 Yukari Okamoto
Yukari Okamoto Sojin Shikano*
Sojin Shikano*- Department of Biochemistry and Molecular Genetics, University of Illinois at Chicago, Chicago, IL, United States
Chemokine receptors play a central role in the maintenance of immune homeostasis and development of inflammation by directing leukocyte migration to tissues. GPR15 is a G protein-coupled receptor (GPCR) that was initially known as a co-receptor for human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV), with structural similarity to other members of the chemoattractant receptor family. Since the discovery of its novel function as a colon-homing receptor of T cells in mice a decade ago, GPR15 has been rapidly gaining attention for its involvement in a variety of inflammatory and immune disorders. The recent identification of its natural ligand C10orf99, a chemokine-like polypeptide strongly expressed in gastrointestinal tissues, has established that GPR15-C10orf99 is a novel signaling axis that controls intestinal homeostasis and inflammation through the migration of immune cells. In addition, it has been demonstrated that C10orf99-independent functions of GPR15 and GPR15-independent activities of C10orf99 also play significant roles in the pathophysiology. Therefore, GPR15 and its ligands are potential therapeutic targets. To provide a basis for the future development of GPR15- or GPR15 ligand-targeted therapeutics, we have summarized the latest advances in the role of GPR15 and its ligands in human diseases as well as the molecular mechanisms that regulate GPR15 expression and functions.
Introduction
The major task of the intestinal immune system is to tolerate innocuous food antigens and commensal microbes while fighting ingested pathogens. Failure to balance tolerogenic and inflammatory reactions can result in diseases, such as inflammatory bowel disease and gastrointestinal infections. Multiple immune mechanisms, including the balanced activities of regulatory T (Treg) cells and effector T (Teff) cells, contribute to the maintenance of intestinal immune homeostasis. Trafficking of these immune cells to the intestines is tightly controlled by a wide array of chemokines, chemokine receptors, and adhesion molecules expressed by leukocytes, the vascular endothelium, and the epithelium (1). For instance, the expression of CCR5, CCR6, CCR9, and CCR10 together with α4β7 integrin has been shown to target lymphocytes in the small intestine and colon (2–5). One of the recent advances in this field is the identification of G-protein coupled receptor 15 (GPR15) as a new chemoattractant receptor that mediates the homing of T cells to the colon in response to the natural ligand C10orf99 produced in the colon (6–8). A growing number of studies have suggested important roles of GPR15 in immune homeostasis and pathology of gastrointestinal (GI) tissues (6, 9–11). Similarly important is the discovery of GPR15-independent activity of C10orf99, as well as additional GPR15 ligands, that appear to be involved in the pathology of a broader range of tissues. Collectively, these findings indicate that GPR15 and its ligands are promising new targets for intervention. However, currently there is no approved drug that can specifically modulate GPR15 or GPR15 ligand activities. This review aimed to provide a basis for the development of GPR15- or GPR15 ligand-targeted interventions by summarizing recent research advances in the roles of GPR15 and its ligands in human pathophysiology, as well as the regulatory mechanisms of GPR15 expression and functions.
GPR15
GPR15 is a member of the Class A GPCR family that was cloned in 1996 (12) and identified in 1997 as a co-receptor for SIV, macrophage-tropic, non-syncytium-inducing HIV type 1 (M-tropic HIV-1), and HIV-2 (13, 14). Although this receptor was found to mediate T cell trafficking to the colon (6) and later “deorphanized” when a chemokine-like protein C10orf99 was identified as a functional endogenous ligand (7, 8), GPR15 is an “orphan” in terms of relatively low sequence similarity to its paralogues; it resembles a probable orphan receptor GPR25 with highest similarity of 36%, which is marginally higher than that to angiotensin II receptors, apelin receptor, and other chemokine receptor members (https://www.ensembl.org). GPR15 is also unique in that it lacks cysteines in the NH2-terminal region and the third extracellular loop, which are thought to form a disulfide bond and are required for optimal ligand binding and/or receptor activation in many GPCRs (15). Nevertheless, the NH2-terminal region of GPR15 carries several Tyr and acidic residues, a feature shared by multiple chemokine receptors (16). Sulfation of Tyr residues is known to promote the receptor binding of HIV/SIV (16, 17) as well as chemokine ligands (18–20), and the sulfated Tyr residues in the GPR15 NH2 terminus were recently shown to be required for optimal binding to C10orf99 (21). In humans, Gpr15 mRNA is highly expressed in the colon and lymphoid tissues, including peripheral blood lymphocytes and the spleen (13), while different studies have reported GPR15 protein expression in the colonic and small bowel mucosa, lymphoid cells, testis, liver, prostate, vascular endothelium, and skin (7, 22, 23). In peripheral blood, GPR15 is expressed in T cells (primarily CD4+) and at lower levels in B cells, monocytes, and neutrophils (11, 24, 25). It is of note that GPR15 expression was not induced by retinoic acid (6), that is known to regulate lymphocyte migration to the small intestine by enhancing the expression of CCR9 and integrin α4β7 (26, 27). This may contribute to the colon-specificity of GPR15+ cell homing, combined with the abundant expression of C10orf99 in the colon compared with the small intestine (7, 8).
Role of GPR15 in disease pathology
GPR15 expression in mouse T cell subsets and colitis
A novel function of GPR15 as a colon-homing receptor was discovered in 2013 by Kim et al. using Gpr15-deficient mice (6). Knock-in mice with Gpr15 gene replaced with the GFP sequence showed preferential expression of GFP in Foxp3+ Tregs in the large intestine lamina propria (LILP); approximately 60–70% of LILP CD4+Foxp3+ cells expressed GPR15, compared to 7–20% of CD4+Foxp3− cells. The results of a cell transfer assay confirmed the efficient homing of GPR15+ cells to LILP in an α4β7-dependent manner. The Gpr15 gene knockout (KO) reduced Treg numbers in LILP and exacerbated colitis induced by Citrobacter rodentium infection. In addition, in a non-infectious colitis model in Rag2-/- mice, where CD40 stimulation induced innate immune cell-mediated colitis (28–30), adoptive transfer of Tregs from wild-type mice reduced colitis severity and tissue damage, but Tregs from Gpr15 KO mice failed to do so (6). These observations suggest that GPR15 is required to dampen the immune response in the large intestine by directing homing of Tregs in mice.
However, a subsequent study by Nguyen et al. using Gpr15 KO mice demonstrated that GPR15 is also important for the colon migration of pathogenic Teff cells that cause inflammation (11). In a T cell-mediated colitis model in which adoptive transfer of CD45RBhi T cells (naïve CD4 T cells depleted of Tregs) to immunodeficient recipient mice resulted in the generation and intestinal trafficking of Teff cells to cause colitis (31–33), Rag2−/− mice that received naïve Gpr15-KO T cells were protected from developing colitis. In addition, GPR15 was found to be induced in in vitro–generated mouse Th17 effector cells under conventional polarizing conditions (11). Moreover, a more recent study by Xiong et al. clearly showed substantial GPR15 expression in all Th1, Th2, and Th17 subsets isolated from mouse LILP, although at a lower frequency compared with that of Treg cells (34). Hence, GPR15 is capable of directing the colon homing of both Treg and Teff CD4+ T cells in mice, and the impact of this receptor in colitis pathology will depend on the experimental settings regarding the relative requirement of Treg and Teff subsets for the development of colitis.
GPR15 expression in human T cell subsets and colitis
How are these mouse studies translatable to humans? The original study by Kim et al., who discovered the colon homing function of GPR15 in mouse Tregs, observed an increased amount of Gpr15 mRNA in the human CD25−CD4+ T cell population than in Treg-enriched CD25+CD4+ T cells from colon tissues of patients with colorectal cancer (6), implying a difference between mice and humans. The following study by Nguyen et al. found that in human colon tissues, GPR15 expression was highly enriched in the IL-5+ or IL-13+ Th2 subset, particularly in patients with ulcerative colitis (UC), and there was little or no GPR15 expression in Treg cells (11). In vitro polarization of human peripheral blood mononuclear cells (PBMCs) and mouse spleen cells by cytokines also revealed disparate GPR15 expression patterns in T cell subsets between the two species; GPR15 was expressed primarily in Th2 cells in humans, whereas GPR15 was expressed in Treg and Th17 subsets in mice (11). Further analyses of Gpr15 gene and master T cell transcription factors for Th2 (GATA3) and Treg cells (Foxp3) led to the conclusion that the following mechanism underlies preferential expression of GPR15 in Th2 cells in humans: (i) GATA3 promotes Gpr15 gene expression in human Th2 cells by binding to the 3′ enhancer of Gpr15, while (ii) Foxp3 binding to the enhancer suppresses Gpr15 expression in human Treg cells, and (iii) this GATA3 binding does not occur in mouse Th2 cells and Foxp3 binding is much weaker in mouse Treg cells because of the sequence difference in the Gpr15 enhancer. These authors concluded that GPR15 is preferentially expressed in Teff cells rather than in Tregs in humans and this is reflected in the inflamed colon of patients with UC.
In another study, Fischer et al. utilized a humanized mouse model to comparatively determine the role of GPR15 and α4β7 integrin, in which human peripheral blood T cells from patients with UC were transferred into mice and examined for migration to the colon in dextran sulfate sodium (DSS)-induced colitis (35). Expression of both GPR15 and α4β7 integrin was elevated in Tregs but not in Teff cells in the colon of patients with UC compared with those in healthy controls and patients with CD. Pre-treatment of these cells with siRNA for GPR15 only affected Teff cell homing but did not affect Treg homing, whereas treatment with an α4β7 antibody (vedolizumab) suppressed both Teff and Treg homing, suggesting that GPR15 is important for Teff homing but not Treg homing in humans (35).
Despite all these findings suggesting species differences, a simplified interpretation such as “GPR15 is expressed by Teff cells in humans, while it is expressed by Treg cells in mice,” requires caution. Indeed, multiple studies from different groups, including later studies from the same group of Nguyen et al., have shown that GPR15 is expressed by human Tregs in the peripheral blood at similar or even higher levels than that in Teff cells (6, 7, 35–37), which is not consistent with the generalized model that GPR15 expression is promoted in Th2 cells and suppressed in Tregs in humans. Furthermore, Adamczyk et al. reported that GPR15 was expressed at an even lower frequency in Teff cells than in Treg cells in colon tissues of either healthy controls or patients with UC (36). Interestingly, they observed significantly increased expression of GPR15 in both Treg and Teff cells from the uninflamed region, but not from the inflamed region, in patients with UC compared with that in tissues from healthy controls. This is also consistent with a more recent study by Xiong et al. that observed substantial expression of GPR15 (nearly 40% on average) in Tregs from uninflamed regions of UC patient colons and a positive correlation between GPR15 and Foxp3 expression in human colonic Tregs (34). These findings are in contrast to the predominant GPR15 expression by Th2 cells observed in UC colon by Nguyen et al. (11). The reason for this discrepancy is not clear, and further human studies are necessary; however, the differential expression of GPR15 in Tregs in the inflamed and uninflamed regions of the UC colon could provide some insights. The fact that GPR15 expression was not increased in Tregs from the inflamed region of the UC colon implies that GPR15 is not an exclusive master regulator for the migration of Tregs into the inflamed colon. On the other hand, the increased GPR15 expression on Tregs in the non-inflamed region also raises the possibility that GPR15 drives Tregs to the colon of patients with UC, but its expression is downregulated in the inflamed environment. For instance, increased local production of the GPR15 ligand might enhance receptor internalization and subsequent degradation. In addition, since dysbiosis in UC is characterized by reduced levels of bacterial metabolites, including short chain fatty acids (SCFAs) (38) and GPR15 expression was found to be upregulated by SCFAs (36, 39, 40), the diminished local production of SCFAs could result in the reduced expression of GPR15 on Tregs in the inflamed colons of patients with UC. Interestingly, the aforementioned study by Adamczyk et al. (36) found that the majority of Teffs and Tregs in the peripheral blood did not co-express GPR15 and α4β7 integrin, and this lack of co-expression was also detectable in colonic biopsies of healthy individuals and patients with UC. This indicates the phenotypic heterogeneity of T cells, especially Tregs (41, 42) and highlights the need for careful analysis of the expression of multiple molecular factors potentially involved in the colonic migration of T cells.
