 Jamie Lees
Jamie Lees Jodie Hay
Jodie Hay Michael W. Moles
Michael W. Moles Alison M. Michie
Alison M. Michie
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 May 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1179101
This article is part of the Research TopicMolecular and Cellular Control of B Cell responses: Germinal Center and Extrafollicular Responses for Cellular OutputsView all 6 articles
Forkhead box (FOX) class O (FOXO) proteins are a dynamic family of transcription factors composed of four family members: FOXO1, FOXO3, FOXO4 and FOXO6. As context-dependent transcriptional activators and repressors, the FOXO family regulates diverse cellular processes including cell cycle arrest, apoptosis, metabolism, longevity and cell fate determination. A central pathway responsible for negative regulation of FOXO activity is the phosphatidylinositol-3-kinase (PI3K)-AKT signalling pathway, enabling cell survival and proliferation. FOXO family members can be further regulated by distinct kinases, both positively (e.g., JNK, AMPK) and negatively (e.g., ERK-MAPK, CDK2), with additional post-translational modifications further impacting on FOXO activity. Evidence has suggested that FOXOs behave as ‘bona fide’ tumour suppressors, through transcriptional programmes regulating several cellular behaviours including cell cycle arrest and apoptosis. However, an alternative paradigm has emerged which indicates that FOXOs operate as mediators of cellular homeostasis and/or resistance in both ‘normal’ and pathophysiological scenarios. Distinct FOXO family members fulfil discrete roles during normal B cell maturation and function, and it is now clear that FOXOs are aberrantly expressed and mutated in discrete B-cell malignancies. While active FOXO function is generally associated with disease suppression in chronic lymphocytic leukemia for example, FOXO expression is associated with disease progression in diffuse large B cell lymphoma, an observation also seen in other cancers. The opposing functions of the FOXO family drives the debate about the circumstances in which FOXOs favour or hinder disease progression, and whether targeting FOXO-mediated processes would be effective in the treatment of B-cell malignancies. Here, we discuss the disparate roles of FOXO family members in B lineage cells, the regulatory events that influence FOXO function focusing mainly on post-translational modifications, and consider the potential for future development of therapies that target FOXO activity.
FOXO transcription factors are a family of proteins belonging to a larger superfamily that contains an evolutionarily conserved forkhead domain ordered alphabetically from FOXA to FOXR (the FOX transcription factor superfamily; reviewed in (1)). The FOXO family comprise four highly related members: FOXO1 (FKHR), FOXO3 (FKHRL1), FOXO4 (AFX) and FOXO6 in mammals, which are orthologs of DAF-16 in Caenorhabditis elegans and dFOXO in Drosophila melanogaster. Mammalian FOXO family members were initially reported as part of pro-oncogenic fusion proteins of paired box protein 3/7 (PAX)-FOXO1 in alveolar rhabdomyosarcoma, whereby FOXO1 trans-activation and the FOXO-dependent TGF-β response was inhibited, thus promoting tumorigenesis (2, 3). Similarly, FOXO3 and FOXO4 form mixed lineage leukemia (MLL) fusion partners, and aggressive paediatric acute leukemia, from translocation of t(6;11)(q21;q23) and t(X;11)(q13.1;q23) respectively (4–6). FOXO family members regulate gene expression through activation or repression. As such, FOXOs are vital for the regulation of a plethora of cellular processes, from cell cycle arrest and apoptosis to metabolism and oxidative damage modulation (7). Structurally, FOXOs differ from the rest of the FOX superfamily, containing a specific amino acid sequence flanking the DNA-binding domain (DBD) (Gly-Asp-Ser-Asn-Ser) enabling interaction with the FHRE (forkhead response element; 5’-GTAAACAA-3’) (8–10). Within the family, FOXOs have shared DBD homology, however the structure of their transactivation domains (TAD) differ; it is this difference in TAD structure that determines the nature of FOXO interactors to define role specificity (11). Partnered with differences in TAD structure are the tissue-specific expression patterns of FOXO isoforms, suggesting FOXOs have cell-specific roles (10). While FOXO1, FOXO3 and FOXO4 family members are ubiquitously expressed, FOXO6 has a more restricted expression pattern. Interestingly, FOXO1-deficient mice are embryonic lethal, dying at day 10.5 of gestation, due to a block in vascular development, while FOXO3- and FOXO4-deficient mice are viable and largely indistinguishable from wildtype mice (12). Therefore, functional redundancy exists between FOXO family members (12–14), with studies suggesting that FOXOs can be overexpressed to fulfil the roles of other family members should they become dysfunctional (15). In addition to TAD sequences, FOXO proteins also contain NLS (nuclear localisation signal), NES (nuclear export signal), all of which are regulated by post-translational modification (Figure 1 (7, 11)). As FOXOs require nuclear DNA binding to produce cellular effects, FOXO activity is partnered with subcellular localisation.
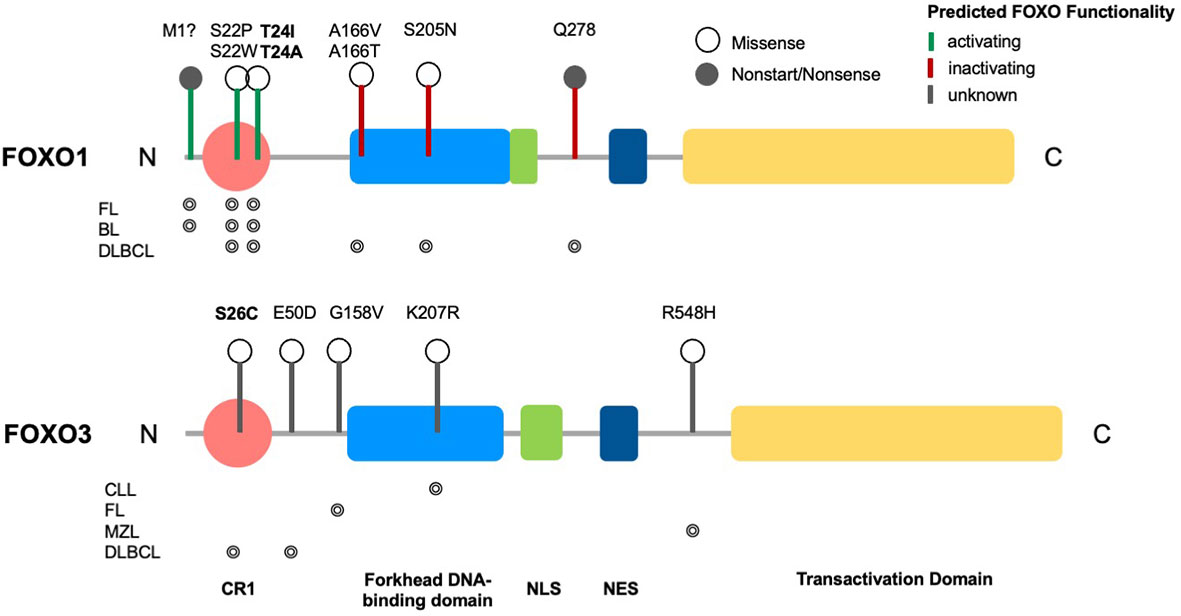
Figure 1 Structure of FOXO transcription factors and common mutations across B cell malignancies. Domain structure of FOXO1 (655 aa) and FOXO3 (673 aa), consisting of a conserved region (CR1), a forkhead DNA-binding domain (DBD), a nuclear localization signal (NLS), a nuclear export sequence (NES) and a C-terminal transactivation domain (TAD). Markers show FOXO1 driver mutations across B cell malignancies (CLL, DLBCL, FL, BL, MZL) and variants of uncertain significance (VUS) in FOXO3, with phosphorylation sites highlighted in bold. Occurrence of respective mutations in FOXO1 and FOXO3 across B cell malignancies are shown below the relevant structural schematic [data from combined studies, 2298 samples (16–18)].
In B lymphocytes, B-cell receptor (BCR) ligation is a key signalling component responsible for inducing cell growth and survival (19, 20). Upon crosslinking the BCR with antigen, ITAM motifs on CD79A/B are phosphorylated by Lyn tyrosine kinase enabling the generation of signalling hubs containing adaptor proteins and tyrosine kinases including BLNK, BCAP and Syk. Activated Syk phosphorylates BCAP, which in turn activates PI3Kδ, resulting in conversion of PIP2 to PIP3 and recruitment of PDK1 and AKT to the plasma membrane. PDK1 phosphorylates and activates AktT308, enabling further phosphorylation/activation of AKTS473 by mechanistic target of rapamycin complex 2 (mTORC2 (19, 21)). AKT regulates a wide array of cell functions via phosphorylation of target proteins including the FOXO family. Multiple serine/threonine RxRxxS/T regions within the N-terminus, NLS and NES of FOXOs are present, which are conserved from the C. elegans protein DAF-16 (FOXO1 - T24, S256, S319; FOXO3 - T32, S253, S315; FOXO4 - T28, S193, S258; FOXO6 - T26, S184. FOXO6 lacks a C-terminal phosphorylation site (7, 15, 22, 23)). AKT-mediated phosphorylation leads to FOXO inactivation by enabling docking of 14-3-3 proteins via RSxpS/TxP and RxxxpSxP motifs (24, 25). 14-3-3 proteins can also affect the binding affinity of DNA for FOXOs by interrupting the DNA binding process at the DBD (26). This leads to a conformational change that exposes more of the NES than the NLS, thus preventing FOXO from returning to the nucleus post-translocation (27, 28). Downstream of AKT, mTORC1 also has a pivotal role in inducing positive cellular effects such as protein translation, cell growth and proliferation (29). FOXOs hinder mTORC1 function by inducing RICTOR expression, thus reducing RAPTOR-mTOR association, in turn mTORC1 function and preventing cell proliferation (30). FOXOs also downregulate mTORC1 via SESN3 (sestrin3) upregulation, which in turn activates tuberous sclerosis 1 and 2 (TSC1 & TSC2) via AMPK (31). Interestingly, TSC1 and TSC2 can also activate mTORC1 activity if stimulated by other proteins (30), demonstrating the effectiveness of FOXOs in overriding proliferative signals.
