 Karthik Chandiran
Karthik Chandiran Linda S. Cauley
Linda S. Cauley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 June 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1199671
This article is part of the Research TopicTGF-β and T Cell BiologyView all 5 articles
Cytotoxic T lymphocytes (CTLs) play an important role in defense against infections with intracellular pathogens and anti-tumor immunity. Efficient migration is required to locate and destroy infected cells in different regions of the body. CTLs accomplish this task by differentiating into specialized subsets of effector and memory CD8 T cells that traffic to different tissues. Transforming growth factor-beta (TGFβ) belongs to a large family of growth factors that elicit diverse cellular responses via canonical and non-canonical signaling pathways. Canonical SMAD-dependent signaling pathways are required to coordinate changes in homing receptor expression as CTLs traffic between different tissues. In this review, we discuss the various ways that TGFβ and SMAD-dependent signaling pathways shape the cellular immune response and transcriptional programming of newly activated CTLs. As protective immunity requires access to the circulation, emphasis is placed on cellular processes that are required for cell-migration through the vasculature.
Transforming growth factor beta (TGFβ) is the signature cytokine for a large family of growth factors. The three isoforms of TGFβ (TGFβ − 1, 2 & 3) are encoded by separate genes and produce structurally similar cytokines (1). These cytokines use autocrine and paracrine signaling pathways to control a diverse range of biological processes that support immune cell function and tissue repair (2). TGFβ is secreted as an inactive complex with the latency associated peptide (LAP), that binds to the extracellular matrix (ECM) or a receptor (glycoprotein A repetitions predominant, GARP) that is expressed on regulatory T cells (Tregs) and platelets (3). Biologically active TGFβ is released from reservoirs in peripheral and lymphoid tissues when proteases or adhesion molecules induce steric changes in the conformation of LAP (4, 5). The active cytokine is a charged peptide with a short half-life that limits biological activity to the local tissues (1). TGFβ interacts with a bivalent transmembrane receptor with serine/threonine kinase activity, that elicits cellular response via a network of interconnected signaling pathways (6). Canonical signals are mediated by a cascade of structurally-related signaling intermediates known as SMAD proteins (7). SMAD4 is an adaptor for multiple pathways within the SMAD cascade (8). Multiple SMAD-dependent signaling pathways coordinate changes in homing receptor expression as cytotoxic T lymphocytes (CTLs) respond to infection (9–11). The various ways that TGFβ impacts the CTL response depends on the timing and context of cytokine exposure. In this review, we examine the influence of TGFβ at different ages during the CTLs response (Figure 1), starting with naïve CD8 T cells, followed by effector CD8 T cells (TEFF) and specialized subsets of memory CD8 T cells that are adapted to circulate in the blood, or enter peripheral tissues.
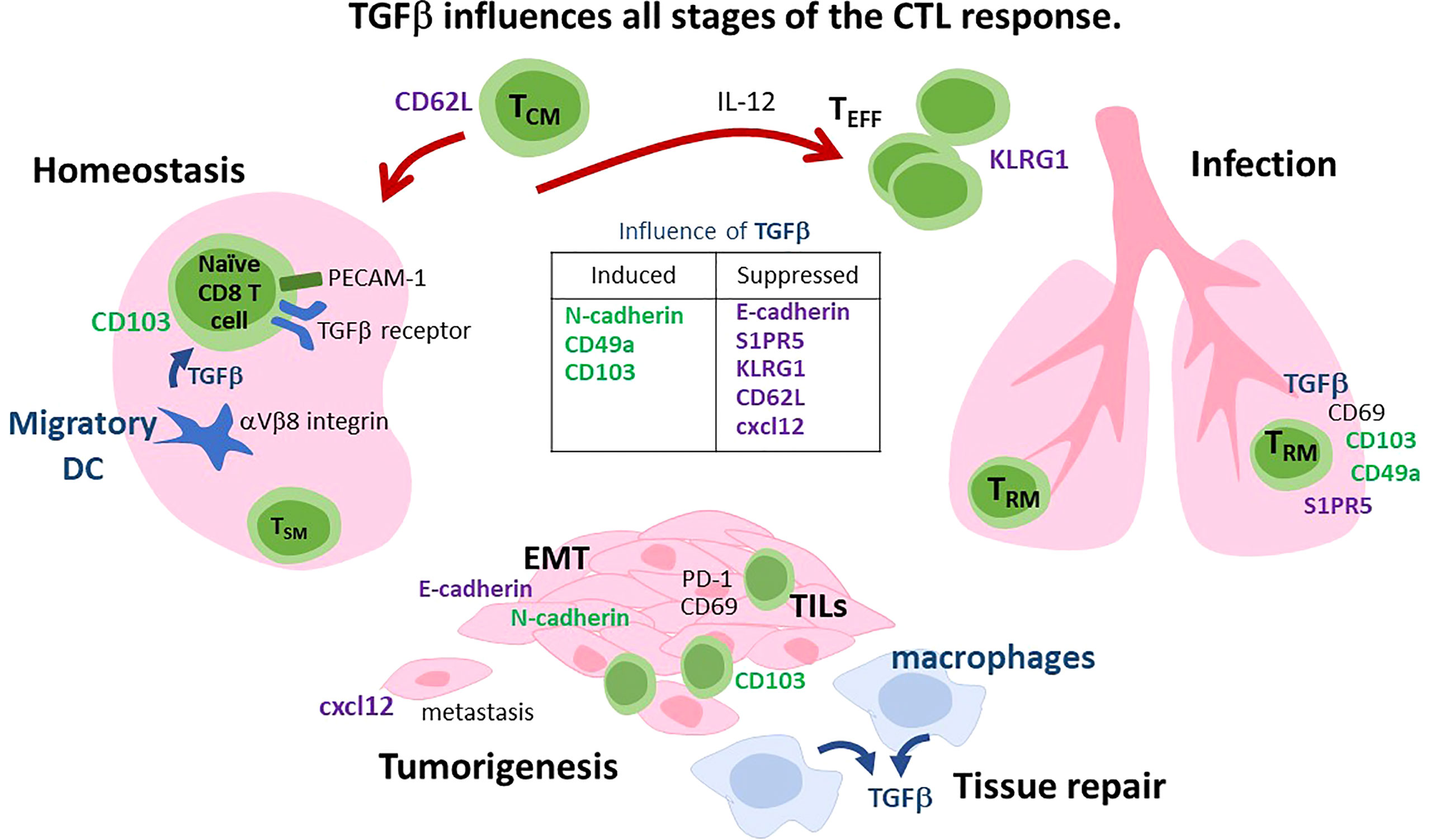
Figure 1 Diverse functions for TGFβ during the CTL response. i) Homeostasis: PECAM-1 is expressed on naïve CD8 T cells and interacts with the TGFβ receptor to inhibit autoimmunity. Some naïve CD8 T cells are preconditioned to become TRM cells by migratory DC that express αVβ8 integrin and activate TGFβ. CD62L is lymphoid homing receptor that can be downregulated by TGFβ. ii) Infection. IL-12 overrides the suppress effects of TGFβ to enhance TEFF and TCM formation. TRM cells express adhesion molecules (CD103 and CD49a) that are induced by TGFβ, while S1PR5 is downregulated. Signaling via CD69 increases TGFβ production in the spleen. iii) Tumorigenesis and tissue repair. TGFβ is an angiogenic factor that promotes tumor growth and tissue repair. During EMT, TGFβ induces a ‘cadherin-switch’ by downregulating E-cadherin and inducing N-cadherin expression on mural cells. Macrophages are an important source of TGFβ during tissue repair. Specialized subsets of CTLs express cadherin-binding proteins (KLRG1 and CD103) and reside at barrier surfaces. CD103+ TRM cells mobilize from draining lymph nodes to the tumor when TGFβ signaling is disrupted. CTLs are excluded from tumors when CXC12 is expressed. Cancer cells undergo EMT in the presence of TGFβ. CXCL12 can be downregulated by TGFβ.
Secondary lymphoid organs (SLO) are surveyed by quiescent populations of naïve CD8 T cells searching for early signs of infection. Although homogeneous surface markers indicate limited functional diversity, transcriptome analysis reveals extensive genetic variability as clonal populations of naïve CD8 T cells respond to cognate antigen. Studies show that environmental stimuli play an important role in transcriptional programming of naïve CD8 T cells during transit around the body. Transient interactions with Major histocompatibility complex class I molecules (MHCI) extend the lifespan of naïve T cells through tonic activation of the T cell receptor (TCR). The circulating cells encounter different assortments of peptides according to the tissue of origin and mechanisms that are used for antigen processing. The impact of TCR stimulation varies according to the affinity of interactions with self-peptides as well as availability of costimulatory signals and exposure to local inflammatory conditions (12, 13). In lymphoid tissues, naïve CD8 T cells are preconditioned to become tissue resident memory CD8 T cells (TRM) during interactions with migratory dendritic cells (DCs) that activate TGFβ (14).
