 Panyin Shu
Panyin Shu Hantian Liang†
Hantian Liang† Yubin Lin
Yubin Lin Dunfang Zhang
Dunfang Zhang- Department of Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China
Reactive oxygen species (ROS) are produced both enzymatically and non-enzymatically in vivo. Physiological concentrations of ROS act as signaling molecules that participate in various physiological and pathophysiological activities and play an important role in basic metabolic functions. Diseases related to metabolic disorders may be affected by changes in redox balance. This review details the common generation pathways of intracellular ROS and discusses the damage to physiological functions when the ROS concentration is too high to reach an oxidative stress state. We also summarize the main features and energy metabolism of CD4+ T-cell activation and differentiation and the effects of ROS produced during the oxidative metabolism of CD4+ T cells. Because the current treatment for autoimmune diseases damages other immune responses and functional cells in the body, inhibiting the activation and differentiation of autoreactive T cells by targeting oxidative metabolism or ROS production without damaging systemic immune function is a promising treatment option. Therefore, exploring the relationship between T-cell energy metabolism and ROS and the T-cell differentiation process provides theoretical support for discovering effective treatments for T cell-mediated autoimmune diseases.
Introduction
Reactive oxygen species (ROS) are oxidants produced during intracellular or extracellular aerobic energy metabolism, and enzymatic reactions have attracted extensive attention from researchers since their discovery (1, 2). The variety of ROS molecules is a collective term for several related molecules. ROS molecules that play physiological roles in the body include H2O2, , and OH-, and different single entities can be converted into one another through interconversion reactions (3–5). Initially, ROS were considered “toxic” molecules that destroy cellular components through oxidation, participate in the pathogenesis of various diseases, and lead to aging (6). Further research has shown that ROS have a dual function. The physiological concentration of ROS plays an important role as a regulatory medium in the signal transduction process, and metabolism-related diseases may be affected by the redox balance; however, when the concentration of ROS is uncontrolled, oxidative stress results in a disturbance in the normal redox state of the cell and/or oxidative damage (7–10). There are several processes through which ROS are generated in vivo, including those within the mitochondria (11), cytoplasm (12, 13), endoplasmic reticulum (ER) (14, 15), and peroxisomes (16, 17). Enzyme complexes can produce ROS, including NADPH oxidases and cytochrome P450-dependent oxygenases (18, 19).
T cell-mediated immune responses are essential for resisting multiple pathogenic microbial infections and antitumor immune responses (20). The core of the T cells activation process is metabolic reprogramming, in which oxidative phosphorylation is transformed into aerobic glycolysis (21–23). Because different CD4+ T-cell subtypes depend on different energy metabolism methods, T helper 1 (Th1), Th2, and Th17 cells rely on aerobic glycolysis to reduce lipid oxidation. Whereas regulatory T cells (Tregs) mainly depend on lipid oxidation as the main source of metabolism, ROS produced during oxidative metabolism affect the differentiation of CD4+ T cells through various mechanisms (24–30). In this study, we describe ROS, the ROS generation pathway in vivo, oxidative stress, and the harmful effects of oxidative stress on physiological functions. The main immune functions of different helper T cells, the energy source on which they depend for differentiation, and the effect of ROS produced during oxidative metabolism on the differentiation of CD4+ T cells are described. This study provides techniques for exploring new methods to treat autoimmune diseases.
ROS and oxidative stress
ROS are byproducts of the redox reactions of oxygen molecules during biological oxidation. Because the ground-state oxygen molecule contains two unpaired electrons, it is readily reduced in redox reactions, and the product after reduction by a single electron can be used as a precursor to producing other ROS molecules. The term “reactive oxygen species” does not refer to a specific chemical molecule; it is a collective term that includes several related molecules with high chemical reactivity because of their unpaired electrons. This broad term ignores the fact that the biology of individual types of ROS is highly diverse, and their chemical reactivity and second-order rate vary significantly (3, 4, 31–33). The abundance of ROS in the body and the inherent duality of their functions have attracted the interest of numerous researchers over the past 50 years. Physiological ROS levels usually act as biological signals that regulate the physiological activities of organisms. However, supraphysiological concentrations of ROS lead to non-specific toxic effects on DNA, proteins, and lipids, causing damage to cellular and genetic structures (1, 2). Additionally, reactive nitrogen species (RNS), another common product of metabolism, have dual physiological functions similar to those of ROS (34). The commonly seen RNS is mainly the less reactive nitric oxide (NO), which can react with to form the highly oxidative peroxynitrite (ONOO-) (35, 36). As the major primary ROS species, H2O2, , and OH- play important roles in the redox regulation of biological activities (18, 37–51). Although each molecule in the ROS species can function biologically as a single entity, molecules can transform into others under certain reaction conditions. is decomposed to H2O2 by the action of superoxide dismutase (SOD), which can be further reduced to H2O or OH-. The process of OH- formation is accompanied by the oxidation of the [4Fe–4S] cluster. The generated iron is repeatedly reduced, allowing this process to continue (5, 52).
“Oxidative stress,” also known as oxidative adversity, describes a series of adaptive responses caused by the inability of the antioxidant system to remove excess oxidants promptly, causing alterations in the intracellular redox status, interfering with normal signaling, and mediating oxidative damage (2, 3). Oxidative stress is an embodiment of the dual functions of ROS (7, 8). When the steady-state concentrations of major ROS molecules, such as H2O2 and , are at a certain threshold, ROS can be used as versatile pleiotropic physiological signaling agents during the physiological activity of higher organisms (9, 38, 42). Oxidative modification of target proteins by ROS alters protein activity and localization, which regulates processes, such as signal transduction and metabolic metabolism, between cells and organs (10). Oxidation of nuclear factor-κB (NF-κB) by H2O2 can lead to its activation, but if T cells are exposed to H2O2 in vitro for a prolonged period, their DNA-binding capacity will be inhibited (53). Therefore, uncontrolled increases in the concentrations of these oxidants may lead to indiscriminate oxidative damage and altered response patterns in proteins, lipids, polysaccharides, and DNA, resulting in growth stagnation and death (8, 54–56). Research has shown that the pathogenesis of many diseases is linked to high concentrations of local ROS and oxidative damage, including cancer, diabetes, and neurodegenerative diseases (9, 10).
The duality of the roles of ROS is also evident in the physiological activity of T cells. Evidence shows that medium or low concentrations of ROS in T cells act as intracellular signaling molecules during homeostasis and antigen recognition. ROS levels and localization can alter the redox status of effector proteins and transcription factors (TFs), which could affect T-cell responses (45). However, high physiological concentrations of ROS can cause reversible and irreversible damage to cellular molecules and participate in the pathogenesis of numerous diseases. For example, physiological concentrations of ROS can participate in the activation and proliferation of T cells by activating TFs, such as NFAT, NF-κB, and AP-1 (57); however, if T cells are exposed to H2O2 for a long time in vitro, the DNA-binding ability of NFAT and NF-κB is selectively inhibited, resulting in the downregulation of IL-2 transcription (53). Long-term or chronic ROS upregulation can also lead to T-cell homeostasis disorders, mitochondrial membrane polarization, and T-cell failure and non-response (58, 59). Therefore, maintaining a stable physiological concentration of ROS in T cells or maintaining the redox balance is essential for the metabolism and function of T cells. Cells express a variety of antioxidant enzymes, such as SODs, catalase, peroxidase reductase, thioredoxin system (Trx), glutathione (GSH), and other small-molecule antioxidants to remove excess ROS, thereby maintaining the redox balance in cells (60–62). The same occurs in T cells. When the T-cell receptor (TCR) is stimulated, it is accompanied by the rapid production of a large amount of ROS. Simultaneously, antigen-presenting cells can secrete cysteine, which is absorbed by T cells to generate GSH, thereby avoiding the oxidative stress caused by the excessive production of ROS over a short period (63, 64). In the body, the main producers of ROS include localized and compartmentalized organelles and related enzymes, and the sources of ROS production in vivo can be divided into enzymatic and non-enzymatic pathways (9, 10, 38) (Figure 1). Mitochondria are the main source of aerobic energy in eukaryotes. The electron transport chain (ETC) is a continuous reaction system consisting of four membrane protein complexes and lipid-soluble electron carriers in a certain order, used to convert the reduction potential into a proton gradient across the membrane, accompanied by electron transfer to bind to oxygen molecules and produce water and generate adenosine triphosphate (ATP) for energy. ROS are produced by the electrons that “leak” from respiratory chain complexes (11). In addition to the mitochondria, ROS are byproducts of other cell compartments, including the cytoplasm, cell membrane, ER, and peroxisomes. Enzymatic sources include NADPH oxidases (nicotinamide adenine dinucleotide phosphate, NOX), located on the cell membrane of polymorphonuclear cells, macrophages, endothelial cells (ECs) (47, 48, 65, 66); cytochrome P450-dependent oxygenases (18, 19); monoaminoxidase (MAO) (67, 68); a-glycerophosphate dehydrogenase (69, 70); electron transfer flavoprotein (ETF), and ETF quinone oxidoreductase (ETF dehydrogenase) (71, 72) (Figure 1). Several major ROS production mechanisms are described in the following section.
