 Hui Zhang1,2,3,4,5Qi-Wei Li1,3,4,5,6
Hui Zhang1,2,3,4,5Qi-Wei Li1,3,4,5,6 Yuan-Yuan Li1,3,4,5Xue Tang1,3,4,5
Yuan-Yuan Li1,3,4,5Xue Tang1,3,4,5 Ling Gu1,3,4,5*
Ling Gu1,3,4,5* Han-Min Liu1,3,4,5,6*
Han-Min Liu1,3,4,5,6*- 1Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, China
- 2The Fifth People’s Hospital of Chengdu, Chengdu, China
- 3Sichuan Birth Defects Clinical Research Center, West China Second University Hospital, Sichuan University, Chengdu, China
- 4Key Laboratory of Chronobiology (Sichuan University), National Health Commission of China, Chengdu, China
- 5The Joint Laboratory for Lung Development and Related Diseases of West China Second University Hospital, Sichuan University and School of Life Sciences of Fudan University, West China Institute of Women and Children’s Health, West China Second University Hospital, Sichuan University, Chengdu, China
- 6Department of Pediatric Pulmonology and Immunology, West China Second University Hospital, Sichuan University, Chengdu, China
Pulmonary hypertension (PH) is a chronic pulmonary vascular disorder characterized by an increase in pulmonary vascular resistance and pulmonary arterial pressure. The detailed molecular mechanisms remain unclear. In recent decades, increasing evidence shows that altered immune microenvironment, comprised of immune cells, mesenchymal cells, extra-cellular matrix and signaling molecules, might induce the development of PH. Myeloid-derived suppressor cells (MDSCs) have been proposed over 30 years, and the functional importance of MDSCs in the immune system is appreciated recently. MDSCs are a heterogeneous group of cells that expand during cancer, chronic inflammation and infection, which have a remarkable ability to suppress T-cell responses and may exacerbate the development of diseases. Thus, targeting MDSCs has become a novel strategy to overcome immune evasion, especially in tumor immunotherapy. Nowadays, severe PH is accepted as a cancer-like disease, and MDSCs are closely related to the development and prognosis of PH. Here, we review the relationship between MDSCs and PH with respect to immune cells, cytokines, chemokines and metabolism, hoping that the key therapeutic targets of MDSCs can be identified in the treatment of PH, especially in severe PH.
Introduction
Pulmonary hypertension (PH) is a rare but severe vascular disorder, with a mean pulmonary arterial pressure (mPAP) ≥ 20 mmHg at rest (1). Pathologically, PH is characterized by pulmonary vascular remodeling (PVR) (2, 3). PVR involves proliferation and endothelial-mesenchymal transition of endothelial cells (ECs), phenotypic transformation and proliferation of pulmonary arterial smooth muscle cells (PASMCs), and proliferation, migration and excellular cell matrix (ECM) deposition of adventitial fibroblasts (AFs), as well as infiltration of inflammatory cells (4–8). In clinical practice, PH is classified into 5 groups based on etiology, pathophysiology and treatment, including group 1 [pulmonary arterial hypertension (PAH)], group 2 (PH associated with left heart disease), group 3 (PH associated with lung diseases and/or hypoxia), group 4 (PH associated with pulmonary artery obstructions) and group 5 (PH with unclear and/or multifactorial mechanisms) (1). Treatment of PH typically begins with primary therapy aimed at the underlying causes, and patients always die due to right heart failure. For nearly two decades, novel therapeutic agents, such as sotatercept and riociguat, have made progressions in the treatment of patients with PH (9, 10). However, the drugs only target to pulmonary vascular tone, and it is still difficult to reverse PVR. Therefore, to explore a new effective therapy for PVR is urgent. Recently, increasing evidence shows that immune microenvironment in adventitia of pulmonary arteriole plays an important role in PH development (11, 12). In healthy organisms, the immune system could balance the persistent equilibrium state between injury and repair by many sophisticated mechanisms, and distinct immunosuppressive cells could protect against excessive tissue damages (13). However, in chronic diseases like tumors, chronic infections and autoimmune diseases, inflammatory cells may establish a strong immunosuppressive microenvironment that suppresses anti-tumor and inflammatory immune responses, and promotes disease progression (14–16). Myeloid-derived suppressor cells (MDSCs) are a group of immunosuppressive cells that play an important role in PH development (17), and MDSCs may serve as a new therapeutic target for reversing PVR. Therefore, this review focuses on the relationship between PH and MDSCs, including cancer-like proliferation and immune microenvironment in PH, the phenotype of MDSCs, and the links between MDSCs and PH.
Immune microenvironment and cancer-like process in severe PH
Severe PH is an incurable disease. Plexiform lesion, also called angiomatous malignant proliferation, is a morphologic hallmark of severe PH (18–20), which is a complex and disorganized pulmonary arterial proliferative lesion, consisted of vascular channels lined by a continuously proliferating endothelium and separated by core cells. The core cells could be myofibroblasts, PASMCs, ECs and undifferentiated cells (21). In 2008, Rai et al. (22) formally introduced the concept of cancer-like features in severe PH. Until now, accumulating evidence have supported that ECs, PASMCs and AFs involved in PVR demonstrate a hyperproliferative and apopotosis-resistant phenotype, and those cells are characterized by abnormally cancer-like growth characteristics (23–26). The cancer-like proliferative characteristics of vascular cells lead to irreversible changes in severe PH, but on the plus side, they also provide a new framework for antiproliferative and antiangiogenic therapy in severe PH. For the treatment of PH, especially severe PH, as Cool et al. (27) suggest, future treatment strategies will target immune mechanisms. If we want to re-open occluded pulmonary arterioles or halt disease progression, we might get some inspiration from cancer research data and concepts. In recent years, tumor immunotherapy has been effectively applied in clinical practice by the virtue of its targeting specificity (28, 29). Investigations of immune microenvironment and new targets for immunotherapy have become a hot topic in the field of tumor treatment, thus leading a new direction for exploring the targets of cancer-like PH.
Pulmonary vascular remodeling and cancer-like growth are summarized in Figure 1.
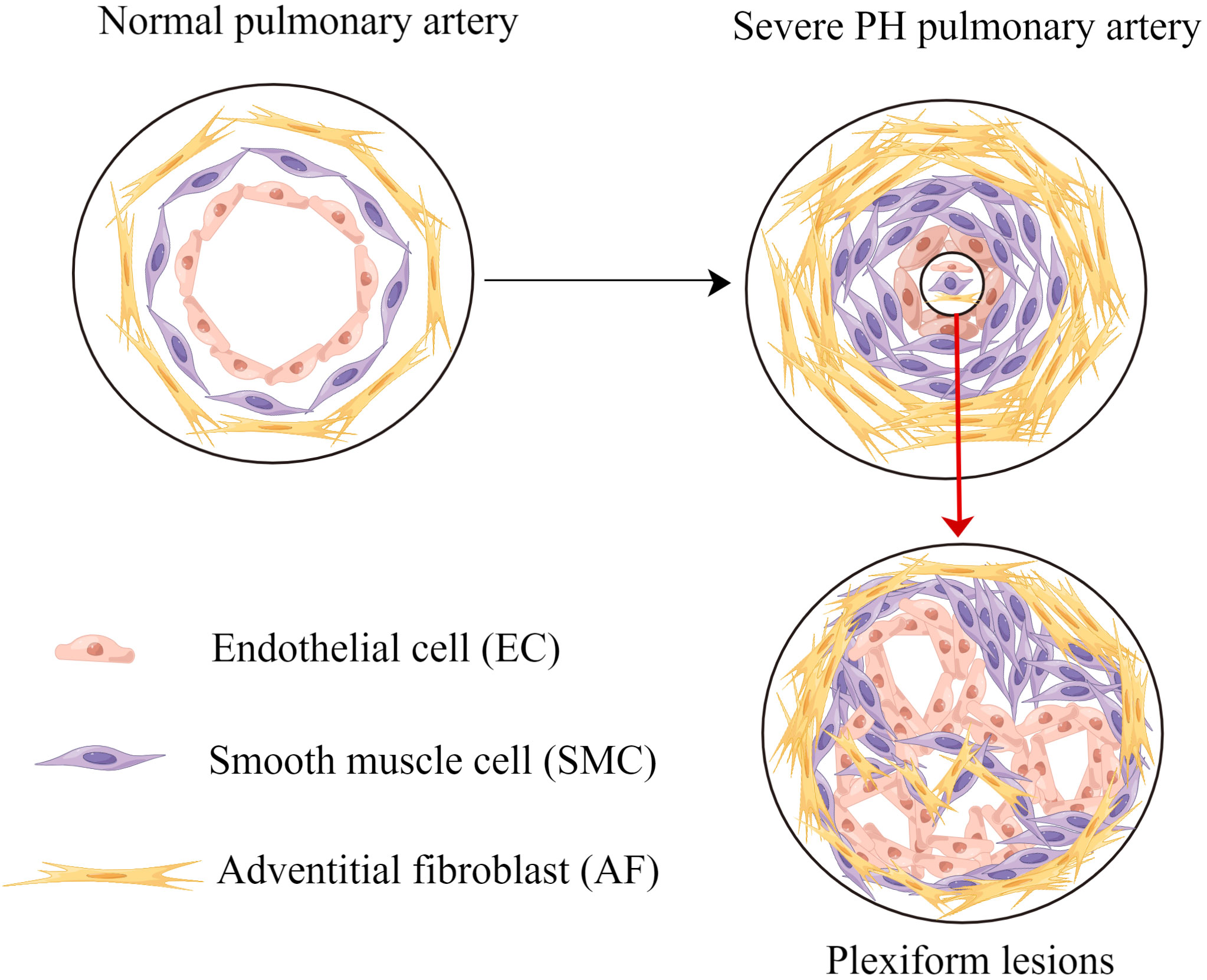
Figure 1 Pulmonary vascular remodeling and cancer-like growth. Pulmonary vascular remodeling causes endothelial cells apoptosis resistance, smooth muscle cells proliferation and adventitial fibroblasts proliferation. Without effective intervention, these processes lead to plexiform lesions and severe PH. PH, pulmonary hypertension.
