 Xinmei Huang
Xinmei Huang Mengshu Cao1,2,3,4*
Mengshu Cao1,2,3,4*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 June 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1195988
Pulmonary alveolar proteinosis (PAP) is a rare pulmonary disorder that is characterized by the abnormal accumulation of surfactant within the alveoli. Alveolar macrophages (AMs) have been identified as playing a pivotal role in the pathogenesis of PAP. In most of PAP cases, the disease is triggered by impaired cholesterol clearance in AMs that depend on granulocyte-macrophage colony-stimulating factor (GM-CSF), resulting in defective alveolar surfactant clearance and disruption of pulmonary homeostasis. Currently, novel pathogenesis-based therapies are being developed that target the GM-CSF signaling, cholesterol homeostasis, and immune modulation of AMs. In this review, we summarize the origin and functional role of AMs in PAP, as well as the latest therapeutic strategies aimed at addressing this disease. Our goal is to provide new perspectives and insights into the pathogenesis of PAP, and thereby identify promising new treatments for this disease.
Pulmonary alveolar proteinosis (PAP) is a rare pulmonary disease with a prevalence of 7 cases per million inhabitants worldwide (1, 2). The pathogenesis of PAP involves dysfunction of alveolar macrophages (AMs), resulting from the accumulation of alveolar cholesterol and phospholipids (3). Based on the underlying pathogenesis, PAP is classified into four main categories: primary (90% of cases), secondary (4% of cases), congenital (1% of cases) or unclassifiable. Primary PAP is led by disruption of granulocyte–macrophage colony-stimulating factor (GM-CSF) signaling, resulting in dysfunction of alveolar macrophages (4, 5). Primary PAP can be further classified as autoimmune (caused by elevated levels of anti-GM-CSF antibodies) or hereditary (due to mutations in genes encoding GM-CSF receptors). Secondary PAP occurs as a consequence of factors that reduce the number of AMs and/or impair the function of AMs. These factors include hematologic disorders, medications, environmental exposure, acute silicosis, or immunodeficiency (6). Congenital PAP is caused by genetic variation, including mutations in genes encoding surfactant proteins (SP-A, SP-B or SP-C) or cationic amino acid transport, which disrupts the production of normal surfactant (7). Finally, in some rare cases, patients who exhibit impaired signaling of GM-CSF without any identifiable evidence will be diagnosed with unclassified PAP (8).
Abnormalities of surfactant clearance and dysfunction of alveolar macrophage (AM) are the main causes of PAP (3). Pulmonary surfactant (PS) is a mixture of phospholipids cholesterol (90%) and surfactant proteins A-D (SPs A-D) (10%), which is secreted by lung alveolar type-II (AT-II) cells (9). The lipids of PS are made of DPPC (36%), unsaturated phosphatidylcholine (PC; 32%), phosphatidylglycerol (PG; 8%), cholesterol (7%), other phospholipids (PL; 4%) and other neutral lipids (NL; 3%) (10). The primary function of PS is to prevent alveolar collapse and protect lungs from infections and inflammation, acting as an opsonin as well as directly killing microbes (11). The balances in production, secretion, uptake, recycling and catabolism of PS are crucial for lung homeostasis (12). Seventy percent of spent PS is taken up by AT-II cells and recycled, while the remaining 30% is taken up and degraded by AMs (13).
The functional role of AMs in PAP is crucial, as evidenced by the excessive PS accumulation and the development of PAP in patients lacking functional AMs due to neutralizing autoantibodies against GM-CSF (14), as well as gene deficiencies in CSF2RA or CSF2RB (4, 15). Further studies clarified that intratracheal transplantation of functional AMs corrected PAP of Csf2rb-/- mice (16, 17). Currently, whole lung lavage (WLL) is still the first-line treatment for PAP (18). However, the effectiveness of WLL is limited due to its highly invasive and not curative, and there are no standardized guidelines for the procedure (19). Therefore, exploring the role of alveolar macrophages in PAP may provide new targets and approaches for the treatment of PAP.
In this review, we mainly discuss the current state of knowledge regarding the process of alveolar macrophage development and its role in the pathogenesis of PAP. Our aim is to provide an update on the mechanisms underlying the initiation and progression of PAP. This, in turn, will hopefully aid in the development of effective therapeutic strategies for PAP in clinical settings.
There has been a longstanding debate regarding the origin of AMs. Previous studies have suggested that all tissue-resident macrophages, including AMs, originate from circulating monocytes (20–22). However, this theory has been revised during the past decade. Under pathological conditions like osteoarthritis, pancreatic cancer, and steatohepatitis, monocytes can infiltrate target tissues by expressing the C-C chemokine receptor type 2 (CCR2). Therefore, researchers hypothesized that mice lacking Ccr2 would have a lower count of AMs compared to control mice. However, the proportions and numbers of AMs were found to be similar between Ccr2 knockout and control mice, suggesting that circulating monocytes make only a minimal contribution to the AMs pool (23). Moreover, transcriptional and functional data revealed that circulating monocytes rarely serve as precursors of AMs, with different origins, functions, gene expression profiles, and strategies to regulate compartment size (24–26). In line with this, recent studies indicated that alveolar macrophages derive from embryonic monocyte progenitors, which originate from the yolk sac or fetal liver during embryonic development and then migrate into the lung, where they differentiate into alveolar macrophages (25, 27–29). However, it is also feasible that interstitial macrophages derived from circulating monocytes may become precursors of AMs during injury, depletion or explant culture (30–33). Taken together, these studies suggested that alveolar macrophages are capable of dividing, self-renewing, and sustaining themselves without relying on circulating monocytes (23–26).
Several cytokines are involved in AMs differentiation and maturation, including granulocyte-macrophage colony-stimulating factor (GM-CSF), transforming growth factor-beta (TGF-β), and interleukin-1α (IL-1α) (Figure 1). GM-CSF serves as a key regulator of AMs terminal differentiation and maintenance, with studies showing that GM-CSF-deficient mice have a significant reduction in the number of mature AMs (15, 34, 35). Further discoveries in patients with rare CSF2RA mutations or anti-GM-CSF autoantibodies confirmed that the unique function of GM-CSF in the lung is fundamentally conserved in humans (4, 36, 37). Notably, GM-CSF triggered the development of AMs by upregulating the expression of PU.1 in AMs (35), and selectively inducing PPAR-γ in fetal monocytes in the lungs (35, 38, 39). In contrast to GM-CSF, although macrophage colony-stimulating factor (M-CSF) serves as a critical cytokine in regulating the development of monocyte and macrophage cells, it has minimal involvement in the differentiation of AMs (40). TGF-β is a multifunctional cytokine that is involved in many biological processes, including immune regulation (41). In contrast to other tissue-resident macrophages, AMs also require TGF-β signaling for development and homeostasis in an autocrine manner (42–44). Further studies indicated that TGF-β signaling leads to transcriptional changes in genes associated with AMs signature (Scgb1a1, Epcam, and Cyp4f18), lipid metabolism (Pparg, Apo-E, Olr1), and the phosphorylation of PPAR-γ (42, 45). IL-1α is another cytokine that has been implicated in AMs differentiation and maturation (46). It was investigated that the lack of IL-1α led to decreased proliferation and maturation of CD11blow alveolar macrophages during granuloma formation, which ultimately resulted in compromised alveolar clearance and the onset of PAP (46).
