 Marie J. Y. Reolo1
Marie J. Y. Reolo1 Masayuki Otsuka1Justine Jia Wen Seow2Joycelyn Lee3
Masayuki Otsuka1Justine Jia Wen Seow2Joycelyn Lee3 Yun Hua Lee1Phuong H. D. Nguyen1Chun Jye Lim1
Yun Hua Lee1Phuong H. D. Nguyen1Chun Jye Lim1 Martin Wasser1Camillus Chua1Tony K. H. Lim4Wei Qiang Leow4Alexander Chung5Brian K. P. Goh5,6Pierce K. H. Chow5,6,7
Martin Wasser1Camillus Chua1Tony K. H. Lim4Wei Qiang Leow4Alexander Chung5Brian K. P. Goh5,6Pierce K. H. Chow5,6,7 Ramanuj DasGupta2
Ramanuj DasGupta2 Joe Poh Sheng Yeong4
Joe Poh Sheng Yeong4 Valerie Chew1*
Valerie Chew1*- 1Translational Immunology Institute (TII), SingHealth-DukeNUS Academic Medical Centre, Singapore, Singapore
- 2Genome Institute of Singapore (GIS), Agency for Science, Technology and Research (A*STAR), Singapore, Singapore
- 3Division of Medical Oncology, National Cancer Centre Singapore, Singapore, Singapore
- 4Department of Anatomical Pathology, Singapore General Hospital, Singapore, Singapore
- 5Department of Hepatopancreatobiliary and Transplant Surgery, Division of Surgery and Surgical Oncology, Singapore General Hospital and National Cancer Centre Singapore, Singapore, Singapore
- 6SingHealth-DukeNUS Academic Surgery Program, Duke-NUS Graduate Medical School, Singapore, Singapore
- 7Division of Medical Science, National Cancer Center, Singapore, Singapore
Introduction: Despite recent advances in immunotherapy for hepatocellular carcinoma (HCC), the overall modest response rate underscores the need for a better understanding of the tumor microenvironment (TME) of HCC. We have previously shown that CD38 is widely expressed on tumor-infiltrating leukocytes (TILs), predominantly on CD3+ T cells and monocytes. However, its specific role in the HCC TME remains unclear.
Methods: In this current study, we used cytometry time-of-flight (CyTOF), bulk RNA sequencing on sorted T cells, and single-cell RNA (scRNA) sequencing to interrogate expression of CD38 and its correlation with T cell exhaustion in HCC samples. We also employed multiplex immunohistochemistry (mIHC) for validating our findings.
Results: From CyTOF analysis, we compared the immune composition of CD38-expressing leukocytes in TILs, non-tumor tissue-infiltrating leukocytes (NIL), and peripheral blood mononuclear cells (PBMC). We identified CD8+ T cells as the dominant CD38-expressing TILs and found that CD38 expression was significantly higher in CD8+ TRM in TILs than in NILs. Furthermore, through transcriptomic analysis on sorted CD8+ TRM from HCC tumors, we observed a higher expression of CD38 along with T cell exhaustion genes, including PDCD1 and CTLA4, compared to the circulating memory CD8 T cells from PBMC. This was validated by scRNA sequencing that revealed co-expression of CD38 with PDCD1, CTLA4, and ITGAE (CD103) in T cells from HCC tumors. The protein co-expression of CD38 and PD-1 on CD8+ T cells was further demonstrated by mIHC on HCC FFPE tissues, marking CD38 as a T cell co-exhaustion marker in HCC. Lastly, the higher proportions of CD38+PD-1+ CD8+ T cells and CD38+PD-1+ TRM were significantly associated with the higher histopathological grades of HCC, indicating its role in the aggressiveness of the disease.
Conclusion: Taken together, the concurrent expression of CD38 with exhaustion markers on CD8+ TRM underpins its role as a key marker of T cell exhaustion and a potential therapeutic target for restoring cytotoxic T cell function in HCC.
1 Introduction
Hepatocellular carcinoma (HCC) accounts for at least 75% of primary liver cancer and is the third leading cause of cancer death globally (1). Inhibitors against programmed cell death protein (PD)-1 and its ligand PD-L1 or collectively known as Immune checkpoint blockades (ICBs), are the most recent therapeutic options for many advanced solid malignancies, including HCC (2, 3). However, the objective response rate (ORR) to ICB in patients with HCC remains modest at 10-20% for monotherapy (4, 5) or less than 30% for combination immunotherapy (6). The variation in clinical outcomes after immunotherapy underscores the need to better understand the immune landscape of HCC and to discover new biomarkers or therapeutic targets to improve clinical outcomes for patients with HCC.
CD38 is a multifunctional type II transmembrane glycoprotein with enzymatic functions that are involved in immune cell activation and regulation in homeostasis, inflammation and various diseases (7–9). Although initially thought to be expressed on T cells, CD38 is also expressed on other lymphoid and myeloid cell populations (7). CD38-expressing immune cells have been detected in the TME of numerous cancer types and are often associated with cancer progression (8, 10, 11). Therefore, there is a growing interest in CD38 as a novel therapeutic target for immune-based therapies, especially in solid tumors. In HCC, CD38 expression on myeloid cells has been associated with better HCC patient survival after surgery (12) and anti-PD1 therapy (13). Collectively, these studies suggest that CD38 has a promising potential as both an immunotherapeutic target and a biomarker for therapeutic response in HCC.
Our previous report has shown an association of CD38+ tumor-infiltrating leukocytes (TIL) with HCC prognosis and identified CD3+ T cells and monocytes as the dominant CD38-expressing immune populations among the TILs (14). While other studies have demonstrated the potential roles of CD38+ tumor-associated macrophages in the clinical outcome of HCC patients (12, 13), the role of CD38 in tumor-infiltrating T cells in the HCC TME remains to be further elucidated. In this study, we investigated the immune composition of CD38-expressing leukocytes in TILs, non-tumor tissue-infiltrating leukocytes (NILs), and peripheral blood mononuclear cells (PBMCs) using cytometry by time-of-flight (CyTOF), bulk and single-cell RNA sequencing as well as validation using multiplex immunohistochemistry (mIHC). We identified CD8+ TRM cells as the predominant CD38-expressing immune cells in TILs, and their association with the T cell exhaustion signature in HCC TME and the higher histopathological tumor grades. Taken altogether, our findings show that CD38 is a marker of exhausted CD8+ TRM cells in HCC that could be a potential therapeutic target in conjunction with other ICBs to restore cytotoxic T cell function.
2 Methods
2.1 Patient samples
Patient samples collection from the National University Hospital (NUH), National Cancer Center Singapore and Singapore General Hospital was approved by the NUH CIRB and SingHealth Central Institution Review Board (CIRB) (CIRB Ref: 2018/2112 and 2016/2626), respectively. Peripheral blood, tumor and adjacent non-tumor liver samples were collected from 17 HCC patients; each provided written informed consent, with demographics and clinical characteristics as described in Table S1. Depending on the tumor size, the tumors collected from each patient were dissected into two to five sectors (total tumor sectors= 60), separated by at least 1 cm to account for intratumoral heterogeneity (15). The adjacent non-tumor liver tissue, which is at least 2 cm away from the tumor, was also harvested. Each tissue sector was allocated for downstream analysis using Cytometry by Time-of-Flight (CyTOF), and bulk tissue RNA sequencing. A subset of them was subjected to single-cell RNA sequencing or mIHC, as described below. The peripheral blood mononuclear cells (PBMC) (n=17) were isolated from the blood using the Ficoll-Paque Plus (GE Healthcare) density gradient centrifugation. The tumor-infiltrating leukocytes (TIL) (from a total of n=60 tumor sectors) and non-tumor tissue-infiltrating leukocytes (NIL) (n=17) were isolated using enzymatic digestion with 500μg/mL collagenase IV (Thermo Fisher Scientific; Cat#: 17104019) and 50μg/mL DNase I (Roche, Indianapolis, IN; Cat#: 4716728001) for 30 min in 37°C. The cells were stored in liquid nitrogen with 10% DMSO in fetal bovine serum (FBS) until further analysis.
