 Sigbjørn Berentsen
Sigbjørn Berentsen Bruno Fattizzo
Bruno Fattizzo Wilma Barcellini
Wilma Barcellini- 1Department of Research and Innovation, Haugesund Hospital, Helse Fonna Hospital Trust, Haugesund, Norway
- 2Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, and Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy
- 3Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
Autoimmune hemolytic anemia (AIHA) is defined by increased erythrocyte turnover mediated by autoimmune mechanisms. While corticosteroids remain first-line therapy in most cases of warm-antibody AIHA, cold agglutinin disease is treated by targeting the underlying clonal B-cell proliferation or the classical complement activation pathway. Several new established or investigational drugs and treatment regimens have appeared during the last 1-2 decades, resulting in an improvement of therapy options but also raising challenges on how to select the best treatment in individual patients. In severe warm-antibody AIHA, there is evidence for the upfront addition of rituximab to prednisolone in the first line. Novel agents targeting B-cells, extravascular hemolysis, or removing IgG will offer further options in the acute and relapsed/refractory settings. In cold agglutinin disease, the development of complement inhibitors and B-cell targeting agents makes it possible to individualize therapy, based on the disease profile and patient characteristics. For most AIHAs, the optimal treatment remains to be found, and there is still a need for more evidence-based therapies. Therefore, prospective clinical trials should be encouraged.
Introduction
We define autoimmune hemolytic anemia (AIHA) as anemia due to increased turnover of erythrocytes, caused by autoimmune mechanisms (1–3). The immune pathogenesis is usually mediated by autoantibodies against erythrocyte surface antigens, but the monocyte-macrophage system and T-lymphocytes are also involved (4, 5). It should be emphasized that AIHA is a collective term for several diseases (Table 1), classified according to findings by the monospecific direct antiglobulin test (DAT), the autoantibody class, the temperature optimum of the antigen-antibody reaction, and the absence or presence of an underlying or associated disease (6–8). The choice of optimal therapy differs between the AIHAs; therefore, exact diagnosis of the subtype is required to select the appropriate treatment (2).

Table 1 Autoimmune hemolytic anemias.
The first known case of what was likely an AIHA was described in the 13th century by the Byzantine court physician Johannis Actuarius, who observed black urine in a patient after exposure to cold temperatures (9). Probably, his patient suffered from paroxysmal cold hemoglobinuria (PCH). Karl Landsteiner discovered cold agglutinins in 1903 (10). It was not until 1938 that Dameshek & Schwartz draw a distinction between congenital and acquired hemolytic anemia and, two years later, postulated an immune mechanism based on the finding of hemolysin in patient sera (11). Until the turn of the millennium, treatment was mostly based on theoretical considerations, clinical experience, and expert opinion, but during the last two decades, several prospective studies have been conducted (1, 12–14). Currently, several new therapies have appeared and quite a few clinical trials are running (15, 16). Furthermore, not only the treatment options but also the clinical landscape of AIHA have changed over time (17, 18). The complexity of the disease group and the growing number of new treatment options underscore the need for a thorough diagnostic workup to provide a basis for selecting “the right therapy to the right patient” (17, 19, 20).
This review will first address the different types of AIHA and then discuss the optimal selection of therapy based on AIHA type, disease features, and patient characteristics, with particular emphasis on novel therapies.
The AIHA landscape and established therapies
Warm-antibody AIHA
Approximately 70% of AIHA cases are warm antibody-mediated (wAIHA). In wAIHA, the autoantibodies are polyclonal, mostly directed at erythrocyte antigens of the Rh system, and have a temperature optimum for antigen binding at 37°C. They are mostly of the immunoglobulin G (IgG) class, but occasionally, IgA or warm-reactive IgM can be involved, alone or in combination with IgG (1, 8, 21, 22). Of the IgG subclasses, involvement of IgG1 is predominant, either alone or in combination with other subclasses (23–25). IgG1 antibodies have complement-activating properties and also high affinity for the neonatal Fc-receptor (FcRn) which affects IgG half-life (26, 27), both of which are thought to contribute to the severity and persistence of hemolysis.
Phagocytosis of immunoglobulin-opsonized erythrocytes by macrophages of the mononuclear phagocytic system, to a large extent in the spleen, is an important mechanism of extravascular hemolysis (28). Complement-mediated red blood cell destruction is involved in about 50% of the patients. The polyspecific (“simple”) DAT is used to confirm autoimmune pathogenesis by detecting antigen-bound immunoglobulin and/or complement on the erythrocyte surface (29, 30). The specific immunoglobulin class(es) and the occurrence of complement on the erythrocytes can be identified by the monospecific (“extended”) DAT, in which diagnostic antibodies specific for IgG, IgM, IgA, complement protein fragment 3c (C3c), and C3d are used as reagents (1, 2, 30, 31).
WAIHA occurs as a primary disease in slightly less than 50% of the cases and secondary to other disorders in the remaining cases (7, 8, 32, 33). The underlying or associated diseases in secondary cases are listed in Table 2. Occasionally, more than one associated disease is present. Evans’ syndrome was originally defined as AIHA with thrombocytopenia (34); now usually defined as the simultaneous or sequential combination of at least two autoimmune cytopenias, most often AIHA with immune thrombocytopenia (35–37). Infection-induced exacerbation of a preexisting primary wAIHA is not regarded as secondary.
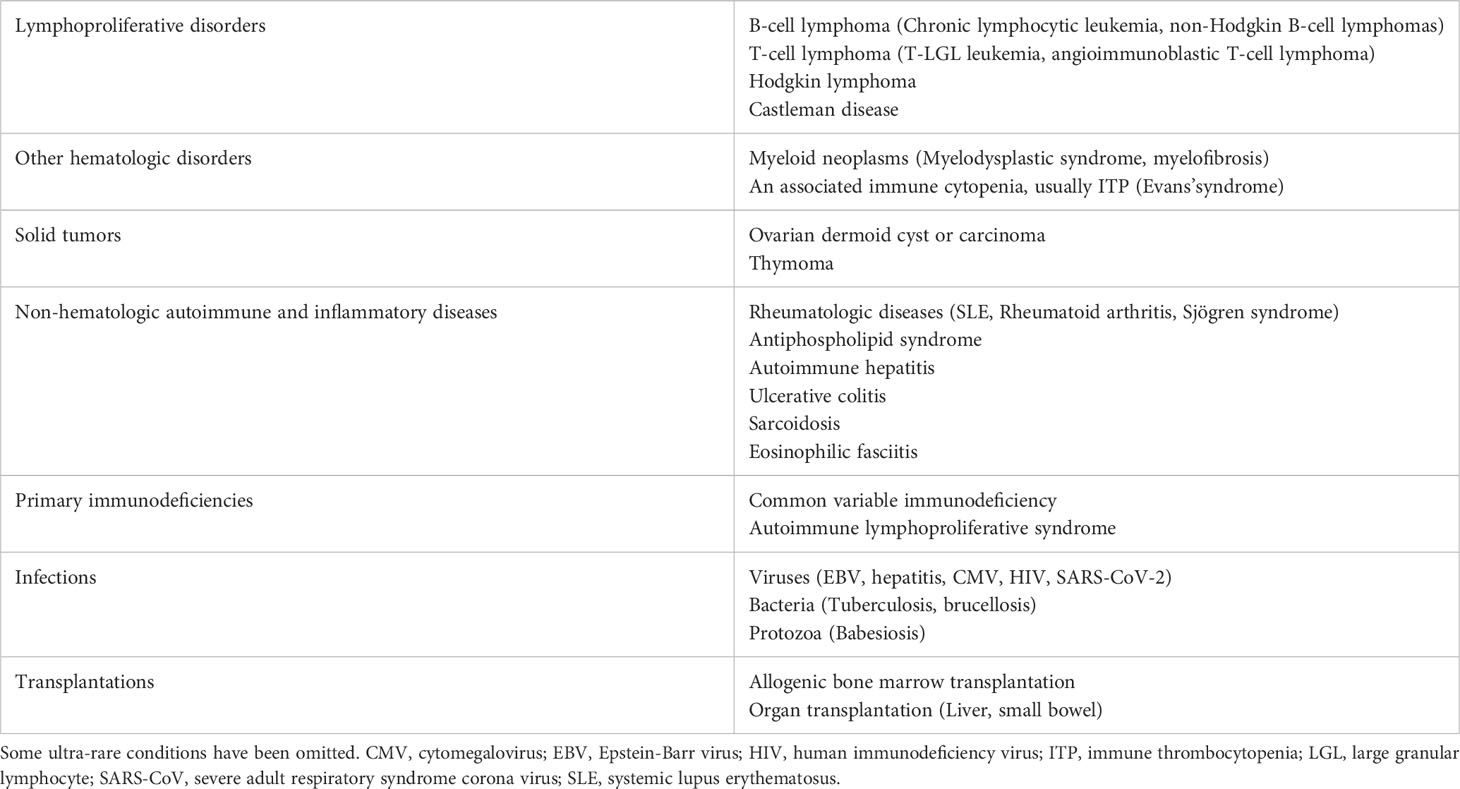
Table 2 Underlying or associated conditions in secondary warm AIHA.
Figure 1 shows an algorithm for diagnostic workup of AIHA including wAIHA. It should be noted that although the absolute reticulocyte count is usually elevated because of increased erythrocyte turnover and bone marrow compensation, some patients have normal counts or even reticulocytopenia (38–40). This is probably explained by autoantibody activity against erythroid precursors (40), but T-cell mediated suppression or inflammatory mechanisms may also contribute. WAIHA carries an increased risk of thrombosis, which, in addition to a risk of infection and other treatment complications, probably contributes to increased mortality (41–48). In most cases, wAIHA is a chronic relapsing rather than a chronic disease. Usually, patients are symptomatic at presentation and require treatment.
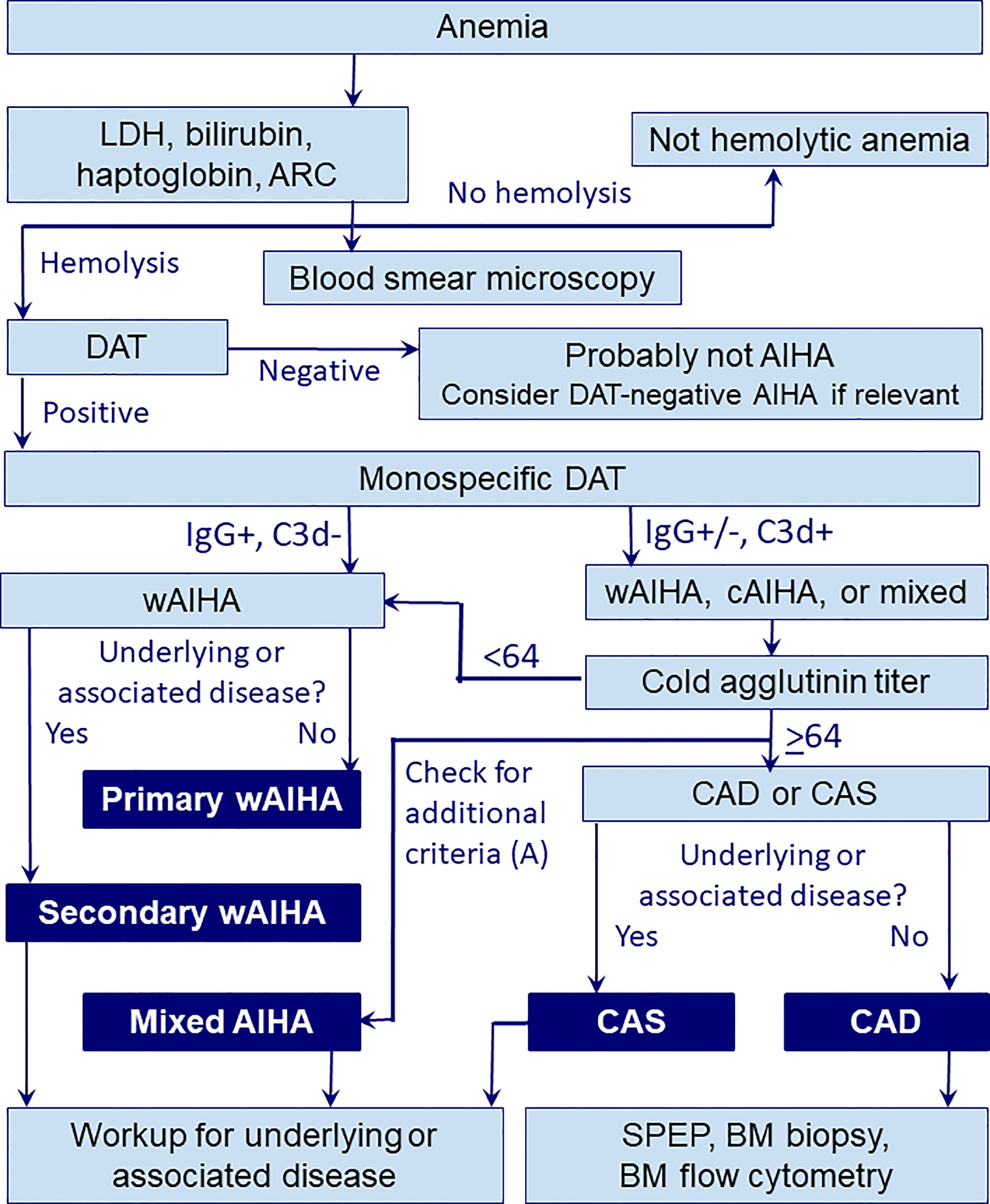
Figure 1 Diagnostic workup in AIHA. A, additional criteria for mixed AIHA (DAT strongly positive for both IgG and C3d, cold agglutinin titer ≥64, indirect antiglobulin test positive for IgG at 37°C); ARC, absolute reticulocyte count; BM, bone marrow; CAD, cold agglutinin disease; cAIHA, cold-antibody autoimmune hemolytic anemia; CAS, cold agglutinin syndrome; DAT, direct antiglobulin test; LDH, lactate dehydrogenase; SPEP, serum protein electrophoresis; wAIHA, warm-antibody autoimmune hemolytic anemia.
