 Hsin-Fang Chang
Hsin-Fang Chang Claudia Schirra
Claudia Schirra Elmar Krause
Elmar Krause Ute Becherer
Ute Becherer- Department of Cellular Neurophysiology, Center for Integrative Physiology and Molecular Medicine (CIPMM), Saarland University, Homburg, Germany
Regulated exocytosis is a central mechanism of cellular communication. It is not only the basis for neurotransmission and hormone release, but also plays an important role in the immune system for the release of cytokines and cytotoxic molecules. In cytotoxic T lymphocytes (CTLs), the formation of the immunological synapse is required for the delivery of the cytotoxic substances such as granzymes and perforin, which are stored in lytic granules and released via exocytosis. The molecular mechanisms of their fusion with the plasma membrane are only partially understood. In this review, we discuss the molecular players involved in the regulated exocytosis of CTL, highlighting the parallels and differences to neuronal synaptic transmission. Additionally, we examine the strengths and weaknesses of both systems to study exocytosis.
1 Introduction
CD8+ cytotoxic T lymphocytes (CTLs) eliminate virus-infected or cancerous cells by releasing pore-forming perforin and proteases such as granzymes that induce apoptosis of target cells. These toxic substances are stored in lytic granules (LG), which undergo exocytosis upon T cell antigen receptor (TCR) signaling, releasing cytotoxic molecules at the contact zone with the target cells. This contact zone, where TCR-mediated signal transduction and secretory events take place, is named the immunological synapse (IS) (1). Exocytosis of LGs is a highly regulated process ensuring that the cytotoxins are delivered only to the target cell. LG exocytosis requires tethering, docking/priming and finally fusion with the plasma membrane. After exocytosis, the membrane of the granule is retrieved and recycled (Figure 1A). These steps are tightly controlled by a complex molecular machinery.
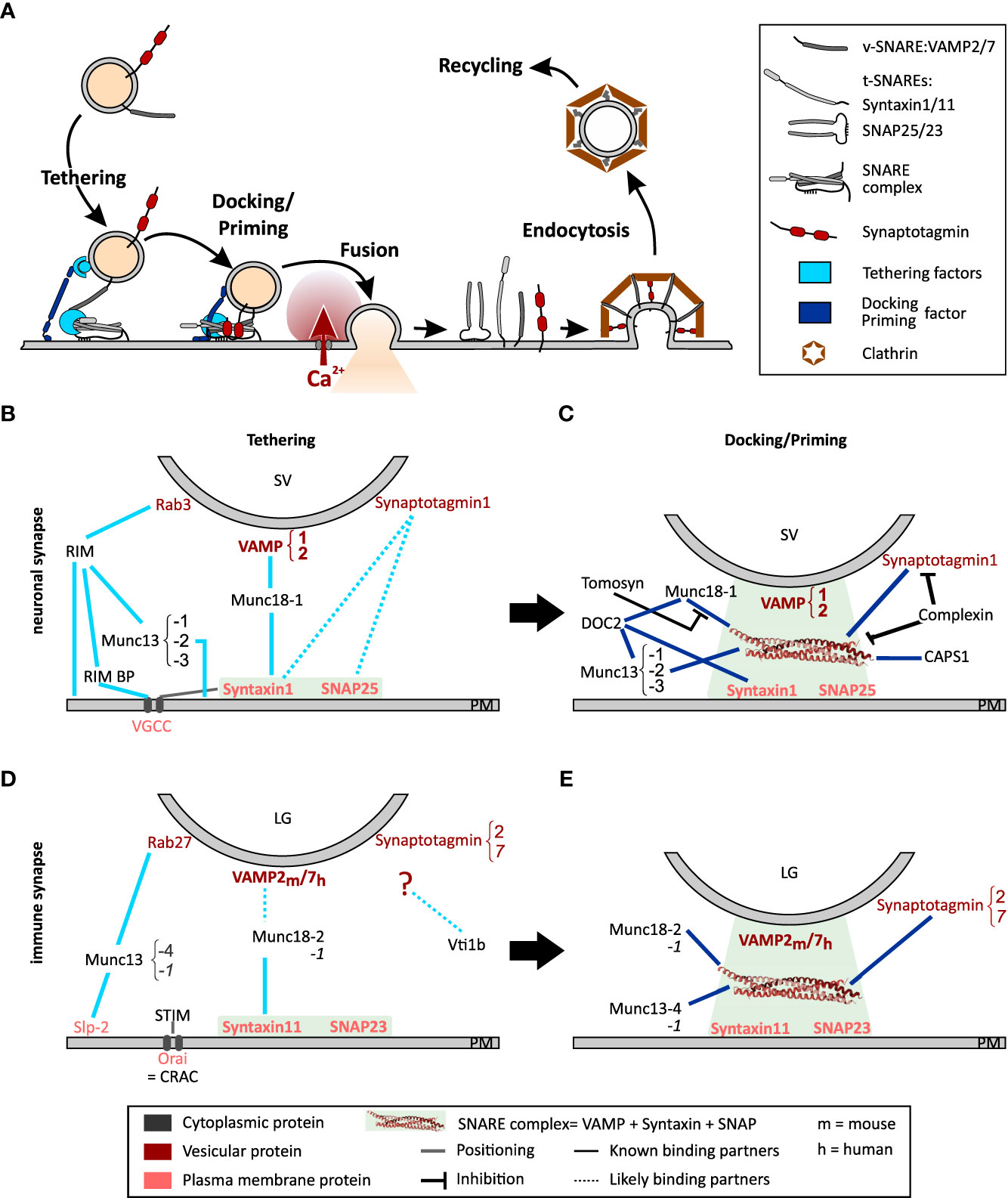
Figure 1 Model of the exocytosis machinery. (A) Schematic representation of the steps that SVs and LGs undergo before and after fusion with the plasma membrane. For sake of clarity only selected proteins involved in this process are shown. More comprehensive protein-protein interaction networks are shown in (B–E). (B, C) Protein Interactions occurring during tethering (B) and docking/priming (C) of SVs in neurons. (D, E) Protein interaction required for tethering (D) and docking/priming (E) of LGs in CTL. Bottom legends applies to panels (B-E) The light green squares indicate the strong interaction of the t-SNAREs during tethering and of the entire SNARE complex during docking/priming. Lines indicate protein interactions, stippled lines show probable protein interactions. Black lines with blunt arrow correspond to inhibitory interactions.
Interestingly, very similar mechanisms govern neuronal synaptic transmission. The release of neurotransmitters by neurons is by far the most highly regulated form of exocytosis known. The speed, accuracy and temporal resolution found at synapses are unmatched. Due to their complexity and importance, the mechanisms involved have been the target of intense study. A variety of the molecules discovered at neuronal synapses have recently been found to have similar roles in LG exocytosis. Furthermore, a number of immunological deficits have been tied to mutations of proteins involved in neuronal synaptic transmission. The mechanisms underlying LG exocytosis in CTLs are not well characterized and their understanding has benefited greatly from the knowledge of synaptic transmission. The aim of this review is to describe the molecular process of neuronal synaptic vesicle release in neurons and compare it with that of LG exocytosis in CTLs. We highlight the similarities and differences between the two systems and identify gaps in our current understanding of these key cellular processes.
2 Regulated exocytosis in neurons
The core molecular machinery for membrane fusion is formed by the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins. They were discovered by R. Schekman in yeast (2) and J. Rothman showed that these proteins also exist in mammals where they perform similar tasks (3–5). Subsequently, their function was elucidated in neurons, by studying the effects of proteolytic clostridial neurotoxins, i.e. tetanus- and botulinum-toxins. These toxins specifically cleave SNARE proteins leading to a complete arrest of neurotransmitter release (6, 7). The resulting work demonstrated that the SNARE complex is a fusion machine, which provides the force that drives the fusion of synaptic vesicles (SV) with the pre-synaptic target membrane. The SNARE complex consists of a vesicular SNARE (v-SNARE), synaptobrevin, and two target SNAREs (t-SNAREs), syntaxin-1 and synaptosomal-associated protein 25 (SNAP-25), located on the plasma membrane. Synaptobrevin and syntaxin-1 contain one SNARE motif, while SNAP25 contains two. These coiled-coil motifs assemble into tight four-helix bundles called “trans”-SNARE complexes, which attach the SV to the presynaptic membrane. Induction of SV fusion with the plasma membrane requires the assistance of many other proteins in a well-coordinated fashion, as described thereafter.
2.1 The SV fate: from the reserve pool to fusion
The molecular mechanism of synaptic vesicle exocytosis is highly complex and is described in detail in excellent reviews (8–11). Therefore, it will not be repeated in this review. Here, we present an outline of the molecular events leading to SV fusion.
SV are maintained in a reserve pool by the mesh-forming synapsin (12). Under activation by Ca2+-calmodulin, this mesh dissolves releasing SVs that move toward the active zone at the pre-synapse along F-actin using myosin II or V as the motor protein (13–15). An overload of SV at the active zone is prevented by a dense cortical F-actin meshwork that functions as a semipermeable barrier, which dynamically regulates plasma membrane accessibility (16–18). This role of F-actin has been studied in great detail in neuroendocrine cells (see Meunier and Gutierrez (19) for review). Upon arrival at the active zone, SVs loosely attach to the plasma membrane by means of a tethering mechanism consisting of multilayered protein interactions (Figures 1A, B). The first tether consists of the active zone proteins RIM, RIM-binding protein, and Munc13-1 or Munc13-2, which attach to the SV via Rab3 (20–22). A second tether is generated by Munc18-1, which bridges the t-SNARE, syntaxin-1, and the v-SNARE, synaptobrevin2 (23, 24). Finally, it was proposed that synaptotagmin1 interacts directly with syntaxin-1 and SNAP25 thereby pulling the SV closer to the plasma membrane. However, while this last tether has been reported to attach secretory vesicles to the plasma membrane in neuroendocrine cells (25), its relevance at neuronal synapses is still under debate (26). SV proximity to voltage dependent Ca2+ channels (VGCC) is promoted by the t-SNAREs and Munc18-1 (27–29).
At this stage the SVs can be docked, i.e. primed. Nowadays, both terms describe the same step but they have been defined by different techniques. While docked SVs are defined by their direct contact with the plasma membrane in electron micrographs, priming of a vesicle corresponds to the ability of the SV to fuse with the plasma membrane as measured by functional assays (see 2.3). As the resolution of electron microscopy has improved, the analysis of electron micrographs has become more precise, and it is now accepted that docking equates to priming, which is why we refer to this process as docking/priming (30). For docking/priming, the v-SNARE synaptobrevin2 interacts with the t-SNAREs, SNAP25 and syntaxin-1 in a Munc18-1 and Munc13-1 or -2 dependent manner (Figures 1A, C). Indeed, while Munc18-1 forms a template for the SNARE complex, Munc13-1 or -2 is required to open syntaxin-1 allowing its interaction with SNAP25 and synaptobrevin2 (31–33). This corresponds to a loose docking/priming step in which the SNARE complex is only partially formed. Now synaptotagmin 1 that is partially bound to syntaxin-1, attaches to the entire SNARE complex, which is stabilized by complexin (34, 35) (Figures 1A, C). Finally, a steep increase of the presynaptic intracellular Ca2+ concentration, due to the opening of the VGCCs, induces the complete zippering of the SNARE complex and the dipping of the C2A domain of synaptotagmin 1 in the plasma membrane (36–38). Both actions pull the vesicle membrane close enough to the plasma membrane to induce fusion. Immediately after fusion the SNARE complex is disassembled by N-ethylmaleimide-sensitive factor (NSF) through its ATPase activity, with the help of SNAPs to allow v-SNARE recycling (39–41).
