 Shengbo Zhang
Shengbo Zhang Cindy Audiger
Cindy Audiger Michaël Chopin
Michaël Chopin Stephen L. Nutt
Stephen L. Nutt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 July 2023
Sec. Immunological Tolerance and Regulation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1182553
This article is part of the Research Topic Transcription Factors in Immunological Disease and Haematological Malignancies View all 12 articles
Dendritic cells (DCs) are sentinel immune cells that form a critical bridge linking the innate and adaptive immune systems. Extensive research addressing the cellular origin and heterogeneity of the DC network has revealed the essential role played by the spatiotemporal activity of key transcription factors. In response to environmental signals DC mature but it is only following the sensing of environmental signals that DC can induce an antigen specific T cell response. Thus, whilst the coordinate action of transcription factors governs DC differentiation, sensing of environmental signals by DC is instrumental in shaping their functional properties. In this review, we provide an overview that focuses on recent advances in understanding the transcriptional networks that regulate the development of the reported DC subsets, shedding light on the function of different DC subsets. Specifically, we discuss the emerging knowledge on the heterogeneity of cDC2s, the ontogeny of pDCs, and the newly described DC subset, DC3. Additionally, we examine critical transcription factors such as IRF8, PU.1, and E2-2 and their regulatory mechanisms and downstream targets. We highlight the complex interplay between these transcription factors, which shape the DC transcriptome and influence their function in response to environmental stimuli. The information presented in this review provides essential insights into the regulation of DC development and function, which might have implications for developing novel therapeutic strategies for immune-related diseases.
Our body is constantly exposed to danger in the form of pathogenic micro-organisms that seek to break through the skin and the mucous membranes that provide the first barrier of defense. The acquisition of mutations in our own cells resulting in their transformation into malignant clones represents another form of danger to which the body must respond in order to avoid the development of cancer. A rare group of heterogeneous immune cells known collectively as dendritic cells (DCs) are central to sensing these dangers and orchestrating the appropriate response, while at the same time ignoring normal healthy cells and commensal micro-organisms.
DCs are a diverse group of cell types that are widely dispersed throughout the body. They act as sentinels to capture exogenous antigens that are processed and presented via either major histocompatibility complex class II (MHC-II) to CD4+ T cells (direct presentation) or shuttled through a specialized pathway to MHC-I to engage CD8+ T cells (cross-presentation) (1–5). Antigen uptake alone is insufficient to fully activate DCs, thus allowing DCs to remain tolerant to harmless antigens derived from healthy tissue or commensal microbes (6–12). However, DCs express an array of pattern-recognition receptor (PRRs) and C-type lectin receptors (CLRs) whose engagement induces maturation and migration, key steps in promoting their interaction with antigen specific T cells and thereby initiating adaptive immunity (13–15).
To face this variety of immune challenges, DCs have evolved into a variety of phenotypically and functionally distinct cellular subsets in both mouse and human (5, 16–19). DCs can be broadly separated into conventional dendritic cells (cDCs), plasmacytoid DCs (pDCs), and monocyte-derived DCs (moDCs), the latter becoming prevalent during inflammation. Conventional DCs can be further divided into type 1 cDC (cDC1s) and type 2 cDCs (cDC2s). Of note Langerhans Cells that were traditionally classified as DCs due to their morphological and phenotypic similarities with DCs and their ability to prime T cell response, are now recognized to be a specialized population of tissue macrophages (20, 21), and therefore their ontogenetic and homeostatic properties differ greatly from DC (22, 23).
Generally, mouse cDCs and moDCs are defined by high cell surface expression of the integrin CD11c (encoded by Itgax) and MHC-II. Beyond the expression of CD11c and MHC-II, additional cell surface markers can be used to distinguish mouse DC subsets. cDC1s co-express the cell surface molecules XCR1, CD24, DEC205, CD8a and CLEC9A (24, 25) (Figure 1). In the peripheral lymphoid and non-lymphoid organs such as the lung, gut and LN, cDC1s also can also be identified as CD103+CD11b- cDCs (26, 27). The splenic cDC2 subset is defined by the presence of CD11b, Sirpα (CD172a) and CD4 on the cell surface (28, 29). Adding to that cDC2s can co-express CD103+CD11b+ in non-lymphoid organs (27, 30). Although the cDC2 compartment has been described as a discrete subset, the advent of single cell technology has revealed a high degree of diversity within this population and some additional markers have been proposed to define the basis of this heterogeneity (discussed later). Under inflammatory conditions, moDCs can respond to the chemokines such as CCL2 and CCL7 and upregulate cell surface expression of MHC-II, CD11c and CD11b, and thus can be easily mistaken as cDC2s (31). Additional markers such as CD64 and MAR-1 can be used to discriminate moDCs from cDC2s (32). pDCs are distinct from the other DC subsets in that they exhibit a lower level of expression of CD11c and MHC-II. pDCs also express a variety of unique markers (compared to cDCs and moDCs), including BST2, B220, and SiglecH (33). Whether pDC belongs to the DC lineage remains at present a matter of debate given that pDCs express some lymphoid markers and overall have a limited capacity to present antigens to T cells compared to the cDC or moDC compartments (34–36).
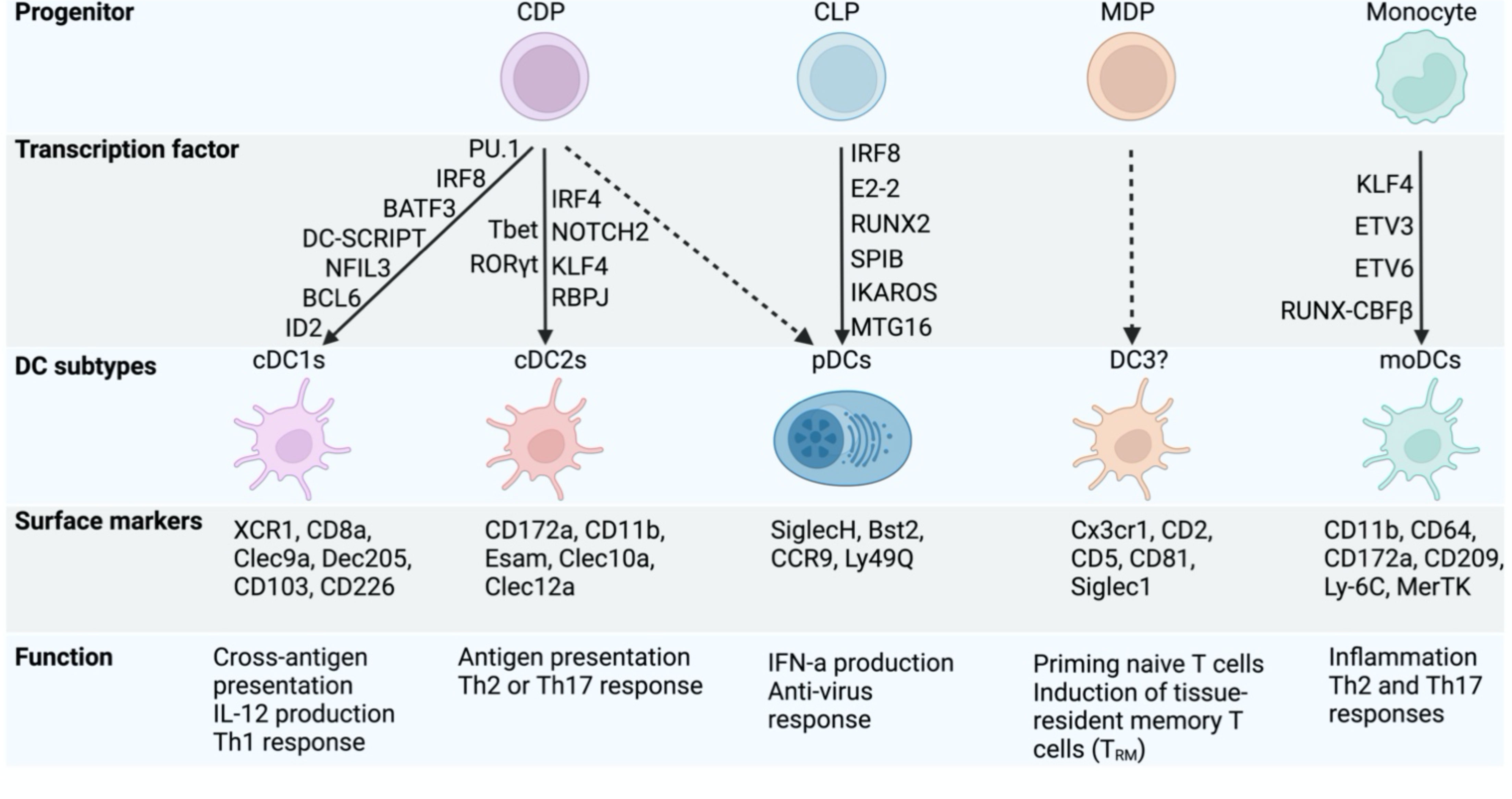
Figure 1 Transcription factors controlling DC specification and function. The figure highlights the development of cDCs subtypes and pDCs from the shared common dendritic cell progenitors (CDP). Some other DCs subtypes (DC3) have also been reported recently in mouse and human and derive from monocyte-dendritic cell progenitors (MDP). The common lymphoid progenitor (CLP) generates pDCs but lack cDC potential. Under inflammation, monocytes can differentiate into monocyte-derived (mono)DCs. Each DC subtype has unique surface markers and attributes in regulating immune response. The transcription factors governing DC lineage specification and function are shown.