Pathological histology in patients and animal models of PH shows various degrees of inflammatory infiltration around the pulmonary vasculature. Inflammatory cells like T cells, macrophages, dendritic cells (DCs) and natural killer (NK) cells are observed around small pulmonary arteries. Abnormally high levels of circulating cytokines and chemokines are detected in peripheral blood (11, 30–32). In the past, most studies focus on macrophages, neutrophils, and DCs, to explore inflammatory microenvironment (33–35). Nowadays, more and more studies suggest that MDSCs could also play an important role in inflammatory microenvironment (17, 36–38). Under physiological conditions, MDSCs are important immune cells for hematopoietic stem cells to maintain homeostasis (39). Under pathological conditions, recruitment of MDSCs from circulating blood to the outer membrane of pulmonary artery may be an important mechanism for PVR and PH progression (17, 38). Thus, there is an imperative need to explore MDSCs in PH, and targeting MDSCs may provide clinical guidance for the treatment of PH.
Myeloid-derived suppressor cells
MDSCs are a heterogeneous population of immature cells derived from myeloid progenitors with potent immunosuppressive activity (40, 41). In healthy mice, MDSCs constitute 20-30% of normal bone marrow (BM) cells and 2-4% of all nucleated splenocytes (42, 43). In normal myelopoiesis, immature myeloid cells differentiate into neutrophils and monocytes (44, 45). However, in pathological and chronic inflammatory conditions, such as tumors, chronic infections, and autoimmune diseases, a block in the normal myeloid differentiation occurs, and the persistent stimulation of myelopoiesis results in the expansion of MDSCs (41, 46). Thus, MDSCs in the BM are increased and released into the peripheral blood, and then migrate into various tissues of the body, especially around the lesions, to exert immunosuppressive functions.
Although MDSCs have been recognized for almost 30 years (47), the functional importance of MDSCs in the immune system is appreciated recently. MDSCs have been shown to be a potentially promising and well-tolerated therapeutic approach in tumor (48–50). The role of MDSCs in the PH process has not been fully illustrated yet. Elucidation of the immunosuppressive function and manipulation of the MDSCs phenotypes are therefore essential to fully understand.
Phenotype of MDSCs
There are two subgroups of MDSCs according to their cell origins: polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). In human, CD11b and CD33 are used as pan markers for MDSCs (41). However, CD11b and CD33 are expressed not only in PMN-MDSCs and M-MDSCs but also in neutrophils and monocytes, so further studies on surface markers are required to distinguish specific cell populations of MDSCs (51, 52). The accepted markers of PMN-MDSCs in human are CD33+CD11b+CD14-CD15+/CD66b+ (41). In 2016, Condamine reported that LOX-1 was practically undetectable in neutrophils in the peripheral blood of healthy donors, whereas 5-15% of total neutrophils in patients with cancer and 15-50% of neutrophils in tumor tissues were LOX-1(+) (53). LOX-1 is a class E scavenger receptor expressed on macrophages, chondrocytes, ECs and smooth muscle cells (54). LOX-1(+) neutrophils have potent immunosuppressive activity, up-regulation of ER stress and other biochemical characteristics of PMN-MDSCs, so LOX-1(+) is proposed as a specific marker for human PMN-MDSCs (53). CD13hi neutrophil-like MDSCs are found to be more immunosuppressive-active than CD13lo neutrophil-like MDSCs in human pancreatic cancer, and CD13hi is associated with poor prognosis, suggesting CD13hi may be a new marker for PMN-MDSCs (55). Human M-MDSCs are described as CD33+CD11b+CD14+CD15-HLA-DR-/lo (41). Monocytes, macrophages and DCs are mononuclear phagocytes that express major histocompatibility complex (MHC) class II molecules, whereas M-MDSCs usually lack MHC class II (56, 57). S100A9 is a new marker for monocytic human MDSCs (58). In humans, CD84 is expressed in both M-MDSCs and PMN-MDSCs, and CD84 has been identified as a marker of MDSCs in human cancer (40)
In mice, CD11b+ and Gr-1+ are used as pan markers, and the marker of PMN-MDSCs is CD11b+Ly6G+Ly6Clow, whereas that of M-MDSCs is CD11b+Ly6G-Ly6Chi (40, 41). In models of inflammatory bowel diseases and tumor-bearing mice, the CD49d+ subset of MDSCs is mainly monocytic and strongly suppress proliferation of antigen-specific T cells via a nitric oxide-dependent mechanism, which are similar to Gr-1(dull/int.) MDSCs. Thus, CD49d may potentially be a new marker to replace Gr-1 (59). Secreted protein acidic and rich in cysteine (SPARC) is a matrix protein, which can specifically control MDSCs suppressive activity, and SPARC is proposed as a new potential marker of MDSCs (60). CD84hi MDSCs exhibit T cell-suppressive capacity and increased reactive oxygen species (ROS) production, thus CD84 has also been identified as a marker of MDSCs in cancer of mice (40).
The human and mouse MDSCs phenotypes are presented in Table 1.
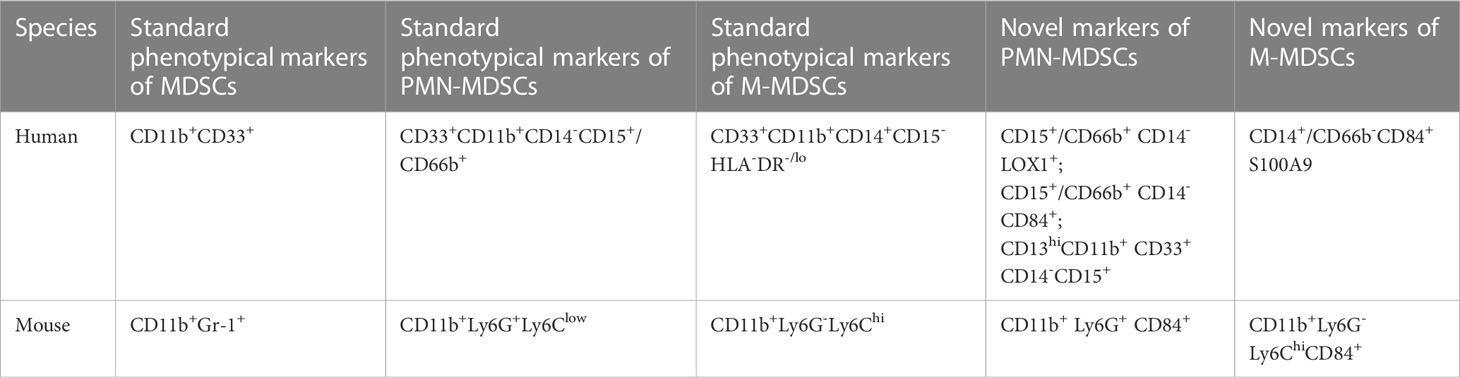
Table 1 Human and mouse phenotypes of MDSCs.