Colorectal cancer
In addition to colitis, GPR15 has also been implicated in the pathogenesis of CRC. Tregs infiltrating the tumor sites create an immunosuppressive tumor microenvironment that prevents the development of effective anti-tumor immune responses (42, 43). Adamczyk et al. compared gene expression in Treg cells from tumors and healthy colonic tissues of colitis-associated CRC mice induced by azoxymethane (AOM)/DSS treatment (10). They identified a specific set of genes that are preferentially expressed in tumor-associated Tregs, including GPR15. Similar to mice, the frequency of GPR15+ Tregs, but not that of GPR15+ Teffs, in the tumor sites of patients with CRC was significantly higher than that in non-tumor sites, suggesting a distinct role for GPR15 in Treg delivery to CRC sites in humans (10). Importantly, genetic deletion of Gpr15 in mice significantly decreased the infiltration of tumor-associated Tregs, reduced the Treg/CD8+ T cell ratio, and diminished tumor development (10), suggesting that GPR15 is responsible for directing the colon migration of Treg cells that support the growth of CRC. Thus, GPR15 represents a promising novel target for modifying T cell-mediated anti-tumor immunity in CRC.
Eosinophilic esophagitis
EoE is an allergic disease characterized by chronic esophageal inflammation with prominent recruitment of eosinophils (44). Inflammation in EoE critically involves Th2 cells that produce cytokines such as IL-5 and IL-13, which promote eosinophil recruitment and activation and exacerbate epithelial barrier dysfunction, respectively (44, 45). In a recent study, Morgan et al. (9) conducted single-cell RNA analysis of T cells in tissues from patients with EoE. They found that Gpr15 expression was increased in highly polarized pathogenic effector Th2 (peTh2) clonotypes detected in both esophageal tissue and peripheral blood of patients with EoE, and Gpr15 was the most significantly upregulated transcript in these cells in the esophagus compared with peripheral blood. While the genes encoding integrin α4β7 were broadly expressed by T cells in both the esophagus and duodenum, GPR15 was only expressed in esophageal T cells, and CCR9, a chemokine receptor known for gut homing (46), was only expressed in duodenal T cells. In addition, the authors detected the expression of C10orf99 and CCR9 ligand CCL25 only in the esophageal epithelium and duodenal epithelium, respectively (9). This is consistent with the fact that the esophagus is one of the tissues that most strongly expresses C10orf99 (https://gtexportal.org). These findings collectively support the model that GPR15 expression promotes esophageal homing of peTh2 cells and exacerbates inflammation during EoE, and in addition, GPR15 may serve as a marker for esophagus-migrating peTh2 cells in the peripheral blood of patients.
Rheumatoid arthritis
An earlier study on RA reported that GPR15 is expressed by macrophages in synovial tissue and monocytes and neutrophils in peripheral blood, and its expression is upregulated in patients with RA compared to non-RA controls (24). A more recent study examined GPR15 expression on T cells from patients with RA and found that the frequency of CD4+/CD8+ GPR15+ T lymphocytes was higher in patients with RA than in healthy subjects (47). In addition, the frequency of CD4+/CD8+ GPR15+ T lymphocytes was higher in the synovial fluid of patients with RA than in that of patients with osteoarthritis. Immunostaining results of synovial tissue sections demonstrated that GPR15 and GPR15L are present in the synovial tissues of patients with RA (47). These findings implicate the GPR15-GPR15L axis in RA pathogenesis, which involves both innate and adaptive immune cells.
Skin inflammation
The mouse skin epithelium contains a specialized population of γδ T cell receptor (TCR)+ cells called dendritic epidermal T cells (DETCs), which exclusively express the monoclonal Vγ3Vδ1 TCR and are implicated in protection of skin homeostasis, host defense, and wound healing (48). DETCs mature in the fetal thymus and migrate to the skin during late embryogenesis, after which they are maintained through self-renewal. An earlier study by Lahl et al. found that GPR15 is highly expressed on fetal thymic DETC precursors and that Gpr15 KO substantially reduces the frequency of epidermal DETCs in neonatal (Day 1) mice compared with Gpr15 -/+ heterozygote neonates (23). This suggested that GPR15 is essential for migration of embryonic DETC to the skin, which is also consistent with the high expression of C10orf99 in keratinocytes of embryonic (day 16) and neonatal skin (7). In a more recent study, Sezin et al. conducted a profiling of T cell populations in the skin of adult (8-16 weeks) Gpr15 KO mice and found that the DETCs were reduced by approximately 60% compared to wild-type littermates (49). In addition, the niche of DETCs in the epidermis was populated by αβ TCR+ cells; approximately 40% of all CD3+ cells in Gpr15 KO mice were αβ TCR+ compared to only 10% in wild-type mice (49). Furthermore, these changes were also associated with shifts in the composition of skin microbiome in Gpr15 KO mice (49). These studies collectively highlighted a pivotal role of GPR15 in the skin homing of DETCs in mice, which appears to impact the composition of T cell populations and microbiome even in the adulthood.
On the other hand, the role of GPR15 in the skin disease settings remain somewhat elusive. Deficiency in Gpr15 did not alter the course of disease neither in the imiquimod-induced psoriasiform dermatitis nor in the IL-23-induced dermatitis model, despite the increased expression of C10orf99 in the inflamed skin (50). However, in the antibody transfer mouse model of bullous pemphigoid-like epidermolysis bullosa acquisita (BP-like EBA), an autoimmune subepithelial and mucocutaneous blistering disease, the Gpr15 KO was found to markedly aggravate the skin pathology (51). Importantly, this was associated with an increased accumulation of γδ TCR+ cells in the dermis (51), suggesting a possibility that GPR15 may counteract antibody-mediated skin inflammation through direct and/or indirect mechanisms that limit the recruitment of γδ TCR+ cells into the dermis.
GPR15 ligands and their roles in pathophysiology
C10orf99 in GPR15 signaling
C10orf99 was reported as a natural GPR15 agonist in 2017 (7, 8). Mature human C10orf99 is a short, 57-amino acid basic protein (pI = 11.28) with two pairs of Cys residues that form intramolecular disulfide bridges, implying that this protein is related to a CC chemokine. The tissues expressing C10orf99 include the digestive tract (particularly the colon and esophagus), skin, tonsils, cervix, and bladder. In the mouse colon, C10orf99 expression did not appear to be regulated by colonic inflammation or the presence of commensal bacteria (7). C10orf99 has several unique features that differ from those of the canonical chemokines. Secondary structures typically found in chemokine family proteins, such as loops, β-strands, and helices, were not identified in C10orf99 by structure prediction programs (8). In contrast to most chemokines that require their N-termini for receptor binding and activation (52), N-terminal deletion of C10orf99 by up to 10 amino acids showed no marked change in GPR15-dependent calcium signaling (8). Instead, the hydrophobic C-terminal region of C10orf99, which is highly conserved among species, is critically required for receptor binding and signaling (8, 21, 53). In addition, unlike canonical chemokines and chemokine receptors, C10orf99 and GPR15 show highly specific interactions; C10orf99 does not cross-activate any of the known 22 chemokine receptors, and GPR15 does not respond to any of the 27 known chemokines (8). Functionally, C10orf99 interaction with GPR15 leads to inhibition of cAMP production, which can be reversed by pertussis toxin, indicating that GPR15 is a Gαi/o-coupled receptor, similar to most chemokine receptors. C10orf99 activates extracellular signal-regulated kinase (ERK)1/2, induces calcium release, promotes β-arrestin recruitment and receptor endocytosis, and induces chemotaxis in GPR15-expressing immune cells (7, 8, 21, 53, 54) (Figure 1A).
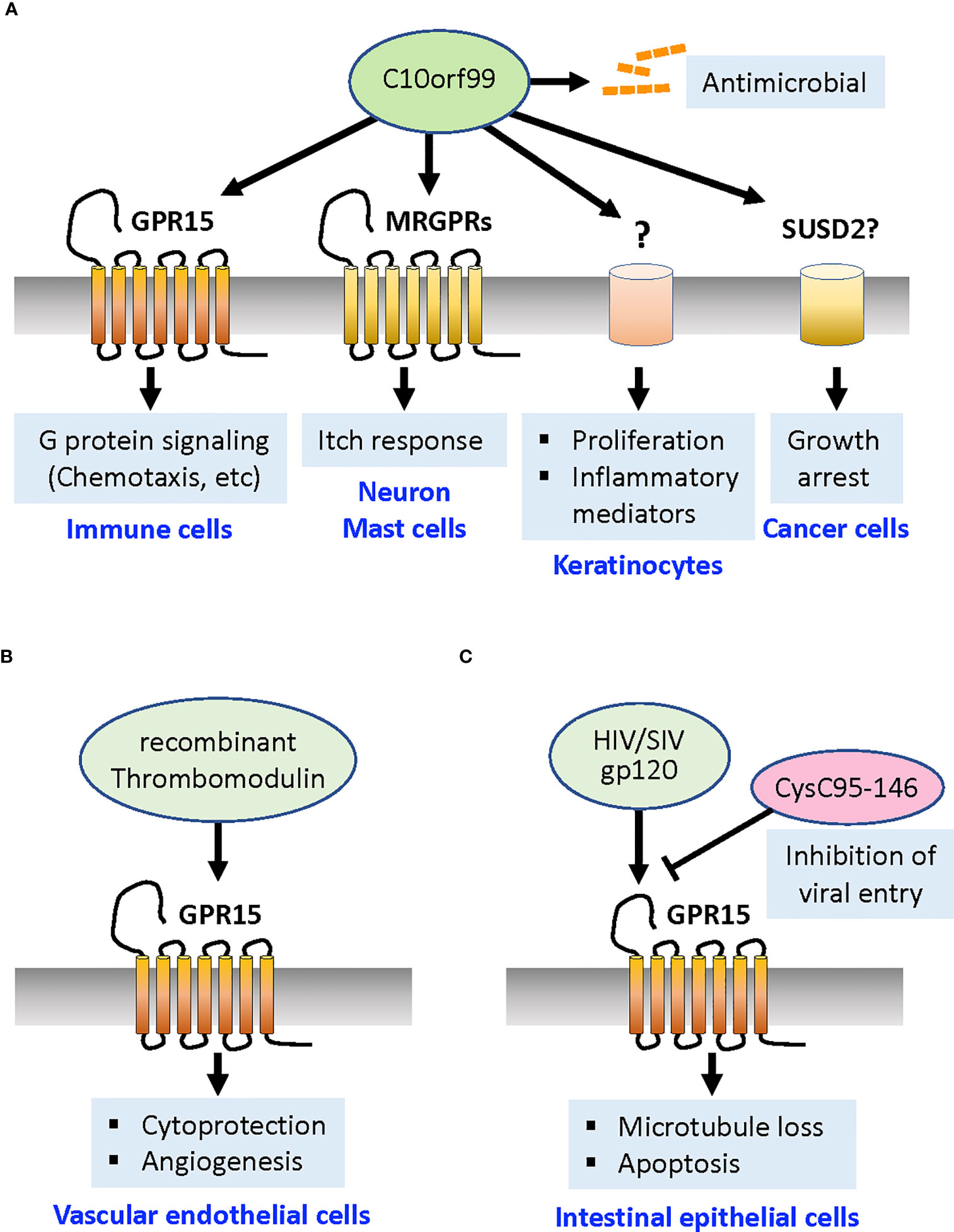
Figure 1 GPR15 ligands and their functions. (A) A natural GPR15 ligand C10orf99 activates the Gαi-mediated signaling pathways that induce chemotaxis of GPR15+ T cells and other immune cells toward this ligand. C10orf99 also functions independently of GPR15 to exert antimicrobial activities. In the psoriatic skin, C10orf99 evokes itch response by activation of MRGPRs on sensory neurons and mast cells, and promote proliferation and production of inflammatory mediators of keratinocytes through unknown receptor/mechanism. C10orf99 also induces growth arrest of cancer cells by activating SUSD2 receptor (although controversial). (B) Recombinant thrombomodulin acts on GPR15 expressed by vascular endothelial cells via its fifth region of EGF-like domain to confer cytoprotection from apoptosis signal and promote angiogenesis. (C) HIV protein gp120 induces loss of microtubule and apoptosis in the intestinal epithelial cells. The C-terminal fragment of cystatin C (CysC95-146) competes for GPR15 and inhibits the entry of HIV and SIV. MRGPRs, Mas-related G protein-coupled receptors; SUSD2, Sushi Domain Containing 2 receptor; HIV, human immunodeficiency virus; SIV, simian immunodeficiency virus.