2.2 Cytometry by Time-of-Flight
TILs, NILs and PBMCs were thawed and rested for 1 h in RPMI medium with 10% FBS and 1% penicillin/streptomycin. The cells were stained as previously described (16) using a panel of 41 heavy-metal conjugated antibodies against surface and intracellular markers, including three anti-human CD45 barcode antibodies (Table S2). The data were obtained using Helios equipped with the CyTOF® 6.7 system control software (Fluidigm).
The generated files were analyzed by FlowJo (v.10.2): live single-cells (cisplatin-negative and DNA-intercalator-positive) were debarcoded to each sample file based on their unique CD45 barcodes as previously described (17). The resulting data was down-sampled to 1x104 cells/sample for subsequent analysis. Two-dimensional t-distributed (t-SNE) plots were generated to represent the expression of individual immune markers or various immune subsets. The data was further validated by manual gating using FlowJo (v.10.2).
2.3 Cell sorting and RNA sequencing
Tissue-resident memory CD8+ T cells (TRM) from TILs and matched circulating memory CD8+ T cells from PBMCs were sorted from seven HCC patients as previously described (16). Briefly, cells were stained with the fluorochrome-conjugated anti-human antibodies (Table S3) for 30 min, followed by DAPI staining (for live/dead cell stain). Then, using the FACS Aria II flow cytometer (BD Biosciences), the stained cells were sorted at an efficiency of 91%–100% into live TRM (DAPI-CD45+CD3+CD8+CD45RO+CD103+) from TILs or circulating memory CD8+ T cells (DAPI-CD45+CD3+CD8+CD45RO+) from PBMCs for bulk RNA sequencing (Figure S1A).
For RNA sequencing, total RNAs from sorted CD8+ TRM cells from TILs (n=7) and matched circulating memory CD8+ T cells from PBMCs (n=6; one sample was omitted due to poor RNA quality) were isolated using Picopure RNA-Isolation kit (Arcturus, Ambion) and cDNA was generated using the SMART-Seq® v4 UltraTM Low Input RNA Kit for Sequencing (Clontech, USA). With the Nextera XT DNA Library Prep Kit (Illumina, USA), indexed libraries were created and multiplexed for 2x 101 bp-sequencing. The raw reads were aligned to the Human Reference Genome hg19 via STAR (18) and the gene-level expected counts were calculated using RSEM (19). The samples with protein-coding genes of more than 0.5 counts were retained. Analyses on differentially expressed genes (DEGs) were performed using R packages DESeq2 and Limma (20) with the false discovery rate (FDR) adjusted for multiple testing using the Benjamini-Hochberg. Functional pathway analyses were performed using DAVID pathway analysis v.6.8, with adjusted p-value < 0.01.
2.4 Single-cell RNA sequencing
Single cells were isolated from tumor and adjacent non-tumor tissues from 14 patients with HCC by enzymatic digestion as described previously (21). Briefly, dead cells were removed using a dead cell removal kit (Miltenyi, Cat#: 130-090-101). CD45+ cells were enriched using CD45 MicroBeads (Miltenyi, Cat#: 130-045-801) before CD45+ and CD45- cells were processed using the Chromium Single Cell 30 (v2 Chemistry) platform (10x Genomics, Pleasanton, CA). All data were then aggregated using cellranger aggr by normalizing all runs to the same sequencing depth. Downstream analysis was performed using Scanpy (version 1.8.1); All genes expressed by a minimum of 30 cells were considered, and cells with fewer than 200 genes and greater than 5% mitochondrial content were excluded from the analysis. A subset of T cells from the whole atlas was obtained. Leiden (scanpy.api.tl.leiden) with the resolution parameters set at 0.35 and 0.25 for NILs and TILs, respectively, was utilized as the clustering algorithm for data visualization and downstream analysis.
2.5 Multiplexed fluorescent immunohistochemistry
mIHC was performed on formalin-fixed paraffin-embedded (FFPE) tissues from 12 HCC patients (CIRB Reference No: 2016/2613). The tissues were stained with anti-human antibodies for CD8, PD-1 and CD38 (Table S4) using the OPAL™ 7-color IHC Kit (Perkin-Elmer) and DAPI. Images were acquired using Vectra 3.0 Pathology Imaging System Microscope (Perkin-Elmer) and analyzed using InForm v2.1 (Perkin-Elmer) and Imaris v9.1.0 (Bitplane).
2.6 Statistics
All statistical analyses were performed using GraphPad Prism (V9.0). Comparisons of cell frequencies between groups were done using the non-parametric one-way ANOVA Kruskal-Wallis test with Dunn’s posthoc multiple comparison test. The paired two-tailed Wilcoxon-matched T-test or unpaired two-sided T-test with Welch’s correction were applied for pairwise comparisons. Spearman’s Regression Analysis was used for correlation analysis.
3 Results
3.1 CD38 expression profile in PBMCs, NILs and TILs from HCC patients
To investigate CD38 expression in HCC, we used CyTOF to profile PBMCs, NILs and TILs obtained from 17 HCC patients (Table S1) with an antibody panel of 41 surface and intracellular immune markers (Table S2). In addition, to account for intratumoral heterogeneity (15), two to five tumor sectors (n=60) were collected from patients along with matched NILs (n = 17) and PBMCs (n = 17) samples.
Two-dimensional tSNE plots were generated from the CyTOF data, showing individual cells clustered based on the similarity in the expression of each immune marker (Figure 1A). The immune cells were clustered into five major immune lineages: CD3+ T cells, CD56+ Natural Killer (NK) cells, CD3+CD56+ NKT cells, CD14+ monocytes and CD19+ B cells (Figure 1A). tSNE plots based on CD38 expression show ubiquitous expression across major lineages from TILs, NILs and PBMCs (Figure 1B). There is no significant difference in the proportions of total immune cells expressing CD38 (CD38+CD45+) across PBMCs, NILs and TILs (Figures 1C, D). However, each compartment exhibited a distinct CD38+ immune profile (Figures 1A, B).
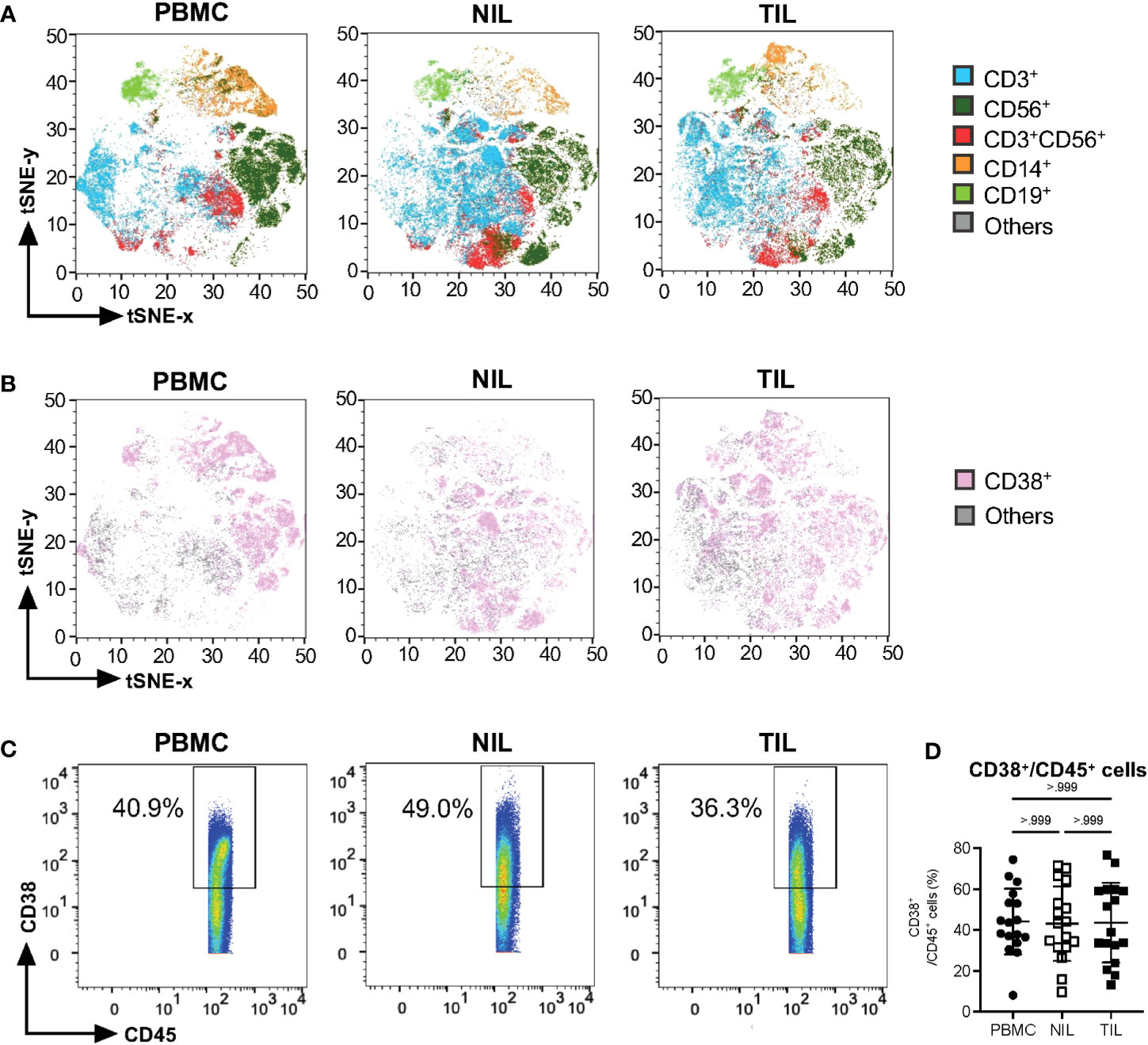
Figure 1 Characterization of CD38 expression profiles in PBMC, NIL and TIL of HCC. (A) tSNE plots showing five major immune lineages from PBMC, NIL and TIL. Each immune cluster is represented by one color. (B) tSNE plots demonstrating ubiquitous CD38 expression on five immune subsets in PBMC, NIL and TIL. (C) Representative manual gating of total CD38+CD45+ immune cells from the PBMC, NIL and TIL. (D) Percentage of CD38+ immune cells in PBMC, NIL and TIL. Data is represented as mean ± SD. Friedman one-way ANOVA test calculated by Dunn’s post-hoc multiple pairwise comparisons was performed. (A, B, D) PBMC, peripheral blood mononuclear cells (n=17); NIL, Non-tumor-infiltrating lymphocyte (n=17); TIL, tumor-infiltrating lymphocyte (n=60 from 2-5 tumor sectors per case).
