 Johannes Hirsch
Johannes Hirsch Günalp Uzun
Günalp Uzun Jan Zlamal1,2
Jan Zlamal1,2 Anurag Singh
Anurag Singh Tamam Bakchoul
Tamam Bakchoul
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 19 May 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1186000
This article is part of the Research Topic The Immunological Role of Platelet Activation in the Pathophysiology of COVID-19 View all 11 articles
Coronavirus disease 2019 (COVID-19) is known to commonly induce a thrombotic diathesis, particularly in severely affected individuals. So far, this COVID-19-associated coagulopathy (CAC) has been partially explained by hyperactivated platelets as well as by the prothrombotic effects of neutrophil extracellular traps (NETs) released from neutrophils. However, precise insight into the bidirectional relationship between platelets and neutrophils in the pathophysiology of CAC still lags behind. Vaccine-induced thrombotic thrombocytopenia (VITT) is a rare autoimmune disorder caused by auto-antibody formation in response to immunization with adenoviral vector vaccines. VITT is associated with life-threatening thromboembolic events and thus, high fatality rates. Our concept of the thrombophilia observed in VITT is relatively new, hence a better understanding could help in the management of such patients with the potential to also prevent VITT. In this review we aim to summarize the current knowledge on platelet-neutrophil interplay in COVID-19 and VITT.
Coronavirus disease 2019 (COVID-19) is caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The disease was initially recognized as a predominantly respiratory illness after its first appearance in the city of Wuhan, China in late 2019, but the presence of the virus at extrapulmonary sites and its fatal effects were subsequently demonstrated (1–3).
COVID-19 patients often suffer from coagulopathy in addition to mortality due to respiratory failure. These are mainly consequences of the prothrombotic state, especially in moderate and severe cases. Venous thromboembolism (VTE), thrombocytopenia and disseminated intravascular coagulation (DIC) were early described as common complications in SARS-CoV-2 infected patients (4–7). Agarwal et al. calculated the overall prevalence of VTE to be as high as 20.7%, with the risk being doubled in COVID-19 cases admitted to the intensive care unit (8). A large retrospective analysis of more than 370,000 cases from England found that 86% of hospitalized COVID-19 patients with VTE also suffered a concomitant pulmonary embolism, highlighting the coagulation-related risks in COVID-19 (9).
The pathophysiology of COVID-19-associated coagulopathy (CAC) is still under investigation and both cellular and plasmatic constituents of the coagulation system appear to be affected by infection with SARS-CoV-2. It has been established that hyperactivated platelets play a major role in CAC (10–14). Recently, the contributions of cells of the immune system during thrombus formation have been discussed in the setting of immunothrombosis. Histopathological examinations of thrombi from COVID-19 patients have demonstrated an increased deposition of neutrophils within the thrombus matrix in the lung vasculature suggesting that platelet-neutrophil interplay may be crucial in initiation and perpetuation of thrombosis in (hyper-)inflammatory diseases such as COVID-19 (13, 15–18). Several studies have already shown an increase in platelet-neutrophil aggregates (PNAs) circulating in blood of COVID-19 patients. However, the exact mechanisms of interaction between platelets and neutrophils remain unclear as little research was conducted yet on how these cells interact in promoting CAC. Next to their ability of phagocytosis and secretion of antimicrobial enzymes, neutrophils are capable of releasing neutrophil extracellular traps (NETs) mainly consisting of DNA (19). NETs serve as attachment structures for enzymes such as myeloperoxidase (MPO) or neutrophil elastase but also trap pathogens and allow their degradation by the antimicrobial substances. NETs have been studied extensively in the last decade for their impact on thrombus formation (20, 21). Various prothrombotic conditions including DIC in septic patients (22), neoplasms (23) and heparin-induced thrombocytopenia (HIT) (24, 25) have been found to be associated with elevated levels of NETs. SARS-CoV-2 was also demonstrated to directly induce NETosis (26–28). Thus, the detrimental consequences of severe COVID-19 have been partially attributed to both direct and indirect effects of NETs.
Up until now several vaccine candidates have been approved worldwide to mitigate the burden on society and healthcare caused by the COVID-19 pandemic. Among the first vaccine platforms authorized in Europe were mRNA-based vaccines and vaccines using adenoviral vectors. Shortly after the rollout of immunization programs, cases of thrombocytopenia accompanied by thrombotic events have been reported in individuals recently vaccinated with Vaxzevria (ChAdOx1 nCoV-19 vaccine, AstraZeneca) or Janssen Covid-19 vaccine (Ad26.COV2.S, Johnson & Johnson) – both vaccines relying on the adenoviral vector technique. For the first (or unknown) immunization with Vaxzevria, the UK´s Medicines and Healthcare products Regulatory Agency (MHRA) calculated the overall reported incidence of thromboembolic events associated with thrombocytopenia to be 15.9 cases/million doses (29). This specific syndrome, termed vaccine-induced thrombotic thrombocytopenia (VITT), is caused by the formation of antibodies against platelet factor 4 (PF4). Thrombosis and particularly cerebral venous sinus thrombosis (CVST) is the key finding of VITT with the case fatality rate estimated to be approximately 18% (30, 31). The diagnosis of VITT usually requires a history of immunization with an adenoviral vector anti-SARS-CoV-2 vaccine (mainly Vaxzevria or Janssen) minimum 4 days prior, detection of anti-PF4 antibodies in serum and additional more specific platelet aggregation tests (32–34). Treatment options in VITT include non-heparin anticoagulants, administration of intravenous immunoglobulins (IVIG) as well as supportive care (35, 36).
Here, we give an overview of the current state of research on the interaction between platelets and neutrophils in CAC and VITT.
Besides a reduction in platelet count, the functional properties of platelets are reportedly deranged during COVID-19. Platelets of patients infected with SARS-CoV-2 were found to have higher expression of activation markers than non-COVID-19 controls (11, 12, 37–40). Table 1 lists markers for platelet and neutrophil activation described in COVID-19. Moreover, platelets in COVID-19 had an increased tendency towards aggregation and showed greater responses to stimuli as ADP, thrombin receptor activator peptide 6 (TRAP-6) or thrombin itself (10, 14, 37, 38). This indicates both a hyper-active and hyper-reactive platelet phenotype during infection with SARS-CoV-2.
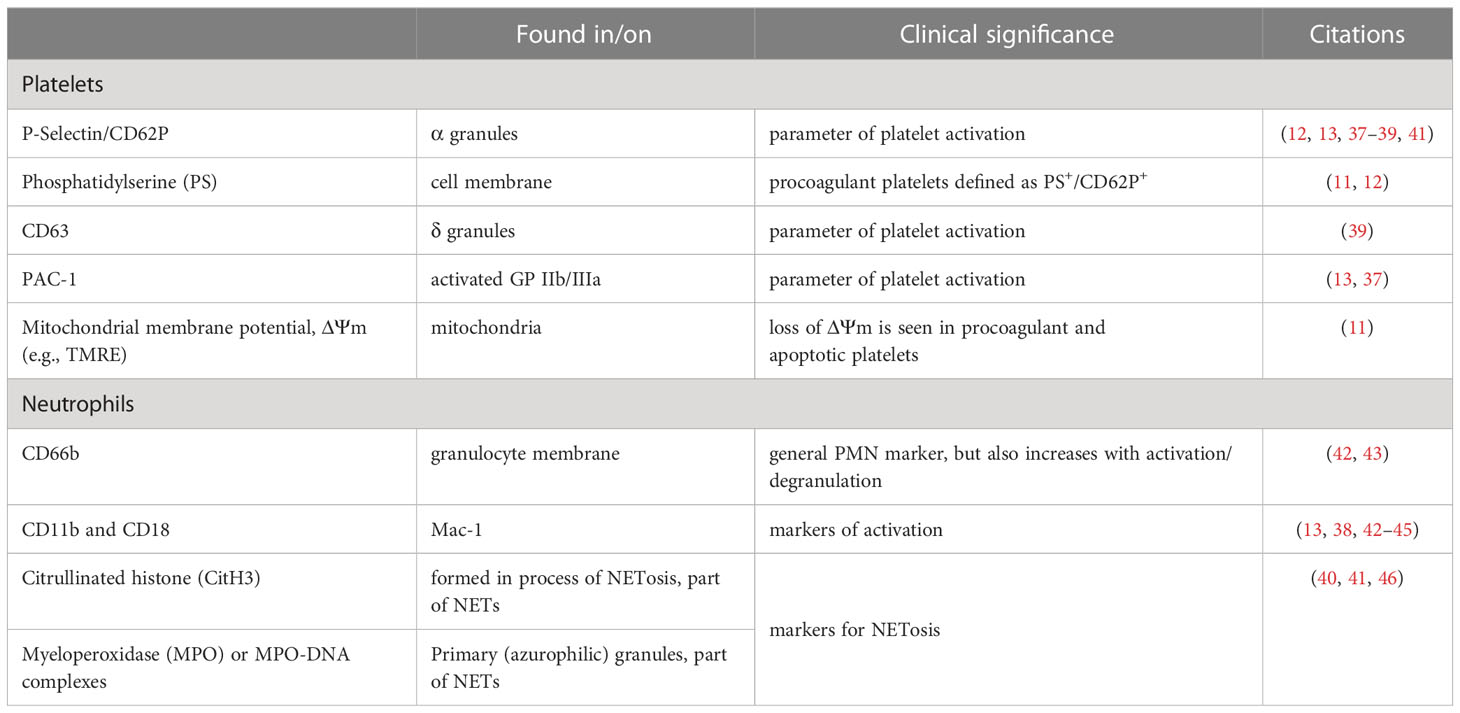
Table 1 Markers of platelet and neutrophil activation described in COVID-19.
