The enteropathogenic escherichia coli effector cif induces delayed apoptosis in epithelial cells. Infect Immun (2009) 77(12):5471–7. doi: 10.1128/IAI.00860-09
9. Eichner M, Augustin C, Fromm A, Piontek A, Walther W, Bucker R, et al. In colon epithelia, clostridium perfringens enterotoxin causes focal leaks by targeting claudins which are apically accessible due to tight junction derangement. J Infect Dis (2017) 217(1):147–57. doi: 10.1093/infdis/jix485
10. Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol (2011) 12(2):79–88. doi: 10.1038/nri3131
11. Patankar JV, Becker C. Cell death in the gut epithelium and implications for chronic inflammation. Nat Rev Gastroenterol Hepatol (2020) 17(9):543–56. doi: 10.1038/s41575-020-0326-4
12. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
13. Shi S, Verstegen MMA, Roest HP, Ardisasmita AI, Cao W, Roos FJM, et al. Recapitulating cholangiopathy-associated necroptotic cell death In vitro using human cholangiocyte organoids. Cell Mol Gastroenterol Hepatol (2022) 13(2):541–64. doi: 10.1016/j.jcmgh.2021.10.009
14. Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond) (2015) 129(8):721–39. doi: 10.1042/CS20140732
15. Afonso MB, Rodrigues PM, Simao AL, Ofengeim D, Carvalho T, Amaral JD, et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis (2016) 7(9):e2390. doi: 10.1038/cddis.2016.280
16. Sisto M, D'Amore M, Caprio S, Mitolo V, Scagliusi P, Lisi S. Tumor necrosis factor inhibitors block apoptosis of human epithelial cells of the salivary glands. Ann N Y Acad Sci (2009) 1171:407–14. doi: 10.1111/j.1749-6632.2009.04688.x
17. Yakymenko O, Schoultz I, Gullberg E, Strom M, Almer S, Wallon C, et al. Infliximab restores colonic barrier to adherent-invasive e. coli in crohn's disease via effects on epithelial lipid rafts. Scand J Gastroenterol (2018) 53(6):677–84. doi: 10.1080/00365521.2018.1458146
18. Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-alpha on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol (2013) 304(11):G970–9. doi: 10.1152/ajpgi.00183.2012
19. Fries W, Muja C, Crisafulli C, Costantino G, Longo G, Cuzzocrea S, et al. Infliximab and etanercept are equally effective in reducing enterocyte APOPTOSIS in experimental colitis. Int J Med Sci (2008) 5(4):169–80. doi: 10.7150/ijms.5.169
20. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol (2018) 15(12):738–52. doi: 10.1038/s41575-018-0065-y
21. Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol (2018) 15(12):721–37. doi: 10.1038/s41575-018-0054-1
22. Chen X, Zeh HJ, Kang R, Kroemer G, Tang D. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol (2021) 18(11):804–23. doi: 10.1038/s41575-021-00486-6
23. Bayir H, Dixon SJ, Tyurina YY, Kellum JA, Kagan VE. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol (2023) 19:315–36. doi: 10.1038/s41581-023-00689-x
24. Miao R, Fang X, Zhang Y, Wei J, Zhang Y, Tian J. Iron metabolism and ferroptosis in type 2 diabetes mellitus and complications: mechanisms and therapeutic opportunities. Cell Death Dis (2023) 14(3):186. doi: 10.1038/s41419-023-05708-0
25. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1(2):112–9. doi: 10.1038/nchembio711
26. Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature (2011) 477(7364):330–4. doi: 10.1038/nature10273
27. Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature (2011) 477(7364):335–9. doi: 10.1038/nature10400
28. Hamon A, Piquet-Pellorce C, Dimanche-Boitrel MT, Samson M, Le Seyec J. Intrahepatocytic necroptosis is dispensable for hepatocyte death in murine immune-mediated hepatitis. J Hepatol (2020) 73(3):699–701. doi: 10.1016/j.jhep.2020.05.016
29. Preston SP, Stutz MD, Allison CC, Nachbur U, Gouil Q, Tran BM, et al. Epigenetic silencing of RIPK3 in hepatocytes prevents MLKL-mediated necroptosis from contributing to liver pathologies. Gastroenterology (2022) 163(6):1643–57.e14. doi: 10.1053/j.gastro.2022.08.040