Prednisolone (or prednisone), administered at high initial doses (100 mg fixed dose or 1-1.5 mg/kg body weight daily), then slowly tapered after 2-3 weeks and discontinued after about 4-6 months, remains first-line therapy in wAIHA and yields responses in approximately 80% of the patients (1, 43, 49–51). Other oral corticosteroids have not shown higher efficacy (1, 52).
Patients who fail to respond to first-line therapy should be considered for a new diagnostic workup to reveal any overlooked underlying or concomitant disease that might be subject to specific therapy, such as associated autoimmune conditions, lymphoproliferative disorders, other neoplasms, or non-immune hemolytic anemias (1, 8, 53). In addition to a thorough clinical evaluation and review of the records and medication history, this assessment should include extensive autoantibody panels, serum protein electrophoresis, bone marrow examination, and exclusion of paroxysmal nocturnal hemoglobinuria and congenital hemolytic anemias (54).
Rituximab at a conventional (375 mg/m2 at 1-week interval for 4 weeks) or low dose (100 mg fixed dose at the same schedule) is currently recommended in the second line in primary wAIHA (1, 51, 55, 56). However, there are no published data to support repeated use of rituximab in wAIHA patients who have received this monoclonal antibody and subsequently relapsed. Third-line therapies include unspecific immune suppressants, such as azathioprine, cyclosporine, mycophenolate, and others (1, 57). Splenectomy, which was previously often recommended in the second line, is now considered an option in the third or subsequent lines (1, 2, 14, 58).
With some exceptions, recommended first-line treatment in secondary wAIHA is generally as for primary wAIHA (1, 2, 51). Treating the underlying or associated disease is indicated if this disease requires treatment by itself or if first-line therapy of the AIHA has failed.
Cold-antibody AIHA
Cold agglutinin disease
Cold agglutinin disease (CAD) accounts for 20-30% of AIHA cases (6, 59). The autoantibodies responsible for hemolysis in CAD are termed cold agglutinins, referring to their ability to agglutinate erythrocytes at temperatures below 37°C; such temperatures are normally found in the acral parts of the body (60). Cold agglutinins in CAD are monoclonal, usually of the IgM class with κ light chain restriction (61, 62). The cold agglutinin-producing pathogenetic process is a clonal B-cell lymphoproliferative disorder of the bone marrow, now recognized as a distinct entity by the World Health Organization classification of hematolymphoid neoplasms, but not considered to be a malignant lymphoma (63–65).
Binding of cold agglutinin to its cell surface antigen results in agglutination of erythrocytes, leading to complement-mediated hemolysis and, often, circulatory symptoms such as acrocyanosis and Raynaud-like phenomena (16, 61). Of note, the agglutination and circulatory symptoms are not complement-mediated. Complement is activated via the classical pathway, triggered by fixation of the C1qrs complex to antigen-bound IgM. Activated C1s cleaves C4 and C2, resulting in the formation of C3 convertase, coating of the erythrocytes with C3b, and phagocytosis of opsonized erythrocytes by macrophages (extravascular hemolysis), mainly in the liver (66–69). Especially in severely affected patients or acute exacerbations, split products may also combine to form C5 convertase on the cell surface, resulting in formation of the C5b-9 complex and intravascular hemolysis. The soluble split products, C3a and C5a, have proinflammatory properties and are thought to result in, or contribute to, fatigue in many patients with CAD (16, 70, 71).
The severity of anemia in CAD is shown in Figure 2 (61, 71). In a multinational, observational study, 146 patients (69.5% of 210 patients with available relevant data) had hemolytic anemia with no or mild peripheral circulatory symptoms, 44 (21%) had hemolytic anemia with circulatory symptoms interfering with daily living, while 20 (9.5%) had circulatory symptoms with compensated hemolysis (61). Patients have an increased risk of thrombosis (61, 72, 73).
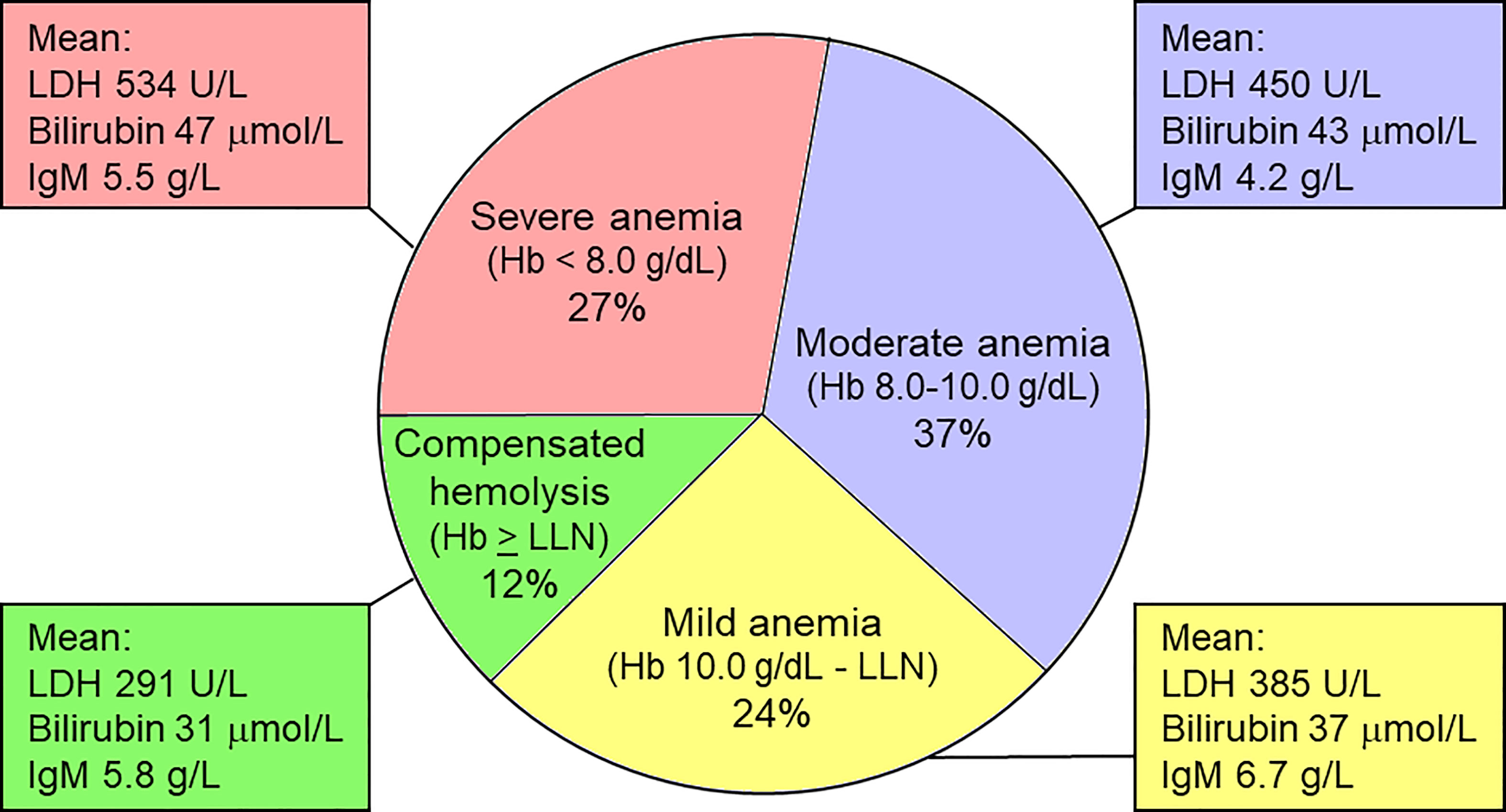
Figure 2 Severity of anemia in CAD. Hemoglobin levels correlate negatively with parameters of hemolysis, but do not correlate with IgM levels. LDH, lactate dehydrogense; LLN, lower limit of normal. Based on data from Berentsen et al. (61). Figure first published in Front Immunol 2020 by Berentsen (71), reused under a Creative Commons CC-BY license 4.0 (https://creativecommons.org/licenses/by/4.0/). © S. Berentsen 2020.
Patients with no or mild symptoms have not been shown to benefit from being treated. Older literature, however, has probably underestimated the symptom burden and need for therapy (61, 72, 74). Taking the increasing number of effective therapies into account, we consider symptomatic anemia, marked fatigue, or bothersome circulatory symptoms as indications for treatment (2, 20, 75). CAD should not be treated with corticosteroids, unspecific immune suppression, or splenectomy (1, 2, 20, 61, 76). Treatments directed at the pathogenic B-cell clone or the classical complement pathway have shown efficacy (61, 70, 77–85), as listed in Table 3. Rituximab at the dose of 375 mg/m2/week for 4 weeks is the most widely used first-line therapy, although response rates are modest and response duration is relatively short (78, 79). Addition of bendamustine highly improves response rates and duration but is also associated with some toxicity (61, 81).