In rats, CD11b/c and His48 are used as pan-MDSC markers (61). Literature on rat MDSCs is limited and MDSCs are not uniformly labeled. We find that approximately 13 papers report MDSCs in rats by searching PubMed and Web of Science (61–73). CD11b/c+ and His48+ cells were first used as markers of MDSCs in T9 glioma rats in 2002 (61). Among the 13 studies, four use CD11b/c+ and His48+ as markers of MDSCs (61, 63, 64, 67), and one use CD11b/cint and His48hi (69). However, CD11b/c and His48 are expressed not only on MDSCs, but on neutrophils, monocytes, macrophages and DCs. Some studies use CD172a as a myeloid cell marker instead of CD11b/c (68), and some use CD161 as an alternative indicator of His48 in MDSCs (71). The phenotypes of rat MDSCs are summarized in Table 2. The studies on MDSCs in rat models are limited, and there are almost no specific markers to distinguish PMN-MDSCs and M-MDSCs. Therefore, further studies are needed to identify the cell markers of PMN-MDSCs and M-MDSCs in rats.
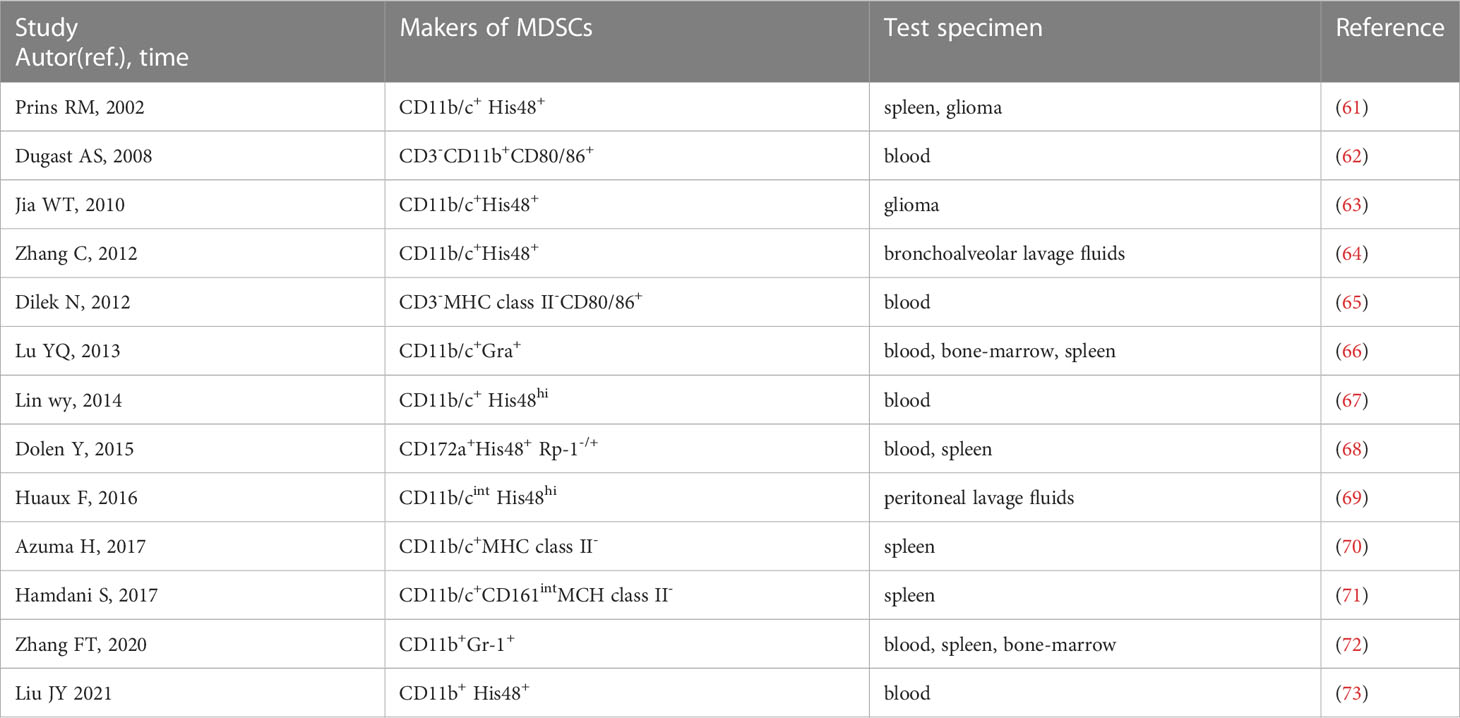
Table 2 Rat phenotypes of MDSCs.
Immunomodulatory functions of MDSCs in PH
The presence of MDSCs is confirmed in clinical patient samples and mouse models of PH (17, 36). MDSCs are significantly increased and positively correlated with mean pulmonary artery pressure in the PH patients group compared to the control group (36). In addition, pulmonary vascular muscularization, right ventricular remodeling and the worsening of PH are associated with the increase in pulmonary MDSCs, particularly PMN-MDSC (37). PMN-MDSCs mediate immunosuppressive effects through upregulation of arginase 1(Arg 1), ROS and prostaglandin E2, whereas M-MDSCs mediate the capacity through increasing expression of nitric oxide (NO), immunosuppressive cytokines including interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), and immune regulatory molecules like programmed death-ligand 1 (PD-L1) (40). Increased levels of Arg 1, ROS, inducible nitric oxide synthase (iNOS) and TGF-β exacerbate the progression of PH (74–78). Group 3 PH is associated with lung diseases and/or hypoxia, and it is found the MDSCs are dominated by PMN-MDSCs, which exert the immune suppressive function through Arg1 and ROS (36, 37, 40). It is suggested that MDSCs are important constituents of immune microenvironment with a pivotal role in PH progression (38). Next, we focus on the chemokines, cytokines, T cells, NK cells and molecular mechanisms to explore the immune-regulatory functions of MDSCs in PH, which may provide us with new strategy for treatment of PH.
MDSCs recruitment and activation: Cytokines and chemokines
Chemokines and cytokines play a pivotal role on MDSCs recruitment and activation during PH progression, which are summarized in Figure 2. High levels of CXCL12 and CXCR4 are found in the lung tissues of PH patients, and PI3K/Akt signaling mediates CXCL12/CXCR4 regulation of proliferation and cell cycle progression in PASMCs, thus leading to PVR (79). Concurrently, the CCL2/CCR2 and CCL5/CCR5 pathways are necessary for cooperation between macrophages and PASMCs to initiate and expand PASMCs in migration and proliferation during PH development (80, 81). MDSCs express a wide range of chemokine receptors, including CCR2, CXCR2 and CXCR4 (82). In PH, the migration of MDSCs from BM to lesions may mediate by chemokines receptor/chemokine signaling, such as CXCL12/CXCR4, CXCL2/CXCR2 and CCL2/CCR2. In PH mice models, Bryant et al. find that PMN-MDSCs are transported to the lung through the chemokine receptor CXCR2, and promote the disease development (17). Deletion of CXCR2 in myeloid cells attenuates the recruitment of PMN-MDSCs to the lung microenvironment, and therefore inhibits PVR, and protects against PH (17, 83). In tumors, CXCR4 overexpression promotes infiltration of MDSCs in lung tissues. It will accelerate lung cancer progression and promote lung metastasis from other primary tumors (84–86). Recently, it has been suggested that targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex enhances anti-PD-L1 immunotherapy (84). Cytokines are significantly increased in patients with PH. IL-6, IL-1β and IL-18 can induce the proliferation, migration, and differentiation of pulmonary vascular cells, thereby promoting PVR (11, 87–89). Meanwhile, cytokines may promote the recruitment of MDSCs, exacerbate the inflammatory response in the blood vessels, and aggravate the disease. Thus, cytokines, chemokines and MDSCs contribute to the formation of the immune microenvironment of PH and play a key role in the pathogenesis of PH. Targeting cytokines and chemokines to inhibit MDSCs infiltration in PH is little studied. Fortunately, there are more and deeper studies in tumors. Targeting CXCR4 or CXCR2 can reduce MDSCs infiltration, which contributes to the inhibition of tumor growth and metastasis (90, 91). Targeting IL-6 or IL-6 receptors for tumor immunotherapy can block MDSCs-mediated immunosuppression (92). Therefore, targeting chemokines and cytokines might be a potential new method for reversing PVR in PH.