Although AKT-mediated FOXO phosphorylation is the most understood post-translational modification of FOXO activity, there are other pathways through which FOXO activity is modulated leading to a broad range of biological effects. FOXO localisation (and subsequent transcriptional activity) is negatively regulated by MAPK/ERK, CDK2 and casein kinase 1-mediated phosphorylation (32–36). Conversely, FOXO activity is upregulated by JNK-mediated phosphorylation in response to oxidative stress or AMPK-mediated phosphorylation in response to reduced intracellular ATP, leading to enhanced nuclear localisation and subsequent transcriptional activity, for example through phosphorylation of FOXO1 at Ser383 and Thr649 (37, 38). In cases such as these, FOXOs respond to intracellular stress stimuli by upregulating genes that maintain homeostasis, such as GPX1 (oxidative damage) and PGC1α (metabolism) (15). FOXOs are also susceptible to post-translational modification via acetylation, methylation and ubiquitination (39, 40). Several acetyltranferases, deacetylases, ubiquitin ligases and methyltransferases can either activate or repress FOXO activity through lysine modification, resulting in subsequent changes in DNA binding efficacy, protein interaction effectiveness and overall stability (41–44), while mono- and poly-ubiquitination of FOXO transcription factors impact on protein stability and subcellular localisation (32, 45). miRNAs also regulate the FOXO family post-transcriptionally, such as miR-27a, miR-96 and miR-182 reducing FOXO1 expression in MCF7 cells by targeting the 3’ untranslated region of FOXO1 (46).
Selective FOXO family members play important roles at distinct stages during B-cell lineage commitment and development (Figure 2). During the initial stages of B cell lineage commitment, FOXO1 enables the differentiation of common lymphoid progenitor (CLP) cells towards the pro-B cell stage. This is initiated by E2A and HeLa E-box binding (HEB) proteins, with the ablation of these proteins diminishing FOXO1 expression and inducing a block at the CLP stage (47, 48). The transition from the pro-B to pre-B cell stage is dependent on the generation of a functionally rearranged immunoglobulin heavy chain µ (µIgH), surrogate light chains (VPREB and IGLL1) and the signalling components CD79A and CD79B, to form the pre-BCR (49). Pre-B cells further divide and rearrange the Ig light chains (Igκ or Igλ) to generate a functional, mature BCR (50).

Figure 2 FOXO expression is critical for early B-cell development. B-cells undergo a specific set of developmental stages in the bone marrow (BM), which are tightly controlled by the expression of particular FOXO transcription factors, enabling differentiation and proliferation at distinct stages of lymphopoiesis. FOXO3 regulates commitment of CLP cells to the B-cell lineage, whereupon FOXO1 expression enables pro-B cell differentiation via E2A and HEB activity. FOXO1, in conjunction with EBF1, promotes B-cell lineage commitment via activation of PAX5, alongside positive regulators such as IL-7R. Cells advancing to the pre-B cell stage are coordinated by SYK activity, which promotes proliferation or differentiation via activation of PI3K or BLNK respectively. PI3K signalling inactivates FOXO1, leading to an upregulation of MYC and CCND2 expression driving pre-B cell proliferation, while BLNK induces cell cycle arrest via FOXO1 and BCL6 upregulation. FOXO1 expression is ablated to allow for differentiation of small pre-B cells into immature B-cells primed to leave the BM to further mature and differentiate in GC reactions. Figure produced in BioRender.
FOXO1 specifically plays a critical role during these stages of development through transcriptional activation of the recombination-activating gene (RAG) proteins RAG1 and RAG2, which are responsible for initiating Ig gene rearrangement (50–52). In addition, positive regulators of early lymphopoiesis (IL7R, SYK, PI3KCα and VPREB1/3) are regulated by and can regulate FOXO1 activity (52, 53). During the pre-B cell stage, SYK inactivates or activates FOXO1 via downstream activation of PI3K or BLNK respectively (53). An absence of FOXO1 during this stage prevents somatic recombination in pro-B cells and reduces Ig light chain rearrangement in pre-B cells, while an absence of FOXO3 has no effect (52). Interestingly, a loss of FOXO1 in pro-B cells also provokes increased levels of apoptosis linked to an increase in the expression of BCL2L11 (BIM) and lowered BCL2L1 (BCL-XL) expression (52). These studies highlight a key role for FOXO1 in driving early B cell differentiation and supporting pro-B cell survival.
FOXO1 can evoke further positive or negative impacts on pre-B cell proliferation through upregulation of CCND3 or BCL6 respectively. CCND3 (Cyclin D3) is a FOXO target and crucial for pre-B cell proliferation (54). BCL6 is a repressor of DNA recombination-induced cell death (induced by p53 downregulation) but also prevents proliferation via MYC/CCND2 repression (55, 56). This temporal regulation can lead to cell cycle arrest of pre-B cells (57). These observations demonstrate the significant role of FOXO1 in early B-cell development. However FOXO3 and FOXO4 are also expressed during development (55). Although the role of FOXO4 expression in B-cell development is unclear, evidence is emerging for the significance of FOXO3, as FOXO3-/- mice exhibit a loss of the pre-B cell population, and a reduction of B cells in the bone marrow (BM) and peripheral blood (58). Although the loss of FOXO1 impaired B cell lineage commitment, studies using a FOXO3 conditional deletion model demonstrated that FOXO3 deletion affected the production of LMPPs, CLPs and B-cell precursors (59). Furthermore, deletion of both FOXO1 and FOXO3 results in a complete block of CLP commitment towards the B-cell lineage (59). These data shed light on the importance of FOXO3, in addition to FOXO1, in normal B-cell development and indicate a predominant switch of FOXO3 to FOXO1 expression is pivotal for B-cell lymphopoiesis (Figure 2).
Mature B-cells express both FOXO1 and FOXO3. Upon BCR crosslinking and downstream PI3K-AKT activation, FOXO1 is downregulated and inactivated (60, 61). There is conflicting evidence regarding the function of FOXO1 in mature B cells. Srinivasan et al., demonstrated that FOXO1 induces apoptosis in response to a lack of BCR signalling, partnered with elevated BCL2L11 and CDKN1B FOXO target expression (62). This was supported by studies in which constitutively active FOXO1 (FOXO1-A3) promoted apoptosis and cell cycle arrest in B-cells (55). However, FOXO1 expression also supports B-cell populations: conditional deletion of FOXO1 (CD21-Cre+) reduced B-cell lymph node populations, presenting FOXO1 expression as being crucial for correct B cell migration (52). FOXO1 deletion resulted in a reduced capacity for B cells to induce BCR signalling and effectively proliferate (52, 61). The role of FOXO3 in mature B cell maintenance and regulation is also conflicting; constitutively active FOXO3 increased levels of cell cycle arrest and apoptosis in B-cells (61), suggesting a pro-apoptotic function for FOXO3 in mature B-cells. On the other hand, FOXO3-/- Eμ-myc transgenic mice were shown to have accelerated levels of B-cell lymphomagenesis (63), suggesting that FOXO3 is required for normal B-cell maintenance and regulation. These data demonstrate the context-specific bimodality of FOXO function, with FOXO activity either hindering or promoting cell growth and proliferation.
Germinal centres (GC) are specialised structures within secondary lymphoid organs (SLO) in which B-cells undergo clonal expansion and somatic hypermutation (SHM), leading to the generation of antibodies that possess a higher affinity for antigen (affinity maturation) and the export of long-lived plasma cells (PC) and memory B cells (Figure 3 (reviewed in 61)). Upon initial recognition of antigen, B cells (pre-GC B cells) congregate at the border between the follicle and the T cell zone and undergo proliferation, which leads to the development of the GC structure. GCs comprise a proliferation-rich dark zone (DZ), in which B-cells undergo SHM and clonal expansion, and a light zone (LZ), in which B-cells (centrocytes) undergo selection processes: the new antibody is tested for interaction with follicular dendritic cell bound antigen and receive help from follicular helper T (Tfh) cells (64, 65). Following positive selection of B cells expressing higher affinity antibodies, the B-cells re-enter the DZ and proliferate further, driven by cyclin D3 (66, 67). Activation-induced cytidine deaminase (AID) is an essential component of affinity maturation due to its ability to initiate SHM (68, 69). While introducing nucleotide changes in the variable regions of Igh, processing of these mutations can induce DNA double stranded breaks which leads to class switch recombination (CSR) resulting in the generation of IgG, IgA and IgE antibodies. However, evidence indicates that CSR mainly occurs earlier in pre-GC cells (70). Bcl-6 also plays a vital role within the GC B cell program, modulating Myc and Prdm1 (encoding Blimp1) expression which, together with FOXO1 activity, assist in normal GC function (71–73). Indeed, FOXO1 is a critical component in the DZ phenotype, as indicated by the finding that deletion of FOXO1 in GC B cells results in a loss of the anatomical structure of the DZ (73, 74). This happens in part because of the absence of CXCR4 assisted B cell migration: FOXO1-mediated CXCR4 expression ensures the retention of B-cells in the DZ (73). While FOXO1 knockdown in GC-B cells prevents DZ formation and inhibits CSR, SHM and clonal expansion are unaffected suggesting that FOXO1 may act to delay the LZ transcription program (52, 73, 74). Supporting these findings, an enhancement in CSR was noted in B cells with nuclear sequestered FOXO1T24A via subsequent AICDA (AID) upregulation, while inactivation of FOXO1 through AKT-mediated signals led to IRF4-driven PC differentiation (75). In addition to FOXO1 function being required for DZ B-cell populations and GC anatomical formation, FOXO1-mediated induction of BATF is required for effective LZ B-cell proliferation (76), demonstrating a need for FOXO1 expression in both GC compartments. Of note, FOXO1 plays a key role in positive GC B-cell selection, as BCR signalling is programmed to signal via FOXO1 activity, while CD40 ligation induces NF-κB activity: both CD40- and BCR-mediated signals are required to induce c-Myc upregulation thus promote B-cell survival (77). Further, FOXO1 induces upregulation of CCND3 in the GC DZ B cells, enabling GC B cell expansion (66). Collectively, these findings demonstrate that FOXO1 plays a central role in regulating GC processes.