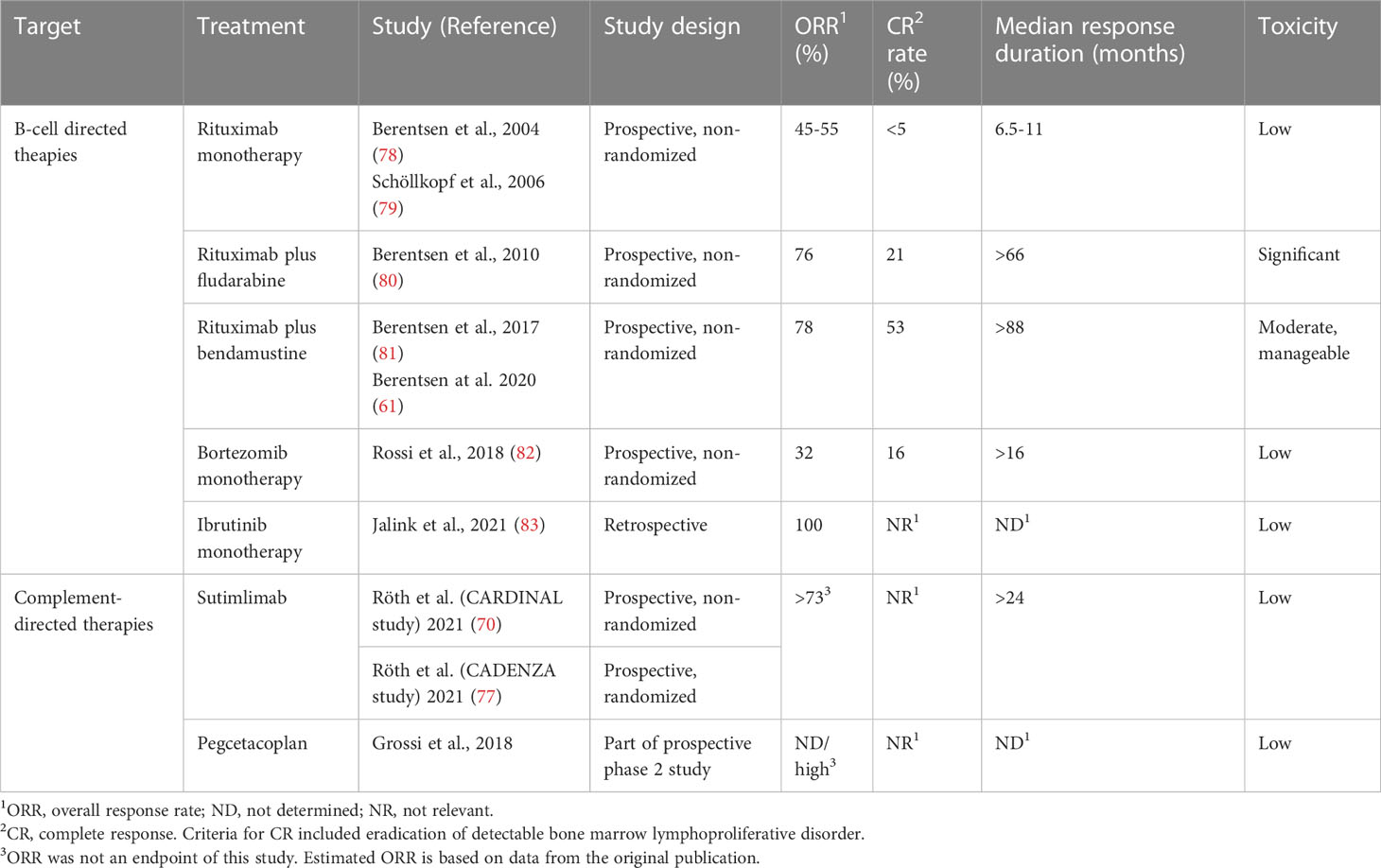
Table 3 Published studies of therapy in CAD.
Secondary cold agglutinin syndrome
In secondary cold agglutinin syndrome (CAS), cold agglutinin-mediated AIHA is caused by, or associated with, another clinical disease, such as infection with Mycoplasma pneumoniae, Epstein-Barr virus, cytomegalovirus, SARS-CoV-2, or other viruses, or a malignant disease, most often an overt B-cell lymphoma (86–92). There is no evidence-based therapy except for treating the underlying disease, if possible. Upstream complement inhibition has a strong theoretical rationale as a temporary measure until treatment of the underlying disorder takes effect, but the benefit remains unproven except for casuistic observations (84, 93).
Paroxysmal cold hemoglobinuria
PCH is an AIHA mediated by a polyclonal, biphasic antibody (Donath-Landsteiner antibody), usually an IgG (18, 94). Classical and terminal complement activation is strong and hemolysis is mainly intravascular. The term biphasic implies that the antigen-antibody reaction and fixation of the early complement components occur at temperatures below 37°C, whereas further complement activation takes place after rewarming to central body temperature (18, 95). Today, most cases of this rare condition occur as an acute, transient hemolytic anemia following a viral or other febrile infection in children (18, 96). The chronic paroxysmal (adult) form of PCH has become extremely rare and can occur in tertiary syphilis, hematologic malignancies, or without any identifiable cause.
Atypical AIHA
Mixed warm and cold-antibody AIHA is characterized by a monospecific DAT strongly positive for both IgG and C3d, high-titer cold agglutinins, and the concomitant presence of IgG warm-antibodies in serum (8, 97). Many patients have severe anemia and receive multiple lines of therapy before responding.
DAT-negative AIHA. In 3-10% of patients with AIHA, the polyspecific DAT is negative, mostly due to warm-autoantibody opsonization below the detection level. The diagnosis of DAT-negative AIHA remains difficult and requires extensive exclusion of differential diagnoses (non-immune causes of hemolysis) (1, 19, 54). More sensitive methods for detecting immunoglobulin on the erythrocyte surface may be helpful, but have lower specificity. A monospecific DAT for IgA is also mandatory, despite the negative polyspecific test (2, 54).
How to select therapy in warm AIHA
When to add rituximab in the first line?
As stated above, prednisolone (or prednisone), monotherapy is recommended in the first line in most patients with wAIHA. However, the low rate of long-sustained remission after tapering and discontinuation (30-40% after 1 year) is a concern, particularly in severely affected patients (98–100). In clinical practice, patients face a risk of being maintained on inappropriately high doses of steroids for years (14).Two prospective, randomized trials evaluated the upfront addition of rituximab (either 4 infusions of 375 mg/m2 at one-week interval or 2 infusions of 1000 mg fixed dose at two-week interval) (99, 100). The results were nearly identical, showing twice the rate of long-term responses in patients who had been given the combination upfront as compared to those treated with prednisolone only. Therefore, the international consensus group has recommended that severely anemic patients (defined as hemoglobin [Hb] < 8 g/dL) should be considered for rituximab plus prednisolone combination therapy in the first line (1). In this context, atypical AIHA (IgA-mediated, DAT-negative, or mixed) and Evans’ syndrome are also regarded as severe (1, 2, 22, 54, 101).
When to use erythropoiesis-stimulating agents?
The use of erythropoiesis-stimulating agents (ESA; erythropoietin [EPO] or its analogs) in AIHA has been studied in two retrospective series (102, 103). In the largest and most recent of these, 51 patients with warm or cold-antibody AIHA and reduced EPO levels or inadequate EPO response had been treated with an ESA, starting at median 24 months after diagnosis (103). Fifty-five percent of the patients responded within 15 days and more than 70% responded within 3 months; most responses seemed to be sustained on continued therapy, and the median increase in Hb was 2.1 g/dL. An attempt of ESA treatment will be worthwhile in patients with inadequate reticulocytosis and/or inadequate endogenous EPO levels at disease onset as well as after failing on corticosteroids, rituximab, and an immunosuppressive agent. For patients in whom corticosteroid maintenance is inevitable, an ESA may be successful as a steroid-sparing drug.
When to use intravenous immunoglobulin in wAIHA?
Administration of intravenous immunoglobulin (IVIG) at a total dose of 2 g/kg body weight (0.4 g/kg day for 5 days or 1 g/kg for 2 days) is supposed to inhibit extravascular hemolysis by saturating the reticuloendothelial system and Fc receptors (FcR) (104, 105). Saturating the FcRn will also promote clearance of the autoantibody (106). IVIG may be considered in hemolytic crisis and may be particularly useful in septic patients and those with an underlying autoimmune disease or primary immune deficiency.
When to recommend splenectomy?
Although splenectomy is far from being a new treatment for wAIHA, its position in the therapeutic armamentarium has changed. The response rates are high; probably 70-80% (8, 107), but long-term data are sparser (108). Disadvantages are a risk of early or late bacterial infection, a further increased risk of thrombosis, and the irreversibility of the procedure (109, 110). Therefore, we only recommend splenectomy in the third or subsequent lines. Although several experts will postpone splenectomy until at least one unspecific immunosuppressant has been tried, patient preferences should be taken into account (14, 111). However, considering the difficulties in treating some wAIHA patients and the relatively high efficacy of removing the spleen, this option should not be withheld in drug-refractory cases (1, 14). Patients should be vaccinated against encapsulated bacteria according to national recommendations or the Advisory Committee on Immunization Practices (ACIP) guidelines (112).
When to use investigational therapies in wAIHA?
Clinical trials
The ideal therapy for wAIHA should be low-toxic, easy to administer, should yield high response rates and durable responses, and should substantially reduce the high risk of relapses. Such a therapy does currently not exist. With this in mind, more prospective clinical trials are needed (12, 14, 15). Patients requiring therapy in any line may be considered for a prospective trial if eligible, provided randomization to the control arm (if relevant) is considered ethically justifiable.
Inhibitors of the neonatal Fc receptor
FcRn is required for physiological recirculation of IgG, which explains the long normal biological half-life of IgG1, IgG2, and IgG4 at approximately 23 days (27, 106). Blockade of this receptor, therefore, results in marked shortening of IgG half-life and lowering of IgG levels to about 10% of normal without inhibiting the production (113). Nipocalimab (M281), rozanolixizumab (UCB7665), and orilanolimab (SYNT001) are FcRn-targeting monoclonal antibodies (114, 115), and the safety and efficacy of nipocalimab is being studied in a prospective trial in wAIHA (ClinicalTrials.gov, NCT04119050). Available clinical data do not permit suggestions for its future role in treatment. However, the very quick and reversible pharmacodynamics observed in preclinical trials may suggest their use in the acute setting, similarly to IVIG employment.
Fostamatinib
Inhibitors of cellular mediators of phagocytosis are currently being investigated for AIHA treatment. One of these drugs, fostamatinib, a splenic tyrosine kinase (syk) inhibitor, was found to increase Hb levels to ≥10 g/dL or by 2 g/dL or more in 11 (46%) of 24 patients with wAIHA in a phase 2 trial (116). Participants had failed at least one previous treatment. Forty-two per cent of the patients experienced diarrhea; and fatigue, hypertension, dizziness, and insomnia were also frequent adverse events. The drug is currently approved in the US and Europe for treatment of immune thrombocytopenia. A phase 3 trial in wAIHA has finished inclusion (117), and an open-label extension study is also being performed (ClinicalTrials.gov, NCT03764618; NCT04138927). While still investigational, fostamatinib may turn out to be an option in the third line.
Other inhibitors of signal transducers
Rilzabrutinib, a reversible, covalent Bruton tyrosine kinase (BTK) inhibitor (118), is currently being studied in a phase 2 trial in wAIHA (ClinicalTrials.gov, NCT05002777). This drug also inhibits phagocytosis via interaction with the syk pathway. Parsaclisib, a phosphatidylinositol 3-kinase δ inhibitor, is a candidate drug in wAIHA as well as CAD (119). Preliminary phase 2 results in relapsed or refractory wAIHA show a 64% response rate, although with some toxicities including diarrhea, cytomegalovirus reactivation, and psoriasis (120). A randomized, controlled phase 3 trial is ongoing (ClinicalTrials.gov, NCT05073458).
Plasma cell-directed therapies
Bortezomib, a potent and selective proteasome inhibitor extensively used for the treatment of multiple myeloma, has showed promising results in autoimmune disorders by targeting the antibody-producing cells, including long-lived plasma cells (121). Beneficial effect of bortezomib in refractory wAIHA has been described in several case reports (122). In a retrospective series of adults who received 6 cycles of bortezomib plus dexamethasone, 6 of 8 patients responded following a median of 2 (range, 1-4) cycles (123). A small, prospective study found response to a combination of bortezomib, low-dose rituximab, and dexamethasone in 6 of 7 patients with refractory wAIHA (124).
At present, the evidence for bortezomib in wAIHA is limited because evaluation of pooled case reports is likely to be influenced by selection and publication bias and because the two systematic studies are small. Despite these reservations, bortezomib-based therapy may accommodate a previously unmet need in refractory or relapsed patients in whom rituximab has failed or is contraindicated.
In a small case series, therapy with the CD38-targeting monoclonal antibody daratumumab was used as a last resort in 3 patients with life-threatening, refractory wAIHA after stem cell transplantation (125). Two patients enjoyed a complete and sustained response with only minor toxicity, while the third patient experienced a transient improvement for 8 months but then suffered a lethal relapse. A systematic study, for example a larger retrospective series, should be performed before the potential role of daratumumab in wAIHA can be determined. The interference of daratumumab with DAT and indirect antiglobulin testing should be observed (126). A phase 1 study of isatuximab, an anti-CD38 monoclonal antibody for subcutaneous administration in wAIHA, is ongoing (ClinicalTrials.gov, NCT04661033).
Sirolimus
Sirolimus, an immunosuppressive ingredient of Streptomyces hygroscopicus, has been found to prevent graft rejection following organ transplantations (127). Administration of this drug was followed by remission in 4 children with wAIHA; 2 of them with overlapping pure red cell aplasia (128). Beneficial effect has also been observed in children with Evans’ syndrome (129). A retrospective study of sirolimus therapy in 45 adults and children with relapsed or refractory autoimmune cytopenias, of whom 14 had primary wAIHA and 12 had Evans’ syndrome, reported favorable response and safety data (127). Sirolimus may find a future role in treatment of refractory wAIHA including Evans’ syndrome in children, possibly also in adults, but should still be considered experimental as prospective clinical studies are lacking. Frequent hematological, gastrointestinal, cardiac, and immunological adverse effects may be a concern. A prospective study would be welcome.