Given their critical role in orchestrating adaptive immune responses, high dimensional and throughput techniques such as single cell RNAseq and Cytometry by time of flight (CyTOF), have been applied to the DC lineages. These approaches have revealed unexpected heterogeneity within the DC subsets in both mouse and human, especially the cDC2s (37–39). Single-cell analysis of human mononuclear phagocytes also identified an inflammatory subset of CD5-CD163+CD14+ inflammatory DC3s that were distinct from cDC2s and able to prime Th2 responses (40). The integration of these newly identified subsets into the overall picture of DC development is a very active area of current research (41–43). In this review we will focus on the recent insights on both the transcriptional programming and the ontogeny of the DC lineages and discuss how these findings inform our understanding of the functional specialization of the DC subsets.
cDC1s differentiate principally from the common dendritic cell progenitor (CDP), a population that also gives rise to cDC2s (44, 45). A CDP subset committed to cDC1 fate has been characterized through the expression of CD11c–MHC-II-/intCD117intZbtb46-GFP+ in the bone marrow (46) and pre-cDC1s (CD11c+MHC-II-/intCD135+CD172-Siglec-H-Ly6C-) (47) in the bone marrow and spleen (44, 48, 49). However, cellular barcoding and fate mapping studies have challenged this linear model of differentiation, given that cDC1 imprinting could be detected as early as the hematopoietic stem cell (HSC) (50–52).
Despite the challenges surrounding their origin, there is a very good understanding of the transcriptional mechanisms controlling cDC1 differentiation. cDC1 commitment is dependent on the expression of specific transcription factors (TFs), including BATF3 (Basic Leucine Zipper ATF-Like Transcription Factor 3) (53), IRF8 (Interferon Regulatory Factor 8) (54), PU.1 (55), NFIL3 (Nuclear Factor, Interleukin 3 Regulated) (56, 57), and ID2 (Inhibitor of DNA Binding 2) (58), where the specific inactivation of any of these TFs is associated with a strong defect in cDC1 development (Figure 1). However, this cDC1 deficiency can be rescued by short-term bone marrow reconstitution (59) or over-expressing IRF8 in absence of BATF3 (60), highlighting the significant role of IRF8 and the fine network of TFs allowing cDC1 differentiation.
cDC1 differentiation is intimately linked to optimal expression of IRF8 which is tightly regulated by the spatio-temporal coordinated action of key TFs (Figure 2A). Indeed, its expression is initiated in early DC progenitors, including Lymphoid Primed Multipotent Progenitors (LMPPs) and is dependent on PU.1-induced chromatin remodelling (61). At the LMPP stage, RUNX and CBFβ induce the activation of the distal +56Kb Irf8 enhancer that is essential for the initiation of IRF8 expression (62). Further down the path toward DC differentiation the activity of two additional enhancers have been shown to be pivotal in dictating cDC1 vs pDC fate: +41Kb and +32kb Irf8 enhancers. In progenitors, E protein controls the activation of +41Kb Irf8 enhancer, which results into the commitment of DC progenitors to the pDC lineage. As alluded to earlier IRF8 expression in progenitors is central for cDC1 differentiation, therefore it has been proposed that the upregulation of ID2 can counteract the action of E protein on the +41Kb Irf8 enhancer, which results in the activation of the +32Kb Irf8 enhancer whose accessibility is tightly regulated by BATF3, DC-SCRIPT and IRF8 itself to maintain adequate IRF8 level in pre-cDC1 and cDC1 (46, 63, 64). This key decisional step is also controlled by additional transcription factors, namely ZEB2 (Zinc finger E-box binding homeobox 2) and NFIL3. ZEB2 inhibits ID2 expression of in CDPs thereby promoting pDC differentiation(65, 66). In contrast, NFIL3 acts upstream of ID2 and ZEB2 to control cDC1 differentiation as its binding in CDPs to the -165Kb Zeb2 enhancer prevents ZEB2 expression in CDPs, promoting the transition from a ZEB2hiID2lo CDPs to ZEB2loID2hi CDPs (57, 63). This concomitant reduction in ZEB2 expression and increase in ID2 expression drive the differentiation of cDC1s (63). Beyond the important role for IRF8 in controlling DC fate in progenitors, a role for IRF8 in maintaining cDC1 survival has been postulated (67). However, recent studies suggested that rather than being essential for their survival, IRF8 as well as BATF3 control cDC1 identity in fully differentiated cells as their deletion, in both cases, enables the appearance of cDC1-like cells expressing cDC2 features (68, 69).
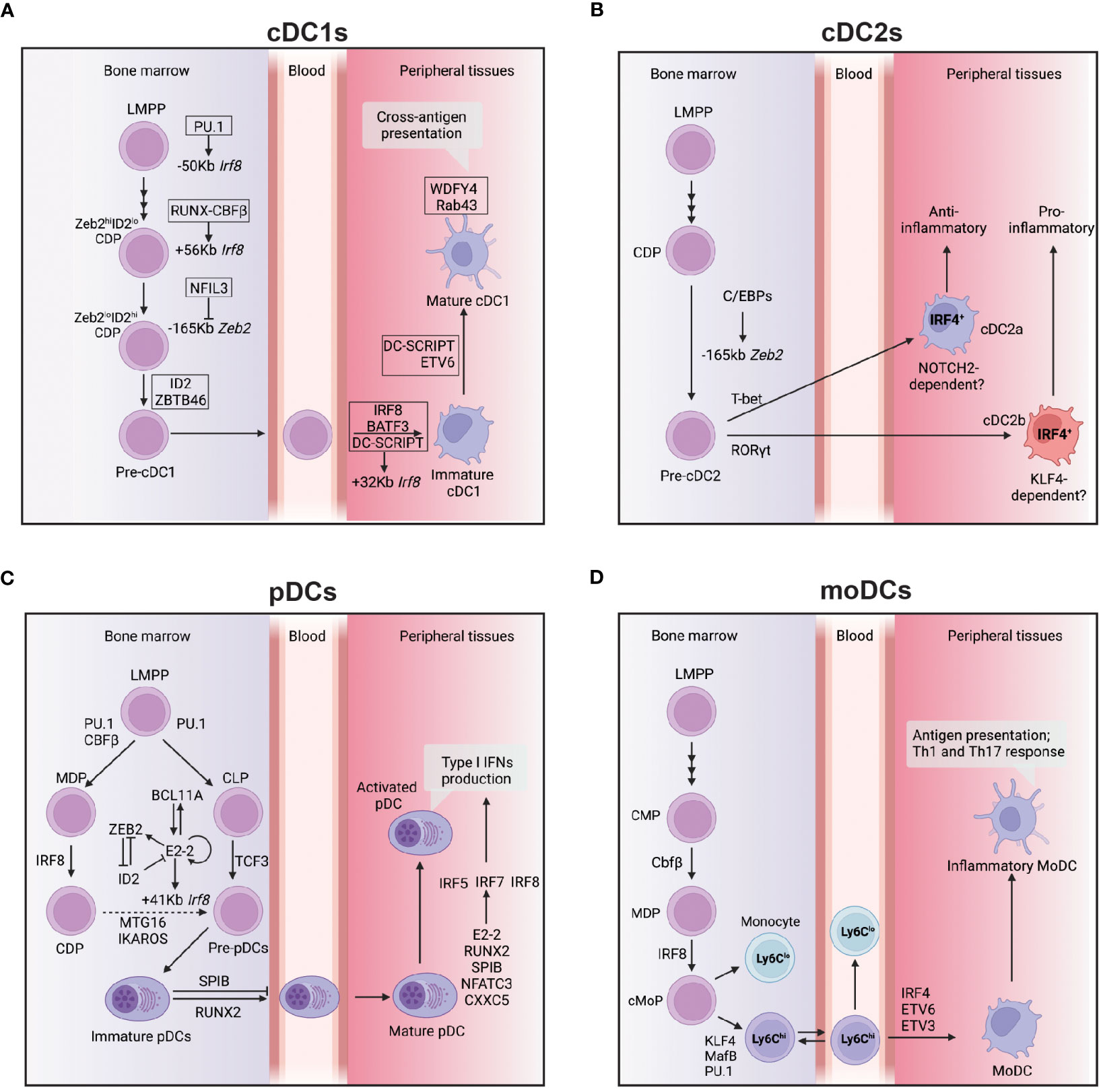
Figure 2 Transcriptional network controlling the development and function of DCs subsets. (A) This figure depicts the transcriptional network that regulates the development and function of cDC1s from bone marrow progenitors to peripheral tissues. The transcription factor IRF8 plays a crucial role in cDC1 development, and its expression is regulated by several enhancers located at -50 kb, +56 kb, and +32 kb relative to the Irf8 gene locus. The transcription factors PU.1, RUNX-CBFβ, BATF3, and DC-SCRIPT activate these enhancers at different stages of cDC1 development. In addition, NFIL3 is required for cDC1 development, and it suppresses ZEB2 expression via binding at-165kb Zeb2 enhancer during the CDP stage. ZBTB46 expression marks the commitment to the cDC1 lineage, while DC-SCRIPT and ETV6 promote the maturation process of cDC1s. WDFY4 is a co-activator that primarily controls the cross-antigen presentation ability of mature cDC1s. (B) cDC2s express IRF4 and can be further divided into two subtypes: cDC2a, which have an anti-inflammatory function, and cDC2b, which have a pro-inflammatory function. The development of cDC2a requires T-bet, while the development of cDC2b requires RoRγt. Both cDC2a and cDC2b develop from a CDP and this process is controlled by C/EBP binding at the -165kb zeb2 enhancer. (C) This figure illustrates the transcriptional network that controls the development and function of pDCs from bone marrow to peripheral tissues. The development of pDCs from multiple lineages requires the transcription factors PU.1, CBFβ, IRF8, and TCF3. The primary regulator of pDC development is E2-2, controlled by a network of transcription factors, including BCL11A, ZEB2, and ID2. E2-2 also controls the expression of IRF8 via binding to the Irf8 + 41kb enhancer region at the CDP stage, possibly through complex formation with other transcription factors such as MTG16. The function of SPIB is to retain immature pDCs in the bone marrow, while RUNX2 expression promotes the egress of pDCs from the bone marrow. Type I IFN production, a significant function of pDCs is mainly controlled by IRF5, IRF7, and IRF8. Other transcription factors, such as E2-2, RUNX2, SPIB, NFATC3, and CXXC5, can directly control IRF7 expression and regulate type 1 IFN production. (D) moDCs develop from Ly6Chi monocytes under the control of several transcription factors, including KLF4, MafB, and PU.1, as well as low levels of IRF8. The final differentiation of moDCs also requires the activity of IRF4, ETV6, and ETV3. Arrows indicate positive regulation, while bars indicate negative regulation.
