
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 June 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1195699
This article is part of the Research TopicUnderstanding How Myeloid Cell Development and Function Meet Tissue Distinct Metabolic RequirementsView all 8 articles
 Bader Alabdulaali1,2‡
Bader Alabdulaali1,2‡ Fatema Al-rashed1
Fatema Al-rashed1 Mohammed Al-Onaizi3,1
Mohammed Al-Onaizi3,1 Anwar Kandari1,2
Anwar Kandari1,2 Joanna Razafiarison4
Joanna Razafiarison4 Dorothy Tonui4
Dorothy Tonui4 Michayla R. Williams1Camille Blériot4,5
Michayla R. Williams1Camille Blériot4,5 Rasheed Ahmad1
Rasheed Ahmad1 Fawaz Alzaid1,4*
Fawaz Alzaid1,4*The liver is the site of first pass metabolism, detoxifying and metabolizing blood arriving from the hepatic portal vein and hepatic artery. It is made up of multiple cell types, including macrophages. These are either bona fide tissue-resident Kupffer cells (KC) of embryonic origin, or differentiated from circulating monocytes. KCs are the primary immune cells populating the liver under steady state. Liver macrophages interact with hepatocytes, hepatic stellate cells, and liver sinusoidal endothelial cells to maintain homeostasis, however they are also key contributors to disease progression. Generally tolerogenic, they physiologically phagocytose foreign particles and debris from portal circulation and participate in red blood cell clearance. However as immune cells, they retain the capacity to raise an alarm to recruit other immune cells. Their aberrant function leads to the development of non-alcoholic fatty liver disease (NAFLD). NAFLD refers to a spectrum of conditions ranging from benign steatosis of the liver to steatohepatitis and cirrhosis. In NAFLD, the multiple hit hypothesis proposes that simultaneous influences from the gut and adipose tissue (AT) generate hepatic fat deposition and that inflammation plays a key role in disease progression. KCs initiate the inflammatory response as resident immune effectors, they signal to neighbouring cells and recruit monocytes that differentiated into recruited macrophages in situ. Recruited macrophages are central to amplifying the inflammatory response and causing progression of NAFLD to its fibro-inflammatory stages. Given their phagocytic capacity and their being instrumental in maintaining tissue homeostasis, KCs and recruited macrophages are fast-becoming target cell types for therapeutic intervention. We review the literature in the field on the roles of these cells in the development and progression of NAFLD, the characteristics of patients with NAFLD, animal models used in research, as well as the emerging questions. These include the gut-liver-brain axis, which when disrupted can contribute to decline in function, and a discussion on therapeutic strategies that act on the macrophage-inflammatory axis.
Among its many functions, the liver detoxifies and metabolizes components in blood arriving from the hepatic portal vein and hepatic artery, stores glycogen, secretes bile to aid digestion, and produces cholesterol as well as major plasma proteins such as albumin and fibronectin. These synthetic and secretory capacities make the liver the largest gland in the human body (1). Anatomically, the liver is in the upper right abdomen, beneath the right hemidiaphragm, and it is protected by the rib cage. It is separated by visible fissures, the most prominent of which is the umbilical fissure, which is lined by the falciform ligament. Such divisions allow identification of different lobes, for example, the large right lobe and a smaller left lobe (2, 3). The right lobe is further divided into a quadrate lobe and a caudate lobe; these are functional sections where the gallbladder and the inferior vena cava reside (4, 5) (Figure 1A).
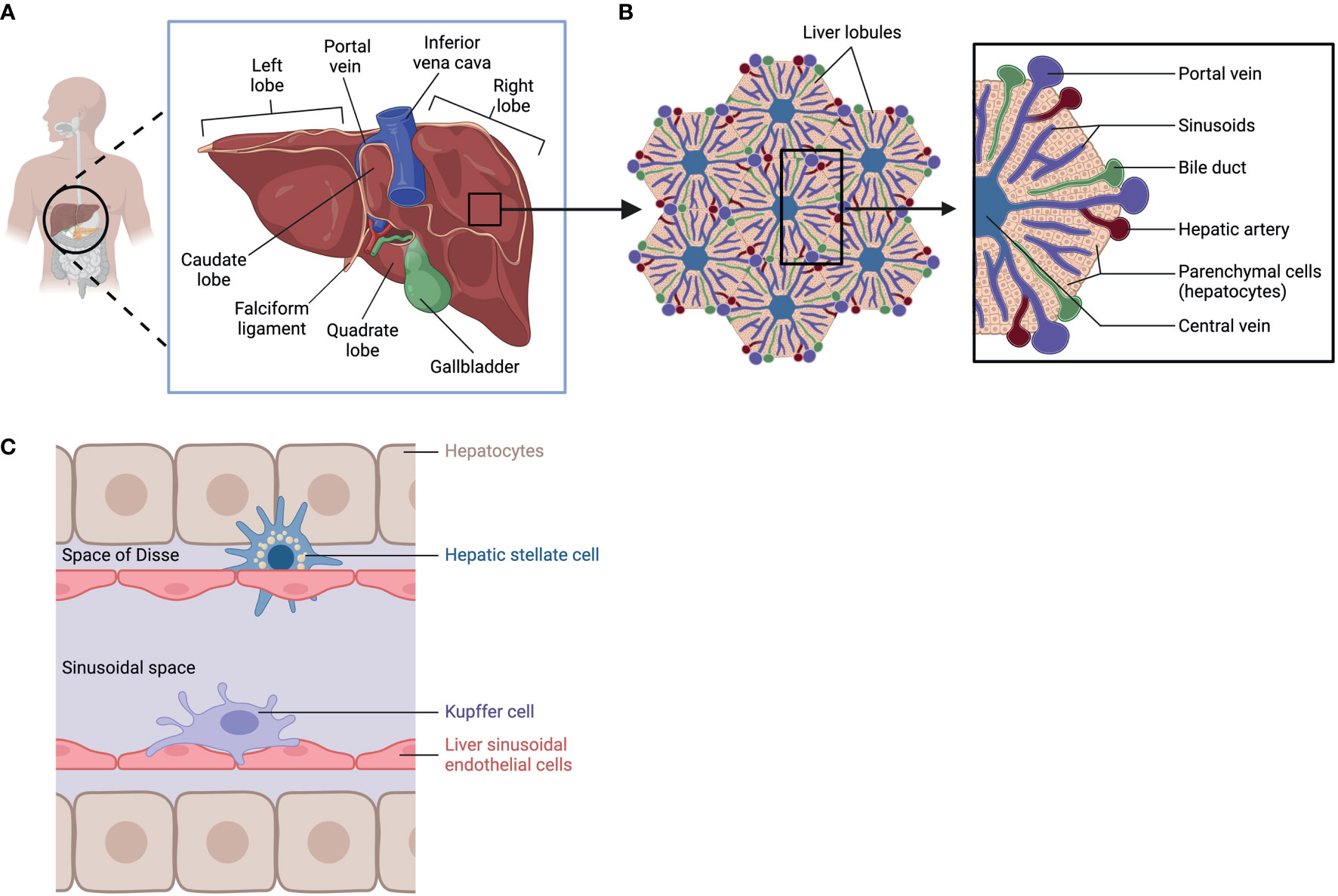
Figure 1 Liver Anatomy, lobular structure, and cell types. (A) anatomic location of the liver. (B) Lobular structure of the liver. (C) Cell types in the liver: hepatocytes, Hepatic stellate cell, Kupffer cell, and Liver sinusoidal endothelial cells. Created with BioRender.com.
To carry out its specialized functions, the liver is made up of a variety of cell types organized in lobular structures. One lobule is roughly hexagonal in shape and formed around the central vein with portal triads that demarcate the six corners of each lobule containing bile ducts, sinusoids, branches of the hepatic portal vein, and the hepatic artery (Figure 1B). Within its lobular structure are several cell types that cooperate to carry out physiological functions and maintain homeostasis in the tissue microenvironment. The different cell types can be generally divided into parenchymal cells, hepatocytes, and non-parenchymal cells which include hepatic stellate cells (HSC), Kupffer cells (KC), and liver sinusoidal endothelial cells (LSEC) (6) (Figure 1C). These cells carry out essential functions in the liver (Table 1) and are crucial in responding to injury (7).
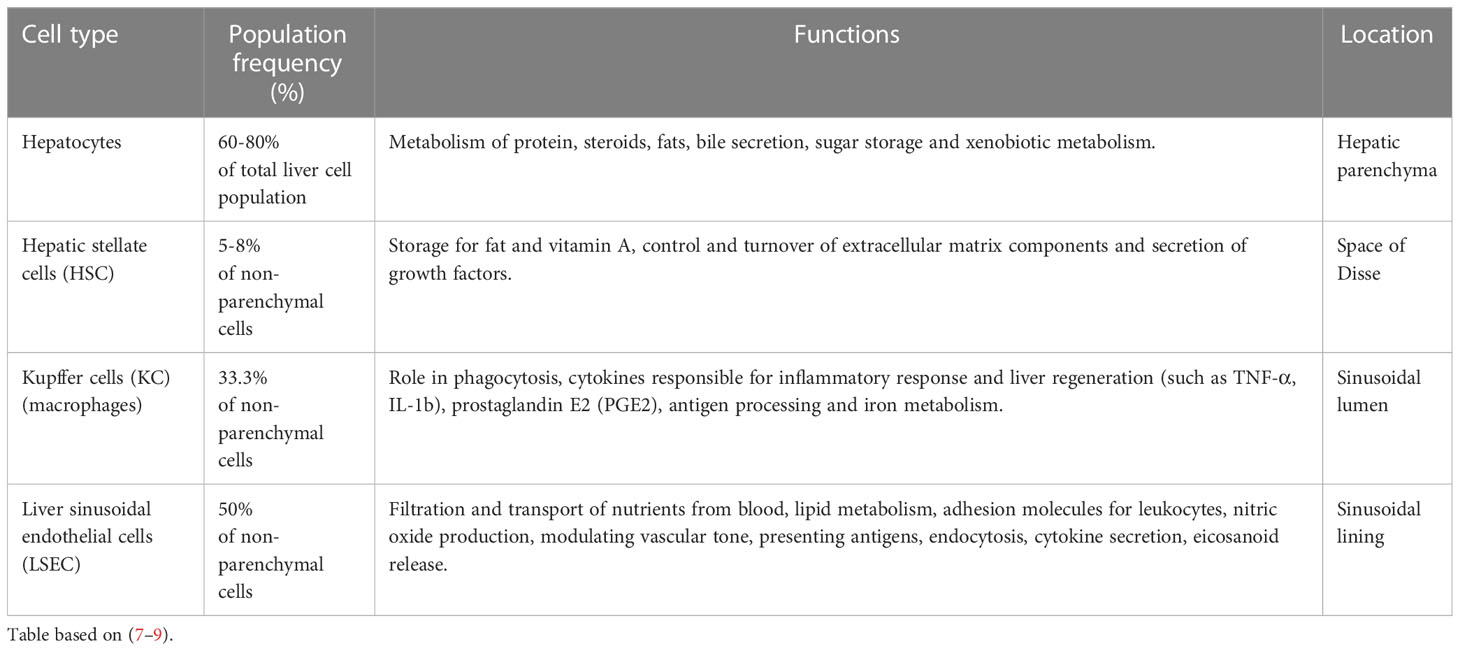
Table 1 Major cell type distribution in the liver and their known functions.
Focusing on the immune compartment, KCs are the main immune cells that populate the liver in steady state. They extensively interact with hepatocytes, HSCs, and LSECs. They can stimulate leukocyte chemotaxis and adherence and produce cytokines that influence their activation (10, 11). KCs phagocytose foreign particles and microorganisms in portal circulation coming from the gut (12). These cells are the first line to defense against pathogens, they remove abnormal cells and cellular debris, participate in recycling erythrocytes, and their signaling can dictate inflammation and the immune response (13). KCs also contribute to liver disease when they are dysfunctional (14), this is discussed in more detail below.
Non-alcoholic fatty liver disease (NAFLD) refers to a spectrum of conditions affecting the liver. These range from benign steatosis to steatohepatitis (NASH), fibrosis, and cirrhosis. Importantly, NASH and fibrosis often occur simultaneously and are the last reversible steps of the condition. At the stage of cirrhosis, liver function is impaired, and patients are at high risk of developing hepatocellular carcinoma (HCC). Liver transplantation is the only possible intervention strategy at or beyond the stage of cirrhosis. Many individuals have the early stages of NAFLD, benign ectopic lipid storage in the form of steatosis, without any signs or symptoms; this is because the presence of fat in the form of simple steatosis does not cause damage to the liver (15–17) (Figure 2A). NAFLD and its progressive form NASH have similar risk factors, including overweight or obesity, insulin resistance, high levels of fats, particularly triglycerides, and hyperglycemia, indicating prediabetes or type 2 diabetes (T2D) (17).