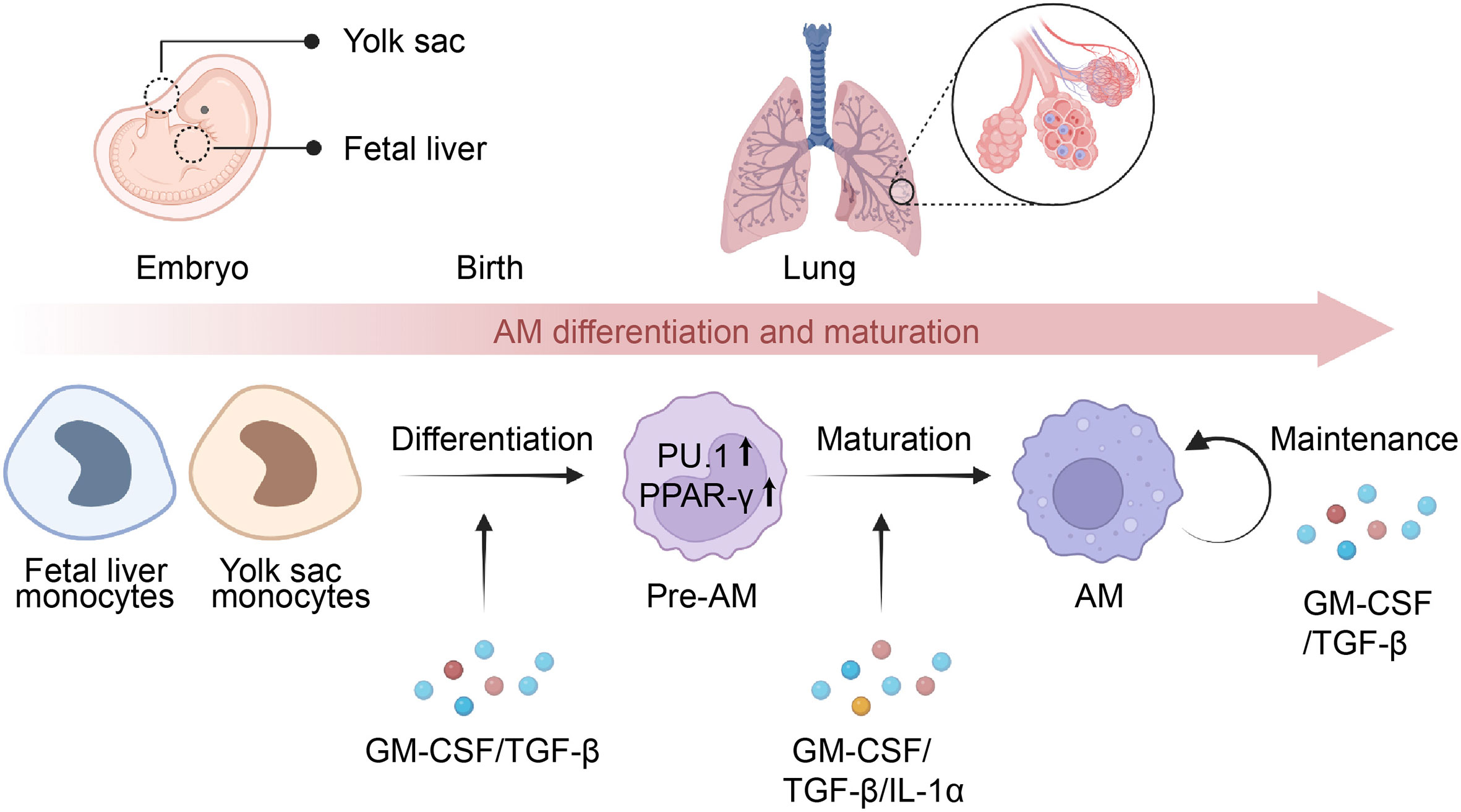
Figure 1 Differentiation and maturation of AMs. Alveolar macrophages (AMs) originate from monocytes present in the yolk sac or fetal liver and later migrate to the lungs, where they develop into pre-AMs and eventually mature into AMs. The process of AM differentiation is facilitated by the presence of GM-CSF and TGF-β, which activate the expression of PU.1 and PPAR-γ, playing a crucial role in determining their identity. Meanwhile, the maturation of pre-AMs relies on the presence of GM-CSF, TGF-β, and IL-1α, leading to transcriptional changes in the signature genes of AMs. Additionally, GM-CSF and TGF-β are essential for the maintenance of mature AMs via an autocrine mechanism.
In addition to cytokines, the transcription factor PU.1 has been shown to be critical for the terminal differentiation of AMs (47–49) (Figure 1). Studies utilizing GM-CSF knockout mice have demonstrated that reduced expression of the PU.1 in AMs is associated with decreased expression of CD32, mannose receptor, and macrophage colony-stimulating factor receptor (M-CSFR), which results in the blockade of maturation and differentiation of AMs (47). Moreover, PU.1 is responsible for regulating the constitutive expression of FcγRs and opsonophagocytosis in AMs (49). Notably, GM-CSF signaling is required for the expression of PU.1 in AMs (47, 50). Kruppel-like factor 4 (KLF4) is another transcription factor that is involved in AM differentiation and maturation (51, 52). Further studies have demonstrated that KLF4 promotes AMs polarizing to M2 phenotype through the activation of the downstream STAT6 signaling (52).
The pulmonary surfactant (PS) is composed of lipids, including phosphatidylcholine, phosphatidylglycerol, and surfactant protein A-D (53). The vital function of PS is to decrease the surface tension of the alveoli during respiration, which is crucial in preventing lung collapse by reducing the pressure required to inflate the lungs during inhalation (54). Maintaining an appropriate size of the surfactant pool is imperative for proper lung function and is carefully regulated by the balanced processes of surfactant production, secretion, reuptake, recycling, and catabolism within the alveoli (55–57).
In healthy individuals, pulmonary surfactant is produced and recycled by type II alveolar epithelial cells (AECs), with around 30% of the surfactant being taken up and catabolized by AMs (13, 58). Low-density lipoprotein (LDL), very low-density lipoprotein (VLDL), and oxidized lipoproteins (Ox-L) are taken up by AMs through various mechanisms, including macropinocytosis, phagocytosis, and scavenger receptor-mediated pathways (59) (Figure 2). The lipids taken up by AMs are degraded into free cholesterol and free fatty acids in lysosomes. Subsequently, the free cholesterol is re-esterified into cholesterol esters in the endoplasmic reticulum (ER) and stored in the cytoplasm as lipid droplets (60). The accumulation of intracellular cholesterol activates transcription factors such as liver X receptor α and β (LXRα/LXRβ), Retinoid X receptor (RXR), and Peroxisome proliferator-activated receptor α and γ (PPARα and PPARγ), which in turn promote the transcription and expression of lipid transporters ATP binding cassette subfamily A member 1(ABCA1) and ATP-binding cassette transporter G1(ABCG1) (61–64). These transporters subsequently regulate the efflux of free cholesterol and the maturation of HDLs (65). In addition, the efflux of free cholesterol is facilitated by simple diffusion or SR-BI-mediated facilitated diffusion (66).
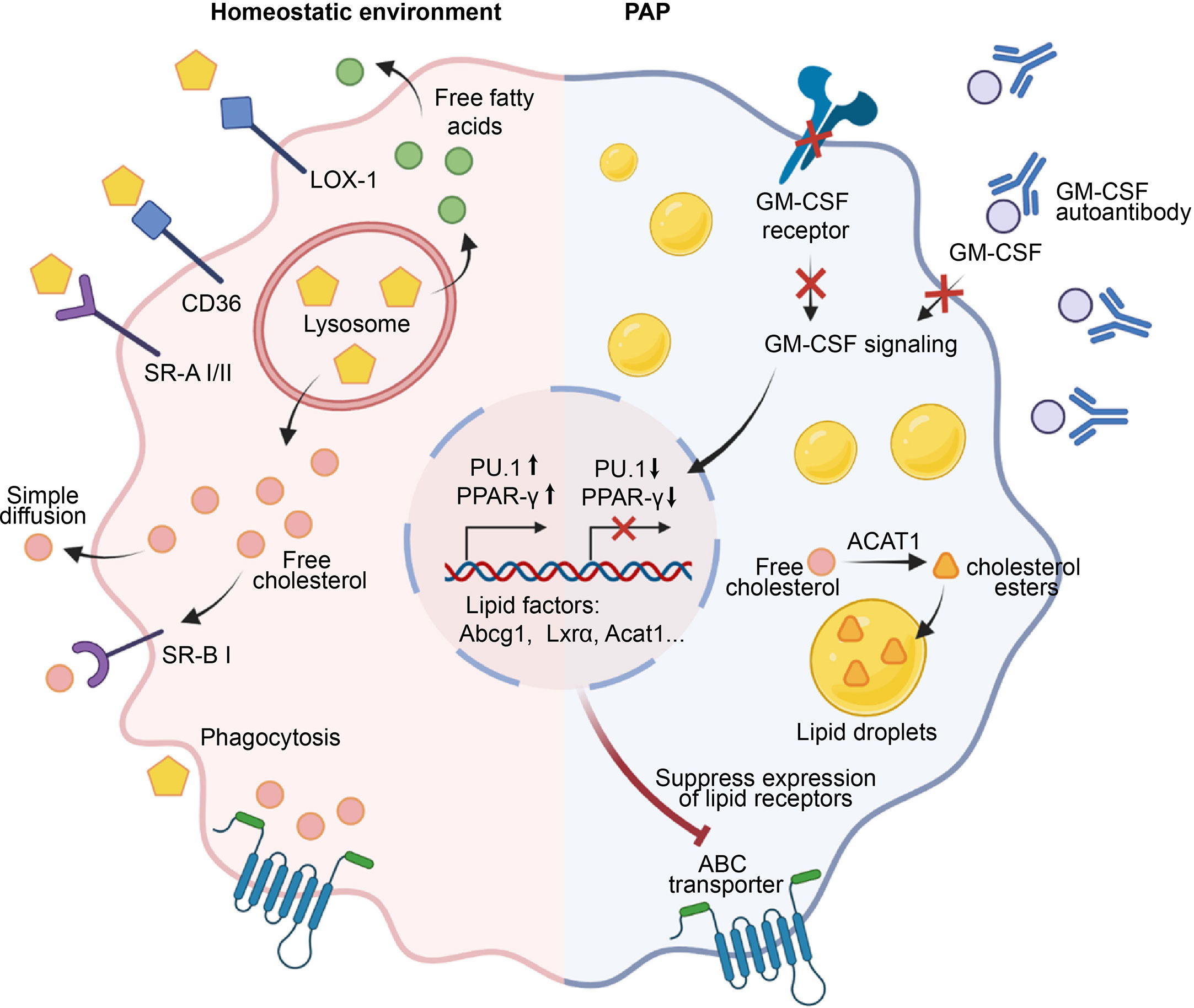
Figure 2 Role of AMs in the maintenance of surfactant homeostasis. Under a homeostatic environment, AMs take up lipids through phagocytosis, macropinocytosis, and scavenger receptors such as LOX-1, SR-A1, SR-B1, and CD36. The engulfed lipids enter lysosomes, where they are degraded into free cholesterol and fatty acids that accumulate in the cell and activate transcription factors such as PPAR-γ, promoting the transcription and expression of downstream lipid factors Abcg1, Lxra, Acat1, etc., and ultimately facilitating the efflux of free cholesterol. In the pathogenesis of PAP, GM-CSF signaling is impaired due to defects in GM-CSF or its receptor, or the presence of anti-GM-CSF autoantibodies. Consequently, this failure to induce the expression of PU.1 and PPAR-γ leads to the downregulation of lipid transporters and dysfunction of AMs. Subsequently, a significant accumulation of free cholesterol occurs within AMs, which is esterified by cholesterol acyltransferase ACAT to form cholesterol esters, stored within lipid droplets. Ultimately, the accumulation of a large number of lipid droplets in the cytoplasm of AMs leads to the generation of foam cells, which results in the deposition of surfactant in the alveoli.