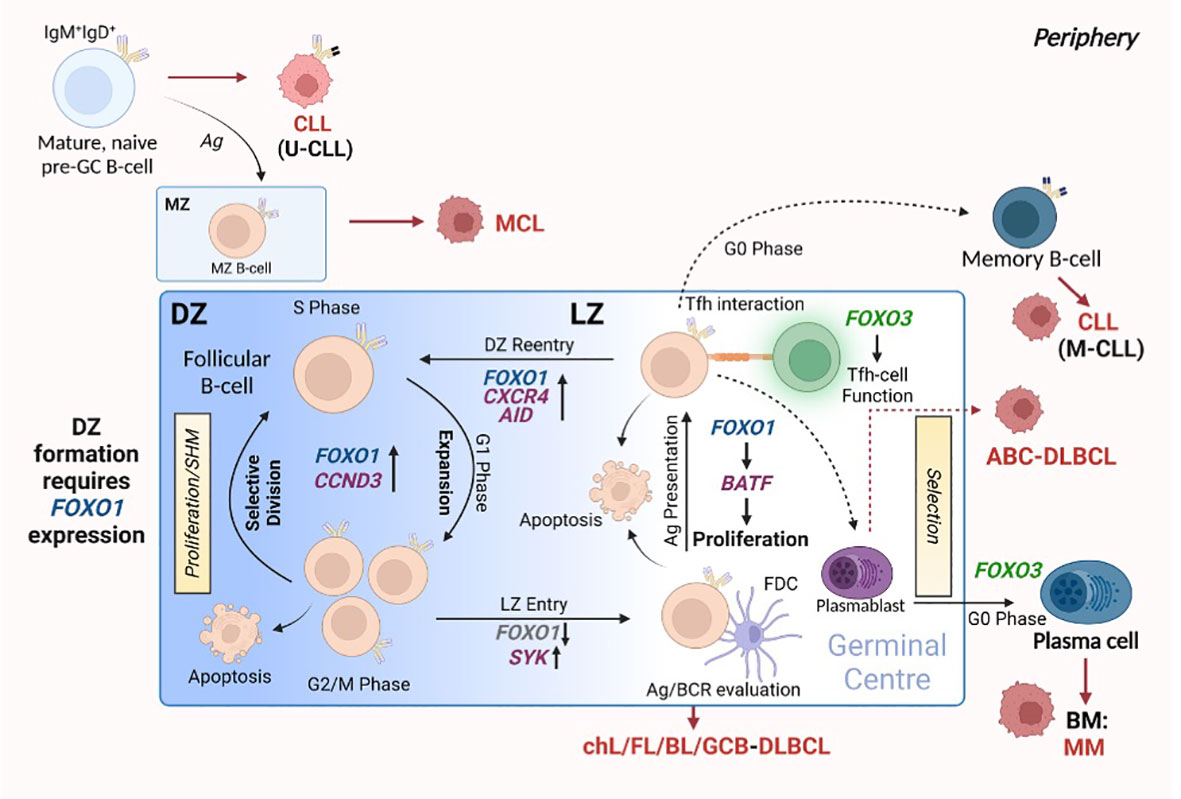
Figure 3 FOXO expression assists in germinal centre (GC) B-cell differentiation. The formation of the GC is crucial for the generation of B-cells that produce high affinity antibodies towards specific antigens and differentiate into plasma cells (PCs) and memory B-cells. This occurs through GC-centralised processes: SHM, affinity maturation and clonal expansion. GCs consist of two distinct compartments: the DZ, in which B-cells cycle between proliferation and SHM, and the LZ, in which cells undergo evaluation and selection processes. FOXO1 expression is critical for forming and retaining cells in the DZ. Cells then enter the LZ, where the modified/mutated antibody is tested for high-affinity towards antigen: this process requires FOXO1 downregulation via SYK. Here, B-cells interact with FDCs to evaluate antigen affinity and BCR function, while Tfh-cell interactions aid in B-cell differentiation and proliferation by providing the appropriate signals. Of note, the LZ requires specific regulation of FOXO1 expression to allow for correct LZ proliferation via BATF induction. B-cells from the LZ gain re-entry into the DZ to undergo clonal expansion regulated by FOXO1 and CCND3. All the while, FOXO3 expression maintains GC Th-cell populations and allows for the differentiation of PCs. Red arrows and malignant-like cells indicate stages in GC B-cell differentiation where B-cell malignancy can originate. Figure produced in BioRender.
FOXO3 is also important in B-cell maturation through Tfh cell function, an essential cellular component of GC formation (65, 78). FOXO3-deficient mice exhibit lower levels of IL-21, anti-ovalbumin antibodies when challenged with ovalbumin, and decreased Tfh and B-cell populations. Additionally, there is a marked reduction in the occurrence of T cell co-stimulator (ICOS)-induced Tfh differentiation (78). FOXO3 is strongly expressed in B cells committed to PC differentiation (65). Thus, FOXO1 and FOXO3 play inverse roles, both being critical in facilitating mature B-cell differentiation as part of the adaptive immune response.
FOXOs are traditionally regarded as tumour suppressors due to their canonical activity being associated with detrimental cellular fate (e.g. cell cycle arrest and apoptosis (15)). Indeed, early studies demonstrated that the conditional triple FOXO1/3/4 deletion in adult mice resulted in the development of thymic lymphomas and hemagiomas (14). However, in line with FOXO family members playing an important physiological role in maintaining self-renewal in stem cell compartments (79), more recent studies revealed that FOXO family members can maintain leukaemia-initiating cells in myeloid leukemias (AML and CML), and promote breast tumour invasion, suggesting that in certain cellular contexts FOXOs play a tumour-promoting role (80–82). The paradox of FOXO proteins driving the inhibition or promotion of cancer development in specific contexts, and the complex regulatory mechanisms that subvert FOXO function in malignant cells identifies the need for a deeper understanding of the cellular and molecular function of these proteins in specific cancers.
FOXO signalling is often compromised and exploited during B-cell development and maturation to promote proliferative and anti-apoptotic signals, providing malignant cells with the means to bypass checkpoints preventing classical cancer hallmarks. Typically, this occurs in a disease-specific context, where FOXOs can exhibit bipartite behaviours, either aiding or preventing tumourigenesis. Of note, somatic point mutations of FOXO1 occur more frequently across B cell malignancies than other cancers, and within B cell malignancies occur predominantly in Burkitt’s lymphoma (BL), follicular lymphoma (FL) and diffuse large B cell lymphoma (DLBCL), with a frequency of ~11%, 6% and ~5% respectively (Figures 4A, B (16–18, 83)). Within these diseases, six point mutations have been identified as driver mutations of oncogenesis, where drivers are defined as mutations, fusions and copy number alterations in OncoKB (84) or CancerHotspots (85), with mutations occurring mostly in the N-terminus or forkhead DBD within exon 1 (Figure 1). All six driver mutations are considered to be oncogenic through either: gain of function, as demonstrated in in vitro studies, which showed escape from PI3K/AKT regulation through loss of AKT phosphorylation; increased nuclear sequestration and increased DNA binding compared to wildtype (86), or; loss-of function, identified through preserved AKT phosphorylation, cytoplasmic retention and decreased transcriptional activity compared to wildtype (87, 88). Additionally, around forty missense and in-frame deletion mutations of FOXO1 have been identified within B cell malignancies, which are considered variants of uncertain significance (VUS). Mutations of the other FOXO family members are less frequent within B cell malignancies (Figure 4C). No driver mutations have been characterised within FOXO3, however five missense VUS mutations have been identified within exon 1, with one mutation affecting a putative phosphorylation site, Ser26 within marginal zone lymphoma, DLBCL, chronic lymphocytic leukemia (CLL) and BL (Figure 1) (89). It should be noted that the 6q21 region encoding FOXO3 is frequently deleted in DLBCL (with deletions of 6q21-q22 occurring in 40% of ABC-DLBCL cases and 22% of GCB-DLBCL cases) (90) and is associated with adverse prognosis (91). Deletions of 6q21 also occur in mantle cell lymphoma (MCL), FL, acute lymphoblastic leukemia (ALL) and CLL (90, 92).