C10orf99 in psoriasis
C10orf99 was implicated in immune regulation prior to the discovery of its GPR15 agonistic activity. Its first implication in human diseases came from global analyses of psoriasis-associated genes in 2009 which reported significant upregulation of C10orf99 in psoriatic skin (55, 56). A large-scale gene knockout study in 2010 showed that disruption of C10orf99 gene leads to an increased ratio of CD4+/CD8+ cells and a decreased serum IgM level in mice (57). Multiple studies have demonstrated the regulated expression of C10orf99 in the epidermis of the skin and its association with psoriasis. Consistent with the epidermal migration of fetal thymic DETCs that express GPR15 (23), C10orf99 mRNA is highly expressed in the fetal and neonatal epidermis of mice, but is low or nearly absent in the uninflamed adult epidermis both in mouse and human (7, 51). However, C10orf99 expression was highly upregulated in wounded skin (7), antibody-mediated model of BP-like EBA (51), and imiquimod-induced models of psoriasis in mice (8, 51, 58), and in patients with psoriasis (8, 58–60) or atopic dermatitis (59, 61). C10orf99 has also been indicated as a reliable marker gene for the classification of psoriasis (60).
A major question is whether C10orf99 plays a determining role in disease progression or whether it merely indicates loss of epithelial integrity. It appears that C10orf99 has GPR15-independent functions. Yang et al. reported that C10orf99 acts as an antimicrobial peptide that exhibits broad-spectrum antimicrobial activity (62) (Figure 1A), as has been reported for some chemokines (63). In addition, local sustained delivery of nanoparticle-encapsulated C10orf99 peptide promoted granulation tissue formation and wound healing in a full-thickness dermal defect rat model (64). Similarly, overexpression of C10orf99 gene in transgenic mouse was reported to reduce skin inflammation and remodeling after an imiquimod challenge in a published patent application (65). These findings implicate a protective role for C10orf99 in the inflammatory skin.
However, a later study using human keratinocyte HaCaT cells under inflammatory conditions showed that C10orf99 knockdown decreased cell proliferation, whereas overexpression of C10orf99 promoted their proliferation (58) (Figure 1A). A more recent study also showed that C10orf99 transfection into normal human epidermal keratinocytes induced the expression of inflammatory mediators and reduced the expression of barrier-related genes such as filaggrin and loricrin (59). The addition of synthetic C10orf99 peptide reduced the expression levels of barrier-related genes in human keratinocyte 3D cultures, suggesting that C10orf99 binding induces specific signaling in keratinocytes (59). Furthermore, local depletion of C10orf99 by lentiviral shRNA vectors or systemic knockout of C10orf99 in mice effectively ameliorated imiquimod-induced psoriatic dermatitis, supporting the proinflammatory role of C10orf99 (58, 59). Collectively, these results indicate that C10orf99 is a primary inducible regulator that reduces barrier formation and induces the inflammatory response of keratinocytes under psoriatic conditions (Figure 1A). As already mentioned, deficiency in the Gpr15 gene did not alter the course of disease in imiquimod-induced psoriasiform dermatitis or the IL-23-induced dermatitis model, suggesting that C10orf99 modulates psoriasiform dermatitis via GPR15-independent pathways (50).
The notion of GPR15-independent activity of C10orf99 is further supported by a recent study by Tseng and Hoon (61) who discovered that C10orf99 can act as an endogenous pruritogen during inflammation that activates Mas-related G protein-coupled receptors (MRGPRs). These authors found that C10orf99 selectively stimulates mouse dorsal root ganglion neurons that express Mrgpra3 and evokes intense itch responses and vasodilation. C10orf99 also caused mast cell degranulation through the stimulation of MRGPRX2 and Mrgprb2, and genetic disruption of C10orf99 expression attenuated scratch responses in an imiquimod-induced psoriasis model (61). Together, these studies suggest that elevated expression of C10orf99 during psoriasis can aggravate the disease by promoting the proliferation and inflammatory response of keratinocytes, reducing barrier formation, and inducing itch responses and vasodilation by acting on neurons and mast cells (Figure 1A).
C10orf99 in cancer cell growth
The above-mentioned studies indicate that C10orf99 has multiple receptors and cellular substrates that are involved in physiologically different reactions. An earlier study by Pang et al. reported another function of C10orf99: the growth inhibition of cancer cells (66). C10orf99 was found to interact with the transmembrane protein Sushi Domain Containing 2 (SUSD2), hence termed a colon-derived SUSD2 binding factor (CSBF) (66). The authors showed that the C-terminally IgG-Fc-tagged recombinant C10orf99/CSFB protein binds to SUSD2 expressed in CRC cell lines and inhibits cell growth through G1 cell cycle arrest (66) (Figure 1A). However, this model requires further investigation because the inhibitory effect of C10orf99 on CRC cells could not be reproduced in a follow-up study by a different group (62) who used the untagged C10orf99 protein and observed its cytotoxic effect only on a specific B-cell lymphoma line.
Thrombomodulin as a ligand to GPR15 in vascular endothelial cells
A novel function of GPR15 in ECs, mediated by a ligand that is completely distinct from C10orf99 in structure, was demonstrated in 2017 (67) (see more comprehensive review (68)). Pan et al. found that the recombinant soluble protein TME5, which encodes the fifth region of the epidermal growth factor (EGF)-like domain of TM, binds to the GPR15 expressed by human vascular ECs in vitro (Figure 1B). TM is a type I transmembrane protein constitutively expressed by ECs and acts as an anticoagulant by binding to thrombin and activated protein C (69). The blood level of soluble TM is known to be elevated under various pathological conditions involving endothelial damage, such as sepsis, COVID-19 infection, progressive systemic sclerosis, and diabetes (70–73). Recombinant human soluble TM (rTM, ART-123), consisting of the extracellular domain of TM, has been used to treat disseminated intravascular coagulation (DIC) (74, 75). Pan et al. demonstrated that recombinant TME5 rescued growth inhibition and apoptosis caused by the calcineurin inhibitor FK506 in vascular ECs isolated from wild-type but not from FK506-treated Gpr15 KO mice (67). This cytoprotective effect was mediated by the activation of ERK1/2 and increased level of anti-apoptotic proteins (67). In addition, TME5 enhanced the migratory activity of ECs and increased their production of nitrogen oxide. Moreover, in vivo Matrigel plug angiogenesis assay revealed that TME5 stimulates angiogenesis in wild-type mice but not in Gpr15 KO mice (67) (Figure 1B). TME5 also ameliorates inflammation in a murine sepsis model in a GPR15-dependent manner through suppression of NF-kB activity and release of pro-inflammatory cytokines in macrophages (76). GPR15 in T cells also appears to mediate TME5-induced anti-inflammatory effects in a murine model of acute graft-versus-host disease (GVHD) caused by allogeneic hematopoietic stem cell transplantation (77).
Interestingly, in a more recent study testing the potential anti-tumor effect of rTM, it was suggested that GPR15 mediates rTM-induced growth inhibition of pancreatic ductal adenocarcinoma (PDAC) (78). The anti-tumor effect of rTM was only observed in PDACs with high GPR15 expression, and rTM suppressed NF-kB and ERK1/2 activation in a GPR15-dependent manner. This inhibition of ERK1/2 activity by rTM is not consistent with its cytoprotective effect in ECs that involves activation of ERK1/2 (67), suggesting that the consequence of rTM-GPR15 interaction is dependent on the cell context. Further investigations in different cell types are necessary to delineate TM-induced GPR15 signaling pathways.
HIV gp120 in enteropathy
Chemokine receptors are expressed in numerous non-hematopoietic cells and play important roles beyond chemotaxis of leukocytes, such as development, angiogenesis, and apoptosis (79–81). GPR15 is abundantly expressed on the basolateral surface of the intestinal epithelium, unlike CXCR4 and CCR5, which are present mainly at or near the luminal surface (22). It was previously reported that HIV-1 surface protein gp120 induces calcium signaling, microtubule loss, and physiological changes, including increased paracellular permeability in the intestinal cell line; these changes resemble HIV enteropathy (82, 83). Fantini et al. found that these gp120-induced effects were inhibited by anti-GPR15 neutralizing antibody or selective G protein inhibitor pertussis toxin (22, 84). They also found that GPR15 mediates viral strain-specific gp120-induced calcium signaling at low, physiologically reasonable gp120 concentrations, which are up to 10,000-fold lower gp120 concentrations than those of the principal HIV co-receptors. These findings suggest that gp120 is involved in HIV enteropathy via its interaction with the GPR15. GPR15 has also been implicated in the apoptosis of intestinal epithelial cells in an SIV infection model (85) (Figure 1C).
Regulation of GPR15 expression
The molecular mechanisms that have been reported to regulate Gpr15 gene expression are depicted in Figure 2.
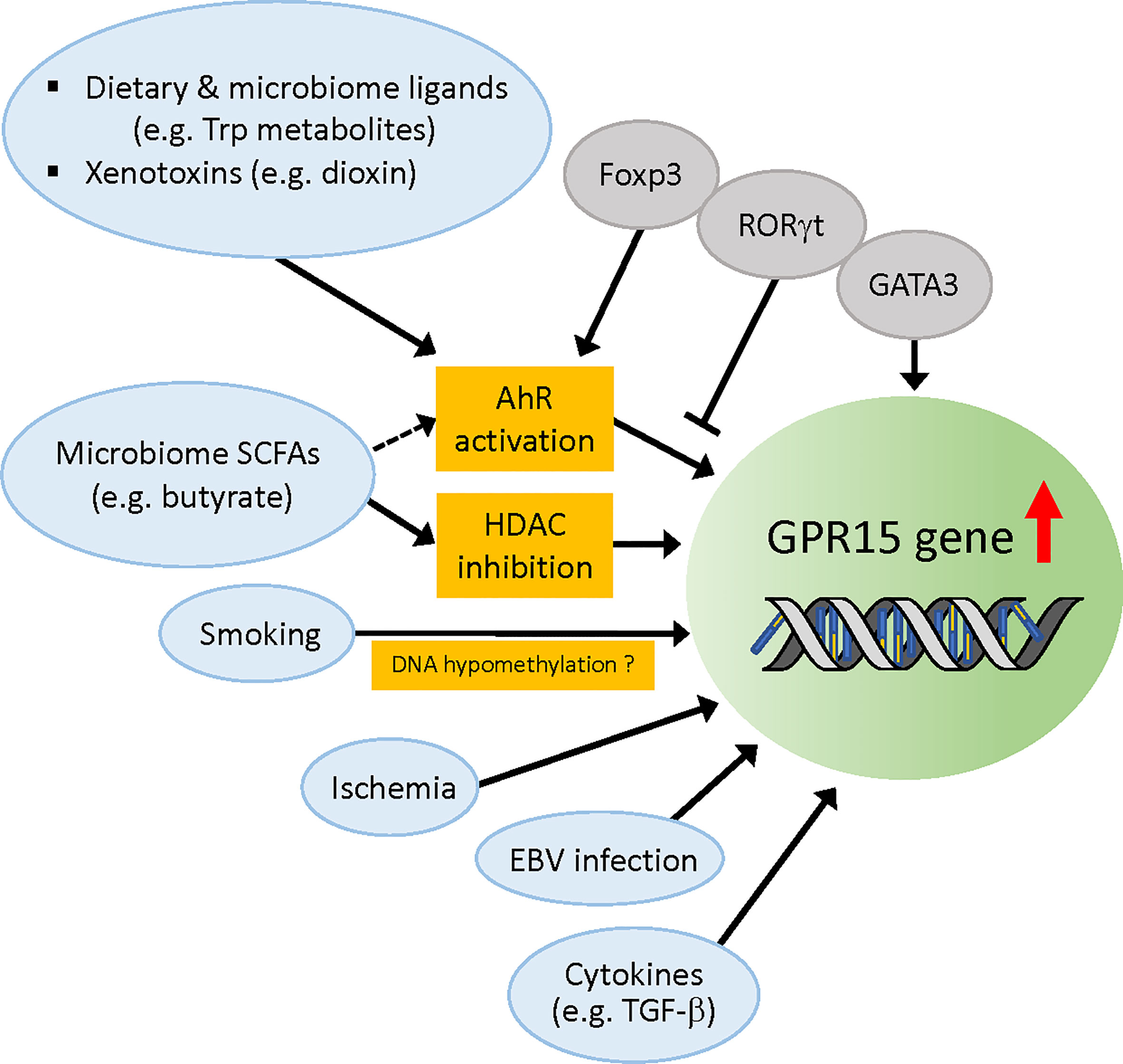
Figure 2 Regulation of Gpr15 gene expression. Molecular factors that have been reported to upregulate Gpr15 expression are depicted. Dietary and microbiome ligands such as tryptophan metabolites activate AhR that directly acts on Gpr15 gene. Foxp3 cooperates with AhR (although contradictory data exist) while RORγt competes with AhR, and GATA3 enhances Gpr15 expression independently of AhR. Foxp3 and GATA3 are reported to differentially bind Gpr15 enhancer and regulate Gpr15 expression in the T cells of human and mouse (not depicted in this figure). SCFAs from microbiota such as butyrate upregulate Gpr15 expression by inhibition of HDAC but might also by activation of AhR. Smoking habit is strongly associated with DNA hypomethylation of Gpr15 and increased mRNA expression of Gpr15 in blood leukocytes, but the evidence for the direct effect of cigarette smoke extract on Gpr15 DNA methylation is lacking. Instead, the increased frequency of peripheral CD3+GPR15+ T cell population appears to account for the apparent hypomethylation of Gpr15 DNA in blood samples from smokers. Infarction-related ischemia in mice and ischemic condition in cell culture induces Gpr15 expression in cardiomyocytes. EBV infection in B cells enhances Gpr15 expression by an unidentified mechanism. TGF-β induces GPR15 expression in vivo. AhR, aryl hydrocarbon receptor; SCFAs, short chain fatty acids; HDAC, histone deacetylase; EBV, Epstein-Barr virus.