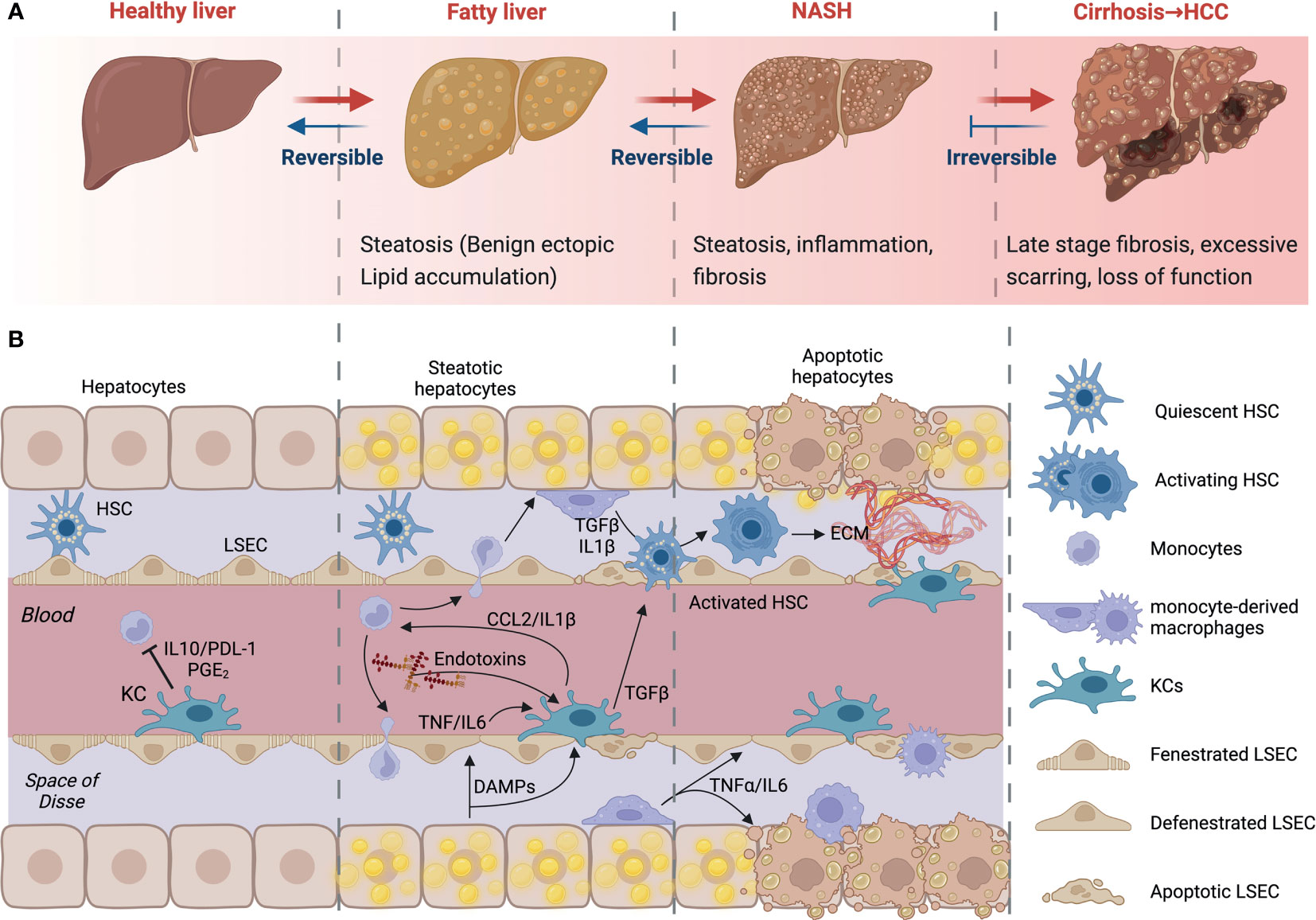
Figure 2 NAFLD progression and cell-to-cell signaling. (A) Development and progression of non-alcoholic fatty liver disease (NAFLD) to non-alcoholic steatohepatitis (NASH) and advanced fibrosis. NAFLD can develop into NASH, liver cirrhosis and/or hepatocellular carcinoma (HCC) due to several factors. (B) Kupffer cells (KCs) recognize imbalances in homeostasis. Their consequent signaling allows the activation of HSCs, contributing to apoptosis and phagocytosis of damaged cells. Early infiltration and differentiation of monocytes contributes to inflammation. KC- and macrophage-derived cytokines induce HSC activation. HSCs will deposit extracellular matrix to create fibrous septa. Created with BioRender.com.
The excessive increase of free fatty acids and triglycerides increases lipid oxidation and the production of reactive oxygen species (ROS) and lipotoxicity. This causes cellular damage and the secretion of cytokines, triggering KC activation and the recruitment of monocytes and other immune cells from the circulation. Ensuing hepatocyte apoptosis and HSC activation results in the deposition of extracellular matrix, which can lead to fibrosis if excessive (Figure 2B). At this stage, there is obvious persistent scar tissue in the liver and in the blood vessels around the liver. Nonetheless, the liver may still work well, and treating the cause of the inflammation may inhibit further development or even partially reverse the damage (18). However, when the replacement of normal liver tissue by fibrous septa exceeds a certain point, this develops into cirrhosis, the late stage of NAFLD. At this stage, there is extensive scarring and loss of function. Symptoms will include variceal bleeding, ascites, fatigue, and jaundice (19, 20). As a result, the liver stops functioning, urgent medical care and liver transplant surgery may be required.
The majority of patients diagnosed with NAFLD are asymptomatic, and this may remain silent until it has developed into cirrhosis (21). Right upper quadrant pain and fatigue are the most noted symptoms among NAFLD patients. Individuals may also have liver fat, based on an imaging assessment or evidence of echogenic liver on ultrasound (22). Serum alanine aminotransferase (ALT) is typically higher than serum aspartate aminotransferase (AST) in liver-related serum tests, indicating hepatocellular stress (23). When advanced fibrosis, cirrhosis, and portal hypertension develop, platelet count gradually declines over time (24).
Globally, almost one billion individuals are diagnosed with NAFLD (25). South America and the Middle East have the highest prevalence, whereas Africa has the lowest (15). In non-obese individuals, the rate is around 10%-30% across Eastern and Western countries (21). There are also differences in gender and ethnic groups in the occurrence of NAFLD in non-obese individuals; for example, the prevalence of NAFLD is much lower in non-obese South Asian women than in non-obese South Asian men (22). Interestingly, NAFLD prevalence in women rises with age, yet does not change with age in men (26). There are several risk factors associated with NAFLD, including primary risk factors related to insulin resistance and metabolic conditions such as T2D, obesity, and dyslipidemia. Secondary risk factors are related to the use of certain medications (corticosteroids, tamoxifen, and amiodarone), metabolically acquired or congenital alterations, surgical operations, and nutritional changes (16). In terms of diagnosis, NAFLD is diagnosed by imaging or by histological assessment of a liver biopsy showing a minimum infiltration of 5% of hepatocytes with steatosis in patients who drink little or no alcohol. Additionally, no other causes of hepatic steatosis are identified, such as Wilson’s disease or drugs such as steroids (27).
Insulin resistance can in part be attributed to abnormal hepatic insulin processing; this would otherwise physiologically regulate glucose and lipid metabolism. The liver produces glucose using glycogen; this happens with the use of glucogenic precursors in the presence of a high glucagon-to-insulin ratio during a fasting period (28). During the fed state, reductions in glucagon and raised insulin levels signal the liver to raise glucose uptake, cease glucose production, and store the remaining nutrients as glycogen and lipids (29). In pathological conditions of insulin resistance, insulin fails to regulate liver metabolism, resulting in excessive glucose production alongside increased lipid synthesis (30); consequently, NAFLD is associated with insulin-resistance (31).
In obese individuals with T2D and NAFLD, dyslipidemia and hyperinsulinemia are more severe than in individuals without NAFLD (32). Insulin resistance develops in overweight or obese individuals when insulin action on its target tissues is compromised. Excess fatty acids, due to lifestyle factors as well as lipogenesis, accumulate in peripheral tissues such as the liver and AT, which play key roles in lipid storage, synthesis, and metabolism (33). Whilst independently affected by insulin resistance, significant crosstalk also occurs between the liver and AT, this crosstalk has been reported to influence the liver’s immune compartment. Indeed, it has been demonstrated that transplanting donor visceral AT (vAT) from obese mice increased high cholesterol diet (HCD)-induced liver macrophage content, worsening liver damage, compared to mice receiving transplants from lean donors. Adipose tissue macrophage (ATM) depletion prior to vAT transplantation abrogated this effect. On normal chow diets, vAT transplantation increased circulating and hepatic neutrophil numbers in obese-transplanted mice, similarly ATM depletion prior to vAT transplantation reversed this effect (34). Microarray analysis of sorted CD11c+ and CD11c− macrophages isolated from donor adipose tissue showed that obesity increased expression of genes involved in chemotaxis from CD11c+ ATMs. CD11c+ ATMs are known to increase in proportion in obese and insulin resistant conditions (34). These findings indicate that secreted molecules from ATMs act in an endocrine manner and can affect the liver’s immune compartment, influencing susceptibility to, and progression of, NAFLD.
AT also secretes adipokines, a special class of cytokine-like messenger molecules, such as leptin and adiponectin (35). Adiponectin regulates lipid accumulation both in the liver and in AT by inhibiting fatty acid oxidation (36). Moreover, it controls glucose homeostasis through regulating hepatic insulin sensitivity (37). Individuals with NAFLD have lower serum adiponectin levels than the healthy population (38). Hypoadiponectinemia promotes chronic inflammation of the liver that results from dysregulation of fatty acid metabolism associated with insulin resistance (39). Therefore, maintaining the adiponectin level in individuals with NAFLD may prevent progression to NASH. In contrast, leptin levels have been reported to be higher in individuals with NAFLD, and higher levels of circulating leptin are associated with increased disease severity (40). However, in vivo studies do not support a causal association since ob/ob mice that lack leptin and develop hyperphagic obesity have severe steatosis (41). Administration of leptin reverses the obese phenotype and improves steatosis. Hence, leptin may play multiple roles in NAFLD (42).
KCs are the dominant immune population in the liver; they are bona fide resident macrophages that stem from the yolk sac and thus have an embryonic origin (43). KCs are self-renewing and are phenotypically distinct from monocyte-derived macrophages that can infiltrate the liver during disease (44). Monocyte-derived macrophages differentiate in situ from circulating monocytes, and thus they originate from bone marrow hematopoiesis (45). Under healthy conditions, rodent models have between 20 and 40 macrophages for every 100 hepatocytes in the liver, making it the organ with the highest proportion of tissue macrophages compared to other tissues. This supports a functional role in homeostasis and normal physiology of the liver (46). They perform a number of tasks, including clearance of metabolic waste and cellular debris (46), regulation of iron homeostasis via red blood cell phagocytosis and iron recycling (47), maintenance of cholesterol homeostasis (48), maintenance of immune tolerance (49), and promotion of antimicrobial defense (50).
Once thought to be a homogenous population, recent studies reveal the heterogeneity of liver macrophages, beyond their origin, and evidence continues to accumulate to indicate that certain subpopulations are necessary in maintaining different aspects of homeostasis. Liver macrophage heterogeneity is increasingly deconvoluted with the emergence of precision sorting, single-cell/-nucleus RNA sequencing (scRNA-seq), and macrophage targeting technologies (51). For example, one study demonstrated that there are distinct populations of liver macrophages that function in an inflammatory and non-inflammatory or regulatory manner (52) Another study reported that diet-induced steatohepatitis impairs differentiation of myeloid cells in the liver and bone marrow (53). The diversity and subtypes of liver macrophages are further discussed in a dedicated section below, here we focus on the physiological distinction and roles of liver macrophages.
Interestingly, liver macrophages constitute a separate immune surveillance niche and form a cellular network distinct from those in the hepatic capsule (54). The capsular macrophages are phenotypically and developmentally different from KCs. They arise from circulating monocytes and express the macrophage markers F4/80 and CD64 yet are negative for the canonical resident macrophage markers Clec4F and T-cell immunoglobulin mucin domain containing 4(Tim4) (55). In addition, they can express markers also associated with dendritic cells or M1-like polarization, such as CD11c and MHCII. These cells sense bacteria in the peritoneum and promote neutrophil recruitment to restrict peritoneal bacteria spread to the liver (44). KCs on the other hand are prone to exposure to nutrients and microbial products in the blood rising from portal circulation. KCs encounter this rich venous blood and blood rich in insulin and oxygen from the hepatic artery in sinusoids (56). Mice deficient in KCs have weakened survival following infection with Listeria monocytogenes, indicating that KCs plays an important role in the control of bloodborne bacteria. Because of their high exposure to microbial products and nutrient-rich blood, KCs are generally tolerogenic and promote immune tolerance in the liver microenvironment (57). This is achieved by the expression of regulatory or anti-inflammatory molecules like interleukin-10 and prostaglandins, such signaling is important in maintaining immune tolerance and can also promote the differentiation of regulatory T cells (57) (Figure 2B).