30. Krishna-Subramanian S, Singer S, Armaka M, Banales JM, Holzer K, Schirmacher P, et al. RIPK1 and death receptor signaling drive biliary damage and early liver tumorigenesis in mice with chronic hepatobiliary injury. Cell Death Differ (2019) 26(12):2710–26. doi: 10.1038/s41418-019-0330-9
31. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42(1):145–58. doi: 10.1016/j.immuni.2014.12.020
32. Tisch N, Mogler C, Stojanovic A, Luck R, Korhonen EA, Ellerkmann A, et al. Caspase-8 in endothelial cells maintains gut homeostasis and prevents small bowel inflammation in mice. EMBO Mol Med (2022) 14(6):e14121. doi: 10.15252/emmm.202114121
33. Contreras CJ, Mukherjee N, Branco RCS, Lin L, Hogan MF, Cai EP, et al. RIPK1 and RIPK3 regulate TNFalpha-induced beta-cell death in concert with caspase activity. Mol Metab (2022) 65:101582. doi: 10.1016/j.molmet.2022.101582
34. Louhimo J, Steer ML, Perides G. Necroptosis is an important severity determinant and potential therapeutic target in experimental severe pancreatitis. Cell Mol Gastroenterol Hepatol (2016) 2(4):519–35. doi: 10.1016/j.jcmgh.2016.04.002
35. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with salmonella enterica serovar typhimurium. Nat Immunol (2012) 13(10):954–62. doi: 10.1038/ni.2397
36. D'Alessio S, Ungaro F, Noviello D, Lovisa S, Peyrin-Biroulet L, Danese S. Revisiting fibrosis in inflammatory bowel disease: the gut thickens. Nat Rev Gastroenterol Hepatol (2022) 19(3):169–84. doi: 10.1038/s41575-021-00543-0
37. Lee EY, Stenson WF, DeSchryver-Kecskemeti K. Thickening of muscularis mucosae in crohn's disease. Mod Pathol (1991) 4(1):87–90.
38. Huang W, Xie W, Gong J, Wang W, Cai S, Huang Q, et al. Heat stress induces RIP1/RIP3-dependent necroptosis through the MAPK, NF-kappaB, and c-jun signaling pathways in pulmonary vascular endothelial cells. Biochem Biophys Res Commun (2020) 528(1):206–12. doi: 10.1016/j.bbrc.2020.04.150
39. Li D, Xu T, Cao Y, Wang H, Li L, Chen S, et al. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA (2015) 112(16):5017–22. doi: 10.1073/pnas.1505244112
40. Yu X, Mao M, Liu X, Shen T, Li T, Yu H, et al. A cytosolic heat shock protein 90 and co-chaperone p23 complex activates RIPK3/MLKL during necroptosis of endothelial cells in acute respiratory distress syndrome. J Mol Med (Berl) (2020) 98(4):569–83. doi: 10.1007/s00109-020-01886-y
41. Zhang W, Fan W, Guo J, Wang X. Osmotic stress activates RIPK3/MLKL-mediated necroptosis by increasing cytosolic pH through a plasma membrane Na(+)/H(+) exchanger. Sci Signal (2022) 15(734):eabn5881. doi: 10.1126/scisignal.abn5881
42. Shindo R, Ohmuraya M, Komazawa-Sakon S, Miyake S, Deguchi Y, Yamazaki S, et al. Necroptosis of intestinal epithelial cells induces type 3 innate lymphoid cell-dependent lethal ileitis. iScience (2019) 15:536–51. doi: 10.1016/j.isci.2019.05.011
43. Lehle AS, Farin HF, Marquardt B, Michels BE, Magg T, Li Y, et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology (2019) 156(1):275–8. doi: 10.1053/j.gastro.2018.09.041
44. Yang ZH, Wu XN, He P, Wang X, Wu J, Ai T, et al. A non-canonical PDK1-RSK signal diminishes pro-caspase-8-Mediated necroptosis blockade. Mol Cell (2020) 80(2):296–310 e6. doi: 10.1016/j.molcel.2020.09.004
45. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
46. Meng Y, Horne CR, Samson AL, Dagley LF, Young SN, Sandow JJ, et al. Human RIPK3 c-lobe phosphorylation is essential for necroptotic signaling. Cell Death Dis (2022) 13(6):565. doi: 10.1038/s41419-022-05009-y
47. O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol (2011) 13(12):1437–42. doi: 10.1038/ncb2362