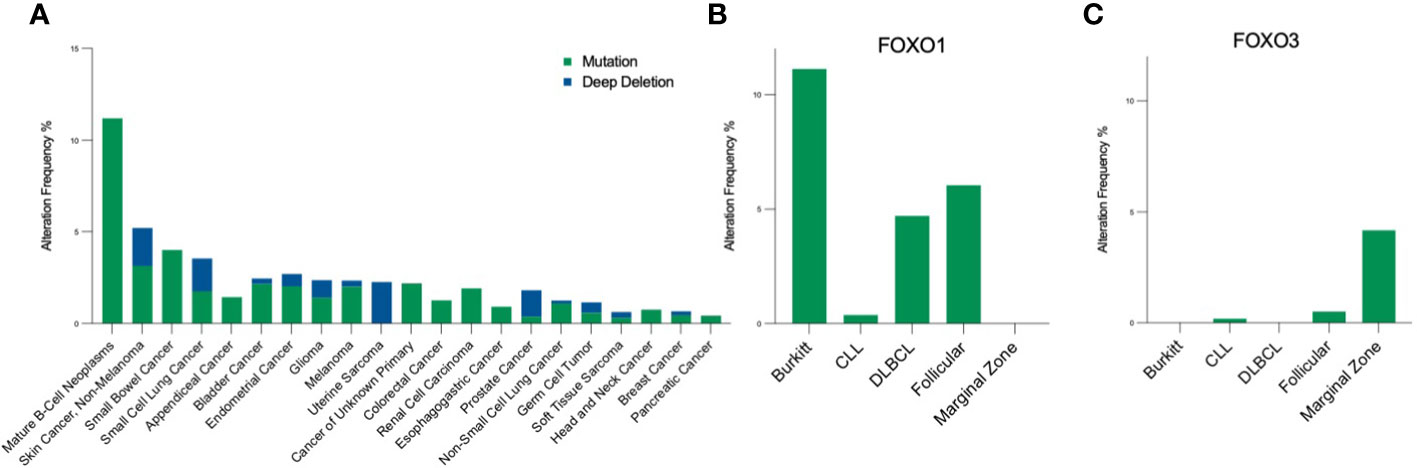
Figure 4 Pan-cancer frequency of FOXO mutations. (A) The frequency of FOXO1 mutations across malignancies [data from MSK-IMPACT, 10945 samples; (83)] highlight that FOXO1 mutations are most prevalent in B-cell neoplasms. Within the B-cell malignancy subset, (B) FOXO1 is most frequently mutated in Burkitt's lymphoma, and (C) FOXO3 is most frequently mutated in marginal zone lymphoma. Mutations encompass, in-frame, missense and truncating mutations, and deep deletion indicates a deep loss, possibly a homozygous deletion.
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common childhood malignancy originating from pro-B and pre-B cells (49, 93). BCP-ALL arises as a result of a B-cell differentiation block, dysregulation of proliferation and the promotion of survival signals (94). FOXO1 expression is not altered in BCP-ALL (95, 96). In BCR+ BCP-ALL cell lines, pharmacological SYK inhibition blocks FOXO1T24 phosphorylation, leading to increased FOXO1 activity and enhanced cell death. Supporting this, constitutively active FOXO1 increases cell death (97). Bhansali et al. demonstrated that the kinase DYRK1A is overexpressed and crucial for BCP-ALL progression; inhibition of DYRK1A and subsequent activation of FOXO1 led to DNA-damage-mediated cell death (98). In contrast, genetic and pharmacological inactivation of FOXO1 produces a tumour suppressive effect by means of caspase-induced cell death in pre-BCR- and pre-BCR+ BCP-ALL cells via CCND3 downregulation (99, 100). FOXO3 is typically deleted in BCP-ALL (90). In mouse models, FOXM1 is negatively regulated by FOXO3 which promotes cell growth via cell cycle progression (101). Interestingly, patient-derived BCP-ALL cells with highly expressed levels of FOXM1 are associated with poor prognostic outcomes; this data coincides with reduced FOXO3 expression (102). This suggests BCP-ALL promotes cell growth and survival by means of bypassing the expression of tumour suppressive components such as FOXO3.
CLL is characterized by expansion of small, monoclonal CD5+ B-lymphocytes in the blood, BM and SLOs (103). FOXO family members play a vital role in CLL biology, with regulation of individual family members impacting on CLL survival. FOXO family mutations occur infrequently in CLL patients, with analysis of the ICGC database (containing data from 551 CLL patients) showing that single base substitutions within FOXO family genes occurred in 4% (22/551) of CLL patients, of which 2% (11/551) occurred in FOXO1, with a functionally “high impact” missense mutation of FOXO1 encoded in just one patient (104). Therefore the tumour microenvironment has a more significant impact on the activity of FOXO family members in CLL. FOXO3 is inactivated by chemokine expression (CCL19, CLL21, CXCL12, CXCL13) leading to subsequent BIM downregulation and increased cell survival. Expression of constitutively active FOXO3 increased levels of cell death whereas a reduction in FOXO3 expression increased cell survival, providing evidence for FOXO3 playing a tumour suppressor role in CLL cells (103). Palacios et al. showed that CLL cells required elevated levels of miR-22 expression for effective proliferation, with miR-22 leading to decreased PTEN and increased PI3K-AKT signalling, particularly in poor prognostic patient cohorts. Subsequent FOXO1 inhibition coincided with reduced levels of CDKN1B (105). We have demonstrated that while FOXO1 expression was upregulated in CLL cells compared with B cells from healthy donors, it appeared to be inactive, perhaps due to tonic CLL-BCR signalling. However, pharmacological inhibition of PI3K-mTOR/AKT signalling reduced FOXO1 phosphorylation, increased FOXO1 nuclear localisation and resulted in an upregulation of FOXO1-mediated genes, ultimately inducing CLL cell death (106). These studies indicate that FOXO1 is being prevented from performing a tumour suppressor role due to tumour microenvironmental factors supporting the CLL cells, similar to that noted for FOXO3 (103). In contrast, in an aggressive CLL mouse model, FOXO1 was shown to be a driver of disease through induction of IGF1R in PI3K-inhibitor-resistant SLO tumours: this was attenuated by pharmacological FOXO1 inhibition (AS1842856) (107). Interestingly, FOXO1 has also been reported to induce GAB1 which aids in maintenance of CLL cell survival through sustained basal AKT phosphorylation (108). These studies suggests that a low level of FOXO1 activity, possibly promoted through tonic BCR signalling, sustains CLL cell survival, while higher FOXO1 activity induced by inhibition of upstream PI3K/AKT signals through drug treatment triggers CLL cell death (106, 108). Interestingly, a recent study demonstrated that hyperactivation of PI3K/AKT activity mediated by SHIP1 inhibition, can induce CLL cell death (109). While this study did not directly address the involvement of the FOXO family in the induction of cell death, it suggests that the strength of signal upstream of FOXO may regulated the threshold of FOXO activity induced, which in turn impacts on cell fate decisions within CLL. Of note, discrete tumour microenvironments are also likely to play a major role in modulating FOXO activity.
MCL is a rare B-cell malignancy caused by the overexpression of cyclin D1 in B-cells inhabiting the mantle zone surrounding GCs. Studies suggest a tumour suppressive role for FOXO3, as pharmacological inhibition of PI3K-AKT-mTOR signalling resulted in dephosphorylation and activation of FOXO3 leading to a reduction in MCL cell survival and proliferation (110). Additionally, FOXO3 is constitutively inactive and cytoplasm-localised in MCL. Recent evidence however suggests that FOXO1, not FOXO3, acts as a “master regulator” driving MCL progression, with FOXO1 inducing the expression of genes governing MCL lineage survival (111). A novel inhibitor of FOXO1 activity cpd10, blocked MCL progression in vivo (111). These data highlight differential roles for FOXO1 and FOXO3 in MCL.
FL originates in the GC from activated centroblast and centrocyte B-cells defined by genomic aberrations including the t(14,18) translocation resulting in the overexpression of BCL-2 (112). Somatic point mutations suppressing FOXO1 DNA binding activity identified in DLBCL are evident in FL (87). The nonsense mutation at the M1 residue, at the N terminus, leads to a truncation of FOXO1 and loss of the AKT recognition motif, resulting in increased nuclear localisation and transcriptional activity (Figure 1 (113)). This in turn promotes pro-proliferative and anti-apoptotic signalling and lymphomagenesis (114). However, the FOXO1 mutations drive altered transcriptional profiles which lead to positive selection of GC B-cells (86). Therefore, GC B-cells expressing FOXO1M1 expand abnormally with a competitive advantage over normal GC B-cells, and the altered signalling identified in the VavP-Bcl2 transgenic mouse model of human FL, promotes lymphomagenesis (86). While FL cells exhibit abnormal function of B-cell epigenetic regulators (EZH2, HIST1H1), they also incorporate genetic defects in multiple pathways affecting B-cell growth and development, such as PI3K/AKT and mTOR, suggesting mutation is a key factor through which FL cells bypass screening for correct growth (112). Downstream of PI3K-AKT/mTOR signalling, Pastore et al. demonstrated an association between shorter failure-free survival and FOXO1 gene mutations, identifying FOXO1 mutation as a potential biomarker for a model determining FL disease risk (M7-FLIPI) (112, 115). Moreover, venetoclax (BCL2i)-resistant FL cells exhibit increased levels of p-FOXO1/p-FOXO3 associated with diminished BIM activity, thereby preventing cell death supporting the role of FOXO family members as tumour suppressors in FL (116).
Activity of the MYC oncogene is the focal point driving BL pathogenesis (117). As FOXOs are known to negatively regulate MYC, FOXOs were considered to have tumour suppressive properties in BL. Supportive work by Bouchard et al. demonstrated an acceleration of BL tumourigenesis following either FOXO3 knockdown or expression of a FOXO3 dominant negative construct in the Eµ-Myc mouse model (118). However, mutations forcing FOXO1 nuclear accumulation can promote BL cell proliferation and survival. Within sporadic BL, mutations of Thr24 are most common, while missense mutations of Ser22 are most frequent in endemic disease (119, 120). Phosphorylation of Ser22 by AMPK directly prohibits the binding of 14-3-3 proteins through steric hindrance and electrostatic repulsion, and indirectly through negative regulation of Thr24, therefore mutations of this residue are likely to lead to inactive FOXO1 function and cytoplasmic localisation (120). This mutation is also prevalent in DLBCL and FL (Figure 1 (16, 17)). CRISPR/Cas9-mediated FOXO1 ablation strongly restricted tumour growth, demonstrating FOXO1 as a key driver of BL (114). Gehringer et al. demonstrated a loss of BL cell proliferative capacity following FOXO1 knockdown, supported by an increase in cell death following AS1842856-mediated FOXO1 inhibition (121). Thus, these data provide interesting evidence for FOXO1 behaving as a driver of disease while FOXO3 behaves as a tumour suppressor in BL.