Although KCs are generally tolerogenic, they retain the capacity to raise an alarm following the detection of danger signals from neighboring cells. KCs will sense disturbances in homeostasis from lipotoxic hepatocytes and endothelial cells; their subsequent signaling places them at the center of an intense cellular crosstalk (Figure 2B). This crosstalk allows the recruitment of other immune cells, the activation of HSCs, and the induction of apoptosis and phagocytosis of damaged cells. NAFLD is characterized by an early infiltration of monocytes; these monocytes differentiate into macrophages in situ and contribute to the inflammatory response. KC and other macrophage-derived cytokines can target HSCs. TNFα, IL-1β and TGFβ can all induce HSC activation. In turn, HSCs up-regulate several ligands, like CCL2, which are able to attract and regulate the activity of macrophages and other immune cells in NASH (57). Fibrosis progression is largely mediated by liver macrophage-HSC crosstalk (58). Once activated, HSCs will deposit extracellular matrix to create fibrous septa. Thus, macrophages amplify inflammation, support fibrogenic phenotypes and facilitate the survival of HSCs through the chemokines and cytokines they release (57–59). The initial inflammatory hit is key in the progression of the disease (60). KCs can also influence inflammation by binding of bacterial products in the liver’s portal vein. Activation of macrophages is also accompanied by the generation of malondialdehyde because of oxidative stress within the liver. However, KC ablation decreased steatosis and insulin resistance in the liver supporting the notion that KCs can also drive the development of early NAFLD (61). Indeed, an increase in CD68+ cells has been reported in biopsies from patients with NASH compared to those with steatosis (62). In animal studies, high-fat diet (HFD) and methionine-choline-deficient (MCD) diet-fed mice have more liver macrophages that produce pro-inflammatory cytokines (63). In addition, mice fed a MCD diet produced these inflammatory mediators at a higher level after 4 weeks and decreased later, suggesting that the release of these mediators may specifically contribute to progression and that macrophages may play different roles once NASH is established (64). Furthermore, clodronate injections, which deplete phagocytes, decrease steatosis and NASH severity (65). In the absence of KCs, fatty acid oxidation genes and peroxisome proliferator-activated receptor (PPAR) expression were increased in the liver. This mechanism was reported to be dependent on interleukin-1β-mediated suppression of PPAR-α (66, 67). Further, Zhang et al (68) demonstrated that p38α expression is increased in biopsies of patients with NAFLD, relative to control individuals. The importance of macrophage expression of p38α was demonstrated using macrophage-specific knockouts, in which mice deficient for p38α developed less severe liver disease and insulin resistance upon multiple dietary models of NAFLD and NASH. Hence, progression to steatohepatitis is promoted in a p38-dependent manner; p38α promotes macrophage polarization and proinflammatory cytokine release (57).
Liver macrophages can be categorized in multiple ways, for example, based on their origin, their polarization states, their functional specificities, or the expression of phenotypic markers. These ways of categorizing liver macrophages are a subject of continuous debate, with new categories being continuously proposed as we learn more about population and subpopulation specificities. When origin is used, broadly speaking, liver macrophages are grouped into resident and recruited cells, resident cells being KCs, that are embryonically derived, and recruited macrophages differentiate from monocytes that originate from bone marrow hematopoiesis (69) (Figure 3A). In mice, KCs are distinguishable by their variable expression of typical macrophage markers as F4/80hi, CD68+, and CD11bint cells, alongside specific liver resident markers Tim4 and Clec4F (70). Recruited macrophages will not carry the latter to markers, and their monocyte progenitors are Cx3cr1+, CD11b+, Ly6c+, and CC-chemokine receptor 2 (CCR2+) (43). CCR2 in particular plays an important role as it regulates the recruitment of monocytes and macrophages. Genetic deficiency of Ccr2 in mice fed a HFD reduced their food intake and decreased their development of obesity compared to wild-type mice. Ccr2 deficiency led to a decrease in AT macrophage content and inflammatory profiles, a substantial rise in adiponectin expression, decreased severity of hepatic steatosis and improved insulin sensitivity in obese mice. An antagonist of CCR2 was found to have significant effects in enhancing insulin sensitivity and lowering macrophages number in adipose tissue in obese mice. In obesity and its related metabolic consequences, CCR2 plays a role in recruiting monocytes to tissues under metabolic stress, sustaining and amplifying inflammation in AT and the liver, and promoting insulin resistance (71). Polarization state can also be used to classify macrophages, it refers to a final stage of differentiation that macrophages reach in response to signals from their immediate environment. Such signals include cytokines, growth factors, exosomes, fatty acids, or various PAMPs and DAMPs. Historically, macrophage polarization states were designated as either M1 or M2 (72). This M1-M2 classification is now considered outdated, macrophages in-fact exhibit enormous diversity, they can adopt intermediate phenotypes that lay on a sliding scale, where M1 and M2 are extremes (Figure 3B). When considering phenotypic extremes, M1 macrophages are proinflammatory, releasing an elevated level of proinflammatory cytokines and generating reactive oxygen species, which stimulate inflammatory responses (57, 59). M1 macrophages are activated by GM-CSF, TNF-α, and IFN-γ (73) and are involved in triggering the Th1 response via the production of cytokines, including TNF-α (74). They are fueled via glycolysis, which has been directly linked to IL-1β production (75). On the other hand, M2 macrophages express molecules that play anti-inflammatory and reparative roles (76). Furthermore, M2 macrophages utilize oxidative metabolism to fuel their functions (75). M2 macrophages are induced by IL-13 or IL-4 (77). They produce polyamines, ornithine, and arginase-1 and induce a Th2 response, promoting immune tolerance and tissue repair (78). M2 macrophages have been found to be protective in the context of inflammatory and metabolic disease, such as insulin resistance; however, they can play deleterious roles in certain conditions with extensive tissue remodeling, such as atherosclerosis and cancer (79). M2 macrophages can be further categorized into four subtypes based on the stimuli and activated transcriptional states: alternatively activated macrophages stimulated by IL-13 or IL-4 (M2a), type 2 macrophages activated via IL-10 or IL-1RA (M2b), deactivated macrophages stimulated via IL-10 or glucocorticoids (M2c), and M2-like macrophages induced by adenosines or IL-6 (M2d) (79). The canonical transcriptional regulators for M1-like and M2-like polarizations are respectively IRF5, NFkb, STAT1 and IRF4, PPARG, and other members of the STAT and SMAD families of transcription factors (80, 81). In terms of phenotypic identification of these subtypes, both M1-like and M2-like macrophages will express typical macrophage markers (e.g., CD68), in addition to CD11c or CD206 to denote polarization as M1 or M2, respectively (75, 77). Double positivity has been reported for these two markers, as well as associating other functional markers to denote subpopulations (82). Depending on stimulus and context, other markers have been employed to denote M1-like and M2-like polarization. These include CD40, CD86 and high expression of MHC-II for M1-like macrophages, and Dectin-1, CD36 and CD163 for M2-like macrophages (76, 83). The most robust phenotyping strategies should include a combination of appropriate surface markers and the use of intracellular markers that indicate function, i.e., transcription factors responsive to the condition studied.
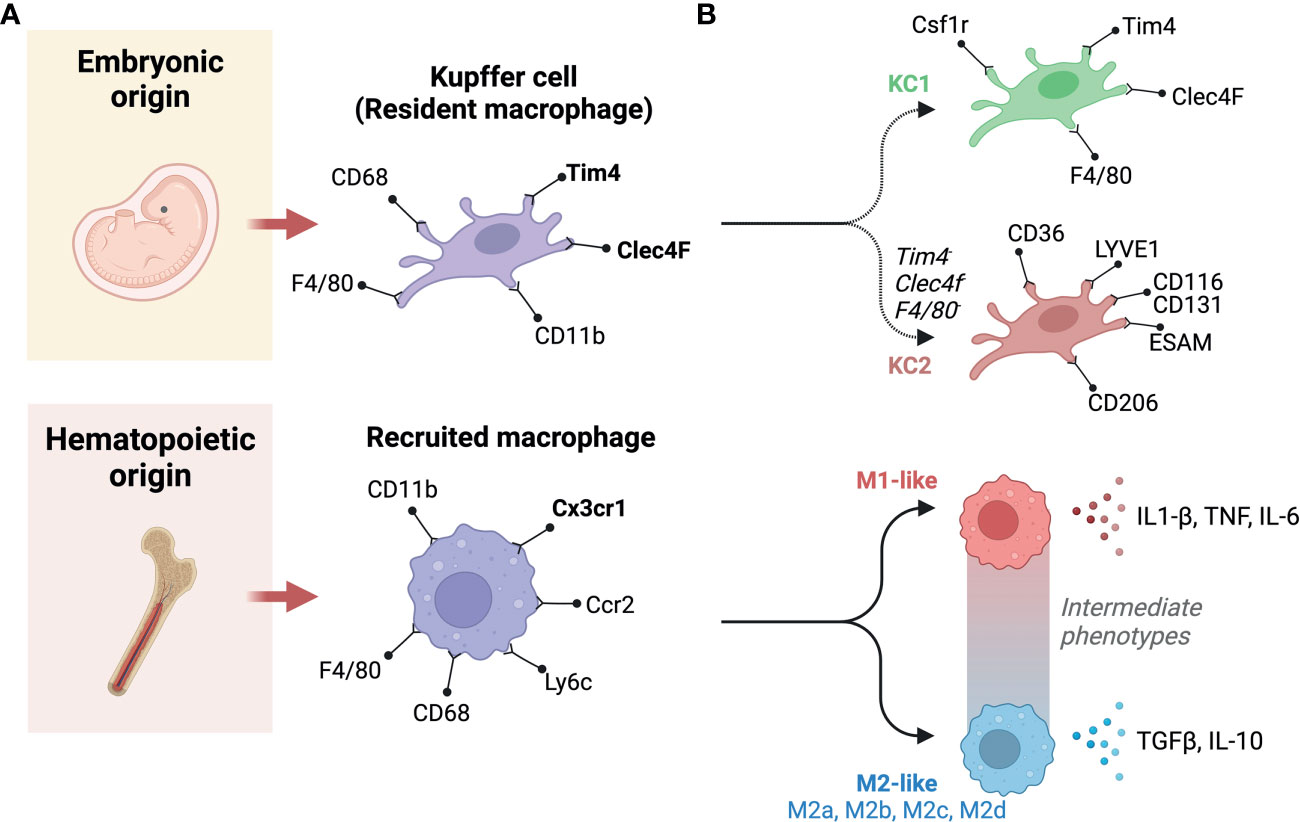
Figure 3 Classification and origins of liver macrophages. (A) Based on origin, macrophages can be classified into two subgroups. One originates from the yolk sac (Embryonic origin) and another derives from circulating monocytes that have hematopoietic origins. (B) There are two populations of liver resident Kupffer cells (KCs), including KC1s which are CD206 low and Endothelial cell-Selective Adhesion Molecule (ESAM) negative; and KC2s which are CD206high and ESAM+. KC1s express the KC markers: colony-stimulating factor-1 receptor (Csf1r), T-cell immunoglobulin and mucin domain containing 4 (Tim4), C-type lectin domain family 4 member F (Clec4F), and F4/80. In contrast, The KC2 express the markers: CD36, lymphatic vessel endothelial hyaluronan receptor-1 (LYVE1), ESAM, and CD206. Recruited macrophages can be classified based on polarity into M1 and M2. M1 macrophages are canonically inducible by lipopolysaccharide (LPS) and interferon-γ (IFN-γ), whereas interleukin (IL)-4 and IL-13 can induce M2 polarization. M1 macrophages secrete pro-inflammatory cytokines, such as IL-1β and tumor necrosis factor-α (TNF-α). Whereas, M2 macrophages primarily produce anti-inflammatory factors, such as IL-10 and transforming growth factor-β (TGF-β). In between these extremes are a number of intermediate phenotypes, and M2 macrophages can be further categorized into M2a, M2b, M2c, and M2d subtypes. Created with BioRender.com.
Using the M1-M2 model, not only are the two extremes functionally distinct, they are also distinct in terms of their cellular metabolic profiles (84). The activation of M1 macrophages leads to the induction of aerobic glycolysis that generates ATP and lactate (85, 86). The pentose phosphate pathway (PPP), that branches from the early stages of glycolysis, is also induced under the same conditions, by IFN-γ or LPS, and produces NADPH and substrates for nucleotide synthesis (87). Additionally, the M1 phenotype is regulated by carbohydrate kinase-like protein (CARKL), implicated in the catalysis of sedoheptulose-7-phosphate (88). CARKL is involved in the metabolic control of pro-inflammatory immune responses by causing a redox shift in M1 macrophages. Consequently, stimulation of an M1-like state by LPS inhibits CARKL and leads to generation of NADH and GSH, while the stimulation of an M2-like state positively regulates CARKL (88). In terms of fatty acid oxidation and oxidative metabolism, IL-4 triggers STAT-6, which results in increased mitochondrial respiration (89, 90). M2 activation facilitates the entrance of pyruvate into the Krebs cycle, supporting the Electron Transport Chain (ETC) and providing the energy required for the remodeling and repair of tissues (84). The activation of M1 macrophages results in increased glycolysis, which aids in microbicidal activity and the management of hypoxia that occurs within the microenvironment of the tissue (91). Glucose, in the cytosol, is converted into L-lactate by glycolysis, in which hexokinase (HK), 6-phosphofructokinase 1 (PFK1) and pyruvate kinase (PK) are key enzymes. During glycolysis, glucose-6-phosphate can be propelled to PPP pathway promoting pentose NADPH and phosphate production. Pentose phosphates are used for the synthesis of amnio acid and nucleotide, while NADPH contributes to the production of ROS and NO (92, 93). Pyruvate is stimulated to lactate in hypoxic conditions, while decarboxylated into acetyl-CoA in the mitochondria in normoxic conditions. Here, acetyl-CoA goes into the TCA cycle, bringing reducing agents to the ETC to generate energy. The TCA metabolite, Citrate, participates in fatty acid synthesis when exported to the cytoplasm, this process can support membrane such functions as membrane synthesis (94). Two breakpoints in M1 macrophages cause the generation of itaconate and the increase of succinate, decreasing pH and stabilizing HIF-1a. HIF-1a results in upregulation of glycolysis and M1-like functions via GLUT1 expression and IL-1b production (84). What’s more, the production of NO inhibits the ETC. Nonetheless, M2 macrophages can acquire sufficient ATP from the ETC and via the TCA cycle (95).