3.2 Elevated CD38 expression on CD3+ T cells in HCC TILs
Despite the lack of significant differences in the proportions of CD38+CD45+ across PBMCs, NILs or TILs, a distinct CD38 expression profile could be observed (Figure 1B). We hence examined the compositions of specific immune subsets among the CD38-expressing immune cells by manual gating (Figure S2A). First, we observed that among the CD38+ immune cells, the proportion of CD19+ B cells is higher in the PBMCs than in the NILs and TILs; while CD56+ NK cells are significantly lower in the TILs as compared to the PBMCs and NILs; and the proportion of CD3+CD56+ NKT cells is significantly higher in the NILs and TILs compared to the PBMCs (Figures 2A, S2B). Consistent with a previous report (14), CD3+ T cells represent the main immune population that expresses CD38, particularly in TILs compared to PBMCs (Figure 2B). The CD14+ myeloid cells, on the other hand, follow the opposite trend, with the lowest proportion among the CD38+ immune cells in TILs as compared to PBMCs (Figure 2B).
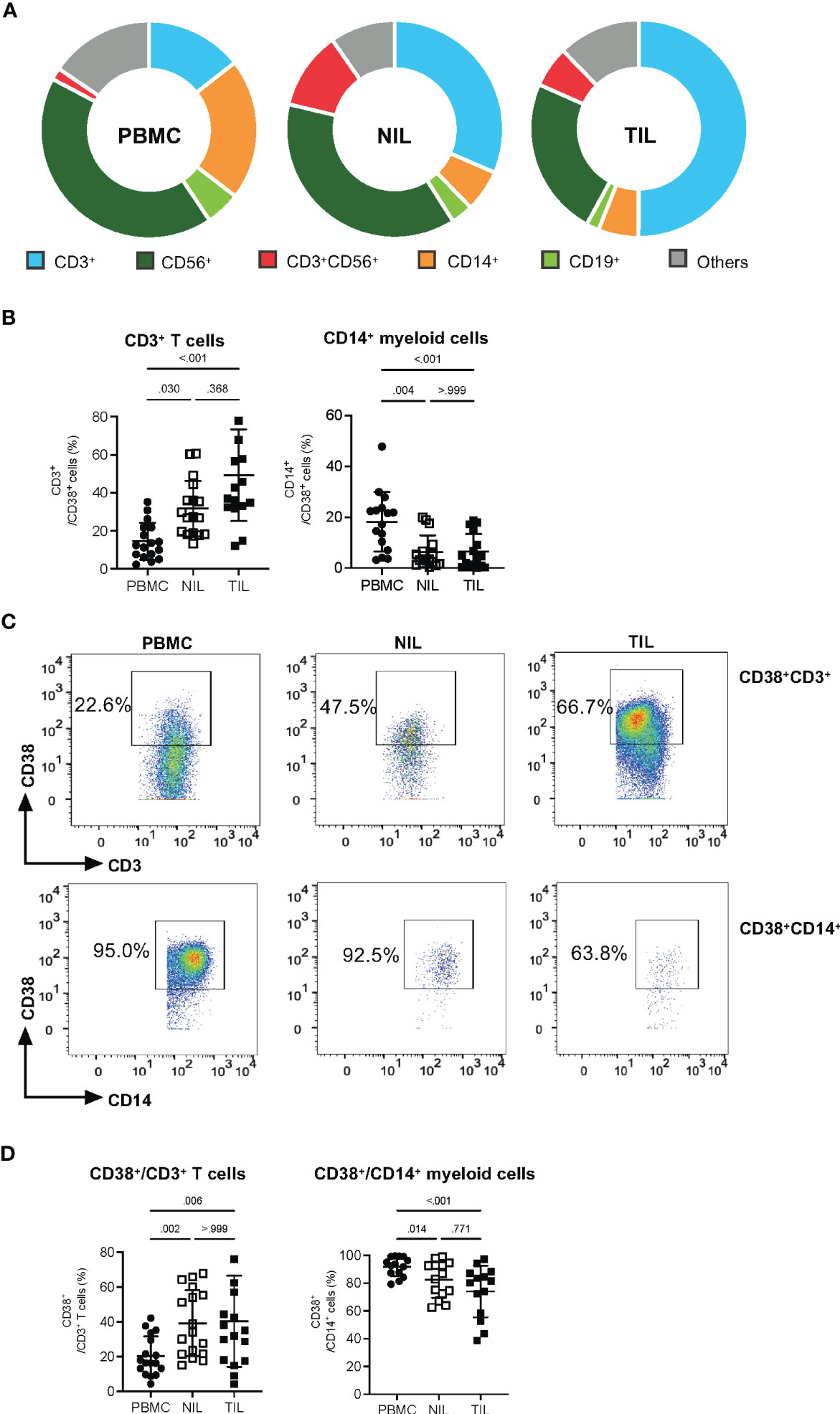
Figure 2 Elevated CD38 expression in CD3+ TILs. (A) CD38+ compositions from PBMC, NIL and TIL of HCC patients: CD3+ T cells, CD14+ myeloid cells, CD56+ Natural Killer (NK) cells, CD3+CD56+ NKT cells and CD19+ B cells. (B) Percentages of CD3+ T cells and CD14+ myeloid cells among the total CD38+ immune cells. (C) Representative manual gating strategy to identify the CD38-expressing CD3+ T cells and CD14+ myeloid cells from PBMC, NIL and TIL. (D) Percentages of CD38+ populations among the CD3+ T cells and CD14+ myeloid cells in PBMC, NIL and TIL. (B, D) Data are represented as mean ± SD. Friedman one-way ANOVA test calculated by Dunn’s post-hoc multiple pairwise comparisons was performed. p-value <0.05 is considered as significant. (A, B, D) PBMC, peripheral blood mononuclear cells (n=17); NIL, Non-tumor-infiltrating lymphocyte (n=17); TIL, tumor-infiltrating lymphocyte (n=60 from 2-5 tumor sectors per case).