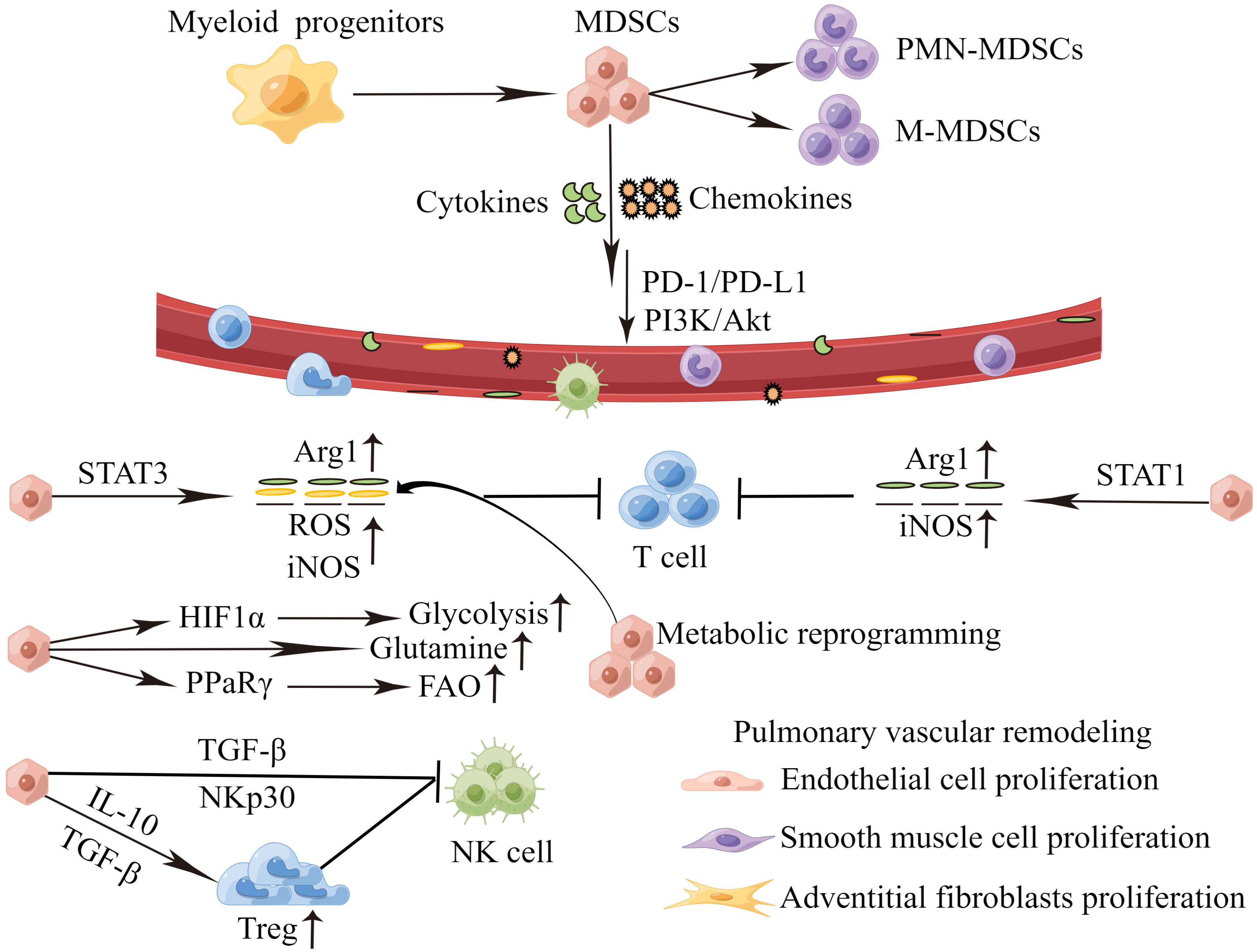
Figure 2 MDSCs recruitment and activation during PH progression. Perivascular immune cells constantly release inflammatory mediators, and the persistent stimulation of myelopoiesis results in the expansion of MDSCs in the BM. The MDSCs migrate to the pulmonary vascular site through the interaction between CCR,CXCR and respective chemokines (CCL,CXCL). In the PH microenvironment, MDSCs are activated and strongly inhibit an anti-inflammation reactivity of T cells and NK cells via various mechanisms. which in turn cause proliferation of endothelial cells, smooth muscle cells and adventitial fibroblasts, inducing pulmonary vascular remodeling. MDSCs, myeloid-derived suppressor cells; PMN-MDSCs, polymorphonuclear MDSCs; M-MDSCs, monocytic MDSCs; PD-1/PD-L1, programmed death 1 and programmed death-ligand 1; PI3K-Akt, phosphatidylinositol 3-kinase/Akt. Arg1, arginase 1; ROS, reactive oxygen species; iNOS, inducible nitric oxide synthase; HIF1α, hypoxia-inducible factor 1α; PPaRγ, peroxisome proliferator-activated receptor γ; FAO, fatty acid oxidation; TGF-β, transforming growth factor-β; IL-10, interleukin-10; NK cell, natural killer cells.
Target cells of MDSCs: T cells and NK cells
The T-cell immune response has a protective effect in PH, and an intact T-cell immune system is important in protecting against pulmonary angioproliferation (93). Athymic nude rats treated with the vascular endothelial growth factor (VEGF) receptor blocker SU5416 developed severe PH and PVR under normoxic conditions, whereas normal rats may not develop severe PH under normoxic conditions (94). Similarly, the reduction and deficiency of CD8+ cytotoxic T cells exacerbate the risk of death in PH patients (95). Regulatory T cells (Tregs) are a naturally occurring subpopulation of T cells with immunosuppressive functions, and Tregs primarily maintain autoantigenic immune tolerance and effectively control the exacerbation of inflammatory responses (96–98). Normally, Tregs regulate immune cells by controlling IL-2 availability, inhibitory receptors, and secretion of cytokines such as IL-10 and TGF-β, which may maintain immune homeostasis and suppress autoimmune damage (99). Tregs are dysfunctional in human patients and animal models of PH and may contribute to the development and progression of the disease (100). In chronic inflammatory diseases of the lungs, MDSCs can attenuate the role of helper T cells and cytotoxic T cells and exacerbate inflammatory cell infiltration around the pulmonary vasculature (101). MDSCs can likewise induce Tregs amplification, but the amplified Tregs exert a negative regulation of immunity (102), which promotes immune escape in tumors and autoimmune diseases. These factors may contribute to the possibility that MDSCs promote PH development.
NK cells are cytotoxic lymphocytes that are critical to the innate immune system and can recognize and rapidly kill target cells (103, 104). Emerging evidence indicates that MDSCs can interact with NK cells and regulate their functions (105). MDSCs can strongly inhibit anti-tumor immune responses of NK cells and promote the progression of tumors (106, 107). MDSCs suppress NK cells activation by inducing Tregs (108). In a mouse model of orthotopic liver cancer-bearing, it is shown that downregulation of NK cells function is inversely correlated with the marked increase in MDSCs in the liver and spleen, and MDSCs inhibit cytotoxicity, NKG2D expression and IFN-γ production of NK cells through membrane-bound TGF-β (109). Alternatively, co-culture of MDSCs and NK cells from hepatocellular carcinoma patients reveals that MDSCs-mediated inhibition of NK cell function is dependent mainly on the NKp30 on NK cells (110). MDSCs have an inhibitory effect on NK cells, mainly promoted by TGF-β or Tregs. NK cells impairment is a feature of PH and contributes to PVR in animal models of the disease (111). A prospective survival study of PH patients has confirmed a positive correlation between the number of NK cells and short-term survival in PH patients, suggesting that deficiencies in NK cells might be associated with an increased risk of death in PH patients (95). However, the cell interaction of MDSCs, NK cells and its molecular mechanisms have not been reported in PH yet, which needs further researches.