Direct and indirect aspects of platelet activation have been proposed. Zhu et al. confirmed the presence of SARS-CoV-2 RNA within platelets. Six out of the seven patients with this finding deceased shortly after. On the contrary, only one out of 24 COVID-19 patients from the survivor group was found to have RNA positive platelets (47). The principal mechanism of cellular uptake of SARS-CoV-2 is assumed to occur via the angiotensin-converting enzyme (ACE2) receptor in combination with the transmembrane serine protease/serine subfamily member 2 (TMPRSS2) (48). However, whether ACE2 and TMPRSS2 are expressed on platelets is still under debate and other mechanisms of viral entry have also been proposed (37, 49, 50). Furthermore, CD147 may serve as a site of direct interaction between platelets and SARS-CoV-2 and was also described to be a mediator of viral entry into cells via endocytosis (51, 52). Additionally, glycoprotein Ib (or CD42b) was identified as a receptor used by the spike protein of SARS-CoV-2 (53). Furthermore, the direct effects of SARS-CoV-2 on platelets appear to be mediated through the upregulation of both caspase-dependent (apoptosis) and caspase-independent pathways (necroptosis) (54, 55).
Examples for indirect mechanisms of platelet activation during SARS-CoV-2 infection include specific immunoglobulins found in sera of COVID-19 patients that induce procoagulant platelets via FcγRIIa signaling (11, 12), endothelial dysfunction with increased expression of von Willebrand factor (vWF) (56, 57) and stimulation of platelets by proinflammatory markers during the cytokine storm complicating severe cases of COVID-19 (58, 59). Tissue factor (TF) secreted from SARS-CoV-2 infected cells such as epithelium also indirectly activates platelets via thrombin-mediated signaling (59, 60). Moreover, thrombopoietin (TPO), which promotes in vitro platelet hyperresponsiveness and platelet-leukocyte interaction, is found to be increased in COVID-19 patients (61, 62) Additionally, platelets also secrete cytokines during SARS-CoV-2 infection themselves and consequently contribute to the hyperinflammatory state increasing the risk of CAC (63, 64).
Leukocytosis and thus, neutrophilia are common laboratory findings in COVID-19 as the mobilization of immune cells from the bone marrow is one of the earliest responses to combat pathogens (65). The phenotype of neutrophils changes during the infection with SARS-CoV-2. As expected, serum levels of typical markers of neutrophil activation (degranulation and NETosis) such as MPO-DNA complexes or citrullinated histone H3 (Cit-H3) in COVID-19 patients were found to be correlating with disease severity (41, 46). Furthermore, TF increases on neutrophils isolated from patients with severe COVID-19 (66). This suggests how among other pathways primed neutrophils potentially promote or even elicit thrombus formation. As mentioned, NETs are composed of DNA, DNA-associated structures (e.g., histones) and contents of neutrophil granules. Noubouossie et al. reported on the ability of neutrophil DNA to induce thrombin generation (TG) in both platelet-rich and platelet-free plasma although histone-mediated TG appeared to require the presence of platelets (67). For the latter, the toll-like receptors (TLR) 2 and 4 on platelets seem to be of importance in mediating the increase in TG (68).
In a mouse model for SARS-CoV-2 infection, Sung et al. demonstrated the importance of TLR2 and C-type lectin domain family 5 member (CLEC5A) in neutrophils for NETosis and the release of proinflammatory cytokines such as interleukin 6 (IL-6). Interestingly, further in vitro experiments with SARS-CoV-2 and mice neutrophils showed accumulation of MPO, Cit-H3 and DNA within the neutrophilic cytoplasm after 5 hours. However, marked NETosis became evident only when incubated with autologous platelets. This suggests that platelet presence could be necessary for further neutrophil activation and NETosis in the case of SARS-CoV-2 infection. Interestingly, in contrast to former evidence with the Dengue virus where NET formation was found to be thread-like, the authors concluded that NETosis elicited by SARS-CoV-2 had a different, more aggregated appearance (69).
Additionally, low-density neutrophils (LDNs) appear to be increasing in number in COVID-19 more than other neutrophil subpopulations (44). These cells - termed CD16int due to their behavior to only intermediately stain with anti-FcγRIII (CD16) – also display an upregulation of genes that are related to NETosis when compared to the CD16high neutrophils. As expected, the authors reported spontaneous in vitro formation of NETs in these LDNs (42). Schulte-Schrepping et al. have further elaborated on the myeloid response in severe COVID-19 giving rise to distinct neutrophil precursor subclasses which are characterized by different gene activation signatures including genes involved in NETosis (70). Previously, LDNs have commonly been described in rheumatological diseases as systemic lupus erythematosus or anti-phospholipid syndrome for their proinflammatory effects although consensus on their precise characterization in terms of origin, function and fate has not been reached (71, 72).
As a side note, neutrophils contribute to CAC by releasing a variety of immune mediators causing a cytokine storm and DIC (73). For instance, Kaiser et al. proposed a vicious cycle of IL-8 released from neutrophils in severe COVID-19 which further attracts and activates additional neutrophils (74).
The first quantifiable endpoint of platelet and neutrophil interaction in CAC is represented by complex formation. As mentioned, such platelet-neutrophil aggregates (PNAs) are abundant in SARS-CoV-2 positive patients (37, 41, 45, 75–77). COVID-19 disease severity correlates with blood levels of PNAs (78). Both normal-density neutrophils (NDNs) and LDNs form PNAs although complexes of platelets and CD16int had significantly higher expression of P-selectin (CD62P) and CD40 than PNAs with CD16high neutrophils (42). This could be an additional hint for the hyperactive properties of LDNs and their dominant role in mediating a potential synergy between activated platelets and neutrophils in thrombosis.
In the following, we provide an outline of the most relevant receptors in the process of aggregate formation. Figure 1 illustrates the direct and indirect aspects of platelet-neutrophil interaction in COVID-19.