What to do in emergencies?
Suggested emergency therapies in critically anemic wAIHA patients with no or slow response to prednisolone have been addressed elsewhere and include high-dose intravenous methylprednisolone, IVIG, plasma exchange, emergency splenectomy, and partial splenic embolization (1, 2, 51, 130, 131). While success has been documented in single cases, the evidence supporting each of these options is limited, and most of them are old and not within the scope of this review.
Regarding newer approaches in the emergency setting, prompt effect of complement inhibition with high, frequently repeated doses of plasma-derived C1-inhibitor was reported in a single case of severe, secondary IgM-mediated wAIHA with complement involvement (132). However, the preliminary results of a prospective trial seem disappointing (133). Using the specific C1s-targeting, monoclonal antibody sutimlimab in this context has a strong mechanistic rationale but has not been systematically studied. Still, based on theoretical considerations, case observations, and immediate effect on the classical complement pathway, C1 inhibition may be considered a low-toxic and potentially efficient option in life-threatening wAIHA with a positive DAT for C3 fragments (1, 132, 134). Furthermore, a phase 2 study of the C3 inhibitor pegcetacoplan showed some efficacy in wAIHA with C3-positive DAT (135).
Bortezomib-based combination regimens, addressed above, might also be used in emergencies based on the high response rates reported in small series and relatively low toxicity (123, 124). Administration of an ESA can be a useful supplement in a hemolytic crisis (1, 103). Potential future agents for the acute setting may also include the already mentioned FcRn inhibitors that may boost the clearance of the autoantibodies, thus limiting the hemolytic crisis (27, 106, 114).
Figure 3 shows a therapeutic algorithm covering the most commonly encountered situations in wAIHA patients.
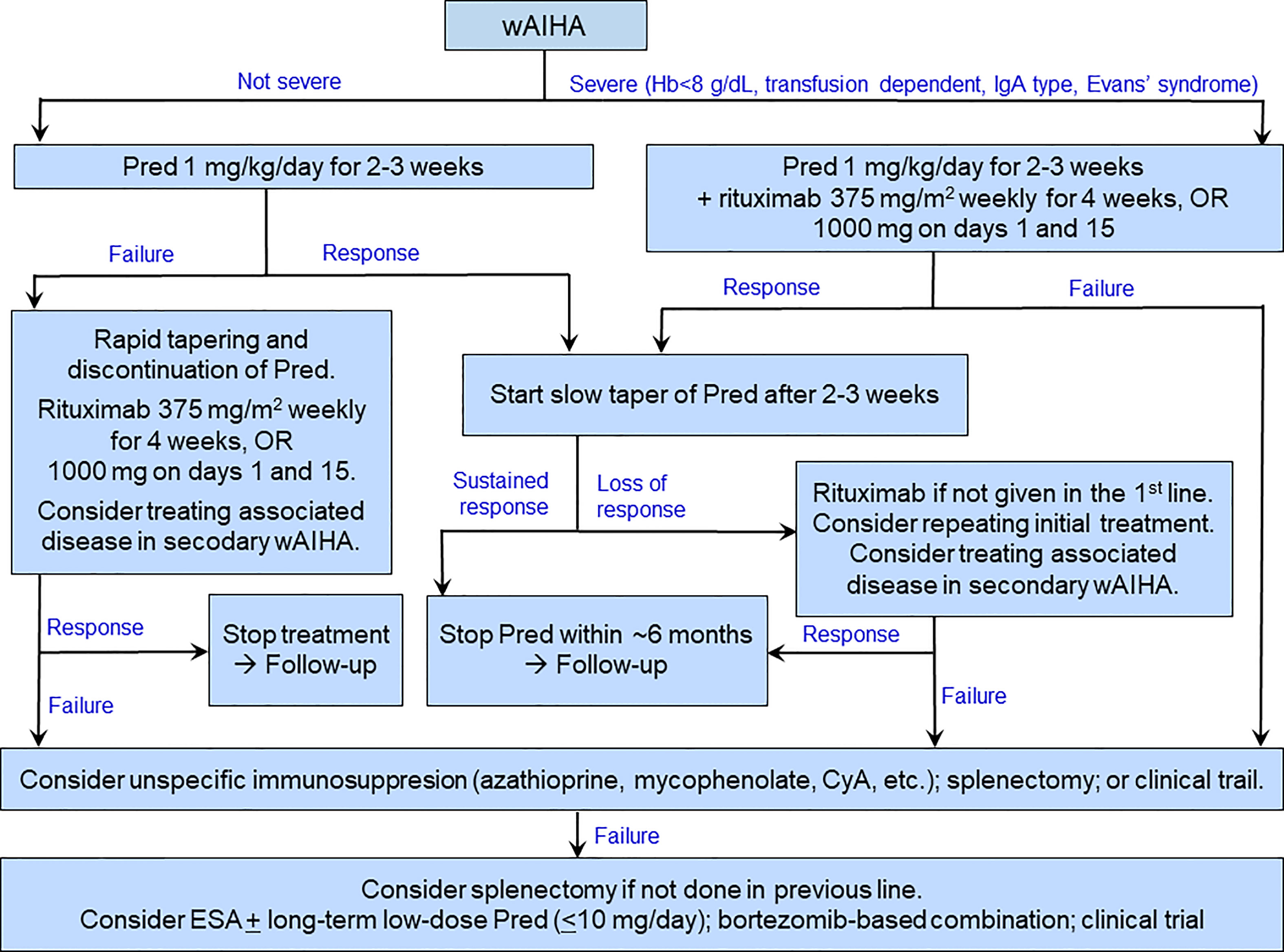
Figure 3 Suggested therapeutic algorithm in warm-antibody AIHA. ESA may be used as a supplement at any stage as well as in the last line. CyA, cyclosporine A; ESA, erythropoiesis-stimulating agent; Pred, prednisolone or prednisone.
How to select therapy in CAD
B-cell or complement directed therapy?
Although comparative trials of B-cell directed therapies versus complement inhibition have not been performed, some conclusions on preferences are justified based on the different profiles of these approaches (16, 136, 137), as indicated in Table 4. Advantages of the existing B-cell directed therapies are the time-limited treatment, the high rate of overall and complete responses and long response duration with bendamustine plus rituximab, the effect on circulatory symptoms as well as hemolytic anemia, and the relatively low toxicity of rituximab monotherapy. Drawbacks are the often-long time to response, the existing although usually manageable toxicity of bendamustine plus rituximab, and the relatively low response rate and short response duration with rituximab monotherapy (61, 78, 81). The most extensively studied complement inhibitor, sutimlimab, has obvious advantages in the very rapid onset of effect, the high response rate, and the low toxicity. Disadvantages of sutimlimab are the lack of effect on circulatory symptoms, the probable need for indefinite treatment duration, the biweekly intravenous infusions, and the very high costs (136, 138). Figure 4 shows a suggested therapeutic algorithm.
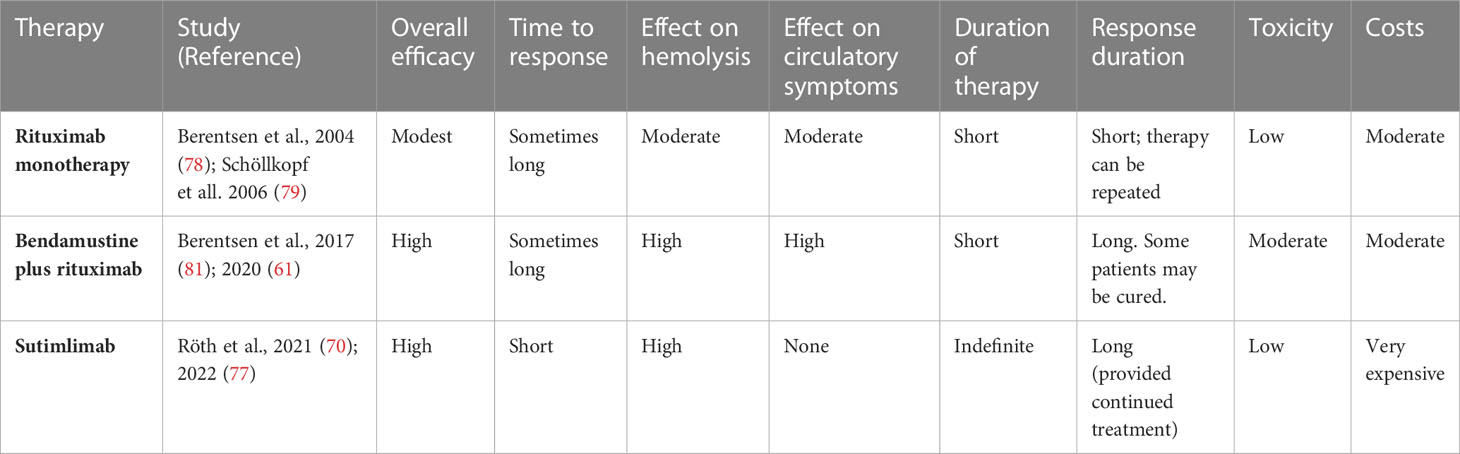
Table 4 B-cell directed versus complement-directed therapies in CAD.
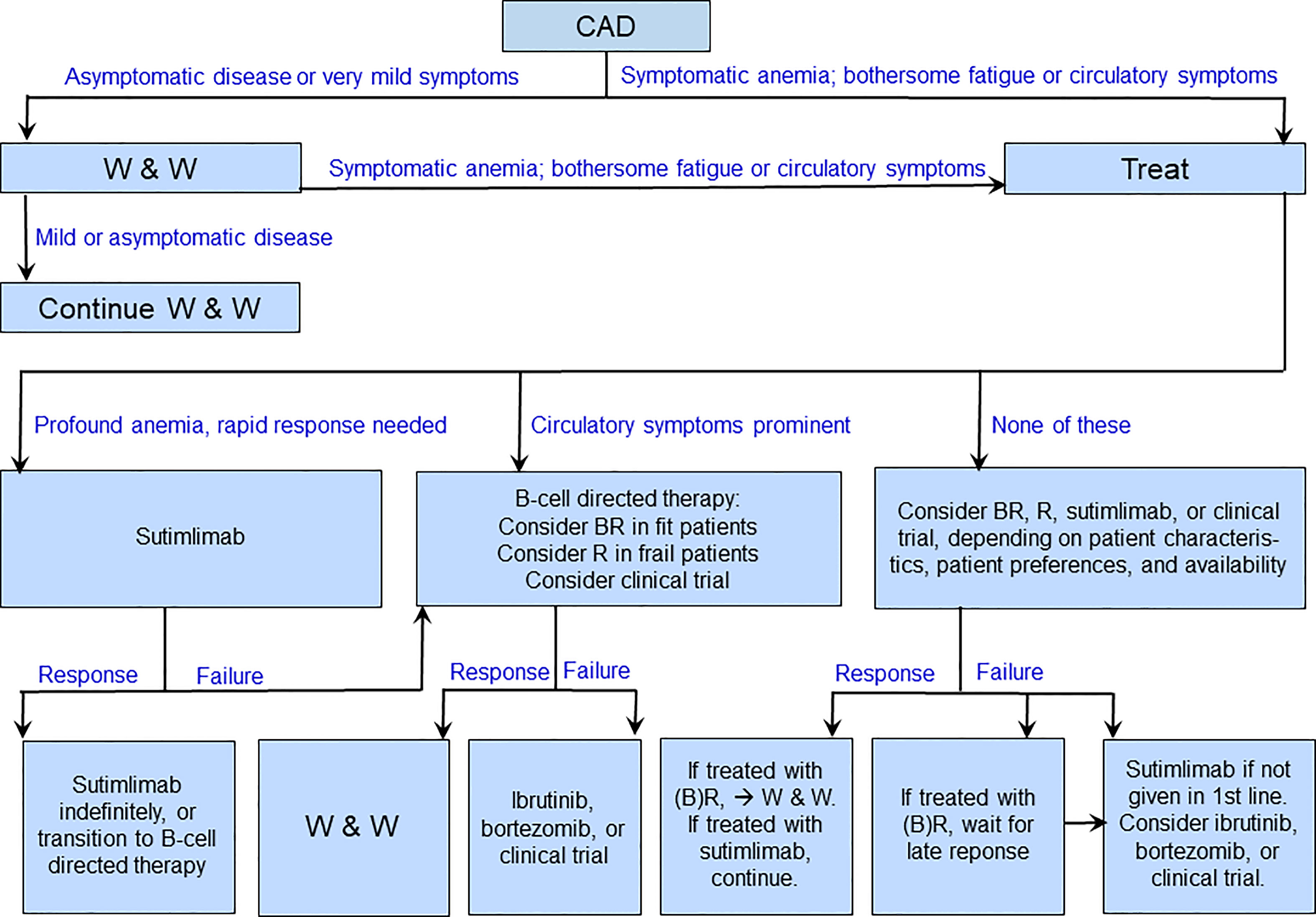
Figure 4 Suggested therapeutic algorithm in cold agglutinin disease. B, bendamustine; R, rituximab; W&W, watch and wait.