One should notice that SV docking/priming is assisted by several additional priming factors (Figure 1C). The Ca2+-dependent activator protein for secretion-1 (CAPS1) promotes priming via interaction with syntaxin-1 probably downstream of Munc13-1 or -2 (42, 43). Similarly, DOC2A and DOC2B interact with syntaxin-1, Munc18-1 and Munc13-1 or -2 to promote exocytosis in a phorbol ester- and Ca2+-dependent manner (reviewed by Pinheiro et al. (44)). Finally, snapin binds to synaptotagmin enhancing its interaction with the SNARE complex, thereby stabilizing SV priming and enhancing exocytosis at low intracellular Ca2+ concentration (45–47). Few proteins inhibit docking/priming. The most prominent is probably tomosyn (STXBP5) that competes with Munc18-1 for its interaction with syntaxin-1 (48) and additionally binds to SNAP25 thereby forming a dead-end tomosyn-SNARE complex (49, 50).
2.2 Vesicle pools and recycling
In order to allow for a sustained high rate of neurotransmitter release, each of the steps leading to SV fusion is carried out simultaneously by numerous synaptic vesicles. These form individual pools of tethered vesicles and/or docked/primed vesicles ready for exocytosis. This ensures that docked/primed vesicles are released within 1 ms after depolarization of the pre-synapse. However, since primed vesicles are fully release-ready, they must be prevented from fusing. This task is performed by complexin, which not only stabilizes the fusion machinery but also has a regulatory function (34, 35). Furthermore, the tethered and the reserve pools allow a steady replenishment of readily releasable vesicles (51–53). A tight coupling between exo- and endocytosis ensures maintenance of these pools to sustain neurotransmitter release, and homeostasis of membrane composition (54–56). In so-called kiss-and-run exocytosis, the fusion of SVs with the plasma membrane is transient and the membrane of the vesicle does not mix with the plasma membrane (57, 58). On the contrary, in the full fusion mode, the SV membrane completely integrates in the plasma membrane. An ATP dependent dynamic assembly of filamentous actin, involving N-WASP and formin, appears to be required for this type of fusion (59). The membrane components of the SVs are recycled via classical clathrin-mediated endocytosis, fast endocytosis or bulk endocytosis. The mode of endocytosis depends on the strength of the stimulus, i.e. the pre-synaptic Ca2+ concentration, and the amount and the speed of SV fusion (60, 61). Finally, SV filling with neurotransmitter occurs on site via specific transmembrane transporters.
2.3 Main methods to study neurotransmission
This complex model of the exocytosis machinery at synapses was resolved through the combination of a wide array of techniques (Table 1). With biochemical techniques it was possible to define the minimum fusion machinery and to tease out direct molecular interactions. The core of these techniques involved in-vitro lipid mixing assays in which donor and acceptor artificial membranes are reconstituted with a variety of lipids and proteins and their fusion kinetics are analyzed [reviewed in Grothe et al. (62)]. Co-immunoprecipitation assays of the proteins involved in exocytosis coupled with site specific mutagenesis allowed precise determination of protein domain function. Additionally, crystallography of the full SNARE complex alone or in association with other proteins such as Munc18-1 have revealed in detail the amino acids involved in these inter-protein interactions [reviewed in Brunger et al. (63)]. The results obtained with these techniques were validated in the cellular environment by testing the effects of gene deletion or mutation on synaptic transmission. The classical approach to detect synaptic transmission is the measurement of postsynaptic currents in patch clamp experiments. This has provided the basis for our current understanding of exocytosis. Alternatively, the fusion of individual vesicles with the plasma membrane can be precisely assessed with high temporal resolution by membrane capacitance measurements (64, 65). However, this method can be applied only to very large synapses in very demanding experiments (66, 67). Therefore, almost all these experiments were performed on neuroendocrine cells (i.e. chromaffin cells) and the results were extrapolated to neurons (68–70). In addition, total internal fluorescence microscopy (TIRFM), which can visualize fluorescently labeled vesicles prior to and during release, allowed for more detailed assessment of tethering and docking/priming (71–73). In parallel, the impact of genetic modifications on the ultrastructure of synapses, i.e. on different vesicle pools, was analyzed by electron microscopy [(26, 74, 75); for review see Zuber and Lučić (76)]. One remaining problem was to understand how the proteins involved in exocytosis are organized at the pre-synapse. This was largely solved with the advent of super-resolution microscopy, such as stimulated emission depletion (STED) microscopy and direct stochastic optical reconstruction microscopy (dSTORM)/photo-activated localization microscopy (PALM) (77, 78), and CLEM experiments, in which super-resolution microscopy is combined with electron microscopy (EM) (79). Finally, live cell imaging of fluorescent markers allowed differentiation between events occurring during exocytosis, endocytosis or vesicle recycling. In early experiments, endocytosis was visualized using fluorescent dyes such as FM1-43, which are taken up in an activity dependent manner (80). Later, these experiments were performed with pH sensitive GFPs such as the super ecliptic pHluorin [SEP, Miesenbock et al. (81)] or RFPs, like pHuji (82), linked to the luminal domain of a SV protein such as synaptophysin [SypHy; Granseth et al. (83)]. In inactive synapses the fluorophore is located in the acidic SV lumen and is therefore quenched. Upon exocytosis the fluorescent protein is exposed to the neutral extracellular medium inducing a strong increase of its fluorescence. Upon endocytosis the pH sensitive fluorescent protein is re-internalized and quenched again by re-acidification of the SV lumen. As a result neuronal activity is visualized by fluorescence intensity variation at the synapses (84, 85).
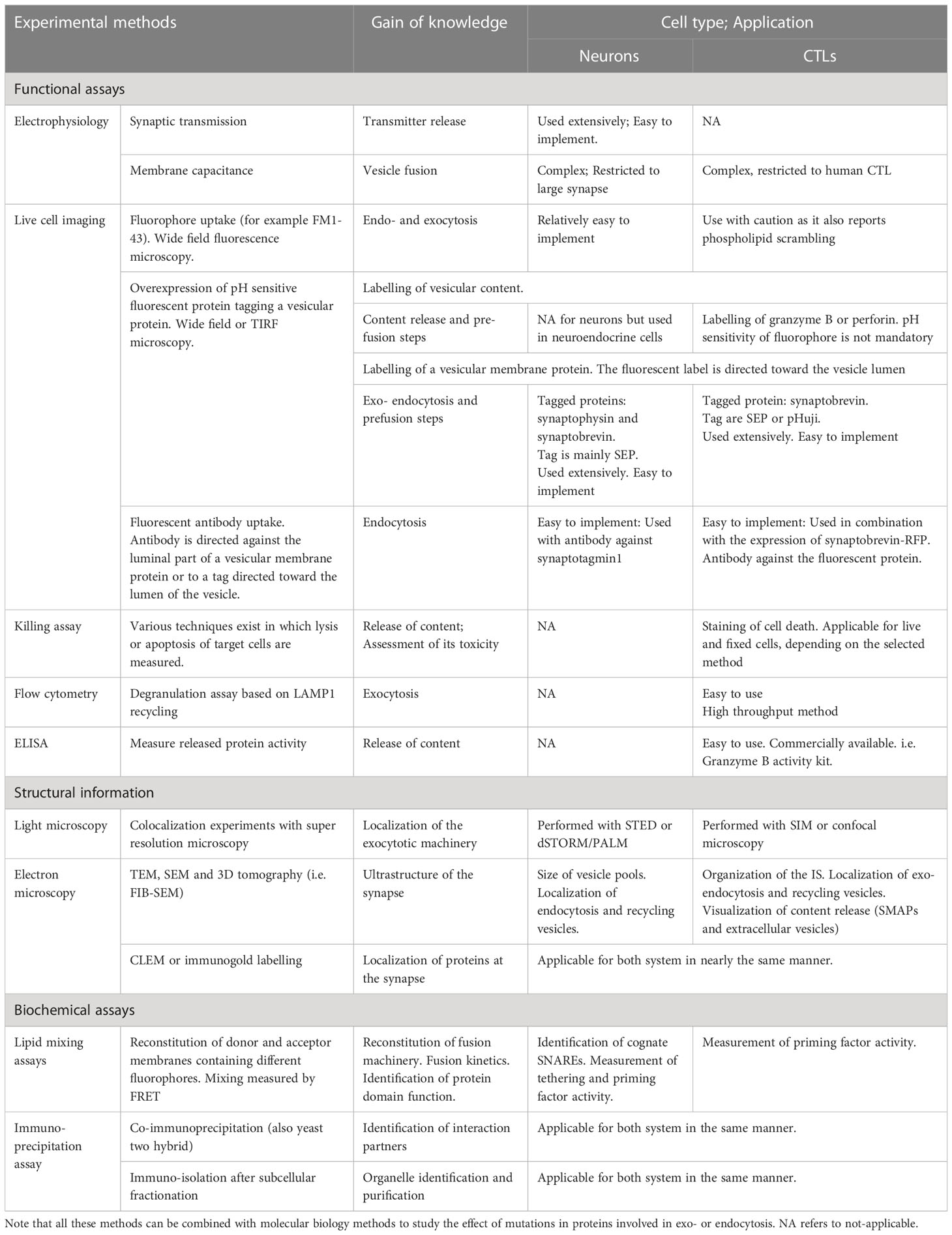
Table 1 Methods to analyze exocytosis.
Taken together, the last 25 years of intensive study of synaptic transmission have revealed in minute detail the components of the exocytotic machinery and the precise timing of protein interactions required for SV exocytosis. These studies of neuronal synapses preceded the description of the IS and resulted in key concepts of the molecular mechanisms of synaptic vesicle docking/priming that appear to be valid for other cellular models. We will now discuss how they can be adapted to describe LG exocytosis at the IS of CTLs.
3 Regulated exocytosis in cytotoxic T cells
How can this detailed knowledge of neurotransmission help us understand LG release at the IS? CTLs are part of the adaptive immune system. They circulate in the blood stream and patrol tissues and organs to detect infected cells and tumor cells. When CTLs detect a target cell for which they express a specific T cell receptor, they form a synaptic interface, i.e. the IS, with the target cell, and deliver cytotoxic molecules to the synaptic cleft via fusion of LG with their plasma membrane. This process is largely identical in CTL and natural killer (NK) cells. The main difference between the two cell types lies in the recognition of the target cells. For this reason, we will discuss the mechanisms of LG exocytosis using data obtained on both cells (86). The release of perforin and granzymes is a highly regulated process that is essential to kill the infected or malignant target cell. Indeed, loss of cytotoxicity in CTLs and NK cells results in an immune deficit, as is the case for the human immune disease, hemophagocytic lymphohistiocytosis (HLH) and familial hemophagocytic lymphohistiocytosis (FHL).
3.1 How a human disease helped to solve LG fusion mechanisms
The first indications that LG exocytosis shared similar features with SV exocytosis did not come from well-planned experiments but rather from clinical observations in patients affected by clinical syndromes such as FHL and HLH, which lead to reduced or abolished cytotoxicity of CTL and NK cells. The underlying causes of these defects are: 1. CTLs or NK cells lack or express a mutated form of the cytotoxic protein perforin (PRF1) causing FHL type 2 (87); 2. their cytokine production is impaired due to the mutations of either SH2D1A/SAP (coding for SLAM-associated protein), ITK (Tyrosine protein kinase) or CD27 (receptor for TNF) inducing FHL type 1 (88–90); 3. the biogenesis of LG is perturbed (gene mutation of LYST, AP3B1 or XIAP/BIRC4) (91–93); 4. Immune cells are unable to release LG content due to defective exocytotic machinery (94, 95). In the latter case, the mutated proteins include the SNARE protein, syntaxin11 (STX11, FHL 4) (96), and three tethering/priming factors Rab27a (RAB27A, Griscelli syndrome type 2), Munc13-4 (UNC13D, FHL 3) and Munc18-2 (STXBP2, FHL 5) (97–100). All these proteins are the same or isoforms of proteins involved in SV exocytosis. By the time they were discovered in CTLs, their function was elucidated to a large degree in neurons. Hence these results not only sparked the interest of immunologists but also of neuroscientists who had access to a large array of genetically modified mice in which these and other genes involved in exocytosis are deleted. Their interdisciplinary collaboration generated considerable advances in the understanding of LG release in CTLs and NK cells.