Fate-mapping techniques underscore the importance of environmental stimuli during CD8 T cell differentiation. A study of naive CD8 T cells isolated from different tissues showed variable capacities to proliferate and differentiate into specialized subsets of memory CD8 T cells (15). Similarly, naive CD8+ T cells that were isolated at different times after birth showed that early microbial exposure has long term effects during the CTL response (15, 16). CTLs that were derived from newborn mice rapidly differentiated into TEFF cells, whereas CTLs from adult animals were more inclined to become memory CD8 T cells under similar conditions (15, 16). The effects of chronic exposure to inflammation also contribute to age-related changes in protective immunity (17). Characteristics of naïve CD8 T cells in elderly people include diminished capacity for TEFF and memory formation (18, 19). Several mechanisms contribute to suboptimal CTL responses during ageing, including immune cell senescence and increased production of TGFβ (20, 21). Responses to TGFβ vary at different stages during the CTL response, as revealed by changes in cell proliferation, survival and memory formation (22–24).
CD8 T cells express a variety of inhibitory receptors (IR) that help maintain diverse repertoires of functional CTLs under homeostatic conditions. Platelet endothelial cell adhesion molecule-1 (PECAM-1 or CD31) is an adhesion molecule with an immunoreceptor tyrosine-based inhibitory (ITIM) motif in the cytoplasmic tail (25) and forms homophilic interactions between endothelial cells to maintain barrier integrity. PECAM-1 is also expressed on cells in the vascular compartment, including naïve CD8 T cells and cooperates with the TGFβ receptor to inhibit autoimmunity during stimulation with self-antigens (24, 26) by enhancing non-canonical (SMAD-independent) signaling (27). PECAM-1 is transcriptionally downregulated during formation of short-lived TEFF cells (SLECs), whereas expression is maintained in memory precursors (28). In the absence of PECAM-1, TEFF cells become resistant to the suppressive effects of TGFβ during production of IFN-γ, and granzyme B, as well as cell-proliferation. Naïve CD8 T cells express other receptors that inhibit responses to self-peptides, including V-domain Ig suppressor of T cell activation (VISTA) (29). A study found that exogenous TGFβ decreased VISTA expression on human CD8 T cells in vitro (30). P-selectin glycoprotein-1 (PSGL-1) is another adhesion molecule that is expressed on endothelial cells and interacts with VISTA to inhibit TCR activation and IL-2 production (31, 32). During LCMV infection, increased numbers of CD4 T cells expressed PSGL-1 in the absence of the TGFβ receptor (33), although the study did not reveal whether a similar change occurred in CD8 T cells.
Blood is transported around the body through an elaborate network of interconnected vessels. Extracellular fluid drains from peripheral tissues into blind-ended lymphatic vessels that join the blood supply at the thoracic duct (34). The vessels are lined with a tightly connected layer of endothelial cells that provide a physical barrier between the circulation and surrounding tissues (35). Cutaneous and mucosal surfaces are covered with similar layers of stromal cells that provide a barrier against infection (36). Contacts between adjacent stromal cells are mediated by protein complexes called tight junctions (TJs) and adherens junctions (AJ). AJs are mediated by calcium-dependent adhesion molecules, known as cadherins (35, 37). Migrating T cells use specialized homing receptors to penetrate the stromal layer and enter the vascular system during immune surveillance.
During infection, barrier integrity can be comprised by high concentrations of proinflammatory cytokines that disrupt connections between adjacent stromal cells (38). In the respiratory tract, damaged bronchial epithelial cells are released from the basement membrane and expelled from the lungs by the mucociliary escalator (39). As inflammation resolves, macrophages produce large quantities of TGFβ to facilitate tissue repair (40, 41). TGFβ endows epithelial cells with migratory properties by triggering epithelial-mesenchymal transition (EMT) (42). A ‘cadherin-switch’ occurs as TGFβ downregulates epithelial (E)-cadherin (43–45) and induces neural (N)-cadherin expression (40). During angiogenesis, TGFβ converts endothelial cells to mural cells via a similar process (35, 46, 47). AJs between endothelial cells are mediated by vascular endothelial (VE)-cadherin, whereas N-cadherin is diffusely expressed over the surface of endothelial and mural cells including pericytes (48, 49). As CTLs mobilize to inflamed tissues, PECAM-1+ TEFF cells disrupt AJs by dephosphorylating VE-cadherin to transit across the blood vessel wall (50). Pericytes that reside between endothelial cells and the basement membrane around capillaries play a central role in angiogenesis (51).
During priming, naïve CD8 T cells participate in serial interactions with DCs (52) that provide costimulatory signals to augment TEFF functions and memory formation (53). Initial interactions with DCs occur in the interfollicular and cortical regions of the draining lymph node (54). After a few rounds of cell-division, activated CTLs join clusters of DCs that express chemokine receptor XCR1 and CD8 (53, 55). TGFβ that is activated by migratory DCs that express αV integrin conditions naive CD8 T cells to become TRM cells (14). After proliferation, activated CTLs enter the circulation and distribute around the body. The properties of migrating T cells can be modified through interactions with endothelial cells that express costimulatory molecules (CD40, ICOSL, 4-1BB, and OX40L), or ligands of IRs such as PD-L1 and PD-L2 (56). Antigen-presentation on endothelial cells promotes extravasation of CTLs from the blood vessels to adjacent tissues.
Cytokines have important regulatory functions during the CTL response (57). During the acute phase of infection, interleukin IL-12 and other inflammatory molecules initiate a transcriptional program that leads to TEFF formation (58). T cell factor 1 (TCF1) is a transcription factor that programs stem-like memory (TSM) cells for multipotency and self-renewal (59) and enables partially exhausted CTLs to maintain some TEFF functions during chronic infection (60). During the TEFF response, IL-12 downregulates TCF1 (61), as Tbet and LFA-1 are induced (62, 63). Most short-lived TEFF cells undergo apoptotic cell death as the infection comes under control, leaving heterogeneous populations of memory CD8 T cells in the circulation and infected tissues. TGFβ has opposing effects on T cell survival at specific stages during the CTL response (24, 64). As inflammation subsides, the IL-12 receptor is down-regulated by TGFβ as TRM cells settle in peripheral tissues (65, 66). T-bet prolongs survival of TRM cells in the skin by upregulating the receptor for IL-15 (67).
TEFF cells produce large quantities of pathogenic cytokines (IFNγ and TNFα) and lytic molecules that contribute to organ damage during severe infections (68–70). Local concentrations of inflammatory molecules must be tightly controlled to restrict immune pathology in the lungs. Killer cell lectin-like receptor G1 (KLRG1) is a membrane-bound adhesion molecule that is expressed some subsets of NK cells, Tregs and CTLs (71). Inflammatory mediators that are released during microbial infection are responsible for massive up-regulation of KLRG1 on CD8+ TEFF cells (72). Although large numbers of newly activated CTLs transiently express KLRG1 during antigen-stimulation (73), stable expression is a feature of terminally-differentiated CTLs that do not convert to a memory phenotype (74). Several studies found long lived KLRG1+ CTLs in the vasculature and spleens after infections with different pathogens (75–77). After transfer, these CTLs provided robust protection against infection with Listeria monocytogenes (LM) through a robust lytic response and IFNγ production (76). Prolonged survival of KLRG1+ CTLs after LCMV infection required Treg-derived IL-15 (78).
PSGL-1 and KLRG1 are IRs with ITIM sequences in the cytoplasmic domain (79). KLRG1 interacts with two members of the cadherin family (E- and N-cadherin) that are expressed on different types of stromal cells and differentially regulated by TGFβ (79, 80). While the function of KLRG1 is poorly understood, crosslinking with anti-KLRG1 antibodies impaired TCR-mediated Ca2+ mobilization and cytolysis in CTLs that were engineered to express KLRG1 at high levels (81). The same study found that cross-linking antibodies also decreased IL-2 production from cell lines that over expressed KLRG1.
Infection with LM elicits a robust inflammatory that favors formation of TEFF that express KLRG1. A study found that the frequency of these terminally differentiated CTLs increased when the TGFβ receptor was not expressed (82). Although KLRG1+ CTLs can produce large quantities of lytic molecules and proinflammatory cytokines, intranasal infection with LM-OVA does not cause lethal pathology in the lungs, irrespective of whether the TGFβ receptor is present (9). It possible that inhibitory signals that are mediated via KLRG1 protect cadherin-positive endothelial cells from cytolysis and thus minimize damage to capillaries during the TEFF response. A specific mechanism that is responsible for retaining KLRG1+ CTLs in the vasculature has been not identified, however these CTLs express chemokine receptor CX3CR1 at high levels which may influence tissue localization (9, 83). Potential functions of blood resident CTLs during protective immunity include recruitment of circulating immune cells to inflamed tissues by altering vascular permeability.