Pulmonary alveolar proteinosis (PAP) is characterized by a reduction in AMs-mediated surfactant clearance, resulting in the formation of dysfunctional foamy AMs and accumulation of alveolar surfactant (67, 68). GM-CSF plays a key role in surfactant clearance of AMs, as evidenced by the development of hereditary PAP in patients with deficiencies in either CSF2RA or CSF2RB resulting in a lack of AMs (4, 69), and the development of autoimmune PAP in patients with non-functional AMs due to neutralizing auto-antibodies against GM-CSF (70). The esterified and free cholesterol are mainly accumulated by AMs in PAP, while the surfactant component displays a higher proportion of cholesterol (71). Mechanistically, the primary defect in cholesterol clearance caused by disruption of GM-CSF signaling leads to a secondary reduction in both surfactant uptake and clearance (71) (Figure 2). Moreover, disruption of GM-CSF signaling also caused a decrease in mRNA levels of several cholesterol homeostasis factors that are critical to AMs, including Abcg1, Lxrα, Acat1, Neutral cholesterol ester hydrolase 1(Nceh1), and Lipase A (Lipa) (71, 72). In line with this, replenishing PPAR-γ in AMs of GM-CSF knockout mice results in a decrease in intracellular lipid accumulation and an increase in cholesterol efflux activity by upregulating ABCG1 (73). Similar studies showed that PPAR-γ is implicated in the mitigation of foamy AMs formation by reducing the expression of ABCG1, CD36, Oxidized low-density lipoprotein receptor 1(Olr1), Fatty acid-binding proteins 1(Fabp1), Fabp4, and Cell Death Inducing DFFA Like Effector C(Cidec), which play crucial roles in the uptake, transport, storage, and processing of lipids (38, 74–76). Interestingly, lipid metabolism factors ABCA1 and LXRβ are inadequate in preventing surfactant accumulation in AMs (75). In addition to PPAR-γ, another crucial transcription factors regulated by GM-CSF is PU.1, which is known to modulate the expression of lipid transporters ABCA1 and ABCG1 (72, 76). Collectively, these studies indicate that GM-CSF signaling is necessary for cholesterol clearance in AMs and identify impaired cholesterol clearance as the main AMs abnormality underlying the development of PAP.
The alveolar immune microenvironment is a complex, multi-cellular system consisting of alveolar macrophages, dendritic cells, lymphocytes, and epithelial cells (77). These cells collaborate to maintain pulmonary homeostasis and defend against inhaled pathogens and environmental insults (78). Alveolar macrophages, as the predominant phagocytic cells in the lungs, play an essential role in the removal of inhaled particles and pathogens (79). Additionally, they release cytokines and chemokines that orchestrate the recruitment and activation of other immune cells at the site of infection (80).
Under physiological conditions, AMs demonstrate heightened phagocytic activity, low inflammatory cytokine production, and overall suppression of inflammation and adaptive immunity (80, 81) (Figure 3). AMs facilitate the resolution of inflammation by phagocytosing apoptotic and necrotic cells (a process known as efferocytosis), which in turn prevents the release of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), Interleukin-1(IL-1), IL-8, and leukotriene C4 in the alveolar microenvironment (82–84). Consistent with this, efferocytosis prompts AMs to secrete anti-inflammatory cytokines such as TGF-β, prostaglandin E2 (PGE2), and platelet-activating factor (PAF), thereby preventing the onset of unnecessary inflammatory responses (85–87). Apart from its role in efferocytosis, an alternative mechanism through which AMs inhibit inflammation is by activating the regulatory T cells (Treg) immune response (88). It was demonstrated that the isolation of mouse-derived AMs, which were subsequently stimulated with ovalbumin and co-cultured with antigen-specific naive CD4+ T cells, led to generation of Foxp3+ Treg cells (89). Notably, this functional role of AMs was partially reversed by either inhibiting the binding of retinoic acid (RA) to its receptor (RAR), or by impeding the signaling of TGF-β (89). In addition to directly affecting Treg cell conversion by influencing the components of RA and TGF-β, AMs may also initiate the generation of Tregs through an indirect pathway (90–92). In the context of respiratory viral infections, AMs generate the immunoregulatory cytokine IL-27 via the induction of IL-6, which in turn recruits Ly6C+ monocytes and facilitates the local differentiation of Tregs (92). Communication with the alveolar microenvironment also modulates the immunological functions of AMs. The binding of CD200 and TGF-β, present on the cell membrane of alveolar epithelial cells, to their respective receptors, CD200R and TGF-βR, expressed on AMs, functions as a negative regulator of AM pro-inflammatory activity (93–95).
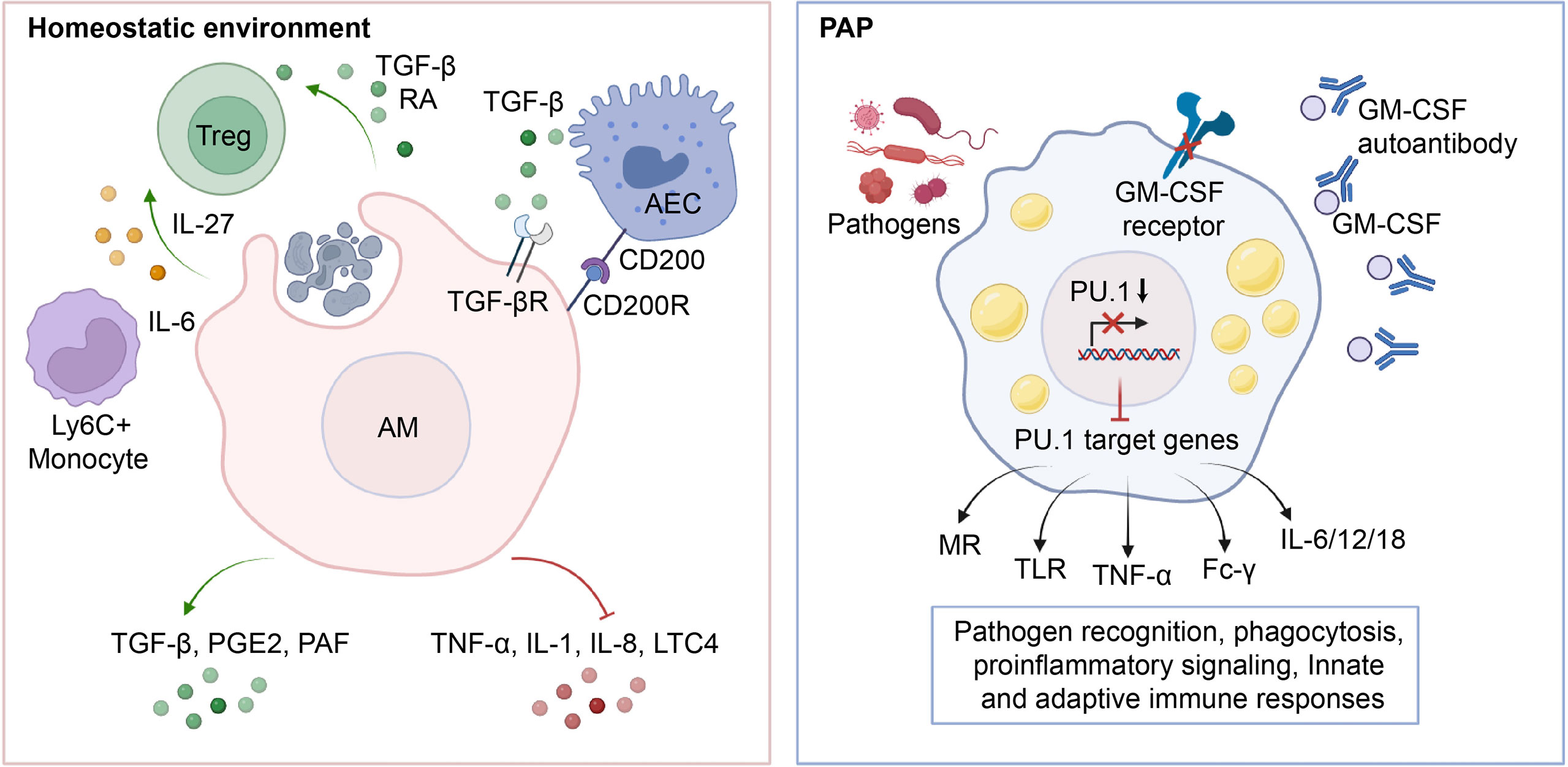
Figure 3 Role of AMs in the maintenance of immunological homeostasis. In physiological conditions, AMs phagocytose apoptotic cells, leading to an increase in the production of anti-inflammatory cytokines such as TGF-β, PGE2, and PAF, which in turn inhibits the release of pro-inflammatory cytokines TNF-α, IL-1, IL-8, and LTC4 into the microenvironment. Additionally, AMs induce Treg differentiation by secreting TGF-β, RA, IL-27, etc., and maintain lung immune homeostasis through crosstalk with AECs. In PAP, the impairment of GM-CSF signaling leads to downregulation of PU.1 expression, which in turn reduces the transcription and expression of downstream target genes such as MR, TLR, TNF-α, Fc-γ, and IL-6/12/18. This inhibition of antigen recognition, phagocytosis, pro-inflammatory signaling, and related innate and adaptive immune responses in AMs ultimately predisposes PAP patients to secondary infections.