DLBCL is a heterogeneous disease which was transcriptionally divided into molecular subtypes based on cell of origin: normal GC B-cells (GCB-DLBCL) or activated B-cells (ABC-DLBCL), with ABC-DLBCL associated with an inferior outcome (122). These subtypes have since been sub-divided into five distinct DLBCL subsets, based on the expression of driver mutations, somatic copy number variations and structural variations, which have enabled a more detailed landscape of DLBCL pathogenesis to be appreciated (123). Patients with relapsed/refractory disease (rrDLBCL) have poor disease outcome, with around 40% of patients achieving a longer term remission due to salvage chemotherapy or stem cell transplant (124). The genetic profile of de novo DLBCL is well characterised, with common genomic alterations, including chromosomal translocations and mutations found between subtypes, contributing to promotion of pro-survival pathways (125). Within DLBCL, FOXO1 missense mutations have been reported in ~9% cases (113). However, between the two subtypes, there is an enrichment of mutation frequency of some genes, including FOXO1 in GCB-DLBCL, which is considered to be a driver mutation (86). Furthermore, in rrDLBCL a significant enrichment of FOXO1 mutations is seen when compared to untreated DLBCL patients (27% vs. 8.6%) (124). FOXO family members have been reported to elicit conflicting tumour promoting or suppressing activities in DLBCL, which may also reflect the mutational burden that is found in DLBCL patient samples. FOXO1 mutations however, have been associated with poor prognosis and diminished treatment response; FOXO1 missense mutations affecting the DBD and the Thr24 mutation (Figure 1), led to poorer overall survival of patients treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) and is associated with poor prognosis in DLBCL (113). This could in part be due to the negative regulation of CD20 expression by FOXO1, which leads to a reduced response to rituximab. Therefore within non-Hodgkin lymphoma, mutations prohibiting AKT-phosphorylation of FOXO1 may represent an important biomarker of rituximab response (126). Within the DBD, Ser205 can be mutated within DLBCL, and in vitro studies show that this impairs DNA binding capacity, while retaining some transcriptional activity. It has been hypothesised that mutations within the DBD may suppress DNA dependent tumour suppressor activities but maintain pro-lymphogenic FOXO1 functions (87). A further truncating mutation, Gln278 residue on exon 2 of FOXO1, has been identified in DLBCL although it is rare (16). The FOXO1Q278 gene product lacks the TAD, rendering it transcriptionally inactive and prevents DNA binding but retains the AKT recognition motif (Figure 1 (127, 128)), suggesting it will be excluded from the nucleus. Of note, Szydlowski et al. confirmed that FOXO1 drives BIM and CDKN1B transcription following SYK inhibition (using R406), while FOXO1 ablation prevented R406-induced apoptosis and cell cycle inhibition (129).
FOXO4 has also been associated with poor prognosis in DLBCL, identified as an inducer of resistance to doxorubicin and phenylbutyrate. Additionally, FOXO4 knockdown reduced colony forming ability of DLBCL cell lines (130). FOXO3, however, has been associated with tumour suppression, as indicated by studies showing that prevention of FOXO3 inactivation due to nuclear export reduces levels of ibrutinib resistance in DLBCL (131). Zheng et al. reported low levels of FOXO3 in DLBCL cells compared to healthy B-cells and high levels of miRNA miR-155. Inhibiting miR-155 induced cell death and reduced proliferation, which coincided with higher expression of FOXO3, suggesting FOXO3 possesses tumour suppressive qualities in DLBCL (132). Collectively, specific FOXOs may differentially contribute to DLBCL disease progression, with FOXO1 and FOXO3 activity assisting in promoting DLBCL cell death and preventing resistance to targeted therapies.
cHL is thought to originate as a result of the deactivation of particular signalling pathways in activated GC-localised B-cells, such as NF-κB and JAK/STAT pathways (133). FOXO1 is downregulated in cHL cell lines and cHL patient samples. Additionally, constitutively active FOXO1 induced apoptosis and cell cycle arrest, partnered with increased expression of FOXO targets CDKN1B and BCL2L11. This study concluded that FOXO1 behaves as a tumour suppressor, and that cHL cells actively reduce FOXO1 levels to aid in disease progression (133). Conversely, FOXO3 levels are higher in cHL than GC B-cells but lower than in differentiated PCs, suggesting FOXO3 expression contributes directly to the cHL phenotype of being an “abortive PC”. Indeed, overexpression of FOXO3 in cell line models activated cHL-inactive PRDM1α (134), a critical transcription factor that drives the PC differentiation program. These findings indicate that FOXO3 downregulation by cHL cells contributes to the cHL phenotype and overall disease progression, identifying FOXO3 as a tumour suppressor in cHL.
MM is a B-cell malignancy characterized by the accumulation of slow proliferating, apoptosis-resistant PCs residing in the BM (135). FOXO1 and FOXO3 have been found to be highly phosphorylated and inactivated, suggesting tumour suppressive functions for FOXOs in MM. As such, targeted inhibition of GSK3 activity increases FOXO1 and FOXO3 activity leading to cell death via upregulation of downstream apoptotic factors including Fas ligand (136). More recently, pharmacological AKT inhibition was demonstrated to induce cell cycle arrest and cell death via FOXO activation, thus further cementing the tumour suppressive qualities of FOXO family members in MM (137, 138). De Bruyne et al. demonstrated that IGF1 signalling induces MM cell survival via downregulation of the pro-apoptotic factor and FOXO target BIM, at least in part through AKT activation and subsequent inactivation of FOXO3 (139). Supporting this, PI3K signal ablation using a selective inhibitor induces cell cycle arrest and apoptosis coinciding with decreases in FOXO phosphorylation (140). In addition to the PI3K-AKT pathway being a viable target for treating MM, previous MM treatments such as bortezomib, a proteosomal inhibitor, has been shown to induce MM apoptosis coinciding with increased FOXO3 expression and activity (141). More recently the nuclear export receptor inhibitor Selinexor has been approved for clinical use for MM and identifies the contribution of FOXO1 and FOXO3 nuclear sequestration as one of its major functions (142–144). Taken together, these data suggest FOXO1 and FOXO3 harbour tumour suppressive qualities in MM.
FOXOs have traditionally been regarded as tumour suppressors not only due to their canonical activity being associated with detrimental cellular fate (e.g. cell cycle arrest and apoptosis) (15), but also due to deletion of FOXO1/3/4 in adult mice leading to tumour formation (14). Supporting this, a number of studies have demonstrated that FOXO function is diminished in B cell malignancies as discussed above, either by reducing FOXO expression (101, 105, 132), or by FOXO inactivation through distinct cellular environments (103, 106, 139). However, FOXO family members can also promote tumorigenesis, and this can occur in a disease- and cell lineage-dependent manner, as observed in B-cell malignancies (107, 111) and in solid tumours; high expression of FOXO3 is associated with glioblastoma progression, pancreatic ductal adenocarcinoma, and poor survival rates in breast and colorectal cancers (CRC) (145). Furthermore, FOXO mutations are associated with B-cell disease progression, particularly in DLBCL and BL (113, 114), impacting on the transcriptional activity of FOXO family members and implicating a plethora of avenues by which disease can exploit FOXO expression to promote cell survival and proliferation. In addition to the well documented roles of FOXO1 and FOXO3 in regulating B-cell malignancies, lower expression levels of FOXO4, and perhaps FOXO6, do not preclude these family members having a role in lymphomagenesis. In DLBCL, FOXO4 expression is important for maintaining colony formation and drug resistance (130). This study aligns with findings that FOXO3 activity is important for maintenance of leukaemia-initiating cells in myeloid leukemias (80–82), and highlights that individual FOXOs may perform specific functions in a lineage-dependent manner.
In the broader context, studies investigating FOXO expression as a prognostic biomarker have contrasting results. In CRC, low expression of FOXO3 is associated with cancer progression in specimens with normal tissue (146), while high FOXO1 expression is associated with good prognosis in prostate and breast cancers (147, 148). In B-cell malignancy, FOXO1 mutations are used as an indicator of FL progression (115). These particular mutations are also evident in BL and DLBCL and are associated with poorer disease outcomes (87) demonstrating a potential use of FOXO1 as biomarker in specific B-cell malignancies. However, in addition to considering the expression levels of specific FOXO family members or their mutational status, as biomarkers, it may be interesting to consider the utility of FOXO activity. While FOXO family members can be inactivated as a result of microenvironmental factors that impinge on the tumour cell, drugs targeting protein/pathways upstream of FOXO can lead to a reactivation and promotion of its tumour suppressor role (97, 106, 110, 129, 149). The resultant change in FOXO activity can be exploited for the development of novel tumour-specific FOXO-gene signatures. Indeed, repression of FOXO1/3-regulated genes in a MM patients has prognostic significance, being associated with reduced overall survival (138). Therefore gaining a deeper understanding of FOXO family regulation, in addition to providing targeting opportunities for the development of novel therapies, may reveal robust FOXO-related pharmacological/prognostic biomarkers to enhance the clinical management and survival prospects of patients.
JL, JH and MM wrote the manuscript and JL and JH prepared figures. AM reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
The research surrounding this review is funded by a project grant from LLR (Blood Cancer UK – Ref. 18003). JL is funded by the Joan Snodgrass Endowment PhD studentship awarded by the Paul O’Gorman Leukaemia Research Centre, University of Glasgow. JH is funded by a Bloodwise (BCUK) project grant (18003). MM was funded by the Hugh Forgie Friends of Paul O’Gorman PhD studentship awarded by the University of Glasgow.