Multiple subsets of memory CD8 T cells use the bloodstream to circulate around the body. Central memory CD8 T cells (TCM) express lymphoid homing receptors [CD62L & chemokine receptor 7 (CCR7)] and enter SLO, whereas effector memory CD8 T cells (TEM) cells express molecules that aid localization to inflamed tissues. Although TEM cells are excluded from resting peripheral lymph nodes (pLN), some cells enter reactive pLN during the resolution of the immune response and help restore homeostasis by destroying antigen-bearing APCs (84). As inflammation declines TGFβ counteracts the effects of IL-12 (82) to promote development of TCM and non-circulating TRM cells (66, 85, 86). Under resting conditions, constant signaling via the TGFβ receptor shapes the memory compartment (87) and limits homeostatic proliferation by increasing sensitivity to IL-7 and IL-15 (88). The circulating pool of memory cells also includes some stem-like memory CD8 T cells (TSM) that express TCF1 and differentiate into mixed populations of TCM and TEM cells after antigen stimulation (59, 89). TCF1 inhibits TRM formation by inhibiting TGFβ-induced CD103 expression (85). During chronic infection, TCF1 induces a transcriptional program that preserves TEFF functions of exhausted CTLs by upregulating FOXO1, ZEB2, Id3, and Eomesodermin (EOMES) (60). TGFβ enforces the stem-like properties of PD-1+ TSM cells in lymphoid tissues by inhibiting TEFF formation (90, 91).
TCM cells follow similar migratory patterns to naïve CD8 T cells and enter pLN from wide blood vessels known as high endothelial venules (HEVs) (34). Selectins are vascular adhesion molecules that aid leukocyte migration to peripheral tissues (92). During transit through HEVs, circulating T cells that express (L)-selectin (CD62L) adhere to peripheral lymph node addressin (PNAd) expressed on cuboidal endothelial cells (34). After T cells are tethered to the blood vessel wall, ligands of CCR7 trigger an adhesion cascade that results in trans-endothelial migration (93, 94). PSGL-1 interacts with selectins expressed on activated endothelial cells and initiates a similar adhesion cascade as TEFF cells extravasate to infected tissues (95, 96). TCM cells (but not naive T cells) are also able to access pLN via a CCR7-independent mechanism that involves CXCL12/CXCR4 (97). Although CD62L is associated with immune surveillance in SLO, this molecule also facilitates recruitment of CTLs to inflamed tissues (98). Endothelial cells that line HEVs express a leucine-rich HEV glycoprotein (LRHG) that binds to latent TGFβ (99). Downregulation of CD62L by TGFβ (9) may limit recruitment of circulating T cells to reactive pLN and inflamed tissues during tissue repair.
Chemokine receptors are dynamically regulated during the CTL response and aid localization in different tissues. Several receptors are regulated by TGFβ including CXCR3 which is expressed on TEFF cells during mobilization to inflamed tissues (100). A study showed that TGFβ reduced CXCR3 expression on TEFF cells and prevented tumor infiltration via a SMAD2-dependent pathway (101). The fractalkine receptor (CX3CR1) is also absent from naïve CD8 T cells and induced during the TEFF response. Whereas TCM, TEM and TRM cells all lack CX3CR1 (102), expression is maintained on long lived KLRG1+ TEFF cells in the vasculature (9). Additional memory CD8 T cells that express CX3CR1 at intermediate levels, exhibit features of both TCM and TEM cells and are primarily responsible for immune surveillance in non-lymphoid tissues (102, 103).
Cutaneous and mucosal surfaces exposed to a diverse array of pathogens that propagate in human cells. Barrier tissues are densely populated with TRM cells that leave the circulation and become lodged in the local tissues where they are poised for a rapid response upon infection (104). TRM cells reside in virtually all tissues in the human body (105) and augment immunity by releasing inflammatory molecules that attract circulating immune cells to the site of infection (106). These cells have limited capacity for self-renewal and proliferation after antigen stimulation and can be divided into subsets based on variations in surface receptor expression. The canonical markers of TRM cells include two members of the integrin family that direct localization in peripheral tissues. α1β1 integrin (CD49a) is induced by stimulation with either IL-12 or TGFβ, and aids localization in tissues that contain collagen (107, 108). Conversely, αEβ7 integrin (CD103) is induced by TGFβ and negatively regulated by IL-12 during the TEFF response (66). Interactions between CD103 and E-cadherin enhance retention of TRM cells in tissues with an epithelial layer and stabilize interactions with target cells during cytolysis (109). Although CD103 is expressed on a majority of TRM cells at barrier surfaces and in glandular tissues, it is not a universal marker of TRM cells in all tissues (110). In the liver, TRM cells develop independently of TGFβ and lack CD103 expression (110). TGFβ upregulates CD103 expression on naïve CD8 T cells, but not TCM cells (111). After reactivation, TCM cells give rise to CD69+ TRM cells that do not express CD103 in the presence of TGFβ (111, 112).
Sphingosine-1-phosphate (S1P) is a bioactive lipid that is present in blood and lymph and attracts migrating leukocytes into the circulation. Different S1P receptors (S1PR) are required for CTLs to leave peripheral and lymphoid tissues and are downregulated on TRM cells to prevent entry into the circulation (113, 114). CD69 is a C-type lectin that is transiently induced during antigen stimulation and prevents newly activated CTLs from leaving SLO during clonal expansion, by modulating S1PR1 from the cell surface (115, 116). Lymphoid TRM cells that express CD69 remain in reactive pLN while S1PR1 is down regulated (114). S1PR5 is required for CTLs to leave peripheral tissues and is downregulated by TGFβ during TRM formation (114). An arthritis model showed that signaling via CD69 reduced immune reactivity by stimulating TGFβ production from mouse splenocytes and transformed CD8 T cells (117). Large numbers of TRM cells in peripheral tissues maintain CD69 expression in the absence of antigen stimulation. While it is unclear how this phenotype is enforced, CTLs acquired properties of TRM cells and upregulated CD69 in vitro during interactions with activated endothelial cells (118). CD69 is not required for TRM development in some tissues (119).
During tissue remodeling, TGFβ induces a ‘cadherin switch’ by altering E- and N-cadherin expression (40, 42). KLRG1 and CD103 are cadherin-binding proteins that are transcriptionally regulated by TGFβ (9). Whereas KLRG1 is down-regulated by TGFβ (9, 82) CD103 is induced (120) and opposing signaling mechanisms prevent dual expression on the same CTLs (9). When a retroviral vector was used to force KLRG1 expression in newly activated CTLs, reduced numbers of CD69+CD103+ TRM cells accumulated in the skin of HSV infected mice (121). It is unclear whether the ITIM sequence in the cytoplasmic domain of KLRG1 prevented TRM development, or whether dysregulated homing receptor expression promoted localization in other tissues.
Therapeutic approaches for cancer treatment include strategies that are designed to elicit robust responses from tumor specific CTLs. Although entry into the tumor microenvironment is a critical step in immune control, only small numbers of circulating CTLs home to solid tumors. Efforts to augment antitumor immunity by vaccination have been hindered by limited knowledge of the mechanisms that determine how CTLs distribute to different tissues.
TGFβ is a multifaceted cytokine that has beneficial and detrimental effects during cancer treatment. The protective effects of TGFβ include suppression of cancer cell-proliferation and retention of CD103+ TRM cells in the tumor micro-environment. The presence of CD103+ TRM cells in tumors is associated with positive prognoses for several types of cancer (122, 123). Murine models show that TRM cells delay tumor progression by releasing cytokines (Interferons and TNFα) that attract other immune cells into the tumor microenvironment.
During cancer treatment, the therapeutic effects of TGFβ are outweighed by its proangiogeneic properties that support tumor growth and capacity to promote metastatic disease through induction of EMT (40, 124). TGFβ also contributes to dysfunctional responses by tumor specific CTLs. Cell mediated immunity is compromised as tumor infiltrating lymphocytes (TILs) respond to continuous antigen stimulation by upregulating IRs and progressively lose the capacity to produce cytokines and destroy tumor cells (125). TGFβ contributes to the functional impairment of TILs by altering the expression levels of several IRs including programmed death protein-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) (122).
A prostate cancer model was recently used to divide the anti-tumor response into stages. During the induction phase tumor specific CTLs were primed by APCs in the surrounding pLN (126, 127). After antigen-stimulation, stem-like CTLs mobilized to the tumors and acquired TEFF functions after receiving additional costimulatory signals (126). Several studies found that tumor draining lymph nodes contained stem-like CD8+ T cells that converted to CD103+ TRM cells in the presence of TGFβ (91, 127, 128). Importantly, disruption of TGFβ signaling enhanced cell mediated immunity by converting lymphoid TRM cells to TEFF cells that localized in the tumor (91). Others induced a robust antiglioma response in mice that were vaccinated with tumor cell-lysate, using a compound that inhibits TGFβ-signaling (129).