Regulation of GPR15 expression by aryl hydrocarbon receptor
The AhR is a ligand-activated transcription factor that has been studied for many decades as a sensor for environmental contaminants, such as dioxins. Research in the past 15 years has led to the emergence of AhR as a key physiological regulator of immune responses, affecting both innate and adaptive systems by sensing a variety of endogenous, dietary, microbial (e.g., tryptophan metabolites), and environmental ligands (86). AhR is known to regulate the differentiation, homing, and immunosuppressive functions of Tregs (87, 88) and numerous studies have shown that AhR ligand activation can reduce inflammation and ameliorate disease (89–92). AhR is also downregulated in the intestinal tissue of patients with IBD (93). Using genetic mouse models, Zhou et al. found in 2017 that GPR15 expression is significantly reduced in AhR-deficient colonic Treg cells (94). A more recent study by the same group (34) demonstrated that the AhR-dependency of GPR15 expression was not confined to Tregs, but was observed for all murine CD4+ T cell subsets in the colon. In addition, when wild-type mice were fed with an AhR-ligand-deficient diet, the addition of the dietary AhR ligand indole-3-carbinol (I3C) (95) significantly enhanced GPR15 expression in intestinal CD4+ T cell subsets, while this effect was abrogated in Tregs (but not in other CD4+ subsets) in engineered mice in which Ahr was specifically ablated in Tregs (34). Moreover, in a short-term homing assay, in which in vitro differentiated Tregs (iTregs) were transferred to Rag1-/- mice, the number of Ahr-/- Tregs migrating to the colon was approximately 3-fold lower than that of Ahr+/+ Tregs. Importantly, forced expression of GPR15 in Ahr-/- Tregs significantly enhanced their homing to the colon but not to other organs, suggesting that AhR promotes colon-specific homing of Tregs by enhancing GPR15 expression in mice (34). Notably, GPR15 expression in iTregs generated from human PBMCs was enhanced or reduced by the AhR agonist or antagonist, respectively, indicating that AhR regulates GPR15 in humans as well (34). This is supported by another study by McAleer et al. (96) who found regulation of GPR15 expression by an AhR agonist and antagonist in human CD4+ T cells. In a separate recent study, Swaminathan et al. reported the regulation of GPR15 expression by AhR in activated human PBMCs, sorted effector/memory CD4+ T cells, and in vitro polarized human Th2 and Treg cells (37). Collectively, these studies have uncovered a novel role of AhR in controlling colon homing of CD4+ T cells by positively regulating GPR15 expression in mice and humans. It is interesting to note that, retrospectively, there was already an indication of AhR regulation of GPR15 in 2007 when Gpr15 was identified as a novel dioxin-inducible gene (97).
A recent study demonstrated that the AhR intrinsically promotes differentiation and function of resident memory CD8+ T cells, including those in the intestinal epithelium (98). Nevertheless, genetic deletion or activation of AhR did not affect GPR15 expression in CD8+ T cells (34), suggesting an AhR-independent mechanism for GPR15 expression in CD8+ T cells. The crucial role of AhR in regulating GPR15 expression in CD4+ but not in CD8+ T cells suggests a potential therapeutic target in intestinal disorders, e.g., colorectal cancer where disruption of Gpr15 gene reduced infiltration of CD4+ Tregs but not CD8+ T cells into tumor sites and inhibited tumor growth (10).
Direct activation of Gpr15 expression by AhR and regulation by other transcription factors
How does AhR regulate GPR15 expression? Chromatin immunoprecipitation sequencing (ChIP-seq) of AhR using iTregs and Th17 cells identified Gpr15 as one of the top genome-wide targets of AhR in both cell types (34). In addition, mutations in the DNA-binding region abolished the ability of AhR to promote Gpr15 expression in iTregs, suggesting that AhR regulates transcription by directly binding to the Gpr15 locus. Consistent with this notion, both aforementioned studies (34, 37) identified AhR binding sites/xenobiotic response elements (XREs, 5′-GCGTG-3′) within the Gpr15 3′ enhancer sequence that are conserved across mammalian species. As expected, the Gpr15 promoter-driven luciferase reporters attached to these XRE-containing enhancer regions exhibited AhR agonist-dependent activity that was inhibited by an AhR antagonist (34, 37). Further analyses by Xiong et al. (34) revealed that Foxp3 cooperates with AhR, potentially via interactions with AhR at the Gpr15 locus, to enhance GPR15 expression in Tregs. In contrast, RORγt, which is frequently expressed in gut Tregs and Th17 cells, negatively regulates GPR15 expression, at least in part, by competing with AhR for binding to the Gpr15 locus (34). In addition, Swaminathan et al. showed that GATA3 and AhR independently and synergistically promoted GPR15 expression (37). Intriguingly, however, these authors also found a significant decrease in GPR15 expression by Foxp3 in the presence of either AhR or GATA3 in the luciferase assay, contrary to the findings of Xiong et al. (34). Although further investigation is necessary to elucidate the crosstalk between AhR and other regulatory factors, the discovery of AhR-mediated regulation of GPR15 expression and colon homing of T cells has significantly advanced our understanding of the roles of GPR15 in the intricate mechanisms of T cell migration to target tissues.
GPR15 expression in T cells can be induced by SCFAs such as butyrate, propionate, and acetate (36, 39, 40), which are the main metabolites produced by the microbiota in colon. These SCFAs synergistically enhance basal and ligand-induced expression of AhR-responsive genes in a gene- and cell context-dependent manner, likely through the inhibition of histone deacetylase (HDAC) (99). Interestingly, a recent study by Marinelli et al. provided evidence suggesting that butyrate acts as an AhR ligand to enhance transcription of AhR ligand-dependent genes, independent of its HDAC inhibitor activity, in intestinal epithelial cell (IEC)-AhR reporter cell lines (100). Although this observation needs to be confirmed in other cell types, the study raises an interesting possibility that SCFA butyrate can upregulate GPR15 expression through activation of AhR in colon-migrating T cells and possibly also in colon epithelial cells.
Regulation of GPR15 expression by pathophysiological factors
Smoking and DNA methylation
A number of genomic analyses of blood cells, mostly methylation analysis, have been performed to identify the molecular targets responsible for smoking-induced reprogramming. Blood T cells and B cells expressing GPR15 have approximately 50% methylation at cg19859270, a CpG site within the single exon of Gpr15, while non-GPR15-expressing cells are nearly 100% methylated at this site (101). Many studies have shown that smoking is associated with decreased methylation of cg19859270 (102–108) (also see review (109)) and increased Gpr15 mRNA expression (105, 107, 108, 110, 111) in blood leukocytes. Smoking was also shown to increase the frequency of peripheral GPR15+CD3+ T cells (107, 112). The effect of smoking is slowly reversible after cessation (102, 110, 113). These data suggest a potential mechanism by which smoking decreases Gpr15 DNA methylation, which leads to an increase in Gpr15 mRNA expression, resulting in increased GPR15-positive T cells in the blood. However, although it is conceivable that hypomethylation of Gpr15 DNA per se contributes to the increased expression of Gpr15 mRNA, the direct causal effect of smoking on Gpr15 DNA methylation is questionable. By analyzing GPR15 protein expression in leukocyte subtypes, Bauer et al. (107) found that the increased proportion of CD3+GPR15+ T cells in the blood of smokers was responsible for the apparent smoking-induced hypomethylation of the GPR15 gene, since cg19859270 hypomethylation was specifically found in GPR15-expressing cells. In addition, treatment of PBMC cultures with aqueous cigarette smoke extract (CSE) did not induce a higher proportion of this T cell subtype, suggesting that DNA hypomethylation at the cg19859270 site observed in smokers did not arise from the direct effect of tobacco smoking compounds on DNA methylation but rather from the enrichment of a smoking-induced GPR15+ T cell population in the peripheral blood (107). This study also indicates that the frequency of GPR15+ T cells in the blood can be effectively used as a highly reliable biomarker for tobacco smoking. It remains to be elucidated how smoking leads to an increased population of GPR15+ T cells independent of CSE, and whether or how the increased GPR15+ T cells in the blood impact immune homeostasis and smoke-related disease pathology.
Ischemia
In a recent study by Haase et al. (114), transcriptome analysis showed upregulated Gpr15 mRNA expression and downregulated Gpr15 DNA methylation in PBMCs from early onset myocardial infarction (MI) individuals compared to controls. The MI risk prediction analysis indicated that the effect of smoking on MI was fully mediated by Gpr15 mRNA expression; however, the associations between Gpr15 mRNA expression and Gpr15 DNA methylation with MI were found to be independent of smoking status. In addition, cardiac Gpr15 expression was significantly upregulated in a mouse model of infarction-related ischemia (>6-fold increase at five days after MI) as well as in an ischemic cardiomyocyte culture model (4-fold after 24 h induction of ischemic stress) (114). These data imply that GPR15 might play a role, independent of smoking, in the pathogenesis of acute MI and conditions of ischemia, such as artery narrowing by plaques. Interestingly, Gpr15 KO mice had reduced survival compared to wild-type mice after MI induction (114), which raises the possibility that MI-induced cardiac GPR15 expression represents a protective response to oxidative stress. Further investigation is warranted to elucidate the pathophysiological role of GPR15 in MI. It will be particularly interesting to determine whether and how GPR15 in cardiomyocytes is activated by known or novel ligands to induce signaling.
Epstein–Barr virus
The RNAseq data in the GTEx portal (www.gtexportal.org) showed the strongest expression of Gpr15 in EBV-transformed lymphocytes (median transcripts per million (TPM) was 252.9, while median TPM of whole blood was 1.0) among diverse human tissues. The mechanism of EBV-induced GPR15 expression is yet to be understood. EBV infects the oropharynx but frequently induces B-cell proliferation, which causes tumors in the gut of immunosuppressed individuals, such as transplant recipients treated with immunosuppressive drugs, namely post-transplantation lymphoproliferative disorders (PTLDs) (115, 116). Delecluse et al. found that EBV infection induces integrin α4β7 expression in tonsillar B cells (117). Since α4β7 is the key for homing of B cells to the gastrointestinal tract (118), this study suggests that the induction of α4β7 is one of the mechanisms through which EBV-infected cells enter the gastrointestinal mucosa-associated lymphoid tissue. It will be interesting to determine whether elevated GPR15 expression in EBV-transformed B cells may contribute to their specific homing to the colon by synergizing with α4β7 to enhance the pathogenesis of PTLDs.