Conversely, we also examined CD38 expression on individual immune subsets across PBMCs, NILs and TILs (Figures 2C, S3A). Consistent with our results above, the proportions of CD38+ populations among the total CD3+ T cells are significantly higher in TILs (Figure 2D). In contrast, the proportion of CD38+CD14+ myeloid cells is lower in TILs as compared to PBMCs (Figure 2D). On the other hand, the proportion of CD38+CD19+ B cells is higher in PBMCs than in NILs and TILs, the proportion of CD38+ NKT cell population is significantly lower in the PBMCs than in the NILs and TILs, and the proportion of CD38+CD56+ NK cells showed no significant differences across PBMCs, NILs and TILs (Figure S3B).
Taken together, the enriched proportion of CD3+ T cells among the CD38-expressing TILs suggests that CD38 may play a significant role in T cell immunity within the TME of HCC.
3.3 Tumor-infiltrating CD8+ tissue-resident memory T cells (TRM) is the dominant immune subset expressing CD38 in the HCC TME
Next, we sought to identify the specific CD3+ subset that expresses CD38. By gating on CD8+ or CD4+ T cells, a higher frequency of CD38+ can be observed on CD8+ T cells compared to that on CD4+ T cells, particularly on TILs, but not on the NILs and PBMCs (Figures 3A, B), suggesting that CD8+ T cells are the dominant CD38-expressing CD3+ T cells among the TILs in HCC tumors.
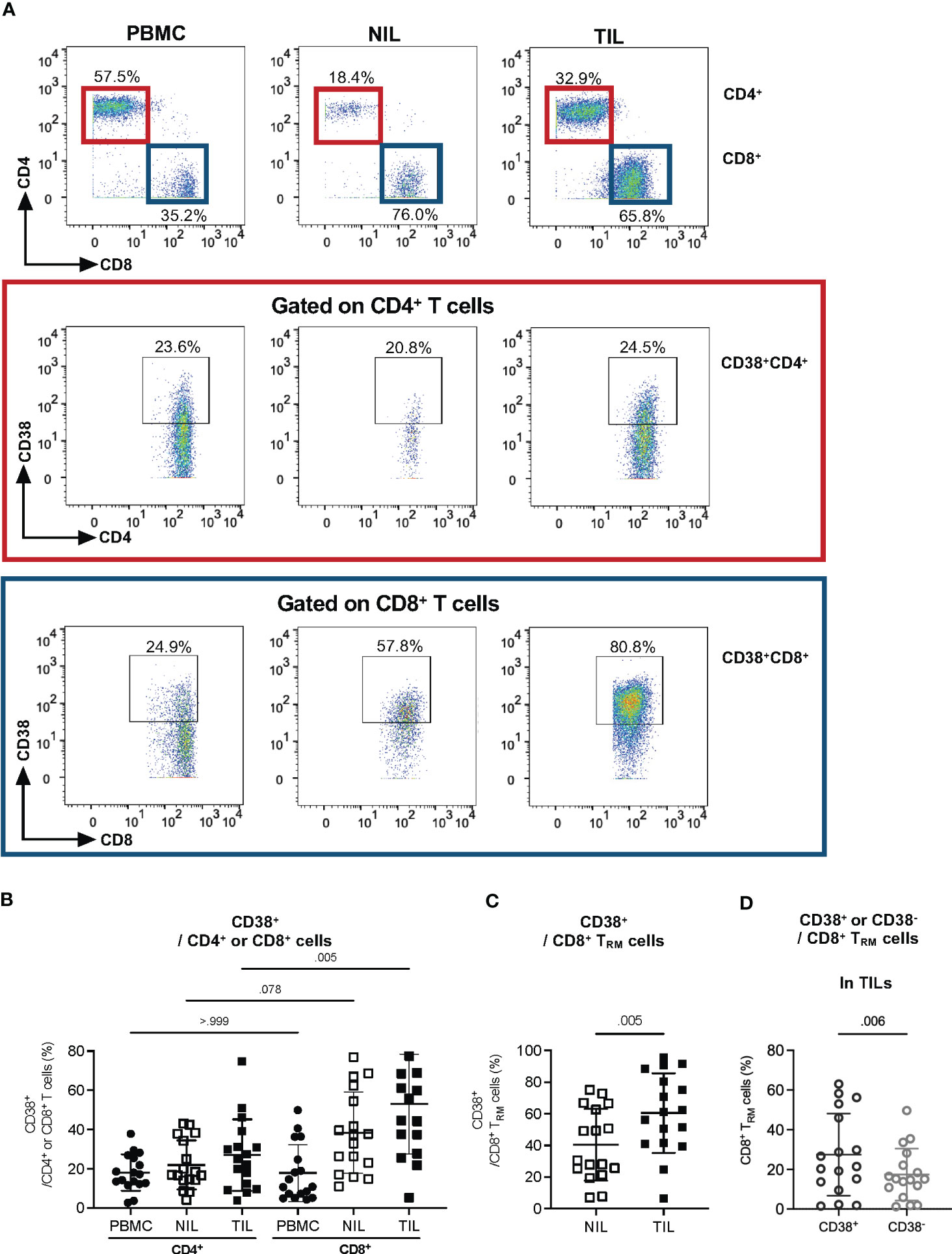
Figure 3 CD38 expression in tumor-infiltrating CD8+ Tissue-resident memory (TRM) cells. (A) Representative manual gating strategy for CD38-expressing CD3+ immune subsets, CD4+ and CD8+ T cells in PBMC, NIL and TIL. (B) Percentages of CD38-expressing CD4+ or CD8+ T cells in PBMC, NIL and TIL. Data is represented as mean ± SD. Friedman one-way ANOVA test calculated by Dunn’s post-hoc multiple pairwise comparisons was performed. p-value <0.05 is considered as significant. (C) The percentages of CD38+ TRM among the CD8+ T cells in NILs and TILs. (D) The percentages of CD38+ and CD38- among CD8+ TRM in TILs. (C, D) Data are represented as mean ± SD. Two-tailed Wilcoxon-matched t-test was performed. p-value <0.05 is considered as significant. (A–D) PBMC, peripheral blood mononuclear cells (n=17); NIL, Non-tumor-infiltrating lymphocyte (n=17); TIL, tumor-infiltrating lymphocyte (n=60 from 2-5 tumor sectors per case).
Since our previous study has shown that the HCC TME is enriched with CD8+ tissue-resident memory cells (TRM) that play an important role in tumor immunity (16), we evaluated CD38 expression on CD8+ TRM (CD103+CD45RO+CD8+) in TILs and NILs (Figure S4A). Indeed, we observed a significantly higher frequency of CD38+ TRM among the CD8+ T cells from TILs than that in the NILs (Figure 3C). Furthermore, within the TILs, the percentage of CD38+ TRM is significantly higher than the CD38- TRM (Figure 3D), indicative of a specific role of CD38 on TRM in the HCC TME.
3.4 Transcriptomic analysis of CD8+ TRM cells from TILs revealed a T cell exhaustion signature associated with CD38
To further assess the role of CD38 in TRM from HCC TILs, we sorted for CD8+CD103+CD45RO+ TRM from TILs and circulating CD8+CD45RO+ memory CD8+ T cells from PBMCs (Figure S1A) and performed bulk RNA sequencing on the sorted populations. Analysis of the differentially expressed genes (DEGs) comparing these two immune populations showed significant enrichment of CD38 in the CD8+ TRM from TILs (Figure 4A, Table S5). In addition, we also observed elevated expression of the genes coding for T cell exhaustion markers, PCDC1, CTLA4, HVCR2, LAG3 and TIGIT in CD8+ TRM from TILs (Figure 4A, Table S5).