3.2 Methods to investigate LG exocytosis
The description of cell biological processes in immunology has historically been performed with a different repertoire of methods allowing high cell throughput (Table 1). Flow cytometry is of great importance for the characterization of heterogeneous cell populations but also for analyzing the expression of cell surface and intracellular molecules, and for the detection of cellular processes, such as exocytosis. A standard method for analyzing the LG exocytotic rate is the degranulation assay, which is based on flow cytometry. This method allows one to quantify the uptake of fluorescence-labeled antibodies raised against the intraluminal domain of lysosome-associated membrane protein 1 (LAMP-1, also named CD107a). When LG fuse with the plasma membrane, the intraluminal domain of LAMP-1 is exposed to the extracellular medium, and the anti-LAMP1 antibody contained in the medium can bind to it (101, 102). Hence, the brightness of the cell is then directly proportional to the number of exocytosed vesicles. This very potent method has been widely used to study LG exocytosis in CTL and NK cells from HLH or other immune-deficient patients (103). This flow cytometry based high throughput analysis generates solid results in a timely manner as thousands of cells can be screened rapidly. However, obtained results need to be interpreted with caution since LGs are only a fraction of the lysosomal compartments, all of which contain LAMP1 (104, 105). Population based functional assays should be used to complement the degranulation assay. One of these approaches is the killing assay, which measures the ability of CTL to kill target cells by quantifying signals from lysed target cells, such as lactate dehydrogenase release, surface phosphatidylserine expression, propidium iodide uptake or decay of fluorescence of intracellular dyes (106–109). The interpretation of these experiments must also take into account the ability of the CTL to kill via Fas-FasL that occurs via a different vesicular trafficking pathway (110).
While these methods were sufficient to identify proteins that are involved in LG release, they do not allow determination of their function during exocytosis. This limitation was solved via high resolution single cell imaging techniques such as confocal microscopy or TIRFM that allow the visualization of LG exocytosis in real time (104, 111, 112). In particular, TIRFM enabled the investigation of IS formation, docking/priming of LGs and their fusion kinetics. In these experiments the cells are seeded on glass slides or on supported lipid bilayers that display adhesion molecules and cognate peptide in the major histocompatibility complex (pMHC) or anti-CD3 antibody that triggers IS formation (113–116). The LGs are labeled with Lysotracker or more specifically via the expression an LG protein bound to a fluorescent protein. For example granzyme B-mTFP was used to monitor the release and diffusion of the LG content, while synaptobrevin2-mRFP allows observation of the fate of the fused LG membrane in the plasma membrane (104). In addition, using a pH sensitive label such as SEP or pHuji enables the precise measurement of the LG with the plasma membrane, fusion time, i.e. of the pore opening for the release of lytic components (81, 82). To obtain more detailed information about LG size and fusion kinetics, membrane capacitance measurement using patch-clamp electrophysiology was applied (117). However, adapting patch clamp electrophysiology to primary human CTLs has been extremely challenging and almost impossible for mouse CTLs. Hence this method will have limited use in the future. Live cell methods were complemented by the analysis of the IS ultrastructure with electron microscopy and CLEM (111, 118, 119). Finally, biochemical assays, such as lipid mixing assays, co-immunoprecipitation assays, or crystallography have been used to better understand the intrinsic properties of each protein involved in LG release (120).
3.3 Fate of LGs from IS formation to fusion
In contrast to neurons, the IS is not a long-lived structure in which vesicles are poised to exocytose. As a correlate the LGs are not organized in vesicle pools and the steps upstream from exocytosis are somewhat different in CTLs as compared to neurons. However, the overall sequence of events is similar, including the final transport of LG to the plasma membrane, the tethering and the docking/priming steps. The proteins mediating these steps are either identical or they are highly conserved paralogs from those proteins involved in synaptic transmission in neurons.
The IS is formed on demand upon recognition by the CTL through the TCR of the antigenic peptide associated to the major histocompatibility complex (pMHC) (1, 121). In NK cells, IS formation with the target cell is initiated by the combination of two signals. The first is a lack of MHC1 recognition (disinhibition), and the second is a positive signal from a variety of germline-encoded activation receptors that bind to proteins such as lectins or hemagglutinins on the target cell (122). Target cell recognition then triggers complex signaling cascade that leads to a rapid realignment of the Golgi complex and microtubule network by shifting the microtubule organizing center (MTOC) toward the IS and polymerizing microtubules toward the distal pole of the cell. Along these microtubule tracks, LGs and a variety of other organelles, such as multi vesicular bodies and mitochondria, move toward the IS. This function of the MTOC is important to ensure LG delivery to the IS but not for their exocytosis as such (123). Once close to the plasma membrane LGs switch their transport pathway from tubulin to F-actin through myosin IIa (124, 125), which is reminiscent of the transport of SVs to the active zone. Their final destination is the secretory domain of the central-supramolecular activation complex (c-SMAC). Similar to neurons, in CTLs actin also appears to form a barrier for LGs that prevents them from joining the IS. In fact, actin clearance at the c-SMAC is required for LG exocytosis. (112, 126). Thus, a fine balance in the density of the F-actin network appears to be required for LG secretion to occur (127).
Concurrent with LG transport, the plasma membrane at the IS adapts to become a platform for LG fusion. One of the modifications consist of an accumulation of Orai Ca2+ channels, that occurs simultaneously to an IP3/Ca2+ dependent activation and translocation of STIM proteins to the ER close to the IS. Activated STIM proteins interact with Orai, forming the store-operated Ca2+ release activated Ca2+ (CRAC) channel complex, which open leading to store operated Ca2+ entry (SOCE) (128–130). The resulting increase in cytoplasmic Ca2+ concentration is enhanced by nearby mitochondria (131, 132) ensuring synaptic activation (133, 134). The second modification ensures that the molecular components of the release machinery are at the right place. For that the t-SNARE syntaxin11, which is required for LG fusion, translocates to the IS and integrates into the plasma membrane in a VAMP8 dependent manner (135, 136). Membrane patches with syntaxin11 accumulation serve as hotspots for LG release. Whether SNAP23, the second t-SNARE, relocates to the plasma membrane during IS formation is not known.
At this stage the IS is ready for LGs to tether to the plasma membrane via two different protein complexes. The first is composed of Rab27a that associates with LG membranes in a GTP dependent fashion, the synaptotagmin like protein-2 (Slp-2) anchored in the plasma membrane and probably Munc13-4 (121, 137–139) (Figure 1D). The second consists of syntaxin11 at the plasma membrane and Munc18-2 as a bridge to the LGs (140). The identity of the LG protein to which Munc18-2 binds at this stage is elusive. By analogy to neuronal tethering of SVs, we speculate that it is a v-SNARE. This tether is likely the gateway for docking/priming in which SNARE complex assembly is initiated (Figure 1E). As indicated above, the t-SNAREs forming this complex are syntaxin11 (135, 141) and SNAP23 (96, 142), while the v-SNARE is VAMP2 in mouse and VAMP7 in human CTLs (118, 142). The SNARE complex assembly is mediated by Munc18-2 and Munc13-4. Similarly to the role of Munc18-1 in neurons, Munc18-2 is required in CTLs as a chaperone for syntaxin11 (140) and favors SNARE complex formation (120). The role of Munc13-4 is still under debate. Early studies showed that it is involved in a step prior to exocytosis in which recycling and late endosomal vesicles fuse to form LGs (143). More recent work demonstrated that Munc13-4 is required for priming (144). Whether Munc13-4 is required for opening syntaxin11 prior to SNARE complex formation is likely but not fully elucidated (145). Interestingly, the neuronal Munc18-1 and Munc13-1 are also expressed in human and mouse CTLs. Both can sustain LG exocytosis and they appear to have a compensatory role when Munc18-2 and Munc13-4 are deficient (140, 144, 146). Finally, upon increased Ca2+ concentration, the SNARE complex fully zippers and LGs fuse with the plasma membrane releasing perforin and granzymes into the synaptic cleft. The SNARE complex disassembly following fusion that is required for LG membrane recycling has not been investigated until now in CTL nor in NK cells.
The Ca2+-dependence of LG exocytosis has been described in many excellent reviews, so we will not address it here. Instead, we will discuss the identity of the Ca2+ sensor that assists the zippering of the SNARE complex. Mouse CTL express synaptotagmin 2, 7, 11 and 16 but only synaptotagmin 2 and 7 are Ca2+ sensitive (147). While synaptotagmin 7 is undoubtedly involved in LG exocytosis whether it is the Ca2+ sensor that triggers LG fusion with the plasma membrane is under debate (147–149). In fact, Sleiman et al. (147) showed that in mouse CTLs synaptotagmin 7 is rather involved in LG trafficking whereas synaptotagmin 2 might be the actual Ca2+ sensor for secretion. An alternative candidate is Munc13-4 as it contains Ca2+ binding N- and C-terminal C2 domains (C2A and C2B). Mutations of these C2 domains that alter their Ca2+ sensitivity, abolished LG exocytosis in NK cells (150). While the C2A domain participates in SNARE complex zippering, the C2B binds to the lipids of the plasma membrane upon increased Ca2+ concentration (145) possibly leading to the final pull on the vesicle membrane to induce fusion. Interestingly, in neurons Munc13-1 also plays an important role in calcium sensing albeit for a different function namely the replenishment of the RRP (151, 152).
Overall, the major steps that LGs undergo before fusing with the plasma membrane are the same as those that SVs must undergo for synaptic transmission. However, the timing of LG exocytosis is not as precise as SV in neurons. Whereas the latter occurs within milliseconds of the Ca2+ trigger, LG exocytosis takes minutes. Therefore, a smaller number of proteins seems to be required to control LG exocytosis.
3.4 Kinetic of fusion and content release
As for SVs, full fusion and kiss-and-run/kiss-and-stay modes can be detected for LG secretion in NK cells (153) while only full fusion was reported in CTL until now (105, 115). Furthermore, the fusion kinetics of full fusion events can vary. Estl et al. (105) found that LG stained through the expression of synaptobrevin2-pHuji showed a fluorescent signal that disappeared after fusion with the plasma membrane according to two different time courses. In 80% of the cases LG fluorescence disappeared in less than a second (300 ms in average, at 20°C). In the remaining 20%, fluorescence decay was much slower with an average time of 308 s. These two different membrane mixing behaviors might be explained by fast vs. very slow fusion pore dilation or by stable clustering of synaptobrevin2 in the plasma membrane at the fusion site. It will be interesting to elucidate whether F-actin, as in neuroendocrine cells, is involved in the expansion of LGs fusion pores (59). In contrast, all exocytosis events that were analyzed, showed a content release (labelled via granzyme B-mTFP expression) in about 300 ms. Events, in which content diffusion was much slower, were ignored from the analysis as it was questioned whether they corresponded to proper LG exocytosis. However, in a groundbreaking work, Bálint et al. (154) decisively expanded the view of LG exocytosis. They showed that granzyme B can be released as a soluble protein or within previously overlooked particles. The insoluble granzyme B particles were coined supramolecular attack particles (SMAPs). Subsequently, Chang et al. (119) showed that LGs can be divided in two different populations. While SMAPs were localized to multicore granules, diffusible granzyme B was found in classical single core lytic granules. The SNAREs involved in release of both types of LGs are probably identical because both harbor synaptobrevin2 on their surface (119). Whether tethering or priming factors are specific for each type of granule remains to be established.