48. Roberts JZ, Crawford N, Longley DB. The role of ubiquitination in apoptosis and necroptosis. Cell Death Differ (2022) 29(2):272–84. doi: 10.1038/s41418-021-00922-9
49. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol (2015) 16(6):618–27. doi: 10.1038/ni.3172
50. Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat Commun (2020) 11(1):1747. doi: 10.1038/s41467-020-15466-8
51. Boutaffala L, Bertrand MJ, Remouchamps C, Seleznik G, Reisinger F, Janas M, et al. NIK promotes tissue destruction independently of the alternative NF-kappaB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ (2015) 22(12):2020–33. doi: 10.1038/cdd.2015.69
52. Dondelinger Y, Delanghe T, Rojas-Rivera D, Priem D, Delvaeye T, Bruggeman I, et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol (2017) 19(10):1237–47. doi: 10.1038/ncb3608
53. Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun (2019) 10(1):1729. doi: 10.1038/s41467-019-09690-0
54. Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell (2017) 66(5):698–710.e5. doi: 10.1016/j.molcel.2017.05.003
55. Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol (2018) 20(12):1389–99. doi: 10.1038/s41556-018-0229-6
56. Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell (2018) 174(6):1477–91 e19. doi: 10.1016/j.cell.2018.07.041
57. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun (2017) 8(1):359. doi: 10.1038/s41467-017-00406-w
58. Tu H, Xiong W, Zhang J, Zhao X, Lin X. Tyrosine phosphorylation regulates RIPK1 activity to limit cell death and inflammation. Nat Commun (2022) 13(1):6603. doi: 10.1038/s41467-022-34080-4
59. Patankar JV, Muller TM, Kantham S, Acera MG, Mascia F, Scheibe K, et al. E-type prostanoid receptor 4 drives resolution of intestinal inflammation by blocking epithelial necroptosis. Nat Cell Biol (2021) 23(7):796–807. doi: 10.1038/s41556-021-00708-8
60. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
61. Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem (2013) 288(23):16247–61. doi: 10.1074/jbc.M112.435545
62. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell (2012) 150(2):339–50. doi: 10.1016/j.cell.2012.06.019
63. Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, et al. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell (2018) 173(5):1244–53.e10. doi: 10.1016/j.cell.2018.03.032
64. Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ (2014) 21(10):1511–21. doi: 10.1038/cdd.2014.76
65. Frank D, Garnish SE, Sandow JJ, Weir A, Liu L, Clayer E, et al. Ubiquitylation of RIPK3 beyond-the-RHIM can limit RIPK3 activity and cell death. iScience (2022) 25(7):104632. doi: 10.1016/j.isci.2022.104632
66. Xie Y, Zhao Y, Shi L, Li W, Chen K, Li M, et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J Clin Invest (2020) 130(4):2111–28. doi: 10.1172/JCI133264
67. Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y, et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci USA (2017) 114(36):E7450–E9. doi: 10.1073/pnas.1707531114
68. Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun (2018) 9(1):2422. doi: 10.1038/s41467-018-04714-7
69. Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell (2019) 75(3):457–68.e4. doi: 10.1016/j.molcel.2019.05.022
70. Lu J, Krepelova A, Rasa SMM, Annunziata F, Husak O, Adam L, et al. Characterization of an in vitro 3D intestinal organoid model by using massive RNAseq-based transcriptome profiling. Sci Rep (2021) 11(1):16668. doi: 10.1038/s41598-021-96321-8
71. Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, et al. A single-cell survey of the small intestinal epithelium. Nature (2017) 551(7680):333–9. doi: 10.1038/nature24489
72. Nault R, Saha S, Bhattacharya S, Dodson J, Sinha S, Maiti T, et al. Benchmarking of a Bayesian single cell RNAseq differential gene expression test for dose-response study designs. Nucleic Acids Res (2022) 50(8):e48. doi: 10.1093/nar/gkac019