Beyond the above standard classifications, new subpopulations of macrophages are constantly being discovered, they can functionally lay along the M1-M2 spectrum or have significant phenotypic differences from previously described populations. For example, the recently described functionally distinct KC1 and KC2 populations (96) or Trem2+ lipid-associated macrophage (LAM) (97, 98). The more abundant KC1s are identifiable by their low expression of CD206 and are endothelial cell-selective adhesion molecule negative (ESAM-), whereas KC2s highly express CD206, ESAM, CD36 and the lymphatic vessel endothelial hyaluronan receptor-1 (LYVE1) (52). LAMs were first described in adipose tissue, they enhance the degradation and processing of lipids via lipoprotein lipase (Lpl), fatty acid transporter Cd36, and fatty acid binding proteins 4 and 5 (Fabp4, Fabp5) (99). LAMs have also been described in other lipid-rich environments such as in atherosclerotic plaques, here LAM expression of Spp1 and Cd9 is correlated with lesion calcification and a downregulation of pro-inflammatory genes (79). With regards to NAFLD, LAMs can localize to fibrotic areas, macrophage aggregates or to hepatic crown-like structures (hCLS) (100, 101), and have been reported to mitigate liver fibrosis (100). Daemen et al (101) revealed a number of LAM specificities, that they express Trem2, Cd63, Cd9 and Gpmnb; and that they are recruited through Ccr2. Their failure to accumulate in the liver of Ccr2-deficient mice increases liver fibrosis upon high-fat, high-sucrose feeding, indicating that they play an important role in tissue remodeling (101). Interestingly, Trem2 that is expressed on LAMs also exists in a soluble form, and levels in plasma reflect recruitment and expansion of LAMs in the liver (98) (Figure 3B).
Whilst significant progress has been made in deciphering the heterogeneity of liver macrophages, under physiological and pathophysiological conditions, most of these studies have been carried out in murine models. More recently, several resources have been developed using novel single cell or single nucleus sequencing to translate findings to humans and shed light on conserved subsets and population features in liver macrophages from human biopsies (e.g., www.livercellatlas.org) (102–104). Thanks to these studies that expand the knowledgebase on human liver macrophages, the translatability of findings from murine studies is increasing with time.
The multiple hit hypothesis, which postulates that simultaneous influences from the gut and adipose tissue produce hepatic fat deposition and inflammation (105, 106), has gained widespread acceptance. Monocytes, recruited and resident macrophages are involved in influencing hepatic lipid accumulation and in triggering inflammation that takes place in NAFLD (107, 108). The role of macrophages in lipid processing is an area of active research, where questions address both cellular metabolism of microenvironmental lipids, as well as macrophage efferent signaling that can influence tissue and systemic lipid homeostasis (109, 110). In a study by Rivera et al (110), it was reported that the depletion of KCs abolished fat accumulation in the liver and delayed the development of NASH induced by an MCD diet. This observation goes hand-in-hand with reduced inflammatory burden (110). This observation was found to be associated with a reduction in two receptors: CD36 and the toll-like receptor 2 (TLR2). This observation is further supported by the requirement of TLR2-dependent fatty acid uptake of diacylated lipoproteins by CD36 for lipid trafficking (67, 99, 110, 111). The innate immune signaling system mediated by Toll-like receptors (TLRs) is implicated in the progression of NASH. However, combination of TLR2 and palmitic acid is required for inflammasome activation, which results in NASH progression. To induce NASH in wild-type (WT) and TLR2-deficient mice, a choline-deficient amino acid-defined (CDAA) deficient diet was fed for 22-weeks. After the recipient mice were lethally irradiated, bone marrow transplanted-TLR2 chimerism was generated. WT mice were treated with TLR2 ligands and/or palmitic acid to stimulate their KCs and HSCs. In response to a CDAA diet, WT mice exhibited severe steatohepatitis and liver fibrosis. Conversely, mice lacking TLR2 did not progress to NASH. Although KCs and HSCs both respond to TLR2 ligands, TLR2 bone marrow chimeric mice demonstrated that KCs play a greater role in the TLR2-mediated progression of NASH than HSCs (112). Conversely, the impact of lipid accumulation in KCs is not always an unwanted outcome. Indeed, in work presented by Leroux and colleagues, it was argued that the upregulation of triglyceride storage in KCs from HFD-fed mice is triggered as part of a normal immune response. In HFD-fed mice, lipidomic and RT-qPCR analysis revealed that KCs become lipid-laden and up-regulate lipogenesis genes Dgat1 and Scd1 (100, 113, 114). This upregulation is associated with a functional adaptation, that is lipid-laden KCs are primed to recruit lymphocytes more efficiently (CD4+ T cells and B cells in their investigation), of note this adaptation is reversible with the inhibition of lipogenesis. In addition, it was discovered that systemic cholesterol levels influence cellular cholesterol homeostasis in lipid-laden KCs (100). During hypercholesterolemia, macrophage cholesterol efflux is impaired, this drives their polarization towards a proinflammatory phenotype (115, 116). The association is strong between dietary cholesterol intake and hepatic cholesterol in the development of NASH in both human and animal models. A study by Bieghs et al (91), showed that the deletion of Cd36 and macrophage scavenger receptor 1 (Msr1) in low-density lipoprotein (LDL) receptor-deficient (Ldlr-/-) mice attenuates liver inflammation, indicating involvement of the cholesterol uptake pathway (117). This is supported by large scale epidemiological studies, where dietary cholesterol consumption was independently associated with the development of NASH and cirrhosis (118, 119). It has also been shown that the beginning of hypercholesterolemia might affect membrane fluidity, which, in turn, can cause changes in phagocytic capacity, a process known to be disrupted in murine models of NASH (115, 119–121). It is speculated that the KCs scavenged free cholesterol (including crystals), cholesterol esters, and triglycerides from the remaining big lipid droplets of dying hepatocytes. Naturally, KCs will hydrolyze cholesterol esters and TG before oxidizing the liberated fatty acids. However, in the case of free cholesterol, it cannot be further metabolized, causing it to be retained in KCs, leading to transformation into foam cells, stimulating inflammatory and fibrotic pathways that cause NAFLD to progress to NASH. Together, these observations provide insight into the importance of KCs in participating in the underlying mechanism of hepatic lipid accumulation and inflammation.
There is a wide range of mouse models for investigating the pathogenesis of NAFLD. Agreed standards in the use of these models would be valuable to enable valid interpretations and comparisons between the many studies being carried out (122). Furthermore, given that the mechanisms for the development of NAFLD in humans are still not fully defined, it follows that it is highly unlikely that the mechanistically ideal mouse model has been developed. An ideal model for this multi-organ, systemic disease would closely replicate the features of the human disease within a predictable time frame while also showing that its use in potential treatment trials is translatable to the human condition. With echoes of Knudson’s two-hit hypothesis for cancer in the early 1970s, James et al (123) proposed a similar concept for NAFLD, with important factors in disease progression being both steatosis and inflammation, and neither alone being sufficient to lead to NAFLD. It is also entirely possible that NAFLD is reached via multiple pathways even though the endpoint definitive features of the disease are similar; this also means that multiple mouse models may be valid for studying different aspects of NAFLD. Here we briefly discuss the main approaches and the features recapitulated by different models (Table 2). Characteristics of NALFD progression in humans fall into three broad categories: firstly, metabolic syndrome (obesity, hyperglycemia, insulin resistance, T2D); secondly, specific liver features (steatosis, hepatocyte ballooning, lobular inflammation, liver fibrosis); and, thirdly, wider systemic inflammatory disease (including adipose tissue inflammation, intestinal inflammation with intestinal barrier dysfunction, and alterations to gut microbiota) (122). Mouse models of NAFLD used in research to date also fall into three common categories, or combinations thereof: dietary, chemical, and genetic. Notably, most models use the C57Bl6 strain because of the propensity of this strain to develop metabolic and inflammatory disease, and several features of NAFLD compared with other strains. Although, the same amount of fibrosis and severity of NASH do not always spontaneously arise in these mice as occurs in human disease (124). In terms of methodology, it would be most ideal to be able to show disease progression during an ongoing study, at multiple timepoints. There is also no standard agreed-upon pathological progression in terms of liver histology in mice as it relates to human NAFLD, though at least one has been suggested (118, 125). Thus, progress has been made with murine models but the field’s translatability to humans is still under active investigation (126).
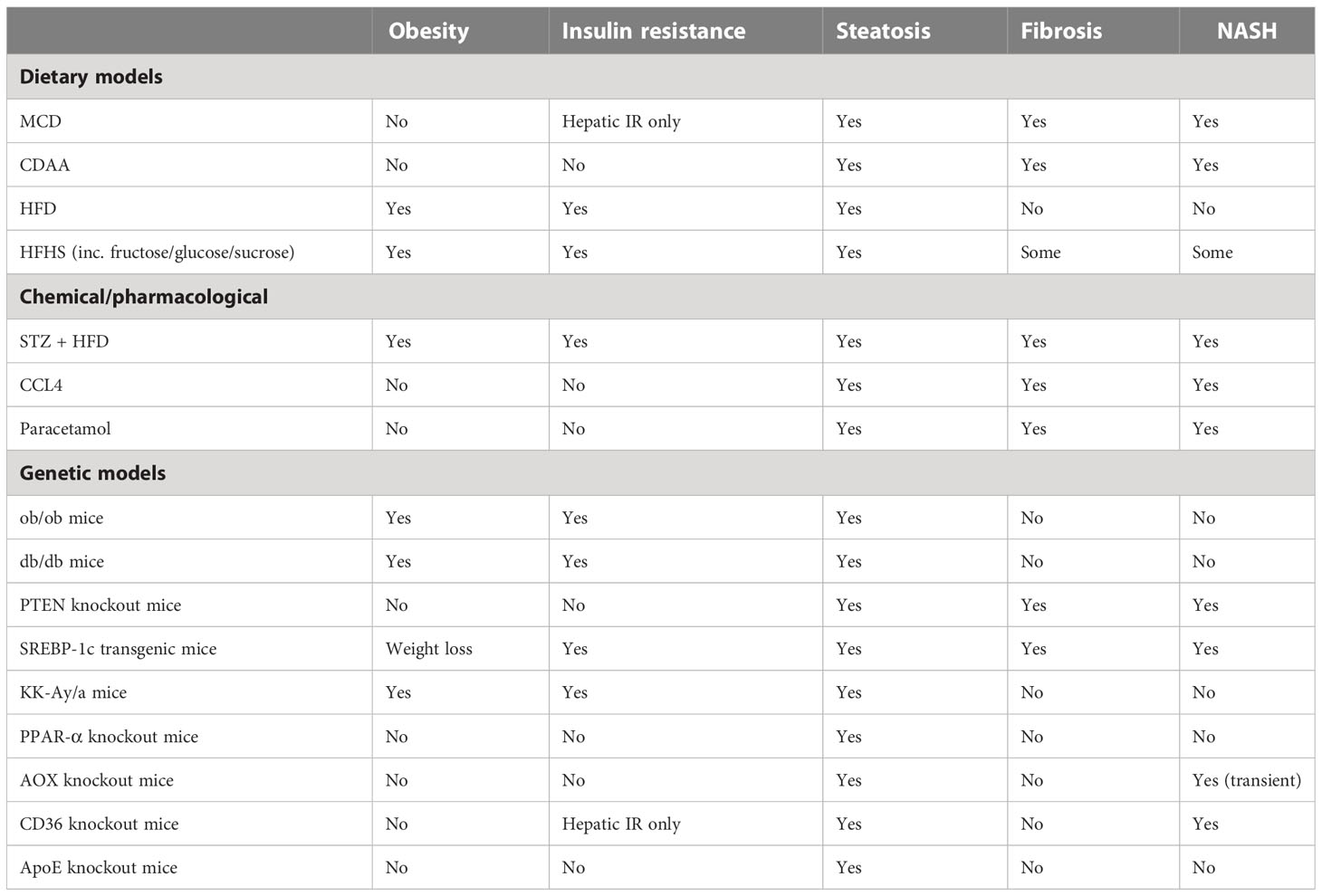
Table 2 Comparison of different rodent models of NAFLD.
Among the dietary models, a common approach has been to use diets deficient in one or more nutrients. The MCD diet has been adopted for some studies and has the advantage of inducing NASH within a few weeks, but it fails to model the human disease in terms of its often-concomitant obesity. Animals on this diet lose weight, but they still develop hepatic insulin resistance (127). A modification of this diet, the choline-deficient-L-amino-acid-defined (CDAA) diet, overcomes the weight loss issue with the MCD diet; though NASH develops later (20-22 weeks), the diet still elicits limited metabolic syndrome-like characteristics compared to NAFLD in humans (128–131). The established atherogenic diet has also been investigated for its usefulness in NAFLD mouse models: the diet induces steatosis, inflammation, and other histological features of NASH and NAFLD in the mouse liver but does not cause weight gain or insulin resistance (132, 133). Also, simply adding fructose to water was found to result in steatosis, increased weight, and hyperglycemia, though no definitive NASH (134). More commonly used currently are high-fat diets (HFD), which can vary widely in the amounts and types of fat used. These generally induce steatosis and inflammation to a lesser degree than in mice fed an MCD diet. However, they have the advantage of inducing a metabolic syndrome-like pathology that is more similar in profile to human NAFLD, with obesity, insulin resistance, and hyperglycemia (135). Modifications of the HFD, including a combination with fructose (High Fat High Fructose Diet) or sucrose (High Fat High Sucrose Diet) added, either to water or food, result in a phenotype more closely resembling human NAFLD, but not the full extent of liver damage (136, 137). Mice on other HFD variations, the High Fat High Cholesterol Diet (111) and the American Lifestyle Induced Obesity Syndrome (ALIOS) diet which is rich in trans fats (138), also showed additional NAFLD compared with mice on a typical HFD. A large disparity exists between individuals in terms of evolution from NAFLD to NASH, and between mouse strains used for in vivo modelling. In one study, an MCD diet was used to induce NASH in C57Bl6/J and Balb/c mice, which exhibit very different responses in terms of innate and adaptive immune responses.C57Bl6/J mice that are more prone to Th1 and M1-like responses, showed more steatosis and lobular inflammation following 4 weeks on the MCD diet than Balb/c mice, which are more prone to Th2 and M2-like responses. Accordingly, neither the Th1/Th2 bias nor IL-4 (interleukin 4) or GATA-3 expression in the liver of either strain is significantly modified by MCD feeding (indicating that innate immune polarization plays a more important role). As a result of MCD feeding, liver mRNA expression of macrophage activation markers M1 (iNOS), MLD (inducible NO synthase), and MCD (CXC chemokine ligand 10) were significantly higher in C57BL6/J mice, however, M2 polarization markers IL-10 and MGL-1 (macrophage galactose-type C-type lectin-1) remained the same. In addition, C57Bl6/J mice fed MCD had a higher level of circulating IL-12 than Balb/c mice fed MCD. Compared with mice fed MCD, macrophages isolated from the livers of C57Bl6/J mice expressed significantly more M1 markers (139).