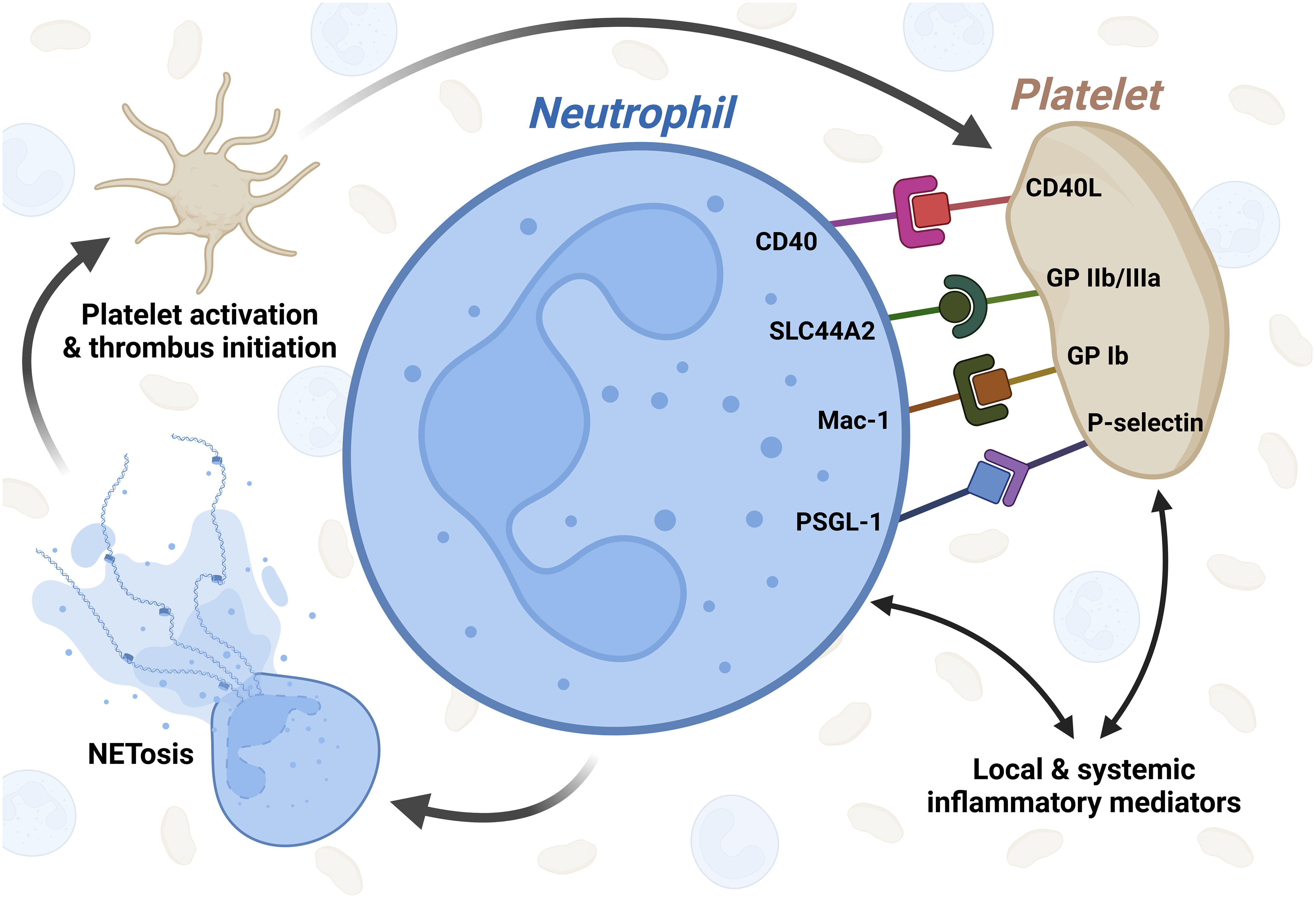
Figure 1 Graphical illustration of platelet-neutrophil interaction in COVID-19. Inflammatory mediators released from immune cells as well as endothelial cells during COVID-19 activate both platelets and neutrophils independently. After activation, several receptors are known to be involved in platelet-neutrophil interaction in COVID-19 which in turn leads to NETosis, subsequent platelet activation and thrombus initiation. CD40L - CD40 ligand; SLC44A2 - solute carrier family 44 member 2; GP IIb/IIIa - glycoprotein IIb/IIIa; Mac-1 - macrophage-1 antigen complex; GP Ib - glycoprotein Ib; PSGL-1 - P-selectin glycoprotein ligand-1. Created with BioRender.com.
Platelet CD62P (P-selectin) and neutrophil P-selectin glycoprotein ligand-1 (PSGL-1, CD162) are a long-known interaction site for platelets and granulocytes (79, 80). Wang et al. demonstrated the major role of PSGL-1 in the coagulopathy associated with systemic inflammation suggesting that CD62P-PSGL-1 coupling also is involved in CAC (81). P-selectin has been identified as the major platelet receptor for monocyte-platelet aggregation in COVID-19 patients (82). Interestingly, platelets in vitro activated by SARS-CoV-2 spike protein were shown to cause activation of monocytes via CD62P-PSGL-1 coupling (53). Non-specific gene signature analysis of whole blood from severe to critical COVID-19 patients additionally has shown an upregulation of SELPG, the gene encoding PSGL-1 (83).
The macrophage-1 antigen (Mac-1) is made up of the two integrins αM (CD11b) and β2 (CD18) and serves several purposes including binding complement and regulation of leukocyte extravasation (84). Despite the name, neutrophils also express Mac-1 and determination of CD11b is considered a typical marker of neutrophil activation beside CD66b (Table 1). Previously, it was shown that Mac-1 interacts with platelet glycoprotein Ibα (vWF receptor) in mediating thrombosis (21, 85). Increased expression of both CD11b and CD18 on neutrophils isolated from COVID-19 patients was noted when compared to healthy volunteers suggesting one potential mechanism of platelet activation via Mac-1 binding of platelet GP Ib (43, 45). Additionally, the behavior of three different neutrophil subpopulations in COVID-19 patients was investigated by Reyes et al. First, neutrophils isolated by density gradient centrifugation from both the PMN and PBMC layer were separated into NDNs and LDNs. LDNs were further characterized for maturity based on expression of CD10 and CD16. Interestingly, mature LDNs (CD16+/CD10+) showed high levels of Mac-1 similar to NDNs but formed more complexes with platelets than NDNs. On the other hand, immature LDNs (CD16-/CD10-) showed lower levels of Mac-1 and appeared to form fewer PNAs (43).
As discussed later, Mac-1 also recognizes several chemokines secreted from platelets including PF4.
The platelet glycoprotein IIb/IIIa (integrin α2bβ3, CD41/CD61) is known to interact with the widely distributed choline transporter-like protein 2 (CTL2, SLC44A2) presenting on neutrophils. The importance of SLC44A2 in hemostasis and particularly VTE has already been established in both genetic and animal studies (86–88). Constantinescu-Bercu et al. highlighted the neutrophil SLC44A2 - platelet integrin α2bβ3 axis as an important communication channel of NETosis. Neutrophils were shown to form NETs when infused through GP IIb/IIIa-coated microchannels although simple incubation without flow resulted in a significant decrease in NETosis (89). This implies that formation of NETs also depends on mechanistic effects. From studies on platelet-monocyte interactions, Hottz et al. reported that in vitro inhibition of GP IIb/IIIa with abciximab limited the ability of platelets from COVID-19 patients to activate TF expression by monocytes (82). Up to this point there is no data available on SLC44A2 in COVID-19 and how it could potentially impact CAC except for fundamental proteomic data which suggests a significant downregulation of SLC44A2 in neutrophils from severe COVID-19 patients (90).
Apart from many immune responses which are regulated by CD40 and its ligand CD40L (CD154), platelets and neutrophils were also demonstrated to use this pathway (91, 92). Both CD40L expressed on the platelet membrane and soluble CD40L released from platelets (sCD40L) were found to be critical for neutrophil activation in animal models (93, 94). It was also established that CD40L is not uniquely limited to bind CD40 as it also interacted with Mac-1 (95). In general, neutrophil adhesion to platelets was shown to be enhanced by CD40L but this effect was dependent on Mac-1 as its inhibition with anti-CD11b reversed the bonding affinity of neutrophils for platelets (96). Blood from COVID-19 patients had significantly higher concentrations of sCD40L than healthy volunteers (97, 98). However, this was not consistent with the report of Blasi et al. where no significant difference in plasma sCD40L was evident between COVID-19 patients and healthy controls (99). Interestingly, Al-Tamimi et al. showed soluble CD40L levels peaking with moderate disease followed by a decline when disease severity increases (100). This may explain, at least in part, the inconsistencies observed at different time points in the course of COVID-19. In vitro stimulation of platelet-rich plasma with the receptor-binding domain of SARS-CoV-2 caused the levels of soluble CD40L to increase (101). This suggests a direct viral effect on platelets causing sCD40L secretion which in turn could induce neutrophil activation. Li et al. showed that spike protein-activated platelets interacted with monocytes using CD40L (53). This clearly highlights the substantiality of platelet presence in fully unfolding the effects of SARS-CoV-2.
