 María Enriqueta Núñez-Núñez1
María Enriqueta Núñez-Núñez1 Juan Carlos Lona-Reyes2,3
Juan Carlos Lona-Reyes2,3 Brenda López-Barragán4
Brenda López-Barragán4 Rosa Margarita Cruz-Osorio3,5
Rosa Margarita Cruz-Osorio3,5 Bricia Melissa Gutiérrez-Zepeda6,7
Bricia Melissa Gutiérrez-Zepeda6,7 Antonio Quintero-Ramos6,8*
Antonio Quintero-Ramos6,8* Denisse Stephania Becerra-Loaiza6,7*
Denisse Stephania Becerra-Loaiza6,7*- 1Departamento de Alergia e Inmunología Clínica Pediátrica, Nuevo Hospital Civil de Guadalajara “Dr. Juan I. Menchaca”, Guadalajara, Mexico
- 2Departamento de Infectología, Nuevo Hospital Civil de Guadalajara “Dr. Juan I. Menchaca”, Guadalajara, Mexico
- 3Clínicas de Pediatría, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 4Departamento de Pediatría, Nuevo Hospital Civil de Guadalajara “Dr. Juan I. Menchaca”, Guadalajara, Mexico
- 5Departamento de Hemato-Oncología Pediátrica, Nuevo Hospital Civil de Guadalajara “Dr. Juan I. Menchaca”, Guadalajara, Mexico
- 6Laboratorio de Inmunología, Departamento de Fisiología, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 7Doctorado en Genética Humana, Departamento de Biología Molecular y Genómica, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara, Guadalajara, Mexico
- 8Unidad de Investigación Biomédica 02, Hospital de Especialidades, Centro Médico Nacional de Occidente, Instituto Mexicano del Seguro Social, Guadalajara, Mexico
The most common causes of congenital neutropenia are mutations in the ELANE (Elastase, Neutrophil Expressed) gene (19p13.3), mostly in exon 5 and the distal portion of exon 4, which result in different clinical phenotypes of neutropenia. Here, we report two pathogenic mutations in ELANE, namely, c.607G>C (p.Gly203Arg) and a novel variant c.416C>G (p.Pro139Arg), found in two Mexican families ascertained via patients with congenital neutropenia who responded positively to the granulocyte colony-stimulating factor (G-CSF) treatment. These findings highlight the usefulness of identifying variants in patients with inborn errors of immunity for early clinical management and the need to rule out mosaicism in noncarrier parents with more than one case in the family.
1 Introduction
Congenital neutropenia (CN) encompasses a family of genetic diseases characterized by 1) low neutrophil counts (≤0.5–1.5 × 109/L) and susceptibility to infection, 2) various organ dysfunctions, and 3) an extraordinarily high risk of leukemic transformation (1). The CN-specific manifestation of neutropenia can be a permanent condition in the patients or can occur in a cyclic/intermittent pattern where periods of normalization alternate with episodes of severe neutropenia (1). These diseases commonly result from mutations in the ELANE gene (19p13.3), which encodes for neutrophil elastase (NE) (EC 3.4.21.37), a pleiotropic enzyme pivotal for host innate defense, tissue remodeling, and local inflammatory responses, mediated by neutrophil and monocyte granules (2–4).
Heterozygous mutations in the ELANE gene cause autosomal dominant CN phenotypes exhibiting extremely high penetrance (5, 6). Mutations in exons 4 and 5 of ELANE may disrupt the disulfide bond domain in the C-terminus of the protease, which is essential for its correct intracellular localization. This disruption could lead to dysregulated vesicular sorting, trafficking, and reduced stability of mutant protein, manifesting as impaired neutrophil maturation and diverse CN phenotypes (2, 7). Identical ELANE mutations can cause severe congenital neutropenia (SCN) or cyclic/intermittent neutropenia (CyN) (8, 9) owing to neutrophil precursor arrest at the promyelocyte or myelocyte stage (4). Therefore, the clinical phenotype and natural history of patients should be assessed in the context of molecular findings.
Here, we report two ELANE mutations in three patients with CN from two families. One of them, a known mutation, formerly named c.607G>C (Gly203Arg), was observed in two sisters with different fathers who were diagnosed with SCN. To our knowledge, for the first time, a c.416C>G (p.Pro139Leu) mutation was associated with the clinical expression of CyN in a male child. We also reviewed literature on the identified mutations for this inborn error of immunity (IEI).
2 Case reports
2.1 Family 1
Patient 1 (P1) (Figures 1A, B) was a female child born in September 2011 to a primiparous mother and an unrelated father from Jalisco, Mexico. P1 was hospitalized at 12 days of age due to irritability, fever, hyaline ear secretions, and suspected sepsis. At 45 days of age, she was hospitalized again with purulent secretions in the umbilical scar and infection in the soft tissues located in the right nostril (Figure 2A).