Molecular mechanisms of MDSCs: Arginine, iNOS and metabolic mechanisms
It is well known that MDSCs regulate immune response through a variety of mechanisms, such as arginase, iNOS activation and energy metabolic dysregulation (112). MDSCs highly express the immunosuppressive molecule Arg 1. L-Arg depletion by MDSCs blocked the re-expression of CD3zeta in stimulated T cells and inhibited antigen-specific proliferation of OT-1CD8+ and OT-2 CD4+ T cells, which will impair T-cell functions and affect T-cell-mediated immune responses (113). MDSCs are detected in the peripheral circulation of patients with PH, and active MDSCs expression increases transcripts for Arg 1 (36). Arginase activity and alterations in arginine metabolic pathways have been implicated in the pathophysiology of PH (114). Arginase activity is elevated almost two-fold (p=0.07) in patients with PH (74). Dysregulation of arginine metabolism contributes to endothelial dysfunction and PH in sickle cell disease and is strongly associated with prospective patient mortality (115). Arginase inhibitors can reduce PVR and collagen deposition, and then prevent bleomycin-induced neonatal PH in rats, and may prevent inflammation and remodeling in a guinea pig model of chronic obstructive pulmonary disease (116, 117). Arginase catalyzes the degradation of L-arginine to L-ornithine and urea. The decrease of L-arginine will impair T-cell functions and decrease NO production, and may exacerbate PVR and accelerate PH development.
Three distinct genes encode three NOS isoforms: neuronal nitric oxide synthase (nNOS), endothelial nitric oxide synthase (eNOS), and iNOS (118, 119). MDSCs exerted their inhibitory function on T cells in an iNOS-dependent manner (120). iNOS is an enzyme that catalyzes the formation of NO and citrulline from L-arginine, which can lead to the block of T-cell synthesis by reducing L-arginine, and can also inhibit the functions of T cells by producing excessive NO (121, 122). In hypoxic PH, the iNOS is found to be upregulated and involved in the formation of PH (123, 124). Mice lacking iNOS are protected against emphysema and PH (125). In a smoke-exposed mice study, iNOS expression in BM-derived cells drives pulmonary vascular remodeling (125). iNOS deletion in myeloid cells confers protection against PH and provides evidence for an iNOS-dependent communication between M2-like macrophages and PASMCs in underlying pulmonary vascular remodeling (126). Treatment of wild-type mice with the iNOS inhibitor N (6)- (1-iminoethyl)-L-lysine (L-NIL) prevents structural and functional alterations in the lung vasculature and alveoli and reverses emphysema and PH (127). In hypoxia-induced PH, iNOS may release large amounts of NO and damage the vascular endothelium, and the endothelial damage will diminish NO bioavailability (75, 128, 129). MDSCs can promote immune suppression by the production of ROS (40). In PH, the overproduction of ROS contributes to pulmonary vasoconstriction, muscularization of pulmonary arterioles, perivascular fibrosis and PVR (130). MDSCs highly express iNOS and produce ROS that may exacerbate PVR through either a direct or an indirect mechanism.
Metabolic dysregulation has emerged as a major area of research in the pathobiology of PH (131). Compared to that of healthy individuals, the microenvironment of PH has different metabolic features, such as excessive intracellular glucose uptake, increased glycolytic metabolism, insulin resistance, and alterations in high-density lipoprotein (HDL), cholesterol and leptin (24, 132). The metabolism of MDSCs, such as glycolysis, fatty acid oxidation and amino acid metabolism, is reprogrammed in the tumor microenvironment (40). In tumor, the MDSCs metabolic reprogramming enhances the immunosuppressive activity of Arg1 and iNOS, which will lead to apoptosis of effector T cells and suppression of cell proliferation, and promote tumor proliferation and metastasis (133, 134). Under hypoxic conditions, the activation of hypoxia- inducible factor 1α (HIF-1α) induces the switch from oxidative phosphorylation to glycolysis in MDSCs (40). The activation of the HIF-1α pathway promotes the immune suppressive activity of MDSCs (135). Blocking lactate production in tumor cells or deleting HIF-1α in MDSCs reverse anti-tumor T-cell responses and effectively inhibite tumor progression after radiotherapy for pancreatic cancer patients (136). Lipid metabolism plays a key role in the differentiation and functions of MDSCs. MDSCs exhibit fatty acid uptake and increase fatty acid oxidation (FAO), which support the immunosuppressive functions of MDSCs (137). Tumor-infiltrating MDSCs in mice may prefer fatty acid oxidation (FAO) as a primary energy source, while treatment with FAO inhibitors improves anti-tumor immunity (138). Mouse and human PMN-MDSCs upregulate fatty acid transport protein 2 (FATP2), and the selective pharmacological inhibition of FATP2 abrogates the activity of PMN-MDSCs and substantially delays tumor progression (139). MDSCs can regulate T cells functions by depriving the essential metabolites, such as arginine, tryptophan and cysteine from the microenvironment (40). MDSCs can deplete arginine through upregulation of Arg1 and reduce tryptophan through upregulation of indoleamine 2,3-dioxygenase, which suppress T-cell proliferation and activation (140, 141). In a triple-negative breast cancer immunotherapy-resistant model, targeting glutamine metabolism also significantly inhibites the production and recruitment of MDSCs and suppresses tumor growth (142). Thus, MDSCs may also promote disease development by suppressing T-cell functions through metabolic pathways in PH.
Conclusion
The complex changes in cytokines, chemokines, and immune cells in PH and their association with PVR suggest that immune mechanisms play an important role in PH. The cancer-like growth characteristic is one of the important features of severe PH. The prognosis of severe PH is even worse than most of the cancers in children. Hence, it is urgent to find a novel therapeutic strategy for the cancer-like PH. Tumor immunotherapy has been effectively applied in clinical practice by virtue of its specificity and targeting. Recently, there is ample evidence to demonstrate that tumor immunotherapy can be effectively improved by targeting MDSCs. Just as in tumors, MDSCs play a crucial role in development and progression of PH. Therefore, targeting MDSCs may be a potential protocol in the treatment of PH to arrest PVR, including blocking the migration, recruitment, activity and metabolism, and promoting the maturation of MDSCs to restore immune homeostasis. We believe that the MDSCs‐targeting treatment can provide a first-line survival opportunity for patients with PH, especially severe PH.
Author contributions
Conceptualization HZ. Original draft preparation HZ, QL, YL, XT. Review LG, HL. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Nature Science Foundation of China (No. 82170067 and No. 82241005), Thanks for the financial support.
Acknowledgments
We sincerely thank the free drawing support provided by the Home for Research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J (2022) 43(38):3618–731. doi: 10.1093/eurheartj/ehac237
2. Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther (2001) 92(1):1–20. doi: 10.1016/s0163-7258(01)00157-7
3. Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med (2013) 91(3):297–309. doi: 10.1007/s00109-013-0998-0
4. Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiology-Lung Cell Mol Physiol (2007) 293(1):L1–8. doi: 10.1152/ajplung.00378.2006
5. Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res (2009) 10(1):1–9. doi: 10.1186/1465-9921-10-95
6. Gomes MT, Bai Y, Potje SR, Zhang L, Lockett AD, Machado RF. Signal transduction during metabolic and inflammatory reprogramming in pulmonary vascular remodeling. Int J Mol Sci (2022) 23(5):2410. doi: 10.3390/ijms23052410
7. Dai Z, Zhu MM, Peng Y, Jin H, Machireddy N, Qian Z, et al. Endothelial and smooth muscle cell interaction via FoxM1 signaling mediates vascular remodeling and pulmonary hypertension. Am J Respir Crit Care Med (2018) 198(6):788–802. doi: 10.1164/rccm.201709-1835oc
8. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J (2018) 53:1801887. doi: 10.1183/13993003.01887-2018
9. Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR, et al. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med (2021) 384(13):1204–15. doi: 10.1056/NEJMoa2024277
10. Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med (2013) 369(4):319–29. doi: 10.1056/nejmoa1209657
11. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res (2014) 115(1):165–75. doi: 10.1161/circresaha.113.301141
12. Wang RR, Yuan TY, Wang JM, Chen YC, Zhao QL, Li MT, et al. Immunity and inflammation in pulmonary arterial hypertension: from pathophysiology mechanisms to treatment perspective. Pharmacol Res (2022) 180:106238. doi: 10.1016/j.phrs.2022.106238
13. Placek K, Schultze JL, Aschenbrenner AC. Epigenetic reprogramming of immune cells in injury, repair, and resolution. J Clin Invest (2019) 129(8):2994–3005. doi: 10.1172/JCI124619
14. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
15. Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc (2015) 12(Suppl 2):S169–75. doi: 10.1513/AnnalsATS.201503-126AW
16. Kim R, Emi M, Tanabe K. Cancer immunosuppression and autoimmune disease: beyond immunosuppressive networks for tumour immunity. Immunology (2016) 119(2):254–64. doi: 10.1111/j.1365-2567.2006.02430.x
17. Bryant AJ, Shenoy V, Fu C, Marek G, Lorentsen KJ, Herzog EL, et al. Myeloid-derived suppressor cells are necessary for development of pulmonary hypertension. Am J Respir Cell Mol Biol (2018) 58(2):170–80. doi: 10.1165/rcmb.2017-0214oc
18. Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol (2001) 195(3):367–74. doi: 10.1002/path.953
19. Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, et al. Plexiform lesions in pulmonary arterial hypertension. Am J Pathol (2011) 179(1):167–79. doi: 10.1016/j.ajpath.2011.03.040
20. Carman BL, Predescu D, Machado R, Predescu SA. Plexiform arteriopathy in rodent models of pulmonary arterial hypertension. Am J Pathol (2019) 189(6):1133–44. doi: 10.1016/j.ajpath.2019.02.005
21. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation (2010) 121(25):2747–54. doi: 10.1161/circulationaha.109.92768
22. Rai PR, Cool CD, King JAC, Stevens T, Burns N, Winn RA, et al. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med (2008) 178(6):558–64. doi: 10.1164/rccm.200709-1369pp
23. Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci (2016) 17(5):761. doi: 10.3390/ijms17050761
24. Guignabert C, Tu Ly, Hiress ML, Ricard N, Sattler C, Seferian A, et al. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev (2013) 22(130):543–51. doi: 10.1183/09059180.00007513
25. Sakao S, Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol (2011) 147(1):4–12. doi: 10.1016/j.ijcard.2010.07.003
26. Kurakula K, Smolders VFED, Tura-Ceide O, Jukema JW, Quax PHA, Goumans MJ. Endothelial dysfunction in pulmonary hypertension: cause or consequence? Biomedicines (2021) 9(1):57. doi: 10.3390/biomedicines9010057
27. Cool CD, Kuebler WM, Bogaard HJ, Spiekerkoetter E, Nicolls MR, Voelkel NF. The hallmarks of severe pulmonary arterial hypertension: the cancer hypothesis-ten years later. Am J Physiology-Lung Cell Mol Physiol (2020) 318(6):L1115–30. doi: 10.1152/ajplung.00476.2019
28. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
29. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/nejmoa1200690
30. Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation (2012) 126(4):455–67. doi: 10.1161/circulationaha.112.10317
31. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med (2012) 186(3):261–72. doi: 10.1164/rccm.201201-0164OC
32. Yoo HHB, Marin FL. Treating inflammation associated with pulmonary hypertension: an overview of the literature. Int J Gen Med (2022) 15:1075–83. doi: 10.2147/IJGM.S295463
33. Chen S, Yan D, Qiu A. The role of macrophages in pulmonary hypertension: pathogenesis and targeting. Int Immunopharmacol (2020) 88:106934. doi: 10.1016/j.intimp.2020.106934
34. Van Uden D, Boomars K, Kool M. Dendritic cell subsets and effector function in idiopathic and connective tissue disease-associated pulmonary arterial hypertension. Front Immunol (2019) 10:11. doi: 10.3389/fimmu.2019.00011
35. Taylor S, Isobe S, Cao AQ, Contrepois K, Benayoun BA, Jiang LH, et al. Endogenous retroviral elements generate pathologic neutrophils in pulmonary arterial hypertension. Am J Respir Crit Care Med (2022) 206(8):1019–34. doi: 10.1164/rccm.202102-0446OC
36. Yeager ME, Nguyen CM, Belchenko DD, Colvin KL, Takatsuki S, Ivy DD, et al. Circulating myeloid-derived suppressor cells are increased and activated in pulmonary hypertension. Chest (2012) 141(4):944–52. doi: 10.1378/chest.11-0205
37. Fu C, Lu Y, Williams MA, Brantly ML, Ventetuolo CE, Morel LM, et al. Emergency myelopoiesis contributes to immune cell exhaustion and pulmonary vascular remodeling. Br J Pharmacol (2019) 178(1):187–202. doi: 10.1111/bph.14945
38. Bryant A, Mehrad B, Brusko T, West J, Moldawer L. Myeloid-derived suppressor cells and pulmonary hypertension. Int J Mol Sci (2018) 19(8):2277. doi: 10.3390/ijms19082277
39. Lee HJ, Ko JH, Kim HJ, Jeong HI, Oh JY. Mesenchymal stromal cells induce distinct myeloid-derived suppressor cells in inflammation. JCI Insight (2020) 5(12):e136059. doi: 10.1172/jci.insight.136059
40. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21(8):485–98. doi: 10.1038/s41577-020-00490-y
41. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
42. Esher SK, Fidel PL, Noverr MC. Candida/Staphylococcal polymicrobial intra-abdominal infection: pathogenesis and perspectives for a novel form of trained innate immunity. J Fungi (2019) 5(2):37. doi: 10.3390/jof5020037
43. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
44. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature (2000) 404(6774):193–7. doi: 10.1038/35004599
45. Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev (2010) 238(1):37–46. doi: 10.1111/j.1600-065x.2010.00963.x
46. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann NY Acad Sci (2014) 1319(1):47–65. doi: 10.1111/nyas.12469
47. Young MR, Newby M, Wepsic HT. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res (1987) 47:100–5.
48. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
49. Ma TM, Renz BW, Ilmer M, Koch D, Yang YH, Werner J, et al. Myeloid-derived suppressor cells in solid tumors. Cells (2022) 11(2):310. doi: 10.3390/cells11020310
50. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol (2018) 9:398. doi: 10.3389/fimmu.2018.00398
51. Zhang Y, Wilt E, Lu X. Human isogenic cell line models for neutrophils and myeloid-derived suppressor cells. Int J Mol Sci (2020) 21(20):7709. doi: 10.3390/ijms21207709
52. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood (2019) 133(20):2159–67. doi: 10.1182/blood-2018-11-844548
53. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
54. Taye A, El-Sheikh AAK. Lectin-like oxidized low-density lipoprotein receptor 1 pathways. Eur J Clin Invest (2013) 43(7):740–5. doi: 10.1111/eci.12092
55. Zhang J, Xu X, Shi M, Chen Y, Yu D, Zhao C, et al. CD13hi neutrophil-like myeloid-derived suppressor cells exert immune suppression through arginase 1 expression in pancreatic ductal adenocarcinoma. OncoImmunology (2017) 6(2):e1258504. doi: 10.1080/2162402x.2016.1258504
56. Cardoso CC, Matiollo C, Pereira CHJ, Fonseca JS, Alves HEL, da Silva OM, et al. Patterns of dendritic cell and monocyte subsets are associated with disease severity and mortality in liver cirrhosis patients. Sci Rep (2021) 11(1):5923. doi: 10.1038/s41598-021-85148-y
57. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
58. Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, et al. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology (2012) 136(2):176–83. doi: 10.1111/j.1365-2567.2012.03566.x
59. Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol (2010) 185(1):203–10. doi: 10.4049/jimmunol.0903573
60. Sangaletti S, Talarico G, Chiodoni C, Cappetti B, Botti L, Portararo P, et al. SPARC Is a new myeloid-derived suppressor cell marker licensing suppressive activities. Front Immunol (2019) 10:1369. doi: 10.3389/fimmu.2019.01369