On the other hand, the expression of CD40 on LDNs (CD16int) correlated with both disease severity and the concentration of D-dimers (42). As described previously, such LDNs are thought to be pro-NETotic. Increased expression of CD40 on LDNs as binding site for platelet surface CD40L and sCD40L released from platelets among other cells (e.g., endothelium) could render these LDNs more susceptible to PNA formation and subsequent platelet-mediated neutrophil activation. For further information on the role of CD40/CD40L in thromboinflammation we refer to the review by Cognasse et al. (102).
Numerous indirect ways of communication between platelets and neutrophils have been reported. Precise dissection of these pathways is challenging and often ambiguous. Most importantly, inflammatory mediators (e.g., cytokines) secreted from both cell types and so-called microvesicles (MVs) are thought to participate in indirect platelet-neutrophil interaction.
Microvesicles are released from cells through membrane budding and usually contain intracellular contents. Platelets are well known to release such extracellular vesicles into circulation in various situations including COVID-19 (63, 64, 103). High levels of platelet-derived MVs expressing TF were found in COVID-19 patients highlighting the thrombotic diathesis of SARS-CoV-2 infection (58, 104). Neutrophils from COVID-19 patients also release MVs which are an important source of TF. Skendros et al. suggested that this platelet-neutrophil-TF axis may be the critical link between immune defense and both plasmatic and cellular hemostasis, leading to CAC (66). Previously, a circular relationship between MVs from neutrophils and platelets has been proposed where direct interaction via P-selectin/PSGL-1 coupling initiated platelet-induced arachidonic release from neutrophils. In turn, after uptake into the platelet interior, thromboxane A2 (TxA2) is generated and released causing endothelial activation and subsequently leukocyte rolling and diapedesis (105). Furthermore, TxA2 has been previously reported to play a role in NET formation in the pathogenesis of transfusion related acute lung injury (TRALI) (106). However, whether this applies to NETosis in COVID-19 as well is yet to determine.
The cytokine response to SARS-CoV-2 viremia is complex and sometimes progresses to an hyperinflammatory state with excessive cytokine release (“cytokine storm”). Multiple cell types participate in this process including neutrophils and platelets. As mentioned, platelets in COVID-19 may secrete soluble CD40L but also other non-cytokine mediators such as the positively charged PF4 in COVID-19 (64). PF4 or CXCL4 is known to interact with the neutrophil Mac-1 receptor and also directly with NETs as by their anionic nature. In general, its effects are diverse but include neutrophil chemotaxis, stimulation of NET formation and NET compaction (107–112). The exact role of PF4-mediated platelet-neutrophil interaction in COVID-19 was not investigated further albeit a single study on COVID-19 patients that reported elevated levels of both PF4 and RANTES (Regulated and Normal T cell Expressed and Secreted), a chemokine released from platelets (76). High-mobility group box 1 (HMGB1) also plays a role among the mediators of platelet-neutrophil interplay and high HMGB1 levels were shown to be associated with COVID-19 mortality (113, 114). HMGB1 is a damage-associated molecular pattern (DAMP) protein which can be released from activated or necrotic cells. The function of HMGB1 in thrombosis has only been superficially covered but findings from acute myocardial infarction patients suggested that platelet-derived HMGB1 acts on neutrophils and stimulates the release of NETs. Here, the RAGE receptor (Receptor for Advanced Glycation End products) is of importance (115). An animal study from Vogel et al. has further elaborated on the essential role of HMGB1 in passing prothrombotic signals from platelets to neutrophils (116).
Another example of a relevant mediator in platelet-neutrophil interplay is represented by IL-6 which has already been identified as a main target in counteracting the hyperinflammatory state observed in severe cases of COVID-19 (58, 63). Interestingly, IL-6 blockade in COVID-19 plasma with tocilizumab significantly reduced the high levels of TF+-platelet MVs and PNAs when compared to control plasma (58).
Additionally, neutrophils may release calprotectin or S100A8/A9 upon activation, a protein with the potential to induce procoagulant platelets via GP Ibα in vitro. COVID-19 patients showed high levels of S100A8/A9 correlating with disease severity (117, 118). Again, differences in upregulation of both S100A8 and S100A9 gene were noted among distinct neutrophil precursor subclasses in severe COVID-19 (70). Additionally, calprotectin deposits have been identified on lung autopsies of COVID-19 deaths (119). The procoagulant effects of neutrophil cathepsin G on the other hand is more certain and direct interaction between this serine protease and platelets is thought to be mediated by protease-activated receptors, PAR-1 and/or PAR-4 (120–123). High levels of cathepsin G are found in COVID-19 but also pneumonia with acute respiratory distress syndrome of different etiologies (124, 125).
Although little is known about platelet-neutrophil interplay in VITT yet, recent evidence suggests the importance of this relationship in initiating and perpetuating vaccine-induced thrombosis. Direct platelet-neutrophil interaction in the form of PNAs was already found to be upregulated in VITT resulting in higher levels of PNAs as compared to control (126). On a single cellular level, both platelets and neutrophils have been demonstrated to be directly activated in vitro by VITT antibodies (126–128). Several case reports have highlighted the presence of NETs within thrombi of VITT patients indicating that NETs are involved in vaccine-induced thrombosis (129, 130). Additionally, increased plasma levels of NET markers (e.g., Cit-H3, MPO-DNA complex) were observed in VITT patients (126, 129). This is in line with recent findings in other prothrombotic conditions such as COVID-19 and HIT. Furthermore, the severity of side effects of the immunization has been correlated to serum histone 3 levels as well (131).
For neutrophils, the proportion of NET-releasing granulocytes was found to be significantly higher in VITT patients compared to control groups (126). Again, LDNs appear to be particularly involved here. NETosis from LDNs in VITT was significantly higher than NET release from NDNs (126, 129). Further research should be directed towards functions and significance of this peculiar neutrophil subpopulation to eventually identify potential pharmaceutical targets in counteracting NET formation in immunothrombosis. Interestingly, Greinacher et al. reported that in vitro incubation of isolated neutrophils with VITT serum and PF4 did not lead to NETosis unless platelets were also added to the experiment (128). Thus, it could be concluded that platelets have a crucial role in neutrophil activation and NET formation also in the setting of VITT. Here, microvesicles released from platelets seem to be of importance for the prothrombotic milieu and could help in explaining the cerebral venous tropism of VITT thrombosis (132). However, future efforts are needed to investigate the exact direct and indirect mechanisms of intercellular communication and interplay of neutrophil and platelets in VITT.
Recent studies have focused on neutrophil-activating peptide 2 (NAP2) or CXCL7 released from platelets stimulated with VITT antibodies which in turn activated neutrophils (133–135). Hundelshausen et al. proposed the use of Bruton tyrosine kinase inhibitors (BTKi) in VITT as they were shown to limit platelet P-selectin expression, reduce neutrophil activation and inhibit platelet-neutrophil aggregation (136, 137). Apart from stored VITT sera mainly from spring 2021, future VITT models could rely on chimeric anti-PF4 antibodies mimicking vaccine-induced thrombotic thrombocytopenia such as 1E12 (138).
In general, both platelets and neutrophils on their own are considered to be major actors in the prothrombotic state seen in COVID-19 and VITT. With our understanding of thromboinflammation still evolving, further efforts should be directed towards dissecting the precise mechanisms of direct and indirect platelet-neutrophil interplay. From our point of view, this relationship should be seen as bidirectional with both types of cells closely interacting and potentiating the thrombotic cascade of initiation, formation and extension of thrombosis. Detailed insights into this interaction and its pathways could be used to design targeted therapies that reduce the occurrence of life-threatening thrombotic complications in COVID-19 and VITT. Additional research on the role of LDNs may help limit the consequences of hyperinflammation associated with COVID-19. In vitro thrombosis models, such as microfluidic systems, might be helpful in this regard to understand the role of different cells in the development of thrombosis in patients with COVID-19 as well as VITT.