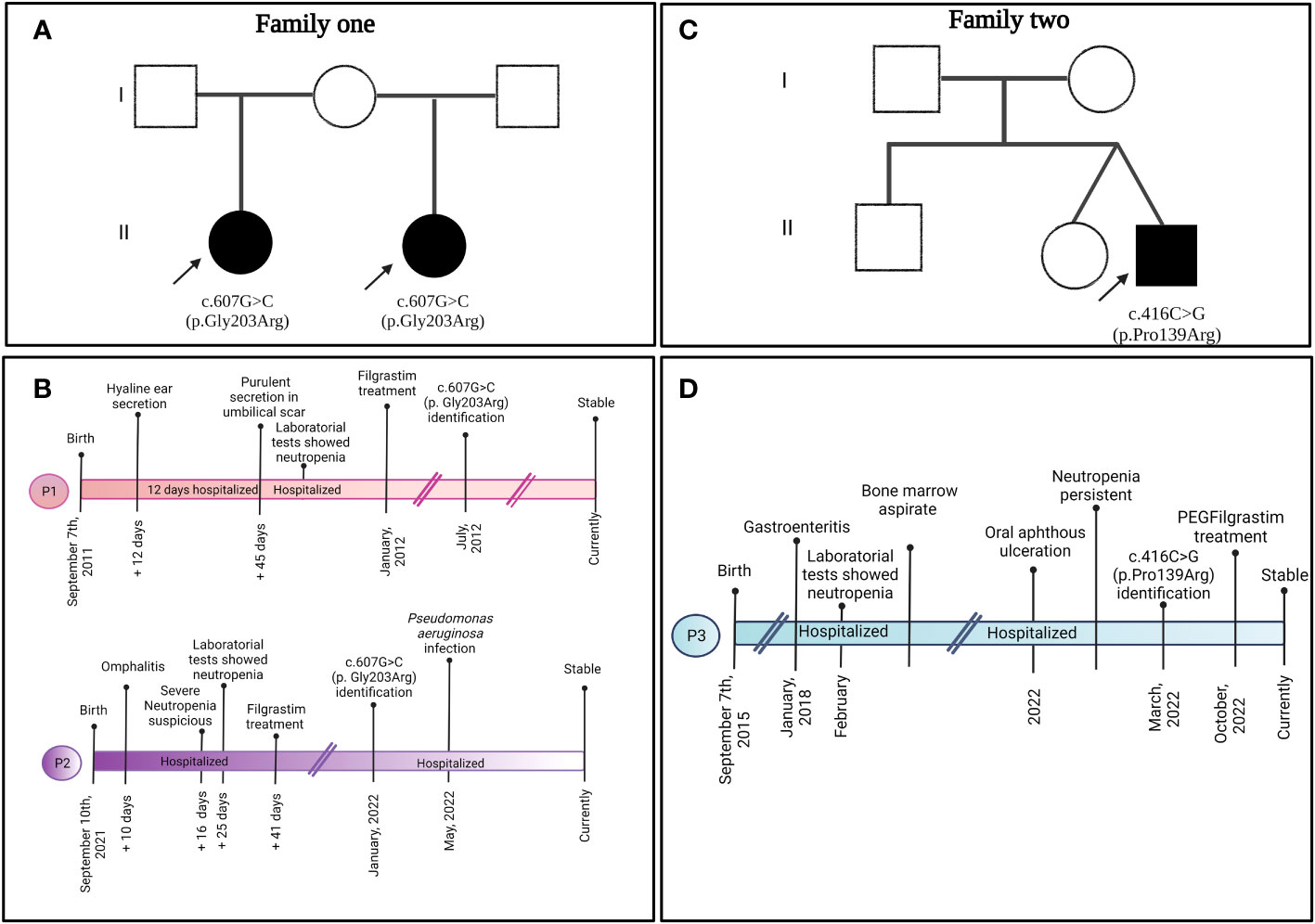
Figure 1 Familial pedigree of (A) family 1 and (C) family 2 depicting the clinical presentation timeline of (B) P1, P2, and (D) P3.