61. Prins RM, Scott GP, Merchant RE, Graf MR. Irradiated tumor cell vaccine for treatment of an established glioma. II. expansion of myeloid suppressor cells that promote tumor progression. Cancer Immunol Immunother (2002) 51(4):190–9. doi: 10.1007/s00262-002-0270-x
62. Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol (2008) 180(12):7898–906. doi: 10.4049/jimmunol.180.12.7898
63. Jia WT, Jackson-Cook C, Graf MR. Tumor-infiltrating, myeloid-derived suppressor cells inhibit T cell activity by nitric oxide production in an intracranial rat glioma + vaccination model. J Neuroimmunol (2010) 223(1-2):20–30. doi: 10.1016/j.jneuroim.2010.03.011
64. Zhang C, Lei GS, Shao S, Jung HW, Durant PJ, Lee CH. Accumulation of myeloid-derived suppressor cells in the lungs during pneumocystis pneumonia. Infect Immun (2012) 80(10):3634–41. doi: 10.1128/iai.00668-12
65. Dilek N, Poirier N, Usal C, Martinet B, Blancho G, Vanhove B. Control of transplant tolerance and intragraft regulatory T cell localization by myeloid-derived suppressor cells and CCL5. J Immunol (2012) 188(9):4209–16. doi: 10.4049/jimmunol.1101512
66. Lu YQ, Gu LH, Zhang Q, Jiang JK, Mou HZ. Hypertonic saline resuscitation contributes to early accumulation of circulating myeloid-derived suppressor cells in a rat model of hemorrhagic shock. Chin Med J (Engl) (2013) 126(7):1317–22. doi: 10.3760/cma.j.issn.0366-6999.20122549
67. Lin WY, Hsieh CC, Yang TY, Chen ML, Huang LY, Lin YP, et al. Transient increase in circulating myeloid-derived suppressor cells after partial bladder outlet obstruction. J Urol (2014) 192(5):1569–73. doi: 10.1016/j.juro.2014.05.045
68. Dolen Y, Gunaydin G, Esendagli G, Guc D. Granulocytic subset of myeloid derived suppressor cells in rats with mammary carcinoma. Cell Immunol (2015) 295(1):29–35. doi: 10.1016/j.cellimm.2015.02.005
69. Huaux F, de Bousies VD, Parent MA, Orsi M, Uwambayinema F, Devosse R, et al. Mesothelioma response to carbon nanotubes is associated with an early and selective accumulation of immunosuppressive monocytic cells. Particle Fibre Toxicology. (2016) 13(1):46. doi: 10.1186/s12989-016-0158-0
70. Azuma H, Yoshida Y, Takahashi H, Ishibazawa E, Kobayashi H, Sakai H, et al. Liposomal microparticle injection can induce myeloid-derived suppressor cells (MDSC)-like cells in vivo. Immunopharmacol Immunotoxicol (2017) 39(3):140–7. doi: 10.1080/08923973.2017.1306867
71. Hamdani S, Thiolat A, Naserian S, Grondin C, Moutereau S, Hulin A, et al. Delayed and short course of rapamycin prevents organ rejection after allogeneic liver transplantation in rats. World J Gastroenterol (2017) 23(38):6962–72. doi: 10.3748/wjg.v23.i38.6962
72. Zhang FT, Liu F, Yu SF, Zhang GH, Sun XJ. Protective effect of curcumin on bone trauma in a rat model via expansion of myeloid derived suppressor cells. Med Sci Monitor: Int Med J Exp Clin Res (2020) 26:e924724. doi: 10.12659/MSM.924724
73. Liu JY, Toy R, Vantucci C, Pradhan P, Zhang ZJ, Kuo KM, et al. Bifunctional janus particles as multivalent synthetic nanoparticle antibodies (SNAbs) for selective depletion of target cells. Nano Lett (2021) 21(1):875–86. doi: 10.1021/acs.nanolett.0c04833
74. Morris CR, Morris SM, Hagar W, Warmerdam JV, Claster S, Kepka-Lenhart D, et al. Arginine therapy a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med (2003) 168(1):63–9. doi: 10.1164/rccm.200208-967OC
75. Lázár Z, Mészáros M, Bikov A. The nitric oxide pathway in pulmonary arterial hypertension: pathomechanism, biomarkers and drug targets. Curr Med Chem (2020) 27(42):7168–88. doi: 10.2174/0929867327666200522215047
76. Huetsch J, Suresh K, Shimoda L. Regulation of smooth muscle cell proliferation by NADPH oxidases in pulmonary hypertension. Antioxidants (2019) 8(3):56. doi: 10.3390/antiox8030056
77. Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, et al. Role of the TGF-β/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med (2008) 177(8):896–905. doi: 10.1164/rccm.200707-1083oc
78. Kurakula K, Hagdorn QAJ, van der Feen DE, Vonk Noordegraaf A, ten Dijke P, de Boer RA, et al. Inhibition of the prolyl isomerase Pin1 improves endothelial function and attenuates vascular remodelling in pulmonary hypertension by inhibiting TGF-β signalling. Angiogenesis (2021) 25(1):99–112. doi: 10.1007/s10456-021-09812-7
79. Wei L, Zhang B, Cao W, Xing H, Yu X, Zhu D. Inhibition of CXCL12/CXCR4 suppresses pulmonary arterial smooth muscle cell proliferation and cell cycle progression via PI3K/Akt pathway under hypoxia. J Recept Signal Transduct (2014) 35(4):329–39. doi: 10.3109/10799893.2014.984308
80. Abid S, Marcos E, Parpaleix A, Amsellem V, Breau M, Houssaini A, et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J (2019) 54(4):1802308. doi: 10.1183/13993003.02308-2018
81. Amsellem V, Lipskaia L, Abid S, Poupel L, Houssaini A, Quarck R, et al. CCR5 as a treatment target in pulmonary arterial hypertension. Circulation (2014) 130(11):880–91. doi: 10.1161/circulationaha.114.01075
82. Seo EH, Namgung JH, Oh CS, Kim SH, Lee SH. Association of chemokines and chemokine receptor expression with monocytic-Myeloid-Derived suppressor cells during tumor progression. Immune Netw (2018) 18(3):e23. doi: 10.4110/in.2018.18.e23
83. Oliveira AC, Fu C, Lu Y, Williams MA, Pi L, Brantly ML, et al. Chemokine signaling axis between endothelial and myeloid cells regulates development of pulmonary hypertension associated with pulmonary fibrosis and hypoxia. Am J Physiology-Lung Cell Mol Physiol (2019) 317(4):L434–44. doi: 10.1152/ajplung.00156.2019
84. Li Z, Wang Y, Shen Y, Qian C, Oupicky D, Sun M. Targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex to enhance anti–PD-L1 immunotherapy. Sci Adv (2020) 6(20):eaaz9240. doi: 10.1126/sciadv.aaz9240
85. Takahashi R, Amano H, Ito Y, Eshima K, Satoh T, Iwamura M, et al. Microsomal prostaglandin e synthase-1 promotes lung metastasis via SDF-1/CXCR4-mediated recruitment of CD11b+Gr1+MDSCs from bone marrow. Biomed Pharmacother (2020) 121:109581. doi: 10.1016/j.biopha.2019.109581
86. Li Z, Shen Y, Wang Y, Zhu L, Zhu C, Qian C, et al. Perfluorocarbon nanoemulsions for combined pulmonary siRNA treatment of lung metastatic osteosarcoma. Adv Ther (2019) 2(7):1900039. doi: 10.1002/adtp.201900039
87. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med (1995) 151(5):1628–31. doi: 10.1164/ajrccm.151.5.7735624
88. Florentin J, Zhao JS, Tai YY, Vasamsetti SB, O’Neil SP, Kumar R, et al. Interleukin-6 mediates neutrophil mobilization from bone marrow in pulmonary hypertension. Cell Mol Immunol (2021) 18(2):374–84. doi: 10.1038/s41423-020-00608-1
89. Udjus C, Cero FT, Halvorsen B, Behmen D, Carlson CR, Bendiksen BA, et al. Caspase-1 induces smooth muscle cell growth in hypoxia-induced pulmonary hypertension. Am J Physiology-Lung Cell Mol Physiol (2019) 316(6):L999–L1012. doi: 10.1152/ajplung.00322.2018
90. Wang J, Tannous BA, Poznansky MC, Chen H. CXCR4 antagonist AMD3100 (plerixafor): from an impurity to a therapeutic agent. Pharmacol Res (2020) 159:105010. doi: 10.1016/j.phrs.2020.105010
91. Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun (2018) 9(1):1685. doi: 10.1038/s41467-018-03966-7
92. Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. Il-6 as a major regulator of mdsc activity and possible target for cancer immunotherapy-sciencedirect. Cell Immunol (2020) 359:104254. doi: 10.1016/j.cellimm.2020.104254
93. Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration (2008) 75(3):272–80. doi: 10.1159/000111548
94. Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, et al. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med (2007) 175(12):1280–9. doi: 10.1164/rccm.200608-1189oc
95. Edwards AL, Gunningham SP, Clare GC, Hayman MW, Smith M, Frampton CM, et al. Professional killer cell deficiencies and decreased survival in pulmonary arterial hypertension. Respirology (2013) 18(8):1271–7. doi: 10.1111/resp.12152
96. Wadwa M, Klopfleisch R, Buer J, Westendorf AM. Targeting antigens to DEC-205 on dendritic cells induces immune protection in experimental colitis in mice. Eur J Microbiol Immunol (2016) 6(1):1–8. doi: 10.1556/1886.2015.00048
97. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
98. Gupta S. Immune homeostasis: regulatory T cells (Treg) and molecules. J Clin Immunol (2008) 28(6):617–8. doi: 10.1007/s10875-008-9259-3
99. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
100. Huertas A, Phan C, Bordenave J, Tu L, Thuillet R, Le Hiress M, et al. Regulatory T cell dysfunction in idiopathic, heritable and connective tissue-associated pulmonary arterial hypertension. Chest (2016) 149(6):1482–93. doi: 10.1016/j.chest.2016.01.004
101. Kolahian S, Öz HH, Zhou B, Griessinger CM, Rieber N, Hartl D. The emerging role of myeloid-derived suppressor cells in lung diseases. Eur Respir J (2016) 47(3):967–77. doi: 10.1183/13993003.01572-2015
102. Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen S. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol/Hematol (2011) 77(1):12–9. doi: 10.1016/j.critrevonc.2010.02.004
103. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
104. Robertson MJ, Ritz J. Biology and clinical relevance of human natural killer cells. Blood (1990) 76(12):2421–38. doi: 10.1182/blood.v76.12.2421.2421
105. Joshi S, Sharabi B. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol Ther (2022) 235:108114. doi: 10.1016/j.pharmthera.2022.108114
106. Pelosi A, Besi F, Tumino N, Merli P, Vacca P. NK cells and pmn-mdscs in the graft from g-csf mobilized haploidentical donors display distinct gene expression profiles from those of the non-mobilized counterpart. Front Immunol (2021) 12:657329. doi: 10.3389/fimmu.2021.657329
107. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol (2019) 10:771. doi: 10.3389/fimmu.2019.00771
108. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
109. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF- 1. J Immunol (2008) 182(1):240–9. doi: 10.4049/jimmunol.182.1.240
110. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50(3):799–807. doi: 10.1002/hep.23054
111. Hiltonn LR, Ratsep MT, VandenBroek MM, Jafri S, Laverty KJ, Mitchell M, et al. Impaired interleukin-15 signaling via BMPR2 loss drives natural killer deficiency and pulmonary hypertension. HYPERTENSION (2022) 79(11):2493–504. doi: 10.1161/HYPERTENSION.122.19178
112. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells (2020) 9(3):561. doi: 10.3390/cells9030561
113. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.can-04-0465
114. Morris CR. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA (2005) 294(1):81. doi: 10.1001/jama.294.1.81
115. Morris C, Gladwin M, Kato G. Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med (2008) 8(7):620–32. doi: 10.2174/156652408786241447
116. Grasemann H, Dhaliwal R, Ivanovska J, Kantores C, McNamara PJ, Scott JA, et al. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am J Physiology-Lung Cell Mol Physiol (2015) 308(6):L503–10. doi: 10.1152/ajplung.00328.2014
117. Pera T, Zuidhof AB, Smit M, Menzen MH, Klein T, Flik G, et al. Arginase inhibition prevents inflammation and remodeling in a Guinea pig model of chronic obstructive pulmonary disease. J Pharmacol Exp Ther (2014) 349(2):229–38. doi: 10.1124/jpet.113.210138
118. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33(7):829–37. doi: 10.1093/eurheartj/ehr304
119. Guix FX, Uribesalgo I, Coma M, Munoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol (2005) 762(2):126–52. doi: 10.1016/j.pneurobio.2005.06.001
120. Zhang W, Li J, Qi G, Tu G, Yang C, Xu M. Myeloid-derived suppressor cells in transplantation: the dawn of cell therapy. J Trans Med (2018) 16(1):19. doi: 10.1186/s12967-018-1395-9
121. Nathan C, Xie Q. Nitric oxide synthases: roles, tolls, and controls. Cell (1994) 78(6):915–8. doi: 10.1016/0092-8674(94)90266-6
122. Wang W, Zou W. Amino acids and their transporters in T cell immunity and cancer therapy. Mol Cell (2020) 80(3):384–95. doi: 10.1016/j.molcel.2020.09.006
123. Hampl V, Bíbová J, Baňasová A, Uhlík J, Miková D, Hniličková O, et al. Pulmonary vascular iNOS induction participates in the onset of chronic hypoxic pulmonary hypertension. Am J Physiology-Lung Cell Mol Physiol (2006) 290(1):L11–20. doi: 10.1152/ajplung.00023.2005
124. Xia X, Xu Z, Hu X, Wu C, Dai Y, Yang L. Impaired iNOS-sGC-cGMP signalling contributes to chronic hypoxic and hypercapnic pulmonary hypertension in rat. Cell Biochem Funct (2012) 30(4):279–85. doi: 10.1002/cbf.2796
125. Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, et al. Inducible NOS inhibition reverses tobacco-Smoke-Induced emphysema and pulmonary hypertension in mice. Cell (2011) 147(2):293–305. doi: 10.1016/j.cell.2011.08.035
126. Gredi C, Wu CY, Hadzic S, Pak O, Savai R, Kojonazarov B, et al. Myeloid-cell-specific deletion of inducible nitric oxide synthase protects against smoke-induced pulmonary hypertension in mice. Eur Respir J (2022) 59(4):2101153. doi: 10.1183/13993003.01153-2021
127. Fysikopoulos A, Seimetz M, Hadzic S, Knoepp F, Wu CY, Malkmus K, et al. Amelioration of elastase-induced lung emphysema and reversal of pulmonary hypertension by pharmacological iNOS inhibition in mice. Br J Pharmacol (2020) 171(1):152–71. doi: 10.1111/bph.15057
128. Bonartsev AP, D’yakonov KB, Postnikov AB, Medvedeva NA. Effect of chronic administration of aminoguanidine on vascular reactivity of the greater circulation in rats with monocrotaline-induced pulmonary hypertension. Biol Bull (2005) 32(3):258–63. doi: 10.1007/s10525-005-0099-x
129. Harrison DG. Endothelial function and oxidant stress. Clin Cardiol (1997) 20(11 Suppl 2):II-11-7. doi: 10.1002/j.1932-8737.1997.tb00007.x
130. Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, et al. Aldosterone inactivates the endothelin-b receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation (2012) 126(8):963–74. doi: 10.1161/circulationaha.112.09472
131. Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res (2014) 115(1):148–64. doi: 10.1161/circresaha.115.301130
132. Maron BA, Leopold JA, Hemnes AR. Metabolic syndrome, neurohumoral modulation and pulmonary arterial hypertension. Br J Pharmacol (2019) 177(7):1457–71. doi: 10.1111/bph.14968
133. Wang Y, Jia A, Bi Y, Wang Y, Liu G. Metabolic regulation of myeloid-derived suppressor cell function in cancer. Cells (2020) 9(4):1011. doi: 10.3390/cells9041011
134. OuYang LY, Wu XJ, Ye SB, Zhang R, Li ZL, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Trans Med (2015) 13(1):47. doi: 10.1186/s12967-015-0410-7
135. Travelli C, Consonni FM, Sangaletti S, Storto M, Morlacchi S, Grolla AA, et al. Nicotinamide phosphoribosyltransferase acts as a metabolic gate for mobilization of myeloid-derived suppressor cells. Cancer Res (2019) 79(8):1938–51. doi: 10.1158/0008-5472.can-18-1544
136. Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res (2020) 8(11):1440–51. doi: 10.1158/2326-6066.cir-20-0111
137. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. OncoImmunology (2017) 6(10):e1344804. doi: 10.1080/2162402x.2017.1344804
138. Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol (2019) 10:1399. doi: 10.3389/fimmu.2019.01399
139. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
140. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222(1):180–91. doi: 10.1111/j.1600-065x.2008.00608.x
141. Li A, Barsoumian HB, Schoenhals JE, Cushman TR, Caetano MS, Wang X, et al. Indoleamine 2,3-dioxygenase 1 inhibition targets anti-PD1-resistant lung tumors by blocking myeloid-derived suppressor cells. Cancer Lett (2018) 431:54–63. doi: 10.1016/j.canlet.2018.05.005
Keywords: myeloid-derived suppressor cells, pulmonary hypertension, immune microenvironment, cancer-like, cytokines, chemokines, metabolism
Citation: Zhang H, Li QW, Li YY, Tang X, Gu L and Liu HM (2023) Myeloid-derived suppressor cells and pulmonary hypertension. Front. Immunol. 14:1189195. doi: 10.3389/fimmu.2023.1189195
Received: 18 March 2023; Accepted: 18 May 2023;
Published: 07 June 2023.
Edited by:
Djuro Kosanovic, I.M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Stefan Hadzic, University of Giessen, GermanyAkylbek Sydykov, University of Giessen, Germany
Copyright © 2023 Zhang, Li, Li, Tang, Gu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Gu, Z3VsaW5nQHNjdS5lZHUuY24=; Han-Min Liu, bGl1aG1Ac2N1LmVkdS5jbg==