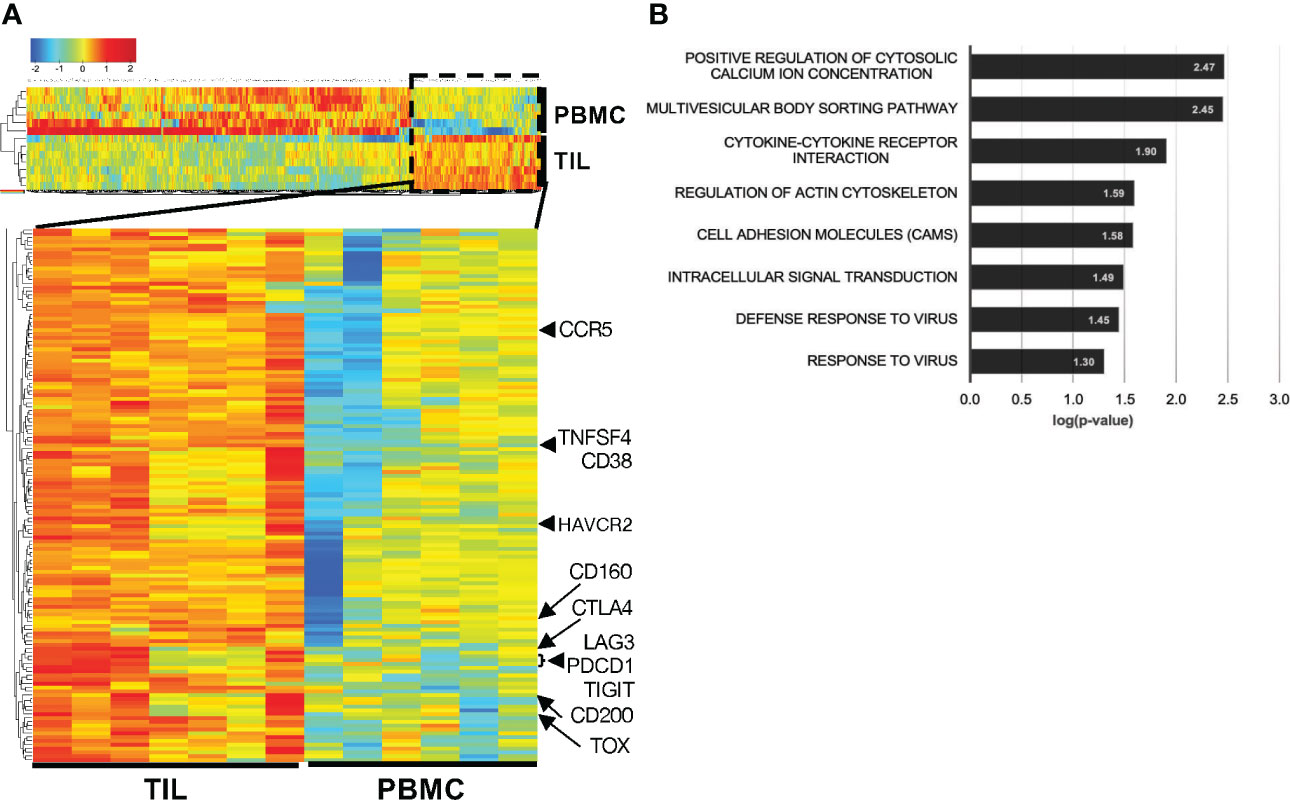
Figure 4 Higher CD38 and exhaustion marker genes expression in the CD8+ TRM from HCC TILs. (A) Differentially expressed genes (DEGs) between the TRM extracted from the TILs (n=7) and circulating memory T cells extracted from the PBMCs (n=6). Cells were isolated from matched patient samples; one PBMC sample was omitted due to poor RNA quality. (B) DAVID Functional pathway analysis of the enriched DEGs from TIL-TRM obtained from (A). The genes of each pathway are listed in Table S6.
To further understand the genes associated with phenotypes of CD8+ TRM from TILs, we interrogated their enriched DEGs by functional pathway analysis. Several significant pathways processes enriched in the tumor-infiltrating CD8+ TRM include positive regulation of cytosolic calcium ion concentration (GO:0007204), multivesicular body sorting pathway (GO:0071985), cell adhesion molecules (CAM; hsa04514), intracellular signal transduction (GO:0035556), defense response to virus (GO:0051607) and response to virus (GO:0009615) (Figure 4B, Table S6). Intracellular calcium level is well known to affect T cell functions and activities (22) and is linked to T cell exhaustion (23). Several genes involved in the cell adhesion molecules pathway, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and programmed cell death protein 1 (PDCD1), are known to control T cell motility (24) and well-known T cell exhaustion markers as well (25).
Collectively, the targeted transcriptomics analyses support an association between CD38 and T cell exhaustion, specifically in TRM from HCC TILs.
3.5 CD8+ TRM cells in TILs co-express CD38 and multiple exhaustion makers
Given the association of CD38 with T cell exhaustion gene signature from the sorted CD8+ TRM in TILs, we next sought to determine if CD38 correlates with exhaustion markers PD-1 and CTLA-4 in CD3 and CD8 TILs at the protein expression level using our CyTOF data. Within the HCC TILs, we observed the correlation of CD38 with PD-1 only on CD3+ T cells, but the correlation of CD38 expression with both PD-1 and CTLA-4 was observed on CD8+ T cells and CD8+ TRM (Figure 5A). Importantly, a similar correlation between CD38 and T cell exhaustion markers was not observed in the NILs or PBMCs (Figures S5A–C), highlighting that the correlation is specific to CD8+ T cells or CD8+ TRM in HCC TME. This data again marks CD38 as a co-exhaustion marker for CD8+ T cells in the TME of HCC.
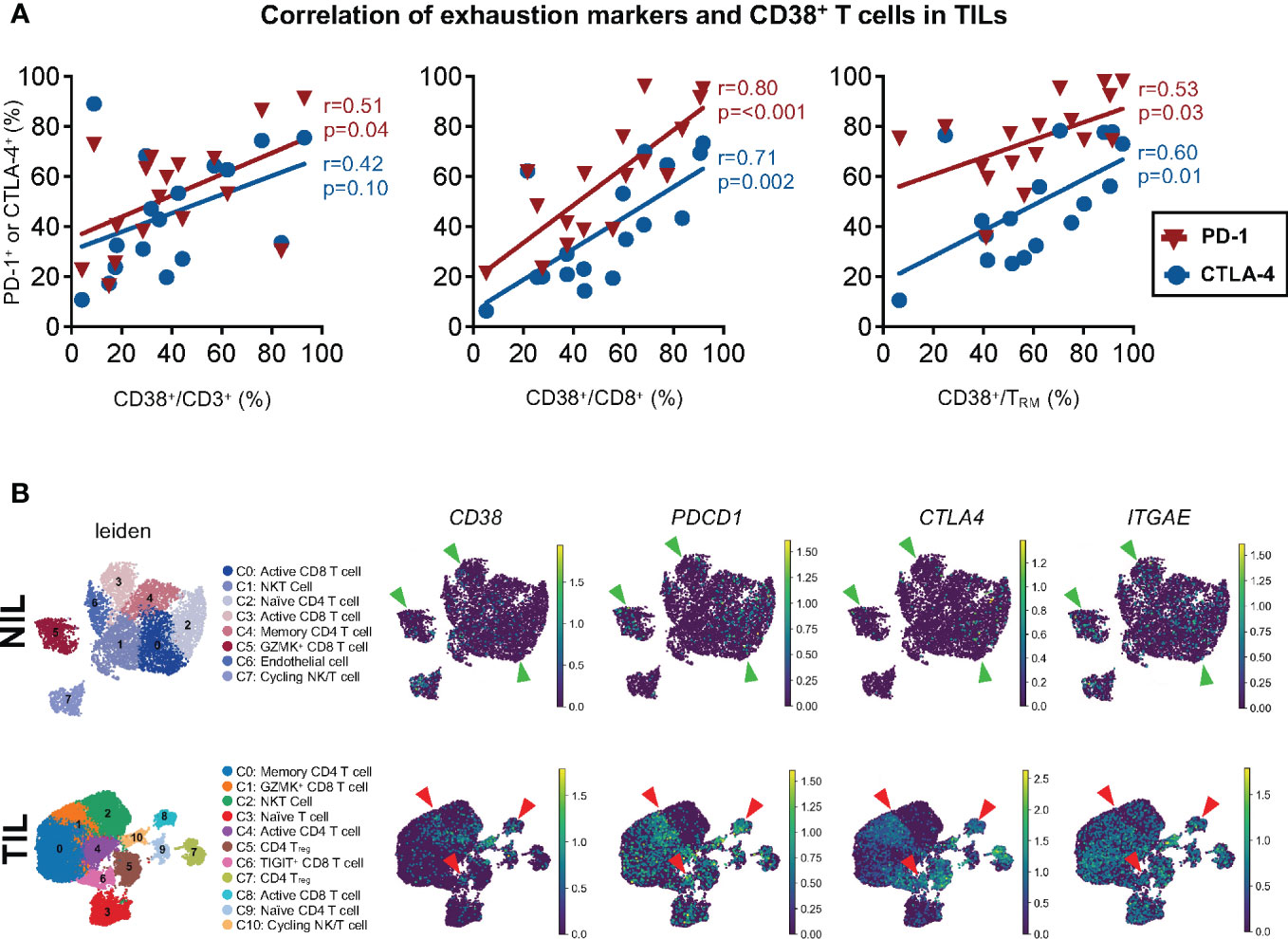
Figure 5 Association between CD38 and exhaustion markers on the tumor-infiltrating T cells. (A) Correlation between PD-1 or CTLA-4 with CD38 expression on CD3+ T cells (left), CD8+ T cells (middle) and CD8+ TRM (right) from TILs. Spearman’s r and p-values are reported. p-value <0.05 denotes significant correlation. TIL, tumor-infiltrating lymphocyte (n=60 from 2-5 tumor sectors per case). (B) Single-cell RNA-seq data demonstrating the co-expression of CD38, PDCD1 (PD-1), and CTLA4 and ITGAE (CD103) in the NILs (top) and TILs (bottom). The green and red arrows indicate the CD8 T cell clusters in the NILs and TILs, respectively. The clusters were identified and annotated using the differentially expressed genes listed in Tables S7 and S8.