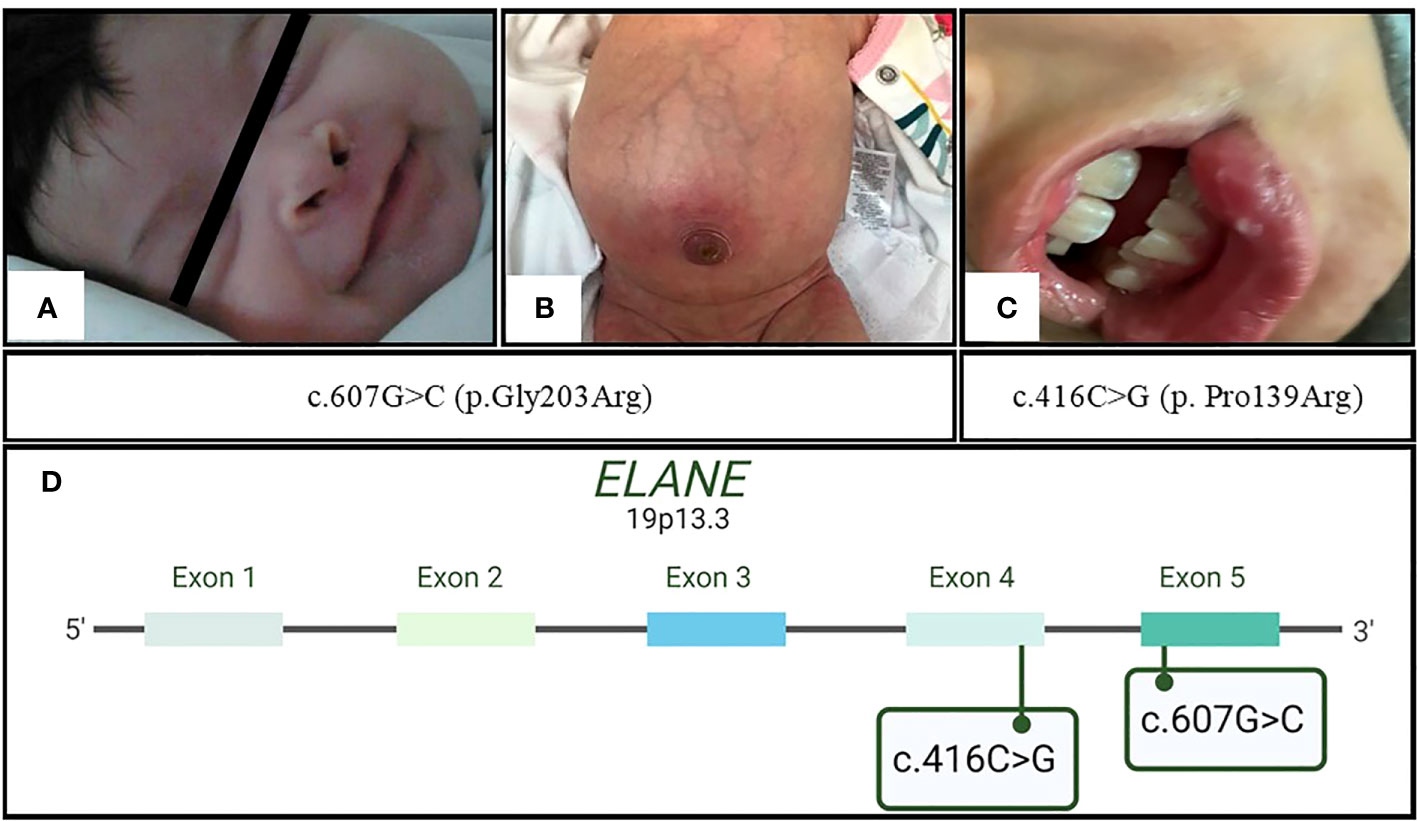
Figure 2 Initial clinical presentation of the patients. In family 1 with c.607G>C (p.Gly203Arg) mutation, (A) P1 with a necrotic lesion located in the right nostril and (B) P2 with omphalitis; in family 2 with novel mutation c.416C>G (p.Pro139Arg), (C) P3 with oral aphthous ulceration. (D) Ubication of the abovementioned ELANE mutations in exons 5 and 4, respectively.
Laboratory tests revealed neutropenia (50 cells/µL), anemia (8.1 g/dL), and monocytosis (3,930 cells/µL). The infection resolved after antimicrobial treatment, but neutropenia persisted with no improvement at 51 days of age. The bone marrow aspirate showed a 17% decrease in the number of monocytes and neutrophils. Immunoglobulins (Igs) and lymphocyte levels were normal: IgM = 132 mg/dL, IgG = 1,045 mg/dL, IgA = 89 mg/dL, IgE = 23.9 UI/dL; lymphocyte subsets CD3+ (3,495 cells/µL), CD4+ (2,522 cells/µL), CD8+ (922 cells/µL), and CD19+ (1,547 cells/µL). A second bone marrow aspirate revealed a decreased neutrophil count (7%), eosinophilia, and monocytosis but normal red blood cell counts. After eliminating other causes of neutropenia and considering the clinical course and persistent neutropenia (70 cells/µL, Figure 3), treatment with granulocyte colony-stimulating factor (G-CSF Filgrastim®; 30 µg/kg) was initiated at 3 months of age.