In patients with PAP, the lack of GM-CSF signaling impairs the immunological functions of AMs and neutrophils, making them more vulnerable to superimposed infections caused by fungal pathogens, nocardia, cytomegalovirus, pneumocystis carinii, anaerobes, and mycobacteria (96–102). Generally, up to 13% of patients with PAP may experience secondary pulmonary infections, with fungal infections being more commonly diagnosed and mycobacterial infections having the lowest mortality rate (99). About 20% of deaths in PAP patients are caused by secondary infections (103). Similarly, AMs from GM-CSF-deficient mice exhibit reduced phagocytosis, pathogen killing, expression and release of pro-inflammatory cytokines (35). Mechanistically, the impaired maturation and function of AMs resulting from GM-CSF signaling deficiency is the primary factor of secondary infections in PAP (50) (Figure 3). It was found that GM-CSF promotes the uptake and clearance of adenovirus by AMs through a PU.1-dependent transcriptional program that redirects virion trafficking from the nucleus to lysosomes (104). Moreover, GM-CSF also regulates the production of genes that facilitate multiple immune and non-immune functions by stimulating the expression of PU.1 in AMs (50). It was demonstrated that the expression of PU.1 in GM-CSF-deficient AMs significantly increase the expression of mannose receptor (MR) and Toll-like receptor 2/4 (TLR-2/4), which are involved in the phagocytic internalization of microorganisms and pro-inflammatory signaling responses (35, 105). In line with this, the expression of PU.1 in GM-CSF-deficient AMs mediates a dose-dependent increase in the release of TNF-α and IL-6 (35). In addition, GM-CSF/PU.1 signaling orchestrates the molecular crosstalk between innate and adaptive immunity by modulating the production of IL-12, IL-18, and IFN-γ, consisting with the regulation of AMs phagocytosis via Fc-γ receptors (49). Together, these studies suggest that AMs have a critical function in preserving pulmonary immunological equilibrium in PAP patients, underscoring the essentiality of meticulous monitoring for infectious complications.
Whole-lung lavage (WLL) is a standard treatment for patients of PAP, which generally improves pulmonary function in the majority of patients (106). However, due to the impaired function of AMs and the recurrence of surfactant accumulation, repeated treatments are often necessary (107). Therefore, restoring normal AM function to maintain pulmonary lipid and immune homeostasis is a fundamental approach for the treatment of primary PAP (108). In the treatment of secondary PAP, the primary approach is to address the underlying primary disease or remove the causative factors, with adjunctive whole lung lavage (WLL) being an additional option to enhance therapeutic efficacy in severe cases (109, 110). Furthermore, gene therapy-induced restoration of normal AM function also serves as a therapeutic option for hereditary PAP (111). In this section, we will discuss the applications of GM-CSF administration, rituximab, pharmacotherapies that restore lipid homeostasis in AMs, AM transplantation, and induced pluripotent stem cell (iPSC)-derived AMs in the treatment of PAP.
The administration of GM-CSF is a promising therapeutic strategy for autoimmune PAP, as it neutralizes the anti-GM-CSF antibodies and restores AM function. However, the effectiveness of GM-CSF therapy in secondary PAP remains to be established (112). A number of case series have reported that administering exogenous GM-CSF therapy has led to clinical improvement in patients with PAP (Table 1). Recombinant GM-CSF is commonly administered through subcutaneous injection or inhalation, with various dosing regimens, including low-dose treatment (125 μg/day) and high-dose treatment (250 or 300 μg/day), etc (113–116, 118, 120–123). Therapeutic response of GM-CSF administration was evaluated via disease severity (alveolar arterial gradient (A-a) Do2, oxygenation, etc.), pulmonary function tests, serum biomarkers of PAP, HRCT score, respiratory symptom scores, etc. The majority of the case series documented a response rate of 30-60%, with remission of respiratory symptoms and improvement in pulmonary function (113–116, 118, 120–123). Intriguingly, Campo et al. demonstrated that inhaled recombinant GM-CSF was well tolerated and effective in a PAP, and combination therapy proved to be more effective than WLL alone (119). Moreover, Tazawa et al. conducted a study in 3 successfully treated patients with idiopathic PAP, which found that the cell number, expressions of surface mannose receptor and PU.1, and phagocytic ability of AMs were all restored to control levels (124). Notably, the treatment resulted in a significant reduction in the neutralizing capacity and the levels of anti-GM-CSF autoantibodies in BALF of patients. Similarly, other studies also revealed that the administration of GM-CSF restored the morphology and adhesive function of AMs, upregulated the expression of GM-CSF receptor and PU.1, and improves lung clearance in PAP (47, 117, 125).

Table 1 Relevant clinical data on therapy of PAP by GM-CSF administration.
Rituximab is a chimeric monoclonal antibody that specifically recognizes the B-lymphocyte antigen CD20 (126). It has been previously used to treat CD20+ B-cell lymphoma, as well as autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis (126, 127). Expanding on this, Rituximab has also been utilized as a B-cell depleting agent in order to eradicate anti-GM-CSF autoantibodies and subsequently serve as a therapeutic intervention for PAP. Multiple studies have reported positive outcomes in terms of improvement in lung function and imaging variables in cases of autoimmune PAP following treatment with Rituximab, as observed in both case series and isolated cases (128–131) (Table 2). In addition, the administration of Rituximab therapy led to a consistent reduction in both CD19+ B-cells in peripheral blood and anti-GM-CSF autoantibodies in BALF (131). Mechanistically, Rituximab therapy ameliorates the lipid metabolism of AM in patients with PAP, as demonstrated by the upregulation of key lipid metabolism molecules, including PPAR-γ, ABCG1, and lysosomal phospholipase A2 (LPLA2), a critical enzyme for surfactant degradation (132). Furthermore, Oil Red O staining indicates a reduction in intracellular lipid droplets in AMs, leading to an increase in free cholesterol in BALF and maintaining lipid homeostasis in the alveoli (132).
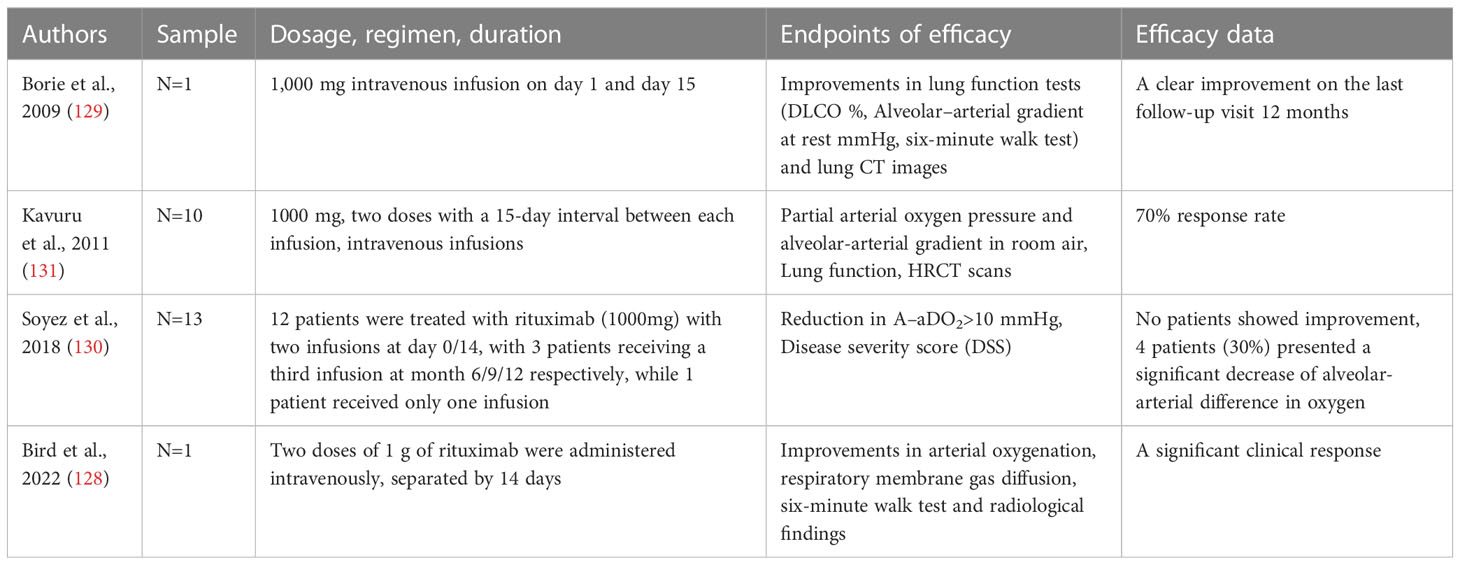
Table 2 Relevant clinical data on therapy of PAP by Rituximab.