To elucidate this further at the single-cell level, we next performed scRNA-seq on 14 NIL and TIL samples. We first sub-gated out the immune population of interest, T cells, from the NILs and TILs as previously described (21). Dimension reduction analysis was performed on a total of 7,628 and 26,441 T cells from NILs and TILs cells, identifying 8 and 11 clusters, respectively (Figure 5B). The clusters were identified and annotated according to the differentially expressed genes (DEGs) (Tables S7, S8). Among the TILs, co-expression of CD38 with exhaustion markers, PDCD1 and CTLA4 (Figure 5B), as well as ENTPD1 (CD39) (Figure S6A), was observed in CD8 T cell clusters 6 and 8, and the margin of cluster 1. However, this co-expression was not observed in NILs (Figures 5B, S6A). Furthermore, we also found that the expression of ITGAE, which encodes for tissue-resident marker CD103, overlapped with the expression of CD38 and the exhaustion markers in clusters 1, 6 and 8 in TILs but not in NILs (Figure 5B). These findings suggest that CD38 co-expression with other known exhaustion markers is a characteristic feature of CD8 T cell, specifically the TRM (Figure 5B).
The scRNA-seq data demonstrated, at the single-cell level, the potential co-expression of CD38 and other exhaustion markers on the same CD8+ T cells or TRM from HCC TME, further validating its role as a co-exhaustion marker in HCC TILs. In support of our data, the co-expression of CD38 with T cells exhaustion markers was also observed on dysfunctional or exhausted CD8+ T cells in other tumor models (26, 27).
3.6 In-situ co-expression of CD38 and exhaustion makers on CD8+ T cells
To visualize that CD38 is indeed expressed by the same exhausted CD8+ T cells, we performed multiplex immunohistochemistry (mIHC) on the formalin-fixed and paraffin-embedded (FFPE) tumor tissues from the HCC patients. We observed the colocalization of CD38, PD-1 and CD8 within HCC tumors (Figure 6A). To further validate that the same CD8+ T cells population does indeed co-express CD38 and PD-1, we manually gated them using our CyTOF data (Figure S7A). In line with our scRNA-seq data, we observed a higher frequency of CD8+ T cells co-expressing CD38 and PD-1 in the TILs compared to the NILs (Figure 6B). Specifically, a significant increase in the co-expression of CD38 and PD-1 was observed on CD8+ TRM in TILs as compared to NILs (Figure 6C). Overall, our result suggests that the co-expression of CD38 with the immune checkpoint markers is specific to the CD8+ TRM cells in the HCC TME.
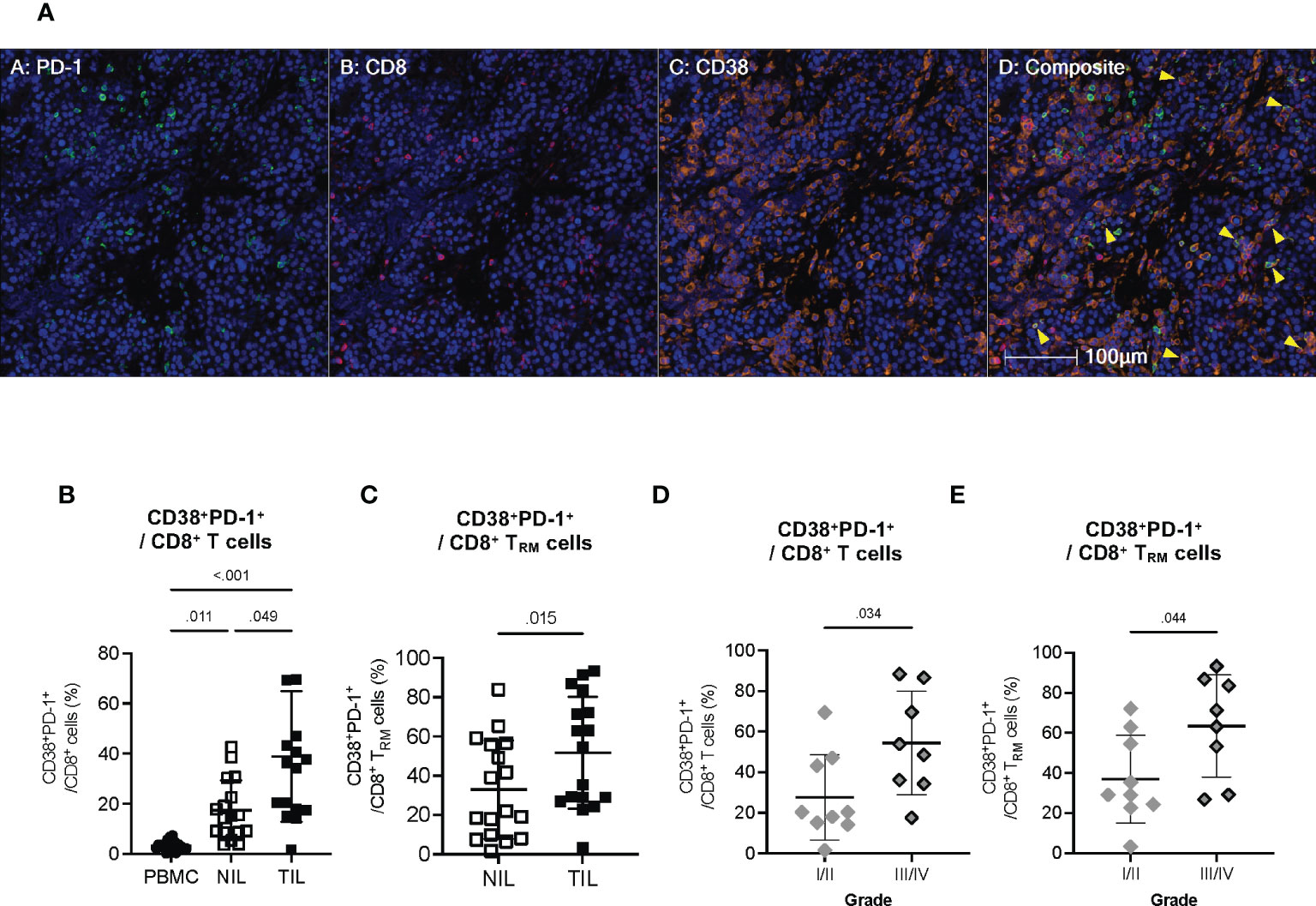
Figure 6 Co-expression of CD38 with exhaustion markers by the CD8+ T cells within TILs (A) Representative multiplex immunohistochemistry (mIHC) images showing co-expression of PD-1 (green), CD8 (red) and CD38 (orange) in the HCC TME. Yellow arrows designate co-expression of PD-1, CD8 and CD38. Cell nuclei are counterstained with DAPI (blue). Images are shown at 200x magnification. (B) Percentages of CD8+ T cells co-expressing CD38 and PD-1 in the PBMC, NIL and TIL. Friedman one-way ANOVA test calculated by Dunn’s post-hoc multiple pairwise comparisons was performed. (C) Proportion of CD8+ TRM cells co-expressing CD38 and PD-1 in the NIL and TIL. Data is Two-tailed Wilcoxon-matched t-test was performed. (D) Percentages of tumor-infiltrating CD38+PD-1+CD8+ T cells in Edmonson-Steiner histopathological grade I or II versus III or IV. (E) Percentages of tumor-infiltrating CD38+PD-1+CD8+ TRM cells in Edmonson-Steiner histopathological grade I or II versus III or IV. (B–E) Data represented as mean ± SD. p-value < 0.05 is considered as significant. PBMC: peripheral blood mononuclear cells (n=17); NIL: Non-tumor-infiltrating lymphocyte (n=17); TIL: tumor-infiltrating lymphocyte (n=60 from 2-5 tumor sectors per case). (D, E) Two-tailed Mann-Whitney t-test was performed. p-value < 0.05 denotes significant association. Clinical data from n=17 patients with HCC.