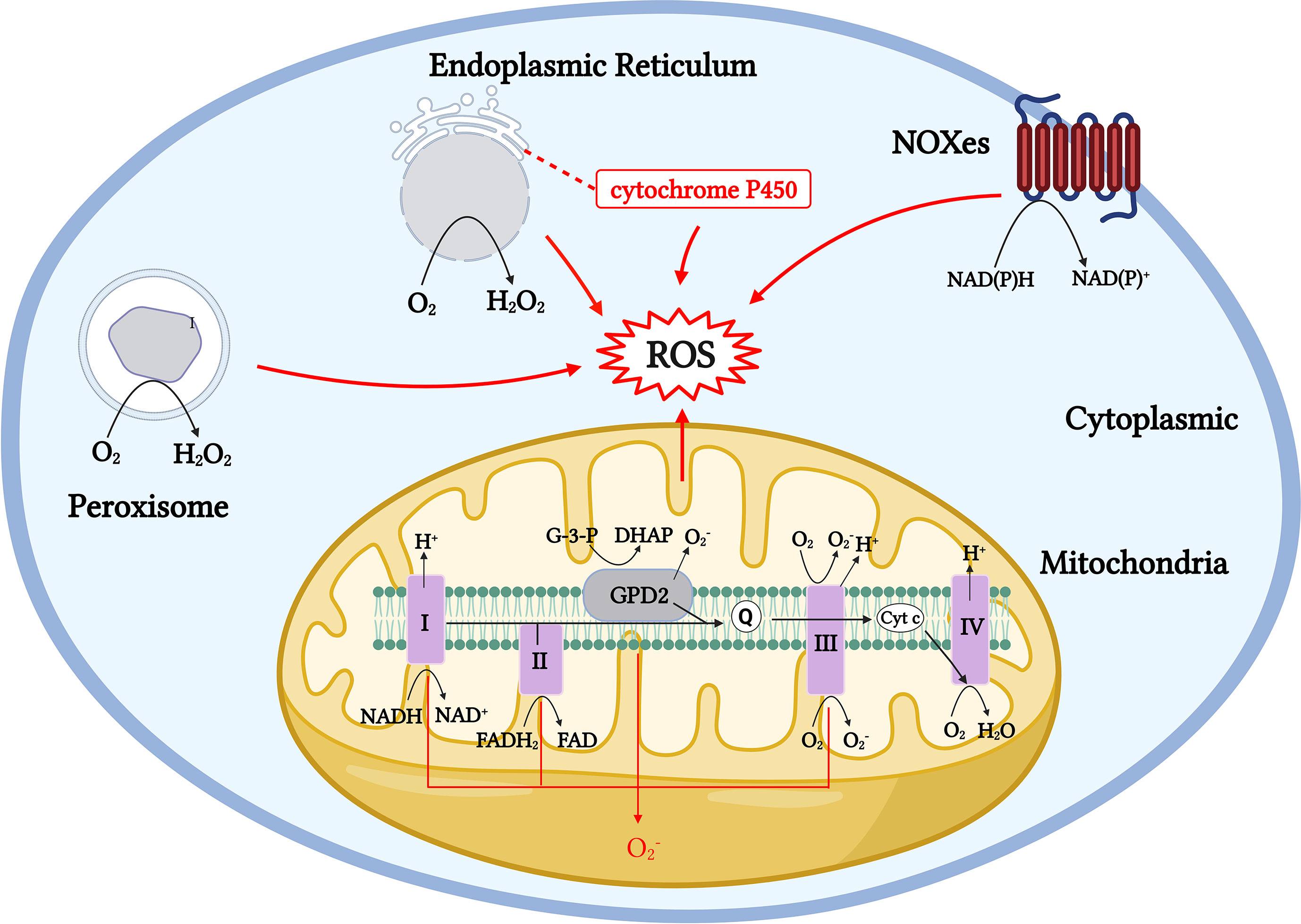
Figure 1 Generation pathway models of ROS production throughout the cell. Mitochondrial ROS are generated by numerous mechanisms, including Complexes I to III. Cytoplasmic ROS production mainly relies on the NADPH oxidase (NOX) family, and NOX proteins produce through NADPH electron exchange. The ER produces H2O2 by transferring acquired electrons through a flavin adenine dinucleotide cofactor to molecular oxygen. The membrane-associated monooxygenase system also produces ROS through cytochrome P450. Peroxisomes produce H2O2 from through oxidases, including ACOX and d-amino acid oxidase.
Generation pathway of ROS in cells
Mitochondria-derived ROS are mainly generated by membrane protein Complexes I and III and lipid-soluble electron carriers through various complex mechanisms in the mitochondria. The electrons used to generate ROS enter the respiratory chain, starting with Complex I (NADH-ubiquinone oxidoreductase) to produce a large amount of . Complex I mainly depends on two mechanisms: when the matrix NADH/NAD+ ratio is high, the flavin mononucleotide (FMN) site on Complex I is reduced, and when the ubiquinone pool is over-reduced, the reduction potential of the ubiquinone/ubiquinol redox pair favors the reduction of Complex I at the Q binding site (site IQ) (73, 74). Complex II and mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH; GPD2) have been shown to drive RET and produce mitochondrial ROS from Complex I (75–78). O2 interacts with reduced FMN to generate and produced in Complex I, which is released into the mitochondrial matrix and converted to H2O2 by manganese superoxide dismutase (MnSOD). As a major component in the production of mitochondrial-derived ROS, the impaired function of Complex I leads to excessive superoxide production and is involved in the pathogenesis of Parkinson’s disease (PD) and various neurodegenerative diseases. Parkinson-related mutations lead to increased production of mitochondrial superoxide and other ROS, in addition to localized high ROS concentrations in this region because of a lack of GSH in the substantia nigra, making it vulnerable to oxidative damage (79–82). The reducing equivalents formed in Complexes I, II, and GPD2 are passed to the Q-cycle of Complex III (ubiquinone-cytochrome c oxidoreductase) for further processing, where GPD2 catalyzes the unidirectional conversion of glycerol-3-phosphate (G-3-P) to dihydroxyacetone phosphate (DHAP) (78) (Figure 1). Complex III emits into the matrix and intermembrane space. The formation of in Complex III can enter the cell membrane from the intermembrane space through voltage-dependent anion channels without prior conversion to H2O2, unlike Complex I, which requires converting to H2O2 before release into the mitochondrial matrix. Studies have shown that the production of in Complex III is much lower than that in Complex I and is therefore negligible under physiological conditions (11, 83, 84). Complex II, often referred to as succinate-coenzyme Q reductase, uses succinate to reduce coenzyme Q to QH2 using covalently bound FAD as a coenzyme to produce reduced flavin adenine nucleotides (FADH2) (Figure 1). In addition to mitochondrial ROS from Complexes I, II, and III, enzymes, such as the α-ketoglutarate dehydrogenase (KGDHC) and pyruvate dehydrogenase (PDC) complexes, involved in mitochondrial metabolism also produce ROS through forward electron transfer, and both act as important sources of ROS in the mitochondria (85–87) (Figure 1).
Cytoplasmic ROS regulates the pentose phosphate pathway (PPP), glycolytic pathways, and other physiological activities. The NADPH oxidase (NOXes) family is a major source of cytoplasmic ROS, and NOX-dependent ROS production is involved in many physiological and metabolic activities and disease pathogeneses (12, 13). ROS produced by NOX are essential for the oxidative burst, in which several innate immune cells kill engulfed pathogens (44). NOX2 and DUOX1 are likely the major NOX isoforms in T cells, and once TCR is stimulated, NOX 2 transfers electrons to oxygen to produce (88) (Figure 2). In the ER, protein folding is highly sensitive to changes in redox homeostasis and is one of the main sources of H2O2 production. H2O2 is the main ROS molecule produced by the ER. In the ER, ER oxidoreductin 1 (ERO1) accepts electrons from peptide substrates via protein disulfide isomerases (PDI) and transfers them for molecular oxygen generation to produce H2O2 (14, 15, 89, 90). A large proportion of aerobic metabolism in the body is conducted with the involvement of the peroxisome, which transfers hydrogen from substrates to O2 to produce H2O2 through a variety of oxidases. The types of oxidases that function in different tissues vary markedly (16, 17, 91) (Figure 1).
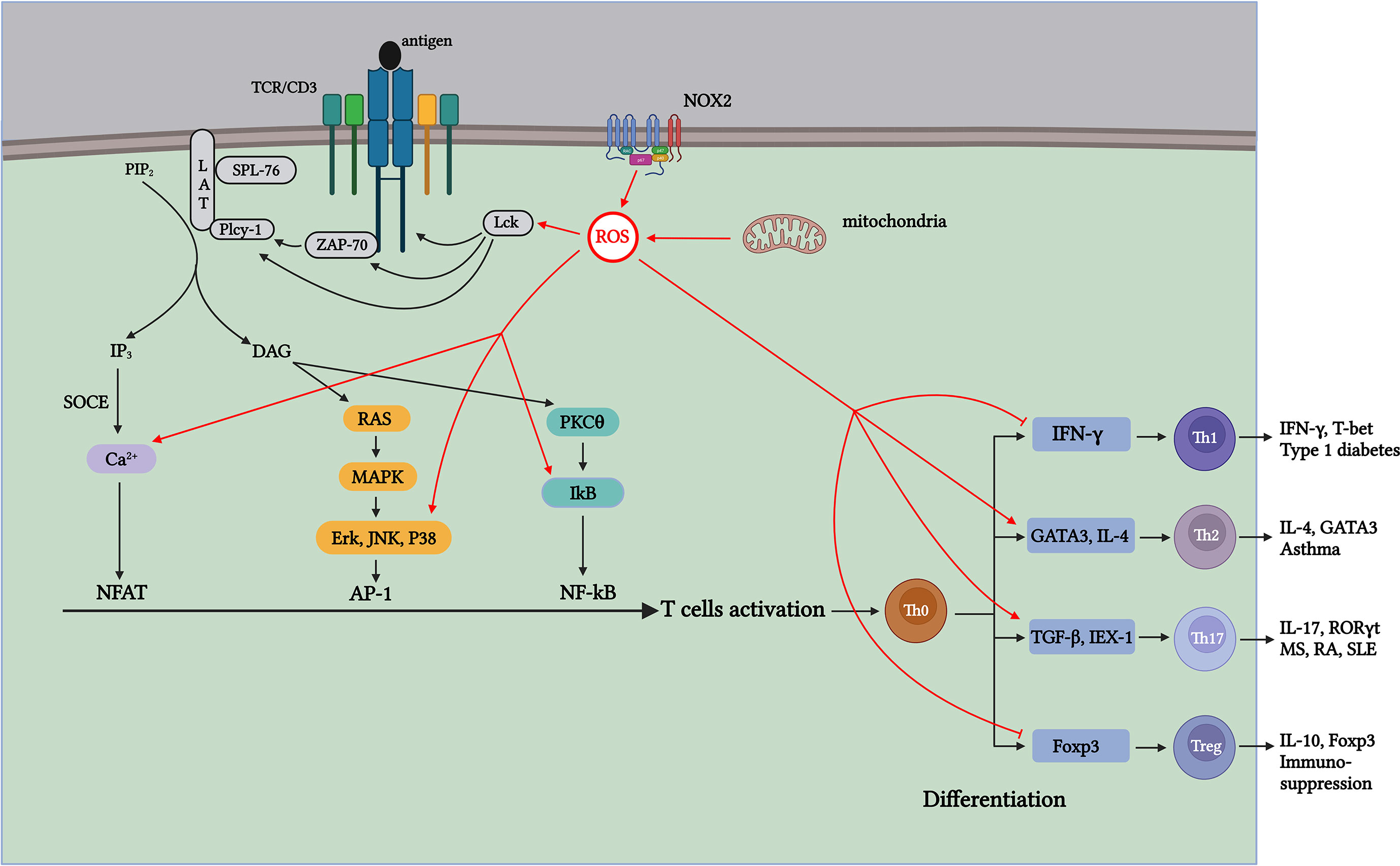
Figure 2 ROS generated during T-cell activation affects TCR signaling and CD4+ T-cell differentiation through distinct mechanisms. Antigen-stimulated TCR induces mitochondria and NOX2 to produce ROS, which promotes T-cell activation and proliferation by further activating signaling molecules in the TCR signaling pathway. Th1 cells contribute to type 1 diabetes (T1D); Th17 cells contribute to pathogenesis, including multiple sclerosis (MS), RA, and systemic lupus erythematosus (SLE); Th2 cells participate in the development of asthma; and Treg cells exert suppressive immune regulation. ROS produced during aerobic glycolysis affects the activation and differentiation of CD4+ T cells through various mechanisms, thus affecting the process of autoimmune diseases mediated by different T-cell subsets.