The importance of cDC1s in the immune system has been highlighted by the interrogation of cDC1-deficient mouse models (53, 70). The absence of cDC1s is associated with a reduction in the control of tumor growth (71–76) and impaired control of viral (53) or parasitic (77) infections. The major role of cDC1s in these contexts is inferred from their capacity to activate naïve CD8+ T cells. Indeed, cDC1s can confer the 3 signals required for the efficient activation of naïve T cells: 1) the presentation of antigen-derived peptides mainly via cross-presentation, 2) co-stimulatory signals and 3) cytokines.
cDC1 are not only important for the activation of naïve CD8+ T cells (78–80) but also for the re-activation of memory CD8+ T cells which confer a faster and higher control of secondary infection, as for example in the case of Listeria monocytogenes (53). In this setting, cDC1s are the main producer of IL-12 and CXCL9 which facilitate the recruitment and activation of memory CD8+ T cells (81). In the tumor context, the production of prostaglandin E2 (PGE2) by tumor cells leads to cDC1 dysfunctionality marked by the downregulation of IRF8, and key effector cytokines such as CXCL9 and IL-12, resulting in poor CD8+ T cell tumor infiltration and ultimately in tumor immune evasion (82, 83). Moreover, cDC1s play a major role in licensing CD4+ T cells for CD8+ T cells activation (84, 85). The cDC1/CD4+ T cell interaction through CD40/CD40L signaling increases expression of CD70 and BCL2L11 in the cDC1, allowing an increase in cDC1 survival and the differentiation and expansion of tumor-specific memory CD8+ T cells (84, 86, 87).
In addition to their role in the initiation of the CD8+ T cell response, cDC1s restrain progenitor of exhausted T (Tpex) cells in the white pulp niche of the spleen in an MHC-I dependent manner. This improves the control of infection by limiting Tpex migration to the red pulp and their differentiation into exhausted T cell (88). How this mechanism can be transposed to the control of tumor growth is still not clear despite evidence of the localization of Tpex in distinct niche in the tumor (89, 90).
Similar to cDC1s, cDC2s also develop from the CDP, although the transcriptional circuitry controlling cDC2 development is less well understood (Figure 2B). As opposed to cDC1s, cDC2s express low amounts of IRF8 and instead highly express IRF4 (Interferon Regulatory Factor 4). Conditional ablation of IRF4 in CD11c+ cells has shown impaired, but not the complete loss of cDC2s (91). A potential explanation for the observation that some cDC2s develop in absence of IRF4 could be that the cDC2 population represents a heterogeneous mix of IRF4-dependent and independent subsets. In line with this possibility, a body of work has highlighted a certain degree of diversity within that compartment and the involvement of different TFs (39, 92).
The first report describing cDC2 diversity revealed that the conditional ablation of NOTCH2 (Neurogenic locus notch homolog protein 2) in CD11c-expressing cells resulted in the reduction of ESAM+ splenic cDC2s and lamina propria CD103+CD11b+ DCs (93). Subsequently, the transcription factor KLF4 (Kruppel-like factor 4) was found to be important for the development of ESAM- cDC2s (43, 94). This evidence indicates that NOTCH2 and KLF4 independently control the development of functionally distinct cDC2 subsets (94, 95).
Yet, a study addressing cDC2 heterogeneity at a single cell level has put forward an alternative model to the one proposed here above (39). Brown et al. suggested that cDC2 could be separated instead into T-BET (T-box expressed in T cells) and RORγt (RAR-related orphan receptor gamma) cDC2s, cDC2a and cDC2b respectively. Importantly, in the aforementioned study, neither the expression of Klf4 or Irf4 enable the discrimination of cDC2a from cDC2b. Instead, the authors proposed the use of additional cell surface markers, namely CLEC10A and CLEC12A, to separate cDC2a and cDC2b. Interestingly, the interrogation of chromatin accessibility revealed that open chromatin regions in cDC2a showed an enrichment for RBPJ (Recombination signal binding protein for immunoglobulin kappa J region) motifs. As RBPJ is the DNA-binding component of the NOTCH TF complex, this finding is compatible with the earlier reported role for NOTCH2 signaling in controlling cDC2 heterogeneity (39, 93).
In addition to the aforementioned role for ZEB2 in controlling pDC differentiation, a role for ZEB2 in controlling cDC2 development has been shown. However, its function remains controversial as conflicting results have been reported. One study showed that conditional deletion of ZEB2 in ItgaxcreZeb2fl/fl mice led to reduced number of splenic cDC2s (65), but a subsequent study failed to confirm this observation (66). This latest study is somewhat contrasting with the development of a novel mouse model lacking cDC2s and other myeloid lineages (57). In this study, a triple mutation of all three NFIL3-C/EBP sites within the -165Kb enhancer of Zeb2 ablated its expression exclusively in the myeloid compartment and led to the complete loss of pre-cDC2 specification and mature cDC2 development in vivo (57). Whilst the nature of this discrepancy warrants further investigation, these studies also highlighted ZEB2 as a critical regulator of pDC development through its repressive activity on ID2, as well as its important role for monocytes commitment as these 2 populations were strongly affected in this mouse model (57).
Compared to cDC1s, cDC2s appear more efficient in presenting antigens via MHC-II molecules to CD4+ T cells (1, 96). However, cDC2s are not equally able to present soluble versus cell associated antigens. CD4+ T cell proliferation in response to soluble antigen was unperturbed in mice lacking cDC1s (Xcr1DTR mice or Batf3-/- mice), demonstrating that cDC2s compensate for the lack of cDC1s in this setting (53, 97). In contrast, cDC2s are far less efficient than cDC1s in the uptake and processing of cell-associated antigens, and thus display a limited capacity to prime CD8+ T cells through this route (98).
As alluded earlier, mice lacking IRF4 were originally used to define the function of cDC2s (91). These studies led to define a key role for cDC2s in the regulation of Th2 and Th17 immune responses aiming to eliminate extracellular pathogens (Nippostrongylus brasiliensis) and parasites (Aspergillus fumigatus), respectively (91, 99). At that time, it remained unclear how cDC2s could direct such distinctive responses. Some clarification for this division of labor came from studies highlighting the distinct roles for NOTCH2 dependent and KLF4 dependent cDC2s. For example, in the gut NOTCH2-dependent cDC2s were the critical source of IL-23 that were required for clearance of extracellular pathogens such as Citrobacter Rodentium though the induction of a Th17 biased immune response (100, 101). In addition, NOTCH2-dependent splenic cDC2s were required to promote T follicular helper (TFH) cell and germinal center (GC) B cell formation in response to Listeria monocytogenes (102, 103). In contrast, it was found that conditional deletion of Klf4 in DCs was detrimental for Th2, but not Th17, immune responses in mice (94). In line with the above, a STAT6/KLF4 dependent CD11blow cDC2 population localized in the skin has been shown to mediate Th2 immune responses (43).
cDC2s are also important for the T cell response to viral infection. Following PV (single-stranded RNA pneumonia virus) infection, cDC2s can acquire a hybrid phenotype characterized by increased IRF8 expression and the capacity to prime both CD4+ and CD8+ T cells. The acquisition of these cDC1-like properties by cDC2s was dependent on the signaling via Toll-like receptors and the type 1 interferon receptor (104). Additionally, the induction of TFH cell differentiation was dependent on the presentation of viral antigens at the T/B border by migratory cDC2s (102). Furthermore, LN resident cDC2s are strategically positioned to capture the influenza A virus (105) and other blood born antigens (106) resulting in the rapid initiation of T cell responses, independent of migratory DCs influx. While moDCs were also reported to activate T cells under similar conditions (107, 108), some studies have suggested that inflammatory cDC2s can acquire moDC like features, such as the expression of MAR-1 and CD64, and the moDCs will express cDC2 signature genes including CD11b and CD172a, suggesting that the antigen presentation capacity of moDCs may actually be due to contamination by inflammatory cDC2s (104, 105). In agreement with this conclusion, the use of CD26 as an additional marker to differentiate inflammatory cDC2 from moDCs, highlighted the limited antigen presentation capacity of CD26- moDCs (104).