Cytokines
GPR15 expression can be induced in vitro by a combination of TGF-β1 and either IL-6 or IL-21 preferentially in mouse Tregs (6). However, Il21r−/−Il6−/− mice crossed with Gpr15gfp/+ mice had a similar level of GFP expression as that of control mice, suggesting that TGF-β1 is a key regulator of GPR15 expression in vivo (6). An earlier study has shown that both TGF-β1 and TGF-β2 can induce de novo Foxp3 expression in CD4+CD25– cells (119). This raises a possibility that the effect of TGF-β on GPR15 expression may involve the increase of Foxp3 expression that would promote Gpr15 transcription through enhancement of the AhR activity (34).
Regulation of GPR15 by posttranslational modifications
Regulation of cell surface expression of GPR15 by phosphorylation
Cell surface expression levels and ligand responsiveness of chemokine receptors are tightly regulated by various post-translational modifications (PTMs). The trafficking of de novo synthesized GPR15 to the plasma membrane is dependent on the phosphorylation of the penultimate Ser359 residue in the cytoplasmic C-terminal tail (120). Phosphorylation of Ser359, which can be mediated by AKT (121), induces binding of 14-3-3 proteins to the receptor (120). The 14-3-3 binding sterically masks the upstream di-Arg (RxR) motif composed of Arg352 and Arg354, which is the binding motif of COPI, a coatomer protein complex that mediates the retrieval of cargo protein-loaded membrane vesicles from the Golgi to the endoplasmic reticulum (ER) (122). Thus, Ser359 phosphorylation promotes the expression of GPR15 at the cell surface (Figure 3A).
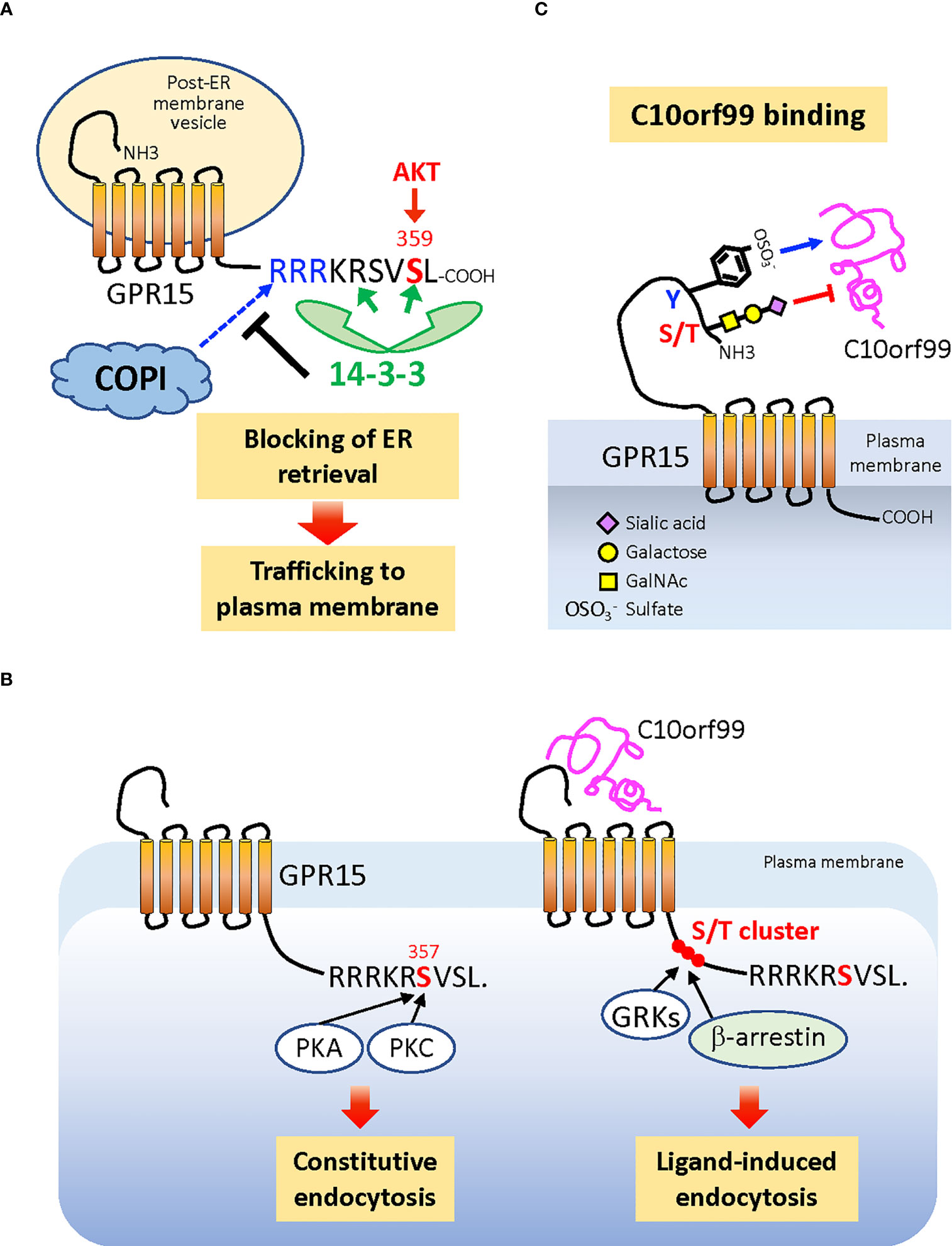
Figure 3 Posttranslational modifications that regulate cell surface expression and ligand binding of GPR15. (A) Phosphorylation of C-terminal penultimate Ser359 of the nascent GPR15 protein, which can be mediated by AKT, recruits dimeric 14-3-3 proteins to the C-terminus, which in turn blocks the binding of COPI to the adjacent di-Arg (RxR) motif. This prevents GPR15-loaded membrane vesicles from being retrieved back from the Golgi to the ER and allows GPR15 trafficking to the plasma membrane. (B) The cell surface level of GPR15 is also regulated by the constitutive (ligand-independent) endocytosis that requires phosphorylation of Ser357, which is the target of PKA and PKC. The more upstream Ser/Thr cluster that is commonly phosphorylated by GRKs and responsible for recruiting β-arrestin to many GPCRs is not required for the constitutive endocytosis of GPR15, while most likely required for the C10orf99-induced endocytosis. (C) The Tyr residues in the N-terminal extracellular region of GPR15 are sulfated (−) while the Ser and/or Thr residues are O-glycosylated and capped with α2,3-sialic acids. Sulfated Tyr residues promote, while sialylated O-glycans inhibit, the binding of C10orf99 to the receptor. ER, endoplasmic reticulum; PKA, protein kinase A; PKC, protein kinase C; AKT, protein kinase B; GRKs, GPCR kinases; COPI, coatomer protein complex I.
In addition to receptor insertion into the plasma membrane, the cell surface expression of GPR15 is also controlled by endocytosis. GPR15 is constitutively internalized in the absence of exogenous ligand (C10orf99) stimulation (123). This constitutive endocytosis requires the phosphorylation of Ser357, which can be mediated by protein kinase A (PKA) or PKC, but does not require the upstream Ser/Thr cluster in the C-terminal tail, which is commonly phosphorylated by G protein-coupled receptor kinases (GRKs) and is critical for recruiting β-arrestin (124) (Figure 3B). Constitutively endocytosed receptors have been successfully utilized for the delivery of therapeutic agents to target cells; for example, drugs conjugated with specific antibodies to receptors (125, 126). Therefore, ligand-independent endocytosis of GPR15 provides a basis for developing interventions targeting this receptor in human inflammatory/immune diseases, where GPR15-expressing cells may play pathogenic roles (9–11, 47).
Regulation of GPR15-C10orf99 interaction by Tyr sulfation and O-glycosylation
PTMs of GPR15 also regulate receptor-ligand interactions. Sulfation of Tyr residues in the extracellular N-terminus of chemokine receptors positively regulates the binding of chemokine ligands through electrostatic interactions (127, 128). GPR15 has also been recently found to be sulfated on Tyr residue(s), which enhances the binding of the endogenous ligand C10orf99 (21). It is interesting to note that latent membrane protein 1 (LMP1), which is encoded by EBV, induces tyrosine sulfation of CXCR4 through upregulation of tyrosylprotein sulfotransferase-1 (TPST-1) and promotes metastasis of cancer cells (129), a mechanism that could also be applied to GPR15.
GPR15-C10orf99 interaction is also regulated by O-linked glycosylation at the receptor N-terminus (21, 120). In contrast to Tyr sulfation, the O-glycans on the N-terminal Ser/Thr cluster negatively regulate ligand binding, which is at least in part due to the α2,3-linked sialic acid that caps O-glycans. This is similar to CCR7, in which sialylated O-glycans add steric hindrance to the receptor N-terminus to limit ligand binding (130). Consistent with their effects on ligand binding, Tyr sulfation and O-glycosylation of GPR15 differentially regulate the magnitude of receptor signaling (21). Thus, GPR15 represents a unique chemoattractant receptor in which two different PTMs on the N-terminus, Tyr sulfation and O-glycosylation, play contrasting roles in ligand binding and consequent signaling (Figure 3C), which is distinct from the reportedly cooperative roles of these two PTMs in the case of CCR5 (18) and CCR8 (20). The highly regulated glycosylation and sialylation during the differentiation and activation of T cells (131, 132) underpins the notion that the strength of the GPR15–GPR15L interaction is dynamically regulated by PTMs in both physiological and pathological conditions.
Inhibitors of GPR15
There is currently no publication reporting small-molecule compounds that inhibit C10orf99-induced GPR15 signaling. Wang et al. (133) predicted the 3D structure of human GPR15 and applied structure-based virtual screening approaches to discover potential antagonists that could bind to the predicted active sites. By screening a chemical database consisting of 62,500 small molecules, they isolated a set of compounds that satisfied the threshold of a high docking score. However, their antagonistic effects on GPR15 signaling have not yet been demonstrated. Hayn et al. screened peptide libraries generated from human hemofiltrate, which essentially represents the entire blood peptidome, to identify novel endogenous ligands of GPR15 (134). Using primate lentiviruses, these authors discovered a C-terminal fragment of cystatin C (CysC95-146) that specifically inhibits GPR15-dependent entry of HIV-1, HIV-2, and SIV, but does not inhibit C10orf99-induced signaling. This indicates that CysC95-146 is an endogenous inhibitor of GPR15-mediated HIV and SIV infections that does not interfere with the physiological function of this GPCR. In another study by Guo et al. (135), Gpr15 was found to be upregulated in CRC tissues, and silencing of Gpr15 by siRNA inhibited the growth, migration, and invasion of CRC cells. These authors found that the expression of GPR15 is post-transcriptionally regulated by microRNA-1225 (miR-1225), the expression of which is significantly downregulated in CRC cells. Overexpression of miR-1225 caused suppression of GPR15 and inhibited the proliferation of CRC cells, suggesting its therapeutic potential (135).
Conclusion and open questions
Over the past decade, a growing number of studies have indicated the involvement of GPR15 and its ligands in a variety of human immune disorders, making them promising therapeutic targets. In the past two decades, 45 drugs targeting chemokine receptors have been tested in clinical trials and only three have been approved by the Food and Drug Administration (FDA) (136). One of the major reasons for the poor success of chemokine receptor inhibitors may be the redundancy of chemokine-chemokine receptor interactions in vivo. In this regard, the unusual lack of cross-reactivity of GPR15 and C10orf99 with known chemokines and chemokine receptors, respectively, may offer an advantage for developing specific and clinically effective inhibitors. GPR15 is expressed in multiple cell types and plays pro-inflammatory or regulatory roles in both humans and mice. Further investigations to determine the relative contribution of specific cellular subsets in individual disease settings are necessary to enable the implementation of GPR15-targeted therapy. In this regard, development of conditional GPR15 KO mice likely facilitates elucidation of cell-intrinsic roles of GPR15. In addition, further discovery and characterization of new GPR15 ligands and regulatory mechanisms of GPR15 expression will expand our understanding of GPR15 biology in broader pathophysiology.