Role of ROS in activation of CD4+ T cells
T cells are central to immune system function. They mediate adaptive immune responses, complement the humoral immune response, and develop immune tolerance to autoantigens. T cells are also essential for specific defense against pathogenic microorganisms. Quiescent lymphocytes and monocytes require minimal energy compared to their activated counterparts, and these demands are met primarily by oxidative phosphorylation. When resting naïve T cells are stimulated by antigens to meet the energy required for T-cell activation, they undergo metabolic reprogramming to enhance activity, biosynthesize intermediates, and construct signaling molecules to spread anabolism, thereby initiating the rapid proliferation and differentiation of CD4+ T cells (92). When an antigen stimulates TCR, it binds to the co-stimulatory molecule CD28 and secretes cytokines, such as IL-2, which induce the activation of TF Akt. Activated Akt initiates the mammalian rapamycin (mTOR) pathway, reprogramming energy metabolism from oxidative phosphorylation to glycolysis. These changes increase nutrient uptake and glucose metabolism rates, promoting the activation and proliferation of naïve T cells (21, 22, 93). Additionally, Myc participates in upregulating aerobic glycolytic metabolism in T cells to meet energy demands for activation and proliferation (93). Therefore, the energy source for CD4+ T-cell differentiation mainly relies on aerobic glycolysis; however, energy sources for differentiating different CD4+ T-cell subsets are also distinct. T helper cells depend on aerobic glycolysis and reduce lipid oxidation, whereas Tregs mainly depend on fatty acid oxidation (FAO) as the main source of energy for metabolism (23–30, 93–99) (Figure 2).
ROS are the byproducts of oxidative metabolism. Owing to the increased demand for energy metabolism during T-cell activation, the local concentration of ROS increases rapidly within a short period (100). ROS produced during T-cell activation have two main physiological sources: mitochondria and the NADPH oxidase complex (88, 101). As a major source of ROS, the mitochondrial ETC transfers electrons from NADH and FADH2 to Complex IV to generate water. During the electron transfer process, a small portion of the “leaked” electrons can react non-enzymatically with O2 to generate (102, 103) (Figure 1). Studies have shown that when the functional activity of the respiratory chain Complex III is specifically inhibited in T cells, the energy generated by mitochondrial respiration is insufficient for the activation of T cells and the secretion of sufficient cytokines IL-2 and IL-4 (100, 104–108). Continuous treatment of pre-activated primary human T cells with ciprofloxacin inhibited TCR-induced ROS production and IL-2 and IL-4 expression, and this inhibitory effect was significantly correlated with the dose of ciprofloxacin. Studies have shown that ROS production in Complex I in resting and pre-activated human T cells is essential for activation-induced IL-2 and IL-4 expression and secretion when TCR is stimulated (109). Studies have shown that the production of mROS is significantly limited when T cells lacking Complexes I and III are activated, resulting in the decreased expression and proliferation of IL-2 and IL-4, which affects the activation process (109). Therefore, the results show that mitochondrial metabolism, especially the production of ROS in mitochondrial Complexes I and III, is important for T-cell activation. In addition to the mitochondria, the NOX protein family is a major ROS producer during CD4+ T-cell activation (88). The NOX enzyme family consists of seven members (NOX 1–5 and two dioxygenases [DUOX], namely 1 and 2). In T cells, TCR-induced ROS production increases mainly depend on two major NOX subtypes: the phagocyte subtype NOX2 and the non-phagocyte subtype DUOX1 (88, 110). The mROS produced by TCR upon antigen stimulation can activate NOX2, which maintains intracellular ROS levels, thereby promoting T-cell activation and proliferation (102). Studies have shown that the lack of NOX2 leads to a significant decrease in and H2O2 in T cells; however, the reduction in NOX2-derived ROS has little effect on T-cell activation and proliferation. Therefore, whether the ROS generated from NOX2 play a crucial role in T-cell activation requires further investigation (111) (Figure 2). DUOX1 is also a component of the redox signal after TCR stimulation, and inhibition of DUOX1 expression significantly reduces anti-CD3-mediated H2-DCFDA oxidation (112, 113).
When stimulated by antigens, TCR signaling is triggered in the plasma membrane, leading to IL-2 production, which further drives the activation and proliferation of T cells. ROS produced during this process, in turn, act as key signaling molecules that regulate the activation of T cells (107, 108, 114). The immunoreceptor tyrosine-based activation motifs (ITAMs) in the cytoplasmic region of the TCR contain two core tyrosines. Tyrosine lymphocyte-specific protein tyrosine kinase (Lck) phosphorylation in ITAM is activated after antigen stimulation (115). Studies have shown that T-cell development is notably blocked in Lck-specific knockout mice (116). Simultaneously, phosphorylated ITAM is further activated by recruiting ZAP-70, and the lack of ZAP-70 can significantly affect signal transduction downstream of the TCR (117, 118). The binding of ZAP-70 to ITAM also activates ZAP-70. Activated ZAP-70 forms a complex signal skeleton by phosphorylating LAT and SLP-76 for signal diversification (119). The formation of the LAT signaling complex activates PLCγ-1 to produce the second messengers diacylglycerol (DAG) and inositol 3,4,5-triphosphate (IP3). DAG activates NF-κB and initiates the Ras-ERK signaling cascade pathway, activating AP-1 (120, 121). Activation of the receptor of the second messenger IP3 leads to the storage of Ca2+ entry (SOCE) in the ER membrane, and the influx of Ca2+ signal transduction activates the TF NFAT (122, 123). At the transcriptional level, the activities of TFs NFAT, AP-1, and NF-κB induce the expression of IL-2 mRNA, which promote T-cell activation and proliferation (124) (Figure 2). Various signaling molecules in the TCR signal transduction pathway contain cysteine residues sensitive to oxidation. Therefore, ROS generated during activation affects TCR signal transduction after antigen stimulation. ROS inducers can promote the formation of lipid rafts on the plasma membrane, which contain important molecules involved in TCR signaling, such as LAT, PLCγ1, and PKCθ (125) (Figure 2). The activation of AP-1 requires the regulation of MAPK members, whereas the phosphorylation of Erk, JNK, and P38, which are important components of the MAPK pathway, depends on H2O2 (126, 127) (Figure 2). Because tyrosine kinases can regulate IκB (an NF-κB inhibitor) (128), H2O2 can indirectly regulate the TF NF-κB activation via tyrosine kinase Lck and ZAP-70, thereby activating gene transcription (129, 130) (Figure 2). In addition, research has shown that ROS produced in the mitochondria during T-cell activation affects Ca2+ homeostasis in a concentration-dependent manner, activating the TF NFAT (107, 131). Therefore, ROS production upon antigen stimulation of TCR promotes TCR signaling and transcriptional activation of IL-2 (Figure 2).
Because CD4+ T cells have different subtypes, the energy metabolism for the proliferation and differentiation of different subtypes also varies. Next, we describe the effects of mitochondrial ROS production on differentiating CD4+ T cells.
Role of ROS in CD4+ T-cell differentiation
Within the CD4+ T-cell subpopulation, Th1 cells were among the first cells identified to be involved in the immune response against foreign antigens, such as microbes. They play an important role in mediating cellular immunity through the secretion of IL-2, TNF-α, and other cytokines and in clearing intracellular pathogens through the secretion of IFN-γ and lymphotoxins. Additionally, Thl cells are involved in the pathogenesis of many autoimmune diseases in vivo, with T1D being the most notable (132). Th1 cells induction begins with antigen-presenting cells (APCs) secreting IL-12, and the IL-12 induces natural killer cells (NK) to produce cytokine IFN-γ. The differentiation process of Th1 cells is tightly regulated by a feedback loop, with Th1 differentiation mainly regulated by TF T-bet, which can promote IFN-γ secretion by upregulating IL-12Rβ2 receptor expression and T-bet expression regulated by signal transducer and activator of transcription 1 (STAT1), which is in turn activated by IFN-γ secreted by TH1 cells (133). When the feedback loop in activated T cells is disrupted, reduced IL-12 secretion decreases that of IFN-γ and T-bet, thereby affecting the Th1 cell-mediated immune response (134–140).