The authors would like to thank all of the fundraisers who generously support the Paul O’Gorman Leukaemia Research Centre at the University of Glasgow.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Herman L, Todeschini AL, Veitia RA. Forkhead transcription factors in health and disease. Trends Genet (2021) 37(5):460–75. doi: 10.1016/j.tig.2020.11.003
2. Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet (1993) 5(3):230–5. doi: 10.1038/ng1193-230
3. Schmitt-Ney M, Camussi G. The PAX3-FOXO1 fusion protein present in rhabdomyosarcoma interferes with normal FOXO activity and the TGF-beta pathway. PloS One (2015) 10(3):e0121474. doi: 10.1371/journal.pone.0121474
4. Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, et al. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23). Oncogene (1997) 14(2):195–202. doi: 10.1038/sj.onc.1200814
5. Parry P, Wei Y, Evans G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer (1994) 11(2):79–84. doi: 10.1002/gcc.2870110203
6. Hillion J, Le Coniat M, Jonveaux P, Berger R, Bernard OA. AF6q21, a novel partner of the MLL gene in t (6,11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood (1997) 90(9):3714–9. doi: 10.1182/blood.V90.9.3714
7. Jiramongkol Y, Lam EW. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev (2020) 39(3):681–709. doi: 10.1007/s10555-020-09883-w
8. Brent MM, Anand R, Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure (2008) 16(9):1207–16. doi: 10.1016/j.str.2008.06.013
9. Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J (2000) 349(Pt 2):629–34. doi: 10.1042/bj3490629
10. Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene (2008) 27(16):2312–9. doi: 10.1038/onc.2008.24
12. Hosaka T, Biggs WH 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci USA (2004) 101(9):2975–80. doi: 10.1073/pnas.0400093101
13. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta (2011) 1813(11):1938–45. doi: 10.1016/j.bbamcr.2011.06.002
14. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell (2007) 128(2):309–23. doi: 10.1016/j.cell.2006.12.029
15. Hornsveld M, Dansen TB, Derksen PW, Burgering BMT. Re-evaluating the role of FOXOs in cancer. Semin Cancer Biol (2018) 50:90–100. doi: 10.1016/j.semcancer.2017.11.017
16. Ma MCJ, Tadros S, Bouska A, Heavican TB, Yang H, Deng Q, et al. Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica (2022) 107(3):690–701. doi: 10.3324/haematol.2020.274258
17. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse Large B cell lymphoma. Cell (2017) 171(2):481–94 e15. doi: 10.1016/j.cell.2017.09.027
18. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature (2015) 526(7574):525–30. doi: 10.1038/nature15395
19. Ten Hacken E, Gounari M, Ghia P, Burger JA. The importance of B cell receptor isotypes and stereotypes in chronic lymphocytic leukemia. Leukemia (2019) 33(2):287–98. doi: 10.1038/s41375-018-0303-x
20. Slupsky JR. Does B cell receptor signaling in chronic lymphocytic leukaemia cells differ from that in other B cell types? Scientifica (Cairo) (2014) 2014:208928. doi: 10.1155/2014/208928
21. Tanaka S, Baba Y. B cell receptor signaling. Adv Exp Med Biol (2020) 1254:23–36. doi: 10.1007/978-981-15-3532-1_2
22. van der Heide LP, Jacobs FM, Burbach JP, Hoekman MF, Smidt MP. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem J (2005) 391(Pt 3):623–9. doi: 10.1042/BJ20050525
23. Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, et al. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem (2002) 277(47):45276–84. doi: 10.1074/jbc.M208063200
24. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell (1999) 96(6):857–68. doi: 10.1016/S0092-8674(00)80595-4
25. Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, et al. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell (1997) 91(7):961–71. doi: 10.1016/S0092-8674(00)80487-0
26. Silhan J, Vacha P, Strnadova P, Vecer J, Herman P, Sulc M, et al. 14-3-3 protein masks the DNA binding interface of forkhead transcription factor FOXO4. J Biol Chem (2009) 284(29):19349–60. doi: 10.1074/jbc.M109.002725
27. Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, et al. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol (2002) 156(5):817–28. doi: 10.1083/jcb.200112059
28. Obsilova V, Vecer J, Herman P, Pabianova A, Sulc M, Teisinger J, et al. 14-3-3 protein interacts with nuclear localization sequence of forkhead transcription factor FoxO4. Biochemistry (2005) 44(34):11608–17. doi: 10.1021/bi050618r
29. Yadav RK, Chauhan AS, Zhuang L, Gan B. FoxO transcription factors in cancer metabolism. Semin Cancer Biol (2018) 50:65–76. doi: 10.1016/j.semcancer.2018.01.004
30. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
31. Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell (2007) 130(3):440–55. doi: 10.1016/j.cell.2007.05.058
32. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol (2008) 10(2):138–48. doi: 10.1038/ncb1676
33. Yang JY, Chang CJ, Xia W, Wang Y, Wong KK, Engelman JA, et al. Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res (2010) 70(11):4709–18. doi: 10.1158/0008-5472.CAN-09-4524
34. Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science (2006) 314(5797):294–7. doi: 10.1126/science.1130512
35. Rena G, Woods YL, Prescott AR, Peggie M, Unterman TG, Williams MR, et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J (2002) 21(9):2263–71. doi: 10.1093/emboj/21.9.2263
36. Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep (2004) 5(1):60–5. doi: 10.1038/sj.embor.7400048
37. Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase ral and JNK. EMBO J (2004) 23(24):4802–12. doi: 10.1038/sj.emboj.7600476
38. Yun H, Park S, Kim MJ, Yang WK, Im DU, Yang KR, et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J (2014) 281(19):4421–38. doi: 10.1111/febs.12949
39. Orea-Soufi A, Paik J, Braganca J, Donlon TA, Willcox BJ, Link W. FOXO transcription factors as therapeutic targets in human diseases. Trends Pharmacol Sci (2022) 43(12):1070–84. doi: 10.1016/j.tips.2022.09.010
40. Brown AK, Webb AE. Regulation of FOXO factors in mammalian cells. Curr Top Dev Biol (2018) 127:165–92. doi: 10.1016/bs.ctdb.2017.10.006
41. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science (2004) 303(5666):2011–5. doi: 10.1126/science.1094637
42. Calnan DR, Webb AE, White JL, Stowe TR, Goswami T, Shi X, et al. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging (Albany NY) (2012) 4(7):462–79. doi: 10.18632/aging.100471
43. Fukuoka M, Daitoku H, Hatta M, Matsuzaki H, Umemura S, Fukamizu A. Negative regulation of forkhead transcription factor AFX (Foxo4) by CBP-induced acetylation. Int J Mol Med (2003) 12(4):503–8. doi: 10.3892/ijmm.12.4.503
44. Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell (2004) 116(4):551–63. doi: 10.1016/S0092-8674(04)00126-6
45. van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol (2006) 8(10):1064–73. doi: 10.1038/ncb1469
46. Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem (2009) 284(35):23204–16. doi: 10.1074/jbc.M109.031427
47. Welinder E, Mansson R, Mercer EM, Bryder D, Sigvardsson M, Murre C. The transcription factors E2A and HEB act in concert to induce the expression of FOXO1 in the common lymphoid progenitor. Proc Natl Acad Sci USA (2011) 108(42):17402–7. doi: 10.1073/pnas.1111766108
48. Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol (2010) 11(7):635–43. doi: 10.1038/ni.1891
49. Ushmorov A, Wirth T. FOXO in B-cell lymphopoiesis and B cell neoplasia. Semin Cancer Biol (2018) 50:132–41. doi: 10.1016/j.semcancer.2017.07.008
50. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131(4):959–71. doi: 10.1016/j.jaci.2013.01.046
51. Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol (2008) 9(6):613–22. doi: 10.1038/ni.1612
52. Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, et al. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol (2008) 9(12):1388–98. doi: 10.1038/ni.1667
53. Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol (2012) 13(3):300–7. doi: 10.1038/ni.2210
54. Cooper AB, Sawai CM, Sicinska E, Powers SE, Sicinski P, Clark MR, et al. A unique function for cyclin D3 in early B cell development. Nat Immunol (2006) 7(5):489–97. doi: 10.1038/ni1324
55. Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol (2011) 23(2):178–83. doi: 10.1016/j.coi.2011.01.001
56. Nahar R, Ramezani-Rad P, Mossner M, Duy C, Cerchietti L, Geng H, et al. Pre-B cell receptor-mediated activation of BCL6 induces pre-B cell quiescence through transcriptional repression of MYC. Blood (2011) 118(15):4174–8. doi: 10.1182/blood-2011-01-331181
57. Duy C, Yu JJ, Nahar R, Swaminathan S, Kweon SM, Polo JM, et al. BCL6 is critical for the development of a diverse primary B cell repertoire. J Exp Med (2010) 207(6):1209–21. doi: 10.1084/jem.20091299
58. Hinman RM, Nichols WA, Diaz TM, Gallardo TD, Castrillon DH, Satterthwaite AB. Foxo3-/- mice demonstrate reduced numbers of pre-b and recirculating B cells but normal splenic B cell sub-population distribution. Int Immunol (2009) 21(7):831–42. doi: 10.1093/intimm/dxp049