73. Nault R, Fader KA, Bhattacharya S, Zacharewski TR. Single-nuclei RNA sequencing assessment of the hepatic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cell Mol Gastroenterol Hepatol (2021) 11(1):147–59. doi: 10.1016/j.jcmgh.2020.07.012
74. Harnik Y, Buchauer L, Ben-Moshe S, Averbukh I, Levin Y, Savidor A, et al. Spatial discordances between mRNAs and proteins in the intestinal epithelium. Nat Metab (2021) 3(12):1680–93. doi: 10.1038/s42255-021-00504-6
75. Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature (2007) 446(7135):557–61. doi: 10.1038/nature05698
76. Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature (2014) 513(7516):90–4. doi: 10.1038/nature13608
77. Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature (2014) 513(7516):95–9. doi: 10.1038/nature13706
78. Vlantis K, Wullaert A, Polykratis A, Kondylis V, Dannappel M, Schwarzer R, et al. NEMO prevents RIP kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-kappaB-Dependent and -independent functions. Immunity (2016) 44(3):553–67. doi: 10.1016/j.immuni.2016.02.020
79. Kattah MG, Shao L, Rosli YY, Shimizu H, Whang MI, Advincula R, et al. A20 and ABIN-1 synergistically preserve intestinal epithelial cell survival. J Exp Med (2018) 215(7):1839–52. doi: 10.1084/jem.20180198
80. Zhou M, He J, Shi Y, Liu X, Luo S, Cheng C, et al. ABIN3 negatively regulates necroptosis-induced intestinal inflammation through recruiting A20 and restricting the ubiquitination of RIPK3 in inflammatory bowel disease. J Crohns Colitis (2021) 15(1):99–114. doi: 10.1093/ecco-jcc/jjaa131
81. Wang R, Li H, Wu J, Cai ZY, Li B, Ni H, et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature (2020) 580(7803):386–90. doi: 10.1038/s41586-020-2127-x
82. Wen S, Ling Y, Yang W, Shen J, Li C, Deng W, et al. Necroptosis is a key mediator of enterocytes loss in intestinal ischaemia/reperfusion injury. J Cell Mol Med (2017) 21(3):432–43. doi: 10.1111/jcmm.12987
83. Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, et al. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis (2020) 11(12):1050. doi: 10.1038/s41419-020-03239-6
84. Kennelly JP, Carlin S, Ju T, van der Veen JN, Nelson RC, Buteau J, et al. Intestinal phospholipid disequilibrium initiates an ER stress response that drives goblet cell necroptosis and spontaneous colitis in mice. Cell Mol Gastroenterol Hepatol (2021) 11(4):999–1021. doi: 10.1016/j.jcmgh.2020.11.006
85. Frank D, Vaux DL, Murphy JM, Vince JE, Lindqvist LM. Activated MLKL attenuates autophagy following its translocation to intracellular membranes. J Cell Sci (2019) 132(5). doi: 10.1242/jcs.220996
86. Pradhan AJ, Lu D, Parisi LR, Shen S, Berhane IA, Galster SL, et al. Protein acylation by saturated very long chain fatty acids and endocytosis are involved in necroptosis. Cell Chem Biol (2021) 28(9):1298–309.e7. doi: 10.1016/j.chembiol.2021.03.012
87. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med (2017) 214(12):3687–705. doi: 10.1084/jem.20170558
88. Liu Y, Xu Q, Wang Y, Liang T, Li X, Wang D, et al. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death Dis (2021) 12(1):62. doi: 10.1038/s41419-020-03365-1
89. Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ (2013) 20(2):185–7. doi: 10.1038/cdd.2012.151
90. Zhang J, Qin D, Yang YJ, Hu GQ, Qin XX, Du CT, et al. MLKL deficiency inhibits DSS-induced colitis independent of intestinal microbiota. Mol Immunol (2019) 107:132–41. doi: 10.1016/j.molimm.2019.01.018
91. Xu J, Li S, Jin W, Zhou H, Zhong T, Cheng X, et al. Epithelial Gab1 calibrates RIPK3-dependent necroptosis to prevent intestinal inflammation. JCI Insight (2023) 8(6). doi: 10.1172/jci.insight.162701