Insulin is a key hormone secreted by B cells that can promote glucose uptake and glycolysis in the liver and muscle cells by converting glucose into glycogen, thereby reducing blood glucose concentration in response to high glucose. A decrease in insulin levels causes excessive blood glucose concentrations, leading to the occurrence and development of diabetes (141). It was demonstrated that Th1 cells with the specific diabetic TCR could induce T1D in NOD mice, and the Th1 cells marker IFN-γ is directly involved in the process of T1D diseases (142). Research shows that IFN-γ can participate in the progress of T1D diseases through several pathways, which include mediating beta cell death by stimulating the cytotoxic CD8 T cells response (142). Therefore, constraining IFN-γ responses (e.g., enhancing Treg function and/or inhibiting Th1 cells differentiation/function) may prevent the onset of diabetes (143–147).
Differentiation of Th1 cells primarily relies on aerobic glycolysis to promote the secretion of T-bet and IFN-γ. Research has shown that in Glut1-deficient Teff cells, owing to their decreased glucose transport capacity, the resulting Th1 cells decreased and showed less pronounced colitis symptoms in mouse models. As a necessary regulator that promotes the differentiation of Th1 cells, studies have revealed that lactate dehydrogenase A (LDHA) is a key factor supporting aerobic glycolysis and promoting IFN-γ expression, driving naïve T cells to differentiate into Th1 cells. In this study, the elimination of LDHA in T cells resulted in the inhibition of IFN-γ overexpression in mice, restoration of normal Treg cell function, and reduction in immunopathological damage (148–150). Another relevant study showed that H2O2 enhances IL-4 production, downregulates IFN-γ production, and promotes the naïve T cells into the Th2 lineage without altering cell proliferation (151, 152). Therefore, reducing autoreactive Th1-type CD4+ T-cell glycolysis and/or inhibiting antioxidant activity could be potential strategies to prevent the development of T1D (Figure 2).
In contrast to the two classical lineages, Th1 and Th2 cells, IL-17-producing T helper 17 (Th17) cells have been classified as an important emerging inflammatory effector CD4+ T-cell subset. Th17 cells cause chronic tissue inflammation and organ failure (153–156). The differentiation of Th17 cells is mainly accomplished by the involvement of IL-6, IL-23, IL-21, and transforming growth factor-β (TGF-β), with TGF-β being the critical cytokine for Th17 differentiation (157–160). Th17 cells stimulate tissue cells to secrete antimicrobial peptides, enhance the immune barrier function of epithelial tissue, and stimulate the local production of cytokines, such as chemokines, which induce an inflammatory response dominated by neutrophils and monocytes (161, 162). In addition, many studies have shown that pro-inflammatory (pathogenic) Th17 cells are involved in the pathogenesis of a variety of inflammatory and autoimmune diseases, such as multiple sclerosis (MS), RA, psoriasis, and inflammatory bowel disease (IBD). Stimulated neoplastic T cells destroy myelin sheaths and axons by generating a pro-inflammatory response, thereby inducing experimental autoimmune encephalomyelitis (EAE), a common experimental model used to study MS (163–166).
Similar to that in Th1 cells, aerobic glycolysis is the energy source for Th17 cell differentiation. The increase in aerobic glycolysis during T-cell activation results in a significant increase in the transport of glucose and amino acids for biosynthesis and energy supply, thus highlighting the importance of Glut 1. Studies have shown that Glut 1 deficiency reduces the efficiency of glycolysis, which in turn affects T-cell activation and proliferation, and a significant number of cells undergo apoptosis (150). IEX-1 affects the proliferation, differentiation, and survival of these cells by accelerating ATP hydrolysis, which in turn impedes ROS production. It has been shown that IEX-1 deficiency promotes the differentiation of Th17 cells. This process is mediated by increased mitochondrial ROS production (167). In addition, research has shown that the upregulation of ROS produced by oxidative metabolism during T-cell activation promotes the differentiation of Th17 cells by activating TF TGF-β. In this study, high glucose-induced upregulation of mitochondrial ROS drives Th17 cell differentiation by activating TGF-β and exacerbating autoimmunity in a mouse model of colitis and EAE (168). Therefore, targeting the immune metabolism to treat Th17 cell-mediated autoimmune diseases has broad application prospects (169) (Figure 2).
Th2 cells, another traditional genealogy in the subset of CD4+ T cells, are naïve T cells that differentiate into Th2 cells by producing IL-4 by inducing the expression of the key TF Gata3. Th2 cells can express lineage-defining TFs, GATA3 and STAT6. GATA3 contributes to the Th2 phenotype by inducing IL-4 to form a positive feedback loop (170–173). However, no known Th2-mediated autoimmune diseases involving Th1 or Th17 cells exist. Th2 cells assist in the proliferation and differentiation of B cells into plasma cells by secreting cytokines, such as IL-4, IL-5, and IL-13, inducing the transformation of macrophages into the M2 phenotype and the recruitment of eosinophils, thereby protecting against worms, poisons, and certain bacteria, and stimulating tissue healing (174–179). In addition, Th2 cells are involved in the immune response and pathogenesis of allergic diseases, including asthma and atopic dermatitis. Disease progression in animal models of RA can be influenced by modulation of the balance between Th1 and Th2 cells. Therefore, the Role of Th2 cells in autoimmune diseases has received increasing attention (180–183).
Differentiation into Th2 cells is dependent on mTOR activity, which is reduced when mTOR activity is inhibited by rapamycin (184, 185). Moreover, the anti-CD3 antibody can reduce ROS production by mitochondrial Complex I and inhibit the expression of IL-2 and IL-4, thereby inhibiting Th2 cell differentiation (109). Research has confirmed that by downregulating the synthesis of superoxide anions and nitric oxide, IL-4 and IL-13 can be reduced, and IL-1β production increased, transforming Th2 into a Th1 response (Figure 2 ). Thus, avoiding airway hyperresponsiveness (AHR) in an asthma model effectively improves the symptoms of patients with asthma (186, 187).
Th1, Th2, and Th17 cells directly or indirectly mediate autoimmune diseases and elicit immune responses against foreign fungi, parasites, and other infections. Tregs are key regulators of inflammation and autoimmunity. They can exert negative immune regulation through various mechanisms important for maintaining self-tolerance and immune homeostasis in multiple tissues, thereby avoiding excessive damage to the body from immune responses. Unlike other CD4+ T-cell subsets, the energy source for Treg cells differentiation rely mainly on FAO rather than aerobic glycolysis (28–30, 188). The differentiation conditions for Treg cells were similar to those for Th17 cells. TGF-β is required for the differentiation of both Th17 and Treg cells. Treg cells can be induced when only TGF-β is present in the culture conditions, while TGF-β and IL-6 preferentially induce Th17 cells. Studies have shown that IL-6 can increase glucose metabolism by promoting glucose and glycogen binding and glucose oxidation in skeletal muscles, whereas the differentiation of Tregs mainly relies on FAO (28–30, 189–193). Foxp3 is a key lineage-defining TF for Treg cells that inhibits the differentiation of naïve T cells toward Th17 cells by limiting RORγt activity and, together with other regulators, maintains the development and function of Treg cells (194–198). Although Tregs are not directly involved in the immune response to foreign antigens, they are necessary for maintaining immune tolerance to autoantigens and immune homeostasis in vivo. The exhaustion of Treg cells in the body can lead to severe autoimmune diseases, and the lack of CTLA-4 is the key molecule causing this problem (199–202). Tregs exert suppressive immune regulation through multiple mechanisms, including the secretion of IL-35, IL-10, and other soluble suppressive immune molecules, to suppress effector signals directly. Tregs can produce high-affinity IL-2 receptors that bind IL-2 competitively with T cells, inhibiting the proliferation and apoptosis of activated T cells. Tregs can also induce apoptosis in a perforin-dependent manner via granzymes A and B (201, 203–207).
Unlike Teffs, which depend on aerobic glycolysis for differentiation, Tregs rely on FAO as their energy source (28–30). Foxp3 expressed by Treg cells can bind to the Myc promoter and suppress Myc gene expression to inhibit glycolysis, thereby stabilizing function and activity of Treg cells (93). Therefore, inhibiting the activity of aerobic glycolysis, such as inhibiting the activity of the glucose transporter Glu1, will seriously impact the differentiation of Teffs but will not affect the activity and function of Treg cells. Tregs can also use this feature to treat autoimmune diseases (208, 209). Studies have shown that inhibiting ROS production during oxidative metabolism reduces the suppressive effect of ROS on Treg cells, thereby regulating Th17/Treg cells and effectively improving psoriasis symptoms in mice (210) (Figure 2).
Conclusion
ROS are byproducts of aerobic metabolism. Studies related to immune metabolism have shown that T cell-derived ROS and immune metabolic reprogramming further affect the outcome of the activation and differentiation of naïve T cells. Studies have shown that the pathogenesis of numerous autoimmune diseases is strongly correlated with CD4+ T cells and mitochondrial dysfunction, which leads to oxidative stress and may affect disease progression by altering CD4+ T-cell status, thereby interfering with normal therapeutic strategies and causing unexpected suffering in patients. Therefore, studying the specific effects of ROS in autoimmune diseases has significant implications for exploring more effective treatments. Current treatment options include global immunosuppression, immune-depleting antibodies, and anti-cytokine therapies. Although these treatments primarily target immune cells involved in pathogenesis, they inevitably damage normal functional cells in the body, leading to increased susceptibility to other diseases and complications. Because CD4+ T-cell activation and T helper cells differentiation are highly dependent on aerobic glycolysis, inhibition of autoreactive T-cell activation and T helper cells differentiation by targeting glycolysis or ROS generation without damaging systemic immune function is a promising direction for solving the problems of current treatment options. Many studies have been conducted using optimized in vitro cell culture media; however, it remains unknown how the metabolic microenvironment in healthy or disease-affected organs affects ROS production in vivo and, consequently, T-cell activation and differentiation. The Role of ROS in the effector function of T cells in vivo needs to be fully explored to exploit this property for better disease treatment. Antioxidants, such as GSH and SOD, can specifically remove oxidants, such as ROS, and maintain the redox balance in the body, thereby effectively preventing the damage caused by oxidative stress. Therefore, using antioxidants as a breakthrough in treating autoimmune diseases by targeting ROS has received extensive attention. However, because GSH cannot cross the blood-brain barrier and has a short half-life, oral administration of GSH does not significantly improve disease progression. Therefore, if the key issue of efficiently using antioxidants can be addressed, it will provide a new immunotherapeutic approach to suppress T cell-mediated autoimmune diseases. Improving self-tolerance by promoting Treg differentiation is an alternative therapy for autoimmune diseases.