JH, GU and TB wrote the manuscript. JH, GU and AS performed the literature review and data collection. JH and JZ designed the Figures. JH, GU, JZ, AS and TB revised the manuscript. All authors contributed to the article and approved the submitted version.
We acknowledge support from the Open Access Publication Fund of the University of Tübingen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med (2020) 383:590–2. doi: 10.1056/NEJMc2011400
2. Yao X-H, Luo T, Shi Y, He Z-C, Tang R, Zhang P-P, et al. A cohort autopsy study defines COVID-19 systemic pathogenesis. Cell Res (2021) 31:836–46. doi: 10.1038/s41422-021-00523-8
3. Basso C, Leone O, Rizzo S, de Gaspari M, van der Wal AC, Aubry M-C, et al. Pathological features of COVID-19-Associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J (2020) 41:3827–35. doi: 10.1093/eurheartj/ehaa664
4. Liao D, Zhou F, Luo L, Xu M, Wang H, Xia J, et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: a retrospective cohort study. Lancet Haematol (2020) 7:e671–8. doi: 10.1016/S2352-3026(20)30217-9
5. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JC, Fogerty AE, Waheed A, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood (2020) 136:489–500. doi: 10.1182/blood.2020006520
6. Wichmann D, Sperhake J-P, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med (2020) 173:268–77. doi: 10.7326/M20-2003
7. Grosse C, Grosse A, Salzer HJ, Dünser MW, Motz R, Langer R. Analysis of cardiopulmonary findings in COVID-19 fatalities: high incidence of pulmonary artery thrombi and acute suppurative bronchopneumonia. Cardiovasc Pathol (2020) 49:107263. doi: 10.1016/j.carpath.2020.107263
8. Agarwal G, Hajra A, Chakraborty S, Patel N, Biswas S, Adler MK, et al. Predictors and mortality risk of venous thromboembolism in patients with COVID-19: systematic review and meta-analysis of observational studies. Ther Adv Cardiovasc Dis (2022) 16:1–22. doi: 10.1177/17539447221105013
9. Roberts LN, Navaratnam AV, Arya R, Briggs TW, Gray WK. Venous thromboembolism in patients hospitalised with COVID-19 in England. Thromb Res (2022) 213:138–44. doi: 10.1016/j.thromres.2022.03.017
10. Jakobs K, Reinshagen L, Puccini M, Friebel J, Wilde A-CB, Alsheik A, et al. Disease severity in moderate-to-Severe COVID-19 is associated with platelet hyperreactivity and innate immune activation. Front Immunol (2022) 13:844701. doi: 10.3389/fimmu.2022.844701
11. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood (2021) 137:1061–71. doi: 10.1182/blood.2020008762
12. Zlamal J, Althaus K, Jaffal H, Häberle H, Pelzl L, Singh A, et al. Upregulation of cAMP prevents antibody-mediated thrombus formation in COVID-19. Blood Adv (2022) 6:248–58. doi: 10.1182/bloodadvances.2021005210
13. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation (2020) 142:1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
14. Barrett TJ, Bilaloglu S, Cornwell M, Burgess HM, Virginio VW, Drenkova K, et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost (2021) 19:3139–53. doi: 10.1111/jth.15534
15. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from new Orleans. Lancet Respir Med (2020) 8:681–6. doi: 10.1016/S2213-2600(20)30243-5
16. Genchi A, Semerano A, Schwarz G, Dell'Acqua B, Gullotta GS, Sampaolo M, et al. Neutrophils predominate the immune signature of cerebral thrombi in COVID-19 stroke patients. Acta Neuropathol Commun (2022) 10:14. doi: 10.1186/s40478-022-01313-y
17. Johnson JE, McGuone D, Xu ML, Jane-Wit D, Mitchell RN, Libby P, et al. Coronavirus disease 2019 (COVID-19) coronary vascular thrombosis: correlation with neutrophil but not endothelial activation. Am J Pathol (2022) 192:112–20. doi: 10.1016/j.ajpath.2021.09.004
18. Tanaka C, Hiraiwa S, Otsuka H, Yamaguchi M. Platelet aggregation with various morphologies of neutrophils in arterial thrombus in a patient with coronavirus disease: a case report. J Surg Case Rep (2022) 2022:1–5. doi: 10.1093/jscr/rjac532
19. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
20. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U.S.A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
21. von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209:819–35. doi: 10.1084/jem.20112322
22. Stiel L, Mayeur-Rousse C, Helms J, Meziani F, Mauvieux L. First visualization of circulating neutrophil extracellular traps using cell fluorescence during human septic shock-induced disseminated intravascular coagulation. Thromb Res (2019) 183:153–8. doi: 10.1016/j.thromres.2019.09.036
23. Zhang Y, Wang C, Yu M, Zhao X, Du J, Li Y, et al. Neutrophil extracellular traps induced by activated platelets contribute to procoagulant activity in patients with colorectal cancer. Thromb Res (2019) 180:87–97. doi: 10.1016/j.thromres.2019.06.005
24. Perdomo J, Leung HH, Ahmadi Z, Yan F, Chong JJ, Passam FH, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun (2019) 10:1322. doi: 10.1038/s41467-019-09160-7
25. Gollomp K, Kim M, Johnston I, Hayes V, Welsh J, Arepally GM, et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight (2018) 3:e99445. doi: 10.1172/jci.insight.99445
26. Arcanjo A, Logullo J, Menezes CC, de Souza Carvalho Giangiarulo TC, Dos Reis MC, de Castro GM, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
27. Youn Y-J, Lee Y-B, Kim S-H, Jin HK, Bae J-S, Hong C-W. Nucleocapsid and spike proteins of SARS-CoV-2 drive neutrophil extracellular trap formation. Immune Netw (2021) 21:e16. doi: 10.4110/in.2021.21.e16
28. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-Triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
29. Medicines and Healthcare products Regulatory Agency. Coronavirus vaccines - summary of yellow card reporting. Available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1128782/Coronavirus_Vaccine_-_Summary_of_Yellow_Card_reporting_23.11.2022_final.pdf.