Collectively, these studies highlight the functional specificities of the various cDC2 subtypes within different organs. Deciphering the molecular mechanisms underpinning this diversity is a prerequisite to define the role of these different subsets of cDC2s in initiating adaptive immune responses in the context of pathogens, virus infection and tumor clearance, as this knowledge will provide a rational framework for their use in clinical settings.
The application of single-cell RNAseq technology to DCs has led to many reports of novel DC subtypes (38, 40, 92, 109). The use of different annotation strategies to define populations with otherwise very similar transcriptomic features has created a good deal of confusion in the field (110). The status of the DC3 population represents an example of this issue.
DC3s were initially identified in the blood of humans through single-cell RNA sequencing (38). The subsequent studies phenotypically characterized the DC3 population as CD163+CD14+ DCs that accumulate in the blood of patients with systemic lupus erythematosus (SLE) (40). DC3s display an intermediate phenotype and function between cDC2s and monocytes and are characterized by low expression of IRF8 (111). Unlike cDC1s and cDC2s, the development of DC3s relies on GM-CSF, but not FLT3L, and it is developmentally independent of the CDP (92). Functionally, these cells have been proposed to promote the differentiation of naïve CD8+ T cells into tissue-homing CD103+ T cells (92).
The AXL+ DC subpopulation was also reported in the blood of humans, alongside the DC3 population, displaying an intermediate phenotype between cDC2s and pDCs (38). This population was characterized by the expression of Siglec6 and AXL. Similarly, in mice, transitional DCs (tDCs), also referred to as “pDC-like” cells, with characteristics spanning between cDC2s and pDCs, were observed during steady-state and influenza infection, and appear to be the equivalent to the AXL+ DCs in humans (109). It has been recently proposed that these “pDC-like” cells are pre-cDC2s and require KLF4 for both their development and function (112).
Other similar single-cell transcriptomic studies have identified another DC population that exhibits an “activated” DC phenotype and is referred to as “DC3” in both mouse and human (113). This DC population lacks the canonical cDC1s and cDC2s gene signature but expresses the matured cDC1 and cDC2 signatures (113). Similar population have also been described as CCR7+LAMP3+ DCs, Mreg DCs or ISG+ DCs within tumors (114–116). It is important to note that these “activated” DC populations represent developmental states of both cDC1s and cDC2s and therebefore they are not to be confounded with CD163+CD14+ DCs (DC3s) reported by Dutertre, Cytlak, Bourdely and Villani et al. Currently, it is recommended to designate this “activated” DC population as “CCR7+ DCs” due to the consistent detection of CCR7, a common marker for DC activation and maturation, in various contexts except ISG+ DCs (110, 116).
Sorting out the cellular relationships between the cDC1, cDC2, DC3 and CCR7+DCs populations is one of the key goals for the DC field moving forward. Regardless of their development origins, identifying the environmental cues and the molecular mechanisms driving DC3 and CCR7+ DC phenotype and functional attributes also warrants further investigation.
pDCs are a distinct cell type first identified through their capacity to rapidly produce large amounts of type I interferons (IFNα/β) (117–120). Whether pDCs developed from lymphoid or myeloid progenitors has remained a controversial question for more than two decades (34, 121). Similar to the development of cDCs, Flt3 signaling is required for optimal pDC development (122). Yet as opposed to cDCs, that can only originate from the myeloid progenitors, Flt3+ CMPs, CDPs and CLPs have all been shown to retain pDC potential both in vitro and in vivo following adoptive transfer (44, 45, 48, 49, 123–125). These findings led to the concept that pDC have a dual origin: myeloid and lymphoid (Figure 2C). However, the myeloid origin of the pDCs is being disputed by different groups (35, 36, 125, 126). This issue has been revisited with IL-7R+ lymphoid progenitors being proposed to be the main source for pDCs in vivo (126). A predominantly lymphoid origin for the pDCs is also supported by their expression history of the recombination activating gene 1 (Rag1) and the rearrangement of the D-J regions of the Igh locus (125, 127). In an effort to distinguish the properties of myeloid- vs lymphoid-derived pDCs, it was found that the myeloid-derived Zbtb46+ pDCs have a distinct transcriptome that resulted in them being more efficient than lymphoid-derived pDCs in their capability to present antigens to T cells (125). While this study is accordance with earlier reports pointing to the dual origin of pDC (127), these findings were subsequently challenged by a study that proposed that a CD115- Ly6D+ lymphoid progenitors are the sole source of pDCs in vivo (126). Crucially, the definition of a lymphoid or myeloid origin of pDCs largely depends on the markers used to track the development history of pDCs. For example, Dress et al. used CD2 as a lymphoid lineage marker to trace the development history of pDCs, and conclude that the pDCs are of lymphoid origin (41, 126). However, CD2 expression is not restricted to the lymphoid lineage as 20% of the cDC are fate mapped in the hCD2-iCre+/–R26-stop-EYFP+ mouse model (128), thus this model cannot completely rule out the participation of myeloid biased progenitor to the pDC pool. Adding to that, clonal tracing of HSC and CX3CR1+ progenitors using FlipJump system and single-cell transcriptome and phenotype analysis (CITE-seq) suggested that cDCs and pDCs share a common progenitor (129). Further characterization of the pDCs specific transcriptional program will be helpful to improve our understanding of pDC ontogeny and the heterogeneity of this population.
The development of pDCs requires the TF E2-2 (E protein encoded by Tcf4) (Figure 2C). E2-2 deficient mice die in utero, but transfer of Tcf4-/- fetal liver cells into irradiated WT recipients results in the complete loss of pDCs from the BM and all peripheral lymphoid organs, but has no impact on the development of other myeloid or lymphoid cell types (33). E2-2 is a member of the basic helix-loop-helix superfamily of TFs that has long (E2-2L) and short (E2-2S) isoforms (130). E2-2S is expressed in all hematopoietic progenitors and different types of mature immune cells, but E2-2L is preferentially expressed in pDCs and binds to the pDC specific 3’ enhancer of Tcf4 to maintain E2-2s expression via a positive feedback loop (130). E2-2s expression initiates in HSCs and is further upregulated during pDC development. E2-2s forms a complex with Mtg16 (myeloid translocation gene on chromosome 16) to directly control the expression of key genes involved in pDC development and function, including CCR9, TLR9, Bst2 and B220 (131). In DC progenitors, ID2 as an E protein inhibitor binds E2-2s preventing its binding to DNA, and thereby inhibits their pDC potential (63). In contrast, ZEB2 expression in progenitors prevents ID2 expression, enabling E2-2s to promote pDC development. In line with the above, constitutive deletion of -165kb Zeb2 enhancer featuring a cluster of E box motifs, results in lack of ZEB2 expression, increased ID2 expression that prevents pDC differentiation (132). Thus, the coordinate action of E2-2L, E2-2s, ID2 and ZEB2 dictates pDCs development at steady state.
Other TFs have been implicated in the cellular fate of BM progenitors. PU.1 is highly expressed in myeloid and lymphoid BM progenitors, but its expression level is substantially reduced following the commitment of progenitors to the pDC lineage (55, 122, 133, 134). High expression of PU.1 in cDC was shown to be essential to maintain their identity as PU.1 deficient cDCs gained pDC like features (55). Thus, it is conceivable that downmodulation of PU.1 in progenitors constitutes a key instrumental step in allowing pDC differentiation (135). In line with this, the expression of PU.1 is negatively regulated by BCL11A (B-cell chronic lymphocytic leukaemia/lymphoma 11A), a critical regulator of pDC development (136). Adding to that, loss of PU.1 in CD11c+ cells resulted in an increased differentiation of progenitor toward the pDC lineage, although PU.1 deficient pDCs were dysfunctional, as IFNα production was reduced in PU.1 deficient pDCs (55). In contrast to the down-modulation of PU.1 following pDCs commitment, IRF8 expression is increased markedly during pDC development (67). Thus, it is somewhat surprising, that IRF8 deficiency in CD11c+ cells has no impact on the development of pDCs. This is in fact due to a compensatory mechanism provided by IRF4 as double knockout mice lack pDCs (67). Although IRF8 is dispensable for pDC differentiation, it is essential for their IFNα production, thus indicating a nonredundant role for IRF8 in controlling pDC function.