The questions that remain open include:
(1) GPR15 is strongly expressed by colon epithelial cells; however, the implication of epithelial GPR15 in disease is limited to HIV enteropathy and the growth of CRC cells. Studies in cell culture systems have demonstrated that chemokine/chemokine receptor signaling, such as CXCL8/CXCR1 and CXCL12/CXCR4, contributes to the maintenance of the epithelial barrier by stimulating the migratory repair process, termed restitution, of wounded epithelium (137, 138). It remains to be determined whether autocrine activation of GPR15 by C10orf99 in colon epithelial cells plays a role in barrier integrity through restitution. Also interesting will be whether GPR15 expression in colon epithelium is regulated by AhR stimulation by dietary and/or commensal metabolites.
(2) It is interesting that C10orf99 functions as an inducible “inflammatory” chemoattractant in psoriatic skin while this protein appears be a “homeostatic” chemoattractant in the colon where its expression was not altered by inflammation or the presence of commensal bacteria (7). It is still unclear what causes the increase in C10orf99 expression at the onset and/or during psoriasis. This is important since such regulatory mechanisms could also be applied to other C10orf99-expressing tissues including colonic and esophageal epithelia. Additionally, it is unknown whether the direct effects of C10orf99 on keratinocyte proliferation and inflammatory response are mediated by MRGPRs or other receptors.
(3) It remains open whether/how the different ligands such as C10orf99 and TME5 activate different signaling pathways (e.g., G-proteins) through GPR15. This may be cell context-dependent. It would also be interesting to investigate whether GPR15 undergoes different PTMs when expressed in different types of cells (e.g., lymphocytes, epithelial cells, and endothelial cells), which could potentially enable preferential interaction with particular ligands.
(4) Many studies indicate a correlation between smoking habit, hypomethylation of GPR15 DNA, and the increase of peripheral blood GPR15+ T cells; however, it is not clear how the smoking leads to the increase of GPR15 expression and whether the increase is specific to any T cell subsets (e.g., Tregs or Teffs). Those questions will be relevant for understanding the potential role of GPR15 in the smoking-related diseases.
Author contributions
YO and SS wrote the manuscript and made figures. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institute of Health (R01GM099974 and R21AI142060 to SS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the small intestine and colon. Gastroenterology (2016) 150(2):340–54. doi: 10.1053/j.gastro.2015.10.046
2. Wang C, Kang SG, Lee J, Sun Z, Kim CH. The roles of Ccr6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol (2009) 2(2):173–83. doi: 10.1038/mi.2008.84
3. Cassani B, Villablanca EJ, Quintana FJ, Love PE, Lacy-Hulbert A, Blaner WS, et al. Gut-tropic T cells that express integrin Alpha4beta7 and Ccr9 are required for induction of oral immune tolerance in mice. Gastroenterology (2011) 141(6):2109–18. doi: 10.1053/j.gastro.2011.09.015
4. Kang SG, Piniecki RJ, Hogenesch H, Lim HW, Wiebke E, Braun SE, et al. Identification of a chemokine network that recruits Foxp3(+) regulatory T cells into chronically inflamed intestine. Gastroenterology (2007) 132(3):966–81. doi: 10.1053/j.gastro.2007.01.008
5. Lazarus NH, Kunkel EJ, Johnston B, Wilson E, Youngman KR, Butcher EC. A common mucosal chemokine (Mucosae-associated epithelial Chemokine/Ccl28) selectively attracts iga plasmablasts. J Immunol (2003) 170(7):3799–805. doi: 10.4049/jimmunol.170.7.3799
6. Kim SV, Xiang WV, Kwak C, Yang Y, Lin XW, Ota M, et al. Gpr15-mediated homing controls immune homeostasis in the Large intestine mucosa. Science (2013) 340(6139):1456–9. doi: 10.1126/science.1237013
7. Ocon B, Pan J, Dinh TT, Chen W, Ballet R, Bscheider M, et al. A mucosal and cutaneous chemokine ligand for the lymphocyte chemoattractant receptor Gpr15. Front Immunol (2017) 8:1111. doi: 10.3389/fimmu.2017.01111
8. Suply T, Hannedouche S, Carte N, Li J, Grosshans B, Schaefer M, et al. A natural ligand for the orphan receptor Gpr15 modulates lymphocyte recruitment to epithelia. Sci Signal (2017) 10(496). doi: 10.1126/scisignal.aal0180
9. Morgan DM, Ruiter B, Smith NP, Tu AA, Monian B, Stone BE, et al. Clonally expanded, Gpr15-expressing pathogenic effector Th2 cells are associated with eosinophilic esophagitis. Sci Immunol (2021) 6(62). doi: 10.1126/sciimmunol.abi5586
10. Adamczyk A, Pastille E, Kehrmann J, Vu VP, Geffers R, Wasmer MH, et al. Gpr15 facilitates recruitment of regulatory T cells to promote colorectal cancer. Cancer Res (2021) 81(11):2970–82. doi: 10.1158/0008-5472.CAN-20-2133
11. Nguyen LP, Pan J, Dinh TT, Hadeiba H, O'Hara E 3rd, Ebtikar A, et al. Role and species-specific expression of colon T cell homing receptor Gpr15 in colitis. Nat Immunol (2015) 16(2):207–13. doi: 10.1038/ni.3079
12. Heiber M, Marchese A, Nguyen T, Heng HH, George SR, O'Dowd BF. A novel human gene encoding a G-Protein-Coupled receptor (Gpr15) is located on chromosome 3. Genomics (1996) 32(3):462–5. doi: 10.1006/geno.1996.0143
13. Deng HK, Unutmaz D, KewalRamani VN, Littman DR. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature (1997) 388(6639):296–300. doi: 10.1038/40894
14. Farzan M, Choe H, Martin K, Marcon L, Hofmann W, Karlsson G, et al. Two orphan seven-transmembrane segment receptors which are expressed in Cd4-positive cells support simian immunodeficiency virus infection. J Exp Med (1997) 186(3):405–11. doi: 10.1084/jem.186.3.405
15. Rana S, Baranski TJ. Third extracellular loop (Ec3)-n terminus interaction is important for seven-transmembrane domain receptor function: implications for an activation microswitch region. J Biol Chem (2010) 285(41):31472–83. doi: 10.1074/jbc.M110.129213
16. Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, et al. Tyrosine sulfation of the amino terminus of Ccr5 facilitates hiv-1 entry. Cell (1999) 96(5):667–76. doi: 10.1016/S0092-8674(00)80577-2
17. Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, et al. The role of post-translational modifications of the Cxcr4 amino terminus in stromal-derived factor 1 alpha association and hiv-1 entry. J Biol Chem (2002) 277(33):29484–9. doi: 10.1074/jbc.M203361200
18. Bannert N, Craig S, Farzan M, Sogah D, Santo NV, Choe H, et al. Sialylated O-glycans and sulfated tyrosines in the Nh2-terminal domain of cc chemokine receptor 5 contribute to high affinity binding of chemokines. J Exp Med (2001) 194(11):1661–73. doi: 10.1084/jem.194.11.1661
19. Zhu JZ, Millard CJ, Ludeman JP, Simpson LS, Clayton DJ, Payne RJ, et al. Tyrosine sulfation influences the chemokine binding selectivity of peptides derived from chemokine receptor Ccr3. Biochemistry (2011) 50(9):1524–34. doi: 10.1021/bi101240v
20. Gutierrez J, Kremer L, Zaballos A, Goya I, Martinez AC, Marquez G. Analysis of post-translational Ccr8 modifications and their influence on receptor activity. J Biol Chem (2004) 279(15):14726–33. doi: 10.1074/jbc.M309689200
21. Okamoto Y, Shikano S. Tyrosine sulfation and O-glycosylation of chemoattractant receptor Gpr15 differentially regulate interaction with Gpr15l. J Cell Sci (2021) 134(8). doi: 10.1242/jcs.247833
22. Clayton F, Kotler DP, Kuwada SK, Morgan T, Stepan C, Kuang J, et al. Gp120-induced Bob/Gpr15 activation: a possible cause of human immunodeficiency virus enteropathy. Am J Pathol (2001) 159(5):1933–9. doi: 10.1016/S0002-9440(10)63040-4
23. Lahl K, Sweere J, Pan J, Butcher E. Orphan chemoattractant receptor Gpr15 mediates dendritic epidermal T-cell recruitment to the skin. Eur J Immunol (2014) 44(9):2577–81. doi: 10.1002/eji.201444628
24. Cartwright A, Schmutz C, Askari A, Kuiper JH, Middleton J. Orphan receptor Gpr15/Bob is up-regulated in rheumatoid arthritis. Cytokine (2014) 67(2):53–9. doi: 10.1016/j.cyto.2014.02.015
25. Kiene M, Marzi A, Urbanczyk A, Bertram S, Fisch T, Nehlmeier I, et al. The role of the alternative coreceptor Gpr15 in siv tropism for human cells. Virology (2012) 433(1):73–84. doi: 10.1016/j.virol.2012.07.012
26. Agace WW. T-Cell recruitment to the intestinal mucosa. Trends Immunol (2008) 29(11):514–22. doi: 10.1016/j.it.2008.08.003
27. Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins a and d take centre stage. Nat Rev Immunol (2008) 8(9):685–98. doi: 10.1038/nri2378
28. Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of il-12 and il-23 in mucosal and systemic innate immune pathology. Immunity (2006) 25(2):309–18. doi: 10.1016/j.immuni.2006.05.017
29. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. Cd4+Cd25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med (2003) 197(1):111–9. doi: 10.1084/jem.20021345
30. Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell (2007) 131(1):33–45. doi: 10.1016/j.cell.2007.08.017
31. Picarella D, Hurlbut P, Rottman J, Shi X, Butcher E, Ringler DJ. Monoclonal antibodies specific for beta 7 integrin and mucosal addressin cell adhesion molecule-1 (Madcam-1) reduce inflammation in the colon of scid mice reconstituted with Cd45rbhigh Cd4+ T cells. J Immunol (1997) 158(5):2099–106. doi: 10.4049/jimmunol.158.5.2099
32. Shigematsu T, Specian RD, Wolf RE, Grisham MB, Granger DN. Madcam mediates lymphocyte-endothelial cell adhesion in a murine model of chronic colitis. Am J Physiol Gastrointest Liver Physiol (2001) 281(5):G1309–15. doi: 10.1152/ajpgi.2001.281.5.G1309
33. Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of Cd4+ T cells induce or protect from chronic intestinal inflammation in c. b-17 scid mice. Int Immunol (1993) 5(11):1461–71. doi: 10.1093/intimm/5.11.1461
34. Xiong L, Dean JW, Fu Z, Oliff KN, Bostick JW, Ye J, et al. Ahr-Foxp3-Rorgammat axis controls gut homing of Cd4(+) T cells by regulating Gpr15. Sci Immunol (2020) 5(48). doi: 10.1126/sciimmunol.aaz7277
35. Fischer A, Zundler S, Atreya R, Rath T, Voskens C, Hirschmann S, et al. Differential effects of Alpha4beta7 and Gpr15 on homing of effector and regulatory T cells from patients with uc to the inflamed gut in vivo. Gut (2016) 65(10):1642–64. doi: 10.1136/gutjnl-2015-310022
36. Adamczyk A, Gageik D, Frede A, Pastille E, Hansen W, Rueffer A, et al. Differential expression of Gpr15 on T cells during ulcerative colitis. JCI Insight (2017) 2(8). doi: 10.1172/jci.insight.90585
37. Swaminathan G, Nguyen LP, Namkoong H, Pan J, Haileselassie Y, Patel A, et al. The aryl hydrocarbon receptor regulates expression of mucosal trafficking receptor Gpr15. Mucosal Immunol (2021) 14(4):852–61. doi: 10.1038/s41385-021-00390-x
38. Parada Venegas D, de la Fuente MK, Landskron G, Gonzalez MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (Scfas)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol (2019) 10:277. doi: 10.3389/fimmu.2019.00277