Currently, the pharmacotherapies utilized to restore lipid homeostasis in AMs comprise Pioglitazone and Statins. In the development of PAP, the absence of key molecules PPAR-γ and ABCG1 in AM is a crucial factor contributing to the dysregulation of cholesterol transport (75). In line with this, restoration of PPAR-γ signaling leads to a subsequent upregulation of ABCG1 expression, which facilitates the efflux of intracellular cholesterol in AMs (73). Pioglitazone is an agonist of PPAR-γ, which has been historically employed for the treatment of type 2 diabetes and Alzheimer’s disease (133, 134). Recent research utilizing Csf2rb-/- PAP mice has demonstrated that pioglitazone can increase the expression of ABCG1, enhance cholesterol efflux in AMs, and alleviate pulmonary symptoms in PAP mice (135). A comparable outcome was documented in an autoimmune PAP patient that was unresponsive to conventional first-line therapies but demonstrated improvement of lung function with pioglitazone treatment (30 mg taken once daily) (136). The clinical study investigating the efficacy of pioglitazone in treating PAP is underway (NCT03231033) (136).
Statin is primarily used to reduce the risk of cardiovascular diseases by lowering cholesterol levels in the blood (137). Notably, a study involving two autoimmune PAP patients who were unresponsive to WLL treatment found that statins could significantly improve the patients’ clinical symptoms, lung function, and radiological features, as well as reduce the intracellular cholesterol levels in AMs (138). In Csf2rb−/− mice, statin treatment also promoted cholesterol efflux in AMs and alleviated disease symptoms (138). In addition, a prospective real-world observational study evaluated the therapeutic efficacy of statins on 40 non-hypercholesterolemic PAP patients (139). 65% of the patients showed a treatment response, characterized by an increase in PaO2, DLCO%, and a decrease in disease severity score (DSS) and radiographic abnormalities (139). The cutoff dose was 67.5 mg daily, with a corresponding specificity of 64.3% and sensitivity of 96.2% (139).
In addition to pharmacological treatments aimed at restoring AM function, direct transplantation of gene-corrected AMs or iPSC-derived AMs has also shown promise in the treatment of hereditary PAP. A study was conducted using lentiviral-mediated overexpression of the Csf2ra gene to prepare gene-corrected AMs, which were then transplanted into Csf2ra-/- mice. The results showed that the transplantation improved AM function, restore lipid homeostasis and surfactant metabolism, and ameliorated the cytological, histological, and biomarker abnormalities associated with PAP (140). To enable the translation of gene-corrected AM therapy to PAP patients, a self-inactivating (SIN) lentiviral vector expressing a codon-optimized human CSF2RA-cDNA driven by an EF1α short promoter (Lv.EFS.CSF2RAcoop) was created. Results indicated that Lv.EFS.CSF2Racoop restored GM-CSF signaling in AMs by reconstituting GM-CSF receptor expression, and did not elicit negative effects in the target cells (141). Similarly, by using TALEN-mediated genetic integration, a codon-optimized CSF2RA transgene was inserted into the AAVS1 locus of iPSCs derived from hereditary PAP patients. This restored GM-CSF receptor functionality and corrected the disease phenotype of monocytes/macrophages derived from hereditary PAP iPSCs in vitro (111). Additionally, it has been demonstrated that human iPSC-derived macrophages can be transplanted into the lungs and differentiate into AMs, leading to a decrease in alveolar proteinosis in a humanized PAP model (142, 143).
AMs are a crucial type of immune cell in the pulmonary microenvironment, which are mainly derived from monocytes present in the yolk sac or fetal liver, and later migrate to the lungs, where they develop into pre-AMs and eventually mature into AMs. GM-CSF and TGF-β, along with their downstream signals PU.1 and PPAR-γ, play a critical role in the differentiation and maintenance of AMs. In the pathogenesis of PAP, GM-CSF signaling is impaired due to defects in GM-CSF or its receptor, or the presence of anti-GM-CSF autoantibodies, which fail to induce the expression of PU.1 and PPAR-γ, resulting in abnormal development and function of AMs. Ultimately, the cytoplasmic accumulation of lipid droplets in AMs results in the development of foam cells, which leads to the deposition of surfactant in the alveoli. Additionally, AMs are also critical in maintaining pulmonary immune homeostasis. In PAP, the impairment of GM-CSF signaling leads to the downregulation of PU.1 expression, which in turn reduces the transcription and expression of downstream immune-related target genes. This leads to abnormal pulmonary innate and adaptive immune responses, ultimately making PAP patients susceptible to secondary infections. Currently, exogenous GM-CSF has been reported as a treatment option for autoimmune PAP, while its utility in other forms of PAP remains unknown. Moreover, rituximab, pharmacotherapies targeting lipid homeostasis in AMs, AM transplantation, and induced pluripotent stem cell (iPSC)-derived AMs have also been utilized for PAP treatment. The objective of these therapies is to directly or indirectly restore normal differentiation and function of AMs.
Although significant progress has been made in comprehending PAP in the last few decades, several crucial questions remain unanswered and will motivate future research on the etiology, pathogenesis, and therapies. Despite notable advancements in the etiology research of PAP, the cause of anti-GM-CSF autoantibody production remains unknown. In addition, although the pathogenic mechanism of abnormal AM function resulting from GM-CSF signaling deficiency has been elucidated, the downstream key effectors and mechanisms involved in lipid and immune homeostasis are still unclear. It is worth noting that currently, AM-targeted PAP therapies, such as GM-CSF administration, rituximab, and AM transplantation, have been demonstrated to be effective and safe. Nonetheless, it is crucial to underscore that these therapies have not yet obtained regulatory approval. In addition, there is still a lack of established therapeutic guidelines and consensus, and some promising treatment strategies are still undergoing clinical validation. We hope that this review will provide perspectives on understanding the function of AMs in PAP and developing new therapeutic strategies.
Manuscript drafting and figures & tables design: XH. Writing-review and editing: MC and YX. Supervision: MC and YX. Funding acquisition: MC and YX. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (82070064, 81670059 and 81200049), Fundings for Clinical Trials from the Affiliated Drum Tower Hospital, Medical School of Nanjing University (2022-LCYJ-MS-11), Special fund project for clinical research of Nanjing Drum Tower Hospital (2021-LCYJ-DBZ-06).
The figure was created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McCarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. Prevalence and healthcare burden of pulmonary alveolar proteinosis. Orphanet J Rare Dis (2018) 13:129. doi: 10.1186/s13023-018-0846-y
2. Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med (2008) 177:752–62. doi: 10.1164/rccm.200708-1271OC
3. Salvaterra E, Campo I. Pulmonary alveolar proteinosis: from classification to therapy. Breathe (Sheff) (2020) 16:200018. doi: 10.1183/20734735.0018-2020
4. Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman S, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med (2008) 205:2703–10. doi: 10.1084/jem.20080990
5. Sakagami T, Uchida K, Suzuki T, Carey BC, Wood RE, Wert SE, et al. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N Engl J Med (2009) 361:2679–81. doi: 10.1056/NEJMc0904077
6. Ishii H, Tazawa R, Kaneko C, Saraya T, Inoue Y, Hamano E, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J (2011) 37:465–8. doi: 10.1183/09031936.00092910
7. Verhasselt-Crinquette M, Franquet-Ansart H, Rakza T, Storme L, Copin MC, Devisme L. Congenital pulmonary alveolar proteinosis related to a surfactant protein b deficiency: report of two cases. Ann Pathol (2009) 29:481–4. doi: 10.1016/j.annpat.2009.10.030
8. Iftikhar H, Nair GB, Kumar A. Update on diagnosis and treatment of adult pulmonary alveolar proteinosis. Ther Clin Risk Manag (2021) 17:701–10. doi: 10.2147/TCRM.S193884
9. Veldhuizen EJ, Haagsman HP. Role of pulmonary surfactant components in surface film formation and dynamics. Biochim Biophys Acta (2000) 1467:255–70. doi: 10.1016/s0005-2736(00)00256-x
10. Sunde M, Pham CLL, Kwan AH. Molecular characteristics and biological functions of surface-active and surfactant proteins. Annu Rev Biochem (2017) 86:585–608. doi: 10.1146/annurev-biochem-061516-044847
11. Wang S, Li Z, Wang X, Zhang S, Gao P, Shi Z. The role of pulmonary surfactants in the treatment of acute respiratory distress syndrome in COVID-19. Front Pharmacol (2021) 12:698905. doi: 10.3389/fphar.2021.698905
12. Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol (2007) 293:L259–271. doi: 10.1152/ajplung.00112.2007
13. Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol (2002) 64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847
14. Uchida K, Nakata K, Trapnell BC, Terakawa T, Hamano E, Mikami A, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood (2004) 103:1089–98. doi: 10.1182/blood-2003-05-1565
15. Paine R 3rd, Morris SB, Jin H, Wilcoxen SE, Phare SM, Moore BB, et al. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. Am J Physiol Lung Cell Mol Physiol (2001) 281:L1210–1218. doi: 10.1152/ajplung.2001.281.5.L1210
16. Happle C, Lachmann N, Skuljec J, Wetzke M, Ackermann M, Brennig S, et al. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci Transl Med (2014) 6:250ra113. doi: 10.1126/scitranslmed.3009750
17. Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, et al. Pulmonary macrophage transplantation therapy. Nature (2014) 514:450–4. doi: 10.1038/nature13807
18. Campo I, Luisetti M, Griese M, Trapnell BC, Bonella F, Grutters J, et al. Whole lung lavage therapy for pulmonary alveolar proteinosis: a global survey of current practices and procedures. Orphanet J Rare Dis (2016) 11:115. doi: 10.1186/s13023-016-0497-9
19. Awab A, Khan MS, Youness HA. Whole lung lavage-technical details, challenges and management of complications. J Thorac Dis (2017) 9:1697–706. doi: 10.21037/jtd.2017.04.10
20. van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med (1968) 128:415–35. doi: 10.1084/jem.128.3.415
21. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46:845–52.