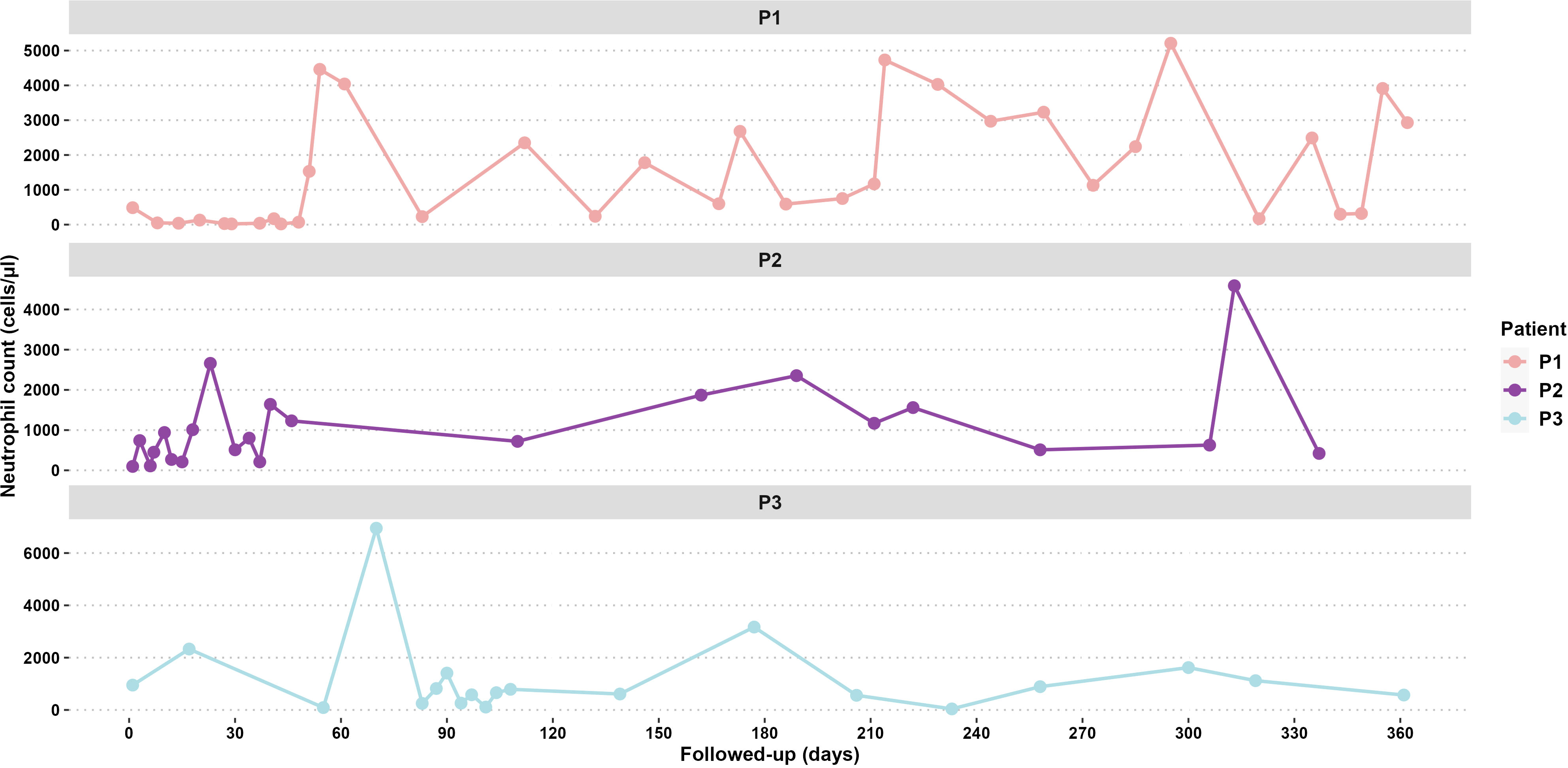
Figure 3 Follow-up neutrophil counts for 365 days of P1, P2, and P3.
Patient 2 (P2) (Figures 1A, B) was the second female child born in September 2021 to the same mother, but from another non-consanguineous father from Jalisco, Mexico. P2 was hospitalized for omphalitis and neonatal sepsis (Figure 2B) at 16 days of age and treated with antibiotics; severe neutropenia was suspected (Figure 3). At 41 days of age, prophylactic antibiotic and Filgrastim® treatments were initiated, but after 1 month, the dose was augmented from 30 µg/kg to 83 µg/kg per day. In May 2022, P2 was hospitalized again for a left inguinal abscess with erythema, high local temperature, and purulent secretions secondary to Pseudomonas aeruginosa infection.
Screening for gene mutations performed by Dr. Kaan Boztug (blood test in P1) or Invitae (buccal swab in P2) via sequencing [Sanger and Next-Generation Sequencing (NGS) panel, respectively] revealed that severe neutropenia in both patients was caused by the same pathogenic mutation, c.607G>C (p.Gly203Arg) (Figure 2D). The same tests on the parental samples revealed that neither parent carried the mutation (electropherograms were not available). At the time of the study, both patients followed their Filgrastim® treatment (16 and 17 µg/kg per day for P1 and P2, respectively; average neutrophil counts: P1 = 2,300 cells/µL, P2 = 2,000 cells/µL), without clinical complications and good treatment response.
2.2 Family 2
Patient 3 (P3) (Figures 1C, D) was a male child born in September 2015 as the second twin of a second pregnancy of non-consanguineous parents (both from Jalisco). At 2 years 4 months of age, P3 was admitted to the emergency department because of severe dehydration secondary to gastroenteritis and was administered intravenous fluids. Laboratory results revealed bicytopenia (Hb = 9.6 g/dL and neutrophils = 100 cells/µL), whereas a bone marrow aspirate discarded a lymphoproliferative disorder. During the hospitalization, repeated neutrophil counts varied considerably (100 cells/µL, 4,850 cells/µL, 440 cells/µL, and 1,750 cells/µL); after 7 days, P3 was discharged.