Author contributions
PS drafted the manuscript. HL edited the manuscript. JZ, YL, and WC revised the manuscript. DZ supervised the study and edited the manuscript. All the authors contributed to the study and approved the manuscript for publication.
Funding
This work was supported by the Key Project of the Science and Technology Department of Sichuan Province (NO. 2022YFH0100), the National Natural Science Foundation of China (NO. 82171829), the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (NO. ZYYC21012), and Fundamental Research Funds for Central Universities (20822041E4084).
Acknowledgments
DZ sincerely wants to commemorate Dr. Sang-A Park, who passed away suddenly on January 22, 2018.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol (2019) 26:101284. doi: 10.1016/j.redox.2019.101284
2. Sies H. Oxidative eustress and oxidative distress: introductory remarks. Oxid Stress. Elsevier (2020), 3–12. doi: 10.1016/B978-0-12-818606-0.00001-8
3. Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem (2017) 86:715–48. doi: 10.1146/annurev-biochem-061516-045037
4. Halliwell B, Gutteridge JM. Free radicals in biology and medicine: Oxford university press, USA. (2015). doi: 10.1093/acprof:oso/9780198717478.001.0001
5. Liochev SI, Fridovich I. Superoxide and iron: partners in crime. IUBMB Life (1999) 48(2):157–61. doi: 10.1080/713803492
6. Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biol Med (2000) 29(3-4):222–30. doi: 10.1016/S0891-5849(00)00317-8
7. Bisht S, Faiq M, Tolahunase M, Dada R. Oxidative stress and Male infertility. Nat Rev Urol (2017) 14(8):470–85. doi: 10.1038/nrurol.2017.69
8. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol (2003) 552(2):335–44. doi: 10.1113/jphysiol.2003.049478
9. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev (2002) 82(1):47–95. doi: 10.1152physrev.00018.2001
10. Sies H, Jones DP. Reactive oxygen species (Ros) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21(7):363–83. doi: 10.1038/s41580-020-0230-3
11. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417(1):1–13. doi: 10.1042/BJ20081386
12. Paik J-Y, Jung K-H, Lee J-H, Park J-W, Lee K-H. Reactive oxygen species-driven Hif1α triggers accelerated glycolysis in endothelial cells exposed to low oxygen tension. Nucl Med Biol (2017) 45:8–14. doi: 10.1016/j.nucmedbio.2016.10.006
13. Baillet A, Hograindleur MA, El Benna J, Grichine A, Berthier S, Morel F, et al. Unexpected function of the phagocyte nadph oxidase in supporting hyperglycolysis in stimulated neutrophils: key role of 6-Phosphofructo-2-Kinase. FASEB J (2017) 31(2):663–73. doi: 10.1096/fj.201600720R
14. Hansen HG, Schmidt JD, Søltoft CL, Ramming T, Geertz-Hansen HM, Christensen B, et al. Hyperactivity of the Ero1α oxidase elicits endoplasmic reticulum stress but no broad antioxidant response. J Biol Chem (2012) 287(47):39513–23. doi: 10.1074/jbc.M112.405050
15. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants Redox Signaling (2014) 21(3):396–413. doi: 10.1089/ars.2014.5851
16. Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors (2009) 35(4):346–55. doi: 10.1002/biof.48
17. Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, et al. Atm functions at the peroxisome to induce pexophagy in response to ros. Nat Cell Biol (2015) 17(10):1259–69. doi: 10.1038/ncb3230
18. Coon MJ, Ding X, Pernecky SJ, Vaz AD. Cytochrome P450: progress and predictions. FASEB J (1992) 6(2):669–73. doi: 10.1096/fasebj.6.2.1537454
19. Manikandan P, Nagini S. Cytochrome P450 structure, function and clinical significance: a review. Curr Drug Targets (2018) 19(1):38–54. doi: 10.2174/1389450118666170125144557
20. Norman PS. Immunobiology: the immune system in health and disease. J Allergy Clin Immunol (1995) 96(2):274. doi: 10.1016/S0091-6749(95)70025-0
21. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259. doi: 10.1146/annurev-immunol-032712-095956
22. Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
23. Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol (2010) 22(3):314–20. doi: 10.1016/j.coi.2010.01.018
24. Wang T, Marquardt C, Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature (1976) 261(5562):702–5. doi: 10.1038/261702a0
25. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by Erk/Mapk during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
26. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory Cd4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
27. Chang C-H, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
28. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi: 10.1038/nm.3704
29. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93. e7. doi: 10.1016/j.cmet.2016.12.018
30. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, et al. Mitochondrial complex iii is essential for suppressive function of regulatory T cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
31. Murphy MP, Holmgren A, Larsson N-G, Halliwell B, Chang CJ, Kalyanaraman B, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab (2011) 13(4):361–6. doi: 10.1016/j.cmet.2011.03.010
32. Hawkins CL, Davies MJ. Detection, identification, and quantification of oxidative protein modifications. J Biol Chem (2019) 294(51):19683–708. doi: 10.1074/jbc.REV119.006217
33. Brandes RP, Rezende F, Schröder K. Redox regulation beyond ros: why ros should not be measured as often. Circ Res (2018) 123(3):326–8. doi: 10.1161/CIRCRESAHA.118.313146
34. Di Meo S, Reed TT, Venditti P, Victor VM. Role of ros and rns sources in physiological and pathological conditions. Oxid Med Cell Longevity (2016) 2016. doi: 10.1155/2016/1245049
35. Bredt D, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem (1994) 63(1):175–95. doi: 10.1146/annurev.bi.63.070194.001135
36. Radi R. Peroxynitrite, a stealthy biological oxidant. J Biol Chem (2013) 288(37):26464–72. doi: 10.1074/jbc.R113.472936
37. Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med (1999) 31(2):53–9. doi: 10.1038/emm.1999.9
38. Hoidal JR. Reactive oxygen species and cell signaling. Am J Respir Cell Mol Biol (2001) 25(6):661–3. doi: 10.1165/ajrcmb.25.6.f213
39. Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem (2001) 11(4):173–86. doi: 10.1159/000047804
40. Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants Redox Signaling (2006) 8(3-4):243–70. doi: 10.1089/ars.2006.8.243
41. D'Autréaux B, Toledano MB. Ros as signalling molecules: mechanisms that generate specificity in ros homeostasis. Nat Rev Mol Cell Biol (2007) 8(10):813–24. doi: 10.1038/nrm2256
42. Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry (2010) 49(5):835–42. doi: 10.1021/bi9020378
43. Sies H. Role of metabolic H2o2 generation: redox signaling and oxidative stress. J Biol Chem (2014) 289(13):8735–41. doi: 10.1074/jbc.R113.544635
44. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol (2014) 15(6):411–21. doi: 10.1038/nrm3801
45. Reczek CR, Chandel NS. Ros-dependent signal transduction. Curr Opin Cell Biol (2015) 33:8–13. doi: 10.1016/j.ceb.2014.09.010
46. WinterbournChristine C. Biological production, detection, and fate of hydrogen peroxide. Antioxidants Redox Signaling (2018) 29(6):541–51. doi: 10.1089/ars.2017.7425
47. Babior BM. The nadph oxidase of endothelial cells. IUBMB Life (2000) 50(4-5):267–9. doi: 10.1080/15216540051080976
48. Vignais P. The superoxide-generating nadph oxidase: structural aspects and activation mechanism. Cell Mol Life Sci CMLS (2002) 59(9):1428–59. doi: 10.1007/s00018-002-8520-9
49. Babior B, Lambeth J, Nauseef W. The neutrophil nadph oxidase. Arch Biochem biophysics (2002) 397(2):342–4. doi: 10.1006/abbi.2001.2642
50. Liochev SI, Fridovich I. Lucigenin as mediator of superoxide production: revisited. Free Radical Biol Med (1998) 25(8):926–8. doi: 10.1016/S0891-5849(98)00121-X
51. Yokoyama Y, Beckman J, Beckman T, Wheat J, Cash T, Freeman B, et al. Circulating xanthine oxidase: potential mediator of ischemic injury. Am J Physiology-Gastrointestinal Liver Physiol (1990) 258(4):G564–G70. doi: 10.1152/ajpgi.1990.258.4.G564
52. Winterbourn CC. The biological chemistry of hydrogen peroxide. Methods enzymology (2013) 528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X
53. Flescher E, Ledbetter JA, Schieven GL, Vela-Roch N, Fossum D, Dang H, et al. Longitudinal exposure of human T lymphocytes to weak oxidative stress suppresses transmembrane and nuclear signal transduction. J Immunol (Baltimore Md: 1950) (1994) 153(11):4880–9. doi: 10.4049/jimmunol.153.11.4880
54. Stadtman ER, Levine RL. Protein oxidation. Ann New York Acad Sci (2000) 899(1):191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x
55. Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, et al. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation Novel Nitrogen-Containing Oxidized Lipid Derivatives. J Biol Chem (1994) 269(42):26066–75. doi: 10.1016S0021-9258(18)47160-8
56. Kaur H, Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett (1994) 350(1):9–12. doi: 10.1016/0014-5793(94)00722-5
57. Oliveira-Marques V, Marinho HS, Cyrne L, Antunes F. Role of hydrogen peroxide in nf-κb activation: from inducer to modulator. Antioxidants Redox Signaling (2009) 11(9):2223–43. doi: 10.1089/ars.2009.2601
58. Zhang B, Liu S-Q, Li C, Lykken E, Jiang S, Wong E, et al. Microrna-23a curbs necrosis during early T cell activation by enforcing intracellular reactive oxygen species equilibrium. Immunity (2016) 44(3):568–81. doi: 10.1016/j.immuni.2016.01.007