Spi-B is another ETS family TF that is highly expressed in pDCs (137). In contrast to the decreased PU.1 expression following pDCs development, Spi-B expression is substantially increased from progenitors to mature pDCs. Germline deletion of SpiB results in decreased pDC numbers in the BM but their numbers are increased in peripheral organs (138). These data suggests that Spi-B is dispensable for pDC differentiation but a critical regulator of pDC homeostasis. Having said that, its role and its mode of action in pDCs remains under investigated. In contrast to BM, the TF RUNX2 (RUNX family transcription factor 2) promotes pDC their egress, as germline ablation or tamoxifen induced deletion of RUNX2 result in reduced number of peripheral pDCs, whilst RUNX2 is dispensable for their differentiation in the BM (139, 140). Two mechanisms were proposed. Sawai et al. showed that RUNX2 was required for the expression of chemokine receptors on the cell surface of pDCs including CCR2 and CCR5 that were required for the migration of pDCs from BM into the periphery in response to their ligands (139). In contrast, Chopin et al. demonstrated that RUNX2 deficiency resulted in increased expression of CXCR4, a key chemokine receptor associated with BM tropism (140). Spi-B and RUNX2 are not only critical regulators of pDC homeostatic in the periphery but also have been both shown to be critical for IFNα production by pDC, though the regulation of Irf7 (138, 140).
BCL11A is a zinc-finger TF and is known to regulate lymphoid development (141). Both BCL11A and PU.1 control Flt3 expression in early hematopoietic progenitors (142), which is required for pDC development and their homeostasis. ChIP-seq data showed that BCL11A bound to the Tcf4 proximal promoter and knockdown of BCL11A strongly reduced E2-2 expression (136). Interestingly, downregulation of Bcl11a occurred after Tcf4 deletion in BM derived pDCs (143), indicating a positive feedforward loop between BCL11A and E2-2 in controlling pDC development.
IKAROS (encoded by IKAROS Family Zinc Finger 1 (Ikzf1)) is a zinc-finger DNA-binding protein that homo- or hetero-dimerizes with other IKAROS family members to suppress the gene expression. IKAROS prevents premature cDC gene expression in CDPs and promotes pDC development (144, 145). The relationship between IKAROS with other TFs that control the development and function of pDCs has not been studied.
Collectively, these studies have revealed a dynamic TF network that regulates the development of pDCs within the hematopoietic system. These studies also highlight a critical point in the current debate about whether pDCs and cDCs share a common ancestor. These findings suggest that the lineage trajectories of DCs are dictated by mutual antagonism between transcription factors (E2.2/ZEB2 vs ID2/NFIL3 or PU.1 vs BCL11A), thus inferring a close relationship between pDCs and cDCs.
Unlike cDCs, pDCs have limited capacity to present antigens. Instead, their key feature is the rapid production of type I IFNs (IFNα/β) after exposure to the ligands for TLR7 (recognize ssRNA) and TLR9 (recognize CpG), especially after the viral infection (33, 146, 147). The early production of type I IFNs by pDCs initiates the anti-viral gene expression program in many cell types and promotes the expansion of NK cells and virus specific CTLs for viral clearance (146, 147). This type I IFN production results in the apoptosis of activated pDCs, potentially limiting the scale of inflammatory response and preventing pathology associated with an overly active anti-viral immune response (148). This control appears important as aberrant type I IFN production by pDCs is strongly linked to the development of autoimmune diseases like SLE and systemic sclerosis in both mouse models and human (149, 150).
The ambiguous nature of moDCs has hampered our capacity to define some of the key TFs associated with their differentiation. Lineage tracing experiments have demonstrated that moDCs derive from a separate myelopoiesis branch distinct from the one producing cDCs and pDCs (151). In contrast to the requirement of high dose IRF8 for cDC1 development, moDCs develop in a relatively low concentration of IRF8. This expression of IRF8 is driven by Irf8 + 56kb enhancer whose activation is controlled by RUNX-CBFβ (62). The differentiation of Ly6C+ monocytes into moDCs or macrophages is controlled by the TFs IRF4 and MafB (MAF BZIP Transcription Factor B), and PU.1 (Figure 2D) (152–154). The differentiation of mouse monocyte into moDCs in presence of GM-CSF and IL-4 requires IRF4. In its absence, the cells differentiate into macrophages (155). It also had been reported that MafB expression will push the human monocytes into the macrophage pathway, while high concentration of PU.1 will suppress MafB and thus promote differentiation into moDCs (152, 156). Apart from PU.1, a most recent study found that ETV3 and ETV6 are able to repress macrophages development potential in monocytes by suppressing MafB expression in both mouse and human (154). Thus, moDCs use a distinct repertoire of TFs compared to those that promote cDC development.
Monocytes represent a major cell population in the circulation, from which they are recruited into the tissues by inflammatory cues and give rise to both macrophage and moDCs. Normally, monocytes express Ly6C and macrophage colony stimulating factor receptor (M-CSFR/CD115) and respond to GM-CSF (157). The moDCs can be easily confounded for cDCs in tissues as they share a variety of cell surface markers including the “canonical DC markers” MHC-II and CD11c, as well as the cDC1 marker CD24 and the cDC2 marker CD172a (158). In addition to sharing cDC phenotypic features, moDCs can present antigen to both CD4+ and CD8+ T cells. Notably, moDCs can cross-present antigen released from certain microorganisms to CD8+ T cells under acute inflammation condition and might replace some (41), but not all anti-infection functions of cDCs (77). As per their cDCs counterpart, moDCs express costimulatory molecules that support the differentiation of CTLs (159) and present antigen directly to CD4+ T cells promoting their differentiation into Th17 cells (160). Furthermore, moDCs are strong producers of proinflammatory cytokines including IL-1β, TNFα, IL-23 (161), and IL-12 in cancer (162). Collectively, although moDCs arise from a distinct myeloid branch compared to cDCs, both subsets share a substantial number of overlapping phenotypic and functional characteristics after activation.
Recent advances in the field of DC research have provided new insights into the heterogeneity and functional diversity of DC subsets. Studies on the transcriptional regulation of DC development and function have led to the identification of key TFs and their targets that shape the transcriptome and function of DCs. In-depth phenotyping of DCs has also identified novel DC subtypes, such as DC3, highlighting the need for continued investigation into the ontogeny of DCs. While much progress has been made, much is still to be learned about the intricate connections between different TFs and their doses regulating the differentiation and activation of DCs.
Whilst we try to build a comprehensive map of the transcriptional network governing DC heterogeneity, which will be essential for their clinical application, there is an urgent need to understand how DC functionalities, independently of their origin, are shaped by environmental signals. To fulfill the long-recognized potential of DC based therapy to treat malignancies, we believe that an in-depth characterization of the signals that drive their diversity and a better under understanding of the environmental cues that shape their functional attributes is urgently required.
SZ, CA and MC contributed to original draft preparation. SZ contributed to the figures. SN, MC, CA and SZ contributed to review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by a Cancer Council Victoria Postdoctoral Research Fellowship to SZ and NHMRC grants and fellowships to CA (2021228), MC (1196235) and SN (1155243 and 2000461).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315(5808):107–11. doi: 10.1126/science.1136080
2. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol (2012) 12(8):557–69. doi: 10.1038/nri3254
3. van Endert P. Intracellular recycling and cross-presentation by MHC class I molecules. Immunol Rev (2016) 272(1):80–96. doi: 10.1111/imr.12424
4. Amigorena S. Editorial overview: usual and unusual ways to antigen presentation. Curr Opin Immunol (2020) 64:iii–iv. doi: 10.1016/j.coi.2020.10.008
5. Roquilly A, Mintern JD, Villadangos JA. Spatiotemporal adaptations of macrophage and dendritic cell development and function. Annu Rev Immunol (2022) 40:525–57. doi: 10.1146/annurev-immunol-101320-031931
6. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi: 10.1038/32588
7. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194(6):769–79. doi: 10.1084/jem.194.6.769
8. Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U.S.A. (2002) 99(1):351–8. doi: 10.1073/pnas.231606698
9. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol (2008) 181(10):6923–33. doi: 10.4049/jimmunol.181.10.6923