Given the potential role of CD38 in T cell exhaustion, we next examined the association of the CD38+PD-1+CD8+ T cell subsets with the clinical parameters of our HCC patient cohort (Table S1). We found a significantly higher frequency of CD38+PD-1+CD8+ T cell and CD38+PD-1+CD8+ TRM to be associated with HCC with higher histopathological grade III and IV (Figures 6D, E), indicating that CD38 as a T cell co-exhaustion marker is also linked to tumor aggressiveness.
Our study provided evidence that CD38 is closely associated with the T cell exhaustion signature, particularly on CD8+ TRM. This suggests that CD38, as another T exhaustion marker, could potentially be the next checkpoint molecule to be targeted for the immunotherapy of HCC.
4 Discussion
Our current study investigated the potential role of CD38 in HCC using an in-depth multi-dimensional immune profiling of the PBMCs, NILs and TILs obtained from patients with HCC. We demonstrated that CD38 is associated with T cell exhaustion in the HCC TME. Mainly, CD38 is co-expressed with PD-1 and CTLA-4 on tumor-infiltrating CD8+ TRM cells, identifying CD38 as a potential immune checkpoint marker that could be harnessed for HCC immunotherapy.
The ubiquitous CD38 expression in different immune cell types shown in our current study is consistent with previous reports (7, 14). Notably in HCC, our current data demonstrated that PBMCs, NILs and TILs each showed a unique CD38 immune profile. Consistent with our earlier study (14), the current data demonstrated CD3+ T cells as the dominant CD38-expressing immune subset in TILs. Furthermore, among the CD3+ T cell subsets, we identified resident memory CD8+ TRM cells (CD103+CD45RO+CD8+) as the specific T cell that expressed a high level of CD38 in the TME of HCC. In a study of non-small-cell lung cancer (NSCLC), CD38+CD8+TRM was also observed within the TME (29) and was shown to be crucial for anti-tumor immunity in NSCLC (28). Here, we report that enrichment of CD38+CD8+TRM in the HCC TME is associated with T cell exhaustion. We showed that CD38 is co-expressed with T cell checkpoint makers like PD-1 and CTLA-4 at both the RNA and protein levels, indicating the role of CD38 in CD8+ TRM exhaustion. Indeed, the expression of CD38 in CD8+ T cells in TME has been associated with an immunosuppressive and dysfunctional phenotype that drives cancer progression (27). Moreover, CD38 could contribute to pro-tumoral TME by acting on other stromal cells and promoting hypoxia and angiogenesis (29, 30). Altogether, these underscore that inhibition of CD38 could potentially be a multi-faceted approach to reinvigorate the exhausted TME for immunotherapy.
Our pathway analysis of the enriched DEGs from isolated HCC TRM shows enrichment of the pathway involved in positive regulation of the cytosolic calcium ion. CD38-mediated regulation of intracellular calcium signaling pathways in T cells has been previously reported (31). CD38 regulates nicotinamide adenine dinucleotide (NAD+) levels involved in intracellular calcium mobilization, resulting in a NAD-mediated suppression of T lymphocytes and poor anti-tumoral immunity in the TME (32), linking CD38 to T cell exhaustion or immune evasion mechanistically. Despite that, we acknowledge the limitation of our current data, which warrants a validation of the detailed mechanism of CD38-mediated T cell exhaustion in HCC. Furthermore, apart from the T cells, we also found other immune cells expressing CD38, including those of the myeloid lineage. A higher density of CD38+ tumor-associated macrophages (TAM) has been associated with improved prognosis (12) and better response to PD-1/PD-L1 therapy (13) in HCC. The association could be attributed to CD38+ TAMs exhibiting the pro-inflammatory M1 phenotype, contributing to anti-tumor immune activity (13). Henceforth, while we show CD38 expression in CD8+ TRM may play a role in T cell exhaustion, the other tumor-infiltrating immune cells could also contribute to the overall tumor immunity of the HCC TME. Interestingly, this suggests a more intricate role of CD38 in the HCC TME.
Our study has limitations that should be considered. The small sample size and variability in the number of tumor sectors obtained from each patient in our cohort may limit the generalizability of our findings. However, we addressed this by validating our findings using an independent HCC cohort and confirming the co-expression of CD38 and PD1 in CD8+ T cells through mIHC. Previous studies support our current findings demonstrating that CD38 is an immunosuppressive molecule and a potential T cell exhaustion marker (26, 27, 33). For instance, expression of CD38 on T-cells is associated with poorer proliferation and reduced pro-inflammatory cytokine secretion (26, 27, 33). The inhibition of CD38 has been shown to reinvigorate the cytotoxic function and proliferative capacity of CD38+CD8+ T cells (26, 27). Notably, a recent study has highlighted an additional role of CD38 in sustaining the survival of exhausted CD8+ T cells (33). These indicate a potential role for CD38 in promoting T cell dysfunction and exhaustion.
Moreover, our data suggest a promising synergistic effect of targeting CD38 and PD-1 to potentially reinvigorate the exhausted T cells in the TME and overcome the resistance to anti-PD-1 therapy. A study in melanoma directly linked the induction of PD1+CD38hiCD8+ T cells to T cell dysfunctionality found in the TME of non-responding patients to PD-1 blockade (27). Interestingly, Chen and colleagues report that CD38 blockade could rescue lung cancer mouse models from acquired anti-PD-1 therapy resistance (26). Although a Phase I/II clinical trial on the concurrent blockade of CD38 and PD-1 in solid-tumor cancers reported poor efficacy without significant anti-tumor activity, post-therapeutic examination revealed about a 40% reduction in the frequency of CD38+ tumor infiltrates in the TME and the reinvigoration of peripheral T-cell activity (34). The outcome of the clinical trial may have been confounded by the limited availability of patient samples and differing treatment histories affecting the baseline CD38 expression of TILs (34). Also, the TME of HCC could differ from that of advanced NSCLC and metastatic castration-resistant prostate cancer. Considering the concurrent expression of CD38 and PD-1 on HCC CD8+ TILs and its association with the higher HCC histological grades in our HCC cohort, targeting CD38 in combination with PD-1 blockade could revive the cytotoxic function of exhausted CD8+ TRM cells. The combination therapy may have a synergistic effect to improve the clinical outcomes of HCC patients. Therefore, our study gives a glimpse of a potential direction for developing novel immunotherapy for HCC. Further studies will be needed to investigate the effectiveness of CD38 blockade with PD-1 in treating HCC in preclinical and clinical settings.
Overall, we provide evidence of CD38 co-expression with other exhaustion markers on CD8+ TRM cells in the HCC TME, underpinning the role of CD38 in T cell exhaustion. Our findings uncover CD38 as a promising immune checkpoint marker as well as a potential target for combination immunotherapy for the treatment of HCC.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI under accession code GSE156625.