Immune checkpoint blockade (ICB) is a strategy that is used to reinvigorate the responses of partially exhausted CTLs during cancer treatment, using antibodies that prevent activation of IRs to restore TEFF functions in TILs (130). Tumors are usually populated with heterogeneous populations of TILs that exhibit varying degrees of dysfunctionality (131). A study that divided TILs into subsets based on their response to antigen stimulation found that the optimal targets of ICB therapy were partially differentiated progenitor cells, that had limited proliferative potential or functional capacity, and CTLs that were programmed for a robust lytic response. Conversely, terminally differentiated CTLs that did not produce cytokines or undergo homeostatic proliferation were poor targets for ICB therapy (132). Development of ICB therapy led to major advances in cancer treatment and improved outcomes for patients with several different types of cancer including metastatic melanoma. The suppressive properties of TGFβ can undermine the effects of ICB therapy by enhancing functional defects in TILs and upregulating hypoxia genes (133, 134). Some cancer treatments use combined strategies to augment CTL responses such as inhibition of PGSL-1 signaling during PD-1 blockade (32). Other cancer treatments utilize agonist antibodies that recognize costimulatory molecules to promote T cell proliferation, survival and memory formation (135). One study found that combined treatment with anti-OX40/anti-CTLA4 reduced IRs expression on tumor specific CTLs and enhanced cytokine production (135), while others found that simultaneous blockade of TGFβ and PD-L1 increased CTL infiltration in tumors of patients with metastatic urothelial cancer (136).
TGFβ alters T cell migration using a variety of direct and indirect mechanisms. A study found that treatment with CXCL12 prompted CTLs to leave cutaneous tumors via lymphatic vessels (137), whereas suppression of CXCL12 expression in senescent tumor cells enhanced T cell infiltration in colonic tumors (138). A third study found that the metastatic properties of mesenchymal cancer cells decreased after CXCL12 was downregulated by TGFβ (139). Others analyzed tumors in breast cancer patients and found that clusters of quiescent cancer cells created an immunosuppressive environment and prevented immune cell infiltration into the tumor by initiating a hypoxia program (140). Another breast cancer study found that hypoxic conditions induced multiple EMT-related pathways including TGFβ (141). CD39 is an ectoenzyme that contributes to immune dysfunction by increasing the concentrations of extracellular adenosine in the tumor microenvironment. Dual CD39 and CD103 expression is a distinguishing feature of TILs in patients with colorectal cancer (142). CD39 and CD73 blockade has been used to enhance CTL responses in tumors. A study found that a TGF-β/SOX4 signaling pathway acted in combination with ROS-driven autophagy to induce CD39 expression on Tregs (143).
Tbet and eomesodermin (EOMES) are homologous Tbox transcription factors that have cooperative functions during lineage-specification of newly activated and exhausted CTLs (58, 144). T-bet is induced by IL-12 soon after antigen stimulation and plays a key role TEFF formation (145), whereas EOMES is expressed with slightly delayed kinetics and cooperates with T-bet to facilitate formation of TCM cells. Both transcription factors are downregulated as TRM cells settle in peripheral tissues (146). Homologous zinc-finger E homeobox-binding proteins (ZEB1 and ZEB2) are also transcription factors that have important functions during EMT and immune cell development (147). During CD8 T cell differentiation, ZEB2 cooperates with T-bet to promote formation of terminally differentiated TEFF cells, while repressing genes that support TCM development (74). KLRG1+ CTLs express ZEB2 at high levels, whereas ZEB1 is induced by TGFβ and promotes formation of TCM cells by counteracting the effects of ZEB2 (148).
During memory formation, TGFβ blocks IL-12 signaling by inhibiting tyrosine phosphorylation and activation of Jak-2 and Tyk-2 kinases (65). EOMES is down regulated by IL-12 during TEFF formation and TGFβ during TRM development (9, 149), whereas expression was maintained in exhausted CTLs (150). EOMES binding sites in the regulatory regions for several genes that encode IRs including PD1, CTLA-4 and CD39 (151). EOMES expression can be upregulated by several different mechanisms during the CTL response, including the antigen receptor (TCR), NF-κB, and selected cytokines. Common gamma chain cytokines are T cell differentiation factors that signal via STAT5 (152). The genes that encode EOMES and TGFβR2 contain STAT5 binding sites. CD8 T cells that were modified to express a constitutively actively form of STAT5 expressed EOMES at increased levels, whereas TGFβR2 was downregulated (153).
Chronic antigen stimulation is largely responsible immune dysfunction during persistent infections and tumorigenesis. Exhausted CTLs can be compartmentalized into subsets based on distinct transcriptional profiles. Definitive markers include T cell factor 1 (TCF-1), which is expressed at high levels in self-renewing CTLs and down regulated during formation of terminally exhausted CTLs that express PD-1 and Tim3 (154). Some TCF-1+ CTLs become functional TEFF cells during ICB (155). Lineage tracing was used to track clonal populations of CTLs during LCMV infection and identified a subset of partially differentiated CTLs that segregated between two bifurcating differentiation pathways to create distinct subsets of terminally differentiated and exhausted CTLs (156). Exhaustion was linked to an IRF7-dependent mechanism that was activated by type I IFN, whereas ZEB2 promoted TEFF formation.
Epigenetic programming limits T cell proliferation during ICB therapy. Thymocyte selection-associated high mobility group box (TOX) is a transcription factor that plays a role in formation of exhausted CTLs and positively correlates with IR expression. A recent study showed that stimulation with TGFβ prolonged survival of chronically activated CTLs by attenuating TCR signaling, while inducing epigenetic changes that accelerated terminal dysfunction and attenuated TOX expression (157). Blocking TGFβ signaling in the presence of BMP4 altered the epigenetic state of dysfunctional T cells and restored some TEFF functions. Similarly, rebalancing TGFβ1/BMP signaling during LCMV infection was sufficient to boost CTL responses during PD-L1 blockade and decrease in viral titers (157).
During recent years, multiple groups used Loxp recombination to prevent TGFβ receptor (TGFβRII) and/or SMAD4 expression in CTLs. Phenotypic changes that were observed after antigen stimulation showed that multiple homing receptors were cooperatively regulated by alternative signaling pathways. Notably, signaling via SMAD4 altered the expression levels several homing receptors via a mechanism that did not involve TGFβ (9, 158, 159). By comparing the transcriptomes of TEFF cells, we showed that TGFβ and SMAD4 coordinate changes in homing receptor expression by altering the expression levels of the same genes in opposite directions (9, 10). The target genes included a collection of adhesion molecules (KLRG1, CD62L, CD103) and transcription factors (Hobit and EOMES) with important functions during memory formation (9). The SMAD4-deficient CTLs expressed EOMES at reduced levels (9), similar to the pattern seen in stem-like memory CD8 T cells during chronic antigen stimulation (9). The regulatory sequences in the EOMES promoter contain multiple SMAD4 binding sites (9, 160). The SMAD4-deficient CTLs expressed CX3CR1 at low/intermediate levels (9), while KLRG1 and CD62L were down regulated indicating a defect during formation of terminally differentiated TEFF cells and TCM cells (10, 11). Ectopic EOMES expression induced phenotypic changes that were consistent with a shift toward TCM formation (9).
Although TGFβ is a key regulator during the CTL response (Table 1), the pleiotropic effects of the cytokine cause complications during immune intervention. The recent discovery that SMAD4 plays a separate role in lineage-specification of newly activated CTLs, via a mechanism that does not involve TGFβ, may reveal new avenues for augmenting CTL responses after vaccination. For example, it may be possible to generate inhibitors that prevent formation of terminally differentiated CTLs and enhance formation of CTLs with stem-like properties that localize to tumors. Since TCR stimulation in the presence of recombinant IL-2 and IL-12 is not sufficient to induce KLRG1 expression on CTLs in vitro (81, 111, 161), current data indicate that multiple stimuli are required for terminal differentiation including inflammatory molecules that signal via SMAD4. Additional work is required to identify of the ligand(s) of this novel regulatory pathway and determine whether signaling induces epigenetic that make terminally differentiated TEFF and TCM refractory to subsequent regulation by TGFβ. Several groups examined the functions of SMAD4 during CD8 T cell differentiation. Variations in the results between studies may reflect the timing of Cre expression. Some studies found that SMAD4-ablation during an early stage of thymic development altered homing receptor expression, as well as cell-proliferation and effector functions (10, 159). Others found that SMAD4-deficient CTLs maintained normal effector functions and proliferation when the mutation occurred immediately before CTLs left the thymus (9, 11). Since KLRG1 is an inhibitory receptor that binds N- cadherin, further work is required to reveal whether interactions between N-cadherin and KLRG1 prevent TEFF cells from extravasating to the periphery during tissue repair.