59. Bettonville M, d'Aria S, Weatherly K, Porporato PE, Zhang J, Bousbata S, et al. Long-term antigen exposure irreversibly modifies metabolic requirements for T cell function. Elife (2018) 7:e30938. doi: 10.7554/eLife.30938
60. Morris G, Anderson G, Dean O, Berk M, Galecki P, Martin-Subero M, et al. The glutathione system: a new drug target in neuroimmune disorders. Mol Neurobiol (2014) 50:1059–84. doi: 10.1007/s12035-014-8705-x
61. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the cuzn-sod (Sod1), Mn-sod (Sod2), and ec-sod (Sod3) gene structures, evolution, and expression. Free Radical Biol Med (2002) 33(3):337–49. doi: 10.1016/S0891-5849(02)00905-X
62. Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem (2012) 287(7):4403–10. doi: 10.1074/jbc.R111.283432
63. Rashida Gnanaprakasam JN, Wu R, Wang R. Metabolic reprogramming in modulating T cell reactive oxygen species generation and antioxidant capacity. Front Immunol (2018) 9:1075. doi: 10.3389/fimmu.2018.01075
64. Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci (2002) 99(3):1491–6. doi: 10.1073/pnas.022630299
65. Panday A, Sahoo MK, Osorio D, Batra S. Nadph oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol (2015) 12(1):5–23. doi: 10.1038/cmi.2014.89
66. Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. Nadph oxidases in cardiovascular health and disease. Antioxidants Redox Signaling (2006) 8(5-6):691–728. doi: 10.1089/ars.2006.8.691
67. Hauptmann N, Grimsby J, Shih JC, Cadenas E. The metabolism of tyramine by monoamine oxidase a/B causes oxidative damage to mitochondrial DNA. Arch Biochem biophysics (1996) 335(2):295–304. doi: 10.1006/abbi.1996.0510
68. Sandri G, Panfili E, Ernster L. Hydrogen peroxide production by monoamine oxidase in isolated rat-brain mitochondria: its effect on glutathione levels and Ca2+ efflux. Biochim Biophys Acta (BBA)-General Subj (1990) 1035(3):300–5. doi: 10.1016/0304-4165(90)90092-B
69. Mráček T, Pecinová A, Vrbacký M, Drahota Z, Houštěk J. High efficiency of ros production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch Biochem biophysics (2009) 481(1):30–6. doi: 10.1016/j.abb.2008.10.011
70. Drahota Z, Chowdhury SK, Floryk D, Mráček T, Wilhelm J, Rauchová H, et al. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J bioenergetics biomembranes (2002) 34(2):105–13. doi: 10.1023/A:1015123908918
71. Beckmann JD, Frerman FE. Reaction of electron-transfer flavoprotein with electron transfer flavoprotein-ubiquinone oxidoreductase. Biochemistry (1985) 24(15):3922–5. doi: 10.1021/bi00336a017
72. Ghisla S, Thorpe C. Acyl-coa dehydrogenases: a mechanistic overview. Eur J Biochem (2004) 271(3):494–508. doi: 10.1046/j.1432-1033.2003.03946.x
73. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol (2013) 1(1):304–12. doi: 10.1016/j.redox.2013.04.005
74. Orr AL, Ashok D, Sarantos MR, Shi T, Hughes RE, Brand MD. Inhibitors of ros production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radical Biol Med (2013) 65:1047–59. doi: 10.1016/j.freeradbiomed.2013.08.170
75. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex ii can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem (2012) 287(32):27255–64. doi: 10.1074/jbc.M112.374629
76. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ros. Nature (2014) 515(7527):431–5. doi: 10.1038/nature13909
77. Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167(2):457–70.e13. doi: 10.1016/j.cell.2016.08.064
78. Langston PK, Nambu A, Jung J, Shibata M, Aksoylar HI, Lei J, et al. Glycerol phosphate shuttle enzyme Gpd2 regulates macrophage inflammatory responses. Nat Immunol (2019) 20(9):1186–95. doi: 10.1038/s41590-019-0453-7
79. Hansford RG, Hogue BA, Mildaziene V. Dependence of H2o2 formation by rat heart mitochondria on substrate availability and donor age. J bioenergetics biomembranes (1997) 29(1):89–95. doi: 10.1023/A:1022420007908
80. Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT. Physiological phenotype and vulnerability in parkinson's disease. Cold Spring Harbor Perspect Med (2012) 2(7):a009290. doi: 10.1101/cshperspect.a009290
81. Kussmaul L, Hirst J. The mechanism of superoxide production by nadh: ubiquinone oxidoreductase (Complex I) from bovine heart mitochondria. Proc Natl Acad Sci (2006) 103(20):7607–12. doi: 10.1073/pnas.0510977103
82. Perry TL, Godin DV, Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency? Neurosci Lett (1982) 33(3):305–10. doi: 10.1016/0304-3940(82)90390-1
83. Trumpower BL. The protonmotive q cycle: energy transduction by coupling of proton translocation to electron transfer by the cytochrome Bc1 complex. J Biol Chem (Print) (1990) 265(20):11409–12. doi: 10.1016/S0021-9258(19)38410-8
84. Bleier L, Dröse S. Superoxide generation by complex iii: from mechanistic rationales to functional consequences. Biochim Biophys Acta (BBA)-Bioenergetics (2013) 1827(11-12):1320–31. doi: 10.1016/j.bbabio.2012.12.002
85. Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, et al. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci (2004) 24(36):7779–88. doi: 10.1523/JNEUROSCI.1899-04.2004
86. Mailloux RJ, Gardiner D, O’Brien M. 2-oxoglutarate dehydrogenase is a more significant source of O2·–/H2o2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radical Biol Med (2016) 97:501–12. doi: 10.1016/j.freeradbiomed.2016.06.014
87. Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce Superoxide/Hydrogen peroxide at much higher rates than complex I. J Biol Chem (2014) 289(12):8312–25. doi: 10.1074/jbc.M113.545301
88. Bedard K, Krause K-H. The nox family of ros-generating nadph oxidases: physiology and pathophysiology. Physiol Rev (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
89. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
90. Tavender TJ, Springate JJ, Bulleid NJ. Recycling of peroxiredoxin iv provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J (2010) 29(24):4185–97. doi: 10.1038/emboj.2010.273
91. Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem (2006) 75(1):295–332. doi: 10.1146/annurev.biochem.74.082803.133329
92. Zhu J, Yamane H, Paul WE. Differentiation of effector Cd4 T cell populations. Annu Rev Immunol (2009) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
93. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
94. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang C-Y, et al. Estrogen-related receptor-α is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci (2011) 108(45):18348–53. doi: 10.1073/pnas.1108856108
95. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity (2007) 27(2):173–8. doi: 10.1016/j.immuni.2007.07.008
96. Krauss S, Brand MD, Buttgereit F. Signaling takes a breath–new quantitative perspectives on bioenergetics and signal transduction. Immunity (2001) 15(4):497–502. doi: 10.1016/S1074-7613(01)00205-9
97. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7(1):11–20. doi: 10.1016/j.cmet.2007.10.002
98. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. Hif1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of Th17 and treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
99. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing Cd8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
100. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195(1):59–70. doi: 10.1084/jem.20010659
101. Lin D-S, Huang Y-W, Ho C-S, Hung P-L, Hsu M-H, Wang T-J, et al. Oxidative insults and mitochondrial DNA mutation promote enhanced autophagy and mitophagy compromising cell viability in pluripotent cell model of mitochondrial disease. Cells (2019) 8(1):65. doi: 10.3390/cells8010065
102. Kaminski M, Kießling M, Söss D, Krammer PH, Gölow K. Novel role for mitochondria: protein kinase cθ-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol (2007) 27(10):3625–39. doi: 10.1128/MCB.02295-06
104. Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, et al. Mitochondrial complex iii ros regulate adipocyte differentiation. Cell Metab (2011) 14(4):537–44. doi: 10.1016/j.cmet.2011.08.007
105. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ros generation are essential for kras-mediated tumorigenicity. Proc Natl Acad Sci (2010) 107(19):8788–93. doi: 10.1073/pnas.1003428107
106. Piganelli JD, Flores SC, Cruz C, Koepp J, Batinic-Haberle I, Crapo J, et al. A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes (2002) 51(2):347–55. doi: 10.2337/diabetes.51.2.347
107. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
108. Murphy MP, Siegel RM. Mitochondrial ros fire up T cell activation. Immunity (2013) 38(2):201–2. doi: 10.1016/j.immuni.2013.02.005
109. Kamiński MM, Sauer SW, Klemke C-D, Süss D, Okun JG, Krammer PH, et al. Mitochondrial reactive oxygen species control T cell activation by regulating il-2 and il-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol (2010) 184(9):4827–41. doi: 10.4049/jimmunol.0901662
110. Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nat Rev Immunol (2004) 4(3):181–9. doi: 10.1038/nri1312
111. Belikov AV, Schraven B, Simeoni L. Tcr-triggered extracellular superoxide production is not required for T-cell activation. Cell Communication Signaling (2014) 12(1):1–10. doi: 10.1186/s12964-014-0050-1
112. Kwon J, Shatynski KE, Chen H, Morand S, De Deken X, Miot F, et al. The nonphagocytic nadph oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci Signaling (2010) 3(133):ra59–ra. doi: 10.1126/scisignal.2000976
113. Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of shp-2 promotes T-cell adhesion through Slp-76–adap. EMBO J (2005) 24(13):2331–41. doi: 10.1038/sj.emboj.7600706
114. Kamiński MM, Röth D, Krammer PH, Gülow K. Mitochondria as oxidative signaling organelles in T-cell activation: physiological role and pathological implications. Archivum immunologiae therapiae experimentalis (2013) 61:367–84. doi: 10.1007/s00005-013-0235-0
115. Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol (2005) 5(1):43–57. doi: 10.1038/nri1530
116. Molina T, Kishihara K, Siderovskid D, Van Ewijk W, Narendran A, Timms E, et al. Profound block in thymocyte development in mice lacking P56lck. Nature (1992) 357(6374):161–4. doi: 10.1038/357161a0
117. Negishi I, Motoyama N, Nakayama K-i, Nakayama K, Senju S, Hatakeyama S, et al. Essential role for zap-70 in both positive and negative selection of thymocytes. Nature (1995) 376(6539):435–8. doi: 10.1038/376435a0
118. Williams BL, Schreiber KL, Zhang W, Wange RL, Samelson LE, Leibson PJ, et al. Genetic evidence for differential coupling of syk family kinases to the T-cell receptor: reconstitution studies in a zap-70-Deficient jurkat T-cell line. Mol Cell Biol (1998) 18(3):1388–99. doi: 10.1128/MCB.18.3.1388
119. Wange RL. Lat, the linker for activation of T cells: a bridge between T cell-specific and general signaling pathways. Science's STKE (2000) 2000(63):re1–re. doi: 10.1126/stke.2000.63.re1
120. Lin X, Wang D. The roles of Carma1, Bcl10, and Malt1 in antigen receptor signaling. In: Seminars in immunology. Elsevier (2004) 16(6):429–35. doi: 10.1016/j.smim.2004.08.022
121. Roose JP, Mollenauer M, Ho M, Kurosaki T, Weiss A. Unusual interplay of two types of ras activators, rasgrp and sos, establishes sensitive and robust ras activation in lymphocytes. Mol Cell Biol (2007) 27(7):2732–45. doi: 10.1128/MCB.01882-06
122. Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. Stim1 is a Ca2+ sensor that activates crac channels and migrates from the Ca2+ store to the plasma membrane. Nature (2005) 437(7060):902–5. doi: 10.1038/nature04147
123. Feske S. Orai1 and Stim1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev (2009) 231(1):189–209. doi: 10.1111/j.1600-065X.2009.00818.x
124. Rao A. Nf-atp: a transcription factor required for the Co-ordinate induction of several cytokine genes. Immunol Today (1994) 15(6):274–81. doi: 10.1016/0167-5699(94)90007-8
125. Lu S-P, Feng M-HL, Huang H-L, Huang Y-C, Tsou W-I, Lai M-Z. Reactive oxygen species promote raft formation in T lymphocytes. Free Radical Biol Med (2007) 42(7):936–44. doi: 10.1016/j.freeradbiomed.2006.11.027
126. Pani G, Colavitti R, Borrello S, Galeotti T. Endogenous oxygen radicals modulate protein tyrosine phosphorylation and jnk-1 activation in lectin-stimulated thymocytes. Biochem J (2000) 347(1):173–81. doi: 10.1042/bj3470173
127. Lee K, Esselman WJ. Inhibition of ptps by H2o2 regulates the activation of distinct mapk pathways. Free Radical Biol Med (2002) 33(8):1121–32. doi: 10.1016/S0891-5849(02)01000-6
128. Hehner SP, Breitkreutz R, Shubinsky G, Unsoeld H, Schulze-Osthoff K, Schmitz ML, et al. Enhancement of T cell receptor signaling by a mild oxidative shift in the intracellular thiol pool. J Immunol (2000) 165(8):4319–28. doi: 10.4049/jimmunol.165.8.4319
129. Los M, Dröge W, Stricker K, Baeuerle PA, Schulze-Osthoff K. Hydrogen peroxide as a potent activator of T lymphocyte functions. Eur J Immunol (2010) 25(1):159–65. doi: 10.1002/eji.1830250127
130. Gloire G, Legrand-Poels S, Piette J. Nf-κb activation by reactive oxygen species: fifteen years later. Biochem Pharmacol (2006) 72(11):1493–505. doi: 10.1016/j.bcp.2006.04.011
131. Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, Niemeyer BA. Redox regulation of calcium ion channels: chemical and physiological aspects. Cell calcium (2011) 50(5):407–23. doi: 10.1016/j.ceca.2011.07.006
132. Ruterbusch M, Pruner KB, Shehata L, Pepper M. In vivo Cd4+ T cell differentiation and function: revisiting the Th1/Th2 paradigm. Annu Rev Immunol (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803
133. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-Bet is a Stat1-induced regulator of il-12r expression in naive Cd4+ T cells. Nat Immunol (2002) 3(6):549–57. doi: 10.1038/ni794
134. Magram J, Connaughton SE, Warrier RR, Carvajal DM, C-Y WU, Ferrante J, et al. Il-12-Deficient mice are defective in ifnγ production and type 1 cytokine responses. Immunity (1996) 4(5):471–81. doi: 10.1016/S1074-7613(00)80413-6
135. Preventing E. In Th1 differentiation: ifn-Γ role of ifn. J Immunol (2002) 168:6165–72. doi: 10.4049/jimmunol.168.12.6165
136. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T Cell responses: naive to memory and everything in between. Adv Physiol Educ (2013) 37(4):273–83. doi: 10.1152/advan.00066.2013
137. Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained il-12 signaling is required for Th1 development. J Immunol (2004) 172(1):61–9. doi: 10.4049/jimmunol.172.1.61
138. Eriksson U, Kurrer MO, Sebald W, Brombacher F, Kopf M. Dual role of the il-12/Ifn-Γ axis in the development of autoimmune myocarditis: induction by il-12 and protection by ifn-Γ. J Immunol (2001) 167(9):5464–9. doi: 10.4049/jimmunol.167.9.5464
139. Nishikomori R, Usui T, Wu C-Y, Morinobu A, O’Shea JJ, Strober W. Activated Stat4 has an essential role in Th1 differentiation and proliferation that is independent of its role in the maintenance of il-12rβ2 chain expression and signaling. J Immunol (2002) 169(8):4388–98. doi: 10.4049/jimmunol.169.8.4388
140. Smeltz RB, Chen J, Hu-Li J, Shevach EM. Regulation of interleukin (Il)-18 receptor α chain expression on Cd4+ T cells during T helper (Th) 1/Th2 differentiation: critical downregulatory role of il-4. J Exp Med (2001) 194(2):143–54. doi: 10.1084/jem.194.2.143
141. Scapin G, Dandey VP, Zhang Z, Prosise W, Hruza A, Kelly T, et al. Structure of the insulin receptor–insulin complex by single-particle cryo-em analysis. Nature (2018) 556(7699):122–5. doi: 10.1038/nature26153
142. Katz JD, Benoist C, Mathis D. T Helper cell subsets in insulin-dependent diabetes. Science (1995) 268(5214):1185–8. doi: 10.1126/science.7761837
143. Walker LS, von Herrath M. Cd4 T cell differentiation in type 1 diabetes. Clin Exp Immunol (2016) 183(1):16–29. doi: 10.1111/cei.12672
144. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi: 10.1172/JCI76238
145. Marrero I, Vong A, Dai Y, Davies JD. T Cell populations in the pancreatic lymph node naturally and consistently expand and contract in nod mice as disease progresses. Mol Immunol (2012) 52(1):9–18. doi: 10.1016/j.molimm.2012.04.004
146. Wachlin G, Augstein P, Schröder D, Kuttler B, Klöting I, Heinke P, et al. Il-1β, ifn-Γ and tnf-α increase vulnerability of pancreatic beta cells to autoimmune destruction. J Autoimmun (2003) 20(4):303–12. doi: 10.1016/S0896-8411(03)00039-8
147. Burrack AL, Martinov T, Fife BT. T Cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (2017) 8:343. doi: 10.3389/fendo.2017.00343
148. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
149. Luckheeram RV, Zhou R, Verma AD, Xia B. Cd4+ T cells: differentiation and functions. Clin Dev Immunol (2012) 2012). doi: 10.1155/2012/925135
150. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for Cd4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
151. Frossi B, De Carli M, Piemonte M, Pucillo C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol Immunol (2008) 45(1):58–64. doi: 10.1016/j.molimm.2007.05.008
152. Murr C, Schroecksnadel K, Winkler C, Ledochowski M, Fuchs D. Antioxidants may increase the probability of developing allergic diseases and asthma. Med Hypotheses (2005) 64(5):973–7. doi: 10.1016/j.mehy.2004.11.011
153. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. In: Seminars in immunopathology. Springer (2019) 41(3):283–97. doi: 10.1007s00281-019-00733-8
154. Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol (2007) 19(3):281–6. doi: 10.1016/j.coi.2007.04.005
155. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing Cd4+ effector T cells develop Via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
156. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of Cd4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
157. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
158. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-β induces development of the Th17 lineage. Nature (2006) 441(7090):231–4. doi: 10.1038/nature04754
159. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. Tgfβ in the context of an inflammatory cytokine milieu supports De novo differentiation of il-17-Producing T cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
160. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. Il-6 programs Th-17 cell differentiation by promoting sequential engagement of the il-21 and il-23 pathways. Nat Immunol (2007) 8(9):967–74. doi: 10.1038/ni1488
161. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell (2010) 140(6):845–58. doi: 10.1016/j.cell.2010.02.021
162. Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of il-17a in inflammatory immune responses and host defense against pathogens. Immunol Rev (2008) 226(1):57–79. doi: 10.1111/j.1600-065X.2008.00699.x
163. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (Eae) as a model for multiple sclerosis (Ms). Br J Pharmacol (2011) 164(4):1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