22. Volkman A, Gowans JL. The origin of macrophages from bone marrow in the rat. Br J Exp Pathol (1965) 46:62–70.
23. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
24. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40:91–104. doi: 10.1016/j.immuni.2013.11.019
25. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med (2012) 209:1167–81. doi: 10.1084/jem.20120340
26. Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol (2008) 38:380–5. doi: 10.1165/rcmb.2007-0224RC
27. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science (2013) 339:156–61. doi: 10.1126/science.1227901
28. Yamane T. Mouse yolk sac hematopoiesis. Front Cell Dev Biol (2018) 6:80. doi: 10.3389/fcell.2018.00080
29. Sugiyama D, Inoue-Yokoo T, Fraser ST, Kulkeaw K, Mizuochi C, Horio Y. Embryonic regulation of the mouse hematopoietic niche. Sci World J (2011) 11:1770–80. doi: 10.1100/2011/598097
30. Liborio-Ramos S, Barbosa-Matos C, Fernandes R, Borges-Pereira C, Costa S. Interstitial macrophages lead early stages of bleomycin-induced lung fibrosis and induce fibroblasts activation. Cells (2023) 12:402. doi: 10.3390/cells12030402
31. Gu Y, Lawrence T, Mohamed R, Liang Y, Yahaya BH. The emerging roles of interstitial macrophages in pulmonary fibrosis: a perspective from scRNA-seq analyses. Front Immunol (2022) 13:923235. doi: 10.3389/fimmu.2022.923235
32. Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, Kuroda MJ. In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. J Immunol (2014) 192:2821–9. doi: 10.4049/jimmunol.1302269
33. Tan SY, Krasnow MA. Developmental origin of lung macrophage diversity. Development (2016) 143:1318–27. doi: 10.1242/dev.129122
34. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med (2013) 210:1977–92. doi: 10.1084/jem.20131199
35. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity (2001) 15:557–67. doi: 10.1016/s1074-7613(01)00218-7
36. Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc Natl Acad Sci USA (2011) 108:2390–5. doi: 10.1073/pnas.1019682108
37. Martinez-Moczygemba M, Doan ML, Elidemir O, Fan LL, Cheung SW, Lei JT, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med (2008) 205:2711–6. doi: 10.1084/jem.20080759
38. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15:1026–37. doi: 10.1038/ni.3005
39. Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol (2012) 189:2614–24. doi: 10.4049/jimmunol.1200495
40. Draijer C, Penke LRK, Peters-Golden M. Distinctive effects of GM-CSF and m-CSF on proliferation and polarization of two major pulmonary macrophage populations. J Immunol (2019) 202:2700–9. doi: 10.4049/jimmunol.1801387
41. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol Cancer (2022) 21:104. doi: 10.1186/s12943-022-01569-x
42. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine TGF-beta promotes the development and homeostasis of alveolar macrophages. Immunity (2017) 47:903–912 e904. doi: 10.1016/j.immuni.2017.10.007
43. Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, et al. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2011) 300:L740–752. doi: 10.1152/ajplung.00146.2010
44. Liu Z, Liao F, Zhu J, Zhou D, Heo GS, Leuhmann HP, et al. Reprogramming alveolar macrophage responses to TGF-beta reveals CCR2+ monocyte activity that promotes bronchiolitis obliterans syndrome. J Clin Invest (2022) 132:e159229. doi: 10.1172/JCI159229
45. Ramirez A, Ballard EN, Roman J. TGFbeta1 controls PPARgamma expression, transcriptional potential, and activity, in part, through Smad3 signaling in murine lung fibroblasts. PPAR Res (2012) 2012:375876. doi: 10.1155/2012/375876
46. Huaux F, Lo Re S, Giordano G, Uwambayinema F, Devosse R, Yakoub Y, et al. IL-1alpha induces CD11b(low) alveolar macrophage proliferation and maturation during granuloma formation. J Pathol (2015) 235:698–709. doi: 10.1002/path.4487
47. Bonfield TL, Raychaudhuri B, Malur A, Abraham S, Trapnell BC, Kavuru MS, et al. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. Am J Physiol Lung Cell Mol Physiol (2003) 285:L1132–1136. doi: 10.1152/ajplung.00216.2003
48. Qian F, Deng J, Lee YG, Zhu J, Karpurapu M, Chung S, et al. The transcription factor PU.1 promotes alternative macrophage polarization and asthmatic airway inflammation. J Mol Cell Biol (2015) 7:557–67. doi: 10.1093/jmcb/mjv042
49. Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage fcgamma r-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood (2002) 100:4193–200. doi: 10.1182/blood-2002-04-1102
50. Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol (2009) 21:514–21. doi: 10.1016/j.coi.2009.09.004
51. Kurotaki D, Osato N, Nishiyama A, Yamamoto M, Ban T, Sato H, et al. Essential role of the IRF8-KLF4 transcription factor cascade in murine monocyte differentiation. Blood (2013) 121:1839–49. doi: 10.1182/blood-2012-06-437863
52. Yang Y, Wei S, Li Q, Chu K, Zhou Y, Xue L, et al. Vitamin d protects silica particles induced lung injury by promoting macrophage polarization in a KLF4-STAT6 manner. J Nutr Biochem (2022) 110:109148. doi: 10.1016/j.jnutbio.2022.109148
53. Autilio C, Perez-Gil J. Understanding the principle biophysics concepts of pulmonary surfactant in health and disease. Arch Dis Child Fetal Neonatal Ed (2019) 104:F443–51. doi: 10.1136/archdischild-2018-315413
54. Zuo YY, Veldhuizen RA, Neumann AW, Petersen NO, Possmayer F. Current perspectives in pulmonary surfactant–inhibition, enhancement and evaluation. Biochim Biophys Acta (2008) 1778:1947–77. doi: 10.1016/j.bbamem.2008.03.021
55. Casals C, Canadas O. Role of lipid ordered/disordered phase coexistence in pulmonary surfactant function. Biochim Biophys Acta (2012) 1818:2550–62. doi: 10.1016/j.bbamem.2012.05.024
56. Vockeroth D, Gunasekara L, Amrein M, Possmayer F, Lewis JF, Veldhuizen RA. Role of cholesterol in the biophysical dysfunction of surfactant in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2010) 298:L117–125. doi: 10.1152/ajplung.00218.2009
57. Discher BM, Maloney KM, Grainger DW, Hall SB. Effect of neutral lipids on coexisting phases in monolayers of pulmonary surfactant. Biophys Chem (2002) 101-102:333–45. doi: 10.1016/s0301-4622(02)00191-6
58. Agudelo CW, Samaha G, Garcia-Arcos I. Alveolar lipids in pulmonary disease. a review. Lipids Health Dis (2020) 19:122. doi: 10.1186/s12944-020-01278-8
59. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res (2016) 118:653–67. doi: 10.1161/CIRCRESAHA.115.306256
60. Yan J, Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol (2020) 30:979–89. doi: 10.1016/j.tcb.2020.09.006
61. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med (2001) 7:53–8. doi: 10.1038/83348
62. Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol (2008) 28:1050–9. doi: 10.1161/ATVBAHA.107.158998
63. Chinetti-Gbaguidi G, Staels B. Lipid ligand-activated transcription factors regulating lipid storage and release in human macrophages. Biochim Biophys Acta (2009) 1791:486–93. doi: 10.1016/j.bbalip.2009.01.009
64. Duplus E, Glorian M, Forest C. Fatty acid regulation of gene transcription. J Biol Chem (2000) 275:30749–52. doi: 10.1074/jbc.R000015200
65. Hutchins PM, Heinecke JW. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr Opin Lipidol (2015) 26:388–93. doi: 10.1097/MOL.0000000000000209
66. Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem (2014) 289:24020–9. doi: 10.1074/jbc.R114.583658
67. Kelly A, McCarthy C. Pulmonary alveolar proteinosis syndrome. Semin Respir Crit Care Med (2020) 41:288–98. doi: 10.1055/s-0039-3402727
68. Tian X, Luo J, Xu KF, Wang L, Zhou J, Feng R, et al. Impaired lipid metabolism in idiopathic pulmonary alveolar proteinosis. Lipids Health Dis (2011) 10:54. doi: 10.1186/1476-511X-10-54