59. Pena-Perez L, Kharazi S, Frengen N, Krstic A, Bouderlique T, Hauenstein J, et al. FOXO dictates initiation of B cell development and myeloid restriction in common lymphoid progenitors. Front Immunol (2022) 13:880668. doi: 10.3389/fimmu.2022.880668
60. Hinman RM, Bushanam JN, Nichols WA, Satterthwaite AB. B cell receptor signaling down-regulates forkhead box transcription factor class O 1 mRNA expression via phosphatidylinositol 3-kinase and bruton's tyrosine kinase. J Immunol (2007) 178(2):740–7. doi: 10.4049/jimmunol.178.2.740
61. Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood (2004) 104(3):784–7. doi: 10.1182/blood-2003-09-3071
62. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell (2009) 139:573–86. doi: 10.1016/j.cell.2009.08.041
63. Vandenberg CJ, Motoyama N, Cory S. FoxO3 suppresses myc-driven lymphomagenesis. Cell Death Dis (2016) 6(1):e2046. doi: 10.1038/cddis.2015.396
64. Vicora GD, Nussenzweig MC. Germinal centers. Ann Rev Immunol (2022) 40:413–42. doi: 10.1146/annurev-immunol-120419-022408
65. Crotty S. T Follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
66. Ramezani-Rad P, Chen C, Zhu Z, Rickert RC. Cyclin D3 governs clonal expansion of dark zone germinal center B cells. Cell Rep (2020) 33(7):108403. doi: 10.1016/j.celrep.2020.108403
67. Pae J, Ersching J, Castro TBR, Schips M, Mesin L, Allon SJ, et al. Cyclin D3 drives inertial cell cycling in dark zone germinal center B cells. J Exp Med (2021) 218(4):e20201699. doi: 10.1084/jem.20201699
68. Muramatsu M, Kinoshita K, Fagarasan S, Tamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102(5):553–63. doi: 10.1016/S0092-8674(00)00078-7
69. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell (2000) 102(5):565–75. doi: 10.1016/S0092-8674(00)00079-9
70. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete P, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity (2019) 51(2):337–50.e7. doi: 10.1016/j.immuni.2019.07.001
71. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic re-entry. Nat Immunol (2012) 13(11):1083–91. doi: 10.1038/ni.2428
72. Tunyaplin C, Shaffer AL, Andelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by bcl-6 inhibits plasmacytic differentiation. J Immunol (2014) 173(2):1158–65. doi: 10.4049/jimmunol.173.2.1158
73. Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, et al. The FOXO1 transcription factor instructs the germinal center dark zone program. Immunity (2015) 43(6):1064–74. doi: 10.1016/j.immuni.2015.10.015
74. Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, et al. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity (2015) 43:1075–86. doi: 10.1016/j.immuni.2015.10.021
75. Zhu Z, Shukla A, Ramezani-Rad P, Apgar JR, Rickert RC. The AKT isoforms 1 and 2 drive B cell fate decisions during the germinal center response. Life Sci Alliance (2019) 2(6):e201900506. doi: 10.26508/lsa.201900506
76. Inoue T, Shinnakasu R, Ise W, Kawai C, Egawa T, Kurosaki T. The transcription factor Foxo1 controls germinal center B cell proliferation in response to T cell help. J Exp Med (2017) 214(4):1181–98. doi: 10.1084/jem.20161263
77. Luo W, Weisel F, Shlomchik MJ. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-myc transcription factor in germinal center B cells. Immunity (2018) 48(2):313–26 e5. doi: 10.1016/j.immuni.2018.01.008
78. Qi H, Tian D, Li M, Zhang C, Jin H, Liu L, et al. Foxo3 promotes the differentiation and function of follicular helper T cells. Cell Rep (2020) 31(6):107621. doi: 10.1016/j.celrep.2020.107621
79. Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell (2007) 128(2):325–39. doi: 10.1016/j.cell.2007.01.003
80. Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature (2010) 463(7281):676–80. doi: 10.1038/nature08734
81. Pellicano F, Scott MT, Helgason GV, Hopcroft LE, Allan EK, Aspinall-O'Dea M, et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells (2014) 32(9):2324–37. doi: 10.1002/stem.1748
82. Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell (2011) 146(5):697–708. doi: 10.1016/j.cell.2011.07.032
83. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23(6):703–13. doi: 10.1038/nm.4333
84. Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol (2017) 1):1–16. doi: 10.1200/PO.17.00011
85. Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discovery (2018) 8(2):174–83. doi: 10.1158/2159-8290.CD-17-0321
86. Roberto MP, Varano G, Vinas-Castells R, Holmes AB, Kumar R, Pasqualucci L, et al. Mutations in the transcription factor FOXO1 mimic positive selection signals to promote germinal center B cell expansion and lymphomagenesis. Immunity (2021) 54(8):1807–24 e14. doi: 10.1016/j.immuni.2021.07.009
87. Sablon A, Bollaert E, Pirson C, Velghe AI, Demoulin JB. FOXO1 forkhead domain mutants in B-cell lymphoma lack transcriptional activity. Sci Rep (2022) 12(1):1309. doi: 10.1038/s41598-022-05334-4
88. Yuan C, Wang L, Zhou L, Fu Z. The function of FOXO1 in the late phases of the cell cycle is suppressed by PLK1-mediated phosphorylation. Cell Cycle (2014) 13(5):807–19. doi: 10.4161/cc.27727
89. Pan C, Olsen JV, Daub H, Mann M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol Cell Proteomics (2009) 8(12):2796–808. doi: 10.1074/mcp.M900285-MCP200
90. Thelander EF, Ichimura K, Corcoran M, Barbany G, Nordgren A, Heyman M, et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk Lymphoma (2008) 49(3):477–87. doi: 10.1080/10428190701817282
91. Offit K, Parsa NZ, Gaidano G, Filippa DA, Louie D, Pan D, et al. 6q deletions define distinct clinico-pathologic subsets of non-hodgkin's lymphoma. Blood (1993) 82(7):2157–62. doi: 10.1182/blood.V82.7.2157.2157
92. Katzenberger T, Kienle D, Stilgenbauer S, Holler S, Schilling C, Mader U, et al. Delineation of distinct tumour profiles in mantle cell lymphoma by detailed cytogenetic, interphase genetic and morphological analysis. Br J Haematol (2008) 142(4):538–50. doi: 10.1111/j.1365-2141.2008.07199.x
93. Eswaran J, Sinclair P, Heidenreich O, Irving J, Russell LJ, Hall A, et al. The pre-B-cell receptor checkpoint in acute lymphoblastic leukaemia. Leukemia (2015) 29(8):1623–31. doi: 10.1038/leu.2015.113
94. Buchner M, Swaminathan S, Chen Z, Muschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol Rev (2015) 263(1):192–209. doi: 10.1111/imr.12235
95. Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol (2015) 12(6):344–57. doi: 10.1038/nrclinonc.2015.38
96. Safavi S, Hansson M, Karlsson K, Biloglav A, Johansson B, Paulsson K. Novel gene targets detected by genomic profiling in a consecutive series of 126 adults with acute lymphoblastic leukemia. Haematologica (2015) 100(1):55–61. doi: 10.3324/haematol.2014.112912
97. Kohrer S, Havranek O, Seyfried F, Hurtz C, Coffey GP, Kim E, et al. Pre-BCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia (2016) 30(6):1246–54. doi: 10.1038/leu.2016.9
98. Bhansali RS, Rammohan M, Lee P, Laurent AP, Wen Q, Suraneni P, et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of FOXO1 and STAT3. J Clin Invest (2021) 131(1):e135937. doi: 10.1172/JCI135937
99. Wang F, Demir S, Gehringer F, Osswald CD, Seyfried F, Enzenmuller S, et al. Tight regulation of FOXO1 is essential for maintenance of B-cell precursor acute lymphoblastic leukemia. Blood (2018) 131(26):2929–42. doi: 10.1182/blood-2017-10-813576
100. Ketzer F, Abdelrasoul H, Vogel M, Marienfeld R, Muschen M, Jumaa H, et al. CCND3 is indispensable for the maintenance of B-cell acute lymphoblastic leukemia. Oncogenesis (2022) 11(1):1. doi: 10.1038/s41389-021-00377-0
101. Chen YJ, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, et al. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem (2009) 284(44):30695–707. doi: 10.1074/jbc.M109.007997
102. Buchner M, Park E, Geng H, Klemm L, Flach J, Passegue E, et al. Identification of FOXM1 as a therapeutic target in B-cell lineage acute lymphoblastic leukaemia. Nat Commun (2015) 6:6471. doi: 10.1038/ncomms7471
103. Ticchioni M, Essafi M, Jeandel PY, Davi F, Cassuto JP, Deckert M, et al. Homeostatic chemokines increase survival of B-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene (2007) 26(50):7081–91. doi: 10.1038/sj.onc.1210519
104. Ramsay AJ, Martinez-Trillos A, Jares P, Rodriguez D, Kwarciak A, Quesada V. Next-generation sequencing reveals the secrets of the chronic lymphocytic leukemia genome. Clin Transl Oncol (2013) 15(1):3–8. doi: 10.1007/s12094-012-0922-z
105. Palacios F, Abreu C, Prieto D, Morande P, Ruiz S, Fernandez-Calero T, et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia (2015) 29(1):115–25. doi: 10.1038/leu.2014.158
106. Cosimo E, Tarafdar A, Moles MW, Holroyd AK, Malik N, Catherwood MA, et al. AKT/mTORC2 inhibition activates FOXO1 function in CLL cells reducing B-cell receptor-mediated survival. Clin Cancer Res (2019) 25(5):1574–87. doi: 10.1158/1078-0432.CCR-18-2036
107. Scheffold A, Jebaraj BMC, Tausch E, Bloehdorn J, Ghia P, Yahiaoui A, et al. IGF1R as druggable target mediating PI3K-delta inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood (2019) 134(6):534–47. doi: 10.1182/blood.2018881029
108. Seda V, Vojackova E, Ondrisova L, Kostalova L, Sharma S, Loja T, et al. FoxO1-GAB1 axis regulates homing capacity and tonic AKT activity in chronic lymphocytic leukemia. Blood (2021) 138(9):758–72. doi: 10.1182/blood.2020008101