As well as dietary fat, the form, content and delivery of sugar can be manipulated to influence susceptibility to NAFLD and a NASH-like phenotypes in rodents (140). The considerations have been whether sugar should be delivered in the form of glucose, fructose or sucrose (or in combination), and whether this is provided as part of the solid diet or in drinking water. There are mechanistic advantages and practical considerations for each of these options, for example quantity consumed or calories per gram are easier to control when incorporated into solid food. Fructose alone making up to 60% (w/v) of drinking water can reliably induce steatosis, when fructose is combined with glucose in a solid diet (30% w/v of each), this also results in steatosis (140). Interestingly, when prolonged the 60% (w/v) fructose in drinking water can induce fibrosis, and the same concentration in a solid diet (kcal/w) can induce inflammation and fibrosis. Up to 50% (w/v) sucrose in drinking water, can induce inflammation and fibrosis. In studies of these diet, fructose-supplemented drinking water causes steatosis after 8 weeks, and leads to a significant rise in body weight, and glucose and plasma triglyceride levels (141). Moreover, intestinal bacterial overgrowth is detected after 8 weeks of treatment along with high levels of endotoxin in the portal blood and activation of KCs (134). However, weight gain, followed by development of fat deposits in the abdomen, is not essentially prognostic of steatosis. It has been revealed that fructose causes greater fat accumulation in the liver than sucrose and glucose, despite of the weight gain from glucose and sucrose (142).
Among the chemical mouse models of NAFLD is the well-used streptozotocin induction of T2D. A low dose of intraperitoneal or subcutaneous streptozotocin shortly after birth leads to inflammation and the destruction of pancreatic islets; when combined with a HFD, animals progress quickly through steatosis, NASH, and increased fibrosis to hepatocellular carcinoma (HCC) around 20 weeks of age (143). The use of carbon tetrachloride (CCl4) in multiple IP doses leads to significant but reversible fibrosis (144); when combined with HFD steatosis, NASH and increasing fibrosis are seen (145);however, the systemic metabolic aspects of NAFLD are absent (146). One-time administration of diethylnitrosamine (DEN) has been used to model HCC; when combined with a HFD, animals do gain weight and develop NASH, as well as some other aspects of the metabolic syndrome associated with NAFLD (147). In each case, the value of toxicity or drug-induced NASH is questionable because it most likely doesn’t parallel the progression of the disease in humans and the mechanisms leading to fibrogenesis may be completely independent.
Leptin is an adipokine that centrally controls appetite. The leptin knockout mice, Lepob/Lepob (ob/ob), are obese, insulin resistant, and hyperglycemic, and show some degree of steatosis. Likewise, leptin receptor knockout mice, Leprdb/Leprdb (db/db), have a similar phenotype to ob/ob, and in both cases, an additional stimulus is required for the development of NASH. Several groups have explored the combination of genetic models with dietary or chemical stimulus to induce NAFLD with varying degrees of success (148–151). Mice with a mutation in Alms1, which encodes a protein involved in control of satiety via the hypothalamus, also require a dietary stimulus (HFD) to induce symptoms similar to NAFLD, with the lipid profile not mirroring the human disease (152). Sterol regulatory element-binding protein (SREBP-1c) transgenic mice also develop insulin resistance and steatosis (123). Better for examining the metabolic syndrome are the established atherosclerosis models such as ApoE-/- mice (apolipoprotein E deficient) and Ldlr-/- mice (LDL receptor deficient); these animals are predisposed to hypercholesterolemia, atherosclerosis, and obesity, and on a HFD or Western diet will also develop NAFLD/NASH (153).
Gut-liver axis refers to the relationship between the gut and liver at the proximal anatomical and physiological level. Bile acids (BA) produced from the liver are secreted into the duodenum to aid in lipid metabolism and bacterial homeostasis. Products of ileal absorption are then transported back to the liver via portal circulation. As such, the liver and the gut are always in contact with each other. Playing an important role in regulating this contact are the KCs and hepatic dendritic cells (DCs) that line sinusoids to filter and remove gut-derived microorganisms, microbial products and microbe-associated molecular patterns (MAMPs) from portal circulation (154).
Similar to the liver, the intestine has its own set of macrophages, mainly in the lamina propria (LP). Intestinal macrophages are most abundant in the colon due to the higher microbial load. Macrophages are constantly replenished in the LP because of extravasated Ly6Clo CX3CR1hi MHCII+ monocytes that develop into mature Ly6Clo CX3CR1int macrophages. LP macrophages are highly phagocytic and typically remain unresponsive towards harmless bacteria and food antigens. They play an important role in the induction of oral tolerance by sampling the gut lumen and presenting antigens to DCs during the induction process. Related self-renewing macrophage are also found near intestinal neurons and blood vessels. Under the LP, macrophages associated with neurons express genes typically enriched in microglia, the specialized central nervous system (CNS) macrophages. As a result, these genes may be essential for maintaining a healthy local neuronal population in a manner similar to microglia in the CNS (154).
The contact of peripheral macrophages with neurons has led to a growing field of research aiming to elucidate the effects of the gut-liver-brain axis in health and disease. The gut-liver-brain axis is complex with several regulators such as the intestinal barrier, gut-vasculature barrier, blood-brain barrier, and the immune system. The gut-brain axis is involved in modulating several physiological and homeostatic functions (155, 156). Namely, the CNS regulates gut function through the hypothalamic-pituitary-adrenal axis (HPA) and the autonomic nervous system. Moreover, the gut also alters CNS function through microbiota-derived molecules, gut hormones, and neurotransmitters (Figure 4A). These molecules enter the CNS via the enteric nervous system, vagus nerve, or the immune system.
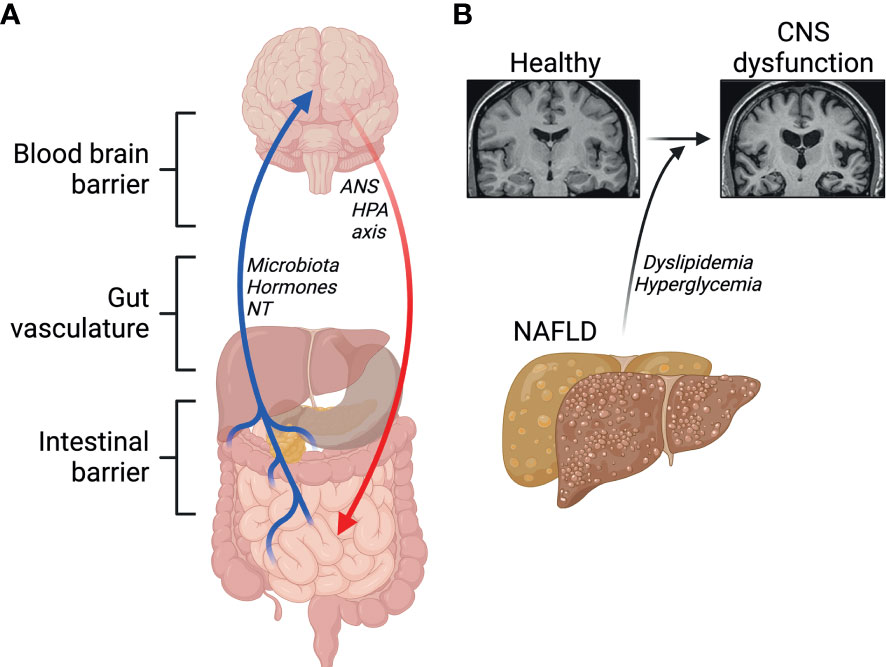
Figure 4 Emerging roles of the gut-liver-brain axis. (A) Barriers and signals between the gut, the liver and the brain. NT, neurotransmitters; ANS, autonomic nervous system; HPA, hypothalamic-pituitary-adrenal. (B) NAFLD and its comorbidities contribute to central nervous system (CNS) dysfunction. Created with BioRender.com.
Gut-liver interaction is regulated by enterohepatic circulation, and a disruption of this axis has been suggested to contribute to NAFLD, which itself has been associated with lower cognitive performance. This suggests a link between abnormal liver metabolism and neurodegenerative diseases, including Alzheimer’s disease (AD) (157–159). Physiologically, circulating Amyloid β peptides (Aβ) are detoxified by the liver. A key feature in AD is the accumulation of Aβ plaques and defective clearance by the liver through lipoprotein receptor-related protein 1 (LRP1) (160). In healthy conditions, insulin promotes LRP1 translocation to the cell membrane in hepatocytes favoring Aβ clearance (161). Low hepatic expression of LRP1 is observed in patients with liver diseases, accompanied with high levels of circulating Aβ, suggesting impaired LRP1-mediated clearance of Aβ (160). Insulin resistance impairs LRP1 translocation, contributing to its functional impairment in clearing Aβ (161). In one study by Więckowska-Gacek et al. (162), long-term feeding of Western diet in mutant amyloid protein precursor (APP) AD mice, which express mutant human APP with the Swedish mutation, accelerated the rate of several abnormalities in the brain, compared to normal diet fed APP AD mice. Interestingly, in the same study, liver damage in Western diet fed mice was correlated with the deposition of Aβ in the brain (162). Indeed, evidence shows that diets high in simple sugars, salt, cholesterol, saturated fatty acids, trans fatty acids and low in fiber and mono- and polyunsaturated fatty acids, as in the Western diet, impair Aβ clearance in the periphery by the liver through increased activity of the receptor for advanced glycation end-products (RAGE) (8, 159, 163). Moreover, mice fed a Western diet exhibit impairments in the blood-brain-barrier (BBB), which has been shown to trigger oxidative stress, leading to enhanced activity of β-secretase and γ-secretase, which promote the generation of Aβ peptides (9, 162). Hypercholesterolemia has also been proposed to play a major role in the progression of neurodegenerative diseases (164) (Figure 4B). Elevated levels of cytochrome P450 CYP27A1 and CYP7A1 isoforms in hepatocytes during NAFLD underlie the conversion of serum cholesterol into 27-hydroxycholesterol, which crosses freely through the BBB (116, 165, 166). High levels of 27-hydroxycholesterol in the brain leads to oxidative stress and the development of AD pathology (165, 167–169). Both dyslipidemia and the systemic inflammation, that often follows, are major candidate contributors to neuroinflammation and compromised cognitive function.
Evidence also shows that the hepatic branch of the vagus nerve, which serves as a major connection between the liver and CNS, plays a pivotal role in the progression of liver cirrhosis, and influences the gut microbiome (145, 146). Another key player in the liver-brain axis is the brain-derived neurotrophic factor (BDNF) (170, 171). In the brain, BDNF plays several roles including, but not limited to regulating synaptic plasticity, neuroinflammation, and neurogenesis (172–175). Furthermore, BDNF can also modulate insulin signaling and liver disease in animal models of cirrhosis and alcohol-induced liver disease (176–178). Interestingly, experiments show that mice with cirrhosis have high levels of the BDNF in the liver, and hepatic vagotomy levels were found to be significantly reduced in the liver but increased in the brain. While these studies clearly indicate that BDNF plays a role in liver and brain diseases, further studies are warranted to elucidate its downstream effects in the CNS and periphery. A common mechanism leading to disruption at the gut, liver or brain levels is inflammation. This inflammation is caused by macrophages that reside in each compartment. The most plausible starting points for dysregulation of the gut-liver-brain-axis are either at the level of the intestine or in the liver itself. These two tissues are populated by specialized macrophages very susceptible to encountering bacteria or bacterial products, they are also strongly affected by metabolic stress and insulin resistance at a very early stage. Subsequent inflammatory signaling may act in an endocrine manner, similar to crosstalk from AT, to influence microglia in the CNS or induce hyper-responsiveness of circulating monocytes, making them more likely to cross the BBB and contribute to neuroinflammation. Other potential mechanisms linking these systems is microbiome-mediated signaling via bacterial metabolites or other products, that can act at the level of each organ; or actions of nerve-associated macrophages (NAM). The study of peripheral NAMs is in its infancy, they have been identified in a limited number of organs and have been attributed physiological roles in few conditions. One relevant example is the role for NAMs in adipose tissue, these have been described to regulate catecholamine release from sympathetic nerves that populate the tissue. Nerve-interactive macrophages have also been identified in the liver and are known to induce neuropathy and promote insulin resistance, however their mechanisms of activation remain unknown (179). In the context of alcohol-associated liver disease however, KCs have been demonstrated to respond to gut-derived catecholamines with hepatoprotective outcomes (180).