10. Jones A, Bourque J, Kuehm L, Opejin A, Teague RM, Gross C, et al. Immunomodulatory functions of BTLA and HVEM govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity (2016) 45(5):1066–77. doi: 10.1016/j.immuni.2016.10.008
11. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol (2017) 198(6):2223–31. doi: 10.4049/jimmunol.1601629
12. Gargaro M, Scalisi G, Manni G, Briseno CG, Bagadia P, Durai V, et al. Indoleamine 2,3-dioxygenase 1 activation in mature cDC1 promotes tolerogenic education of inflammatory cDC2 via metabolic communication. Immunity (2022) 55(6):1032–1050 e14. doi: 10.1016/j.immuni.2022.05.013
13. Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and langerhans cells. Nat Rev Immunol (2002) 2(2):77–84. doi: 10.1038/nri723
14. Manicassamy S, Pulendran B. Modulation of adaptive immunity with toll-like receptors. Semin Immunol (2009) 21(4):185–93. doi: 10.1016/j.smim.2009.05.005
15. Canton J, Blees H, Henry CM, Buck MD, Schulz O, Rogers NC, et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat Immunol (2021) 22(2):140–53. doi: 10.1038/s41590-020-00824-x
16. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi: 10.1038/nri3712
17. Amon L, Lehmann CHK, Baranska A, Schoen J, Heger L, Dudziak D. Transcriptional control of dendritic cell development and functions. Int Rev Cell Mol Biol (2019) 349:55–151. doi: 10.1016/bs.ircmb.2019.10.001
18. Nutt SL, Chopin M. Transcriptional networks driving dendritic cell differentiation and function. Immunity (2020) 52(6):942–56. doi: 10.1016/j.immuni.2020.05.005
19. Park HY, Ashayeripanah M, Chopin M. Harnessing dendritic cell diversity in cancer immunotherapy. Curr Opin Immunol (2023) 82:102341. doi: 10.1016/j.coi.2023.102341
20. Chopin M, Nutt SL. Establishing and maintaining the langerhans cell network. Semin Cell Dev Biol (2015) 41:23–9. doi: 10.1016/j.semcdb.2014.02.001
21. Doebel T, Voisin B, Nagao K. Langerhans cells - the macrophage in dendritic cell clothing. Trends Immunol (2017) 38(11):817–28. doi: 10.1016/j.it.2017.06.008
22. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity (2016) 45(3):669–84. doi: 10.1016/j.immuni.2016.08.015
23. Zhan Y, Zhang Y, Zhang S, Coughlan H, Baldoni PL, Jacquelot N, et al. Differential requirement for the polycomb repressor complex 2 in dendritic cell and tissue-resident myeloid cell homeostasis. Sci Immunol (2021) 6(63):eabf7268. doi: 10.1126/sciimmunol.abf7268
24. Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L, et al. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med (1992) 176(1):47–58. doi: 10.1084/jem.176.1.47
25. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
26. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity (2012) 37(1):60–73. doi: 10.1016/j.immuni.2012.04.012
27. Watchmaker PB, Lahl K, Lee M, Baumjohann D, Morton J, Kim SJ, et al. Comparative transcriptional and functional profiling defines conserved programs of intestinal DC differentiation in humans and mice. Nat Immunol (2014) 15(1):98–108. doi: 10.1038/ni.2768
28. Vremec D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol (2000) 164(6):2978–86. doi: 10.4049/jimmunol.164.6.2978
29. Gurka S, Hartung E, Becker M, Kroczek RA. Mouse conventional dendritic cells can be universally classified based on the mutually exclusive expression of XCR1 and SIRPalpha. Front Immunol (2015) 6:35. doi: 10.3389/fimmu.2015.00035
30. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol (2012) 13(9):888–99. doi: 10.1038/ni.2370
31. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against leishmania. Immunity (2007) 26(4):519–31. doi: 10.1016/j.immuni.2007.01.017
32. Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity (2013) 38(2):322–35. doi: 10.1016/j.immuni.2012.10.016
33. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell (2008) 135(1):37–48. doi: 10.1016/j.cell.2008.09.016
34. Reizis B, Idoyaga J, Dalod M, Barrat F, Naik S, Trinchieri G, et al. Reclassification of plasmacytoid dendritic cells as innate lymphocytes is premature. Nat Rev Immunol (2023) 23(5):336–7. doi: 10.1038/s41577-023-00864-y
35. Ziegler-Heitbrock L, Ohteki T, Ginhoux F, Shortman K, Spits H. Reclassifying plasmacytoid dendritic cells as innate lymphocytes. Nat Rev Immunol (2023) 23(1):1–2. doi: 10.1038/s41577-022-00806-0
36. Ziegler-Heitbrock L, Ohteki T, Ginhoux F, Shortman K, Spits H. Reply to 'Reclassification of plasmacytoid dendritic cells as innate lymphocytes is premature'. Nat Rev Immunol (2023) 23(5):338–9. doi: 10.1038/s41577-023-00866-w
37. Alcantara-Hernandez M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, et al. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity (2017) 47(6):1037–1050 e6. doi: 10.1016/j.immuni.2017.11.001
38. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science (2017) 356(6335). doi: 10.1126/science.aah4573
39. Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallee VP, Mendoza A, et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell (2019) 179(4):846–863 e24. doi: 10.1016/j.cell.2019.09.035
40. Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, et al. Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity (2019) 51(3):573–589 e8. doi: 10.1016/j.immuni.2019.08.008
41. Anderson DA, Dutertre CA, Ginhoux F, Murphy KM. Genetic models of human and mouse dendritic cell development and function. Nat Rev Immunol (2021) 21(2):101–15. doi: 10.1038/s41577-020-00413-x
42. Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis ESC. Dendritic cells revisited. Annu Rev Immunol (2021) 39:131–66. doi: 10.1146/annurev-immunol-061020-053707
43. Mayer JU, Hilligan KL, Chandler JS, Eccles DA, Old SI, Domingues RG, et al. Homeostatic IL-13 in healthy skin directs dendritic cell differentiation to promote TH2 and inhibit TH17 cell polarization. Nat Immunol (2021) 22(12):1538–50. doi: 10.1038/s41590-021-01067-0
44. Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol (2007) 8(11):1217–26. doi: 10.1038/ni1522
45. Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol (2007) 8(11):1207–16. doi: 10.1038/ni1518
46. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol (2015) 16(7):708–17. doi: 10.1038/ni.3197
47. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol (2015) 16(7):718–28. doi: 10.1038/ni.3200
48. Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol (2007) 8(6):578–83. doi: 10.1038/ni1462
49. Waskow C, Liu K, Darrasse-Jeze G, Guermonprez P, Ginhoux F, Merad M, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol (2008) 9(6):676–83. doi: 10.1038/ni.1615
50. Poltorak MP, Schraml BU. Fate mapping of dendritic cells. Front Immunol (2015) 6:199. doi: 10.3389/fimmu.2015.00199
51. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8α(+) conventional DC clonogenic progenitor. Nat Immunol (2015) 16(7):708–17. doi: 10.1038/ni.3197
52. Naik SH. Dendritic cell development at a clonal level within a revised 'continuous' model of haematopoiesis. Mol Immunol (2020) 124:190–7. doi: 10.1016/j.molimm.2020.06.012
53. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322(5904):1097–100. doi: 10.1126/science.1164206
54. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med (2002) 196(11):1415–25. doi: 10.1084/jem.20021263
55. Chopin M, Lun AT, Zhan Y, Schreuder J, Coughlan H, D'Amico A, et al. Transcription factor PU.1 promotes conventional dendritic cell identity and function via induction of transcriptional regulator DC-SCRIPT. Immunity (2019) 50(1):77–90 e5. doi: 10.1016/j.immuni.2018.11.010
56. Kashiwada M, Pham NL, Pewe LL, Harty JT, Rothman PB. NFIL3/E4BP4 is a key transcription factor for CD8alpha(+) dendritic cell development. Blood (2011) 117(23):6193–7. doi: 10.1182/blood-2010-07-295873
57. Liu TT, Kim S, Desai P, Kim DH, Huang X, Ferris ST, et al. Ablation of cDC2 development by triple mutations within the Zeb2 enhancer. Nature (2022) 607(7917):142–8. doi: 10.1038/s41586-022-04866-z
58. Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol (2003) 4(4):380–6. doi: 10.1038/ni903
59. Seillet C, Jackson JT, Markey KA, Brady HJ, Hill GR, Macdonald KP, et al. CD8alpha+ DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood (2013) 121(9):1574–83. doi: 10.1182/blood-2012-07-445650
60. Theisen DJ, Ferris ST, Briseno CG, Kretzer N, Iwata A, Murphy KM, et al. Batf3-dependent genes control tumor rejection induced by dendritic cells independently of cross-presentation. Cancer Immunol Res (2019) 7(1):29–39. doi: 10.1158/2326-6066.CIR-18-0138
61. Schonheit J, Kuhl C, Gebhardt ML, Klett FF, Riemke P, Scheller M, et al. PU.1 level-directed chromatin structure remodeling at the Irf8 gene drives dendritic cell commitment. Cell Rep (2013) 3(5):1617–28. doi: 10.1016/j.celrep.2013.04.007
62. Murakami K, Sasaki H, Nishiyama A, Kurotaki D, Kawase W, Ban T, et al. A RUNX-CBFbeta-driven enhancer directs the Irf8 dose-dependent lineage choice between DCs and monocytes. Nat Immunol (2021) 22(3):301–11. doi: 10.1038/s41590-021-00871-y
63. Bagadia P, Huang X, Liu TT, Durai V, Grajales-Reyes GE, Nitschke M, et al. An Nfil3-Zeb2-Id2 pathway imposes Irf8 enhancer switching during cDC1 development. Nat Immunol (2019) 20(9):1174–85. doi: 10.1038/s41590-019-0449-3