3.5 LG recycling
CTLs actively move from one target cell to another, eventually killing a large numbers of target cells within minutes to hours. Killing one target cell, which requires the release of only few LGs, does not appear to need efficient LG recycling. However, this is not the case for CTLs that engage in serial/simultaneous killing of multiple target cells (155–157). Accordingly, the importance of LG endocytosis and recycling to sustain molecular signaling has been demonstrated (157, 158). The endocytic pathway was unraveled by following the clathrin- and dynamin-dependent endocytosis of the LG membrane protein synaptobrevin2. Like in neurons and neuroendocrine cells, LG endocytosis is promoted by the F-actin MyosinII complex (16, 159). Re-acidification of the endocytosed vesicle occurred within two minutes at 37°C (157). They were then recycled through early and late endosomes where they were refilled with granzyme B, and most likely with all other LG components, such as perforin and serglycin, to generate fully functional LGs. The endocytosed synaptobrevin2 containing granules rapidly mature by acidification of their lumen and reacquisition of cytotoxic proteins via late endosomes. Full recycling of LGs requires 30 to 60 minutes in mouse CTLs before they can be used for the next round of exocytosis (157). De-novo synthesis of key cytotoxic proteins perforin and granzymes in addition to interferon gamma and TNF-alpha is supported by mitochondria. In fact their depletion resulted in a significant reduction in the serial killing ability of CTLs (160). In NK92 cells some lytic components of the LGs, such as granzyme B and perforin, can be captured after exocytosis, recycled and reused for a second round of exocytosis, contributing to NK cell cytotoxicity (161, 162). Finally, as in neurons the coupling between LG exo- and endocytosis is mediated by Ca2+. In this context the calcium channel flower domain-containing protein 1, in short Flower, plays a major function (163). Flower deficient CTLs display a time-dependent block of LG endocytosis that is rescued by reintroduction of Flower in CTLs or by raising the extracellular Ca2+ concentration. Interestingly, a similar role for Flower has been demonstrated in neurons (164).
These findings show how CTLs, upon TCR triggering, combine several mechanisms, such as tight coupling of LG exo- endocytosis and mitochondrial-dependent cytotoxic protein translation, to maintain a constant supply of LGs during killing. This may be one of the many ways in which CTLs achieve a high per-capita killing capacity and therefore function as efficient serial killers (165). This feature may have tremendous importance in vivo for efficient clearance of tumors and viral infections.
4 Differences of LG fusion machinery compared to neurotransmission
Our understanding of LG exocytosis in CTLs has increased rapidly over the last two decades. As shown in the protein-protein network diagram (Figures 1C-E), a complex molecular machinery is required for LG exocytosis at the IS. However, the level of complexity is lower than in neurons. One reason for the differences between LG release at the IS and synaptic transmission in neurons could be the different time scale of each process. While synaptic transmission requires millisecond precision for SV exocytosis, LGs are released within minutes after IS formation. Thus, many proteins required to halt SV release and maintain SVs in a readily releasable state are probably not required in CTLs. In addition, the transient nature of the IS itself prevents unwanted LG release and controls the timing of exocytosis.
4.1 Requirement for docking/priming in LG exocytosis
As indicated above release of neurotransmitter requires a coordinated sequence of tethering and docking/priming factor interactions. Until now Munc18-2 and Munc13-4 have been identified in CTLs. To date no systematic study of other docking/priming factor expression has been performed in CTLs. We detected several additional putative docking/priming in CTL at the mRNA level (Figure 2). CAPS2e, appears to be the only CAPS isoform expressed in activated CTLs (Figure 2A). When examining CAPS2e function, CAPS2 knock-out CTLs did not show any changes in lysosomal compartment degranulation (data not shown). Since LGs are a subpopulation of lysosomes, presumably CAPS2e does not contribute to LG exocytosis. We further detected DOC2B mRNA in CTLs. This protein is of particular interest as it has been identified as a priming factor for lysosome secretion in mast cells (166). It is possible that it plays a similar function in CTLs. We also found mRNA of the transmembrane protein IA2 and the SNARE associated protein snapin at the mRNA level but have not investigated their function in CTLs (Figures 2B, C). Snapin is an interesting candidate as it has been shown to interact with SNAP23 (167), but it could be involved in lysosome recycling as well (168).
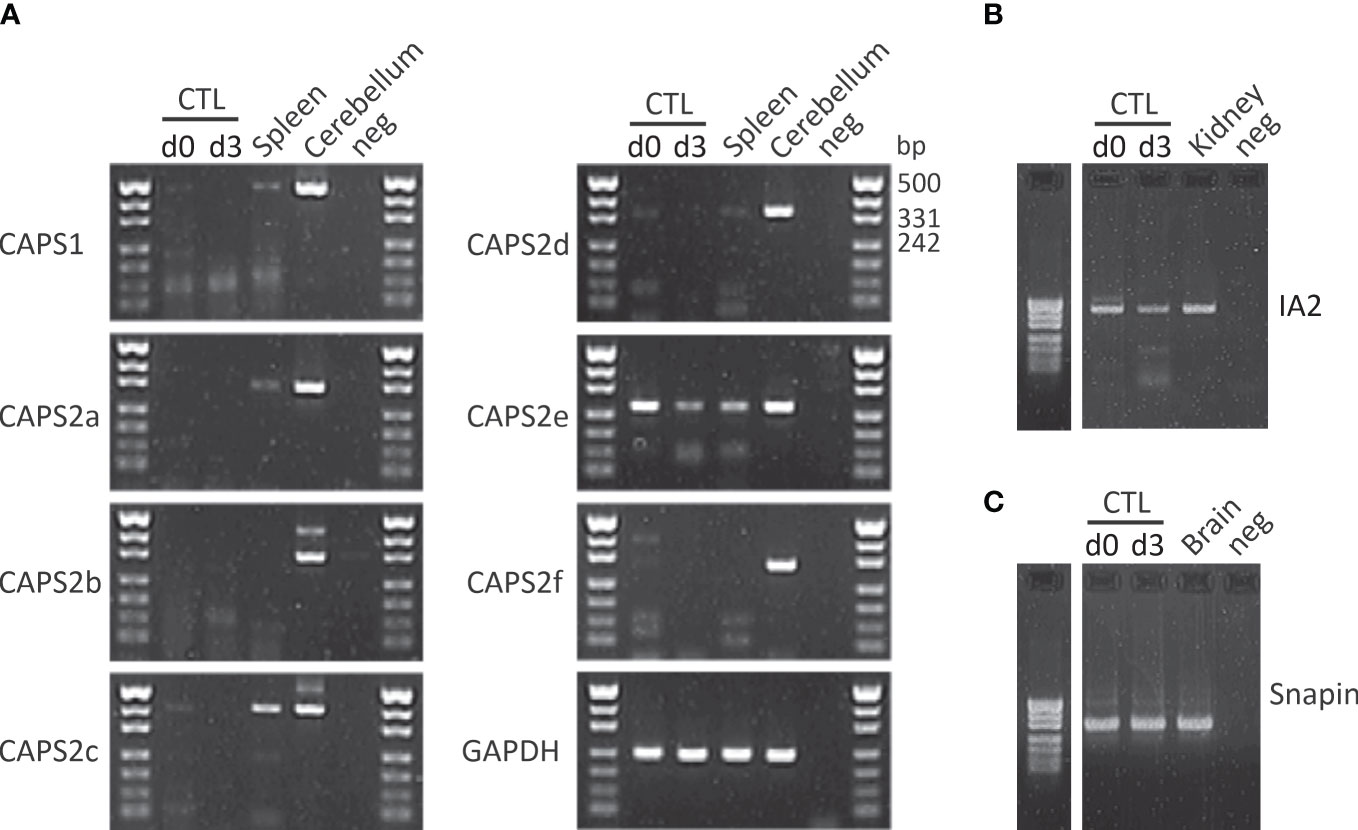
Figure 2 CAPS2e, IA2 and snapin mRNA are found in mouse CD8+ T cells. (A) RT-PCR of murine naïve (d0) and day3 (d3) stimulated CTLs and spleen using primers specific for CAPS1 and all known CAPS2 splice variant as described in Nguyen Truong et al. (2014). Total RNA isolated from cerebellum was used as positive control, water was used as negative control and the housekeeping gene GAPDH as loading control. Note that only CAPS2e was detected in CTL and spleen. Data are representative of two independent experiments from two mice. (B, C) CTL mRNA expression profile of IA2 (B) and snapin (C) before and after 3 days of stimulation with anti-CD3/CD28 antibody-coated beads (d3). Unstimulated CTLs (d0) were harvested directly after CTLs isolation. Total RNA of adult mouse brain and kidney were used as positive control for snapin and IA2, respectively and H2O was used as the negative control for PCR. Data are representative of two independent experiments from two mice. See supplementary material file for materials and methods.
Whether LG exocytosis requires other docking/priming factors is not known. We speculate that this is not the case for 3 reasons. 1. The exocytosis process in CTLs is relatively slow. 2-3 minutes are needed between the time of IS formation and the release of the first LGs (105, 158). An ultra-rapid release of lytic granules has been described in human CTL as a key molecular mechanism of multiple target cell killing (149, 169). However, in this context lytic granule secretion, that precedes microtubule re-organization (123, 149), occurs within tens of second after CTL/target cell contact, a time frame still delayed when compared to neuronal synaptic transmission. 2. The timing of LG exocytosis is dictated by IS formation and the subsequent polarized accumulation of t-SNAREs at the plasma membrane (135, 136). 3. None of the individual vesicle pools have been detected in CTLs (117, 170). Therefore, presumably no protein is required to halt or maintain LGs in an intermediate release-ready state as is the case in neurons. As a consequence, proteins such as complexin or tomosyn might be unnecessary in this context.
4.2 Specific molecular players for LG release
An intriguing point is that LG exocytosis involves proteins with a function that is not required in neuron synapses. One example is Vti1b. This SNARE protein is involved in endosomal fusion events (171–173). Qu and colleagues demonstrated that docking of LG at the IS, requires tethering of LG with CD3-containing endosomes via Vti1b in human CTLs through an unknown interaction partner (174) (Figure 1D). They showed that in comparison to untethered LGs, LG tethering increased their dwell time at the IS and their release probability. Accordingly, downregulation of Vti1b reduced LG tethering, their docking at the IS, and target cell killing. However, Vti1b does not seem to directly mediate the final fusion step of LGs. It plays a role upstream of fusion, clearly affecting CTL cytotoxicity (174, 175). This is reminiscent of the function of VAMP8 or syntaxin7 in CTL (116, 136, 176).
Our interaction diagram clearly shows that many open questions remain (Figure 1). For example: is an interaction between CRAC channels and t-SNAREs required for perfect LG positioning prior to exocytosis? What is the Ca2+ sensor for LG exocytosis? What are the interaction partners for Vti1b? Systematic analysis of protein-protein interactions with pull-down assays or lipid mixing assays should be applied to CTLs (120) to shed light on these and other outstanding questions.