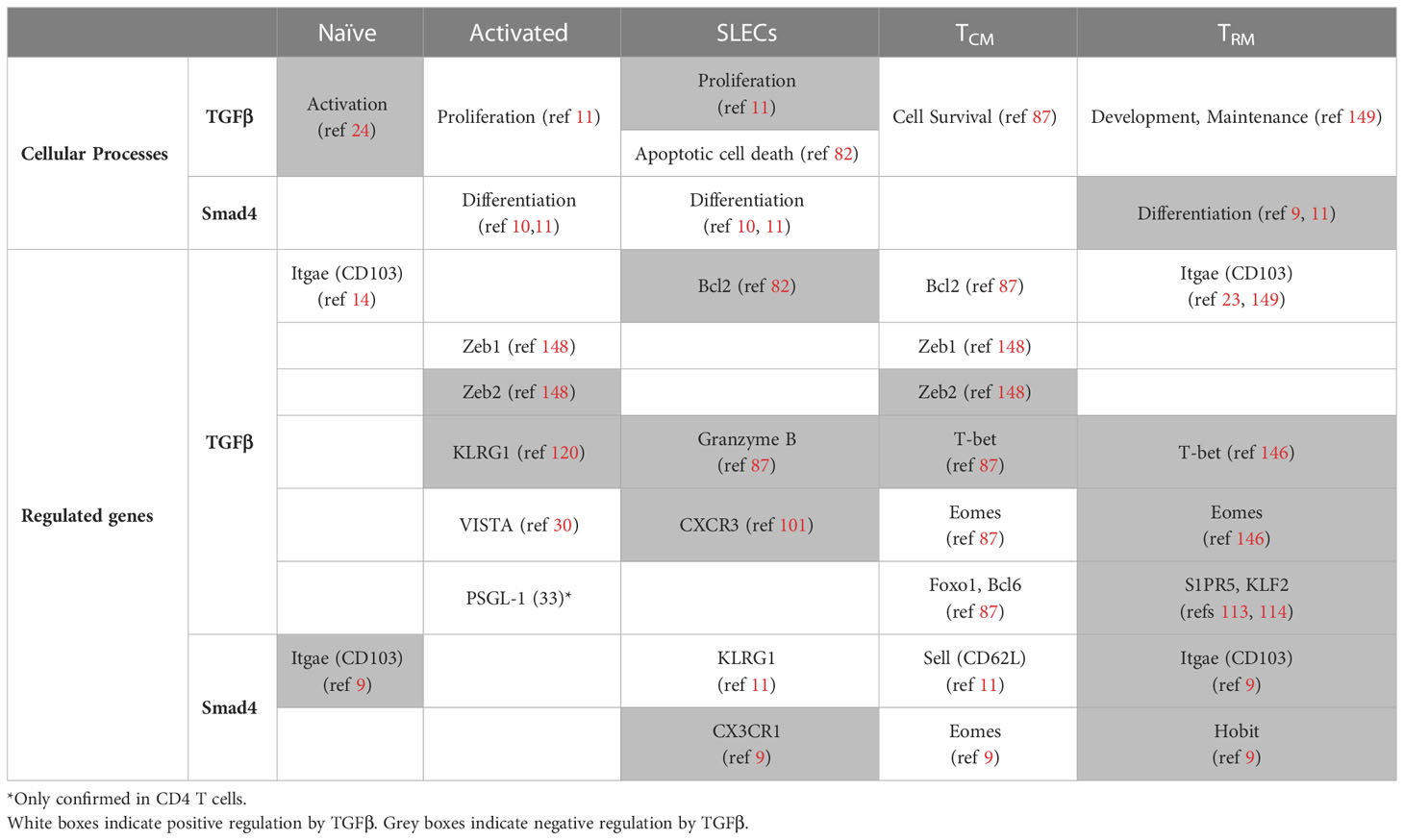
Table 1 Table of regulatory pathways.
This review was written by LC and KC. All authors contributed to the article and approved the submitted version.
KC is supported by a DST-SERB Ramanujan Fellowship, Govt. of India.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gilbert RWD, Vickaryous MK, Viloria-Petit AM. Signalling by transforming growth factor beta isoforms in wound healing and tissue regeneration. J Dev Biol (2016) 4. doi: 10.3390/jdb4020021
2. Horbelt D, Denkis A, Knaus P. A portrait of transforming growth factor beta superfamily signalling: background matters. Int J Biochem Cell Biol (2012) 44:469–74. doi: 10.1016/j.biocel.2011.12.013
3. Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol (2017) 2. doi: 10.1126/sciimmunol.aai7911
4. Jenkins G. The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol (2008) 40:1068–78. doi: 10.1016/j.biocel.2007.11.026
5. Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol (2008) 87:601–15. doi: 10.1016/j.ejcb.2008.01.012
6. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol (2006) 24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737
7. Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev (2005) 19:2783–810. doi: 10.1101/gad.1350705
8. Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol (2007) 8:970–82. doi: 10.1038/nrm2297
9. Chandiran K, Suarez-Ramirez JE, Hu Y, Jellison ER, Ugur Z, Low JS, et al. SMAD4 and TGFbeta are architects of inverse genetic programs during fate determination of antiviral CTLs. Elife (2022) 11. doi: 10.7554/eLife.76457
10. Cao J, Zhang X, Wang Q, Qiu G, Hou C, Wang J, et al. Smad4 represses the generation of memory-precursor effector T cells but is required for the differentiation of central memory T cells. Cell Death Dis (2015) 6:e1984. doi: 10.1038/cddis.2015.337
11. Hu Y, Lee YT, Kaech SM, Garvy B, Cauley LS. Smad4 promotes differentiation of effector and circulating memory CD8 T cells but is dispensable for tissue-resident memory CD8 T cells. J Immunol (2015) 194:2407–14. doi: 10.4049/jimmunol.1402369
12. This S, Rogers D, Mallet Gauthier E, Mandl JN, Melichar HJ. What’s self got to do with it: sources of heterogeneity among naive T cells. Semin Immunol (2023) 65:101702. doi: 10.1016/j.smim.2022.101702
13. Mehal WZ, Sheikh SZ, Gorelik L, Flavell RA. TGF-beta signaling regulates CD8+ T cell responses to high- and low-affinity TCR interactions. Int Immunol (2005) 17:531–8.
14. Mani V, Bromley SK, Aijo T, Mora-Buch R, Carrizosa E, Warner RD, et al. Migratory DCs activate TGF-beta to precondition naive CD8(+) T cells for tissue-resident memory fate. Science (2019) 366. doi: 10.1126/science.aav5728
15. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental origin governs CD8(+) T cell fate decisions during infection. Cell (2018) 174:117–130 e14. doi: 10.1016/j.cell.2018.05.029
16. Tabilas C, Iu DS, Daly CWP, Yee Mon KJ, Reynaldi A, Wesnak SP, et al. Early microbial exposure shapes adult immunity by altering CD8+ T cell development. Proc Natl Acad Sci USA (2022) 119:e2212548119. doi: 10.1073/pnas.2212548119
17. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
18. Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med (2017) 214:1593–606. doi: 10.1084/jem.20161760
19. Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol (2015) 194:4073–80. doi: 10.4049/jimmunol.1500046
20. Shive C, Pandiyan P. Inflammation, immune senescence, and dysregulated immune regulation in the elderly. Front Aging (2022) 3:840827. doi: 10.3389/fragi.2022.840827
21. Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, et al. Integrin beta 3 regulates cellular senescence by activating the TGF-beta pathway. Cell Rep (2017) 18:2480–93. doi: 10.1016/j.celrep.2017.02.012
22. Chen W, Jin W, Tian H, Sicurello P, Frank M, Orenstein JM, et al. Requirement for transforming growth factor beta1 in controlling T cell apoptosis. J Exp Med (2001) 194:439–53. doi: 10.1084/jem.194.4.439
23. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi: 10.1016/j.immuni.2013.08.019
24. Filippi CM, Juedes AE, Oldham JE, Ling E, Togher L, Peng Y, et al. Transforming growth factor-beta suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: a two-faced impact on autoimmunity. Diabetes (2008) 57:2684–92. doi: 10.2337/db08-0609
25. Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med (1993) 178:449–60. doi: 10.1084/jem.178.2.449
26. Zhang N, Bevan MJ. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol (2012) 13:667–73. doi: 10.1038/ni.2319
27. Newman DK, Fu G, Adams T, Cui W, Arumugam V, Bluemn T, et al. The adhesion molecule PECAM-1 enhances the TGF-beta-mediated inhibition of T cell function. Sci Signal (2016) 9:ra27. doi: 10.1126/scisignal.aad1242
28. Newman DK, Fu G, McOlash L, Schauder D, Newman PJ, Cui W, et al. Frontline science: PECAM-1 (CD31) expression in naive and memory, but not acutely activated, CD8(+) T cells. J Leukoc Biol (2018) 104:883–93. doi: 10.1002/JLB.2HI0617-229RRR
29. ElTanbouly MA, Zhao Y, Nowak E, Li J, Schaafsma E, Le Mercier I, et al. VISTA is a checkpoint regulator for naive T cell quiescence and peripheral tolerance. Science (2020) 367. doi: 10.1126/science.aay0524
30. Schlichtner S, Yasinska IM, Ruggiero S, Berger SM, Aliu N, Prunk M, et al. Expression of the immune checkpoint protein VISTA is differentially regulated by the TGF-beta1 - Smad3 signaling pathway in rapidly proliferating human cells and T lymphocytes. Front Med (Lausanne) (2022) 9:790995. doi: 10.3389/fmed.2022.790995
31. Tinoco R, Otero DC, Takahashi AA, Bradley LM. PSGL-1: a new player in the immune checkpoint landscape. Trends Immunol (2017) 38:323–35. doi: 10.1016/j.it.2017.02.002
32. Viramontes KM, Neubert EN, DeRogatis JM, Tinoco R. PD-1 immune checkpoint blockade and PSGL-1 inhibition synergize to reinvigorate exhausted T cells. Front Immunol (2022) 13:869768. doi: 10.3389/fimmu.2022.869768
33. Lewis GM, Wehrens EJ, Labarta-Bajo L, Streeck H, Zuniga EI. TGF-beta receptor maintains CD4 T helper cell identity during chronic viral infections. J Clin Invest (2016) 126:3799–813. doi: 10.1172/JCI87041
34. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12:762–73. doi: 10.1038/nri3298
35. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci (2008) 121:2115–22. doi: 10.1242/jcs.017897
36. Weitnauer M, Mijosek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol (2016) 9:287–98. doi: 10.1038/mi.2015.126
37. Colas-Algora N, Millan J. How many cadherins do human endothelial cells express? Cell Mol Life Sci (2019) 76:1299–317. doi: 10.1007/s00018-018-2991-9
38. Ferrero E, Villa A, Ferrero ME, Toninelli E, Bender JR, Pardi R, et al. Tumor necrosis factor alpha-induced vascular leakage involves PECAM1 phosphorylation. Cancer Res (1996) 56:3211–5.