39. Perrigoue J, Das A, Mora JR. Interplay of nutrients and microbial metabolites in intestinal immune homeostasis: distinct and common mechanisms of immune regulation in the small bowel and colon. Nestle Nutr Inst Workshop Ser (2014) 79:57–71. doi: 10.1159/000360685
40. Song J, Zheng H, Xue J, Liu J, Sun Q, Yang W, et al. Gpr15-C10orf99 functional pairing initiates colonic treg homing in amniotes. EMBO Rep (2022) 23(3):e53246. doi: 10.15252/embr.202153246
41. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
42. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two Foxp3(+)Cd4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med (2016) 22(6):679–84. doi: 10.1038/nm.4086
43. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity (2016) 45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021
44. Davis BP, Rothenberg ME. Mechanisms of disease of eosinophilic esophagitis. Annu Rev Pathol (2016) 11:365–93. doi: 10.1146/annurev-pathol-012615-044241
45. Hill DA, Spergel JM. The immunologic mechanisms of eosinophilic esophagitis. Curr Allergy Asthma Rep (2016) 16(2):9. doi: 10.1007/s11882-015-0592-3
46. Kunkel EJ, Campbell DJ, Butcher EC. Chemokines in lymphocyte trafficking and intestinal immunity. Microcirculation (2003) 10(3-4):313–23. doi: 10.1038/sj.mn.7800196
47. Fernandez-Ruiz JC, Ochoa-Gonzalez FL, Zapata-Zuniga M, Mondragon-Marin E, Lara-Ramirez EE, Ruiz-Carrillo JL, et al. Gpr15 expressed in T lymphocytes from Ra patients is involved in leukocyte chemotaxis to the synovium. J Leukoc Biol (2022) 112(5):1209–21. doi: 10.1002/JLB.3MA0822-263RR
48. Thelen F, Witherden DA. Get in touch with dendritic epithelial T cells! Front Immunol (2020) 11:1656. doi: 10.3389/fimmu.2020.01656
49. Sezin T, Jegodzinski L, Meyne LM, Gupta Y, Mousavi S, Ludwig RJ, et al. The G protein-coupled receptor 15 (Gpr15) regulates cutaneous immunology by maintaining dendritic epidermal T cells and regulating the skin microbiome. Eur J Immunol (2021) 51(6):1390–8. doi: 10.1002/eji.202048887
50. Sezin T, Kempen L, Meyne LM, Mousavi S, Zillikens D, Sadik CD. Gpr15 is not critically involved in the regulation of murine psoriasiform dermatitis. J Dermatol Sci (2019) 94(1):196–204. doi: 10.1016/j.jdermsci.2019.01.008
51. Jegodzinski L, Sezin T, Loser K, Mousavi S, Zillikens D, Sadik CD. The G protein-coupled receptor (Gpr) 15 counteracts antibody-mediated skin inflammation. Front Immunol (2020) 11:1858. doi: 10.3389/fimmu.2020.01858
52. Loetscher P, Clark-Lewis I. Agonistic and antagonistic activities of chemokines. J Leukoc Biol (2001) 69(6):881–4. doi: 10.1189/jlb.69.6.881
53. Foster SR, Hauser AS, Vedel L, Strachan RT, Huang XP, Gavin AC, et al. Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell (2019) 179(4):895–908.e21. doi: 10.1016/j.cell.2019.10.010
54. Deng Y, Moo EV, Almeria CVP, Gentry PR, Vedel L, Mathiesen JM, et al. Delineation of the Gpr15 receptor-mediated galpha protein signalling profile in recombinant mammalian cells. Basic Clin Pharmacol Toxicol (2022) 131(2):104–13. doi: 10.1111/bcpt.13738
55. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with il-23 and nf-kappab pathways. Nat Genet (2009) 41(2):199–204. doi: 10.1038/ng.311
56. Gudjonsson JE, Ding J, Li X, Nair RP, Tejasvi T, Qin ZS, et al. Global gene expression analysis reveals evidence for decreased lipid biosynthesis and increased innate immunity in uninvolved psoriatic skin. J Invest Dermatol (2009) 129(12):2795–804. doi: 10.1038/jid.2009.173
57. Tang T, Li L, Tang J, Li Y, Lin WY, Martin F, et al. A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol (2010) 28(7):749–55. doi: 10.1038/nbt.1644
58. Chen C, Wu N, Duan Q, Yang H, Wang X, Yang P, et al. C10orf99 contributes to the development of psoriasis by promoting the proliferation of keratinocytes. Sci Rep (2018) 8(1):8590. doi: 10.1038/s41598-018-26996-z
59. Dainichi T, Nakano Y, Doi H, Nakamizo S, Nakajima S, Matsumoto R, et al. C10orf99/Gpr15l regulates proinflammatory response of keratinocytes and barrier formation of the skin. Front Immunol (2022) 13:825032. doi: 10.3389/fimmu.2022.825032
60. Guo P, Luo Y, Mai G, Zhang M, Wang G, Zhao M, et al. Gene expression profile based classification models of psoriasis. Genomics (2014) 103(1):48–55. doi: 10.1016/j.ygeno.2013.11.001
61. Tseng PY, Hoon MA. Gpr15l is an epithelial inflammation-derived pruritogen. Sci Adv (2022) 8(24):eabm7342. doi: 10.1126/sciadv.abm7342
62. Yang M, Tang M, Ma X, Yang L, He J, Peng X, et al. Ap-57/C10orf99 is a new type of multifunctional antimicrobial peptide. Biochem Biophys Res Commun (2015) 457(3):347–52. doi: 10.1016/j.bbrc.2014.12.115
63. Yung SC, Murphy PM. Antimicrobial chemokines. Front Immunol (2012) 3:276. doi: 10.3389/fimmu.2012.00276
64. Li X, Fan R, Tong A, Yang M, Deng J, Zhou L, et al. In situ gel-forming ap-57 peptide delivery system for cutaneous wound healing. Int J Pharm (2015) 495(1):560–71. doi: 10.1016/j.ijpharm.2015.09.005
65. Bassilana F, Baumgarten Bu, Carte N-T, Detheux M, Falchetto R, Hannedouche S, et al. Organic compounds. Google Patents (2015) WO2015069459A1.
66. Pan W, Cheng Y, Zhang H, Liu B, Mo X, Li T, et al. Csbf/C10orf99, a novel potential cytokine, inhibits colon cancer cell growth through inducing G1 arrest. Sci Rep (2014) 4:6812. doi: 10.1038/srep06812
67. Pan B, Wang X, Nishioka C, Honda G, Yokoyama A, Zeng L, et al. G-Protein coupled receptor 15 mediates angiogenesis and cytoprotective function of thrombomodulin. Sci Rep (2017) 7(1):692. doi: 10.1038/s41598-017-00781-w
68. Bauer M. The role of Gpr15 function in blood and vasculature. Int J Mol Sci (2021) 22(19). doi: 10.3390/ijms221910824
69. Sadler JE. Thrombomodulin structure and function. Thromb Haemost (1997) 78(1):392–5. doi: 10.1055/s-0038-1657558
70. Akwiwu E, Edem M, Akpotuzor J, Asemota E, Isong I. Glycated haemogloin, fasting plasma glucose, plasminogen activator inhibitor type-1, and soluble thrombomodulin levels in patients with type 2 diabetes mellitus. Niger J Physiol Sci (2021) 36(2):159–64. doi: 10.54548/njps.v36i2.3
71. Dhainaut JF, Yan SB, Cariou A, Mira JP. Soluble thrombomodulin, plasma-derived unactivated protein c, and recombinant human activated protein c in sepsis. Crit Care Med (2002) 30(5 Suppl):S318–24. doi: 10.1097/00003246-200205001-00023
72. Bouck EG, Denorme F, Holle LA, Middelton EA, Blair AM, de Laat B, et al. Covid-19 and sepsis are associated with different abnormalities in plasma procoagulant and fibrinolytic activity. Arterioscler Thromb Vasc Biol (2021) 41(1):401–14. doi: 10.1161/ATVBAHA.120.315338
73. Mizutani H, Hayashi T, Nouchi N, Inachi S, Suzuki K, Shimizu M. Increased endothelial and epidermal thrombomodulin expression and plasma thrombomodulin level in progressive systemic sclerosis. Acta Med Okayama (1996) 50(6):293–7. doi: 10.18926/AMO/30479
74. Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, et al. Efficacy and safety of recombinant human soluble thrombomodulin (Art-123) in disseminated intravascular coagulation: results of a phase iii, randomized, double-blind clinical trial. J Thromb Haemost (2007) 5(1):31–41. doi: 10.1111/j.1538-7836.2006.02267.x
75. Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, et al. A randomized, double-blind, placebo-controlled, phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, art-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med (2013) 41(9):2069–79. doi: 10.1097/CCM.0b013e31828e9b03
76. Pan B, Wang X, Kojima S, Nishioka C, Yokoyama A, Honda G, et al. The fifth epidermal growth factor like region of thrombomodulin alleviates lps-induced sepsis through interacting with Gpr15. Thromb Haemost (2017) 117(3):570–9. doi: 10.1160/TH16-10-0762
77. Pan B, Wang X, Kojima S, Nishioka C, Yokoyama A, Honda G, et al. The fifth epidermal growth factor-like region of thrombomodulin alleviates murine graft-Versus-Host disease in a G-protein coupled receptor 15 dependent manner. Biol Blood Marrow Transplant (2017) 23(5):746–56. doi: 10.1016/j.bbmt.2017.02.001
78. Furukawa K, Ling J, Sun Y, Lu YU, Fu J, Marukuchi R, et al. Recombinant thrombomodulin has anti-tumor effects and enhances the effects of gemcitabine for pancreatic cancer through G-protein coupled receptor 15. Anticancer Res (2022) 42(10):4679–87. doi: 10.21873/anticanres.15972
79. Lu W, Gersting JA, Maheshwari A, Christensen RD, Calhoun DA. Developmental expression of chemokine receptor genes in the human fetus. Early Hum Dev (2005) 81(6):489–96. doi: 10.1016/j.earlhumdev.2004.10.022
80. Mehrad B, Keane MP, Strieter RM. Chemokines as mediators of angiogenesis. Thromb Haemost (2007) 97(5):755–62. doi: 10.1160/TH07-01-0040
81. Wornle M, Schmid H, Merkle M, Banas B. Effects of chemokines on proliferation and apoptosis of human mesangial cells. BMC Nephrol (2004) 5:8. doi: 10.1186/1471-2369-5-8
82. Delezay O, Yahi N, Tamalet C, Baghdiguian S, Boudier JA, Fantini J. Direct effect of type 1 human immunodeficiency virus (Hiv-1) on intestinal epithelial cell differentiation: relationship to hiv-1 enteropathy. Virology (1997) 238(2):231–42. doi: 10.1006/viro.1997.8829
83. Dayanithi G, Yahi N, Baghdiguian S, Fantini J. Intracellular calcium release induced by human immunodeficiency virus type 1 (Hiv-1) surface envelope glycoprotein in human intestinal epithelial cells: a putative mechanism for hiv-1 enteropathy. Cell Calcium (1995) 18(1):9–18. doi: 10.1016/0143-4160(95)90041-1
84. Maresca M, Mahfoud R, Garmy N, Kotler DP, Fantini J, Clayton F. The virotoxin model of hiv-1 enteropathy: involvement of Gpr15/Bob and galactosylceramide in the cytopathic effects induced by hiv-1 Gp120 in the ht-29-D4 intestinal cell line. J BioMed Sci (2003) 10(1):156–66. doi: 10.1159/000068089
85. Li Q, Estes JD, Duan L, Jessurun J, Pambuccian S, Forster C, et al. Simian immunodeficiency virus-induced intestinal cell apoptosis is the underlying mechanism of the regenerative enteropathy of early infection. J Infect Dis (2008) 197(3):420–9. doi: 10.1086/525046
86. Prasad Singh N, Nagarkatti M, Nagarkatti P. From suppressor T cells to regulatory T cells: how the journey that began with the discovery of the toxic effects of tcdd led to better understanding of the role of ahr in immunoregulation. Int J Mol Sci (2020) 21(21). doi: 10.3390/ijms21217849
87. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi: 10.1038/nri1806
88. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(Reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi: 10.1038/nature06880
89. Abron JD, Singh NP, Mishra MK, Price RL, Nagarkatti M, Nagarkatti PS, et al. An endogenous aryl hydrocarbon receptor ligand, ite, induces regulatory T cells and ameliorates experimental colitis. Am J Physiol Gastrointest Liver Physiol (2018) 315(2):G220–G30. doi: 10.1152/ajpgi.00413.2017
90. Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U.S.A. (2010) 107(48):20768–73. doi: 10.1073/pnas.1009201107