64. Zhang S, Coughlan HD, Ashayeripanah M, Seizova S, Kueh AJ, Brown DV, et al. Type 1 conventional dendritic cell fate and function are controlled by DC-SCRIPT. Sci Immunol (2021) 6(58):eabf4432. doi: 10.1126/sciimmunol.abf4432
65. Scott CL, Soen B, Martens L, Skrypek N, Saelens W, Taminau J, et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J Exp Med (2016) 213(6):897–911. doi: 10.1084/jem.20151715
66. Wu X, Briseno CG, Grajales-Reyes GE, Haldar M, Iwata A, Kretzer NM, et al. Transcription factor Zeb2 regulates commitment to plasmacytoid dendritic cell and monocyte fate. Proc Natl Acad Sci U.S.A. (2016) 113(51):14775–80. doi: 10.1073/pnas.1611408114
67. Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity (2016) 45(3):626–40. doi: 10.1016/j.immuni.2016.08.013
68. Lukowski SW, Rodahl I, Kelly S, Yu M, Gotley J, Zhou C, et al. Absence of Batf3 reveals a new dimension of cell state heterogeneity within conventional dendritic cells. iScience (2021) 24(5):102402. doi: 10.1016/j.isci.2021.102402
69. Lanca T, Ungerback J, Da Silva C, Joeris T, Ahmadi F, Vandamme J, et al. IRF8 deficiency induces the transcriptional, functional, and epigenetic reprogramming of cDC1 into the cDC2 lineage. Immunity (2022) 55(8):1431–1447 e11. doi: 10.1016/j.immuni.2022.06.006
70. Dalod M, Scheu S. Dendritic cell functions in vivo: a user's guide to current and next- generation mutant mouse models. Eur J Immunol (2022) 52(11):1712–49. doi: 10.1002/eji.202149513
71. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell (2017) 31(5):711–723 e4. doi: 10.1016/j.ccell.2017.04.003
72. Zhou Y, Slone N, Chrisikos TT, Kyrysyuk O, Babcock RL, Medik YB, et al. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103(+) conventional dendritic cells. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2019-000474
73. Cueto FJ, Del Fresno C, Brandi P, Combes AJ, Hernandez-Garcia E, Sanchez-Paulete AR, et al. DNGR-1 limits Flt3L-mediated antitumor immunity by restraining tumor-infiltrating type I conventional dendritic cells. J Immunother Cancer (2021) 9(5). doi: 10.1136/jitc-2020-002054
74. Ghislat G, Cheema AS, Baudoin E, Verthuy C, Ballester PJ, Crozat K, et al. NF-kappaB-dependent IRF1 activation programs cDC1 dendritic cells to drive antitumor immunity. Sci Immunol (2021) 6(61). doi: 10.1126/sciimmunol.abg3570
75. Zhang S, Chopin M, Nutt SL. Type 1 conventional dendritic cells: ontogeny, function, and emerging roles in cancer immunotherapy. Trends Immunol (2021) 42(12):1113–27. doi: 10.1016/j.it.2021.10.004
76. Svensson-Arvelund J, Cuadrado-Castano S, Pantsulaia G, Kim K, Aleynick M, Hammerich L, et al. Expanding cross-presenting dendritic cells enhances oncolytic virotherapy and is critical for long-term anti-tumor immunity. Nat Commun (2022) 13(1):7149. doi: 10.1038/s41467-022-34791-8
77. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by toxoplasma gondii tachyzoites. Immunity (2011) 35(2):249–59. doi: 10.1016/j.immuni.2011.08.008
78. Bevan MJ. Cross-priming for a secondary cytotoxic response to minor h antigens with h-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med (1976) 143(5):1283–8. doi: 10.1084/jem.143.5.1283
79. den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med (2000) 192(12):1685–96. doi: 10.1084/jem.192.12.1685
80. Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci U.S.A. (2006) 103(28):10729–34. doi: 10.1073/pnas.0601956103
81. Alexandre YO, Ghilas S, Sanchez C, Le Bon A, Crozat K, Dalod M. XCR1+ dendritic cells promote memory CD8+ T cell recall upon secondary infections with listeria monocytogenes or certain viruses. J Exp Med (2016) 213(1):75–92. doi: 10.1084/jem.20142350
82. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172(5):1022–1037 e14. doi: 10.1016/j.cell.2018.01.004
83. Felix Bayerl PM, Donakonda S, Veit R. Buchholz BUS, ttcher JPB. Tumor-derived prostaglandin E2 programs cDC1 dysfunction to impair intratumoral orchestration of anti-cancer T cell responses. Immunity (2023) 56(6):P1341–1358. doi: 10.1016/j.immuni.2023.05.011
84. Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature (2020) 584(7822):624–9. doi: 10.1038/s41586-020-2611-3
85. Wu R, Murphy KM. DCs at the center of help: origins and evolution of the three-cell-type hypothesis. J Exp Med (2022) 219(7). doi: 10.1084/jem.20211519
86. Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, et al. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med (2007) 204(5):1095–106. doi: 10.1084/jem.20070176
87. Wu R, Ohara RA, Jo S, Liu TT, Ferris ST, Ou F, et al. Mechanisms of CD40-dependent cDC1 licensing beyond costimulation. Nat Immunol (2022) 23(11):1536–50. doi: 10.1038/s41590-022-01324-w
88. Dahling S, Mansilla AM, Knopper K, Grafen A, Utzschneider DT, Ugur M, et al. Type 1 conventional dendritic cells maintain and guide the differentiation of precursors of exhausted T cells in distinct cellular niches. Immunity (2022) 55(4):656–670 e8. doi: 10.1016/j.immuni.2022.03.006
89. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature (2019) 576(7787):465–70. doi: 10.1038/s41586-019-1836-5
90. Gueguen P, Metoikidou C, Dupic T, Lawand M, Goudot C, Baulande S, et al. Contribution of resident and circulating precursors to tumor-infiltrating CD8(+) T cell populations in lung cancer. Sci Immunol (2021) 6(55). doi: 10.1126/sciimmunol.abd5778
91. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38(5):970–83. doi: 10.1016/j.immuni.2013.04.011
92. Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, et al. Transcriptional and functional analysis of CD1c(+) human dendritic cells identifies a CD163(+) subset priming CD8(+)CD103(+) T cells. Immunity (2020) 53(2):335–352 e8. doi: 10.1016/j.immuni.2020.06.002
93. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity (2011) 35(5):780–91. doi: 10.1016/j.immuni.2011.08.013
94. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity (2015) 42(5):916–28. doi: 10.1016/j.immuni.2015.04.017
95. Tussiwand R, Gautier EL. Transcriptional regulation of mononuclear phagocyte development. Front Immunol (2015) 6:533. doi: 10.3389/fimmu.2015.00533
96. Kamphorst AO, Guermonprez P, Dudziak D, Nussenzweig MC. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J Immunol (2010) 185(6):3426–35. doi: 10.4049/jimmunol.1001205
97. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell (2019) 177(3):556–571 e16. doi: 10.1016/j.cell.2019.02.005
98. Theisen DJ, Davidson JT, Briseno CG, Gargaro M, Lauron EJ, Wang Q, et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science (2018) 362(6415):694–9. doi: 10.1126/science.aat5030
99. Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity (2013) 39(4):733–43. doi: 10.1016/j.immuni.2013.08.029
100. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med (2015) 21(7):719–29. doi: 10.1038/nm.3895
101. Arnold IC, Mathisen S, Schulthess J, Danne C, Hegazy AN, Powrie F. CD11c(+) monocyte/macrophages promote chronic helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol (2016) 9(2):352–63. doi: 10.1038/mi.2015.65
102. Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, et al. Migratory CD11b(+) conventional dendritic cells induce T follicular helper cell-dependent antibody responses. Sci Immunol (2017) 2(18). doi: 10.1126/sciimmunol.aam9169
103. Briseno CG, Satpathy AT, Davidson JT, Ferris ST, Durai V, Bagadia P, et al. Notch2-dependent DC2s mediate splenic germinal center responses. Proc Natl Acad Sci U.S.A. (2018) 115(42):10726–31. doi: 10.1073/pnas.1809925115
104. Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity (2020) 52(6):1039–56. doi: 10.1016/j.immuni.2020.04.005
105. Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim YA, et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol (2010) 11(5):427–34. doi: 10.1038/ni.1856
106. Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity (2015) 42(1):172–85. doi: 10.1016/j.immuni.2014.12.024
107. Cruz JL, Perez-Giron JV, Ludtke A, Gomez-Medina S, Ruibal P, Idoyaga J, et al. Monocyte-derived dendritic cells enhance protection against secondary influenza challenge by controlling the switch in CD8(+) T-cell immunodominance. Eur J Immunol (2017) 47(2):345–52. doi: 10.1002/eji.201646523
108. Harvey AG, Graves AM, Uppalapati CK, Matthews SM, Rosenberg S, Parent EG, et al. Dendritic cell-natural killer cell cross-talk modulates T cell activation in response to influenza a viral infection. Front Immunol (2022) 13:1006998. doi: 10.3389/fimmu.2022.1006998
109. Leylek R, Alcantara-Hernandez M, Lanzar Z, Ludtke A, Perez OA, Reizis B, et al. Integrated cross-species analysis identifies a conserved transitional dendritic cell population. Cell Rep (2019) 29(11):3736–3750 e8. doi: 10.1016/j.celrep.2019.11.042
110. Ginhoux F, Guilliams M, Merad M. Expanding dendritic cell nomenclature in the single-cell era. Nat Rev Immunol (2022) 22(2):67–8. doi: 10.1038/s41577-022-00675-7
111. Cytlak U, Resteu A, Pagan S, Green K, Milne P, Maisuria S, et al. Differential IRF8 transcription factor requirement defines two pathways of dendritic cell development in humans. Immunity (2020) 53(2):353–370 e8. doi: 10.1016/j.immuni.2020.07.003