30. Lane S, Shakir S. Assessing case fatality on cases of thrombosis with concurrent thrombocytopenia following COVID-19 vaccine AstraZeneca (Vaxzevria) in the united kingdom: a review of spontaneously reported data. Drug Saf (2022) 45:1003–8. doi: 10.1007/s40264-022-01217-9
31. Krzywicka K, Heldner MR, van Sánchez Kammen M, van Haaps T, Hiltunen S, Silvis SM, et al. Post-SARS-CoV-2-Vaccination cerebral venous sinus thrombosis: an analysis of cases notified to the European medicines agency. Eur J Neurol (2021) 28:3656–62. doi: 10.1111/ene.15029
32. Oldenburg J, Klamroth R, Langer F, Albisetti M, von AC, Ay C, et al. Diagnosis and management of vaccine-related thrombosis following AstraZeneca COVID-19 vaccination: guidance statement from the GTH. Hamostaseologie (2021) 41:184–9. doi: 10.1055/a-1469-7481
33. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med (2021) 384:2092–101. doi: 10.1056/NEJMoa2104840
34. Nazy I, Sachs UJ, Arnold DM, McKenzie SE, Choi P, Althaus K, et al. Recommendations for the clinical and laboratory diagnosis of VITT against COVID-19: communication from the ISTH SSC subcommittee on platelet immunology. J Thromb Haemost (2021) 19:1585–8. doi: 10.1111/jth.15341
35. Cines DB, Greinacher A. Spotlight on vaccine-induced thrombosis and thrombocytopenia (VITT). Blood (2023) 141:1659–65. doi: 10.1182/blood.2022017696
36. Gabarin N, Arnold DM, Nazy I, Warkentin TE. Treatment of vaccine-induced immune thrombotic thrombocytopenia (VITT). Semin Hematol (2022) 59:89–96. doi: 10.1053/j.seminhematol.2022.03.002
37. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood (2020) 136:1317–29. doi: 10.1182/blood.2020007214
38. Chao Y, Rebetz J, Bläckberg A, Hovold G, Sunnerhagen T, Rasmussen M, et al. Distinct phenotypes of platelet, monocyte, and neutrophil activation occur during the acute and convalescent phase of COVID-19. Platelets (2021) 32:1092–102. doi: 10.1080/09537104.2021.1921721
39. Bongiovanni D, Klug M, Lazareva O, Weidlich S, Biasi M, Ursu S, et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis (2021) 12:50. doi: 10.1038/s41419-020-03333-9
40. Dechamps M, de Poortere J, Martin M, Gatto L, Daumerie A, Bouzin C, et al. Inflammation-induced coagulopathy substantially differs between COVID-19 and septic shock: a prospective observational study. Front Med (2021) 8:780750. doi: 10.3389/fmed.2021.780750
41. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Association of neutrophil activation, more than platelet activation, with thrombotic complications in coronavirus disease 2019. J Infect Dis (2021) 223:933–44. doi: 10.1093/infdis/jiaa756
42. Morrissey SM, Geller AE, Hu X, Tieri D, Ding C, Klaes CK, et al. A specific low-density neutrophil population correlates with hypercoagulation and disease severity in hospitalized COVID-19 patients. JCI Insight (2021) 6:e148435. doi: 10.1172/jci.insight.148435
43. Reyes L, Sanchez-Garcia M A, Morrison T, Howden AJ, Watts ER, Arienti S, et al. Prothrombotic hyperinflammatory neutrophil signature is distinct for COVID-19 ARDS. Wellcome Open Res (2021) 6:38. doi: 10.12688/wellcomeopenres.16584.2
44. Cabrera LE, Pekkarinen PT, Alander M, Nowlan KH, Nguyen NA, Jokiranta S, et al. Characterization of low-density granulocytes in COVID-19. PloS Pathog (2021) 17:e1009721. doi: 10.1371/journal.ppat.1009721
45. Rieder M, Baldus N, Stallmann D, Jeserich M, Goller I, Wirth L, et al. Early SARS-CoV-2 infection: platelet-neutrophil complexes and platelet function. Res Pract Thromb Haemost (2022) 7:100025. doi: 10.1016/j.rpth.2022.100025
46. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight (2020) 5:e138999. doi: 10.1172/jci.insight.138999
47. Zhu A, Real F, Capron C, Rosenberg AR, Silvin A, Dunsmore G, et al. Infection of lung megakaryocytes and platelets by SARS-CoV-2 anticipate fatal COVID-19. Cell Mol Life Sci (2022) 79:365. doi: 10.1007/s00018-022-04318-x
48. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell (2020) 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052
49. Shen S, Zhang J, Fang Y, Lu S, Wu J, Zheng X, et al. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J Hematol Oncol (2021) 14:72. doi: 10.1186/s13045-021-01082-6
50. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol (2020) 13:120. doi: 10.1186/s13045-020-00954-7
51. Maugeri N, de Lorenzo R, Clementi N, Antonia Diotti R, Criscuolo E, Godino C, et al. Unconventional CD147-dependent platelet activation elicited by SARS-CoV-2 in COVID-19. J Thromb Haemost (2022) 20:434–48. doi: 10.1111/jth.15575
52. Wang K, Chen W, Zhang Z, Deng Y, Lian J-Q, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Sig Transduct Target Ther (2020) 5:283. doi: 10.1038/s41392-020-00426-x
53. Li T, Yang Y, Li Y, Wang Z, Ma F, Luo R, et al. Platelets mediate inflammatory monocyte activation by SARS-CoV-2 spike protein. J Clin Invest (2022) 132:e150101. doi: 10.1172/JCI150101
54. Trugilho MR, Azevedo-Quintanilha IG, Gesto JS, Moraes EC, Mandacaru SC, Campos MM, et al. Platelet proteome reveals features of cell death, antiviral response and viral replication in covid-19. Cell Death Discovery (2022) 8:324. doi: 10.1038/s41420-022-01122-1
55. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res (2021) 129:631–46. doi: 10.1161/CIRCRESAHA.121.319117
56. Won T, Wood MK, Hughes DM, Talor MV, Ma Z, Schneider J, et al. Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. EBioMedicine (2022) 75:103812. doi: 10.1016/j.ebiom.2022.103812
57. Birnhuber A, Fließer E, Gorkiewicz G, Zacharias M, Seeliger B, David S, et al. Between inflammation and thrombosis: endothelial cells in COVID-19. Eur Respir J (2021) 58:2100377. doi: 10.1183/13993003.00377-2021
58. Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Trans Sci (2021) 6:202–18. doi: 10.1016/j.jacbts.2020.12.009
59. Puhm F, Allaeys I, Lacasse E, Dubuc I, Galipeau Y, Zaid Y, et al. Platelet activation by SARS-CoV-2 implicates the release of active tissue factor by infected cells. Blood Adv (2022) 6:3593–605. doi: 10.1182/bloodadvances.2022007444
60. Francischetti IM, Toomer K, Zhang Y, Jani J, Siddiqui Z, Brotman DJ, et al. Upregulation of pulmonary tissue factor, loss of thrombomodulin and immunothrombosis in SARS-CoV-2 infection. eClinicalMedicine (2021) 39:101069. doi: 10.1016/j.eclinm.2021.101069
61. Lupia E, Capuano M, Vizio B, Schiavello M, Bosco O, Gelardi M, et al. Thrombopoietin participates in platelet activation in COVID-19 patients. EBioMedicine (2022) 85:104305. doi: 10.1016/j.ebiom.2022.104305
62. Doi T, Hori T, Onuma T, Mizutani D, Ueda K, Enomoto Y, et al. Thrombopoietin and collagen in low doses cooperatively induce human platelet activation. Acute Med Surg (2022) 9:e769. doi: 10.1002/ams2.769
63. Taus F, Salvagno G, Canè S, Fava C, Mazzaferri F, Carrara E, et al. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol (2020) 40:2975–89. doi: 10.1161/ATVBAHA.120.315175
64. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res (2020) 127:1404–18. doi: 10.1161/CIRCRESAHA.120.317703
65. Zinellu A, Mangoni AA. A systematic review and meta-analysis of the association between the neutrophil, lymphocyte, and platelet count, neutrophil-to-Lymphocyte ratio, and platelet-to-Lymphocyte ratio and COVID-19 progression and mortality. Expert Rev Clin Immunol (2022) 18:1187–202. doi: 10.1080/1744666X.2022.2120472
66. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
67. Noubouossie DF, Whelihan MF, Yu Y-B, Sparkenbaugh E, Pawlinski R, Monroe DM, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood (2017) 129:1021–9. doi: 10.1182/blood-2016-06-722298
68. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
69. Sung P-S, Yang S-P, Peng Y-C, Sun C-P, Tao M-H, Hsieh S-L. CLEC5A and TLR2 are critical in SARS-CoV-2-Induced NET formation and lung inflammation. J BioMed Sci (2022) 29:52. doi: 10.1186/s12929-022-00832-z
70. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell (2020) 182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001
71. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
72. Mauracher L-M, Krall M, Roiß J, Hell L, Koder S, Hofbauer TM, et al. Neutrophil subpopulations and their activation potential in patients with antiphospholipid syndrome and healthy individuals. Rheumatology (2021) 60:1687–99. doi: 10.1093/rheumatology/keaa532
73. Chan L, Karimi N, Morovati S, Alizadeh K, Kakish JE, Vanderkamp S, et al. The roles of neutrophils in cytokine storms. Viruses (2021) 13:2318. doi: 10.3390/v13112318
74. Kaiser R, Leunig A, Pekayvaz K, Popp O, Joppich M, Polewka V, et al. Self-sustaining IL-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19. JCI Insight (2021) 6:e150862. doi: 10.1172/jci.insight.150862
75. Loyer C, Lapostolle A, Urbina T, Elabbadi A, Lavillegrand J-R, Chaigneau T, et al. Impairment of neutrophil functions and homeostasis in COVID-19 patients: association with disease severity. Crit Care (2022) 26:155. doi: 10.1186/s13054-022-04002-3
76. Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
77. Yasseen BA, Elkhodiry AA, El-Messiery RM, El-sayed H, Elbenhawi MW, Kamel AG, et al. Platelets' morphology, metabolic profile, exocytosis, and heterotypic aggregation with leukocytes in relation to severity and mortality of COVID-19-Patients. Front Immunol (2022) 13:1022401. doi: 10.3389/fimmu.2022.1022401
78. Le Joncour A, Biard L, Vautier M, Bugaut H, Mekinian A, Maalouf G, et al. Neutrophil-platelet and monocyte-platelet aggregates in COVID-19 patients. Thromb Haemost (2020) 120:1733–5. doi: 10.1055/s-0040-1718732