Four years later, P3 developed intermittent fever accompanied by monthly oral aphthous ulceration and was rehospitalized (Figure 2C). On this occasion, the hematological parameters were normal, except for the neutrophil count, which ranged from 60 to 610 cells/µL with two peaks of 6,950 cells/µL and 1,410 cells/µL. Neutropenia persisted from November 2021 until August 2022, as diagnosed in serial blood counts performed twice per week during April and May (Figure 3), which also showed normal lymphocyte subpopulation counts and Ig levels. To corroborate the diagnosis of CyN, sequence analysis and deletion/duplication testing of a panel of 574 genes were kindly provided by Invitae; unfortunately, the electropherograms were not available.
P3 was heterozygous for variants of uncertain significance (VUSs) in genes for ATP-binding cassette transporters G5 [ABCG8 (c.255G>T, p.Glu85Asp)], Ankyrin repeat and zinc peptidyl TRNA hydrolase [ANKZF1 (c.1053A>C, p.Glu351Asp)], Centrosomal protein 164 [CEP164 (c.1152A>G, silent)], Cystic fibrosis transmembrane conductance regulator [CFTR (c.335A>G, p.Asp112Gly)], Forkhead box N1 [FOXN1 (c.724C>T, p.Pro242Ser)], and SKI2 subunit of superkiller complex [SKIV2L (c.2165G>A, p.Arg722Gln)]. Furthermore, a novel pathogenic variant, c.416C>G (p.Pro139Arg) (Figure 2D), in ELANE was found. The same tests on the parental samples revealed that neither parent carried these variants. In September 2022, Filgrastim® treatment (5 µg/kg per day) was initiated; however, since October 2022, P3 followed subcutaneous PEGFilgrastim® treatment (1 mg) every 21 days. Currently, P3 presents an average neutrophil count of 1,350 cells/µL, without clinical manifestations and good treatment response.
3 Discussion
In this report, P1 and P2 exhibited the same pathogenic mutation, namely, autosomal dominant c.607G>C (p.Gly203Arg) (rs201139487) located in exon 5 of ELANE, which could account for the nonfamilial SCN of the patients. This mutation was reported for the first time in a Korean girl with SCN (10), with an initial clinical presentation of omphalitis, like that observed in P1 and P2. However, in our case, the clinical diagnosis of P1 and P2 was done earlier (at 4 months of age) than in the case of the Korean girl (at 9 months of age).
The existence of genetically distinct populations of cells in a particular organism, called mosaicism, is an important cause of genetic disease and can appear as de novo DNA mutations (11). Accordingly, Shigemura et al. (12) reported a c.607G>C ELANE mutation in an asymptomatic Japanese mother, who exhibited mosaicism, whose daughter had SCN; however, this report did not provide a clinical description of the patient. Similarly, paternal mosaicism for other ELANE mutations causing SCN has been reported (13–15). Accordingly, in the present study, germline mosaicism was suspected in the unaffected mother of both patients carrying the c.607G>C mutation, which is relevant considering only a few families have presented mosaicism of mutation in the ELANE gene (13).
Sporadic CyN is caused by de novo mutations in the ELANE gene (16). Clinically, patients exhibit fever, infections, and neutropenia, with blood neutrophil counts fluctuating approximately every 14–35 days and profound neutropenia for 3–10 days (17, 18). The clinical evaluation of these events in CyN can be challenging, as observed with P3, whose neutropenia showed irregular fluctuations, and responded to therapy (19). In P3, the neutrophil counts represented the intermittent neutropenia (CyN) pattern with profound neutropenia alternating with peaks of elevated neutrophil counts in a period of 6 weeks; this sufficiently confirmed the diagnosis based on the pediatric hematology protocol of Hospital Civil Nuevo “Dr Juan I Menchaca” and according to the literature (20–22).
The novel mutation in the distal portion of exon 4, c.416C>G (p.Pro139Arg), found in P3 resembles the c.416C>T (p.Pro139Leu) (rs137854448) (23) pathogenic single-nucleotide variant (SNV) in the same codon that causes both CyN and SCN (24–26). The major concern in CN mortality is the progression and development of leukemia (27), which is related to certain germline mutations (26). In this respect, the previously reported SNV c.416C>T (p.Pro139Leu) (rs137854448) (24) has been associated with a good prognosis without an increased risk of leukemia development (28). On the other hand, the mechanism by which defects in NE lead to leukemia is unknown and there is still a lack of hallmark predictors (29). G-CSF treatment is known to increase the risk of leukemia development by 22% after 15 years of treatment, and a higher dose or diminished response augments these probabilities (30, 31). Previously, Freedman et al. (32) disregarded any relationship between age, sex, dosage or duration of treatment, and malignant transformation; thus, the associations are still unclear. However, during treatment, marrow bone cells acquire somatic mutations related to myelodysplastic syndromes/acute myeloid leukemia (MDS/AML) (33–35). Therefore, we suggest that some germline mutations can interact with acquired somatic mutations and moderate the clinical presentation.