69. Suzuki T, Trapnell BC. Pulmonary alveolar proteinosis syndrome. Clin Chest Med (2016) 37:431–40. doi: 10.1016/j.ccm.2016.04.006
70. Byun MK, Kim DS, Kim YW, Chung MP, Shim JJ, Cha SI, et al. Clinical features and outcomes of idiopathic pulmonary alveolar proteinosis in Korean population. J Korean Med Sci (2010) 25:393–8. doi: 10.3346/jkms.2010.25.3.393
71. Sallese A, Suzuki T, McCarthy C, Bridges J, Filuta A, Arumugam P, et al. Targeting cholesterol homeostasis in lung diseases. Sci Rep (2017) 7:10211. doi: 10.1038/s41598-017-10879-w
72. Thomassen MJ, Barna BP, Malur AG, Bonfield TL, Farver CF, Malur A, et al. ABCG1 is deficient in alveolar macrophages of GM-CSF knockout mice and patients with pulmonary alveolar proteinosis. J Lipid Res (2007) 48:2762–8. doi: 10.1194/jlr.P700022-JLR200
73. Malur A, Baker AD, McCoy AJ, Wells G, Barna BP, Kavuru MS, et al. Restoration of PPARgamma reverses lipid accumulation in alveolar macrophages of GM-CSF knockout mice. Am J Physiol Lung Cell Mol Physiol (2011) 300:L73–80. doi: 10.1152/ajplung.00128.2010
74. Ye Y, Liu J, Guo Y, Gao Y, Rao J, Su R, et al. PPARgamma ameliorates mycobacterium tuberculosis H37Ra-induced foamy macrophage formation via the ABCG1-dependent cholesterol efflux pathway in THP-1 macrophages. Front Microbiol (2022) 13:829870. doi: 10.3389/fmicb.2022.829870
75. Baker AD, Malur A, Barna BP, Ghosh S, Kavuru MS, Malur AG, et al. Targeted PPARgamma deficiency in alveolar macrophages disrupts surfactant catabolism. J Lipid Res (2010) 51:1325–31. doi: 10.1194/jlr.M001651
76. Malur A, Huizar I, Wells G, Barna BP, Malur AG, Thomassen MJ. Lentivirus-ABCG1 instillation reduces lipid accumulation and improves lung compliance in GM-CSF knock-out mice. Biochem Biophys Res Commun (2011) 415:288–93. doi: 10.1016/j.bbrc.2011.10.043
77. Didierlaurent A, Goulding J, Hussell T. The impact of successive infections on the lung microenvironment. Immunology (2007) 122:457–65. doi: 10.1111/j.1365-2567.2007.02729.x
78. Lloyd CM, Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. Immunity (2017) 46:549–61. doi: 10.1016/j.immuni.2017.04.005
79. Joshi N, Walter JM, Misharin AV. Alveolar macrophages. Cell Immunol (2018) 330:86–90. doi: 10.1016/j.cellimm.2018.01.005
80. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol (2014) 14:81–93. doi: 10.1038/nri3600
81. Rubins JB. Alveolar macrophages: wielding the double-edged sword of inflammation. Am J Respir Crit Care Med (2003) 167:103–4. doi: 10.1164/rccm.2210007
82. Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med (2013) 5:661–74. doi: 10.1002/emmm.201202382
83. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol (2016) 38:409–23. doi: 10.1007/s00281-016-0555-3
84. Chung EY, Liu J, Homma Y, Zhang Y, Brendolan A, Saggese M, et al. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and prep-1. Immunity (2007) 27:952–64. doi: 10.1016/j.immuni.2007.11.014
85. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi: 10.1172/JCI1112
86. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest (2002) 109:41–50. doi: 10.1172/JCI11638
87. Hoffmann PR, Kench JA, Vondracek A, Kruk E, Daleke DL, Jordan M, et al. Interaction between phosphatidylserine and the phosphatidylserine receptor inhibits immune responses. Vivo J Immunol (2005) 174:1393–404. doi: 10.4049/jimmunol.174.3.1393
88. Rossaint J, Thomas K, Mersmann S, Skupski J, Margraf A, Tekath T, et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J Exp Med (2021) 218:e20201353. doi: 10.1084/jem.20201353
89. Coleman MM, Ruane D, Moran B, Dunne PJ, Keane J, Mills KH. Alveolar macrophages contribute to respiratory tolerance by inducing FoxP3 expression in naive T cells. Am J Respir Cell Mol Biol (2013) 48:773–80. doi: 10.1165/rcmb.2012-0263OC
90. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol (2008) 181:2277–84. doi: 10.4049/jimmunol.181.4.2277
91. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204:1757–64. doi: 10.1084/jem.20070590
92. Pyle CJ, Uwadiae FI, Swieboda DP, Harker JA. Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PloS Pathog (2017) 13:e1006640. doi: 10.1371/journal.ppat.1006640
93. Mayer AK, Bartz H, Fey F, Schmidt LM, Dalpke AH. Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur J Immunol (2008) 38:1689–99. doi: 10.1002/eji.200737936
94. Koning N, van Eijk M, Pouwels W, Brouwer MS, Voehringer D, Huitinga I, et al. Expression of the inhibitory CD200 receptor is associated with alternative macrophage activation. J Innate Immun (2010) 2:195–200. doi: 10.1159/000252803
95. Martin FP, Jacqueline C, Poschmann J, Roquilly A. Alveolar macrophages: adaptation to their anatomic niche during and after inflammation. Cells (2021) 10:2720. doi: 10.3390/cells10102720
96. Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med (2007) 356:567–79. doi: 10.1056/NEJMoa062505
97. Ranchod M, Bissell M. Pulmonary alveolar proteinosis and cytomegalovirus infection. Arch Pathol Lab Med (1979) 103:139–42.
98. Rubin E, Weisbrod GL, Sanders DE. Pulmonary alveolar proteinosis: relationship to silicosis and pulmonary infection. Radiology (1980) 135:35–41. doi: 10.1148/radiology.135.1.7360977
99. Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect (2012) 65:173–9. doi: 10.1016/j.jinf.2012.03.020
100. Witty LA, Tapson VF, Piantadosi CA. Isolation of mycobacteria in patients with pulmonary alveolar proteinosis. Med (Baltimore) (1994) 73:103–9. doi: 10.1097/00005792-199403000-00003
101. Pascual J, Gomez Aguinaga MA, Vidal R, Maudes A, Sureda A, Gomez Mampaso E, et al. Alveolar proteinosis and nocardiosis: a patient treated by bronchopulmonary lavage. Postgrad Med J (1989) 65:674–7. doi: 10.1136/pgmj.65.767.674
102. Tran Van Nhieu J, Vojtek AM, Bernaudin JF, Escudier E, Fleury-Feith J. Pulmonary alveolar proteinosis associated with pneumocystis carinii. ultrastructural identification in bronchoalveolar lavage in AIDS and immunocompromised non-AIDS patients. Chest (1990) 98:801–5. doi: 10.1378/chest.98.4.801
103. Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis. Chron Respir Dis (2006) 3:149–59. doi: 10.1191/1479972306cd101rs
104. Carey B, Staudt MK, Bonaminio D, van der Loo JC, Trapnell BC. PU.1 redirects adenovirus to lysosomes in alveolar macrophages, uncoupling internalization from infection. J Immunol (2007) 178:2440–7. doi: 10.4049/jimmunol.178.4.2440
105. Rehli M, Poltorak A, Schwarzfischer L, Krause SW, Andreesen R, Beutler B. PU.1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human toll-like receptor 4 gene. J Biol Chem (2000) 275:9773–81. doi: 10.1074/jbc.275.13.9773
106. Beccaria M, Luisetti M, Rodi G, Corsico A, Zoia MC, Colato S, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. Eur Respir J (2004) 23:526–31. doi: 10.1183/09031936.04.00102704
107. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med (2002) 166:215–35. doi: 10.1164/rccm.2109105
108. Antoniu SA, Rajnoveanu R, Grigore M, Antohe I. Pharmacotherapy options in pulmonary alveolar proteinosis. Expert Opin Pharmacother (2020) 21:1359–66. doi: 10.1080/14656566.2020.1757650
109. Tsukahara K, Lindell RB, Newman H, Lerman BJ, Kersun LS, Piccione J. Successful whole lung lavage in a child with pulmonary alveolar proteinosis secondary to hematologic malignancy. Pediatr Pulmonol (2022) 57:308–10. doi: 10.1002/ppul.25718
110. Papiris SA, Tsirigotis P, Kolilekas L, Papadaki G, Papaioannou AI, Triantafillidou C, et al. Pulmonary alveolar proteinosis: time to shift? Expert Rev Respir Med (2015) 9:337–49. doi: 10.1586/17476348.2015.1035259
111. Kuhn A, Ackermann M, Mussolino C, Cathomen T, Lachmann N, Moritz T. TALEN-mediated functional correction of human iPSC-derived macrophages in context of hereditary pulmonary alveolar proteinosis. Sci Rep (2017) 7:15195. doi: 10.1038/s41598-017-14566-8