79. Yang J, Furie BC, Furie B. The biology of p-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost (1999) 81:1–7.
80. Palabrica T, Lobb R, Furie BC, Aronovitz M, Benjamin C, Hsu YM, et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by p-selectin on adherent platelets. Nature (1992) 359:848–51. doi: 10.1038/359848a0
81. Wang X-L, Deng H-F, Tan C-Y, Xiao Z-H, Liu M-D, Liu K, et al. The role of PSGL-1 in pathogenesis of systemic inflammatory response and coagulopathy in endotoxemic mice. Thromb Res (2019) 182:56–63. doi: 10.1016/j.thromres.2019.08.019
82. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood (2020) 136:1330–41. doi: 10.1182/blood.2020007252
83. Yatim N, Boussier J, Chocron R, Hadjadj J, Philippe A, Gendron N, et al. Platelet activation in critically ill COVID-19 patients. Ann Intensive Care (2021) 11:113. doi: 10.1186/s13613-021-00899-1
84. Hyun Y-M, Choe YH, Park SA, Kim M. LFA-1 (CD11a/CD18) and mac-1 (CD11b/CD18) distinctly regulate neutrophil extravasation through hotspots I and II. Exp Mol Med (2019) 51:1–13. doi: 10.1038/s12276-019-0227-1
85. Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, A Soloviev D, et al. Leukocyte integrin mac-1 regulates thrombosis via interaction with platelet GPIbα. Nat Commun (2017) 8:15559. doi: 10.1038/ncomms15559
86. Bennett JA, Mastrangelo MA, Ture SK, Smith CO, Loelius SG, Berg RA, et al. The choline transporter Slc44a2 controls platelet activation and thrombosis by regulating mitochondrial function. Nat Commun (2020) 11:3479. doi: 10.1038/s41467-020-17254-w
87. Tilburg J, Coenen DM, Zirka G, Dólleman S, van Oeveren-Rietdijk AM, Karel MF, et al. SLC44A2 deficient mice have a reduced response in stenosis but not in hypercoagulability driven venous thrombosis. J Thromb Haemost (2020) 18:1714–27. doi: 10.1111/jth.14835
88. Germain M, Chasman DI, de Haan H, Tang W, Lindström S, Weng L-C, et al. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet (2015) 96:532–42. doi: 10.1016/j.ajhg.2015.01.019
89. Constantinescu-Bercu A, Grassi L, Frontini M, Salles-Crawley II, Woollard K, Crawley JT. Activated αIIbβ3 on platelets mediates flow-dependent NETosis via SLC44A2. eLife (2020) 9:e53353. doi: 10.7554/eLife.53353
90. Kaiser, et al. The human neutrophil proteome in pneumonia and COVID-19 (2021). Available at: https://neuprocov.mdc-berlin.de/.
91. Vanichakarn P, Blair P, Wu C, Freedman JE, Chakrabarti S. Neutrophil CD40 enhances platelet-mediated inflammation. Thromb Res (2008) 122:346–58. doi: 10.1016/j.thromres.2007.12.019
92. Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix IC, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood (2010) 116:4317–27. doi: 10.1182/blood-2010-01-261206
93. Rahman M, Zhang S, Chew M, Ersson A, Jeppsson B, Thorlacius H. Platelet-derived CD40L (CD154) mediates neutrophil upregulation of mac-1 and recruitment in septic lung injury. Ann Surg (2009) 250:783–90. doi: 10.1097/SLA.0b013e3181bd95b7
94. Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, et al. Soluble CD40 ligand stimulates CD40-dependent activation of the β2 integrin mac-1 and protein kinase c zeda (PKCζ) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PloS One (2013) 8:e64631. doi: 10.1371/journal.pone.0064631
95. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with mac-1. Circulation (2007) 115:1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201
96. Li G, Sanders JM, Bevard MH, Sun Z, Chumley JW, Galkina EV, et al. CD40 ligand promotes mac-1 expression, leukocyte recruitment, and neointima formation after vascular njury. Am J Pathol (2008) 172:1141–52. doi: 10.2353/ajpath.2008.070633
97. Campo G, Contoli M, Fogagnolo A, Vieceli Dalla Sega F, Zucchetti O, Ronzoni L, et al. Over time relationship between platelet reactivity, myocardial injury and mortality in patients with SARS-CoV-2-Associated respiratory failure. Platelets (2021) 32:560–7. doi: 10.1080/09537104.2020.1852543
98. Nossent EJ, Schuurman AR, Reijnders TD, Saris A, Jongerius I, Blok SG, et al. Pulmonary procoagulant and innate immune responses in critically ill COVID-19 patients. Front Immunol (2021) 12:664209. doi: 10.3389/fimmu.2021.664209
99. Blasi A, von Meijenfeldt FA, Adelmeijer J, Calvo A, Ibañez C, Perdomo J, et al. In vitro hypercoagulability and ongoing in vivo activation of coagulation and fibrinolysis in COVID-19 patients on anticoagulation. J Thromb Haemost (2020) 18:2646–53. doi: 10.1111/jth.15043
100. Al-Tamimi AO, Yusuf AM, Jayakumar MN, Ansari AW, Elhassan M, AbdulKarim F, et al. SARS-CoV-2 infection induces soluble platelet activation markers and PAI-1 in the early moderate stage of COVID-19. Int J Lab Hematol (2022) 44:712–21. doi: 10.1111/ijlh.13829
101. Cano-Mendez A, García-Larragoiti N, Damian-Vazquez M, Guzman-Cancino P, Lopez-Castaneda S, Ochoa-Zarzosa A, et al. Platelet reactivity and inflammatory phenotype induced by full-length spike SARS-CoV-2 protein and its RBD domain. Int J Mol Sci (2022) 23:15191. doi: 10.3390/ijms232315191
102. Cognasse F, Duchez AC, Audoux E, Ebermeyer T, Arthaud CA, Prier A, et al. Platelets as key factors in inflammation: focus on CD40L/CD40. Front Immunol (2022) 13:825892. doi: 10.3389/fimmu.2022.825892
103. Zahran AM, El-Badawy O, Ali WA, Mahran ZG, Mahran EE, Rayan A. Circulating microparticles and activated platelets as novel prognostic biomarkers in COVID-19; relation to cancer. PloS One (2021) 16:e0246806. doi: 10.1371/journal.pone.0246806
104. Camera M, Canzano P, Brambilla M, Rovati GE. Montelukast inhibits platelet activation induced by plasma from COVID-19 patients. Front Pharmacol (2022) 13:784214. doi: 10.3389/fphar.2022.784214
105. Rossaint J, Kühne K, Skupski J, van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun (2016) 7:13464. doi: 10.1038/ncomms13464
106. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122:2661–71. doi: 10.1172/JCI61303
107. Lishko VK, Yakubenko VP, Ugarova TP, Podolnikova NP. Leukocyte integrin mac-1 (CD11b/CD18, αMβ2, CR3) acts as a functional receptor for platelet factor 4. J Biol Chem (2018) 293:6869–82. doi: 10.1074/jbc.RA117.000515
108. Deuel TF, Senior RM, Chang D, Griffin GL, Heinrikson RL, Kaiser ET. Platelet factor 4 is chemotactic for neutrophils and monocytes. Proc Natl Acad Sci U.S.A. (1981) 78:4584–7. doi: 10.1073/pnas.78.7.4584
109. Bdeir K, Gollomp K, Stasiak M, Mei J, Papiewska-Pajak I, Zhao G, et al. Platelet-specific chemokines contribute to the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol (2017) 56:261–70. doi: 10.1165/rcmb.2015-0245OC
110. Ngo AT, Yarovoi I, Zhao G, Sarkar A, Rauova L, Kowalska MA, et al. Platelet factor 4 (PF4) modulates the prothrombotic nature of neutrophil-extracellular traps (NETs): therapeutic implications of a NET-stabilization strategy. Blood (2021) 138:2096. doi: 10.1182/blood-2021-153352