109. Ecker V, Stumpf M, Branmeier L, Neumayer T, Pfeuffer L, Engleitner T, et al. Targeted PI3K/AKT-hyperactivation induces cell death in chronic lymphocytic leukemia. Nat Commun (2021) 12(1):3526. doi: 10.1038/s41467-021-23752-2
110. Gupta M, Hendrickson AW, Han JJ, Stenson M, Wellik L, Barr S, et al. Dual inhibition of mTORC1/mTORC2 induces apoptosis of mantle cell lymphoma by preventing rictor mediated AKTS473 phosphorylation by potentiating AKT2-PHLPP1 association. Blood (2010) 116(21):772. doi: 10.1182/blood.V116.21.772.772
111. Jang JY, Hwang I, Pan H, Yao J, Alinari L, Imada E, et al. A FOXO1-dependent transcription network is a targetable vulnerability of mantle cell lymphomas. J Clin Invest (2022) 132(24):e160767. doi: 10.1172/JCI160767
112. Devan J, Janikova A, Mraz M. New concepts in follicular lymphoma biology: from BCL2 to epigenetic regulators and non-coding RNAs. Semin Oncol (2018) 45(5-6):291–302. doi: 10.1053/j.seminoncol.2018.07.005
113. Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJ, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood (2013) 121(18):3666–74. doi: 10.1182/blood-2013-01-479865
114. Kabrani E, Chu VT, Tasouri E, Sommermann T, Bassler K, Ulas T, et al. Nuclear FOXO1 promotes lymphomagenesis in germinal center B cells. Blood (2018) 132(25):2670–83. doi: 10.1182/blood-2018-06-856203
115. Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol (2015) 16(9):1111–22. doi: 10.1016/S1470-2045(15)00169-2
116. Bodo J, Zhao X, Durkin L, Souers AJ, Phillips DC, Smith MR, et al. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget (2016) 7(43):70000–10. doi: 10.18632/oncotarget.12132
117. Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature (2012) 490(7418):116–20. doi: 10.1038/nature11378
118. Bouchard C, Lee S, Paulus-Hock V, Loddenkemper C, Eilers M, Schmitt CA. FoxO transcription factors suppress myc-driven lymphomagenesis via direct activation of arf. Genes Dev (2007) 21(21):2775–87. doi: 10.1101/gad.453107
119. Zhou P, Blain AE, Newman AM, Zaka M, Chagaluka G, Adlar FR, et al. Sporadic and endemic Burkitt lymphoma have frequent FOXO1 mutations but distinct hotspots in the AKT recognition motif. Blood Adv (2019) 3(14):2118–27. doi: 10.1182/bloodadvances.2018029546
120. Saline M, Badertscher L, Wolter M, Lau R, Gunnarsson A, Jacso T, et al. AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14-3-3 proteins. J Biol Chem (2019) 294(35):13106–16. doi: 10.1074/jbc.RA119.008649
121. Gehringer F, Weissinger SE, Swier LJ, Moller P, Wirth T, Ushmorov A. FOXO1 confers maintenance of the dark zone proliferation and survival program and can be pharmacologically targeted in Burkitt lymphoma. Cancers (Basel) (2019) 11(10):1427. doi: 10.3390/cancers11101427
122. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med (2002) 346(25):1937–47. doi: 10.1056/NEJMoa012914
123. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24(5):679–90. doi: 10.1038/s41591-018-0016-8
124. Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, et al. Genetic landscapes of relapsed and refractory diffuse Large B-cell lymphomas. Clin Cancer Res (2016) 22(9):2290–300. doi: 10.1158/1078-0432.CCR-15-2123
125. Pasqualucci L, Dalla-Favera R. Genetics of diffuse large B-cell lymphoma. Blood (2018) 131(21):2307–19. doi: 10.1182/blood-2017-11-764332
126. Pyrzynska B, Dwojak M, Zerrouqi A, Morlino G, Zapala P, Miazek N, et al. FOXO1 promotes resistance of non-Hodgkin lymphomas to anti-CD20-based therapy. Oncoimmunology (2018) 7(5):e1423183. doi: 10.1080/2162402X.2017.1423183
127. Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest (2001) 108(9):1359–67. doi: 10.1172/JCI200112876
128. Nakae J, Kitamura T, Kitamura Y, Biggs WH 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell (2003) 4(1):119–29. doi: 10.1016/S1534-5807(02)00401-X
129. Szydlowski M, Kiliszek P, Sewastianik T, Jablonska E, Bialopiotrowicz E, Gorniak P, et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B-cell lymphomas. Blood (2016) 127(6):739–48. doi: 10.1182/blood-2015-06-654111
130. Ryu KJ, Park C, Hong M, Ko YH, Kim WS, Kim SJ. FOXO4 expression is related to stem cell-like properties and resistance to treatment in diffuse large B-cell lymphoma. Oncotarget (2017) 8(2):2466–76. doi: 10.18632/oncotarget.13690
131. Kapoor I, Li Y, Sharma A, Zhu H, Bodo J, Xu W, et al. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis (2019) 10(12):924. doi: 10.1038/s41419-019-2158-0
132. Zheng X, Rui H, Liu Y, Dong J. Proliferation and apoptosis of B-cell lymphoma cells under targeted regulation of FOXO3 by miR-155. Mediterr J Hematol Infect Dis (2020) 12(1):e2020073. doi: 10.4084/mjhid.2020.073
133. Xie L, Ushmorov A, Leithauser F, Guan H, Steidl C, Farbinger J, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood (2012) 119(15):3503–11. doi: 10.1182/blood-2011-09-381905
134. Osswald CD, Xie L, Guan H, Herrmann F, Pick SM, Vogel MJ, et al. Fine-tuning of FOXO3A in cHL as a survival mechanism and a hallmark of abortive plasma cell differentiation. Blood (2018) 131(14):1556–67. doi: 10.1182/blood-2017-07-795278
135. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer (2007) 7(8):585–98. doi: 10.1038/nrc2189
136. Zhou Y, Uddin S, Zimmerman T, Kang JA, Ulaszek J, Wickrema A. Growth control of multiple myeloma cells through inhibition of glycogen synthase kinase-3. Leuk Lymphoma (2008) 49(10):1945–53. doi: 10.1080/10428190802304966
137. Rizk M, Rizq O, Oshima M, Nakajima-Takagi Y, Koide S, Saraya A, et al. Akt inhibition synergizes with polycomb repressive complex 2 inhibition in the treatment of multiple myeloma. Cancer Sci (2019) 110(12):3695–707. doi: 10.1111/cas.14207
138. Bloedjes TA, de Wilde G, Maas C, Eldering E, Bende RJ, van Noesel CJM, et al. AKT signaling restrains tumor suppressive functions of FOXO transcription factors and GSK3 kinase in multiple myeloma. Blood Adv (2020) 4(17):4151–64. doi: 10.1182/bloodadvances.2019001393
139. De Bruyne E, Bos TJ, Schuit F, Van Valckenborgh E, Menu E, Thorrez L, et al. IGF-1 suppresses bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood (2010) 115(12):2430–40. doi: 10.1182/blood-2009-07-232801
140. Munugalavadla V, Mariathasan S, Slaga D, Du C, Berry L, Del Rosario G, et al. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene (2014) 33(3):316–25. doi: 10.1038/onc.2012.594
141. Jagani Z, Song K, Kutok JL, Dewar MR, Melet A, Santos T, et al. Proteasome inhibition causes regression of leukemia and abrogates BCR-ABL-induced evasion of apoptosis in part through regulation of forkhead tumor suppressors. Cancer Res (2009) 69(16):6546–55. doi: 10.1158/0008-5472.CAN-09-0605
142. Kalakonda N, Maerevoet M, Cavallo F, Follows G, Goy A, Vermaat JSP, et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): a single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol (2020) 7(7):e511–e22. doi: 10.1016/S2352-3026(20)30120-4
143. Corno C, Stucchi S, De Cesare M, Carenini N, Stamatakos S, Ciusani E, et al. FoxO-1 contributes to the efficacy of the combination of the XPO1 inhibitor selinexor and cisplatin in ovarian carcinoma preclinical models. Biochem Pharmacol (2018) 147:93–103. doi: 10.1016/j.bcp.2017.11.009
144. Wang J, Sun T, Meng Z, Wang L, Li M, Chen J, et al. XPO1 inhibition synergizes with PARP1 inhibition in small cell lung cancer by targeting nuclear transport of FOXO3a. Cancer Lett (2021) 503:197–212. doi: 10.1016/j.canlet.2021.01.008
145. Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, et al. Beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med (2012) 18(6):892–901. doi: 10.1038/nm.2772
146. Bullock MD, Bruce A, Sreekumar R, Curtis N, Cheung T, Reading I, et al. FOXO3 expression during colorectal cancer progression: biomarker potential reflects a tumour suppressor role. Br J Cancer (2013) 109(2):387–94. doi: 10.1038/bjc.2013.355
147. Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED, et al. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res (2011) 71(9):3257–67. doi: 10.1158/0008-5472.CAN-10-2603
148. Wu Y, Elshimali Y, Sarkissyan M, Mohamed H, Clayton S, Vadgama JV. Expression of FOXO1 is associated with GATA3 and annexin-1 and predicts disease-free survival in breast cancer. Am J Cancer Res (2012) 2(1):104–15. doi: 10.1158/1538-7445.AM2012-704
Keywords: FOXO transcription factor, B cell malignancy, tumor suppressor, BCR signalling, PI3K/AKT, leukemia, lymphoma
Citation: Lees J, Hay J, Moles MW and Michie AM (2023) The discrete roles of individual FOXO transcription factor family members in B-cell malignancies. Front. Immunol. 14:1179101. doi: 10.3389/fimmu.2023.1179101
Received: 03 March 2023; Accepted: 05 May 2023;
Published: 18 May 2023.
Edited by:
Dessi Malinova, Queen’s University Belfast, United KingdomReviewed by:
Parham Ramezani-Rad, La Jolla Institute for Immunology (LJI), United StatesCopyright © 2023 Lees, Hay, Moles and Michie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alison M. Michie, QWxpc29uLk1pY2hpZUBnbGFzZ293LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.