92. Alvarez-Diaz S, Preaudet A, Samson AL, Nguyen PM, Fung KY, Garnham AL, et al. Necroptosis is dispensable for the development of inflammation-associated or sporadic colon cancer in mice. Cell Death Differ (2021) 28(5):1466–76. doi: 10.1038/s41418-020-00673-z
93. Dong W, Zhang M, Zhu Y, Chen Y, Zhao X, Li R, et al. Protective effect of NSA on intestinal epithelial cells in a necroptosis model. Oncotarget (2017) 8(49):86726–35. doi: 10.18632/oncotarget.21418
94. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin d inhibits inflammatory cell death and sepsis. Sci Immunol (2018) 3(26). doi: 10.1126/sciimmunol.aat2738
95. Rashidi M, Simpson DS, Hempel A, Frank D, Petrie E, Vince A, et al. The pyroptotic cell death effector gasdermin d is activated by gout-associated uric acid crystals but is dispensable for cell death and IL-1beta release. J Immunol (2019) 203(3):736–48. doi: 10.4049/jimmunol.1900228
96. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell (2009) 137(6):1112–23. doi: 10.1016/j.cell.2009.05.037
97. Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe (2010) 7(4):302–13. doi: 10.1016/j.chom.2010.03.006
98. Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology (2013) 58(6):2099–108. doi: 10.1002/hep.26547
99. Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H, et al. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett (2014) 225(3):445–53. doi: 10.1016/j.toxlet.2014.01.005
100. Koschel J, Nishanth G, Just S, Harit K, Kroger A, Deckert M, et al. OTUB1 prevents lethal hepatocyte necroptosis through stabilization of c-IAP1 during murine liver inflammation. Cell Death Differ (2021) 28(7):2257–75. doi: 10.1038/s41418-021-00752-9
101. Shi S, Bonaccorsi-Riani E, Schurink I, van den Bosch T, Doukas M, Lila KA, et al. Liver ischemia and reperfusion induce periportal expression of necroptosis executor pMLKL which is associated with early allograft dysfunction after transplantation. Front Immunol (2022) 13:890353. doi: 10.3389/fimmu.2022.890353
102. Rosentreter D, Funken D, Reifart J, Mende K, Rentsch M, Khandoga A. RIP1-dependent programmed necrosis is negatively regulated by caspases during hepatic ischemia-reperfusion. Shock (2015) 44(1):72–6. doi: 10.1097/SHK.0000000000000371
103. Zhong W, Wang X, Rao Z, Pan X, Sun Y, Jiang T, et al. Aging aggravated liver ischemia and reperfusion injury by promoting hepatocyte necroptosis in an endoplasmic reticulum stress-dependent manner. Ann Transl Med (2020) 8(14):869. doi: 10.21037/atm-20-2822
104. Mohammed S, Thadathil N, Selvarani R, Nicklas EH, Wang D, Miller BF, et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell (2021) 20(12):e13512. doi: 10.1111/acel.13512
105. Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology (2013) 57(5):1773–83. doi: 10.1002/hep.26200
106. Bao C, Shao Y, Li X. Hepatocyte necroptosis induced by ischemic acute kidney injury in rats. Ultrastruct Pathol (2014) 38(3):217–23. doi: 10.3109/01913123.2014.895788
107. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med (2014) 6(8):1062–74. doi: 10.15252/emmm.201403856
108. Zhang B, Li MD, Yin R, Liu Y, Yang Y, Mitchell-Richards KA, et al. O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight (2019) 4(21). doi: 10.1172/jci.insight.127709
109. Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov (2016) 2:16089. doi: 10.1038/cddiscovery.2016.89
110. Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis (2015) 6(5):e1759. doi: 10.1038/cddis.2015.126
111. Weinlich R, Oberst A, Dillon CP, Janke LJ, Milasta S, Lukens JR, et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep (2013) 5(2):340–8. doi: 10.1016/j.celrep.2013.08.045
112. Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest (2016) 126(11):4346–60. doi: 10.1172/JCI87545
113. Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun (2015) 6:8371. doi: 10.1038/ncomms9371