4.3 Calcium signaling
Ca2+ signaling differs significantly between the two synaptic types. First, different Ca2+ channels are involved. While in neuronal synapse Ca2+ enters the presynapse through VGCCs that inactivate relatively rapidly, in the immunological synapse Ca2+ permeates the plasma membrane via CRAC channel complexes which do not inactivate thanks to the buffering function of mitochondria (see section 3.3). This leads to distinct intracellular Ca2+ concentration ([Ca2+]i) increases. Stimulation of a neuronal synapse by one action potential induces a very short (> 10 ms), extremely localized (1-2 µm diameter) but steep (> 10 µM) [Ca2+]i increase, which is sufficient for the fusion of one to three SVs (177, 178). In addition, a typical action potential train at 10 Hz elicits a prolonged Ca2+ influx that phases out after a few seconds due to inactivation of VGCCs. The ensuing [Ca2+]i increase spreads from the active zone to the back of the synapse. In contrast, stimulation of CTL by a target cell induces a biphasic [Ca2+]i increase. First IS formation induces an IP3 mediated Ca2+ release from the endoplasmic reticulum raising the [Ca2+]i transiently. The Ca2+ concentration depletion in the endoplasmic reticulum then activates the CRAC channel complex causing a long lasting (minutes to hours) increases in [Ca2+]i (see section 3.3). The rise of [Ca2+]i is relatively uniform along the IS and can reach values of about 2 µM (133). The Ca2+ then spreads throughout the cytoplasm of the CTL. Some functions of Ca2+ are conserved in both cell types. In neurons it is undisputed that Ca2+ triggers the fusion of SV with the plasma membrane. Although this function is still under debate in CTLs, a large body of work indicate that this is also the case (see Kaschek et al. (179) for review). Additionally, Ca2+ promotes endocytosis allowing a tight coupling between exo- and endocytosis in both cell types. Other functions such as regulating cytoskeleton remodeling or vesicle transport are probably different.
4.4 Variability of LG ultrastructure and composition
The diversity of secreted organelles is an important difference between neuronal cells and CTL secretion. Neurons secrete SVs at synapses and large dense core vesicles (LDCVs) along the entire plasma membrane. Both organelles have well-defined shapes and sizes. The release machinery of LDCV is not fully understood, but appears to be very similar to that of SV exocytosis with some differences in docking/priming factors (180, 181). This is very different for CTLs. Not only do they release very different types of organelles via regulated exocytosis at the IS (Rab11-positive vesicles, LGs and MVBs), but even the LGs are diverse. As mentioned above, LGs can be divided into single-core granules, containing diffusible granzyme B, and multi-core granules, containing SMAPs (119, 154). The SCGs have a well-defined round shape with a diameter of 293 ± 8 nm, whereas the MCG are spheres with a more or less elongated shape of and their size is quite variable with an average diameter of 364 ± 12 nm. These shape and size differences could affect their fusion kinetics (182, 183). Furthermore, it is completely unclear whether they are released in parallel, at different times after IS formation, or upon specific stimuli. If the latter is the case, then different tethering or docking/priming factors should control the exocytosis of single vs. multi-core granules. Understanding the specificity of different LG release would be particularly important for fine-tuning the duration and intensity of lytic function of killer cells (119, 154).
5 Concluding remarks
Both the nervous and immune systems use synaptic contacts to transmit key intercellular information through regulated secretion of intracellular vesicles. They use a similar molecular machinery, with SNAREs at the core and Munc18 and Munc13 as the major facilitators. The protein paralogs in CTLs are different from those in neurons, but their individual domain structures are nearly identical, with the exception of Munc13. Thus, advancing knowledge of exocytosis in one system benefits the study of the other.
A great advantage of studying secretion in immune synapses over neuronal synapse is that human CTL are readily available and can be easily handled. Accumulated research findings in CTL reveal subtle differences in the mouse and human T cell secretory machinery. One example for this diversity is the v-SNARE of LG that is required for exocytosis: VAMP7 is used in human CTL, while VAMP2/synaptobrevin2 is used by mouse CTL (118, 142). VAMP7 is also expressed in mouse CTLs but its function remains to be determined. Similar interspecies differences have not yet been identified for synaptic transmission in the nervous system probably because human neurons are nearly inaccessible. This might change in the near future as human neurons can now be derived from induced pluripotent stem cells (184, 185). In the meantime research performed on human CTLs might be instrumental to shed new light on human neuronal synapses. Understanding interspecies differences is especially important when studying the effect of mutations like those found in FHL. Until now, mutations in Munc13-4, Munc18-2, Rab27a, syntaxin 11, SNAP23 have been regarded as knock out or knock down phenotype. However, the situation might be more complex as subtle alterations in the function of these proteins might contribute to multiple shades of synaptic transmission in immune cells and neurons. Other advantages of working with immune cells is the easier molecular manipulation of blood cells as opposed to nervous system tissue and the possibility to inspect lymphocytes using a panel of high-resolution imaging techniques difficult to apply to human neurons.
In conclusion, in our review, we discussed how “synaptic inspection” in neuroscience and immunology can learn from each other and how important it is to define the precise identity and function of the multiple proteins involved in both SV and LG secretion.
Author contributions
UB, H-FC, and CS conceived and wrote most of the paper. VP and EK wrote some parts of the paper. CS and VP generated the data shown Figure 2. UB prepared the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 894 to EK and UB), and the European Commission (ERC -2021-SyG_951329 to the Department of Cellular Neurophysiology, Saarland University).
Acknowledgments
We thank C.T. Baldari, S. Valitutti and D. Stevens for critical comments on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1177670/full#supplementary-material
References
1. Dustin ML. The immunological synapse. Cancer Immunol Res (2014) 2(11):1023–33. doi: 10.1158/2326-6066.Cir-14-0161
2. Kaiser CA, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell (1990) 61(4):723–33. doi: 10.1016/0092-8674(90)90483-u
3. Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell (1990) 61(4):709–21. doi: 10.1016/0092-8674(90)90482-t
4. Rothman JE, Orci L. Molecular dissection of the secretory pathway. Nature (1992) 355(6359):409–15. doi: 10.1038/355409a0
5. Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, et al. SNAREpins: minimal machinery for membrane fusion. Cell (1998) 92(6):759–72. doi: 10.1016/S0092-8674(00)81404-X
6. Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, et al. Tetanus and botulinum-b neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature (1992) 359(6398):832–5. doi: 10.1038/359832a0
7. Niemann H, Blasi J, Jahn R. Clostridial neurotoxins: new tools for dissecting exocytosis. Trends Cell Biol (1994) 4(5):179–85. doi: 10.1016/0962-8924(94)90203-8
8. Rosenmund C, Rettig J, Brose N. Molecular mechanisms of active zone function. Curr Opin Neurobiol (2003) 13(5):509–19. doi: 10.1016/j.conb.2003.09.011
9. Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron (2013) 80(3):675–90. doi: 10.1016/j.neuron.2013.10.022
10. Rizo J. Mechanism of neurotransmitter release coming into focus. Protein Sci (2018) 27(8):1364–91. doi: 10.1002/pro.3445
11. Rizo J. Molecular mechanisms underlying neurotransmitter release. Annu Rev Biophys (2022) 51:377–408. doi: 10.1146/annurev-biophys-111821-104732
12. Zhang M, Augustine GJ. Synapsins and the synaptic vesicle reserve pool: floats or anchors? Cells (2021) 10(3):658. doi: 10.3390/cells10030658
13. Wöllert T, Patel A, Lee YL, Provance DW Jr., Vought VE, Cosgrove MS, et al. Myosin5a tail associates directly with Rab3A-containing compartments in neurons. J Biol Chem (2011) 286(16):14352–61. doi: 10.1074/jbc.M110.187286
14. Maschi D, Gramlich MW, Klyachko VA. Myosin V regulates spatial localization of different forms of neurotransmitter release in central synapses. Front Synaptic Neurosci (2021) 13:650334 doi: 10.3389/fnsyn.2021.650334
15. Wang X, Sun X, Zhou HC, Luo F. Activation of beta3-adrenoceptor increases the number of readily releasable glutamatergic vesicle via activating Ca(2+)/calmodulin/MLCK/myosin II pathway in the prefrontal cortex of juvenile rats. Sci Rep (2021) 11(1):18300. doi: 10.1038/s41598-021-97769-4
16. Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci (2008) 9(5):344–56. doi: 10.1038/nrn2373
17. Bleckert A, Photowala H, Alford S. Dual pools of actin at presynaptic terminals. J Neurophysiol (2012) 107(12):3479–92. doi: 10.1152/jn.00789.2011
18. Miki T, Malagon G, Pulido C, Llano I, Neher E, Marty A. Actin- and myosin-dependent vesicle loading of presynaptic docking sites prior to exocytosis. Neuron (2016) 91(4):808–23. doi: 10.1016/j.neuron.2016.07.033
19. Meunier FA, Gutierrez LM. Captivating new roles of f-actin cortex in exocytosis and bulk endocytosis in neurosecretory cells. Trends Neurosci (2016) 39(9):605–13. doi: 10.1016/j.tins.2016.07.003
20. Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, et al. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron (2001) 30(1):183–96. doi: 10.1016/s0896-6273(01)00272-0
21. Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, et al. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J (2005) 24(16):2839–50. doi: 10.1038/sj.emboj.7600753
22. Camacho M, Basu J, Trimbuch T, Chang S, Pulido-Lozano C, Chang SS, et al. Heterodimerization of Munc13 C(2)A domain with RIM regulates synaptic vesicle docking and priming. Nat Commun (2017) 8:15293. doi: 10.1038/ncomms15293
23. Stepien KP, Xu J, Zhang X, Bai XC, Rizo J. SNARE assembly enlightened by cryo-EM structures of a synaptobrevin-Munc18-1-syntaxin-1 complex. Sci Adv (2022) 8(25):eabo5272. doi: 10.1126/sciadv.abo5272
24. Xu Y, Zhu L, Wang S, Ma C. Munc18 - Munc13-dependent pathway of SNARE complex assembly is resistant to NSF and α-SNAP. FEBS J (2022) 20:6367–84. doi: 10.1111/febs.16528