Invitae catalogs the missense mutation c.416C>G (p.Pro139Arg) (SCV003915652) as pathogenic, derived from algorithms that predict the disruptive effect of Pro/Arg changes on the protein structure and function. However, these are limiting in silico speculative conjunctions. Follow-up of P3 might demonstrate whether this novel mutation is related to leukemia. The clinical expression of CN (SCN or CyN), depending on the mutation, is homogeneous or variable, suggesting different pathogenic mechanisms (36). Previous in silico analyses showed a potentially high risk of altered protein structure and function for c.607G>C (rs201139487) and c.416C>G (rs137854448) (37) located in the same codon of SNV reported here for the first time. Additionally, Shinwari et al. (37) reported that these SNVs were related to SCN and CyN (24, 25).
Moreover, the synergistic effect of concurrent mutations (38–40) explains the distinct pathomechanisms of phenotype determination aside from modifier genes (41). Significantly, the concomitant mutations in P3 affected the interacting molecules, namely, ABCG8, ANKZF1, CEP164, CFTR, FOXN1, SKIV2L, and ELANE (42, 43). According to Invitae’s report, none of these has been found in CyN or SCN; in the absence of sufficient information to determine their role in CyN, they have been classified as tolerated variants by bioinformatic predictions (SIFT, Polyphen-2, and Align-GVGD). Moreover, the present case of sporadic CyN with multiple mutations could have resulted from a higher germline mutation rate in older males than those in females (44). In addition, ELANE is prone to somatic mutagenesis (40). Thus, our report of the concurrence of these VUSs with ELANE, interacting and co-expressing with many genes (42, 43), enhances the data of new mutations in patients with IEI and may enable a more effective classification of clinical features and complications.
When there is insufficient evidence linking the variant to disease and a VUS is reported, it creates a problem for the clinician in interpretation and management of the patient (45). However, reporting the VUS in different clinical presentations could support the reclassification and genetic testing strategies, thus helping physicians understand how to incorporate these results into clinical care (46, 47). Therefore, it is essential to review the literature on common clinical features and phenotypic variations among affected individuals (48). Our findings highlight the usefulness of identifying variants in patients with IEI, such as CN, for early clinical management and the need to rule out mosaicism in apparently noncarrier parents with more than one case in the family, although for that, this report is speculative. Furthermore, we report a novel c.416C>G (p.Pro139Arg) mutation in a male child diagnosed with CyN. This report enriches the clinical mutation database and can also help further and improve management of patients worldwide with similar clinical manifestations.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SCV003915652 (ClinVar).
Ethics statement
The studies involving humans were approved by Comité de ética e investigación Hospital Civil de Guadalajara Dr. Juan I. Menchaca. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JL-R, BL-B, and RC-O contributed to the conception of the case report and acquired data. MN-N, DB-L, BG-Z, and AQ-R acquired, analyzed, and interpreted the patient data. DB-L and MN-N drafted the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the patients and their parents for their participation as well as Dr. Horacio Rivera-Ramírez and Dr. Luis Antonio Ochoa-Ramírez for their comments. We are also grateful to Dr. Kaan Boztug and their group for the molecular diagnosis of P1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Donadieu J, Beaupain B, Fenneteau O, Bellanné-Chantelot C. Congenital neutropenia in the era of genomics: classification, diagnosis, and natural history. Br J Haematol (2017) 179(4):557–74. doi: 10.1111/bjh.14887
2. Rydzynska Z, Pawlik B, Krzyzanowski D, Mlynarski W, Madzio J. Neutrophil elastase defects in congenital neutropenia. Front Immunol (2021) 12:653932. doi: 10.3389/fimmu.2021.653932
3. Online Mendelian Inheritance in Man, OMIM® . Baltimore, MD: Johns Hopkins University. Available at: https://omimg.org/ (Accessed December 28,2022). MIM Number: #130130: 07/19/2018.
4. Liu Q, Sundqvist M, Li W, Holdfeldt A, Zhang L, Björkman L, et al. Functional characteristics of circulating granulocytes in severe congenital neutropenia caused by ELANE mutations. BMC Pediatr (2019) 19(1):189. doi: 10.1186/s12887-019-1556-x
5. Online Mendelian Inheritance in Man, OMIM® . Baltimore, MD: Johns Hopkins University. Available at: https://omimg.org/ (Accessed December 28,2022). MIM Number: #162800: 06/02/2016.
6. Online Mendelian Inheritance in Man, OMIM® . Baltimore, MD: Johns Hopkins University. Available at: https://omimg.org/ (Accessed December 28,2022). MIM Number: #202700: 04/11/2022.