114. Suda J, Dara L, Yang L, Aghajan M, Song Y, Kaplowitz N, et al. Knockdown of RIPK1 markedly exacerbates murine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NF-kappaB. J Immunol (2016) 197(8):3120–9. doi: 10.4049/jimmunol.1600690
115. Zhao Y, Scott NA, Fynch S, Elkerbout L, Wong WW, Mason KD, et al. Autoreactive T cells induce necrosis and not BCL-2-regulated or death receptor-mediated apoptosis or RIPK3-dependent necroptosis of transplanted islets in a mouse model of type 1 diabetes. Diabetologia (2015) 58(1):140–8. doi: 10.1007/s00125-014-3407-5
116. Hildebrand JM, Lo B, Tomei S, Mattei V, Young SN, Fitzgibbon C, et al. A family harboring an MLKL loss of function variant implicates impaired necroptosis in diabetes. Cell Death Dis (2021) 12(4):345. doi: 10.1038/s41419-021-03636-5
117. Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol (2015) 70:5.47.1–5.20. doi: 10.1002/0471141755.ph0547s70
118. Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc (2021) 1(4):e78. doi: 10.1002/cpz1.78
119. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell (2009) 137(6):1100–11. doi: 10.1016/j.cell.2009.05.021
120. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (2009) 325(5938):332–6. doi: 10.1126/science.1172308
121. Zhao Q, Yu X, Zhang H, Liu Y, Zhang X, Wu X, et al. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in fadd-deficient mice. Cell Rep (2017) 19(4):798–808. doi: 10.1016/j.celrep.2017.04.011
122. Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ (2016) 23(9):1565–76. doi: 10.1038/cdd.2016.46
123. Boonchan M, Arimochi H, Otsuka K, Kobayashi T, Uehara H, Jaroonwitchawan T, et al. Necroptosis protects against exacerbation of acute pancreatitis. Cell Death Dis (2021) 12(6):601. doi: 10.1038/s41419-021-03847-w
124. Liang QQ, Shi ZJ, Yuan T, Chen SY, Li YP, Zhang HR, et al. Celastrol inhibits necroptosis by attenuating the RIPK1/RIPK3/MLKL pathway and confers protection against acute pancreatitis in mice. Int Immunopharmacol (2023) 117:109974. doi: 10.1016/j.intimp.2023.109974
125. Shi FL, Yuan LS, Wong TS, Li Q, Li YP, Xu R, et al. Dimethyl fumarate inhibits necroptosis and alleviates systemic inflammatory response syndrome by blocking the RIPK1-RIPK3-MLKL axis. Pharmacol Res (2023) 189:106697. doi: 10.1016/j.phrs.2023.106697
126. Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol (2011) 29(11):1046–51. doi: 10.1038/nbt.1990
127. Wang Q, Zorn JA, Kuriyan J. A structural atlas of kinases inhibited by clinically approved drugs. Methods Enzymol (2014) 548:23–67. doi: 10.1016/B978-0-12-397918-6.00002-1
128. Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, et al. Discovery of a first-in-Class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem (2017) 60(4):1247–61. doi: 10.1021/acs.jmedchem.6b01751
129. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol (2008) 4(5):313–21. doi: 10.1038/nchembio.83
130. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol (2013) 14(11):727–36. doi: 10.1038/nrm3683
131. Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci (2020) 41(3):209–24. doi: 10.1016/j.tips.2020.01.002
132. Weisel K, Scott N, Berger S, Wang S, Brown K, Powell M, et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol (2021) 8(1). doi: 10.1136/bmjgast-2021-000680
133. Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:238. doi: 10.3389/fcimb.2020.00238
134. Xu G, Li Y, Zhang S, Peng H, Wang Y, Li D, et al. SARS-CoV-2 promotes RIPK1 activation to facilitate viral propagation. Cell Res (2021) 31(12):1230–43. doi: 10.1038/s41422-021-00578-7
135. Li S, Zhang Y, Guan Z, Ye M, Li H, You M, et al. SARS-CoV-2 z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res (2023) 33(3):201–14. doi: 10.1038/s41422-022-00775-y