25. de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, et al. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell (2009) 138(5):935–46. doi: 10.1016/j.cell.2009.07.027
26. Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Südhof TC, et al. The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron (2014) 84(2):416–31. doi: 10.1016/j.neuron.2014.10.009
27. Chan AW, Khanna R, Li Q, Stanley EF. Munc18: a presynaptic transmitter release site n type (CaV2.2) calcium channel interacting protein. Channels (Austin) (2007) 1(1):11–20. doi: 10.4161/chan.3694
28. Toft-Bertelsen TL, Ziomkiewicz I, Houy S, Pinheiro PS, Sørensen JB. Regulation of Ca2+ channels by SNAP-25 via recruitment of syntaxin-1 from plasma membrane clusters. Mol Biol Cell (2016) 27(21):3329–41. doi: 10.1091/mbc.E16-03-0184
29. Sajman J, Trus M, Atlas D, Sherman E. The l-type voltage-gated calcium channel co-localizes with syntaxin 1A in nano-clusters at the plasma membrane. Sci Rep (2017) 7(1):11350. doi: 10.1038/s41598-017-10588-4
30. Neher E, Brose N. Dynamically primed synaptic vesicle states: key to understand synaptic short-term plasticity. Neuron (2018) 100(6):1283–91. doi: 10.1016/j.neuron.2018.11.024
31. Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, et al. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U.S.A. (2002) 99(13):9037–42. doi: 10.1073/pnas.122623799
32. Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, et al. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron (2017) 95(3):591–607.e510. doi: 10.1016/j.neuron.2017.07.004
33. Lipstein N, Chang S, Lin KH, López-Murcia FJ, Neher E, Taschenberger H, et al. Munc13-1 is a Ca(2+)-phospholipid-dependent vesicle priming hub that shapes synaptic short-term plasticity and enables sustained neurotransmission. Neuron (2021) 109(24):3980–4000.e3987. doi: 10.1016/j.neuron.2021.09.054
34. Dhara M, Yarzagaray A, Schwarz Y, Dutta S, Grabner C, Moghadam PK, et al. Complexin synchronizes primed vesicle exocytosis and regulates fusion pore dynamics. J Cell Biol (2014) 204(7):1123–40. doi: 10.1083/jcb.201311085
35. Mohrmann R, Dhara M, Bruns D. Complexins: small but capable. Cell Mol Life Sci (2015) 72(22):4221–35. doi: 10.1007/s00018-015-1998-8
36. Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell (1994) 79(4):717–27. doi: 10.1016/0092-8674(94)90556-8
37. Fernández-Chacón R, Shin OH, Königstorfer A, Matos MF, Meyer AC, Garcia J, et al. Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. J Neurosci (2002) 22(19):8438–46. doi: 10.1523/jneurosci.22-19-08438.2002
38. Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci (2009) 12(6):759–66. doi: 10.1038/nn.2320
39. Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell (1993) 75(3):409–18. doi: 10.1016/0092-8674(93)90376-2
40. Bombardier JP, Munson M. Three steps forward, two steps back: mechanistic insights into the assembly and disassembly of the SNARE complex. Curr Opin Chem Biol (2015) 29:66–71. doi: 10.1016/j.cbpa.2015.10.003
41. Huang X, Sun S, Wang X, Fan F, Zhou Q, Lu S, et al. Mechanistic insights into the SNARE complex disassembly. Sci Adv (2019) 5(4):eaau8164. doi: 10.1126/sciadv.aau8164
42. Jockusch WJ, Speidel D, Sigler A, Sørensen JB, Varoqueaux F, Rhee JS, et al. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell (2007) 131(4):796–808. doi: 10.1016/j.cell.2007.11.002
43. Liu Y, Schirra C, Edelmann L, Matti U, Rhee J, Hof D, et al. Two distinct secretory vesicle-priming steps in adrenal chromaffin cells. J Cell Biol (2010) 190(6):1067–77. doi: 10.1083/jcb.201001164
44. Pinheiro PS, Houy S, Sørensen JB. C2-domain containing calcium sensors in neuroendocrine secretion. J Neurochem (2016) 139(6):943–58. doi: 10.1111/jnc.13865
45. Thakur P, Stevens DR, Sheng ZH, Rettig J. Effects of PKA-mediated phosphorylation of snapin on synaptic transmission in cultured hippocampal neurons. J Neurosci (2004) 24(29):6476–81. doi: 10.1523/jneurosci.0590-04.2004
46. Tian JH, Wu ZX, Unzicker M, Lu L, Cai Q, Li C, et al. The role of snapin in neurosecretion: snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J Neurosci (2005) 25(45):10546–55. doi: 10.1523/jneurosci.3275-05.2005
47. Yu SC, Klosterman SM, Martin AA, Gracheva EO, Richmond JE. Differential roles for snapin and synaptotagmin in the synaptic vesicle cycle. PloS One (2013) 8(2):e57842. doi: 10.1371/journal.pone.0057842
48. Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, et al. Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron (1998) 20(5):905–15. doi: 10.1016/s0896-6273(00)80472-9
49. Hatsuzawa K, Lang T, Fasshauer D, Bruns D, Jahn R. The r-SNARE motif of tomosyn forms SNARE core complexes with syntaxin 1 and SNAP-25 and down-regulates exocytosis. J Biol Chem (2003) 278(33):31159–66. doi: 10.1074/jbc.M305500200
50. Bielopolski N, Lam AD, Bar-On D, Sauer M, Stuenkel EL, Ashery U. Differential interaction of tomosyn with syntaxin and SNAP25 depends on domains in the WD40 β-propeller core and determines its inhibitory activity. J Biol Chem (2014) 289(24):17087–99. doi: 10.1074/jbc.M113.515296
51. Becherer U, Rettig J. Vesicle pools, docking, priming, and release. Cell Tissue Res (2006) 326(2):393–407. doi: 10.1007/s00441-006-0243-z
52. Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic (2008) 9(9):1414–24. doi: 10.1111/j.1600-0854.2008.00759.x
53. Neher E. Merits and limitations of vesicle pool models in view of heterogeneous populations of synaptic vesicles. Neuron (2015) 87(6):1131–42. doi: 10.1016/j.neuron.2015.08.038
54. Gundelfinger ED, Kessels MM, Qualmann B. Temporal and spatial coordination of exocytosis and endocytosis. Nat Rev Mol Cell Biol (2003) 4(2):127–39. doi: 10.1038/nrm1016
55. Schweizer FE, Ryan TA. The synaptic vesicle: cycle of exocytosis and endocytosis. Curr Opin Neurobiol (2006) 16(3):298–304. doi: 10.1016/j.conb.2006.05.006
56. Maritzen T, Haucke V. Coupling of exocytosis and endocytosis at the presynaptic active zone. Neurosci Res (2018) 127:45–52. doi: 10.1016/j.neures.2017.09.013
57. Rizzoli SO, Jahn R. Kiss-and-run, collapse and 'readily retrievable' vesicles. Traffic (2007) 8(9):1137–44. doi: 10.1111/j.1600-0854.2007.00614.x
58. Alabi AA, Tsien RW. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol (2013) 75:393–422. doi: 10.1146/annurev-physiol-020911-153305
59. Wen PJ, Grenklo S, Arpino G, Tan X, Liao HS, Heureaux J, et al. Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat Commun (2016) 7:12604. doi: 10.1038/ncomms12604
60. Watanabe S, Boucrot E. Fast and ultrafast endocytosis. Curr Opin Cell Biol (2017) 47:64–71. doi: 10.1016/j.ceb.2017.02.013
61. Chanaday NL, Cousin MA, Milosevic I, Watanabe S, Morgan JR. The synaptic vesicle cycle revisited: new insights into the modes and mechanisms. J Neurosci (2019) 39(42):8209–16. doi: 10.1523/jneurosci.1158-19.2019
62. Grothe T, Nowak J, Jahn R, Walla PJ. Selected tools to visualize membrane interactions. Eur Biophys J (2021) 50(2):211–22. doi: 10.1007/s00249-021-01516-6
63. Brunger AT, Choi UB, Lai Y, Leitz J, White KI, Zhou Q. The pre-synaptic fusion machinery. Curr Opin Struct Biol (2019) 54:179–88. doi: 10.1016/j.sbi.2019.03.007
64. Neher E, Marty A. Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci U.S.A. (1982) 79(21):6712–6. doi: 10.1073/pnas.79.21.6712
65. Lindau M. High resolution electrophysiological techniques for the study of calcium-activated exocytosis. Biochim Biophys Acta (2012) 1820(8):1234–42. doi: 10.1016/j.bbagen.2011.12.011
66. Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc (2006) 1(4):2075–81. doi: 10.1038/nprot.2006.312
67. Schneggenburger R, Forsythe ID. The calyx of held. Cell Tissue Res (2006) 326(2):311–37. doi: 10.1007/s00441-006-0272-7
68. Rettig J, Neher E. Emerging roles of presynaptic proteins in ca++-triggered exocytosis. Science (2002) 298(5594):781–5. doi: 10.1126/science.1075375
69. Neher E. A comparison between exocytic control mechanisms in adrenal chromaffin cells and a glutamatergic synapse. Pflugers Arch (2006) 453(3):261–8. doi: 10.1007/s00424-006-0143-9
70. Neher E. Neurosecretion: what can we learn from chromaffin cells. Pflugers Arch (2018) 470(1):7–11. doi: 10.1007/s00424-017-2051-6
71. Nofal S, Becherer U, Hof D, Matti U, Rettig J. Primed vesicles can be distinguished from docked vesicles by analyzing their mobility. J Neurosci (2007) 27(6):1386–95. doi: 10.1523/JNEUROSCI.4714-06.2007
72. Pasche M, Matti U, Hof D, Rettig J, Becherer U. Docking of LDCVs is modulated by lower intracellular [Ca2+] than priming. PloS One (2012) 7(5):e36416. doi: 10.1371/journal.pone.0036416
73. Hugo S, Dembla E, Halimani M, Matti U, Rettig J, Becherer U. Deciphering dead-end docking of large dense core vesicles in bovine chromaffin cells. J Neurosci (2013) 33(43):17123–37. doi: 10.1523/JNEUROSCI.1589-13.2013
74. Borges-Merjane C, Kim O, Jonas P. Functional electron microscopy, "Flash and freeze," of identified cortical synapses in acute brain slices. Neuron (2020) 105(6):992–1006.e1006. doi: 10.1016/j.neuron.2019.12.022
75. Maus L, Lee C, Altas B, Sertel SM, Weyand K, Rizzoli SO, et al. Ultrastructural correlates of presynaptic functional heterogeneity in hippocampal synapses. Cell Rep (2020) 30(11):3632–3643.e3638. doi: 10.1016/j.celrep.2020.02.083
76. Zuber B, Lučić V. Molecular architecture of the presynaptic terminal. Curr Opin Struct Biol (2019) 54:129–38. doi: 10.1016/j.sbi.2019.01.008
77. Carvalhais LG, Martinho VC, Ferreiro E, Pinheiro PS. Unraveling the nanoscopic organization and function of central mammalian presynapses with super-resolution microscopy. Front Neurosci (2020) 14:578409. doi: 10.3389/fnins.2020.578409
78. Nosov G, Kahms M, Klingauf J. The decade of super-resolution microscopy of the presynapse. Front Synaptic Neurosci (2020) 12:32. doi: 10.3389/fnsyn.2020.00032
79. Watanabe S, Punge A, Hollopeter G, Willig KI, Hobson RJ, Davis MW, et al. Protein localization in electron micrographs using fluorescence nanoscopy. Nat Methods (2011) 8(1):80–4. doi: 10.1038/nmeth.1537
80. Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci (1999) 22:1–10. doi: 10.1146/annurev.neuro.22.1.1
81. Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature (1998) 394(6689):192–5. doi: 10.1038/28190
82. Shen Y, Rosendale M, Campbell RE, Perrais D. pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J Cell Biol (2014) 207(3):419–32. doi: 10.1083/jcb.201404107
83. Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis: the physiological mechanism of vesicle retrieval at hippocampal synapses. J Physiol (2007) 585(Pt 3):681–6. doi: 10.1113/jphysiol.2007.139022
84. Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J (2000) 79(4):2199–208. doi: 10.1016/s0006-3495(00)76468-x
85. Ariel P, Ryan TA. Optical mapping of release properties in synapses. Front Neural Circuits (2010) 4:18. doi: 10.3389/fncir.2010.00018
86. Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood (2013) 121(8):1345–56. doi: 10.1182/blood-2012-07-442558
87. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286(5446):1957–9. doi: 10.1126/science.286.5446.1957
88. Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1(5):433–40. doi: 10.1038/80877
89. Cannons JL, Yu LJ, Jankovic D, Crotty S, Horai R, Kirby M, et al. SAP regulates T cell-mediated help for humoral immunity by a mechanism distinct from cytokine regulation. J Exp Med (2006) 203(6):1551–65. doi: 10.1084/jem.20052097
90. Lechner KS, Neurath MF, Weigmann B. Role of the IL-2 inducible tyrosine kinase ITK and its inhibitors in disease pathogenesis. J Mol Med (Berl) (2020) 98(10):1385–95. doi: 10.1007/s00109-020-01958-z
91. Rigaud S, Fondanèche M-C, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444(7115):110–4. doi: 10.1038/nature05257
92. Kaplan J, De Domenico I, Ward DM. Chediak-higashi syndrome. Curr Opin Hematol (2008) 15(1):22–9. doi: 10.1097/MOH.0b013e3282f2bcce
93. Jessen B, Bode SF, Ammann S, Chakravorty S, Davies G, Diestelhorst J, et al. The risk of hemophagocytic lymphohistiocytosis in hermansky-pudlak syndrome type 2. Blood (2013) 121(15):2943–51. doi: 10.1182/blood-2012-10-463166
94. de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol (2010) 10(8):568–79. doi: 10.1038/nri2803
95. Becherer U, Medart MR, Schirra C, Krause E, Stevens D, Rettig J. Regulated exocytosis in chromaffin cells and cytotoxic T lymphocytes: how similar are they? Cell Calcium (2012) 52(3-4):303–12. doi: 10.1016/j.ceca.2012.04.002
96. Ye S, Karim ZA, Al Hawas R, Pessin JE, Filipovich AH, Whiteheart SW. Syntaxin-11, but not syntaxin-2 or syntaxin-4, is required for platelet secretion. Blood (2012) 120(12):2484–92. doi: 10.1182/blood-2012-05-430603
97. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause griscelli syndrome associated with haemophagocytic syndrome. Nat Genet (2000) 25(2):173–6. doi: 10.1038/76024
98. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73. doi: 10.1016/s0092-8674(03)00855-9
99. Yamamoto K, Ishii E, Sako M, Ohga S, Furuno K, Suzuki N, et al. Identification of novel MUNC13-4 mutations in familial haemophagocytic lymphohistiocytosis and functional analysis of MUNC13-4-deficient cytotoxic T lymphocytes. J Med Genet (2004) 41(10):763–7. doi: 10.1136/jmg.2004.021121
100. Cetica V, Santoro A, Gilmour KC, Sieni E, Beutel K, Pende D, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet (2010) 47(9):595–600. doi: 10.1136/jmg.2009.075341
101. Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods (2003) 281(1-2):65–78. doi: 10.1016/s0022-1759(03)00265-5
102. Florian AE, Lepensky CK, Kwon O, Haynes MK, Sklar LA, Zweifach A. Flow cytometry enables a high-throughput homogeneous fluorescent antibody-binding assay for cytotoxic T cell lytic granule exocytosis. J Biomol Screen (2013) 18(4):420–9. doi: 10.1177/1087057112466697
103. Chiang SCC, Bleesing JJ, Marsh RA. Current flow cytometric assays for the screening and diagnosis of primary HLH. Front Immunol (20191740) 10:1740. doi: 10.3389/fimmu.2019.01740
104. Chitirala P, Chang HF, Martzloff P, Harenberg C, Ravichandran K, Abdulreda MH, et al. Studying the biology of cytotoxic T lymphocytes in vivo with a fluorescent granzyme b-mTFP knock-in mouse. Elife (2020) 9:e58065. doi: 10.7554/eLife.58065
105. Estl M, Blatt P, Li X, Becherer U, Chang HF, Rettig J, et al. Various stages of immune synapse formation are differently dependent on the strength of the TCR stimulus. Int J Mol Sci (2020) 21(7):2475. doi: 10.3390/ijms21072475
106. Korzeniewski C, Callewaert DM. An enzyme-release assay for natural cytotoxicity. J Immunol Methods (1983) 64(3):313–20. doi: 10.1016/0022-1759(83)90438-6
107. Kroesen BJ, Mesander G, ter Haar JG, The TH, de Leij L. Direct visualisation and quantification of cellular cytotoxicity using two colour flourescence. J Immunol Methods (1992) 156(1):47–54. doi: 10.1016/0022-1759(92)90009-i
108. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods (1995) 184(1):39–51. doi: 10.1016/0022-1759(95)00072-i
109. Kummerow C, Schwarz EC, Bufe B, Zufall F, Hoth M, Qu B. A simple, economic, time-resolved killing assay. Eur J Immunol (2014) 44(6):1870–2. doi: 10.1002/eji.201444518
110. Cassioli C, Baldari CT. The expanding arsenal of cytotoxic T cells. Front Immunol (2022) 13:883010. doi: 10.3389/fimmu.2022.883010
111. Pattu V, Halimani M, Ming M, Schirra C, Hahn U, Bzeih H, et al. In the crosshairs: investigating lytic granules by high-resolution microscopy and electrophysiology. Front Immunol (2013) 4:411 doi: 10.3389/fimmu.2013.00411
112. Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NM, Chen BC, Gawden-Bone C, et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity (2015) 42(5):864–76. doi: 10.1016/j.immuni.2015.04.013
113. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285(5425):221–7. doi: 10.1126/science.285.5425.221
114. Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in jurkat T cells. Proc Natl Acad Sci U.S.A. (2007) 104(51):20296–301. doi: 10.1073/pnas.0710258105
115. Martina JA, Wu XS, Catalfamo M, Sakamoto T, Yi C, Hammer JA. Imaging of lytic granule exocytosis in CD8+ cytotoxic T lymphocytes reveals a modified form of full fusion. Cell Immunol (2011) 271(2):267–79. doi: 10.1016/j.cellimm.2011.07.004
116. Pattu V, Qu B, Marshall M, Becherer U, Junker C, Matti U, et al. Syntaxin7 is required for lytic granule release from cytotoxic T lymphocytes. Traffic (2011) 12(7):890–901. doi: 10.1111/j.1600-0854.2011.01193.x
117. Ming M, Schirra C, Becherer U, Stevens DR, Rettig J. Behavior and properties of mature lytic granules at the immunological synapse of human cytotoxic T lymphocytes. PloS One (2015) 10(8):e0135994. doi: 10.1371/journal.pone.0135994
118. Matti U, Pattu V, Halimani M, Schirra C, Krause E, Liu Y, et al. Synaptobrevin2 is the v-SNARE required for cytotoxic T-lymphocyte lytic granule fusion. Nat Commun (2013) 4:1439. doi: 10.1038/ncomms2467
119. Chang HF, Schirra C, Ninov M, Hahn U, Ravichandran K, Krause E, et al. Identification of distinct cytotoxic granules as the origin of supramolecular attack particles in T lymphocytes. Nat Commun (20221029) 13(1):1029. doi: 10.1038/s41467-022-28596-y
120. Spessott WA, Sanmillan ML, McCormick ME, Patel N, Villanueva J, Zhang K, et al. Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood (2015) 125(10):1566–77. doi: 10.1182/blood-2014-11-610816
121. Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol (2001) 152(4):825–34. doi: 10.1083/jcb.152.4.825
122. Bryceson YT, Chiang SC, Darmanin S, Fauriat C, Schlums H, Theorell J, et al. Molecular mechanisms of natural killer cell activation. J Innate Immun (2011) 3(3):216–26. doi: 10.1159/000325265
123. Tamzalit F, Tran D, Jin W, Boyko V, Bazzi H, Kepecs A, et al. Centrioles control the capacity, but not the specificity, of cytotoxic T cell killing. Proc Natl Acad Sci U.S.A. (2020) 117(8):4310–9. doi: 10.1073/pnas.1913220117
124. Sanborn KB, Rak GD, Maru SY, Demers K, Difeo A, Martignetti JA, et al. Myosin IIA associates with NK cell lytic granules to enable their interaction with f-actin and function at the immunological synapse. J Immunol (2009) 182(11):6969–84. doi: 10.4049/jimmunol.0804337
125. Sanborn KB, Mace EM, Rak GD, Difeo A, Martignetti JA, Pecci A, et al. Phosphorylation of the myosin IIA tailpiece regulates single myosin IIA molecule association with lytic granules to promote NK-cell cytotoxicity. Blood (2011) 118(22):5862–71. doi: 10.1182/blood-2011-03-344846
126. Ritter AT, Kapnick SM, Murugesan S, Schwartzberg PL, Griffiths GM, Lippincott-Schwartz J. Cortical actin recovery at the immunological synapse leads to termination of lytic granule secretion in cytotoxic T lymphocytes. Proc Natl Acad Sci U.S.A. (2017) 114(32):E6585–94. doi: 10.1073/pnas.1710751114
127. Li P, Bademosi AT, Luo J, Meunier FA. Actin remodeling in regulated exocytosis: toward a mesoscopic view. Trends Cell Biol (2018) 28(9):685–97. doi: 10.1016/j.tcb.2018.04.004
128. Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol (2006) 174(6):815–25. doi: 10.1083/jcb.200604015
129. Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, Sen D, et al. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci U.S.A. (2008) 105(6):2011–6. doi: 10.1073/pnas.0706122105
130. Vaeth M, Kahlfuss S, Feske S. CRAC channels and calcium signaling in T cell-mediated immunity. Trends Immunol (2020) 41(10):878–901. doi: 10.1016/j.it.2020.06.012
131. Quintana A, Kummerow C, Junker C, Becherer U, Hoth M. Morphological changes of T cells following formation of the immunological synapse modulate intracellular calcium signals. Cell Calcium (2009) 45(2):109–22. doi: 10.1016/j.ceca.2008.07.003
132. Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria positioning controls local calcium influx in T cells. J Immunol (2010) 184(1):184–90. doi: 10.4049/jimmunol.0902872
133. Quintana A, Pasche M, Junker C, Al-Ansary D, Rieger H, Kummerow C, et al. Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J (2011) 30(19):3895–912. doi: 10.1038/emboj.2011.289
134. Babich A, Burkhardt JK. Coordinate control of cytoskeletal remodeling and calcium mobilization during T-cell activation. Immunol Rev (2013) 256(1):80–94. doi: 10.1111/imr.12123
135. Halimani M, Pattu V, Marshall MR, Chang HF, Matti U, Jung M, et al. Syntaxin11 serves as a t-SNARE for the fusion of lytic granules in human cytotoxic T lymphocytes. Eur J Immunol (2014) 44(2):573–84. doi: 10.1002/eji.201344011
136. Marshall MR, Pattu V, Halimani M, Maier-Peuschel M, Müller ML, Becherer U, et al. VAMP8-dependent fusion of recycling endosomes with the plasma membrane facilitates T lymphocyte cytotoxicity. J Cell Biol (2015) 210(1):135–51. doi: 10.1083/jcb.201411093
137. Neeft M, Wieffer M, de Jong AS, Negroiu G, Metz CH, van Loon A, et al. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell (2005) 16(2):731–41. doi: 10.1091/mbc.e04-10-0923
138. Ménasché G, Ménager MM, Lefebvre JM, Deutsch E, Athman R, Lambert N, et al. A newly identified isoform of Slp2a associates with Rab27a in cytotoxic T cells and participates to cytotoxic granule secretion. Blood (2008) 112(13):5052–62. doi: 10.1182/blood-2008-02-141069
139. Elstak ED, Neeft M, Nehme NT, Voortman J, Cheung M, Goodarzifard M, et al. The munc13-4-rab27 complex is specifically required for tethering secretory lysosomes at the plasma membrane. Blood (2011) 118(6):1570–8. doi: 10.1182/blood-2011-02-339523