112. Rodrigues PF, Kouklas A, Cvijetic G, Bouladoux N, Mitrovic M, Desai JV, et al. pDC-like cells are pre-DC2 and require KLF4 to control homeostatic CD4 T cells. Sci Immunol (2023) 8(80):eadd4132. doi: 10.1126/sciimmunol.add4132
113. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity (2019) 50(5):1317–1334 e10. doi: 10.1016/j.immuni.2019.03.009
114. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell (2019) 179(4):829–845 e20. doi: 10.1016/j.cell.2019.10.003
115. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature (2020) 580(7802):257–62. doi: 10.1038/s41586-020-2134-y
116. Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity (2021) 55(2):308–23. doi: 10.1016/j.immuni.2021.10.020
117. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science (1996) 272(5270):1947–50. doi: 10.1126/science.272.5270.1947
118. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med (1999) 5(8):919–23. doi: 10.1038/11360
119. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284(5421):1835–7. doi: 10.1126/science.284.5421.1835
120. Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol (2001) 31(10):3026–37. doi: 10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H
121. Shigematsu H, Reizis B, Iwasaki H, Mizuno S, Hu D, Traver D, et al. Plasmacytoid dendritic cells activate lymphoid-specific genetic programs irrespective of their cellular origin. Immunity (2004) 21(1):43–53. doi: 10.1016/j.immuni.2004.06.011
122. Carotta S, Dakic A, D'Amico A, Pang SH, Greig KT, Nutt SL, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity (2010) 32(5):628–41. doi: 10.1016/j.immuni.2010.05.005
123. Schlitzer A, Loschko J, Mair K, Vogelmann R, Henkel L, Einwachter H, et al. Identification of CCR9- murine plasmacytoid DC precursors with plasticity to differentiate into conventional DCs. Blood (2011) 117(24):6562–70. doi: 10.1182/blood-2010-12-326678
124. Onai N, Kurabayashi K, Hosoi-Amaike M, Toyama-Sorimachi N, Matsushima K, Inaba K, et al. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity (2013) 38(5):943–57. doi: 10.1016/j.immuni.2013.04.006
125. Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol (2018) 19(7):711–22. doi: 10.1038/s41590-018-0136-9
126. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat Immunol (2019) 20(7):852–64. doi: 10.1038/s41590-019-0420-3
127. Sathe P, Vremec D, Wu L, Corcoran L, Shortman K. Convergent differentiation: myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood (2013) 121(1):11–9. doi: 10.1182/blood-2012-02-413336
128. Siegemund S, Shepherd J, Xiao C, Sauer K. hCD2-iCre and vav-iCre mediated gene recombination patterns in murine hematopoietic cells. PloS One (2015) 10(4):e0124661. doi: 10.1371/journal.pone.0124661
129. Feng J, Pucella JN, Jang G, Alcantara-Hernandez M, Upadhaya S, Adams NM, et al. Clonal lineage tracing reveals shared origin of conventional and plasmacytoid dendritic cells. Immunity (2022) 55(3):405–422 e11. doi: 10.1016/j.immuni.2022.01.016
130. Grajkowska LT, Ceribelli M, Lau CM, Warren ME, Tiniakou I, Nakandakari Higa S, et al. Isoform-specific expression and feedback regulation of e protein TCF4 control dendritic cell lineage specification. Immunity (2017) 46(1):65–77. doi: 10.1016/j.immuni.2016.11.006
131. Ghosh HS, Ceribelli M, Matos I, Lazarovici A, Bussemaker HJ, Lasorella A, et al. ETO family protein Mtg16 regulates the balance of dendritic cell subsets by repressing Id2. J Exp Med (2014) 211(8):1623–35. doi: 10.1084/jem.20132121
132. Huang X, Ferris ST, Kim S, Choudhary MNK, Belk JA, Fan C, et al. Differential usage of transcriptional repressor Zeb2 enhancers distinguishes adult and embryonic hematopoiesis. Immunity (2021) 54(7):1417–1432 e7. doi: 10.1016/j.immuni.2021.04.015
133. Nutt SL, Metcalf D, D'Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med (2005) 201(2):221–31. doi: 10.1084/jem.20041535
134. Chopin M, Seillet C, Chevrier S, Wu L, Wang H, Morse HC, et al. Langerhans cells are generated by two distinct PU.1-dependent transcriptional networks. J Exp Med (2013) 210(13):2967–80. doi: 10.1084/jem.20130930
135. Carotta S, Wu L, Nutt SL. Surprising new roles for PU.1 in the adaptive immune response. Immunol Rev (2010) 238(1):63–75. doi: 10.1111/j.1600-065X.2010.00955.x
136. Ippolito GC, Dekker JD, Wang YH, Lee BK, Shaffer AL, Lin J, et al. Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci U.S.A. (2014) 111(11):E998–1006. doi: 10.1073/pnas.1319228111
137. Schotte R, Rissoan MC, Bendriss-Vermare N, Bridon JM, Duhen T, Weijer K, et al. The transcription factor spi-b is expressed in plasmacytoid DC precursors and inhibits T-, b-, and NK-cell development. Blood (2003) 101(3):1015–23. doi: 10.1182/blood-2002-02-0438
138. Sasaki I, Hoshino K, Sugiyama T, Yamazaki C, Yano T, Iizuka A, et al. Spi-b is critical for plasmacytoid dendritic cell function and development. Blood (2012) 120(24):4733–43. doi: 10.1182/blood-2012-06-436527
139. Sawai CM, Sisirak V, Ghosh HS, Hou EZ, Ceribelli M, Staudt LM, et al. Transcription factor Runx2 controls the development and migration of plasmacytoid dendritic cells. J Exp Med (2013) 210(11):2151–9. doi: 10.1084/jem.20130443
140. Chopin M, Preston SP, Lun ATL, Tellier J, Smyth GK, Pellegrini M, et al. RUNX2 mediates plasmacytoid dendritic cell egress from the bone marrow and controls viral immunity. Cell Rep (2016) 15(4):866–78. doi: 10.1016/j.celrep.2016.03.066
141. Fell HP, Smith RG, Tucker PW. Molecular analysis of the t(2;14) translocation of childhood chronic lymphocytic leukemia. Science (1986) 232(4749):491–4. doi: 10.1126/science.3961491
142. Wu X, Satpathy AT, Kc W, Liu P, Murphy TL, Murphy KM. Bcl11a controls Flt3 expression in early hematopoietic progenitors and is required for pDC development in vivo. PloS One (2013) 8(5):e64800. doi: 10.1371/journal.pone.0064800
143. Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity (2010) 33(6):905–16. doi: 10.1016/j.immuni.2010.11.023
144. Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, et al. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood (2006) 108(13):4025–34. doi: 10.1182/blood-2006-03-007757
145. Mastio J, Simand C, Cova G, Kastner P, Chan S, Kirstetter P. Ikaros cooperates with notch activation and antagonizes TGFbeta signaling to promote pDC development. PloS Genet (2018) 14(7):e1007485. doi: 10.1371/journal.pgen.1007485
146. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity (2010) 33(6):955–66. doi: 10.1016/j.immuni.2010.11.020
147. Cervantes-Barragan L, Lewis KL, Firner S, Thiel V, Hugues S, Reith W, et al. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci U.S.A. (2012) 109(8):3012–7. doi: 10.1073/pnas.1117359109
148. Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med (2011) 208(12):2367–74. doi: 10.1084/jem.20110654
149. Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med (2014) 211(10):1969–76. doi: 10.1084/jem.20132522
150. Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med (2018) 10(423). doi: 10.1126/scitranslmed.aam8458
151. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell (2019) 178(6):1509–1525 e19. doi: 10.1016/j.cell.2019.08.009
152. Bakri Y, Sarrazin S, Mayer UP, Tillmanns S, Nerlov C, Boned A, et al. Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood (2005) 105(7):2707–16. doi: 10.1182/blood-2004-04-1448
153. Wu X, Briseno CG, Durai V, Albring JC, Haldar M, Bagadia P, et al. Mafb lineage tracing to distinguish macrophages from other immune lineages reveals dual identity of langerhans cells. J Exp Med (2016) 213(12):2553–65. doi: 10.1084/jem.20160600
154. Villar J, Cros A, De Juan A, Alaoui L, Bonte PE, Lau CM, et al. ETV3 and ETV6 enable monocyte differentiation into dendritic cells by repressing macrophage fate commitment. Nat Immunol (2023) 24(1):84–95. doi: 10.1038/s41590-022-01374-0
155. Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep (2016) 15(11):2462–74. doi: 10.1016/j.celrep.2016.05.025
156. Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, et al. Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity (2017) 47(3):582–596 e6. doi: 10.1016/j.immuni.2017.08.016
157. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity (2013) 39(5):925–38. doi: 10.1016/j.immuni.2013.10.004
158. Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol (2012) 12(2):101–13. doi: 10.1038/nri3149
159. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol (2017) 17(6):349–62. doi: 10.1038/nri.2017.28
160. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38(2):336–48. doi: 10.1016/j.immuni.2012.10.018
161. Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev (2008) 19(1):41–52. doi: 10.1016/j.cytogfr.2007.10.004
Keywords: dendritic cells, transcription factor, IRF8, cDCs, pDCs
Citation: Zhang S, Audiger C, Chopin M and Nutt SL (2023) Transcriptional regulation of dendritic cell development and function. Front. Immunol. 14:1182553. doi: 10.3389/fimmu.2023.1182553
Received: 08 March 2023; Accepted: 28 June 2023;
Published: 14 July 2023.
Edited by:
Karsten Kretschmer, Technical University Dresden, GermanyReviewed by:
Diana Dudziak, University Hospital Erlangen, GermanyCopyright © 2023 Zhang, Audiger, Chopin and Nutt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengbo Zhang, emhhbmcuc2hAd2VoaS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.