How to treat critically anemic patients with CAD?
Acute exacerbations of CAD are often triggered by cold exposure, febrile infection, major trauma, or major surgery (61, 139, 140). In these situations, it is reasonable to treat the cause of exacerbation when relevant, transfuse the patient if required and, in many cases, wait several weeks for improvement of the hemolytic anemia. In CAD patients who present with profound anemia and need a rapid effect of therapy, first-line use of sutimlimab will often be successful, given the often-long time to response to B-cell directed therapies (70, 77, 136). After a stable response has been achieved, highly selected patients might be considered for transition to a time-limited, B-cell targeting therapy as an alternative to indefinite continuation of sutimlimab, depending on patient characteristics (fit or unfit), clinical disease features (marked circulatory symptoms or not), and patient preferences (136, 141). However, this “bridging” approach is currently not evidence-based.
How to treat patients with prominent circulatory symptoms?
As explained above, peripheral circulatory symptoms are absent or mild in approximately two-thirds of patients with CAD, while about one-fifth have hemolytic anemia with circulatory symptoms interfering with daily living (61). Approximately one-tenth of patients have circulatory symptoms with compensated hemolysis, i.e. as the only or main clinical manifestation. Thus, circulatory symptoms may constitute, or substantially contribute to, the indication for treatment in up to 30% of the patients. As these symptoms are not complement-mediated, they are not expected to improve on complement-inhibiting therapy, which is in accordance with the findings in a clinical trial (142). Patients in this subgroup, therefore, should be treated with B-cell directed therapies, which have the potential to resolve circulatory symptoms as well as hemolysis (16, 78, 81).
How to choose the best therapy in the “typical” CAD patient?
A high proportion of CAD patients requiring therapy have moderate anemia, often fatigue, and no or mild circulatory symptoms (Figure 2) (61). In such patients, current evidence does not allow any hard, general recommendation, and therapy should be individualized. In the first line, a B-cell directed approach will be appropriate in most of these patients (16, 137). Moderate to severe anemia in an otherwise fit patient will favor rituximab plus bendamustine, whereas mild clinical disease and/or unfit patient will favor rituximab monotherapy. In this setting, it is reasonable to consider sutimlimab as a second-line option, which may be used in the first line in patients who have contraindications to chemoimmunotherapy (136). Patient preferences should be taken into consideration. Second-line options, apart from sutimlimab, may include bortezomib (82) or inclusion in a clinical trial, while the use of rituximab plus fludarabine should be restricted to carefully selected patients (61, 80).
When to consider inclusion in a clinical trial?
Even after the recent, major progress in the treatment of CAD, there are several unmet needs. The ideal therapy should be highly efficacious against hemolytic anemia and fatigue as well as circulatory symptoms, rapidly acting, low-toxic, and affordable worldwide. Since such therapies do not exist, clinical trials could be considered in any line of therapy provided randomization (if relevant) is deemed ethically justifiable.
Regarding future B-cell directed approaches, treatment with the BTK inhibitor ibrutinib has shown highly promising results in a retrospective series (83). In this study of 13 patients with CAD or CAS secondary to low-grade non-Hodgkin lymphoma, all patients responded; responses seemed sustained on continued medication, and the drug was generally well tolerated. This option should be considered investigational until completion of a prospective BTK inhibitor trial, which is warranted.
Complement-directed therapies should also be further explored. The C3 inhibitor pegcetacoplan (APL-2), a pegylated cyclic peptide designed for subcutaneous infusion, has yielded favorable response rates with low toxicity in a phase 2 study (85). Pegcetacoplan in CAD is currently being further investigated in a randomized, placebo-controlled phase 3 trial (ClinicalTrials.gov, NCT05096403). Other complement-targeting agents that may find a role in the treatment of CAD are ANX005, a C1q inhibitor (143, 144), BIVV020, which targets C1s (145) (ClinicalTrials.gov, NCT04269551), and ARGX-117, which inhibits C2 (146). Relevant clinical data have not been published for these three monoclonal antibodies, but prospective trials would be welcome.
What to do in emergencies?
The traditional emergency therapy in CAD is plasma exchange (147–149). As 80% of IgM is located in the blood stream, the theoretical chance of success should be high. However, while favorable results have been confirmed in several cases, unsuccessful attempts may be underreported, and response rates have not been determined. The response duration is short, and concomitant initiation of pharmacological therapy is mandatory. For substitution, albumin should be used instead of plasma in order to avoid infusion of exogenous complement (51, 139).
Today, considering the very rapid effect of sutimlimab on hemolytic anemia in CAD, complement C1 inhibition may be preferred as an emergency therapy (1, 20, 70, 136). Even C5 inhibition with eculizumab may be helpful, as C5b-9 mediated, intravascular hemolysis can be prominent in critically hemolytic patients (1, 84, 150).
How to select therapy in other AIHAs
Secondary cold agglutinin syndrome
CAS caused by Mycoplasma pneumoniae or viruses will resolve spontaneously after elimination of the infection. In Mycoplasma infection, however, hemolytic anemia can occasionally be profound and prolonged, and the onset of the hemolytic complication typically occurs in the second week, when antibiotic therapy has often already been initiated or even completed (86, 88, 90). Transfusion is safe provided the same precautions are observed as in CAD (See below). Complement inhibition has a strong theoretical rationale, but clinical evidence is lacking except for a couple of case reports (93).
Paroxysmal cold hemoglobinuria
In children with PCH, the use of corticosteroids has recently been questioned based on a literature study of all 230 published PCH cases, in which there was no difference in length of hospital stay between patients who had received corticosteroids and those who had not (18). In most patients, thermal protection and, if necessary, transfusion will be appropriate until spontaneous resolution occurs (151, 152). Terminal complement blockade has a strong mechanistic rationale; was followed by immediate resolution in a case report, and might be justified in selected cases as an attempt to relieve critical hemolytic anemia in pediatric PCH patients (96). In the extremely rare cases of adult PCH, there is no evidence-based therapy apart from thermal protection and treatment of the underlying disease, when identifiable.
How to treat mixed AIHA?
Mixed warm and cold-antibody AIHA is often difficult to treat, and prospective trials specific for this rare AIHA have not been conducted. In our clinical experience, patients should receive corticosteroids at high doses along with rituximab in the first line (1, 2, 8, 153). If cold agglutinin-related clinical symptoms are prominent, it may be advisable to treat this subtype like CAD (2). In some cases, the same patient may display alternate clinical and laboratory features of wAIHA and CAD, requiring specific treatments. Splenectomy is discouraged.
Which AIHA patients should receive erythrocyte transfusions?
Transfusion in AIHA requires specific precautions, depending on the temperature range of the autoantibodies (1, 2). In wAIHA, pre-transfusion screening for irregular antibodies will be positive, and crossmatching of type-identical blood will show incompatibility. Transfusion may hold a risk of acute or delayed hemolytic transfusion reaction and a risk of alloimmunization. When transfusion is clearly needed, however, this supportive therapy must not be withheld and is generally safe provided the required precautions are observed (154, 155). The old notion to use “the least incompatible donor blood” should be abandoned (156, 157). Instead, an extended phenotyping should be performed if time permits, and phenotype-identical erythrocyte concentrate should be preferred when possible (157, 158). A good communication between the clinician and the transfusion center is of vital importance.
In CAD, precautions are different. Transfusion is generally safer than in wAIHA and should not be omitted when indicated. However, there is a potential risk of agglutination and hemolysis of patient as well as donor erythrocytes because of cooling. Pre-transfusion screening and crossmatching will usually be unremarkable if performed at 37°C, but autoadsorption techniques may be required if the thermal amplitude of the cold agglutinin approaches 37°C (16). The patient and the extremity chosen for transfusion should be kept warm, and the use of an in-line blood warmer is recommended (1, 2, 20, 53).
Conclusion
Prednisolone (or prednisone) at high initial doses remains the first-line treatment of wAIHA, but upfront addition of rituximab should be considered in severe cases. In CAD, effective therapies are directed at the pathogenic B-cell clone or the classical complement pathway. In both warm and cold-antibody mediated AIHA, the last decade has seen an increasing number of established and investigational treatment options, and individualization should be part of the therapeutic considerations. Above, we have outlined established or tentative advice for the choice of therapy in specific situations. In any type of AIHA, the ideal treatment remains to be developed, and some recommendations are still based on relatively weak evidence. Therefore, prospective clinical trials are important for future improvement of therapy in warm as well as cold-antibody mediated AIHA.
Author contributions
SB collected data, prepared the tables and figures, and drafted the manuscript. BF and WB participated in data collection, and reviewed and commented the manuscript. All authors reviewed and approved the submitted version.
Conflict of interest
Outside this work, SB has received advisory board honoraria from Annexon, Momenta, Sanofi, and Sobi, and lecture honoraria from Apellis, BeiGene, Janssen-Cilag, Sanofi, and Sobi. BF declares consultancies with Amgen, Alexion, Annexon, Apellis, Momenta, Novartis, and Sobi. WB has received consultancy and advisory board honoraria from Agios, Alexion, Biocryst, Incyte, Novartis, and Sanofi; lecture honoraria and congress support from Alexion, Incyte, Novartis, and Sanofi; and research support from Alexion.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jäger U, Barcellini W, Broome CM, Gertz MA, Hill A, Hill QA, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev (2020) 41:100648. doi: 10.1016/j.blre.2019.100648:100648
2. Berentsen S, Barcellini W. Autoimmune hemolytic anemias. N Engl J Med (2021) 385(15):1407–19. doi: 10.1056/NEJMra2033982
3. Hill QA, Hill A, Berentsen S. Defining autoimmune hemolytic anemia: a systematic review of the terminology used for diagnosis and treatment. Blood Adv (2019) 3(12):1897–906. doi: 10.1182/bloodadvances.2019000036
4. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
5. Fatone MC, Pavone F, Lauletta G, Russi S. Features of peripheral Cd8(+)Cd57(+) lymphocytes in patients with autoimmune hemolytic anemia. Autoimmunity (2018) 51(4):166–74. doi: 10.1080/08916934.2018.1477132
6. Sokol RJ, Hewitt S, Stamps BK. Autoimmune haemolysis: an 18-year study of 865 cases referred to a regional transfusion centre. Br Med J (Clin Res Ed) (1981) 282(6281):2023–7. doi: 10.1136/bmj.282.6281.2023
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol (2011) 4(6):607–18. doi: 10.1586/EHM.11.60
8. Barcellini W, Fattizzo B, Zaninoni A, Radice T, Nichele I, Di Bona E, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a gimema study of 308 patients. Blood (2014) 124(19):2930–6. doi: 10.1182/blood-2014-06-583021
9. Packman CH. The spherocytic haemolytic anaemias. Br J Haematol (2001) 112(4):888–99. doi: 10.1046/j.1365-2141.2001.02440.x