164. Fu G, Xu Q, Qiu Y, Jin X, Xu T, Dong S, et al. Suppression of Th17 cell differentiation by Misshapen/Nik-related kinase Mink1. J Exp Med (2017) 214(5):1453–69. doi: 10.1084/jem.20161120
165. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic Th17 cells. Nature (2013) 496(7446):518–22. doi: 10.1038/nature11868
166. Heidker RM, Emerson MR, LeVine SM. Metabolic pathways as possible therapeutic targets for progressive multiple sclerosis. Neural Regeneration Res (2017) 12(8):1262. doi: 10.4103/1673-5374.213542
167. Zhi L, Ustyugova IV, Chen X, Zhang Q, Wu MX. Enhanced Th17 differentiation and aggravated arthritis in Iex-1–deficient mice by mitochondrial reactive oxygen species-mediated signaling. J Immunol (2012) 189(4):1639–47. doi: 10.4049/jimmunol.1200528
168. Zhang D, Jin W, Wu R, Li J, Park S-A, Tu E, et al. High glucose intake exacerbates autoimmunity through reactive-Oxygen-Species-Mediated tgf-β cytokine activation. Immunity (2019) 51(4):671–81. e5. doi: 10.1016/j.immuni.2019.08.001
169. Scavuzzi BM, Simão ANC, Iriyoda TMV, Lozovoy MAB, Stadtlober NP, Franchi Santos L, et al. Increased lipid and protein oxidation and lowered anti-oxidant defenses in systemic lupus erythematosus are associated with severity of illness, autoimmunity, increased adhesion molecules, and Th1 and Th17 immune shift. Immunologic Res (2018) 66:158–71. doi: 10.1007/s12026-017-8960-9
170. Walker JA, McKenzie AN. Th2 cell development and function. Nat Rev Immunol (2018) 18(2):121–33. doi: 10.1038/nri.2017.118
171. Romagnani S. T-Cell subsets (Th1 versus Th2). Ann allergy Asthma Immunol (2000) 85(1):9–21. doi: 10.1016/S1081-1206(10)62426-X
172. Zhang Y, Zhang Y, Gu W, Sun B. Th1/Th2 cell differentiation and molecular signals. T helper Cell differentiation their Funct (2014) 841:15–44. doi: 10.1007/978-94-017-9487-9_2
173. Stark JM, Tibbitt CA, Coquet JM. The metabolic requirements of Th2 cell differentiation. Front Immunol (2019) 10:2318. doi: 10.3389/fimmu.2019.02318
174. Kopf M, Gros GL, Bachmann M, Lamers MC, Bluethmann H, Köhler G. Disruption of the murine il-4 gene blocks Th2 cytokine responses. Nature (1993) 362(6417):245–8. doi: 10.1038/362245a0
175. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/Type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of l-arginine metabolism. J Immunol (2001) 167(11):6533–44. doi: 10.4049/jimmunol.167.11.6533
176. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med (1992) 176(1):287–92. doi: 10.1084/jem.176.1.287
177. Coffman RL, Seymour BW, Hudak S, Jackson J, Rennick D. Antibody to interleukin-5 inhibits helminth-induced eosinophilia in mice. Science (1989) 245(4915):308–10. doi: 10.1126/science.2787531
178. Galli SJ, Starkl P, Marichal T, Tsai M. Mast cells and ige in defense against venoms: possible “Good side” of allergy? Allergology Int (2016) 65(1):3–15. doi: 10.1016/j.alit.2015.09.002
179. Paul WE, Zhu J. How are Th2-type immune responses initiated and amplified? Nat Rev Immunol (2010) 10(4):225–35. doi: 10.1038/nri2735
180. Joosten LA, Lubberts E, Helsen M, Saxne T, Coenen-de Roo CJ, Heinegård D, et al. Protection against cartilage and bone destruction by systemic interleukin-4 treatment in established murine type ii collagen-induced arthritis. Arthritis Res Ther (1999) 1(1):1–11. doi: 10.1186/ar14
181. Caramori G, Groneberg D, Ito K, Casolari P, Adcock IM, Papi A. New drugs targeting Th2 lymphocytes in asthma. J Occup Med Toxicol (2008) 3(1):1–29. doi: 10.1186/1745-6673-3-S1-S6
182. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol (2015) 16(1):45–56. doi: 10.1038/ni.3049
183. Masoli M, Fabian D, Holt S, Beasley R, Program G. The global burden of asthma: executive summary of the Gina dissemination committee report. Allergy (2004) 59(5):469–78. doi: 10.1111/j.1398-9995.2004.00526.x
184. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14(7):435–46. doi: 10.1038/nri3701
185. Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets Via distinct signaling pathways. Immunity (2010) 32(6):743–53. doi: 10.1016/j.immuni.2010.06.002
186. Nesi RT, Barroso MV, Souza Muniz VD, de Arantes AC, Martins MA, Brito Gitirana LD, et al. Pharmacological modulation of reactive oxygen species (Ros) improves the airway hyperresponsiveness by shifting the Th1 response in allergic inflammation induced by ovalbumin. Free Radical Res (2017) 51(7-8):708–22. doi: 10.1080/10715762.2017.1364377
187. Mangge H, Becker K, Fuchs D, Gostner JM. Antioxidants, inflammation and cardiovascular disease. World J Cardiol (2014) 6(6):462. doi: 10.4330/wjc.v6.i6.462
188. Göschl L, Scheinecker C, Bonelli M. Treg cells in autoimmunity: from identification to treg-based therapies. In: Seminars in immunopathology. Springer (2019) 41(3):301–14. doi: 10.1007s00281-019-00741-8
189. Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic potential of targeting the Th17/Treg axis in autoimmune disorders. Molecules (2017) 22(1):134. doi: 10.3390/molecules22010134
190. Wang W, Wang X, Lu S, Lv H, Zhao T, Xie G, et al. Metabolic disturbance and Th17/Treg imbalance are associated with progression of gingivitis. Front Immunol (2021) 12:670178. doi: 10.3389/fimmu.2021.670178
191. Wen S, He L, Zhong Z, Zhao R, Weng S, Mi H, et al. Stigmasterol restores the balance of Treg/Th17 cells by activating the butyrate-pparγ axis in colitis. Front Immunol (2021) 4201. doi: 10.3389fimmu.2021.741934
192. Glund S, Deshmukh A, Long YC, Moller T, Koistinen HA, Caidahl K, et al. Interleukin-6 directly increases glucose metabolism in resting human skeletal muscle. Diabetes (2007) 56(6):1630–7. doi: 10.2337/db06-1733
193. Bhaumik S, Basu R. Cellular and molecular dynamics of Th17 differentiation and its developmental plasticity in the intestinal immune response. Front Immunol (2017) 8:254. doi: 10.3389/fimmu.2017.00254
194. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling Via Stat5 constrains T helper 17 cell generation. Immunity (2007) 26(3):371–81. doi: 10.1016/j.immuni.2007.02.009
195. Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. A multiply redundant genetic switch'locks in'the transcriptional signature of regulatory T cells. Nat Immunol (2012) 13(10):972–80. doi: 10.1038/ni.2420
196. Rudra D, Deroos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol (2012) 13(10):1010–9. doi: 10.1038/ni.2402
197. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of Cd4+ Cd25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
198. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, et al. Disruption of a new Forkhead/Winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
199. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. Ctla-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
200. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of tfh cell number and humoral immune responses Via the coreceptor ctla-4. Immunity (2014) 41(6):1013–25. doi: 10.1016/j.immuni.2014.12.006
201. Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-Dependent down-modulation of costimulatory molecules on dendritic cells in Cd4+ Cd25+ regulatory T-Cell-Mediated suppression. Immunology (2006) 118(2):240–9. doi: 10.1111/j.1365-2567.2006.02362.x
202. Walker LS. Treg and ctla-4: two intertwining pathways to immune tolerance. J Autoimmun (2013) 45:49–57. doi: 10.1016/j.jaut.2013.06.006
203. Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions. In: Seminars in immunology. Elsevier (2011) 23(6):424–30. doi: 10.1016/j.smim.2011.10.002
204. Shevach EM. Foxp3+ T regulatory cells: still many unanswered questions–a perspective after 20 years of study. Front Immunol (2018) 9:1048. doi: 10.3389/fimmu.2018.01048
205. Gondek DC, DeVries V, Nowak EC, Lu L-F, Bennett KA, Scott ZA, et al. Transplantation survival is maintained by granzyme b+ regulatory cells and adaptive regulatory T cells. J Immunol (2008) 181(7):4752–60. doi: 10.4049/jimmunol.181.7.4752
206. Shi H, Chi H. Metabolic control of treg cell stability, plasticity, and tissue-specific heterogeneity. Front Immunol (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
207. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2020) 10:3100. doi: 10.3389/fimmu.2019.03100
208. Kawano Y, Edwards M, Huang Y, Bilate AM, Araujo LP, Tanoue T, et al. Microbiota imbalance induced by dietary sugar disrupts immune-mediated protection from metabolic syndrome. Cell (2022) 185(19):3501–19.e20. doi: 10.1016/j.cell.2022.08.005
209. Wing JB, Tanaka A, Sakaguchi S. Human Foxp3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity (2019) 50(2):302–16. doi: 10.1016/j.immuni.2019.01.020
Keywords: reactive oxygen species, CD4+ T cells, inflammation, Treg cells, effector T cells (Teffs), Th17 cells
Citation: Shu P, Liang H, Zhang J, Lin Y, Chen W and Zhang D (2023) Reactive oxygen species formation and its effect on CD4+ T cell-mediated inflammation. Front. Immunol. 14:1199233. doi: 10.3389/fimmu.2023.1199233
Received: 04 April 2023; Accepted: 16 May 2023;
Published: 25 May 2023.
Edited by:
Ulrike Lorenz, Washington University in St. Louis, United StatesReviewed by:
Sascha Kahlfuss, Universitätsklinikum Magdeburg, GermanyPaul Kent Langston, Harvard Medical School, United States
Copyright © 2023 Shu, Liang, Zhang, Lin, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dunfang Zhang, aXpkZkAxNjMuY29t
†These authors have contributed equally to this work