140. Hackmann Y, Graham SC, Ehl S, Höning S, Lehmberg K, Aricò M, et al. Syntaxin binding mechanism and disease-causing mutations in Munc18-2. Proc Natl Acad Sci U.S.A. (2013) 110(47):E4482–4491. doi: 10.1073/pnas.1313474110
141. D'Orlando O, Zhao F, Kasper B, Orinska Z, Müller J, Hermans-Borgmeyer I, et al. Syntaxin 11 is required for NK and CD8+ T-cell cytotoxicity and neutrophil degranulation. Eur J Immunol (2013) 43(1):194–208. doi: 10.1002/eji.201142343
142. Chitirala P, Ravichandran K, Galgano D, Sleiman M, Krause E, Bryceson YT, et al. Cytotoxic granule exocytosis from human cytotoxic T lymphocytes is mediated by VAMP7. Front Immunol (20191855) 10:1855. doi: 10.3389/fimmu.2019.01855
143. Ménager MM, Ménasché G, Romao M, Knapnougel P, Ho CH, Garfa M, et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol (2007) 8(3):257–67. doi: 10.1038/ni1431
144. Dudenhöffer-Pfeifer M, Schirra C, Pattu V, Halimani M, Maier-Peuschel M, Marshall MR, et al. Different Munc13 isoforms function as priming factors in lytic granule release from murine cytotoxic T lymphocytes. Traffic (2013) 14(7):798–809. doi: 10.1111/tra.12074
145. Boswell KL, James DJ, Esquibel JM, Bruinsma S, Shirakawa R, Horiuchi H, et al. Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J Cell Biol (2012) 197(2):301–12. doi: 10.1083/jcb.201109132
146. Lopez JA, Noori T, Minson A, Li Jovanoska L, Thia K, Hildebrand MS, et al. Bi-allelic mutations in STXBP2 reveal a complementary role for STXBP1 in cytotoxic lymphocyte killing. Front Immunol (2018) 9:529. doi: 10.3389/fimmu.2018.00529
147. Sleiman M, Stevens DR, Chitirala P, Rettig J. Cytotoxic granule trafficking and fusion in Synaptotagmin7-deficient cytotoxic T lymphocytes. Front Immunol (20201080) 11:1080. doi: 10.3389/fimmu.2020.01080
148. Fowler KT, Andrews NW, Huleatt JW. Expression and function of synaptotagmin VII in CTLs. J Immunol (2007) 178(3):1498–504. doi: 10.4049/jimmunol.178.3.1498
149. Filali L, Puissegur MP, Cortacero K, Cussat-Blanc S, Khazen R, Van Acker N, et al. Ultrarapid lytic granule release from CTLs activates Ca(2+)-dependent synaptic resistance pathways in melanoma cells. Sci Adv (2022) 8(7):eabk3234. doi: 10.1126/sciadv.abk3234
150. Bin NR, Ma K, Tien CW, Wang S, Zhu D, Park S, et al. C2 domains of Munc13-4 are crucial for Ca(2+)-dependent degranulation and cytotoxicity in NK cells. J Immunol (2018) 201(2):700–13. doi: 10.4049/jimmunol.1800426
151. Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, et al. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell (2004) 118(3):389–401. doi: 10.1016/j.cell.2004.06.029
152. Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, et al. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca(2+)-calmodulin-Munc13-1 signaling. Neuron (2013) 79(1):82–96. doi: 10.1016/j.neuron.2013.05.011
153. Liu D, Martina JA, Wu XS, Hammer JA 3rd, Long EO. Two modes of lytic granule fusion during degranulation by natural killer cells. Immunol Cell Biol (2011) 89(6):728–38. doi: 10.1038/icb.2010.167
154. Bálint Š., Müller S, Fischer R, Kessler BM, Harkiolaki M, Valitutti S, et al. Supramolecular attack particles are autonomous killing entities released from cytotoxic T cells. Science (2020) 368(6493):897–901. doi: 10.1126/science.aay9207
155. Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol (1995) 25(4):1071–9. doi: 10.1002/eji.1830250432
156. Wiedemann A, Depoil D, Faroudi M, Valitutti S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc Natl Acad Sci U.S.A. (2006) 103(29):10985–90. doi: 10.1073/pnas.0600651103
157. Chang HF, Bzeih H, Schirra C, Chitirala P, Halimani M, Cordat E, et al. Endocytosis of cytotoxic granules is essential for multiple killing of target cells by T lymphocytes. J Immunol (2016) 197(6):2473–84. doi: 10.4049/jimmunol.1600828
158. Chang HF, Bzeih H, Chitirala P, Ravichandran K, Sleiman M, Krause E, et al. Preparing the lethal hit: interplay between exo- and endocytic pathways in cytotoxic T lymphocytes. Cell Mol Life Sci (2017) 74(3):399–408. doi: 10.1007/s00018-016-2350-7
159. Kumari A, Pineau J, Saez PJ, Maurin M, Lankar D, San Roman M, et al. Actomyosin-driven force patterning controls endocytosis at the immune synapse. Nat Commun (20192870) 10(1):2870. doi: 10.1038/s41467-019-10751-7
160. Lisci M, Barton PR, Randzavola LO, Ma CY, Marchingo JM, Cantrell DA, et al. Mitochondrial translation is required for sustained killing by cytotoxic T cells. Science (2021) 374(6565):eabe9977. doi: 10.1126/science.abe9977
161. Li P, Zheng G, Yang Y, Zhang C, Xiong P, Xu Y, et al. Granzyme b is recovered by natural killer cells via clathrin-dependent endocytosis. Cell Mol Life Sci (2010) 67(18):3197–208. doi: 10.1007/s00018-010-0377-8
162. Wang L, Sun R, Li P, Han Y, Xiong P, Xu Y, et al. Perforin is recaptured by natural killer cells following target cells stimulation for cytotoxicity. Cell Biol Int (2012) 36(2):223–8. doi: 10.1042/CBI20110242
163. Chang HF, Mannebach S, Beck A, Ravichandran K, Krause E, Frohnweiler K, et al. Cytotoxic granule endocytosis depends on the flower protein. J Cell Biol (2018) 217(2):667–83. doi: 10.1083/jcb.201706053
164. Yao CK, Lin YQ, Ly CV, Ohyama T, Haueter CM, Moiseenkova-Bell VY, et al. A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell (2009) 138(5):947–60. doi: 10.1016/j.cell.2009.06.033
165. Vasconcelos Z, Muller S, Guipouy D, Yu W, Christophe C, Gadat S, et al. Individual human cytotoxic T lymphocytes exhibit intraclonal heterogeneity during sustained killing. Cell Rep (2015) 11(9):1474–85. doi: 10.1016/j.celrep.2015.05.002
166. Higashio H, Nishimura N, Ishizaki H, Miyoshi J, Orita S, Sakane A, et al. Doc2 alpha and Munc13-4 regulate Ca(2+) -dependent secretory lysosome exocytosis in mast cells. J Immunol (2008) 180(7):4774–84. doi: 10.4049/jimmunol.180.7.4774
167. Buxton P, Zhang XM, Walsh B, Sriratana A, Schenberg I, Manickam E, et al. Identification and characterization of snapin as a ubiquitously expressed SNARE-binding protein that interacts with SNAP23 in non-neuronal cells. Biochem J (2003) 375(Pt 2):433–40. doi: 10.1042/bj20030427
168. Lu L, Cai Q, Tian JH, Sheng ZH. Snapin associates with late endocytic compartments and interacts with late endosomal SNAREs. Biosci Rep (2009) 29(4):261–9. doi: 10.1042/bsr20090043
169. Bertrand F, Muller S, Roh KH, Laurent C, Dupre L, Valitutti S. An initial and rapid step of lytic granule secretion precedes microtubule organizing center polarization at the cytotoxic T lymphocyte/target cell synapse. Proc Natl Acad Sci U.S.A. (2013) 110(15):6073–8. doi: 10.1073/pnas.1218640110
170. Sleiman M, Stevens DR, Rettig J. Simultaneous membrane capacitance measurements and TIRF microscopy to study granule trafficking at immune synapses. Methods Mol Biol (2017) 1584:157–69. doi: 10.1007/978-1-4939-6881-7_11
171. Offenhauser C, Lei N, Roy S, Collins BM, Stow JL, Murray RZ. Syntaxin 11 binds Vti1b and regulates late endosome to lysosome fusion in macrophages. Traffic (2011) 12(6):762–73. doi: 10.1111/j.1600-0854.2011.01189.x
172. Emperador-Melero J, Huson V, van Weering J, Bollmann C, Fischer von Mollard G, Toonen RF, et al. Vti1a/b regulate synaptic vesicle and dense core vesicle secretion via protein sorting at the golgi. Nat Commun (2018) 9(1):3421. doi: 10.1038/s41467-018-05699-z
173. Emperador-Melero J, Toonen RF, Verhage M. Vti proteins: beyond endolysosomal trafficking. Neuroscience (2019) 420:32–40. doi: 10.1016/j.neuroscience.2018.11.014
174. Qu B, Pattu V, Junker C, Schwarz EC, Bhat SS, Kummerow C, et al. Docking of lytic granules at the immunological synapse in human CTL requires Vti1b-dependent pairing with CD3 endosomes. J Immunol (2011) 186(12):6894–904. doi: 10.4049/jimmunol.1003471
175. Dressel R, Elsner L, Novota P, Kanwar N, Fischer von Mollard G. The exocytosis of lytic granules is impaired in Vti1b- or Vamp8-deficient CTL leading to a reduced cytotoxic activity following antigen-specific activation. J Immunol (2010) 185(2):1005–14. doi: 10.4049/jimmunol.1000770
176. Loo LS, Hwang LA, Ong YM, Tay HS, Wang CC, Hong W. A role for endobrevin/VAMP8 in CTL lytic granule exocytosis. Eur J Immunol (2009) 39(12):3520–8. doi: 10.1002/eji.200939378
177. DiGregorio DA, Peskoff A, Vergara JL. Measurement of action potential-induced presynaptic calcium domains at a cultured neuromuscular junction. J Neurosci (1999) 19(18):7846–59. doi: 10.1523/JNEUROSCI.19-18-07846.1999
178. Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol (2005) 15(3):266–74. doi: 10.1016/j.conb.2005.05.006
179. Kaschek L, Zöphel S, Knörck A, Hoth M. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. Semin Cell Dev Biol (2021) 115:10–8. doi: 10.1016/j.semcdb.2020.12.002
180. Dean C, Liu H, Staudt T, Stahlberg MA, Vingill S, Bückers J, et al. Distinct subsets of syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. J Neurosci (2012) 32(16):5398–413. doi: 10.1523/jneurosci.4515-11.2012
181. Shaib AH, Staudt A, Harb A, Klose M, Shaaban A, Schirra C, et al. Paralogs of the calcium-dependent activator protein for secretion differentially regulate synaptic transmission and peptide secretion in sensory neurons. Front Cell Neurosci (2018) 12:304 doi: 10.3389/fncel.2018.00304
182. Flašker A, Jorgačevski J, Calejo AI, Kreft M, Zorec R. Vesicle size determines unitary exocytic properties and their sensitivity to sphingosine. Mol Cell Endocrinol (2013) 376(1-2):136–47. doi: 10.1016/j.mce.2013.06.012
183. Kreutzberger AJB, Kiessling V, Stroupe C, Liang B, Preobraschenski J, Ganzella M, et al. In vitro fusion of single synaptic and dense core vesicles reproduces key physiological properties. Nat Commun (20193904) 10(1):3904. doi: 10.1038/s41467-019-11873-8
184. Okita K, Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos Trans R Soc Lond B Biol Sci (2011) 366(1575):2198–207. doi: 10.1098/rstb.2011.0016
Keywords: synapse, CD8+ cells, cytotoxic T lymphocytes, neuron, exocytosis, endocytosis, SNARE proteins
Citation: Chang H-F, Schirra C, Pattu V, Krause E and Becherer U (2023) Lytic granule exocytosis at immune synapses: lessons from neuronal synapses. Front. Immunol. 14:1177670. doi: 10.3389/fimmu.2023.1177670
Received: 01 March 2023; Accepted: 09 May 2023;
Published: 18 May 2023.
Edited by:
Michael Loran Dustin, University of Oxford, United KingdomReviewed by:
Jeremie Rossy, Biotechnology Institute Thurgau, SwitzerlandKaushik Choudhuri, University of Michigan, United States
Copyright © 2023 Chang, Schirra, Pattu, Krause and Becherer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ute Becherer, VXRlLkJlY2hlcmVyQHVrcy5ldQ==