7. Borregaard N. Severe congenital neutropenia: new lane for ELANE. Blood (2014) 123(4):462–3. doi: 10.1182/blood-2013-11-537068
8. Horwitz MS. Neutrophil elastase: Nonsense lost in translation. Cell Stem Cell (2021) 28(5):790–2. doi: 10.1016/j.stem.2021.04.007
9. Vandenberghe P, Beel K. Severe congenital neutropenia, a genetically heterogeneous disease group with an increased risk of AML/MDS. Pediatr Rep (2011) 3 Suppl 2(Suppl 2):e9. doi: 10.4081/pr.2011.s2.e9
10. Shim YJ, Kim HJ, Suh JS, Lee KS. Novel ELANE gene mutation in a Korean girl with severe congenital neutropenia. J Korean Med Sci (2011) 26(12):1646–9. doi: 10.3346/jkms.2011.26.12.1646
11. Mohiuddin M, Kooy RF, Pearson CE. De novo mutations, genetic mosaicism and human disease. Front Genet (2022) 26:983668. doi: 10.3389/fgene.2022.983668
12. Shigemura T, Kobayashi N, Agematsu K, Ohara O, Nakazawa Y. Mosaicism of an ELANE mutation in an asymptomatic mother. J Clin Immunol (2019) 39(1):106–11. doi: 10.1007/s10875-018-0580-1
13. Liu Q, Zhang L, Shu Z, Ding Y, Tang XM, Zhao XD. Two paternal mosaicism of mutation in ELANE causing severe congenital neutropenia exhibit normal neutrophil morphology and ROS production. Clin Immunol (2019) 203:53–8. doi: 10.1016/j.clim.2019.04.008
14. Kim HJ, Song MJ, Lee KO, Kim SH, Kim HJ. Paternal somatic mosaicism of a novel frameshift mutation in ELANE causing severe congenital neutropenia. Pediatr Blood Cancer (2015) 62(12):2229–31. doi: 10.1002/pbc.25654
15. Ancliff PJ, Gale RE, Watts MJ, Liesner R, Hann IM, Strobel S, et al. Paternal mosaicism proves the pathogenic nature of mutations in neutrophil elastase in severe congenital neutropenia. Blood (2002) 100(2):707–9. doi: 10.1182/blood-2002-01-0060
16. ClinVar. ELANE (2023). Available at: https://www.ncbi.nlm.nih.gov/clinvar/?term=ELANE%5Bgene%5D&redir=gene (Accessed August 9, 2023).
17. Walkovich K, Connelly JA. Congenital neutropenia and rare functional phagocyte disorders in children. Hematol Oncol Clin North Am (2019) 33(3):533–51. doi: 10.1016/j.hoc.2019.01.004
18. Dale DC, Welte K. Cyclic and chronic neutropenia. Cancer Treat Res (2011) 157:97–108. doi: 10.1007/978-1-4419-7073-2_6
19. Horwitz MS, Corey SJ, Grimes HL, Tidwell T. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am (2013) 27(1):19–41, vii. doi: 10.1016/j.hoc.2012.10.004
20. Dale DC, Makaryan V. ELANE-Related neutropenia, in: GeneReviews® . Seattle (WA: University of Washington, Seattle (Accessed April 30, 2023). 1993–2023: 23/08/2018.
21. Dale DC, Bolyard AA, Aprikyan A. Cyclic neutropenia. Semin Hematol (2002) 39(2):89–94. doi: 10.1053/shem.2002.31917
22. Palmer SE, Stephens K, Dale DC. Genetics, phenotype, and natural history of autosomal dominant cyclic hematopoiesis. Am J Med Genet (1996) 66(4):413–22. doi: 10.1002/(SICI)1096-8628(19961230)66:4<413::AID-AJMG5>3.0.CO;2-L
23. National Center for Biotechnology Information. ClinVar; [VCV000016743.20] . Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000016743.20 (Accessed May 2, 2023).
24. Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Rodger E, Kelley ML, et al. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol (2015) 22(1):3–11. doi: 10.1097/MOH.0000000000000105
25. Germeshausen M, Deerberg S, Peter Y, Reimer C, Kratz CP, Ballmaier M. The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum Mutat (2013) 34(6):905–14. doi: 10.1002/humu.22308
26. Kurnikova M, Maschan M, Dinova E, Shagina I, Finogenova N, Mamedova E, et al. Four novel ELANE mutations in patients with congenital neutropenia. Pediatr Blood Cancer (2011) 57(2):332–5. doi: 10.1002/pbc.23104
27. Shahrabi S, Maleknia M, Tavakolifar Y, D Zayeri Z, Saki N. Neutropenia and leukemia development: genetic risk factors and prognosis. Leuk Lymphoma (2019) 60(14):3363–74. doi: 10.1080/10428194.2019.1630622
28. Mangaonkar AA, Patnaik MM. Hereditary predisposition to hematopoietic neoplasms: when bloodline matters for blood cancers. Mayo Clin Proc (2020) 95(7):1482–98. doi: 10.1016/j.mayocp.2019.12.013
29. Link DC. Mechanisms of leukemic transformation in congenital neutropenia. Curr Opin Hematol (2019) 26(1):34–40. doi: 10.1097/MOH.0000000000000479
30. Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, et al. Severe Chronic Neutropenia International Registry. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood (2006) 107(12):4628–35. doi: 10.1182/blood-2005-11-4370
31. Rosenberg PS, Zeidler C, Bolyard AA, Alter BP, Bonilla MA, Boxer LA, et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol (2010) 150(2):196–9. doi: 10.1111/j.1365-2141.2010.08216.x
32. Freedman MH, Bonilla MA, Fier C, Bolyard AA, Scarlata D, Boxer LA, et al. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood (2000) 96(2):429–36.
33. Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood (2012) 119(22):5071–7. doi: 10.1182/blood-2012-01-406116
34. Touw IP, van de Geijn GJ. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci (2007) 12:800–15. doi: 10.2741/2103
35. Freedman MH, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol (2002) 39(2):128–33. doi: 10.1053/shem.2002.31912
36. Bellanné-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood (2004) 103(11):4119–25. doi: 10.1182/blood-2003-10-3518
37. Shinwari K, Bolkov MA, Yasir Akbar M, Guojun L, Deryabina SS, Tuzankina IA, et al. In Silico analysis revealed five novel high-risk single-nucleotide polymorphisms (rs200384291, rs201163886, rs193141883, rs201139487, and rs201723157) in ELANE gene causing autosomal dominant severe congenital neutropenia 1 and cyclic hematopoiesis. ScientificWorldJournal (2022) 2022:3356835. doi: 10.1155/2022/3356835
38. Germeshausen M, Zeidler C, Stuhrmann M, Lanciotti M, Ballmaier M, Welte K. Digenic mutations in severe congenital neutropenia. Haematologica (2010) 95(7):1207–10. doi: 10.3324/haematol.2009.017665
39. Lundén L, Boxhammer S, Carlsson G, Ellström KG, Nordenskjöld M, Lagerstedt-Robinson K, et al. Double de novo mutations of ELANE (ELA2) in a patient with severe congenital neutropenia requiring high-dose G-CSF therapy. Br J Haematol (2009) 147(4):587–90. doi: 10.1111/j.1365-2141.2009.07866.x
40. Salipante SJ, Benson KF, Luty J, Hadavi V, Kariminejad R, Kariminejad MH, et al. Double de novo mutations of ELA2 in cyclic and severe congenital neutropenia. Hum Mutat (2007) 28(9):874–81. doi: 10.1002/humu.20529
41. Newburger PE, Pindyck TN, Zhu Z, Bolyard AA, Aprikyan AA, Dale DC, et al. Cyclic neutropenia and severe congenital neutropenia in patients with a shared ELANE mutation and paternal haplotype: evidence for phenotype determination by modifying genes. Pediatr Blood Cancer (2010) 55(2):314–7. doi: 10.1002/pbc.22537
42. Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res (2010) 38(Web Server issue):W214–20. doi: 10.1093/nar/gkq537
43. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res (2009) 37(Database issue):D412–6. doi: 10.1093/nar/gkn760
44. Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet (2000) 1(1):40–7. doi: 10.1038/35049558
45. Nicolosi P, Heald B, Esplin ED. What is a variant of uncertain significance in genetic testing? Eur Urol Focus (2022) 8(3):654–6. doi: 10.1016/j.euf.2022.03.018
46. Macklin SK, Jackson JL, Atwal PS, Hines SL. Physician interpretation of variants of uncertain significance. Fam Cancer (2019) 18(1):121–6. doi: 10.1007/s10689-018-0086-2
47. Newey PJ. Approach to the patient with a variant of uncertain significance on genetic testing. Clin Endocrinol (Oxf) (2022) 97(4):400–8. doi: 10.1111/cen.14818
Keywords: ELANE gene mutation, severe neutropenia, novel mutation, cyclic neutropenia (CyN), Mexican, c.416C>G, c.607G>C, case report
Citation: Núñez-Núñez ME, Lona-Reyes JC, López-Barragán B, Cruz-Osorio RM, Gutiérrez-Zepeda BM, Quintero-Ramos A and Becerra-Loaiza DS (2023) Case Report: Characterization of known (c.607G>C) and novel (c.416C>G) ELANE mutations in two Mexican families with congenital neutropenia. Front. Immunol. 14:1194262. doi: 10.3389/fimmu.2023.1194262
Received: 26 March 2023; Accepted: 30 August 2023;
Published: 18 September 2023.
Edited by:
Elissa Deenick, Garvan Institute of Medical Research, AustraliaReviewed by:
Julia Skokowa, University of Tübingen, GermanyRevathi Raj, Apollo Specialty Hospitals, India
Sushree Sahoo, St. Jude Children’s Research Hospital, United States
Copyright © 2023 Núñez-Núñez, Lona-Reyes, López-Barragán, Cruz-Osorio, Gutiérrez-Zepeda, Quintero-Ramos and Becerra-Loaiza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denisse Stephania Becerra-Loaiza, ZGVuaXNzZS5iZWNlcnJhQGFsdW1ub3MudWRnLm14; Antonio Quintero-Ramos, YW50b25pby5xcmFtb3NAYWNhZGVtaWNvcy51ZGcubXg=