111. Matsumoto K, Yasuoka H, Yoshimoto K, Suzuki K, Takeuchi T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci Rep (2021) 11:222. doi: 10.1038/s41598-020-80685-4
112. Gollomp K, Johnston I, Kim M, Zhai L, Zhao G, Kowalska MA, et al. Platelet factor 4 (PF4)-mediated neutrophil extracellular trap compaction limits endothelial injury and promotes survival following lipopolysaccharide challenge. Blood (2017) 130:997. doi: 10.1182/blood.V130.Suppl_1.997.997
113. Onuk S, Sipahioğlu H, Karahan S, Yeşiltepe A, Kuzugüden S, Karabulut A, et al. Cytokine levels and severity of illness scoring systems to predict mortality in COVID-19 infection. Healthcare (2023) 11:387. doi: 10.3390/healthcare11030387
114. Chen R, Huang Y, Quan J, Liu J, Wang H, Billiar TR, et al. HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon (2020) 6:e05672. doi: 10.1016/j.heliyon.2020.e05672
115. Maugeri N, Campana L, Gavina M, Covino C, de Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12:2074–88. doi: 10.1111/jth.12710
116. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest (2015) 125:4638–54. doi: 10.1172/JCI81660
117. Silvin A, Chapuis N, Dunsmore G, Goubet A-G, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell (2020) 182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002
118. Infantino M, Manfredi M, Alessio MG, Previtali G, Grossi V, Benucci M, et al. Clinical utility of circulating calprotectin to assist prediction and monitoring of COVID-19 severity: an Italian study. J Med Virol (2022) 94:5758–65. doi: 10.1002/jmv.28056
119. Colicchia M, Schrottmaier WC, Perrella G, Reyat JS, Begum J, Slater A, et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbα. Blood (2022) 140:2626–43. doi: 10.1182/blood.2021014966
120. Faraday N, Schunke K, Saleem S, Fu J, Wang B, Zhang J, et al. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PloS One (2013) 8:e71447. doi: 10.1371/journal.pone.0071447
121. Nemmar A, Hoylaerts MF. Neutrophil cathepsin G enhances thrombogenicity of mildly injured arteries via ADP-mediated platelet sensitization. Int J Mol Sci (2022) 23:744. doi: 10.3390/ijms23020744
122. Stoller ML, Basak I, Denorme F, Rowley JW, Alsobrooks J, Parsawar K, et al. Neutrophil cathepsin G proteolysis of protease-activated receptor 4 generates a novel, functional tethered ligand. Blood Adv (2022) 6:2303–8. doi: 10.1182/bloodadvances.2021006133
123. Elaskalani O, Abdol Razak NB, Metharom P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal (2018) 16:24. doi: 10.1186/s12964-018-0235-0
124. Seren S, Derian L, Keleş I, Guillon A, Lesner A, Gonzalez L, et al. Proteinase release from activated neutrophils in mechanically ventilated patients with non-COVID-19 and COVID-19 pneumonia. Eur Respir J (2021) 57:2003755. doi: 10.1183/13993003.03755-2020
125. Beloglazov V, Yatskov I, Nikolaeva A, Lavrenchuk E, DuBuske L. Cathepsin G in patients with SARS-Cov-2 infection of various degrees of severity. J Allergy Clin Immunol (2022) 149:AB59. doi: 10.1016/j.jaci.2021.12.223
126. Leung HH, Perdomo J, Ahmadi Z, Zheng SS, Rashid FN, Enjeti A, et al. NETosis and thrombosis in vaccine-induced immune thrombotic thrombocytopenia. Nat Commun (2022) 13:5206. doi: 10.1038/s41467-022-32946-1
127. Althaus K, Möller P, Uzun G, Singh A, Beck A, Bettag M, et al. Antibody-mediated procoagulant platelets in SARS-CoV-2-Vaccination associated immune thrombotic thrombocytopenia. Haematologica (2021) 106:2170–9. doi: 10.3324/haematol.2021.279000
128. Greinacher A, Selleng K, Palankar R, Wesche J, Handtke S, Wolff M, et al. Insights in ChAdOx1 nCoV-19 vaccine-induced immune thrombotic thrombocytopenia. Blood (2021) 138:2256–68. doi: 10.1182/blood.2021013231
129. Holm S, Kared H, Michelsen AE, Kong XY, Dahl TB, Schultz NH, et al. Immune complexes, innate immunity, and NETosis in ChAdOx1 vaccine-induced thrombocytopenia. Eur Heart J (2021) 42:4064–72. doi: 10.1093/eurheartj/ehab506
130. de Buhr N, Baumann T, Werlein C, Fingerhut L, Imker R, Meurer M, et al. Insights into immunothrombotic mechanisms in acute stroke due to vaccine-induced immune thrombotic thrombocytopenia. Front Immunol (2022) 13:879157. doi: 10.3389/fimmu.2022.879157
131. Hetland G, Fagerhol MK, Wiedmann MK, Søraas AV, Mirlashari MR, Nissen-Meyer LS, et al. Elevated NETs and calprotectin levels after ChAdOx1 nCoV-19 vaccination correlate with the severity of side effects. Vaccines (2022) 10:1267. doi: 10.3390/vaccines10081267
132. Marchandot B, Carmona A, Trimaille A, Curtiaud A, Morel O. Procoagulant microparticles: a possible link between vaccine-induced immune thrombocytopenia (VITT) and cerebral sinus venous thrombosis. J Thromb Thrombolysis (2021) 52:689–91. doi: 10.1007/s11239-021-02505-4
133. Field C, Kim H, Kowalska M, Weitzman M, Arepally G, Cines D, et al. Available at: https://abstracts.isth.org/abstract/in-vivo-murine-studies-demonstrate-that-neutrophil-activation-by-anti-nap2-antibodies-contributes-to-vaccine-induced-immune-thrombocytopenia-and-thrombosis-vitt/.
134. Rauova L, Wang A, Yarovoi S, Khandelwal S, Padmanabhan A, Oleshko O, et al. Vaccine-induced thrombocytopenia and thrombosis (VITT) antibodies recognize neutrophil-activating peptide 2 (NAP2) as well as platelet factor 4 (PF4): mechanistic and clinical implications. Blood (2021) 138:292. doi: 10.1182/blood-2021-151685
135. Brown AJ, Sepuru KM, Sawant KV, Rajarathnam K. Platelet-derived chemokine CXCL7 dimer preferentially exists in the glycosaminoglycan-bound form: implications for neutrophil-platelet crosstalk. Front Immunol (2017) 8:1248. doi: 10.3389/fimmu.2017.01248
136. von Hundelshausen P, Lorenz R, Siess W, Weber C. Vaccine-induced immune thrombotic thrombocytopenia (VITT): targeting pathomechanisms with bruton tyrosine kinase inhibitors. Thromb Haemost (2021) 121:1395–9. doi: 10.1055/a-1481-3039
137. Goldmann L, Duan R, Kragh T, Wittmann G, Weber C, Lorenz R, et al. Oral bruton tyrosine kinase inhibitors block activation of the platelet fc receptor CD32a (FcγRIIA): a new option in HIT? Blood Adv (2019) 3:4021–33. doi: 10.1182/bloodadvances.2019000617
Keywords: COVID-19, platelet activation, coagulopathy, VITT, neutrophil extracellular traps (NET), immunothrombosis
Citation: Hirsch J, Uzun G, Zlamal J, Singh A and Bakchoul T (2023) Platelet-neutrophil interaction in COVID-19 and vaccine-induced thrombotic thrombocytopenia. Front. Immunol. 14:1186000. doi: 10.3389/fimmu.2023.1186000
Received: 14 March 2023; Accepted: 04 May 2023;
Published: 19 May 2023.
Edited by:
Theresa Marie Rossouw, University of Pretoria, South AfricaReviewed by:
Tanya Augustine, University of the Witwatersrand, South AfricaCopyright © 2023 Hirsch, Uzun, Zlamal, Singh and Bakchoul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tamam Bakchoul, VGFtYW0uYmFrY2hvdWxAbWVkLnVuaS10dWViaW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.