NAFLD has become one of the most common liver diseases worldwide. Its complicated pathogenesis is related to disorders such as obesity, insulin resistance, T2D, and hypertension. All these conditions carry the risk of initiating or aggravating the development of steatosis, fibrosis, cirrhosis, and hepatocellular carcinoma. Oxidative stress and changes in chemokines, adipokines, and anti- or pro-inflammatory cytokines are key factors in the development of NAFLD. KCs and monocyte-derived macrophages are phagocytes in the liver that execute several metabolic and immune-related functions under homeostasis and in disease. Macrophages play a vital role at all stages of NAFLD and contribute to pathology. Therefore, targeting liver macrophages is promising as a therapeutic avenue. Focusing on macrophages to treat NAFLD in its early stages may be an ideal method to reduce the damage or likelihood of progressing to the later stages of NASH. Also, further interpretation of the crosstalk between macrophages and other immune cells similarly remains a promising area of exploration, as it will not only improve our understanding of NAFLD pathogenesis but also help us discover novel circulating biomarkers or therapeutic interventions for NAFLD (181). As such, additional studies are required to further investigate the function of specific macrophage subpopulations and their specific markers or receptors in NAFLD progression and target them for treatment (182).
Regulating macrophage subpopulation abundance, or their specific functions, is an area of active research. To meet this research need, in-depth mechanistic studies at the transcriptional or epigenetic levels must be carried out. Epigenetic regulation plays an important role in dictating macrophage fate. Targeting transcriptional machinery, via transcription factor activity or epigenetic modifications is another encouraging strategy (183). Carotenoids, for example, are also promising anti-inflammatory and antioxidant molecules; in preclinical studies, they impede inflammation, steatosis, and fibrosis. However, there is no clinical evidence to date that carotenoids have beneficial effects against NAFLD in patients. Further studies are warranted to demonstrate the potential role of carotenoids, or carotenoid-related molecules, in the prevention and treatment of NAFLD (184).
Inflammation and fibrosis resulting from NASH have also been associated with galectin-3 proteins. Galectin-3 proteins are members of a family of glycoproteins that bind to galactose-containing oligosaccharides. Their expression on macrophages suppresses the pro-inflammatory phenotype and upregulates expression of type-2 molecules, such as TGFb (185). Pre-clinical outcomes of a galectin-3 inhibitor, the GR-MD-02, indicated its success in reversing NASH with cirrhosis, supporting clinical development trials in future that target advanced fibrosis/cirrhosis with NASH. GR-MD-02 is derived from a natural plant compound that contains galactose residues and binds to galectin-3 (186). Another strategy targeting macrophages has been use of a dual inhibitor of CCR2 and CCR5, Cenicriviroc. Cenicriviroc acts by decreasing recruitment of pro-inflammatory macrophages in mice with liver fibrosis (187). Cenicriviroc was proven effective in a phase II clinical trial (NCT02217475) of NASH patients with liver fibrosis. Fibrosis was ameliorated in patients who were administered 150 mg of Cenicriviroc for 2 years, with a good safety profile (188). However, a phase III trial (NCT03028740) with the same compound, in individuals with advanced fibrosis and cirrhosis, was terminated due to lack of efficacy (189). Therefore, a strategy targeting CCR2/CCR5 may only be effective in early stages of NASH, but not in advanced fibrosis or cirrhosis.
Other targetable molecules include those secreted from macrophages, that may have paracrine or endocrine effects. For example, enhanced production of IP-10 and MCP-1 by the intrahepatic KCs can trigger the development of NASH. In addition, silencing of TNFα in myeloid cells abrogated the production of these chemokines and prevented the development of NASH. Thus, Inhibition of TNFα might delineate a novel therapeutic target in NASH. Therefore, blockade of TNFα might represent a novel therapeutic target in NASH with the potential to limit tissue injury and possibly prevent the progression to severe liver disease (190). With a low-dose dexamethasone conjugate, selective anti-CD163 targeting of KCs inhibited fructose-induced steatohepatitis in rats without systemic side effects. Therefore, CD163 positive macrophages could be a potential therapeutic target to inhibit the progression of further liver damage in NASH patients (191). Targeting specific KC subsets, like the recently identified KC2s may also be an avenue for future intervention. The CD206hi ESAM+ KC2 regulate liver metabolism in the obese murine model of obesity by expressing the fatty acid transporter CD36. Silencing CD36 improves glucose homeostasis and based on a mechanism that improves oxidative stress in KC2 cells. In obesity, the reduction of oxidative stress in KCs has been shown to enhance liver metabolism and reduce ROS levels within the liver. As such, strategies that affect KC2 metabolic function can be considered for the regulation of liver metabolic diseases (52).
Innovative technological developments and studies in appropriate preclinical models will further help elucidate the complex pathophysiology of NAFLD and the roles of macrophages. Further basic and clinical research is essential for better understanding the molecular mechanisms by which the chemokine system mediates hepatic and adipose inflammation, as well as their interaction in the progression of NAFLD (192). To improve the status of patients with NAFLD, combination approaches are required that include lifestyle modifications personally tailored to the patient’s disease drivers (obesity, T2D) as well as specific pharmacological interventions that target influential pathways (193). Furthermore, classifying patients at specific disease stages might provide a more individualized treatment strategy, which could improve treatment responses. In conclusion, combinational treatment approaches could lead to beneficial effects. Specifically, combining anti-inflammatory and metabolic approaches is a promising strategy for future clinical studies (194).
BA and FAlz contributed to conception and design; BA, JR, DT and FAlz contributed to making figures; BA, FAl-r, MA-O, AK, JR, DT, MW, CB, RA and FAlz contributed to writing the manuscript and wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
FAlz is supported by KFAS grants RA AM-2022-009 and RA AM-2023-007; and by ANR-JCJC grant for the MitoFLAME Project ANR-19-CE14-0005.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dhingra B, Sharma S, Mishra D, Kumari R, Pandey RM, Aggarwal S, et al. Normal values of liver and spleen size by ultrasonography in Indian children. Indian Pediatr (2010) 47:487–92. doi: 10.1007/s13312-010-0090-6
2. Bismuth H. Surgical anatomy and anatomical surgery of the liver. World J Surg (1982) 6:3–9. doi: 10.1007/BF01656368
4. Joshi SD, Joshi SS, Athavale SA. Some interesting observations on the surface features of the liver and their clinical implications. Singapore Med J (2009) 50:715–9.
5. Chiang JYL. Liver physiology: metabolism and detoxification. In: McManus LM, Mitchell RN, editors. Pathobiology of Human Disease. (2014), p. 1770–82. doi: 10.1016/B978-0-12-386456-7.04202-7
6. Weiskirchen R, Tacke F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells (2019) 8. doi: 10.3390/cells8010016
7. Damania A, Jain E, Kumar A. Advancements in in vitro hepatic models: application for drug screening and therapeutics. Hepatol Int (2014) 8:23–38. doi: 10.1007/s12072-013-9490-8
8. Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling alzheimer’s amyloid β-peptide elimination from the brain. J Neurochem (2010) 115:1077–89. doi: 10.1111/j.1471-4159.2010.07002.x
9. Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of blood-brain barrier in alzheimer’s disease. J Alzheimers Dis (2018) 63:1223–34. doi: 10.3233/JAD-180098
10. Bilzer M, Roggel F, Gerbes AL. Role of kupffer cells in host defense and liver disease. Liver Int Off J Int Assoc Study Liver (2006) 26:1175–86. doi: 10.1111/j.1478-3231.2006.01342.x
11. Wang R, Sheng M, Shi F, Zhao Y, Zhao L, Wu J, et al. Dysfunctional phagocytosis capacity, granulocyte recruitment and inflammatory factor secretion of kupffer cells in diabetes mellitus reversed by lidocaine. Diabetes Metab Syndr Obes Targets Ther (2018) 11:827–34. doi: 10.2147/DMSO.S186695
12. Bouwens L, De Bleser P, Vanderkerken K, Geerts B, Wisse E. Liver cell heterogeneity: functions of non-parenchymal cells. Enzyme (1992) 46:155–68. doi: 10.1159/000468782
13. Jiang Y, Que W, Zhu P, Li X-K. The role of diverse liver cells in liver transplantation tolerance. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01203
14. Amarapurkar DN, Hashimoto E, Lesmana LA, Sollano JD, Chen PJ, Goh KL, et al. How common is non-alcoholic fatty liver disease in the Asia–pacific region and are there local differences? J Gastroenterol Hepatol (2007) 22:788–93. doi: 10.1111/j.1440-1746.2007.05042.x
15. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (2018) 67:123–33. doi: 10.1002/hep.29466
16. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell (2021) 184:2537–64. doi: 10.1016/j.cell.2021.04.015
17. Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin z and reduces hepatic fibrosis. Science (2010) 329:229–32. doi: 10.1126/science.1190354
18. Peng J-K, Hepgul N, Higginson IJ, Gao W. Symptom prevalence and quality of life of patients with end-stage liver disease: a systematic review and meta-analysis. Palliat Med (2019) 33:24–36. doi: 10.1177/0269216318807051
19. Pinter M, Trauner M, Peck-Radosavljevic M, Sieghart W. Cancer and liver cirrhosis: implications on prognosis and management. ESMO Open (2016) 1:e000042–e000042. doi: 10.1136/esmoopen-2016-000042
20. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease–meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64:73–84. doi: 10.1002/hep.28431
21. Kim D, Kim WR. Nonobese fatty liver disease. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc (2017) 15:474–85. doi: 10.1016/j.cgh.2016.08.028
22. Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci USA (2006) 103:18273–7. doi: 10.1073/pnas.0608537103
23. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med (2002) 346:1221–31. doi: 10.1056/NEJMra011775
24. Gotlieb N, Schwartz N, Zelber-Sagi S, Chodick G, Shalev V, Shibolet O. Longitudinal decrease in platelet counts as a surrogate marker of liver fibrosis. World J Gastroenterol (2020) 26:5849–62. doi: 10.3748/wjg.v26.i38.5849
25. de Silva MHAD, Hewawasam RP, Kulatunge CR, Chamika RMA. The accuracy of fatty liver index for the screening of overweight and obese children for non-alcoholic fatty liver disease in resource limited settings. BMC Pediatr (2022) 22:511. doi: 10.1186/s12887-022-03575-w
26. Hamaguchi M, Kojima T, Takeda N, Nakagawa T, Taniguchi H, Fujii K, et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med (2005) 143:722–8. doi: 10.7326/0003-4819-143-10-200511150-00009
27. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American association for the study of liver diseases. Hepatol Baltim. Md (2018) 67:328–57. doi: 10.1002/hep.29367
28. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev (2018) 98:2133–223. doi: 10.1152/physrev.00063.2017
29. Sharabi K, Tavares CDJ, Rines AK, Puigserver P. Molecular pathophysiology of hepatic glucose production. Mol Aspects Med (2015) 46:21–33. doi: 10.1016/j.mam.2015.09.003
30. Santoleri D, Titchenell PM. Resolving the paradox of hepatic insulin resistance. Cell Mol Gastroenterol Hepatol (2019) 7:447–56. doi: 10.1016/j.jcmgh.2018.10.016
31. Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des (2010) 16:1941–51. doi: 10.2174/138161210791208875
32. Lomonaco R, Bril F, Portillo-Sanchez P, Ortiz-Lopez C, Orsak B, Biernacki D, et al. Metabolic impact of nonalcoholic steatohepatitis in obese patients with type 2 diabetes. Diabetes Care (2016) 39:632–8. doi: 10.2337/dc15-1876
33. Méndez-Sánchez N, Arrese M, Zamora-Valdés D, Uribe M. Current concepts in the pathogenesis of nonalcoholic fatty liver disease. Liver Int (2007) 27:423–33. doi: 10.1111/j.1478-3231.2007.01483.x
34. Bijnen M, Josefs T, Cuijpers I, Maalsen CJ, van de Gaar J, Vroomen M, et al. Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut (2018) 67:1317–27. doi: 10.1136/gutjnl-2016-313654
35. Kitade H, Chen G, Ni Y, Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients (2017) 9. doi: 10.3390/nu9040387
36. Angulo P. NAFLD. Obesity, and bariatric surgery. Gastroenterology (2006) 130:1848–52. doi: 10.1053/j.gastro.2006.03.041
37. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med (2001) 7:947–53. doi: 10.1038/90992
38. Masarone M, Federico A, Abenavoli L, Loguercio C, Persico M. Non alcoholic fatty liver: epidemiology and natural history. Rev Recent Clin Trials (2014) 9:126–33. doi: 10.2174/1574887109666141216111143
39. Bugianesi E, Pagotto U, Manini R, Vanni E, Gastaldelli A, de Iasio R, et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J Clin Endocrinol Metab (2005) 90:3498–504. doi: 10.1210/jc.2004-2240
40. Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia (2016) 59:30–43. doi: 10.1007/s00125-015-3769-3