39. Whitsett JA. Airway epithelial differentiation and mucociliary clearance. Ann Am Thorac Soc (2018) 15:S143–8. doi: 10.1513/AnnalsATS.201802-128AW
40. Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, et al. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer (2011) 105:1885–93. doi: 10.1038/bjc.2011.452
41. Weber CE, Li NY, Wai PY, Kuo PC. Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J Burn Care Res (2012) 33:311–8. doi: 10.1097/BCR.0b013e318240541e
42. Yu H, Shen Y, Hong J, Xia Q, Zhou F, Liu X. The contribution of TGF-beta in epithelial-mesenchymal transition (EMT): down-regulation of e-cadherin via snail. Neoplasma (2015) 62:1–15. doi: 10.4149/neo_2015_002
43. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI200522675
44. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med (1989) 170:727–37. doi: 10.1126/science.1213368
45. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
46. Sun X, Nkennor B, Mastikhina O, Soon K, Nunes SS. Endothelium-mediated contributions to fibrosis. Semin Cell Dev Biol (2020) 101:78–86. doi: 10.1016/j.semcdb.2019.10.015
47. Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P. TGF-beta-Induced endothelial-mesenchymal transition in fibrotic diseases. Int J Mol Sci (2017) 18. doi: 10.3390/ijms18102157
48. Jones M, Sabatini PJ, Lee FS, Bendeck MP, Langille BL. N-cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler Thromb Vasc Biol (2002) 22:1972–7. doi: 10.1161/01.ATV.0000036416.14084.5A
49. Cao ZQ, Wang Z, Leng P. Aberrant n-cadherin expression in cancer. BioMed Pharmacother (2019) 118:109320. doi: 10.1016/j.biopha.2019.109320
50. Arif N, Zinnhardt M, Nyamay’Antu A, Teber D, Bruckner R, Schaefer K, et al. PECAM-1 supports leukocyte diapedesis by tension-dependent dephosphorylation of VE-cadherin. EMBO J (2021) 40:e106113. doi: 10.15252/embj.2020106113
51. Dabravolski SA, Andreeva ER, Eremin II, Markin AM, Nadelyaeva II, Orekhov AN, et al. The role of pericytes in regulation of innate and adaptive immunity. Biomedicines (2023) 11. doi: 10.3390/biomedicines11020600
52. Mempel TR, Henrickson SE, Von Andrian UH. T-Cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427:154–9. doi: 10.1038/nature02238
53. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity (2009) 31:823–33. doi: 10.1016/j.immuni.2009.08.027
54. Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol (2008) 9:155–65. doi: 10.1038/ni1557
55. Crozat K, Tamoutounour S, Vu Manh TP, Fossum E, Luche H, Ardouin L, et al. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8alpha+ type. J Immunol (2011) 187:4411–5. doi: 10.4049/jimmunol.1101717
56. Carman CV, Martinelli R. T Lymphocyte-endothelial interactions: emerging understanding of trafficking and antigen-specific immunity. Front Immunol (2015) 6:603. doi: 10.3389/fimmu.2015.00603
57. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell (2011) 146:980–91. doi: 10.1016/j.cell.2011.08.015
58. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6:1236–44. doi: 10.1038/ni1268
59. Pais Ferreira D, Silva JG, Wyss T, Fuertes Marraco SA, Scarpellino L, Charmoy M, et al. Central memory CD8(+) T cells derive from stem-like Tcf7(hi) effector cells in the absence of cytotoxic differentiation. Immunity (2020) 53:985–1000.e11. doi: 10.1016/j.immuni.2020.09.005
60. Wang Y, Hu J, Li Y, Xiao M, Wang H, Tian Q, et al. The transcription factor TCF1 preserves the effector function of exhausted CD8 T cells during chronic viral infection. Front Immunol (2019) 10:169. doi: 10.3389/fimmu.2019.00169
61. Danilo M, Chennupati V, Silva JG, Siegert S, Held W. Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep (2018) 22:2107–17. doi: 10.1016/j.celrep.2018.01.072
62. Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood (2011) 118:3890–900. doi: 10.1182/blood-2011-05-357111
63. Pritchard GH, Phan AT, Christian DA, Blain TJ, Fang Q, Johnson J, et al. Early T-bet promotes LFA1 upregulation required for CD8+ effector and memory T cell development. J Exp Med (2023) 220. doi: 10.1084/jem.20191287
64. McKarns SC, Schwartz RH. Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: a role for T cell intrinsic Smad3. J Immunol (2005) 174:2071–83. doi: 10.4049/jimmunol.174.4.2071
65. Bright JJ, Sriram S. TGF-beta inhibits IL-12-induced activation of jak-STAT pathway in T lymphocytes. J Immunol (1998) 161:1772–7. doi: 10.4049/jimmunol.161.4.1772
66. Marth T, Strober W, Seder RA, Kelsall BL. Regulation of transforming growth factor-beta production by interleukin-12. Eur J Immunol (1997) 27:1213–20. doi: 10.1002/eji.1830270524
67. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10:595–602. doi: 10.1038/ni.1731
68. Hunter CA, Reiner SL. Cytokines and T cells in host defense. Curr Opin Immunol (2000) 12:413–8. doi: 10.1016/s0952-7915(00)00110-2
69. Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol (2001) 158:119–30. doi: 10.1016/s0002-9440(10)63950-8
70. Xu L, Yoon H, Zhao MQ, Liu J, Ramana CV, Enelow RI. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-alpha by antiviral CD8+ T cells. J Immunol (2004) 173:721–5. doi: 10.4049/jimmunol.173.2.721
71. Meinicke H, Bremser A, Brack M, Schrenk K, Pircher H, Izcue A. KLRG1 impairs regulatory T-cell competitive fitness in the gut. Immunology (2017) 152:65–73. doi: 10.1111/imm.12749
72. Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, et al. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J Immunol (2011) 187:4967–78. doi: 10.4049/jimmunol.1102335
73. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1(+) effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity (2018) 48:716–729 e8. doi: 10.1016/j.immuni.2018.03.015
74. Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, et al. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med (2015) 212:2041–56. doi: 10.1084/jem.20150186
75. Ye F, Turner J, Flano E. Contribution of pulmonary KLRG1high and KLRG1low CD8 T cells to effector and memory responses during influenza virus infection. J Immunol (2012) 189:5206–11. doi: 10.4049/jimmunol.1200137
76. Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity (2013) 38:1250–60. doi: 10.1016/j.immuni.2013.05.009
77. Renkema KR, Huggins MA, Borges da Silva H, Knutson TP, Henzler CM, Hamilton SE. KLRG1(+) memory CD8 T cells combine properties of short-lived effectors and long-lived memory. J Immunol (2020) 205:1059–69. doi: 10.4049/jimmunol.1901512
78. Madi A, Wu J, Ma S, Weisshaar N, Mieg A, Hering M, et al. Regulatory T cell-derived interleukin-15 promotes the diversity of immunological memory. Eur J Immunol (2023) 53:e2149400. doi: 10.1002/eji.202149400
79. Tessmer MS, Fugere C, Stevenaert F, Naidenko OV, Chong HJ, Leclercq G, et al. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol (2007) 19:391–400. doi: 10.1093/intimm/dxm004
80. Rosshart S, Hofmann M, Schweier O, Pfaff AK, Yoshimoto K, Takeuchi T, et al. Interaction of KLRG1 with e-cadherin: new functional and structural insights. Eur J Immunol (2008) 38:3354–64. doi: 10.1002/eji.200838690
81. Beyersdorf NB, Ding X, Karp K, Hanke T. Expression of inhibitory “killer cell lectin-like receptor G1” identifies unique subpopulations of effector and memory CD8 T cells. Eur J Immunol (2001) 31:3443–52. doi: 10.1002/1521-4141(200112)31:12<3443::AID-IMMU3443>3.0.CO;2-J
82. Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity (2009) 31:131–44. doi: 10.1016/j.immuni.2009.04.020
83. Nishimura M, Umehara H, Nakayama T, Yoneda O, Hieshima K, Kakizaki M, et al. Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin+/granzyme b+ cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J Immunol (2002) 168:6173–80. doi: 10.4049/jimmunol.168.12.6173
84. Guarda G, Hons M, Soriano SF, Huang AY, Polley R, Martin-Fontecha A, et al. L-selectin-negative CCR7- effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat Immunol (2007) 8:743–52. doi: 10.1038/ni1469
85. Wu J, Madi A, Mieg A, Hotz-Wagenblatt A, Weisshaar N, Ma S, et al. T Cell factor 1 suppresses CD103+ lung tissue-resident memory T cell development. Cell Rep (2020) 31:107484. doi: 10.1016/j.celrep.2020.03.048
86. Obar JJ, Lefrancois L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol (2010) 185:263–72. doi: 10.4049/jimmunol.1000492
87. Ma C, Zhang N. Transforming growth factor-beta signaling is constantly shaping memory T-cell population. Proc Natl Acad Sci USA (2015) 112:11013–7. doi: 10.1073/pnas.1510119112
88. Johnson LD, Jameson SC. TGF-beta sensitivity restrains CD8+ T cell homeostatic proliferation by enforcing sensitivity to IL-7 and IL-15. PLoS One (2012) 7:e42268. doi: 10.1371/journal.pone.0042268
89. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17:1290–7. doi: 10.1038/nm.2446
90. Hu Y, Hudson WH, Kissick HT, Medina CB, Baptista AP, Ma C, et al. TGF-beta regulates the stem-like state of PD-1+ TCF-1+ virus-specific CD8 T cells during chronic infection. J Exp Med (2022) 219. doi: 10.1084/jem.20211574
91. Li G, Srinivasan S, Wang L, Ma C, Guo K, Xiao W, et al. TGF-beta-dependent lymphoid tissue residency of stem-like T cells limits response to tumor vaccine. Nat Commun (2022) 13:6043. doi: 10.1038/s41467-022-33768-x
92. Tedder TF, Steeber DA, Chen A, Engel P. The selectins: vascular adhesion molecules. FASEB J (1995) 9:866–73. doi: 10.1096/fasebj.9.10.7542213
93. Steeber DA, Tang ML, Zhang XQ, Muller W, Wagner N, Tedder TF. Efficient lymphocyte migration across high endothelial venules of mouse peyer’s patches requires overlapping expression of l-selectin and beta7 integrin. J Immunol (1998) 161:6638–47.
94. Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell (1999) 99:23–33. doi: 10.1016/S0092-8674(00)80059-8
95. Xie H, Lim YC, Luscinskas FW, Lichtman AH. Acquisition of selectin binding and peripheral homing properties by CD4(+) and CD8(+) T cells. J Exp Med (1999) 189:1765–76. doi: 10.1084/jem.189.11.1765
96. Sako D, Comess KM, Barone KM, Camphausen RT, Cumming DA, Shaw GD. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for p-selectin binding. Cell (1995) 83:323–31. doi: 10.1016/0092-8674(95)90173-6
97. Scimone ML, Felbinger TW, Mazo IB, Stein JV, Von Andrian UH, Weninger W. CXCL12 mediates CCR7-independent homing of central memory cells, but not naive T cells, in peripheral lymph nodes. J Exp Med (2004) 199:1113–20. doi: 10.1084/jem.20211574
98. Tedder TF, Steeber DA, Pizcueta P. L-selectin-deficient mice have impaired leukocyte recruitment into inflammatory sites. J Exp Med (1995) 181:2259–64. doi: 10.1084/jem.181.6.2259
99. Saito K, Tanaka T, Kanda H, Ebisuno Y, Izawa D, Kawamoto S, et al. Gene expression profiling of mucosal addressin cell adhesion molecule-1+ high endothelial venule cells (HEV) and identification of a leucine-rich HEV glycoprotein as a HEV marker. J Immunol (2002) 168:1050–9. doi: 10.4049/jimmunol.168.3.1050
100. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
101. Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat Commun (2020) 11:1749. doi: 10.1038/s41467-020-15404-8
102. Bottcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Hochst B, et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat Commun (2015) 6:8306. doi: 10.1038/ncomms9306
103. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity (2016) 45:1270–84. doi: 10.1016/j.immuni.2016.10.018
104. Cauley LS, Lefrancois L. Guarding the perimeter: protection of the mucosa by tissue-resident memory T cells. Mucosal Immunol (2013) 6:14–23. doi: 10.1038/mi.2012.96
105. Poon MML, Caron DP, Wang Z, Wells SB, Chen D, Meng W, et al. Tissue adaptation and clonal segregation of human memory T cells in barrier sites. Nat Immunol (2023) 24:309–19. doi: 10.1038/s41590-022-01395-9
106. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8 T cells. Nat Immunol (2013) 14:509–13. doi: 10.1038/ni.2568
107. Cheuk S, Schlums H, Gallais Serezal I, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity (2017) 46:287–300. doi: 10.1016/j.immuni.2017.01.009
108. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity (2004) 20:167–79. doi: 10.1016/S1074-7613(04)00021-4
109. Franciszkiewicz K, F.h.A. Le M, Vergnon I, Schmitt A, Mami-Chouaib F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res (2013) 73:617–28. doi: 10.1158/0008-5472.CAN-12-2569
110. Christo SN, Evrard M, Park SL, Gandolfo LC, Burn TN, Fonseca R, et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat Immunol (2021) 22:1140–51. doi: 10.1038/s41590-021-01004-1
111. Suarez-Ramirez JE, Chandiran K, Brocke S, Cauley LS. Immunity to respiratory infection is reinforced through early proliferation of lymphoid TRM cells and prompt arrival of effector CD8 T cells in the lungs. Front Immunol (2019) 10:1370. doi: 10.3389/fimmu.2019.01370
112. Osborn JF, Hobbs SJ, Mooster JL, Khan TN, Kilgore AM, Harbour JC, et al. Central memory CD8+ T cells become CD69+ tissue-residents during viral skin infection independent of CD62L-mediated lymph node surveillance. PloS Pathog (2019) 15:e1007633. doi: 10.1371/journal.ppat.1007633
113. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14:1285–93. doi: 10.1038/ni.2745
114. Evrard M, Wynne-Jones E, Peng C, Kato Y, Christo SN, Fonseca R, et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J Exp Med (2022) 219. doi: 10.1084/jem.20210116
115. Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol (2012) 30:69–94. doi: 10.1146/annurev-immunol-020711-075011
116. Sancho D, Santis AG, Alonso-Lebrero JL, Viedma F, Tejedor R, Sanchez-Madrid F. Functional analysis of ligand-binding and signal transduction domains of CD69 and CD23 c-type lectin leukocyte receptors. J Immunol (2000) 165:3868–75. doi: 10.4049/jimmunol.165.7.3868
117. Sancho D, Gomez M, Viedma F, Esplugues E, Gordon-Alonso M, Garcia-Lopez MA, et al. CD69 downregulates autoimmune reactivity through active transforming growth factor-beta production in collagen-induced arthritis. J Clin Invest (2003) 112:872–82. doi: 10.1172/JCI200319112
118. Wienke J, Veldkamp SR, Struijf EM, Yousef Yengej FA, van der Wal MM, van Royen-Kerkhof A, et al. T Cell interaction with activated endothelial cells primes for tissue-residency. Front Immunol (2022) 13:827786. doi: 10.3389/fimmu.2022.827786
119. Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, et al. The functional requirement for CD69 in establishment of resident memory CD8(+) T cells varies with tissue location. J Immunol (2019) 203:946–55. doi: 10.4049/jimmunol.1900052
120. Schwartzkopff S, Woyciechowski S, Aichele U, Flecken T, Zhang N, Thimme R, et al. TGF-beta downregulates KLRG1 expression in mouse and human CD8(+) T cells. Eur J Immunol (2015) 45:2212–7. doi: 10.1002/eji.201545634
121. Hochheiser K, Wiede F, Wagner T, Freestone D, Enders MH, Olshansky M, et al. Ptpn2 and KLRG1 regulate the generation and function of tissue-resident memory CD8+ T cells in skin. J Exp Med (2021) 218. doi: 10.1084/jem.20200940
122. Plunkett KR, Armitage JD, Inderjeeth AJ, McDonnell AM, Waithman J, Lau PKH. Tissue-resident memory T cells in the era of (Neo) adjuvant melanoma management. Front Immunol (2022) 13:1048758. doi: 10.3389/fimmu.2022.1048758
123. Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F. The emerging role of CD8(+) tissue resident memory T (TRM) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol (2018) 9:1904. doi: 10.3389/fimmu.2018.01904
124. Trelford CB, Dagnino L, Di Guglielmo GM. Transforming growth factor-beta in tumour development. Front Mol Biosci (2022) 9:991612. doi: 10.3389/fmolb.2022.991612
125. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog (2010) 6:e1000947. doi: 10.1371/journal.ppat.1000947
126. Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, et al. CD8(+) T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity (2023) 56:107–124 e5. doi: 10.1016/j.immuni.2022.12.002
127. Liu Q, Ran L, Yue Z, Su X, Wang L, Wen S, et al. Tumor-specific memory CD8(+) T cells are strictly resident in draining lymph nodes during tumorigenesis. Cell Mol Immunol (2023) 20:423–6. doi: 10.1038/s41423-023-01025-w
128. Sievers C, Craveiro M, Friedman J, Robbins Y, Yang X, Bai K, et al. Phenotypic plasticity and reduced tissue retention of exhausted tumor-infiltrating T cells following neoadjuvant immunotherapy in head and neck cancer. Cancer Cell (2023) 41:887–902 e5. doi: 10.1016/j.ccell.2023.03.014
129. Tu L, Wang Z, Yang L, Sun X, Yao Y, Zhang P, et al. Incorporation of a TGF-beta2-inhibiting oligodeoxynucleotide molecular adjuvant into a tumor cell lysate vaccine to enhance antiglioma immunity in mice. Front Immunol (2023) 14:1013342. doi: 10.3389/fimmu.2023.1013342