112. Chung JH, Pipavath SJ, Myerson DH, Godwin JD. Secondary pulmonary alveolar proteinosis: a confusing and potentially serious complication of hematological malignancy. J Thorac Imaging (2009) 24:115. doi: 10.1097/RTI.0b013e3181930ed6
113. Seymour JF, Presneill JJ, Schoch OD, Downie GH, Moore PE, Doyle IR, et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med (2001) 163:524–31. doi: 10.1164/ajrccm.163.2.2003146
114. Venkateshiah SB, Yan TD, Bonfield TL, Thomassen MJ, Meziane M, Czich C, et al. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest (2006) 130:227–37. doi: 10.1378/chest.130.1.227
115. Wylam ME, Ten R, Prakash UB, Nadrous HF, Clawson ML, Anderson PM. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J (2006) 27:585–93. doi: 10.1183/09031936.06.00058305
116. Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med (2010) 181:1345–54. doi: 10.1164/rccm.200906-0978OC
117. Ohashi K, Sato A, Takada T, Arai T, Nei T, Kasahara Y, et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respir Med (2012) 106:284–93. doi: 10.1016/j.rmed.2011.10.019
118. Papiris SA, Tsirigotis P, Kolilekas L, Papadaki G, Papaioannou AI, Triantafillidou C, et al. Long-term inhaled granulocyte macrophage-colony-stimulating factor in autoimmune pulmonary alveolar proteinosis: effectiveness, safety, and lowest effective dose. Clin Drug Investig (2014) 34:553–64. doi: 10.1007/s40261-014-0208-z
119. Campo I, Mariani F, Paracchini E, Kadija Z, Zorzetto M, Tinelli C, et al. Inhaled sargramostim and whole lung lavage (WLL) as therapy of autoimmune pulmonary alveolar proteinosis (aPAP). Eur Respir J (2016). doi: 10.1183/13993003.congress-2016.PA3870
120. Tazawa R, Ueda T, Abe M, Tatsumi K, Eda R, Kondoh S, et al. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med (2019) 381:923–32. doi: 10.1056/NEJMoa1816216
121. Tian X, Yang Y, Chen L, Sui X, Xu W, Li X, et al. Inhaled granulocyte-macrophage colony stimulating factor for mild-to-moderate autoimmune pulmonary alveolar proteinosis - a six month phase II randomized study with 24 months of follow-up. Orphanet J Rare Dis (2020) 15:174. doi: 10.1186/s13023-020-01450-4
122. Zhang F, Weng D, Su Y, Yin C, Shen L, Zhang Y, et al. Therapeutic effect of subcutaneous injection of low dose recombinant human granulocyte-macrophage colony-stimulating factor on pulmonary alveolar proteinosis. Respir Res (2020) 21:1. doi: 10.1186/s12931-019-1261-1
123. Trapnell BC, Inoue Y, Bonella F, Morgan C, Jouneau S, Bendstrup E, et al. Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. N Engl J Med (2020) 383:1635–44. doi: 10.1056/NEJMoa1913590
124. Tazawa R, Nakata K, Inoue Y, Nukiwa T. Granulocyte-macrophage colony-stimulating factor inhalation therapy for patients with idiopathic pulmonary alveolar proteinosis: a pilot study; and long-term treatment with aerosolized granulocyte-macrophage colony-stimulating factor: a case report. Respirology (2006) 11 Suppl:S61–64. doi: 10.1111/j.1440-1843.2006.00811.x
125. Schoch OD, Schanz U, Koller M, Nakata K, Seymour JF, Russi EW, et al. BAL findings in a patient with pulmonary alveolar proteinosis successfully treated with GM-CSF. Thorax (2002) 57:277–80. doi: 10.1136/thorax.57.3.277
126. Shanafelt TD, Wang XV, Kay NE, Hanson CA, O'Brien S, Barrientos J, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med (2019) 381:432–43. doi: 10.1056/NEJMoa1817073
127. Kaegi C, Wuest B, Schreiner J, Steiner UC, Vultaggio A, Matucci A, et al. Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front Immunol (2019) 10:1990. doi: 10.3389/fimmu.2019.01990
128. Bird D, Evans J, Pahoff C. Rituximab rescue therapy for autoimmune pulmonary alveolar proteinosis. Respir Med Case Rep (2022) 37:101637. doi: 10.1016/j.rmcr.2022.101637
129. Borie R, Debray MP, Laine C, Aubier M, Crestani B. Rituximab therapy in autoimmune pulmonary alveolar proteinosis. Eur Respir J (2009) 33:1503–6. doi: 10.1183/09031936.00160908
130. Soyez B, Borie R, Menard C, Cadranel J, Chavez L, Cottin V, et al. Rituximab for auto-immune alveolar proteinosis, a real life cohort study. Respir Res (2018) 19:74. doi: 10.1186/s12931-018-0780-5
131. Kavuru MS, Malur A, Marshall I, Barna BP, Meziane M, Huizar I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J (2011) 38:1361–7. doi: 10.1183/09031936.00197710
132. Malur A, Kavuru MS, Marshall I, Barna BP, Huizar I, Karnekar R, et al. Rituximab therapy in pulmonary alveolar proteinosis improves alveolar macrophage lipid homeostasis. Respir Res (2012) 13:46. doi: 10.1186/1465-9921-13-46
133. de Boer RA, Martens FM, Kuipers I, Boomsma F, Visseren FL. The effects of the PPAR-gamma agonist pioglitazone on plasma concentrations of circulating vasoactive factors in type II diabetes mellitus. J Hum Hypertens (2010) 24:74–6. doi: 10.1038/jhh.2009.84
134. Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging (2011) 32:1626–33. doi: 10.1016/j.neurobiolaging.2009.10.009
135. Ozasa H, Ayaori M, Iizuka M, Terao Y, Uto-Kondo H, Yakushiji E, et al. Pioglitazone enhances cholesterol efflux from macrophages by increasing ABCA1/ABCG1 expressions via PPARgamma/LXRalpha pathway: findings from in vitro and ex vivo studies. Atherosclerosis (2011) 219:141–50. doi: 10.1016/j.atherosclerosis.2011.07.113
136. Dupin C, Hurtado M, Cazes A, Taille C, Debray MP, Guenee C, et al. Pioglitazone in pulmonary alveolar proteinosis: promising first clinical experience. Respir Med Res (2020) 78:100756. doi: 10.1016/j.resmer.2020.100756
137. Thompson PD, Panza G, Zaleski A, Taylor B. Statin-associated side effects. J Am Coll Cardiol (2016) 67:2395–410. doi: 10.1016/j.jacc.2016.02.071
138. McCarthy C, Lee E, Bridges JP, Sallese A, Suzuki T, Woods JC, et al. Statin as a novel pharmacotherapy of pulmonary alveolar proteinosis. Nat Commun (2018) 9:3127. doi: 10.1038/s41467-018-05491-z
139. Shi S, Gui X, Ding J, Yang S, Xin X, Xu K, et al. Assessment of statin treatment for pulmonary alveolar proteinosis without hypercholesterolemia: a 12-month prospective, longitudinal, and observational study. BioMed Res Int (2022) 2022:1589660. doi: 10.1155/2022/1589660
140. Arumugam P, Suzuki T, Shima K, McCarthy C, Sallese A, Wessendarp M, et al. Long-term safety and efficacy of gene-pulmonary macrophage transplantation therapy of PAP in Csf2ra(-/-) mice. Mol Ther (2019) 27:1597–611. doi: 10.1016/j.ymthe.2019.06.010
141. Hetzel M, Suzuki T, Hashtchin AR, Arumugam P, Carey B, Schwabbauer M, et al. Function and safety of lentivirus-mediated gene transfer for CSF2RA-deficiency. Hum Gene Ther Methods (2017) 28:318–29. doi: 10.1089/hgtb.2017.092
142. Happle C, Lachmann N, Ackermann M, Mirenska A, Gohring G, Thomay K, et al. Pulmonary transplantation of human induced pluripotent stem cell-derived macrophages ameliorates pulmonary alveolar proteinosis. Am J Respir Crit Care Med (2018) 198:350–60. doi: 10.1164/rccm.201708-1562OC
Keywords: alveolar macrophage (AM), pulmonary alveolar proteinosis (PAP), granulocyte-macrophage colony-stimulating factor (GM-CSF), pulmonary homeostasis, therapeutic strategies
Citation: Huang X, Cao M and Xiao Y (2023) Alveolar macrophages in pulmonary alveolar proteinosis: origin, function, and therapeutic strategies. Front. Immunol. 14:1195988. doi: 10.3389/fimmu.2023.1195988
Received: 29 March 2023; Accepted: 31 May 2023;
Published: 14 June 2023.
Edited by:
Matthijs Moerland, Centre for Human Drug Research, NetherlandsReviewed by:
Brenna Carey, Cincinnati Children’s Research Foundation, United StatesCopyright © 2023 Huang, Cao and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonglong Xiao, eW9uZ2xvbmcxMWFAMTYzLmNvbQ==; Mengshu Cao, bWVuZ3NodWNhb0BuanUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.