91. Alrafas HR, Busbee PB, Nagarkatti M, Nagarkatti PS. Resveratrol modulates the gut microbiota to prevent murine colitis development through induction of tregs and suppression of Th17 cells. J Leukoc Biol (2019) 106(2):467–80. doi: 10.1002/JLB.3A1218-476RR
92. Al-Ghezi ZZ, Singh N, Mehrpouya-Bahrami P, Busbee PB, Nagarkatti M, Nagarkatti PS. Ahr activation by tcdd (2,3,7,8-Tetrachlorodibenzo-P-Dioxin) attenuates pertussis toxin-induced inflammatory responses by differential regulation of tregs and Th17 cells through specific targeting by microrna. Front Microbiol (2019) 10:2349. doi: 10.3389/fmicb.2019.02349
93. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate il-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology (2011) 141(1):237–48. doi: 10.1053/j.gastro.2011.04.007
94. Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep (2017) 21(8):2277–90. doi: 10.1016/j.celrep.2017.10.114
95. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes Via aryl hydrocarbon receptor activation. Cell (2011) 147(3):629–40. doi: 10.1016/j.cell.2011.09.025
96. McAleer JP, Fan J, Roar B, Primerano DA, Denvir J. Cytokine regulation in human Cd4 T cells by the aryl hydrocarbon receptor and gq-coupled receptors. Sci Rep (2018) 8(1):10954. doi: 10.1038/s41598-018-29262-4
97. Rivera SP, Saarikoski ST, Sun W, Hankinson O. Identification of novel dioxin-responsive genes by representational difference analysis. Xenobiotica (2007) 37(3):271–9. doi: 10.1080/00498250601169816
98. Dean JW, Helm EY, Fu Z, Xiong L, Sun N, Oliff KN, et al. The aryl hydrocarbon receptor cell intrinsically promotes resident memory Cd8(+) T cell differentiation and function. Cell Rep (2023) 42(1):111963. doi: 10.1016/j.celrep.2022.111963
99. Jin UH, Cheng Y, Park H, Davidson LA, Callaway ES, Chapkin RS, et al. Short chain fatty acids enhance aryl hydrocarbon (Ah) responsiveness in mouse colonocytes and caco-2 human colon cancer cells. Sci Rep (2017) 7(1):10163. doi: 10.1038/s41598-017-10824-x
100. Marinelli L, Martin-Gallausiaux C, Bourhis JM, Beguet-Crespel F, Blottiere HM, Lapaque N. Identification of the novel role of butyrate as ahr ligand in human intestinal epithelial cells. Sci Rep (2019) 9(1):643. doi: 10.1038/s41598-018-37019-2
101. Bauer M, Hackermuller J, Schor J, Schreiber S, Fink B, Pierzchalski A, et al. Specific induction of the unique Gpr15 expression in heterogeneous blood lymphocytes by tobacco smoking. Biomarkers (2019) 24(3):217–24. doi: 10.1080/1354750X.2018.1539769
102. Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet (2012) 21(13):3073–82. doi: 10.1093/hmg/dds135
103. Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African americans. Hum Genet (2013) 132(9):1027–37. doi: 10.1007/s00439-013-1311-6
104. Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics (2014) 15:151. doi: 10.1186/1471-2164-15-151
105. Koks G, Uudelepp ML, Limbach M, Peterson P, Reimann E, Koks S. Smoking-induced expression of the Gpr15 gene indicates its potential role in chronic inflammatory pathologies. Am J Pathol (2015) 185(11):2898–906. doi: 10.1016/j.ajpath.2015.07.006
106. Bauer M, Fink B, Thurmann L, Eszlinger M, Herberth G, Lehmann I. Tobacco smoking differently influences cell types of the innate and adaptive immune system-indications from cpg site methylation. Clin Epigenet (2015) 7:83. doi: 10.1186/s13148-016-0249-7
107. Bauer M, Linsel G, Fink B, Offenberg K, Hahn AM, Sack U, et al. A varying T cell subtype explains apparent tobacco smoking induced single cpg hypomethylation in whole blood. Clin Epigenet (2015) 7:81. doi: 10.1186/s13148-015-0113-1
108. Ohmomo H, Harada S, Komaki S, Ono K, Sutoh Y, Otomo R, et al. DNA Methylation abnormalities and altered whole transcriptome profiles after switching from combustible tobacco smoking to heated tobacco products. Cancer Epidemiol Biomarkers Prev (2022) 31(1):269–79. doi: 10.1158/1055-9965.EPI-21-0444
109. Koks S, Koks G. Activation of Gpr15 and its involvement in the biological effects of smoking. Exp Biol Med (Maywood) (2017) 242(11):1207–12. doi: 10.1177/1535370217703977
110. Vink JM, Jansen R, Brooks A, Willemsen G, van Grootheest G, de Geus E, et al. Differential gene expression patterns between smokers and non-smokers: cause or consequence? Addict Biol (2017) 22(2):550–60. doi: 10.1111/adb.12322
111. Tsaprouni LG, Yang TP, Bell J, Dick KJ, Kanoni S, Nisbet J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics (2014) 9(10):1382–96. doi: 10.4161/15592294.2014.969637
112. Andersen AM, Lei MK, Beach SRH, Philibert RA, Sinha S, Colgan JD. Cigarette and cannabis smoking effects on Gpr15+ helper T cell levels in peripheral blood: relationships with epigenetic biomarkers. Genes (Basel) (2020) 11(2). doi: 10.3390/genes11020149
113. Haase T, Muller C, Krause J, Rothemeier C, Stenzig J, Kunze S, et al. Novel DNA methylation sites influence Gpr15 expression in relation to smoking. Biomolecules (2018) 8(3). doi: 10.3390/biom8030074
114. Haase T, Muller C, Stoffers B, Kirn P, Waldenberger M, Kaiser FJ, et al. G Protein-coupled receptor 15 expression is associated with myocardial infarction. Int J Mol Sci (2022) 24(1). doi: 10.3390/ijms24010180
115. Evens AM, David KA, Helenowski I, Nelson B, Kaufman D, Kircher SM, et al. Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol (2010) 28(6):1038–46. doi: 10.1200/JCO.2009.25.4961
116. Inayat F, Hassan GU, Tayyab GUN, Saif MW. Post-transplantation lymphoproliferative disorder with gastrointestinal involvement. Ann Gastroenterol (2018) 31(2):248–51. doi: 10.20524/aog.2018.0226
117. Delecluse S, Tsai MH, Shumilov A, Bencun M, Arrow S, Beshirova A, et al. Epstein-Barr Virus induces expression of the lpam-1 integrin in b cells in vitro and in vivo. J Virol (2019) 93(5). doi: 10.1128/JVI.01618-18
118. Salmi M, Jalkanen S. Lymphocyte homing to the gut: attraction, adhesion, and commitment. Immunol Rev (2005) 206:100–13. doi: 10.1111/j.0105-2896.2005.00285.x
119. Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, et al. Tgf-beta induces Foxp3 + T-regulatory cells from Cd4 + Cd25 - precursors. Am J Transplant (2004) 4(10):1614–27. doi: 10.1111/j.1600-6143.2004.00566.x
120. Okamoto Y, Shikano S. Phosphorylation-dependent c-terminal binding of 14-3-3 proteins promotes cell surface expression of hiv Co-receptor Gpr15. J Biol Chem (2011) 286(9):7171–81. doi: 10.1074/jbc.M110.199695
121. Chung JJ, Okamoto Y, Coblitz B, Li M, Qiu Y, Shikano S. Pi3k/Akt signalling-mediated protein surface expression sensed by 14-3-3 interacting motif. FEBS J (2009) 276(19):5547–58. doi: 10.1111/j.1742-4658.2009.07241.x
122. Spang A. Retrograde traffic from the golgi to the endoplasmic reticulum. Cold Spring Harb Perspect Biol (2013) 5(6). doi: 10.1101/cshperspect.a013391
123. Okamoto Y, Shikano S. Differential phosphorylation signals control endocytosis of Gpr15. Mol Biol Cell (2017) 28(17):2267–81. doi: 10.1091/mbc.E16-09-0627
124. Bahouth SW, Nooh MM. Barcoding of gpcr trafficking and signaling through the various trafficking roadmaps by compartmentalized signaling networks. Cell Signal (2017) 36:42–55. doi: 10.1016/j.cellsig.2017.04.015
125. Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery Via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev (2002) 54(4):561–87. doi: 10.1124/pr.54.4.561
126. Muro S, Schuchman EH, Muzykantov VR. Lysosomal enzyme delivery by icam-1-Targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther (2006) 13(1):135–41. doi: 10.1016/j.ymthe.2005.07.687
127. Ludeman JP, Stone MJ. The structural role of receptor tyrosine sulfation in chemokine recognition. Br J Pharmacol (2014) 171(5):1167–79. doi: 10.1111/bph.12455
128. Szpakowska M, Fievez V, Arumugan K, van Nuland N, Schmit JC, Chevigne A. Function, diversity and therapeutic potential of the n-terminal domain of human chemokine receptors. Biochem Pharmacol (2012) 84(10):1366–80. doi: 10.1016/j.bcp.2012.08.008
129. Xu J, Deng X, Tang M, Li L, Xiao L, Yang L, et al. Tyrosylprotein sulfotransferase-1 and tyrosine sulfation of chemokine receptor 4 are induced by Epstein-Barr virus encoded latent membrane protein 1 and associated with the metastatic potential of human nasopharyngeal carcinoma. PloS One (2013) 8(3):e56114. doi: 10.1371/journal.pone.0056114
130. Hauser MA, Kindinger I, Laufer JM, Spate AK, Bucher D, Vanes SL, et al. Distinct Ccr7 glycosylation pattern shapes receptor signaling and endocytosis to modulate chemotactic responses. J Leukoc Biol (2016) 99(6):993–1007. doi: 10.1189/jlb.2VMA0915-432RR
131. Pereira MS, Alves I, Vicente M, Campar A, Silva MC, Padrao NA, et al. Glycans as key checkpoints of T cell activity and function. Front Immunol (2018) 9:2754. doi: 10.3389/fimmu.2018.02754
132. Bi S, Baum LG. Sialic acids in T cell development and function. Biochim Biophys Acta (2009) 1790:1599–610. doi: 10.1016/j.bbagen.2009.07.027
133. Wang Y, Wang X, Xiong Y, Li CD, Xu Q, Shen L, et al. An integrated pan-cancer analysis and structure-based virtual screening of Gpr15. Int J Mol Sci (2019) 20(24). doi: 10.3390/ijms20246226
134. Hayn M, Blotz A, Rodriguez A, Vidal S, Preising N, Standker L, et al. Natural cystatin c fragments inhibit Gpr15-mediated hiv and siv infection without interfering with Gpr15l signaling. Proc Natl Acad Sci U.S.A. (2021) 118(3). doi: 10.1073/pnas.2023776118
135. Guo Y, Zhu Q, Chen S, Li Y, Fu D, Qiao D, et al. Post-transcriptional suppression of G protein-coupled receptor 15 (Gpr15) by microrna-1225 inhibits proliferation, migration, and invasion of human colorectal cancer cells. 3 Biotech (2021) 11(3):139. doi: 10.1007/s13205-021-02682-2
136. Lai WY, Mueller A. Latest update on chemokine receptors as therapeutic targets. Biochem Soc Trans (2021) 49(3):1385–95. doi: 10.1042/BST20201114
137. Smith JM, Johanesen PA, Wendt MK, Binion DG, Dwinell MB. Cxcl12 activation of Cxcr4 regulates mucosal host defense through stimulation of epithelial cell migration and promotion of intestinal barrier integrity. Am J Physiol Gastrointest Liver Physiol (2004) 288:G316–G26. doi: 10.1152/ajpgi.00208.2004
Keywords: GPR15, C10orf99, GPCR, chemokine, homing, inflammation
Citation: Okamoto Y and Shikano S (2023) Emerging roles of a chemoattractant receptor GPR15 and ligands in pathophysiology. Front. Immunol. 14:1179456. doi: 10.3389/fimmu.2023.1179456
Received: 04 March 2023; Accepted: 19 June 2023;
Published: 30 June 2023.
Edited by:
James Edward Pease, Imperial College London, United KingdomReviewed by:
Gabriela Fonseca-Camarillo, National Institute of Medical Sciences and Nutrition Salvador Zubirán, MexicoChristian David Sadik, University of Lübeck, Germany
Cheng Deng, Sichuan University, China
Liang Zhou, University of Florida, United States
Copyright © 2023 Okamoto and Shikano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sojin Shikano, c3NoaWthbm9AdWljLmVkdQ==