Ethics statement
The studies involving human participants were reviewed and approved by SingHealth Central Institution Review Board (CIRB) and the National University Hospital (NUH) CIRB (CIRB Ref: 2016/2626, 2016/2613 and 2018/2112). The patients/participants provided their written informed consent to participate in this study.
Author contributions
All authors listed have contributed to this study. MR and MO are involved in data curation and analysis, investigation and manuscript writing. JL, TL, WL, AC, BG and PC recruited patients and provided patient specimens and discussed the data. YL, PN, CL, MW and CC assisted in data acquisition and analysis and discussed the paper. JS and RD obtained and analyzed the scRNA seq data. JY prepared and provided tissue samples, data analysis and discussed the data. VC designed and led the study, performed the analysis and prepared the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Medical Research Council (NMRC), Singapore (ref numbers: NMRC/TCR/015-NCC/2016, NMRC/CSA-SI/0013/2017, NMRC/CSA-SI/0018/2017, NMRC/OFLCG/003/2018) as well as Duke-NUS Khoo Bridge Funding Award (Duke-NUS-KBrFA/2022/0058), International Gilead Sciences Research Scholars Program in Liver Disease – Asia and the National Research Foundation (NRF), Singapore (NRF-CRP26-2021-0005) - RD.
Acknowledgments
The authors would like to thank Dr. David Tai, all members of TII, all participating patients, the clinical research coordinators from NCCS and SGH for their contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1182016/full#supplementary-material
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
3. Sangro B, Sarobe P, Hervas-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol (2021) 18(8):525–43. doi: 10.1038/s41575-021-00438-0
4. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol (2022) 23(1):77–90. doi: 10.1016/S1470-2045(21)00604-5
5. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/S1470-2045(18)30351-6
6. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
7. Glaria E, Valledor AF. Roles of CD38 in the immune response to infection. Cells (2020) 9(1):228. doi: 10.3390/cells9010228
8. Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front Immunol (2019) 10:1187. doi: 10.3389/fimmu.2019.01187
9. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-de-Arellano M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol (2020) 11:597959. doi: 10.3389/fimmu.2020.597959
10. Dwivedi S, Rendon-Huerta EP, Ortiz-Navarrete V, Montano LF. CD38 and regulation of the immune response cells in cancer. J Oncol (2021) 2021:6630295. doi: 10.1155/2021/6630295
11. Wo YJ, Gan ASP, Lim X, Tay ISY, Lim S, Lim JCT, et al. The roles of CD38 and CD157 in the solid tumor microenvironment and cancer immunotherapy. Cells (2019) 9(1):26. doi: 10.3390/cells9010026
12. Lam JH, Ng HHM, Lim CJ, Sim XN, Malavasi F, Li H, et al. Expression of CD38 on macrophages predicts improved prognosis in hepatocellular carcinoma. Front Immunol (2019) 10:2093. doi: 10.3389/fimmu.2019.02093
13. Ng HHM, Lee RY, Goh S, Tay ISY, Lim X, Lee B, et al. Immunohistochemical scoring of CD38 in the tumor microenvironment predicts responsiveness to anti-PD-1/PD-L1 immunotherapy in hepatocellular carcinoma. J Immunother Cancer (2020) 8(2):e000987. doi: 10.1136/jitc-2020-000987
14. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating b cells and T cells controls the progression of hepatocellular carcinoma. Gut (2017) 66(2):342–51. doi: 10.1136/gutjnl-2015-310814
15. Nguyen PHD, Ma S, Phua CZJ, Kaya NA, Lai HLH, Lim CJ, et al. Intratumoural immune heterogeneity as a hallmark of tumour evolution and progression in hepatocellular carcinoma. Nat Commun (2021) 12(1):227. doi: 10.1038/s41467-020-20171-7
16. Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci USA\ (2017) 114(29):E5900–9. doi: 10.1073/pnas.1706559114
17. Lai L, Ong R, Li J, Albani S. A CD45-based barcoding approach to multiplex mass-cytometry (CyTOF). Cytometry A (2015) 87(4):369–74. doi: 10.1002/cyto.a.22640
18. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (2013) 29(1):15–21. doi: 10.1093/bioinformatics/bts635
19. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinf (2011) 12:323. doi: 10.1186/1471-2105-12-323
20. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res (2015) 43(7):e47. doi: 10.1093/nar/gkv007
21. Sharma A, Seow JJW, Dutertre CA, Pai R, Blériot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell (2020) 183(2):377–394 e21. doi: 10.1016/j.cell.2020.08.040
22. Trebak M, Kinet JP. Calcium signalling in T cells. Nat Rev Immunol (2019) 19(3):154–69. doi: 10.1038/s41577-018-0110-7
23. Shao M, Teng X, Guo X, Zhang H, Huang Y, Cui J, et al. Inhibition of calcium signaling prevents exhaustion and enhances anti-leukemia efficacy of CAR-T cells via SOCE-Calcineurin-NFAT and glycolysis pathways. Adv Sci (Weinh). (2022) 9(9):e2103508. doi: 10.1002/advs.202103508
24. Brunner-Weinzierl MC, Rudd CE. CTLA-4 and PD-1 control of T-cell motility and migration: implications for tumor immunotherapy. Front Immunol (2018) 9:2737. doi: 10.3389/fimmu.2018.02737
25. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol Nov (2005) 25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
26. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov (2018) 8(9):1156–75. doi: 10.1158/2159-8290.CD-17-1033
27. Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol (2019) 20(9):1231–43. doi: 10.1038/s41590-019-0441-y
28. Wu P, Zhao L, Chen Y, Xin Z, Lin M, Hao Z, et al. CD38 identifies pre-activated CD8+ T cells which can be reinvigorated by anti-PD-1 blockade in human lung cancer. Cancer Immunol Immunother (2021) 70(12):3603–16. doi: 10.1007/s00262-021-02949-w
29. Boslett J, Hemann C, Christofi FL, Zweier JL. Characterization of CD38 in the major cell types of the heart: endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am J Physiol Cell Physiol (2018) 314(3):C297–309. doi: 10.1152/ajpcell.00139.2017
30. Ben Baruch B, Mantsur E, Franco-Barraza J, Blacher E, Cukierman E, Stein R. CD38 in cancer-associated fibroblasts promotes pro-tumoral activity. Lab Invest (2020) 100(12):1517–31. doi: 10.1038/s41374-020-0458-8
31. Kar A, Mehrotra S, Chatterjee S. CD38: T cell immuno-metabolic modulator. Cells (2020) 9(7):1716. doi: 10.3390/cells9071716
32. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology (2013) 2(9):e26246. doi: 10.4161/onci.26246
33. DeRogatis JM, Neubert EN, Viramontes KM, Henriquez ML, Nicholas DA, Tinoco R. Cell-intrinsic CD38 expression sustains exhausted CD8(+) T cells by regulating their survival and metabolism during chronic viral infection. J Virol (2023) 97(4):e00225-23. doi: 10.1128/jvi.00225-23
Keywords: CD38, PD-1, T cell exhaustion, immunotherapy, HCC, immune checkpoint, tissue resident T cells
Citation: Reolo MJY, Otsuka M, Seow JJW, Lee J, Lee YH, Nguyen PHD, Lim CJ, Wasser M, Chua C, Lim TKH, Leow WQ, Chung A, Goh BKP, Chow PKH, DasGupta R, Yeong JPS and Chew V (2023) CD38 marks the exhausted CD8+ tissue-resident memory T cells in hepatocellular carcinoma. Front. Immunol. 14:1182016. doi: 10.3389/fimmu.2023.1182016
Received: 08 March 2023; Accepted: 22 May 2023;
Published: 12 June 2023.
Edited by:
Jesse Haramati, University of Guadalajara, MexicoReviewed by:
Dainius Characiejus, Vilnius University, LithuaniaFabio Malavasi, University of Turin, Italy
Gabriele Missale, University of Parma, Italy
Copyright © 2023 Reolo, Otsuka, Seow, Lee, Lee, Nguyen, Lim, Wasser, Chua, Lim, Leow, Chung, Goh, Chow, DasGupta, Yeong and Chew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valerie Chew, dmFsZXJpZS5jaGV3QGR1a2UtbnVzLmVkdS5zZw==