136. Raju S, Whalen DM, Mengistu M, Swanson C, Quinn JG, Taylor SS, et al. Kinase domain dimerization drives RIPK3-dependent necroptosis. Sci Signal (2018) 11(544). doi: 10.1126/scisignal.aar2188
137. Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, et al. The b-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis (2014) 5(6):e1278. doi: 10.1038/cddis.2014.241
138. Rheault TR, Stellwagen JC, Adjabeng GM, Hornberger KR, Petrov KG, Waterson AG, et al. Discovery of dabrafenib: a selective inhibitor of raf kinases with antitumor activity against b-Raf-Driven tumors. ACS Med Chem Lett (2013) 4(3):358–62. doi: 10.1021/ml4000063
139. Martens S, Jeong M, Tonnus W, Feldmann F, Hofmans S, Goossens V, et al. Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis (2017) 8(6):e2904. doi: 10.1038/cddis.2017.298
140. Xia K, Zhu F, Yang C, Wu S, Lin Y, Ma H, et al. Discovery of a potent RIPK3 inhibitor for the amelioration of necroptosis-associated inflammatory injury. Front Cell Dev Biol (2020) 8:606119. doi: 10.3389/fcell.2020.606119
141. Li W, Ni H, Wu S, Han S, Chen C, Li L, et al. Targeting RIPK3 oligomerization blocks necroptosis without inducing apoptosis. FEBS Lett (2020) 594(14):2294–302. doi: 10.1002/1873-3468.13812
142. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of mlkl in necroptosis. Cell Res (2013) 23(8):994–1006. doi: 10.1038/cr.2013.91
143. Hildebrand JM, Kauppi M, Majewski IJ, Liu Z, Cox AJ, Miyake S, et al. A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat Commun (2020) 11(1):3150. doi: 10.1038/s41467-020-16819-z
144. Yang W, Tao K, Wang Y, Huang Y, Duan C, Wang T, et al. Necrosulfonamide ameliorates intestinal inflammation via inhibiting GSDMD-medicated pyroptosis and MLKL-mediated necroptosis. Biochem Pharmacol (2022) 206:115338. doi: 10.1016/j.bcp.2022.115338
145. He F, Zheng G, Hu J, Ge W, Ji X, Bradley JL, et al. Necrosulfonamide improves post-resuscitation myocardial dysfunction via inhibiting pyroptosis and necroptosis in a rat model of cardiac arrest. Eur J Pharmacol (2022) 926:175037. doi: 10.1016/j.ejphar.2022.175037
146. Zhang X, Zhang Y, Wang F, Liu Y, Yong VW, Xue M. Necrosulfonamide alleviates acute brain injury of intracerebral hemorrhage via inhibiting inflammation and necroptosis. Front Mol Neurosci (2022) 15:916249. doi: 10.3389/fnmol.2022.916249
147. Wu YL, Ou WJ, Zhong M, Lin S, Zhu YY. Gasdermin d inhibitor necrosulfonamide alleviates Lipopolysaccharide/D-galactosamine-induced acute liver failure in mice. J Clin Transl Hepatol (2022) 10(6):1148–54. doi: 10.14218/JCTH.2021.00560
148. Cui B, Yan B, Wang K, Li L, Chen S, Zhang Z. Discovery of a new class of uracil derivatives as potential mixed lineage kinase domain-like protein (MLKL) inhibitors. J Med Chem (2022) 65(19):12747–80. doi: 10.1021/acs.jmedchem.2c00548
149. Rubbelke M, Fiegen D, Bauer M, Binder F, Hamilton J, King J, et al. Locking mixed-lineage kinase domain-like protein in its auto-inhibited state prevents necroptosis. Proc Natl Acad Sci USA (2020) 117(52):33272–81. doi: 10.1073/pnas.2017406117
Keywords: necroptosis, inflammatory bowel disease, MLKL, caspase 8, RIPK3, therapeutic
Citation: Patankar JV, Bubeck M, Acera MG and Becker C (2023) Breaking bad: necroptosis in the pathogenesis of gastrointestinal diseases. Front. Immunol. 14:1203903. doi: 10.3389/fimmu.2023.1203903
Received: 11 April 2023; Accepted: 07 June 2023;
Published: 20 June 2023.
Edited by:
Liming Sun, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Andre Laval Samson, The University of Melbourne, AustraliaRksubbarao Malireddi, St. Jude Children’s Research Hospital, United States
Copyright © 2023 Patankar, Bubeck, Acera and Becker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Becker, Y2hyaXN0b3BoLmJlY2tlckB1ay1lcmxhbmdlbi5kZQ==