41. Perakakis N, Farr OM, Mantzoros CS. Leptin in leanness and obesity: JACC state-of-the-Art review. J Am Coll Cardiol (2021) 77:745–60. doi: 10.1016/j.jacc.2020.11.069
42. Polyzos SA, Kountouras J, Mantzoros CS. Leptin in nonalcoholic fatty liver disease: a narrative review. Metab - Clin Exp (2015) 64:60–78. doi: 10.1016/j.metabol.2014.10.012
43. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
44. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
45. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
46. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17:306–21. doi: 10.1038/nri.2017.11
47. Anderson ER, Shah YM. Iron homeostasis in the liver. Compr Physiol (2013) 3:315–30. doi: 10.1002/cphy.c120016
48. Trapani L, Segatto M, Pallottini V. Regulation and deregulation of cholesterol homeostasis: the liver as a metabolic ‘power station’. World J Hepatol (2012) 4:184–90. doi: 10.4254/wjh.v4.i6.184
49. Zheng M, Tian Z. Liver-mediated adaptive immune tolerance. Front Immunol (2019) 10:2525. doi: 10.3389/fimmu.2019.02525
50. Hendrikx T, Schnabl B. Antimicrobial proteins: intestinal guards to protect against liver disease. J Gastroenterol (2019) 54:209–17. doi: 10.1007/s00535-018-1521-8
51. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
52. Blériot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. A subset of kupffer cells regulates metabolism through the expression of CD36. Immunity (2021) 54:2101–2116.e6. doi: 10.1016/j.immuni.2021.08.006
53. Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut (2020) 69:551. doi: 10.1136/gutjnl-2019-318382
54. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity (2017) 47:374–388.e6. doi: 10.1016/j.immuni.2017.07.018
55. Blériot C, Ginhoux F. Understanding the heterogeneity of resident liver macrophages. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02694
56. Sanz-García C, Fernández-Iglesias A, Gracia-Sancho J, Arráez-Aybar LA, Nevzorova YA, Cubero FJ. The space of disse: the liver hub in health and disease. Livers (2021) 1:3–26. doi: 10.3390/livers1010002
57. Thibaut R, Gage MC, Pineda-Torra I, Chabrier G, Venteclef N, Alzaid F. Liver macrophages and inflammation in physiology and physiopathology of non-alcoholic fatty liver disease. FEBS J (2022) 289:3024–57. doi: 10.1111/febs.15877
58. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis (2010) 30:245–57. doi: 10.1055/s-0030-1255354
59. Lichtnekert J, Kawakami T, Parks WC, Duffield JS. Changes in macrophage phenotype as the immune response evolves. Cancer • Immunomodulation (2013) 13:555–64. doi: 10.1016/j.coph.2013.05.013
60. Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM Int J Med (2010) 103:71–83. doi: 10.1093/qjmed/hcp158
61. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep Innov Hepatol (2019) 1:30–43. doi: 10.1016/j.jhepr.2019.02.004
62. Park J-W, Jeong G, Kim SJ, Kim MK, Park SM. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol (2007) 22:491–7. doi: 10.1111/j.1440-1746.2006.04758.x
63. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 kupffer cells promote M1 kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology (2014) 59:130–42. doi: 10.1002/hep.26607
64. Jindal A, Bruzzì S, Sutti S, Locatelli I, Bozzola C, Paternostro C, et al. Fat-laden macrophages modulate lobular inflammation in nonalcoholic steatohepatitis (NASH). Exp Mol Pathol (2015) 99:155–62. doi: 10.1016/j.yexmp.2015.06.015
65. Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB, Delzenne NM. Critical role of kupffer cells in the management of diet-induced diabetes and obesity. Biochem Biophys Res Commun (2009) 385:351–6. doi: 10.1016/j.bbrc.2009.05.070
66. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1β–dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology (2010) 51:511–22. doi: 10.1002/hep.23337
67. Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, et al. Aberrant hepatic expression of PPARγ2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis*. J Biol Chem (2006) 281:37603–15. doi: 10.1074/jbc.M604709200
68. Zhang X, Fan L, Wu J, Xu H, Leung WY, Fu K, et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J Hepatol (2019) 71:163–74. doi: 10.1016/j.jhep.2019.03.014
69. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
70. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol (2016) 65:758–68. doi: 10.1016/j.jhep.2016.05.037
71. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116:115–24. doi: 10.1172/JCI24335
72. Yao Y, Xu X-H, Jin L. Macrophage polarization in physiological and pathological pregnancy. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00792
73. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity (2005) 23:344–6. doi: 10.1016/j.immuni.2005.10.001
74. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5:953–64. doi: 10.1038/nri1733
75. Galván-Peña S, O’Neill LAJ. Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420. doi: 10.3389/fimmu.2014.00420
76. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19061801
77. Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet–induced obesity in mice. Diabetes (2010) 59:1171–81. doi: 10.2337/db09-1402
78. Thorp E, Subramanian M, Tabas I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol (2011) 41:2515–8. doi: 10.1002/eji.201141719
79. Avila-Ponce de León U, Vázquez-Jiménez A, Matadamas-Guzman M, Pelayo R, Resendis-Antonio O. Transcriptional and microenvironmental landscape of macrophage transition in cancer: a boolean analysis. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.642842
80. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell (2019) 178:686–698.e14. doi: 10.1016/j.cell.2019.05.054
81. Alzaid F, Lagadec F, Albuquerque M, Ballaire R, Orliaguet L, Hainault I, et al. IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI Insight (2016) 1. doi: 10.1172/jci.insight.88689
82. Orliaguet L, Ejlalmanesh T, Alzaid F. Metabolic and molecular mechanisms of macrophage polarisation and adipose tissue insulin resistance. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21165731
83. Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis (2012) 225:461–8. doi: 10.1016/j.atherosclerosis.2012.09.013
84. O’Neill LAJ, Kishton RJ. & rathmell, j. a guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16:553–65. doi: 10.1038/nri.2016.70
85. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
86. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages. via GLUT1 Infect Immun (1996) 64:108–12. doi: 10.1128/iai.64.1.108-112.1996
87. Koo S-J, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose phosphate shunt modulates reactive oxygen species and nitric oxide production controlling trypanosoma cruzi in macrophages. Front Immunol (2018) 9:202. doi: 10.3389/fimmu.2018.00202
88. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
89. Waqas SFH, Ampem G, Röszer T. Analysis of IL-4/STAT6 signaling in macrophages. Methods Mol Biol Clifton NJ (2019) 1966:211–24. doi: 10.1007/978-1-4939-9195-2_17
90. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
91. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-Macrophage differentiation and polarization: new molecules and patterns of gene Expression1. J Immunol (2006) 177:7303–11. doi: 10.4049/jimmunol.177.10.7303
92. Reactive oxygen species in macrophages: sources and targets - PMC . Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515906/.
93. Palmieri EM, Gonzalez-Cotto M, Baseler WA, Davies LC, Ghesquière B, Maio N, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun (2020) 11:698. doi: 10.1038/s41467-020-14433-7
94. Castoldi A, Monteiro LB, van Teijlingen Bakker N, Sanin DE, Rana N, Corrado M, et al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nat Commun (2020) 11:4107. doi: 10.1038/s41467-020-17881-3
95. Sun J-X, Xu X-H, Jin L. Effects of metabolism on macrophage polarization under different disease backgrounds. Front Immunol (2022) 13:880286. doi: 10.3389/fimmu.2022.880286
96. De Simone G, Andreata F, Bleriot C, Fumagalli V, Laura C, Garcia-Manteiga JM, et al. Identification of a kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity (2021) 54:2089–2100.e8. doi: 10.1016/j.immuni.2021.05.005
97. Rai V, Rao VH, Shao Z, Agrawal DK. Dendritic cells expressing triggering receptor expressed on myeloid cells-1 correlate with plaque stability in symptomatic and asymptomatic patients with carotid stenosis. PloS One (2016) 11:e0154802. doi: 10.1371/journal.pone.0154802
98. Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Miličević N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2+ macrophages that localize to fibrotic areas and limit NASH. J Hepatol (2022) 77:1373–85. doi: 10.1016/j.jhep.2022.06.004
99. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature (2005) 433:523–7. doi: 10.1038/nature03253
100. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic lipids stored by kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol (2012) 57:141–9. doi: 10.1016/j.jhep.2012.02.028
101. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep (2021) 34:108626. doi: 10.1016/j.celrep.2020.108626
102. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
103. Brancale J, Vilarinho S. A single cell gene expression atlas of 28 human livers. J Hepatol (2021) 75:219–20. doi: 10.1016/j.jhep.2021.03.005
104. Liver cell atlas. Available at: https://www.livercellatlas.org/.
105. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
106. Tilg H, Adolph TE, Moschen AR. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: revisited after a decade. Hepatology (2021) 73:833–42. doi: 10.1002/hep.31518
107. Yao J-M, Ying HZ, Zhang HH, Qiu FS, Wu JQ, Yu CH. Exosomal RBP4 potentiated hepatic lipid accumulation and inflammation in high-fat-diet-fed mice by promoting M1 polarization of kupffer cells. Free Radic Biol Med (2023) 195:58–73. doi: 10.1016/j.freeradbiomed.2022.12.085
108. Tada Y, Kasai K, Makiuchi N, Igarashi N, Kani K, Takano S, et al. Roles of macrophages in advanced liver fibrosis, identified using a newly established mouse model of diet-induced non-alcoholic steatohepatitis. Int J Mol Sci (2022) 23. doi: 10.3390/ijms232113251
109. Wallace SJ, Tacke F, Schwabe RF, Henderson NC. Understanding the cellular interactome of non-alcoholic fatty liver disease. JHEP Rep Innov Hepatol (2022) 4:100524–4. doi: 10.1016/j.jhepr.2022.100524
110. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M, et al. Toll-like receptor-4 signaling and kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
111. Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, et al. Increased expression of PPARγ in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun (2005) 336:215–22. doi: 10.1016/j.bbrc.2005.08.070
112. Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes (2015) 64:1120–30. doi: 10.2337/db14-1238
113. Lounis MA, Escoula Q, Veillette C, Bergeron KF, Ntambi JM, Mounier C. SCD1 deficiency protects mice against ethanol-induced liver injury. Biochim Biophys Acta BBA - Mol Cell Biol Lipids (2016) 1861:1662–70. doi: 10.1016/j.bbalip.2016.07.012
114. Aljohani A, Khan MI, Bonneville A, Guo C, Jeffery J, O'Neill L, et al. Hepatic stearoyl CoA desaturase 1 deficiency increases glucose uptake in adipose tissue partially through the PGC-1α-FGF21 axis in mice. J Biol Chem (2019) 294:19475–85. doi: 10.1074/jbc.RA119.009868
115. Maretti-Mira AC, Salomon MP, Hsu AM, Kanel GC, Golden-Mason L. Hepatic damage caused by long-term high cholesterol intake induces a dysfunctional restorative macrophage population in experimental NASH. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.968366
116. Chen Y, Tian Z. Roles of hepatic innate and innate-like lymphocytes in nonalcoholic steatohepatitis. Front Immunol (2020) 11:1500–0. doi: 10.3389/fimmu.2020.01500
117. Bieghs V, Wouters K, van Gorp PJ, Gijbels MJ, de Winther MP, Binder CJ, et al. Role of scavenger receptor a and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology (2010) 138:2477–2486.e24863. doi: 10.1053/j.gastro.2010.02.051
118. Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun (2018) 9:4490. doi: 10.1038/s41467-018-06931-6
119. Horn CL, Morales. AL, Savard C, Farrell GC, Ioannou GN. Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol Commun (2022) 6:12–35. doi: 10.1002/hep4.1801
120. Johnston JM, Angyal A, Bauer RC, Hamby S, Suvarna SK, Baidžajevas K, et al. Myeloid tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci Adv (2020) 5:eaax9183. doi: 10.1126/sciadv.aax9183
121. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res (2017) 58:1067–79. doi: 10.1194/jlr.M072454
122. Jahn D, Kircher S, Hermanns HM, Geier A. Animal models of NAFLD from a hepatologist’s point of view. Anim Models Liver Dis (2019) 1865:943–53. doi: 10.1016/j.bbadis.2018.06.023
123. Day CP, James OF. Steatohepatitis: a tale of two ‘hits’? Gastroenterology (1998) 114:842–5. doi: 10.1016/S0016-5085(98)70599-2
124. Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Rev Ingestive Sci (2004) 81:243–8. doi: 10.1016/j.physbeh.2004.02.006
125. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatol Baltim. Md (2005) 41:1313–21. doi: 10.1002/hep.20701
126. Fang T, Wang H, Pan X, Little PJ, Xu S, Weng J. Mouse models of nonalcoholic fatty liver disease (NAFLD): pathomechanisms and pharmacotherapies. Int J Biol Sci (2022) 18:5681–97. doi: 10.7150/ijbs.65044
127. Itagaki H, Shimizu K, Morikawa S, Ogawa K, Ezaki T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int J Clin Exp Pathol (2013) 6:2683–96.
128. Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, et al. C-jun n-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology (2009) 137:1467–1477.e5. doi: 10.1053/j.gastro.2009.06.045
129. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology (2010) 139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052
130. Ishioka M, Miura K, Minami S, Shimura Y, Ohnishi H. Altered gut microbiota composition and immune response in experimental steatohepatitis mouse models. Dig Dis Sci (2017) 62:396–406. doi: 10.1007/s10620-016-4393-x
131. Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int J Exp Pathol (2013) 94:93–103. doi: 10.1111/iep.12008
132. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatol Baltim Md (2007) 46:1392–403. doi: 10.1002/hep.21874
133. Chusyd DE, Wang D, Huffman DM, Nagy TR. Relationships between rodent white adipose fat pads and human white adipose fat depots. Front Nutr (2016) 3:10–0. doi: 10.3389/fnut.2016.00010
134. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology (2009) 50:1094–104. doi: 10.1002/hep.23122
135. Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res (2007) 37:50–7. doi: 10.1111/j.1872-034X.2007.00008.x
136. Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatol Baltim Md (2013) 58:1632–43. doi: 10.1002/hep.26594
137. Sellmann C, Priebs J, Landmann M, Degen C, Engstler AJ, Jin CJ, et al. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J Nutr Biochem (2015) 26:1183–92. doi: 10.1016/j.jnutbio.2015.05.011
138. Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol 295 G987–G995 (2008). doi: 10.1152/ajpgi.90272.2008
139. Maina V, Sutti S, Locatelli I, Vidali M, Mombello C, Bozzola C, et al. Bias in macrophage activation pattern influences non-alcoholic steatohepatitis (NASH) in mice. Clin Sci Lond Engl 1979 (2012) 122:545–53. doi: 10.1042/CS20110366
140. Eng JM, Estall JL. Diet-induced models of non-alcoholic fatty liver disease: food for thought on sugar, fat, and cholesterol. Cells (2021) 10:1805. doi: 10.3390/cells10071805
141. Mamikutty N, Thent ZC, Haji Suhaimi F. Fructose-drinking water induced nonalcoholic fatty liver disease and ultrastructural alteration of hepatocyte mitochondria in Male wistar rat. BioMed Res Int (2015) 2015:895961. doi: 10.1155/2015/895961
142. Bergheim I, Weber S, Vos M, Krämer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol (2008) 48:983–92. doi: 10.1016/j.jhep.2008.01.035
143. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol (2013) 46:141–52. doi: 10.1007/s00795-013-0016-1
144. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today (2017) 22:1707–18. doi: 10.1016/j.drudis.2017.06.007
145. Kim YO, Popov Y, Schuppan D. Optimized mouse models for liver fibrosis. Methods Mol Biol (2017) 1559:279–96. doi: 10.1007/978-1-4939-6786-5_19
146. Kubota N, Kado S, Kano M, Masuoka N, Nagata Y, Kobayashi T, et al. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin Exp Pharmacol Physiol (2013) 40:422–30. doi: 10.1111/1440-1681.12102
147. Thompson KJ, Swan RZ, Walling TL, Iannitti DA, McKillop IH, Sindram D. Obesity, but not ethanol, promotes tumor incidence and progression in a mouse model of hepatocellular carcinoma in vivo. Surg Endosc (2013) 27:2782–91. doi: 10.1007/s00464-013-2808-8
148. Kristiansen MNB, Veidal SS, Rigbolt KT, Tølbøl KS, Roth JD, Jelsing J, et al. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J Hepatol (2016) 8:673–84. doi: 10.4254/wjh.v8.i16.673
149. Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V, Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp Pathol (2011) 92:413–21. doi: 10.1111/j.1365-2613.2011.00793.x
150. Sanches SCL, Ramalho LNZ, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic steatohepatitis: a search for factual animal models. BioMed Res Int (2015) 2015:574832. doi: 10.1155/2015/574832
151. Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci (2013) 14:11963–80. doi: 10.3390/ijms140611963
152. Liu X, Wang K, Wang L, Kong L, Hou S, Wan Y, et al. Hepatocyte leukotriene B4 receptor 1 promotes NAFLD development in obesity. Hepatol n/a (2022). doi: 10.1002/hep.32708
153. Jacobs A, Warda A-S, Verbeek J, Cassiman D, Spincemaille P. An overview of mouse models of nonalcoholic steatohepatitis: from past to present. Curr Protoc Mouse Biol (2016) 6:185–200. doi: 10.1002/cpmo.3
154. De Muynck K, Vanderborght B, Van Vlierberghe H, Devisscher L. The gut-liver axis in chronic liver disease: a macrophage perspective. Cells (2021) 10. doi: 10.3390/cells10112959
155. Morais LH, Schreiber HL4, Mazmanian SK. The gut microbiota-brain axis in behaviour and brain disorders. Nat Rev Microbiol (2021) 19:241–55. doi: 10.1038/s41579-020-00460-0
156. Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol (2015) 28:203–9.
157. George ES, Georgousopoulou EN, Mellor DD, Chrysohoou C, Pitsavos C, Panagiotakos DB. Exploring the path of Mediterranean diet, non-alcoholic fatty liver disease (NAFLD) and inflammation towards 10-year cardiovascular disease (CVD) risk: the ATTICA study 10-year follow-up (2002-2012). Nutrients (2022) 14:2367. doi: 10.3390/nu14122367
158. Seo SW, Gottesman RF, Clark JM, Hernaez R, Chang Y, Kim C, et al. Nonalcoholic fatty liver disease is associated with cognitive function in adults. Neurology (2016) 86:1136. doi: 10.1212/WNL.0000000000002498
159. Kim JY, Lee C, Oh M, Im JA, Lee JW, Chu SH, et al. Relationship between non-alcoholic fatty liver disease, metabolic syndrome and insulin resistance in Korean adults: a cross-sectional study. Clin Chim Acta (2016) 458:12–7. doi: 10.1016/j.cca.2016.03.018
160. Wang G, Manaenko A, Shao A, Ou Y, Yang P, Budbazar E, et al. Low-density lipoprotein receptor-related protein-1 facilitates heme scavenging after intracerebral hemorrhage in mice. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2017) 37:1299–310. doi: 10.1177/0271678X16654494
161. Tamaki C, Ohtsuki S, Terasaki T. Insulin facilitates the hepatic clearance of plasma amyloid β-peptide (1–40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol Pharmacol (2007) 72:850–5. doi: 10.1124/mol.107.036913
162. Więckowska-Gacek A, Mietelska-Porowska A, Wydrych M, Wojda U. Western Diet as a trigger of alzheimer’s disease: from metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res Rev (2021) 70:101397. doi: 10.1016/j.arr.2021.101397
163. Gali S, Rukmangadha N, Prayaga A, Kumar V. Hepatic mucormycosis in a renal transplant recipient: a rare presentation. Indian J Transplant vol (2019) 13:38–41. doi: 10.4103/ijot.ijot_48_18
164. Xue-Shan Z, Juan P, Qi W, Zhong R, Li-Hong P, Zhi-Han T, Zhi-Sheng J, et al, et al. Imbalanced cholesterol metabolism in alzheimer’s disease. Clin Chim Acta (2016) 456:107–14. doi: 10.1016/j.cca.2016.02.024
165. Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G, et al. Changes in brain oxysterols at different stages of alzheimer’s disease: their involvement in neuroinflammation. Redox Biol (2016) 10:24–33. doi: 10.1016/j.redox.2016.09.001
166. Zhang SL, Yue Z, Arnold DM, Artiushin G. & sehgal, a. a circadian clock in the blood-brain barrier regulates xenobiotic efflux. Cell (2018) 173:130–139.e10. doi: 10.1016/j.cell.2018.02.017
167. Mast N, Anderson KW, Lin JB, Li Y, Turko IV, Tatsuoka C, et al. Cytochrome P450 27A1 deficiency and regional differences in brain sterol metabolism cause preferential cholestanol accumulation in the cerebellum*. J Biol Chem (2017) 292:4913–24. doi: 10.1074/jbc.M116.774760
168. Loera-Valencia R, Cedazo-Minguez A, Kenigsberg PA, Page G, Duarte AI, Giusti P, et al. Current and emerging avenues for alzheimer’s disease drug targets. J Intern Med (2019) 286:398–437. doi: 10.1111/joim.12959
169. Chew H, Solomon VA, Fonteh AN. Involvement of lipids in alzheimer’s disease pathology and potential therapies. Front Physiol (2020) 11. doi: 10.3389/fphys.2020.00598
170. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic KATP channels control hepatic glucose production. Nature (2005) 434:1026–31. doi: 10.1038/nature03439
171. Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging (2017) 49:60–8. doi: 10.1016/j.neurobiolaging.2016.08.019
172. Miranda M, Morici JF, Zanoni MB, Bekinschtein P. Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci (2019) 13:363–3. doi: 10.3389/fncel.2019.00363
173. Numakawa T, Odaka H, Adachi N. Actions of brain-derived neurotrophin factor in the neurogenesis and neuronal function, and its involvement in the pathophysiology of brain diseases. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19113650
174. Harley J, Hagemann C, Serio A, Patani R. TDP-43 and FUS mislocalization in VCP mutant motor neurons is reversed by pharmacological inhibition of the VCP D2 ATPase domain. Brain Commun (2021) 3(3): fcab166. doi: 10.1093/braincomms/fcab166
175. Lima Giacobbo B, Doorduin J, Klein HC, Dierckx RAJO, Bromberg E, de Vries EFJ. Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol Neurobiol (2019) 56:3295–312. doi: 10.1007/s12035-018-1283-6
176. Teillon S, Calderon GA, Rios M. Diminished diet-induced hyperglycemia and dyslipidemia and enhanced expression of PPARα and FGF21 in mice with hepatic ablation of brain-derived neurotropic factor. J Endocrinol (2010) 205:37–47. doi: 10.1677/JOE-09-0405
177. Gao Y, Su J, Guo W, Polich ED, Magyar DP, Xing Y, et al. Inhibition of miR-15a promotes BDNF expression and rescues dendritic maturation deficits in MeCP2-deficient neurons. Stem Cells Dayt Ohio (2015) 33:1618–29. doi: 10.1002/stem.1950
178. Amir M, Yu M, He P, Srinivasan S. Hepatic autonomic nervous system and neurotrophic factors regulate the pathogenesis and progression of non-alcoholic fatty liver disease. Front Med (2020) 7. doi: 10.3389/fmed.2020.00062
179. Liu S, Fu S, Wang G, Cao Y, Li L, Li X, et al. Glycerol-3-phosphate biosynthesis regenerates cytosolic NAD+ to alleviate mitochondrial disease. Cell Metab (2021) 33:1974–87. doi: 10.1016/j.cmet.2021.06.013
180. Kim HH, Shim YR, Choi SE, Kim MH, Lee G, You HJ, et al. Catecholamine induces kupffer cell apoptosis via growth differentiation factor 15 in alcohol-associated liver disease. Exp Mol Med (2023) 55:158–70. doi: 10.1038/s12276-022-00921-x
181. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, et al. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol (2020) 11:1169–9. doi: 10.3389/fimmu.2020.01169
182. Nati M, Chung K-J, Chavakis T. The role of innate immune cells in nonalcoholic fatty liver disease. J Innate Immun (2021) 14:31–41. doi: 10.1159/000518407
183. Chen S, Yang J, Wei Y, Wei X. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol (2020) 17:36–49. doi: 10.1038/s41423-019-0315-0
184. Lee Y, Hu S, Park Y-K, Lee J-Y. Health benefits of carotenoids: a role of carotenoids in the prevention of non-alcoholic fatty liver disease. Prev Nutr Food Sci (2019) 24:103–13. doi: 10.3746/pnf.2019.24.2.103
185. Di Gregoli K, Somerville M, Bianco R, Thomas AC, Frankow A, Newby AC, et al. Galectin-3 identifies a subset of macrophages with a potential beneficial role in atherosclerosis. Arterioscler Thromb Vasc Biol (2020) 40:1491–509. doi: 10.1161/ATVBAHA.120.314252
186. Harrison SA, Marri SR, Chalasani N, Kohli R, Aronstein W, Thompson GA, et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharmacol Ther (2016) 44:1183–98. doi: 10.1111/apt.13816
187. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PloS One (2016) 11:e0158156. doi: 10.1371/journal.pone.0158156
188. Friedman S, Sanyal A, Goodman Z, Lefebvre E, Gottwald M, Fischer L, et al. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR phase 2b study design. Contemp Clin Trials (2016) 47:356–65. doi: 10.1016/j.cct.2016.02.012
189. Anstee QM, Neuschwander-Tetri BA, Wong VW, Abdelmalek MF, Younossi ZM, Yuan J, et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA phase 3 study design. Contemp Clin Trials (2020) 89:105922. doi: 10.1016/j.cct.2019.105922
190. Tosello-Trampont A-C, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
191. Svendsen P, Graversen JH, Etzerodt A, Hager H, Røge R, Grønbæk H, et al. Antibody-directed glucocorticoid targeting to CD163 in M2-type macrophages attenuates fructose-induced liver inflammatory changes. Mol Ther - Methods Clin Dev (2017) 4:50–61. doi: 10.1016/j.omtm.2016.11.004
192. Ramos MJ, Bandiera L, Menolascina F, Fallowfield JA. In vitro models for non-alcoholic fatty liver disease: emerging platforms and their applications. iScience (2022) 25:103549. doi: 10.1016/j.isci.2021.103549
193. Makri ES, Makri E, Polyzos SA. Combination therapies for nonalcoholic fatty liver disease. J Pers Med (2022) 12:1166. doi: 10.3390/jpm12071166
Keywords: non-alcoholic fatty liver disease, fibrosis, macrophages, inflammation, Kupffer cells
Citation: Alabdulaali B, Al-rashed F, Al-Onaizi M, Kandari A, Razafiarison J, Tonui D, Williams MR, Blériot C, Ahmad R and Alzaid F (2023) Macrophages and the development and progression of non-alcoholic fatty liver disease. Front. Immunol. 14:1195699. doi: 10.3389/fimmu.2023.1195699
Received: 28 March 2023; Accepted: 31 May 2023;
Published: 12 June 2023.
Edited by:
Erika M. Palmieri, National Cancer Institute at Frederick (NIH), United StatesReviewed by:
Federico Virga, Spanish National Centre for Cardiovascular Research, SpainCopyright © 2023 Alabdulaali, Al-rashed, Al-Onaizi, Kandari, Razafiarison, Tonui, Williams, Blériot, Ahmad and Alzaid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fawaz Alzaid, ZmF3YXouYWx6YWlkQGRhc21hbmluc3RpdHV0ZS5vcmc=; ZmF3YXouYWx6YWlkQGluc2VybS5mcg==
‡ORCID: Bader Alabdulaali, orcid.org/0000-0003-2203-7456
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.