130. Luoma AM, Suo S, Wang Y, Gunasti L, Porter CBM, Nabilsi N, et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell (2022) 185:2918–2935 e29. doi: 10.1016/j.cell.2022.06.018
131. Yang B, Li X, Zhang W, Fan J, Zhou Y, Li W, et al. Spatial heterogeneity of infiltrating T cells in high-grade serous ovarian cancer revealed by multi-omics analysis. Cell Rep Med (2022) 3:100856. doi: 10.1016/j.xcrm.2022.100856
132. Chakravarti M, Dhar S, Bera S, Sinha A, Roy K, Sarkar A, et al. Terminally exhausted CD8+ T cells resistant to PD-1 blockade promote generation and maintenance of aggressive cancer stem cells. Cancer Res (2023) 83:1815–33. doi: 10.1158/0008-5472.22929972
133. Liikanen I, Lauhan C, Quon S, Omilusik K, Phan AT, Bartroli LB, et al. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8+ T cells. J Clin Invest (2021) 131. doi: 10.1172/JCI143729
134. Deng B, Zhu JM, Wang Y, Liu TT, Ding YB, Xiao WM, et al. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-beta1 in gastric cancer. PloS One (2013) 8:e63777. doi: 10.1371/journal.pone.0063777
135. Emerson DA, Rolig AS, Redmond WL. Enhancing the generation of eomes(hi) CD8(+) T cells augments the efficacy of OX40- and CTLA-4-Targeted immunotherapy. Cancer Immunol Res (2021) 9:430–40. doi: 10.1158/2326-6066.CIR-20-0338
136. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
137. Steele MM, Jaiswal A, Delclaux I, Dryg ID, Murugan D, Femel J, et al. T Cell egress via lymphatic vessels is tuned by antigen encounter and limits tumor control. Nat Immunol (2023) 24:664–75. doi: 10.1038/s41590-023-01491-4
138. Choi YW, Kim YH, Oh SY, Suh KW, Kim YS, Lee GY, et al. Senescent tumor cells build a cytokine shield in colorectal cancer. Adv Sci (Weinh) (2021) 8:2002497. doi: 10.1002/advs.202002497
139. Yu PF, Huang Y, Xu CL, Lin LY, Han YY, Sun WH, et al. Downregulation of CXCL12 in mesenchymal stromal cells by TGFbeta promotes breast cancer metastasis. Oncogene (2017) 36:840–9. doi: 10.1038/onc.2016.252
140. Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho JW, et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell (2022) 185:1694–1708 e19. doi: 10.1016/j.cell.2022.03.033
141. Mallini P, Chen M, Mahkamova K, Lennard TWJ, Pan Y, Wei D, et al. Hypoxia-driven TGFbeta modulation of side population cells in breast cancer: the potential role of ERalpha. Cancers (Basel) (2023) 15. doi: 10.1371/journal.pone.0063777
142. van den Bulk J, van der Ploeg M, Ijsselsteijn ME, Ruano D, van der Breggen R, Duhen R, et al. CD103 and CD39 coexpression identifies neoantigen-specific cytotoxic T cells in colorectal cancers with low mutation burden. J Immunother Cancer (2023) 11. doi: 10.1136/jitc-2022-005887
143. Gerner MC, Ziegler LS, Schmidt RLJ, Krenn M, Zimprich F, Uyanik-Unal K, et al. The TGF-b/SOX4 axis and ROS-driven autophagy co-mediate CD39 expression in regulatory T-cells. FASEB J (2020) 34:8367–84. doi: 10.1096/fj.201902664
144. McLane LM, Ngiow SF, Chen Z, Attanasio J, Manne S, Ruthel G, et al. Role of nuclear localization in the regulation and function of T-bet and eomes in exhausted CD8 T cells. Cell Rep (2021) 35:109120. doi: 10.1016/j.celrep.2021.109120
145. Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol (2006) 177:7515–9. doi: 10.4049/jimmunol.177.11.7515
146. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-Box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43:1101–11. doi: 10.1016/j.immuni.2015.11.008
147. Scott CL, Omilusik KD. ZEBs: novel players in immune cell development and function. Trends Immunol (2019) 40:431–46. doi: 10.1016/j.it.2019.03.001
148. Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, et al. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8(+) T cell fates. J Exp Med (2018) 215:1153–68. doi: 10.1084/jem.20171352
149. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14:1294–301. doi: 10.1038/ni.2744
150. Li J, He Y, Hao J, Ni L, Dong C. High levels of eomes promote exhaustion of anti-tumor CD8(+) T cells. Front Immunol (2018) 9:2981. doi: 10.3389/fimmu.2018.02981
151. He H, Yi Y, Cai X, Wang J, Ni X, Fu Y, et al. Down-regulation of EOMES drives T-cell exhaustion via abolishing EOMES-mediated repression of inhibitory receptors of T cells in liver cancer. J Cell Mol Med (2021) 25:161–9. doi: 10.1111/jcmm.15898
152. Leonard WJ, Lin JX, O’Shea JJ. The gamma(c) family of cytokines: basic biology to therapeutic ramifications. Immunity (2019) 50:832–50. doi: 10.1016/j.immuni.2019.03.028
153. Grange M, Verdeil G, Arnoux F, Griffon A, Spicuglia S, Maurizio J, et al. Active STAT5 regulates T-bet and eomesodermin expression in CD8 T cells and imprints a T-bet-dependent Tc1 program with repressed IL-6/TGF-beta1 signaling. J Immunol (2013) 191:3712–24. doi: 10.4049/jimmunol.1300319
154. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity (2016) 45:415–27. doi: 10.1016/j.immuni.2016.07.021
155. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
156. Kasmani MY, Zander R, Chung HK, Chen Y, Khatun A, Damo M, et al. Clonal lineage tracing reveals mechanisms skewing CD8+ T cell fate decisions in chronic infection. J Exp Med (2023) 220. doi: 10.1084/jem.20220679
157. Saadey AA, Yousif A, Osborne N, Shahinfar R, Chen YL, Laster B, et al. Rebalancing TGFbeta1/BMP signals in exhausted T cells unlocks responsiveness to immune checkpoint blockade therapy. Nat Immunol (2022) 24:280–94. doi: 10.1038/s41590-022-01384-y
158. Igalouzene R, Hernandez-Vargas H, Benech N, Guyennon A, Bauche D, Barrachina C, et al. SMAD4 TGF-beta-independent function preconditions naive CD8+ T cells to prevent severe chronic intestinal inflammation. J Clin Invest (2022) 132. doi: 10.1172/JCI151020
159. Gu AD, Zhang S, Wang Y, Xiong H, Curtis TA, Wan YY. A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor beta receptor signaling. Immunity (2015) 42:68–79. doi: 10.1016/j.immuni.2014.12.019
160. Wu B, Zhang G, Guo Z, Wang G, Xu X, Li JL, et al. The SKI proto-oncogene restrains the resident CD103(+)CD8(+) T cell response in viral clearance. Cell Mol Immunol (2021) 18:2410–21. doi: 10.1038/s41423-020-0495-7
Keywords: cytotoxic T lymphocytes (CTL), adhesion molecules, transforming growth factor beta, CD8 memory T lymphocytes +, SMAD4, CD8 T cell differentiation
Citation: Chandiran K and Cauley LS (2023) The diverse effects of transforming growth factor-β and SMAD signaling pathways during the CTL response. Front. Immunol. 14:1199671. doi: 10.3389/fimmu.2023.1199671
Received: 03 April 2023; Accepted: 12 June 2023;
Published: 23 June 2023.
Edited by:
Nu Zhang, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Linda M. Wakim, The University of Melbourne, AustraliaCopyright © 2023 Chandiran and Cauley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda S. Cauley, bGNhdWxleUB1Y2hjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.