10. Landsteiner K. Über beziehungen zwischen dem blutserum und den körperzellen. Münch Med Wschr (1903) 50:1812–4.
11. Dameshek W, Rosenthal MC, Schwartz LI. The treatment of acquired hemolytic anemia with adrenocorticotrophic hormone (Acth). N Engl J Med (1950) 244(4):117–27. doi: 10.1056/NEJM195101252440401
12. Hill QA, Horan M, Charlton A, Bullock T, Massey E, Hill A, et al. Developing the evidence base for the management of autoimmune haemolytic anaemia (Aiha): the uk experience. Br J Haematol (2021) 192(2):e54–e7. doi: 10.1111/bjh.17217
13. Barcellini W, Fattizzo B, Zaninoni A. Current and emerging treatment options for autoimmune hemolytic anemia. Expert Rev Clin Immunol (2018) 14(10):857–72. doi: 10.1080/1744666X.2018.1521722
14. Kuter DJ. Warm autoimmune hemolytic anemia and the best treatment strategies. Hematol Am Soc Hematol Educ Program (2022) 2022(1):105–13. doi: 10.1182/hematology.2022000405
15. Fattizzo B, Barcellini W. New therapies for the treatment of warm autoimmune hemolytic anemia. Transfus Med Rev (2022) 36(4):175–80. doi: 10.1016/j.tmrv.2022.08.001
16. Berentsen S, D’Sa S, Randen U, Malecka A, Vos JMI. Cold agglutinin disease: improved understanding of pathogenesis helps define targets for therapy. Hemato (2022) 3:574–94. doi: 10.3390/hemato3040040
17. Barcellini W, Fattizzo B. The changing landscape of autoimmune hemolytic anemia. Front Immunol (2020) 11:946. doi: 10.3389/fimmu.2020.00946
18. Jacobs JW, Villalba CAF, Booth GS, Woo JS, Stephens LD, Adkins BD. Clinical and epidemiological features of paroxysmal cold hemoglobinuria: a systematic review. Blood Adv (2023). doi: 10.1182/bloodadvances.2022009516
19. Barcellini W, Fattizzo B. How I treat warm autoimmune hemolytic anemia. Blood (2021) 137(10):1283–94. doi: 10.1182/blood.2019003808
20. Berentsen S. How I treat cold agglutinin disease. Blood (2021) 137(10):1295–303. doi: 10.1182/blood.2019003809
21. Meulenbroek EM, de Haas M, Brouwer C, Folman C, Zeerleder SS, Wouters D. Complement deposition in autoimmune hemolytic anemia is a footprint for difficult-to-Detect igm autoantibodies. Haematologica (2015) 100(11):1407–14. doi: 10.3324/haematol.2015.128991
22. Bardill B, Mengis C, Tschopp M, Wuillemin WA. Severe IgA-mediated auto-immune haemolytic anaemia in a 48-Yr-Old woman. Eur J Haematol (2003) 70(1):60–3. doi: 10.1034/j.1600-0609.2003.02846.x
23. Abramson N, Gelfand EW, Jandl JH, Rosen FS. The interaction between human monocytes and red cells. specificity for IgG subclasses and IgG fragments. J Exp Med (1970) 132(6):1207–15.
24. Zhang Y, Chu Y, Shao Z. [the clinical implications of IgG subclass in 84 patients with autoimmune hemolytic anemia]. Zhonghua Xue Ye Xue Za Zhi (1999) 20(10):524–6.
25. Li TX, Sun FT, Ji BJ. [Correlation of IgG subclass with blood cell parameters in patients with autoimmune hemolytic anemia]. Zhongguo Shi Yan Xue Ye Xue Za Zhi (2019) 27(1):197–201. doi: 10.7534/j.issn.1009-2137.2019.01.032
26. Li Z, Shao Z, Xu Y, Shen L, Chen G, Zhang Y, et al. Subclasses of warm autoantibody IgG in patients with autoimmune hemolytic anemia and their clinical implications. Chin Med J (Engl) (1999) 112(9):805–8.
27. Saxena A, Wu D. Advances in therapeutic fc engineering - modulation of IgG-associated effector functions and serum half-life. Front Immunol (2016) 7:580. doi: 10.3389/fimmu.2016.00580
28. LoBuglio AF, Cotran RS, Jandl JH. Red cells coated with immunoglobulin G: binding and sphering by mononuclear cells in man. Science (1967) 158(3808):1582–5. doi: 10.1126/science.158.3808.1582
29. Coombs RR, Mourant AE, Race RR. A new test for the detection of weak and incomplete Rh agglutinins. Br J Exp Pathol (1945) 26:255–66. doi: 10.1007/978-94-011-6138-1_10
30. Zantek ND, Koepsell SA, Tharp DR Jr., Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol (2012) 87(7):707–9. doi: 10.1002/ajh.23218
31. Barcellini W, Revelli N, Imperiali FG, Villa MA, Manera MC, Paccapelo C, et al. Comparison of traditional methods and mitogen-stimulated direct antiglobulin test for detection of anti-red blood cell autoimmunity. Int J Hematol (2010) 91(5):762–9. doi: 10.1007/s12185-010-0578-9
32. Hansen DL, Møller S, Andersen K, Gaist D, Frederiksen H. Increasing incidence and prevalence of acquired hemolytic anemias in Denmark, 1980-2016. Clin Epidemiol (2020) 12:497–508. doi: 10.2147/CLEP.S250250
33. Fattizzo B, Barcellini W. Autoimmune hemolytic anemia: causes and consequences. Expert Rev Clin Immunol (2022) 18(7):731–45. doi: 10.1080/1744666X.2022.2089115
34. Evans RS, Duane RT. Acquired hemolytic anemia; the relation of erythrocyte antibody production to activity of the disease; the significance of thrombocytopenia and leukopenia. Blood (1949) 4(11):1196–213. doi: 10.1182/blood.V4.11.1196.1196
35. Fattizzo B, Michel M, Giannotta JA, Hansen DL, Arguello M, Sutto E, et al. Evans Syndrome in adults: an observational multicenter study. Blood Adv (2021) 5(24):5468–78. doi: 10.1182/bloodadvances.2021005610
36. Hansen DL, Møller S, Andersen K, Gaist D, Frederiksen H. Evans Syndrome in adults - incidence, prevalence, and survival in a nationwide cohort. Am J Hematol (2019) 94(10):1081–90. doi: 10.1002/ajh.25574
37. Michel M. Adult evans’ syndrome. Hematol Oncol Clin North Am (2022) 36(2):381–92. doi: 10.1016/j.hoc.2021.12.004
38. Liesveld JL, Rowe JM, Lichtman MA. Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood (1987) 69(3):820–6. doi: 10.1182/blood.V69.3.820.820
39. Conley CL, Lippman SM, Ness PM, Petz LD, Branch DR, Gallagher MT. Autoimmune hemolytic anemia with reticulocytopenia and erythroid marrow. N Engl J Med (1982) 306(5):281–6. doi: 10.1056/NEJM198202043060507
40. Branch DR, Shulman IA, Sy Siok Hian AL, Petz LD. Two distinct categories of warm autoantibody reactivity with age-fractionated red cells. Blood (1984) 63(1):177–80. doi: 10.1182/blood.V63.1.177.177
41. Allgood JW, Chaplin H Jr. Idiopathic acquired autoimmune hemolytic anemia. a review of forty-seven cases treated from 1955 through 1965. Am J Med (1967) 43(2):254–73.
42. Pullarkat V, Ngo M, Iqbal S, Espina B, Liebman HA. Detection of lupus anticoagulant identifies patients with autoimmune haemolytic anaemia at increased risk for venous thromboembolism. Br J Haematol (2002) 118(4):1166–9. doi: 10.1046/j.1365-2141.2002.03729.x
43. Roumier M, Loustau V, Guillaud C, Languille L, Mahevas M, Khellaf M. Characteristics and outcome of warm antibody autoimmune hemolytic anemia in adults: new insights based on single-center experience with 60 patients. Am J Hematol (2014) 89(9):E150–E5. doi: 10.1002/ajh.23767
44. Yusuf HR, Hooper WC, Grosse SD, Parker CS, Boulet SL, Ortel TL. Risk of venous thromboembolism occurrence among adults with selected autoimmune diseases: a study among a U.S. cohort of commercial insurance enrollees. Thromb Res (2015) 135(1):50–7. doi: 10.1016/j.thromres.2014.10.012
45. Lecouffe-Desprets M, Neel A, Graveleau J, Leux C, Perrin F, Visomblain B, et al. Venous thromboembolism related to warm autoimmune hemolytic anemia: a case-control study. Autoimmun Rev (2015) 14(11):1023–8. doi: 10.1016/j.autrev.2015.07.001
46. Audia S, Bach B, Samson M, Lakomy D, Bour JB, Burlet B, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. PloS One (2018) 13(11):e0207218. doi: 10.1371/journal.pone.0207218
47. Giannotta JA, Fattizzo B, Cavallaro F, Barcellini W. Infectious complications in autoimmune hemolytic anemia. J Clin Med (2021) 10(1):164. doi: 10.3390/jcm10010164
48. Fattizzo B, Bortolotti M, Giannotta JA, Zaninoni A, Consonni D, Barcellini W. Intravascular hemolysis and multitreatment predict thrombosis in patients with autoimmune hemolytic anemia. J Thromb Haemost (2022) 20(8):1852–8. doi: 10.1111/jth.15757
49. Murphy S, LoBuglio AF. Drug therapy of autoimmune hemolytic anemia. Semin Hematol (1976) 13(4):323–34.
50. Pirofsky B. Immune haemolytic disease: the autoimmune haemolytic anaemias. Clin Haematol (1975) 4(1):167–80. doi: 10.1016/S0308-2261(21)00630-5
51. Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A, et al. The diagnosis and management of primary autoimmune haemolytic anaemia. Br J Haematol (2017) 176(3):395–411. doi: 10.1111/bjh.14478
52. Meyer O, Stahl D, Beckhove P, Huhn D, Salama A. Pulsed high-dose dexamethasone in chronic autoimmune haemolytic anaemia of warm type. Br J Haematol (1997) 98(4):860–2. doi: 10.1046/j.1365-2141.1997.3203137.x
53. Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A, et al. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol (2017) 177(2):208–20. doi: 10.1111/bjh.14654
54. Fattizzo B, Giannotta JA, Serpenti F, Barcellini W. Difficult cases of autoimmune hemolytic anemia: a challenge for the internal medicine specialist. J Clin Med (2020) 9(12):948. doi: 10.3390/jcm9123858
55. Murakhovskaya I. Rituximab use in warm and cold autoimmune hemolytic anemia. J Clin Med (2020) 9(12):4034. doi: 10.3390/jcm9124034
56. Fattizzo B, Zaninoni A, Pettine L, Cavallaro F, Di Bona E, Barcellini W. Low-dose rituximab in autoimmune hemolytic anemia: 10 years after. Blood (2019) 133(9):996–8. doi: 10.1182/blood-2018-12-885228
57. Fattizzo B, Cantoni S, Giannotta JA, Bandiera L, Zavaglia R, Bortolotti M, et al. Efficacy and safety of cyclosporine a treatment in autoimmune cytopenias: the experience of two Italian reference centers. Ther Adv Hematol (2022) 13:20406207221097780. doi: 10.1177/20406207221097780
58. Ho G, Brunson A, Keegan THM, Wun T. Splenectomy and the incidence of venous thromboembolism and sepsis in patients with autoimmune hemolytic anemia. Blood Cells Mol Dis (2020) 81:102388. doi: 10.1016/j.bcmd.2019.102388
59. Dacie J. The auto-immune haemolytic anaemias: introduction. In: Dacie J, editor. The haemolytic anaemias, vol. 3 . London: Churchill Livingstone (1992). p. 1–5.
60. Ulvestad E, Berentsen S, Bø K, Shammas FV. Clinical immunology of chronic cold agglutinin disease. Eur J Haematol (1999) 63(4):259–66. doi: 10.1111/j.1600-0609.1999.tb01887.x
61. Berentsen S, Barcellini W, D’Sa S, Randen U, Tvedt THA, Fattizzo B, et al. Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood (2020) 136(4):480–8. doi: 10.1182/blood.2020005674
62. Harboe M, van Furth R, Schubothe H, Lind K, Evans RS. Exclusive occurrence of K chains in isolated cold haemagglutinins. Scand J Haematol (1965) 2(3):259–66. doi: 10.1111/j.1600-0609.1965.tb01303.x
63. Randen U, Trøen G, Tierens A, Steen C, Warsame A, Beiske K, et al. Primary cold agglutinin-associated lymphoproliferative disease: a b-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica (2014) 99(3):497–504. doi: 10.3324/haematol.2013.091702
64. Naresh KN, Rossi D, Chen X, Berentsen S, Randen U. Cold agglutinin disease. In press: Who classification of haematolymphoid tumours, 5th Edition. Lyon: International Agency for Research on Cancer (2022). p. 349.
65. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
66. Jaffe CJ, Atkinson JP, Frank MM. The role of complement in the clearance of cold agglutinin-sensitized erythrocytes in man. J Clin Invest (1976) 58(4):942–9. doi: 10.1172/JCI108547
67. Shi J, Rose EL, Singh A, Hussain S, Stagliano NE, Parry GC, et al. Tnt003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood (2014) 123(26):4015–22. doi: 10.1182/blood-2014-02-556027
68. Berentsen S. Complement activation and inhibition in autoimmune hemolytic anemia: focus on cold agglutinin disease. Semin Hematol (2018) 55(3):141–9. doi: 10.1053/j.seminhematol.2018.04.002
69. Berentsen S, Hill A, Hill QA, Tvedt THA, Michel M. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther Adv Hematol (2019) 10:2040620719873321. doi: 10.1177/2040620719873321
70. Röth A, Barcellini W, D’Sa S, Miyakawa Y, Broome CM, Michel M, et al. Sutimlimab in cold agglutinin disease. N Engl J Med (2021) 384:1323–34. doi: 10.1056/NEJMoa2027760
71. Berentsen S. New insights in the pathogenesis and therapy of cold agglutinin-mediated autoimmune hemolytic anemia. Front Immunol (2020) 11:590. doi: 10.3389/fimmu.2020.00590
72. Broome CM, Cunningham JM, Mullins M, Jiang X, Bylsma LC, Fryzek JP, et al. Increased risk of thrombotic events in cold agglutinin disease: a 10-year retrospective analysis. Res Pract Thromb Haemost (2020) 4(4):628–35. doi: 10.1002/rth2.12333:928
73. Kamesaki T, Nishimura JI, Wada H, Yu E, Tsao E, Morales J, et al. Demographic characteristics, thromboembolism risk, and treatment patterns for patients with cold agglutinin disease in Japan. Int J Hematol (2020) 112(3):307–15. doi: 10.1007/s12185-020-02899-6
74. Röth A, Barcellini W, Tvedt THA, Miyakawa Y, Kuter DJ, Su J, et al. Sutimlimab improves quality of life in patients with cold agglutinin disease: results of patient-reported outcomes from the cardinal study. Ann Hematol (2022) 101(10):2169–77. doi: 10.1007/s00277-022-04948-y
75. Patriquin CJ, Pavenski KO. Wind, if winter comes … will symptoms be far behind?: exploring the seasonality (or lack thereof) and management of cold agglutinin disease. Transfusion (2022) 62(1):2–10. doi: 10.1111/trf.16765
76. Dacie J. Treatment and prognosis of cold-antibody aiha. In: Dacie J, editor. The haemolytic anaemias, vol. 3 . London: Churchill Livingstone (1992). p. 502–20.
77. Röth A, Berentsen S, Barcellini W, D’Sa S, Jilma B, Michel M, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 cadenza trial. Blood (2022) 140(9):980–91. doi: 10.1182/blood.2021014955
78. Berentsen S, Ulvestad E, Gjertsen BT, Hjorth-Hansen H, Langholm R, Knutsen H, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood (2004) 103(8):2925–8. doi: 10.1182/blood-2003-10-3597
79. Schöllkopf C, Kjeldsen L, Bjerrum OW, Mourits-Andersen HT, Nielsen JL, Christensen BE, et al. Rituximab in chronic cold agglutinin disease: a prospective study of 20 patients. Leuk Lymphoma (2006) 47(2):253–60. doi: 10.1080/10428190500286481
80. Berentsen S, Randen U, Vågan AM, Hjorth-Hansen H, Vik A, Dalgaard J, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood (2010) 116(17):3180–4. doi: 10.1182/blood-2010-06-288647
81. Berentsen S, Randen U, Oksman M, Birgens H, Tvedt THA, Dalgaard J, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood (2017) 130(4):537–41. doi: 10.1182/blood-2017-04-778175
82. Rossi G, Gramegna D, Paoloni F, Fattizzo B, Binda F, D’Adda M, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective gimema study. Blood (2018) 132(5):547–50. doi: 10.1182/blood-2018-03-835413
83. Jalink M, Berentsen S, Castillo JJ, Treon SP, Cruijsen M, Fattizzo B, et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin Disease/Cold agglutinin syndrome. Blood (2021) 138(20):2002–5. doi: 10.1182/blood.2021012039
84. Röth A, Bommer M, Hüttmann A, Herich-Terhurne D, Kuklik N, Rekowski J, et al. Eculizumab in cold agglutinin disease (Decade): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv (2018) 2(19):2543–9. doi: 10.1182/bloodadvances.2018024190
85. Grossi F, Shum MK, Gertz MA, Roman E, Deschatelets P, Hamdani M, et al. Inhibition of C3 with apl-2 results in normalisation of markers of intravascular and extravascular hemolysis in patients with autoimmune hemolytic anemia (Aiha). 60th annual meeting of the American society of hematology, San Diego, Ca. Blood (2018) 132(Suppl 1):3623. doi: 10.1182/blood-2018-99-119468
86. Berentsen S, Tjønnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev (2012) 26(3):107–15. doi: 10.1016/j.blre.2012.01.002
87. Crisp D, Pruzanski W. B-cell neoplasms with homogeneous cold-reacting antibodies (Cold agglutinins). Am J Med (1982) 72(6):915–22. doi: 10.1016/0002-9343(82)90852-X
88. Dacie J. Auto-immune haemolytic anaemia (Aiha): cold-antibody syndromes III: haemolytic anaemia following mycoplasma pneumonia. In: Dacie J, editor. The haemolytic anaemias, vol. 3 . London: Churchill Livingstone (1992). p. 296–312.
89. Schubothe H, Merz KP, Weber S, Dahm K, Schmitz W, Altmann HW. [Acute autoimmune hemolytic anemia caused by cold antibodies following mycoplasma pneumonia with fatal ending]. Acta Haematol (1970) 44(2):111–23. doi: 10.1159/000208668
90. Linz DH, Tolle SW, Elliot DL. Mycoplasma pneumoniae pneumonia. experience at a referral center. West J Med (1984) 140(6):895–900.
91. Dacie J. Auto-immune haemolytic anaemia (Aiha): cold-antibody syndromes IV: haemolytic anemia following infectious mononucleosis annd other viral infections. In: Dacie J, editor. The haemolytic anaemias, vol. 3. London: Churchill Livingstone (1992). p. 313–28.
92. Zagorski E, Pawar T, Rahimian S, Forman D. Cold agglutinin autoimmune haemolytic anaemia associated with novel coronavirus (Covid-19). Br J Haematol (2020) 190(4):e183–e4. doi: 10.1111/bjh.16892
93. Tesfaye A, Broome C. A novel approach for treatment of cold agglutinin syndrome-related severe hemolysis. J Hematol (2016) 5(1):30–3. doi: 10.14740/jh242w
94. Donath J, Landsteiner K. Ueber paroxysmale hämoglobinurie. Münchener medizinische Wochenschrift (1904) 36:1590–3.
95. Rosse WF, Adams J, Logue G. Hemolysis by complement and cold-reacting antibodies: time and temperature requirements. Am J Hematol (1977) 2(3):259–70. doi: 10.1002/ajh.2830020308
96. Lau-Braunhut SA, Stone H, Collins G, Berentsen S, Braun BS, Zinter MS. Paroxysmal cold hemoglobinuria successfully treated with complement inhibition. Blood Adv (2019) 3(22):3575–8. doi: 10.1182/bloodadvances.2019000897
97. Mayer B, Yurek S, Kiesewetter H, Salama A. Mixed-type autoimmune hemolytic anemia: differential diagnosis and a critical review of reported cases. Transfusion (2008) 48(10):2229–34. doi: 10.1111/j.1537-2995.2008.01805.x
98. Kulpa J, Skrabs C, Simanek R, Valent P, Panzer S, Lechner K, et al. Probability of remaining in unsustained complete remission after steroid therapy withdrawal in patients with primary warm-antibody reactive autoimmune hemolytic anemia. Wien Klin Wochenschr (2016) 128(7-8):234–7. doi: 10.1007/s00508-015-0863-y
99. Birgens H, Frederiksen H, Hasselbalch HC, Rasmussen IH, Nielsen OJ, Kjeldsen L, et al. A phase iii randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol (2013) 163(3):393–9. doi: 10.1111/bjh.12541
100. Michel M, Terriou L, Roudot-Thoraval F, Hamidou M, Ebbo M, Le Guenno G, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the raiha study). Am J Hematol (2017) 92(1):23–7. doi: 10.1002/ajh.24570
101. Janvier D, Sellami F, Missud F, Fenneteau O, Vilmer E, Cartron J, et al. Severe autoimmune hemolytic anemia caused by a warm iga autoantibody directed against the third loop of band 3 (Rbc anion-exchange protein 1). Transfusion (2002) 42(12):1547–52. doi: 10.1046/j.1537-2995.2002.00235.x
102. Salama A, Hartnack D, Lindemann HW, Lange HJ, Rummel M, Loew A. The effect of erythropoiesis-stimulating agents in patients with therapy-refractory autoimmune hemolytic anemia. Transfus Med Hemother (2014) 41(6):462–8. doi: 10.1159/000366244
103. Fattizzo B, Michel M, Zaninoni A, Giannotta J, Guillet S, Frederiksen H, et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: a multicenter international study. Haematologica (2021) 106(2):622–5. doi: 10.3324/haematol.2020.250522
104. Bussel JB, Cunningham-Rundles C, Abraham C. Intravenous treatment of autoimmune hemolytic anemia with very high dose gammaglobulin. Vox Sang (1986) 51(4):264–9. doi: 10.1111/j.1423-0410.1986.tb01967.x
105. Flores G, Cunningham-Rundles C, Newland AC, Bussel JB. Efficacy of intravenous immunoglobulin in the treatment of autoimmune hemolytic anemia: results in 73 patients. Am J Hematol (1993) 44(4):237–42. doi: 10.1002/ajh.2830440404
106. Roopenian DC, Akilesh S. Fcrn: the neonatal fc receptor comes of age. Nat Rev Immunol (2007) 7(9):715–25. doi: 10.1038/nri2155
107. Giudice V, Rosamilio R, Ferrara I, Seneca E, Serio B, Selleri C. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars) (2016) 11(1):374–80. doi: 10.1515/med-2016-0068
108. Pincez T, Aladjidi N, Heritier S, Garnier N, Fahd M, Abou Chahla W, et al. Determinants of long-term outcomes of splenectomy in pediatric autoimmune cytopenias. Blood (2022) 140(3):253–61. doi: 10.1182/blood.2022015508
109. Patel NY, Chilsen AM, Mathiason MA, Kallies KJ, Bottner WA. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg (2012) 204(6):1014–9; discussion 9-20. doi: 10.1016/j.amjsurg.2012.05.030
110. Thomsen RW, Schoonen WM, Farkas DK, Riis A, Fryzek JP, Sorensen HT. Risk of venous thromboembolism in splenectomized patients compared with the general population and appendectomized patients: a 10-year nationwide cohort study. J Thromb Haemost (2010) 8(6):1413–6. doi: 10.1111/j.1538-7836.2010.03849.x
111. Yui JC, Brodsky RA. Updates in the management of warm autoimmune hemolytic anemia. Hematol Oncol Clin North Am (2022) 36(2):325–39. doi: 10.1016/j.hoc.2021.11.005
112. ACIP. Altered immunocompetence: general best practice guidelines for immunization: best practices guidance of the advisory committee on immunization practices (Acip) . Available at: https://wwwcdcgov/vaccines/hcp/acip-recs/general-recs/immunocompetencehtml#ref-02 (Accessed March 10, 2023).
113. Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, et al. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal fcr blockade. J Immunol (2007) 178(8):5390–8. doi: 10.4049/jimmunol.178.8.5390
114. Robak T, Kazmierczak M, Jarque I, Musteata V, Trelinski J, Cooper N, et al. Phase 2 multiple-dose study of an fcrn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv (2020) 4(17):4136–46. doi: 10.1182/bloodadvances.2020002003
115. Castleman JS, Moise KJ Jr., Kilby MD. Medical therapy to attenuate fetal anaemia in severe maternal red cell alloimmunisation. Br J Haematol (2021) 192(3):425–32. doi: 10.1111/bjh.17041
116. Kuter DJ, Rogers KA, Boxer MA, Choi M, Agajanian R, Arnold D, et al. Fostamatinib for the treatment of warm antibody autoimmune hemolytic anemia: phase 2, multicenter, open-label study. Am J Hematol (2022) 97(6):691–9. doi: 10.1002/ajh.26508
117. Kuter DJ, Piatek CI, Saikali K, Dummer W. Phase 3, randomized, double-blind, placebo-controlled, global study (Forward) of fostamatinib for the treatment of warm antibody autoimmune hemolytic anemia. Blood (2022) 140(Supplement 1):2438–9. doi: 10.1182/blood-2022-169652
118. Langrish CL, Bradshaw JM, Francesco MR, Owens TD, Xing Y, Shu J, et al. Preclinical efficacy and anti-inflammatory mechanisms of action of the bruton tyrosine kinase inhibitor rilzabrutinib for immune-mediated disease. J Immunol (2021) 206(7):1454–68. doi: 10.4049/jimmunol.2001130
119. Forero-Torres A, Ramchandren R, Yacoub A, Wertheim MS, Edenfield WJ, Caimi P, et al. Parsaclisib, a potent and highly selective Pi3kdelta inhibitor, in patients with relapsed or refractory b-cell malignancies. Blood (2019) 133(16):1742–52. doi: 10.1182/blood-2018-08-867499
120. Barcellini W, Murakhovskaya I, Terriou L, Pane F, Patriarca A, Butler K, et al. Long-term efficacy and safety results from an ongoing open-label phase 2 study of parsaclisib for the treatment of autoimmune hemolytic anemia (Aiha). HemaSphere (2022) 6(Supplement 3):186–7. doi: 10.1097/01.HS9.0000844036.02998.15
121. Pasquale R, Giannotta JA, Barcellini W, Fattizzo B. Bortezomib in autoimmune hemolytic anemia and beyond. Ther Adv Hematol (2021) 12:20406207211046428. doi: 10.1177/20406207211046428
122. Ratnasingam S, Walker PA, Tran H, Kaplan ZS, McFadyen JD, Tran H, et al. Bortezomib-based antibody depletion for refractory autoimmune hematological diseases. Blood Adv (2016) 1(1):31–5. doi: 10.1182/bloodadvances.2016001412
123. Fadlallah J, Michel M, Crickx E, Limal N, Costedoat N, Malphettes M, et al. Bortezomib and dexamethasone, an original approach for treating multi-refractory warm autoimmune haemolytic anaemia. Br J Haematol (2019) 187(1):124–8. doi: 10.1111/bjh.16009
124. Yao M, Zhang J, Li Y, Lv L, Jia L, Yang C, et al. Combination of low-dose rituximab, bortezomib and dexamethasone for the treatment of autoimmune hemolytic anemia. Med (Baltimore) (2022) 101(4):e28679. doi: 10.1097/MD.0000000000028679
125. Schuetz C, Hoenig M, Moshous D, Weinstock C, Castelle M, Bendavid M, et al. Daratumumab in life-threatening autoimmune hemolytic anemia following hematopoietic stem cell transplantation. Blood Adv (2018) 2(19):2550–3. doi: 10.1182/bloodadvances.2018020883
126. Dizon MF. The challenges of daratumumab in transfusion medicine. Lab Med (2017) 48(1):6–9. doi: 10.1093/labmed/lmw055
127. Li H, Ji J, Du Y, Huang Y, Gu H, Chen M, et al. Sirolimus is effective for primary Relapsed/Refractory autoimmune cytopenia: a multicenter study. Exp Hematol (2020) 89:87–95. doi: 10.1016/j.exphem.2020.08.001
128. Miano M, Calvillo M, Palmisani E, Fioredda F, Micalizzi C, Svahn J, et al. Sirolimus for the treatment of multi-resistant autoimmune haemolytic anaemia in children. Br J Haematol (2014) 167(4):571–4. doi: 10.1111/bjh.13010
129. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (Itp) and Evans syndrome (Es): a single institution’s experience. J Pediatr Hematol Oncol (2017) 39(6):420–4. doi: 10.1097/MPH.0000000000000818
130. Thabet AF, Faisal M. Pulse cyclophosphamide therapy in refractory warm autoimmune hemolytic anemia: a new perspective. Indian J Hematol Blood Transfus (2014) 30(4):313–8. doi: 10.1007/s12288-013-0290-z
131. Karlsson C, Hansson L, Celsing F, Lundin J. Treatment of severe refractory autoimmune hemolytic anemia in b-cell chronic lymphocytic leukemia with alemtuzumab (Humanized Cd52 monoclonal antibody). Leukemia (2007) 21(3):511–4. doi: 10.1038/sj.leu.2404512
132. Wouters D, Stephan F, Strengers P, de Haas M, Brouwer C, Hagenbeek A, et al. C1-esterase inhibitor concentrate rescues erythrocytes from complement-mediated destruction in autoimmune hemolytic anemia. Blood (2013) 121(7):1242–4. doi: 10.1182/blood-2012-11-467209
133. de Boer ECV, Jalink M, Delvasto-Nuñes L, Meulenbroek EM, Baas I, Janssen SR, et al. Peritransfusional C1-inhibitor in patients with severe complement-mediated autoimmune hemolytic anemia: an open-label phase 2 trial. Presented at the European hematology association 2022 congress. HemaSphere (2022) 6(S3):2690.
134. Jäger U, D’Sa S, Schörgenhofer C, Bartko J, Derhaschnig U, Sillaber C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-Human trial. Blood (2019) 133(9):893–901. doi: 10.1182/blood-2018-06-856930
135. Gertz M, Roman E, Fattizzo B, Shum M, Hanna W, Ortega G, et al. Inhibition of C3 with apl-2 controlshaemolysis and increases haemoglobin levelsin subjects with autoimmune haemolytic anaemia (Aiha). HemaSphere (2019) 3(Supplement 1):405. doi: 10.1097/01.HS9.0000561876.96057.48
136. Berentsen S, Barcellini W, D’Sa S, Jilma B. Sutimlimab for treatment of cold agglutinin disease: why, how and for whom? Immunotherapy (2022) 14(15):1191–204. doi: 10.2217/imt-2022-0085
137. Berentsen S, Tjønnfjord GE. Current treatment options in cold agglutinin disease: b-cell directed or complement directed therapy? Transfus Med Rev (2022) 36:181–7. doi: 10.1016/j.tmrv.2022.05.001
138. Röth A, Barcellini W, D’Sa S, Miyakawa Y, Broome CM, Michel M, et al. Complement C1s inhibition with sutimlimab results in durable response in cold agglutinin disease: cardinal study 1-year interim follow-up results. Haematologica (2022) 107(7):1698–702. doi: 10.3324/haematol.2021.279812
139. Ulvestad E, Berentsen S, Mollnes TE. Acute phase haemolysis in chronic cold agglutinin disease. Scand J Immunol (2001) 54(1-2):239–42. doi: 10.1046/j.1365-3083.2001.00960.x
140. Tvedt THA, Steien E, Øvrebø B, Haaverstad R, Hobbs W, Wardecki M, et al. Sutimlimab, an investigational C1s inhibitor, effectively prevents exacerbation of hemolytic anemia in a patient with cold agglutinin disease undergoing major surgery. Am J Hematol (2022) 97(2):E51–E4. doi: 10.1002/ajh.26409
141. Berentsen S. Cold agglutinins: fending off the attack. Blood (2019) 133(9):885–6. doi: 10.1182/blood-2019-01-894303
142. Röth A, Berentsen S, Barcellini W, D’Sa S, Jilma B, Michel M, et al. Sustained complement C1s inhibition with sutimlimab in patients with cold agglutinin disease results in continued efficacy during part b of the randomized placebo-controlled phase 3 cadenza study (Nct03347422). Blood (2022) 140(Supplement 1):2825–7. doi: 10.1182/blood-2022-159562
143. Gertz MA, Qiu H, Kendall L, Saltarelli M, Yednock T, Sankaranarayanan S. Anx005, an inhibitory antibody against C1q, blocks complement activation triggered by cold agglutinins in human disease. 58th meeting of the American society of hematology, San Diego, Ca, USA. Blood (2016) 128:1265. doi: 10.1182/blood.V128.22.1265.1265
144. Lansita JA, Mease KM, Qiu H, Yednock T, Sankaranarayanan S, Kramer S. Nonclinical development of Anx005: a humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol (2017) 36(6):449–62. doi: 10.1177/1091581817740873
145. Jalink M, de Boer ECW, Evers D, Havinga MQ, Vos JMI, Zeerleder S, et al. Halting targeted and collateral damage to red blood cells by the complement system. Semin Immunopathol (2021) 43(6):799–816. doi: 10.1007/s00281-021-00859-8
146. Van de Walle I, Silence K, Budding K, Van de Ven L, Dijkxhoorn K, de Zeeuw E, et al. Argx-117, a therapeutic complement inhibiting antibody targeting C2. J Allergy Clin Immunol (2021) 147(4):1420–9 e7. doi: 10.1016/j.jaci.2020.08.028
147. von Baeyer H. Plasmapheresis in immune hematology: review of clinical outcome data with respect to evidence-based medicine and clinical experience. Ther Apher Dial (2003) 7(1):127–40. doi: 10.1046/j.1526-0968.2003.00004.x
148. Zoppi M, Oppliger R, Althaus U, Nydegger U. Reduction of plasma cold agglutinin titers by means of plasmapheresis to prepare a patient for coronary bypass surgery. Infusionsther Transfusionsmed (1993) 20(1-2):19–22. doi: 10.1159/000222800
149. Schwartz J, Padmanabhan A, Aqui N, Balogun RA, Connelly-Smith L, Delaney M, et al. Guidelines on the use of therapeutic apheresis in clinical practice-Evidence-Based approach from the writing committee of the American society for apheresis: the seventh special issue. J Clin Apher (2016) 31(3):149–62. doi: 10.1002/jca.21470
150. Röth A, Hüttmann A, Rother RP, Dührsen U, Philipp T. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood (2009) 113(16):3885–6. doi: 10.1182/blood-2009-01-196329
151. Kvistad SAS, Gunnes MW, Hagen KG, Berentsen S. A three year-old boy with back pain, fever and cola-coloured urine. Tidsskr Nor Laegeforen (2019) 139(6). doi: 10.4045/tidsskr.18.0532
152. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am (2015) 29(3):473–8. doi: 10.1016/j.hoc.2015.01.004
153. Barcellini W, Zaninoni A, Fattizzo B, Giannotta JA, Lunghi M, Ferrari A, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol (2018) 93(9):E243–E6. doi: 10.1002/ajh.25212
154. Petz LD. A physician’s guide to transfusion in autoimmune haemolytic anaemia. Br J Haematol (2004) 124(6):712–6. doi: 10.1111/j.1365-2141.2004.04841.x
155. Chen C, Wang L, Han B, Qin L, Ying B. Autoimmune hemolytic anemia in hospitalized patients: 450 patients and their red blood cell transfusions. Med (Baltimore) (2020) 99(2):e18739. doi: 10.1097/MD.0000000000018739
156. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? a commentary for clinicians and transfusion medicine professionals. Transfusion (2003) 43(11):1503–7. doi: 10.1046/j.1537-2995.2003.00583.x
157. Johnson ST, Puca KE. Evaluating patients with autoimmune hemolytic anemia in the transfusion service and immunohematology reference laboratory: pretransfusion testing challenges and best transfusion-management strategies. Hematol Am Soc Hematol Educ Program (2022) 2022(1):96–104. doi: 10.1182/hematology.2022000406
Keywords: clinical trials, complement inhibitors, cold agglutinin disease, corticosteroids, immune suppression, rituximab, therapy, autoimmune hemolytic anemia
Citation: Berentsen S, Fattizzo B and Barcellini W (2023) The choice of new treatments in autoimmune hemolytic anemia: how to pick from the basket? Front. Immunol. 14:1180509. doi: 10.3389/fimmu.2023.1180509
Received: 06 March 2023; Accepted: 13 April 2023;
Published: 24 April 2023.
Edited by:
Attila Kumanovics, Mayo Clinic, United StatesReviewed by:
Howard Allen Liebman, University of Southern California, United StatesMilen Minkov, Sigmund Freud Private University Vienna, Austria
Copyright © 2023 Berentsen, Fattizzo and Barcellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigbjørn Berentsen, c2lnYmpvcm4uYmVyZW50c2VuQGhhdWduZXR0Lm5v