 Durga Prasanna Misra
Durga Prasanna Misra Kritika Singh
Kritika Singh Aman Sharma
Aman Sharma Vikas Agarwal
Vikas Agarwal- 1Department of Clinical Immunology and Rheumatology, Sanjay Gandhi Postgraduate Institute of Medical Sciences (SGPGIMS), Lucknow, India
- 2Clinical Immunology and Rheumatology Services, Department of Internal Medicine, Post Graduate Institute of Medical Education and Research (PGIMER), Chandigarh, India
Arterial wall damage in Takayasu arteritis (TAK) can progress despite immunosuppressive therapy. Vascular fibrosis is more prominent in TAK than in giant cell arteritis (GCA). The inflamed arterial wall in TAK is infiltrated by M1 macrophages [which secrete interleukin-6 (IL-6)], which transition to M2 macrophages once the inflammation settles. M2 macrophages secrete transforming growth factor beta (TGF-β) and glycoprotein non-metastatic melanoma protein B (GPNMB), both of which can activate fibroblasts in the arterial wall adventitia. Mast cells in the arterial wall of TAK also activate resting adventitial fibroblasts. Th17 lymphocytes play a role in both TAK and GCA. Sub-populations of Th17 lymphocytes, Th17.1 lymphocytes [which secrete interferon gamma (IFN-γ) in addition to interleukin-17 (IL-17)] and programmed cell death 1 (PD1)-expressing Th17 (which secrete TGF-β), have been described in TAK but not in GCA. IL-6 and IL-17 also drive fibroblast activation in the arterial wall. The Th17 and Th1 lymphocytes in TAK demonstrate an activation of mammalian target organ of rapamycin 1 (mTORC1) driven by Notch-1 upregulation. A recent study reported that the enhanced liver fibrosis score (derived from serum hyaluronic acid, tissue inhibitor of metalloproteinase 1, and pro-collagen III amino-terminal pro-peptide) had a moderate-to-strong correlation with clinically assessed and angiographically assessed vascular damage. In vitro experiments suggest the potential to target arterial wall fibrosis in TAK with leflunomide, tofacitinib, baricitinib, or mTORC1 inhibitors. Since arterial wall inflammation is followed by fibrosis, a strategy of combining immunosuppressive agents with drugs that have an antifibrotic effect merits exploration in future clinical trials of TAK.
Introduction
Tissue injury resulting from transmural arterial inflammation in Takayasu arteritis (TAK) is consequent to the infiltration of different immune cells, including macrophages, dendritic cells, neutrophils, mast cells, and T lymphocytes. Such arterial wall inflammation heals with fibrosis, resulting in stenosed arteries with a distorted microarchitecture (1–3). For reasons not yet clear, vascular inflammation in TAK heals with a greater degree of fibrosis (resulting in arterial stenosis) than in the counterpart large vessel vasculitis (LVV) of giant cell arteritis (GCA), where arterial dilatation occurs more often (4–6). Stenosis in TAK can result in critical downstream ischemia (7) leading to myocardial infarction or ischemic stroke (7) which can occur even during inactive disease (8).
Relapses frequently occur when corticosteroids are tapered in TAK (9). Therefore, maintenance immunosuppressive therapy with disease-modifying anti-rheumatic drugs (DMARDs), whether conventional, biologic, or targeted synthetic, is usually initiated along with corticosteroids (10–14). However, quite unlike GCA, no immunosuppressive therapy has been proven to be beneficial against a placebo in a randomized controlled trial of TAK to date (13–15). To date, there are no validated clinical measures of damage (which may reflect vascular fibrosis) in TAK (15). However, angiographic scoring systems are available for TAK, which reflect vascular damage well (16). Angiographic extent of TAK might increase over time despite having inactive disease clinically (17). This necessitates lateral thinking to explore newer therapeutic avenues for TAK to target both arterial wall inflammation and vascular remodeling (18). Given the prominence of arterial wall fibrosis following the resolution of inflammation in TAK (2, 4–6), we review the literature regarding the pathogenic mechanisms driving arterial fibrosis in TAK and their potential for therapeutic modulation.
Cellular populations contributing towards arterial fibrosis in TAK
Macrophages
Infiltration of inflammatory M1 and reparative M2 macrophages (19) into the inflamed arterial wall is recognized in TAK. Kong et al. identified a preponderance of M1 macrophages and localization of the macrophage chemoattractant C-C motif ligand 2 (CCL2) in the adventitia of the arterial wall of TAK in immunosuppressive-naïve TAK. After immunosuppression, M2 macrophages were more prevalent in the media of the arterial wall where CCL-2 was now majorly expressed (as opposed to the adventitial layer previously) (20). Similarly, Cui et al. also reported a dominance of M1 macrophages coexistent with arterial wall inflammation and M2 macrophages coexistent with arterial wall fibrosis in TAK. The addition of leflunomide (but not corticosteroids) to peripheral blood monocytes of TAK cultured in vitro with monocyte colony-stimulating factor (M-CSF) reduced M2 macrophage polarization relative to M1 macrophages. Methotrexate only at higher doses inhibited M2 polarization to a similar extent as leflunomide. Cultured M2 macrophages in the presence of leflunomide secreted lesser CCL22 and transforming growth factor beta (TGF-β) into the culture supernatant. Leflunomide also induced M2 macrophage apoptosis. The same authors further used the THP-1 monocyte cell line polarized towards M2 macrophages using IL-4 and IL-13. Upon treatment with leflunomide, the cultured macrophages showed a reduced expression of IL10 and IRF4 genes (associated with M2 polarization). Pro-fibrotic gene expression (TGFB1, PDGFB, and LAGLS3) was also reduced with leflunomide, mediated by decreased STAT6 phosphorylation (21). Thus, leflunomide reduced M2 macrophage polarization and inhibited their pro-fibrotic phenotype in vitro (21). The role of macrophages in the vascular fibrosis of TAK is summarized in Figure 1.

Figure 1 Immune processes resulting in vascular fibrosis in Takayasu arteritis. Mast cells, M1 macrophages, and T lymphocytes infiltrate the arterial wall in TAK. Activation of Notch-1 with downstream activation of the mammalian target organ of rapamycin complex 1 (mTORC1) is a key mechanism driving the activation of Th1 and Th17 lymphocytes in the inflamed arterial wall of TAK. Mast cells, PD1+ Th17 lymphocytes (which secrete TGF-β), macrophages (source of IL-6), and Th17 and Th17.1 lymphocytes (through IL-17 secretion) activate the resting fibroblast population. Th17 lymphocytes also recruit neutrophils to the inflamed arterial wall resulting in tissue destruction. The activated fibroblast population secretes greater amounts of extracellular matrix and also releases further TGF-β into the inflamed arterial wall. Over time, the inflammatory M1 macrophage population transitions to an M2 phenotype that further secretes TGF-β. M2 macrophages also secrete glycoprotein non-metastatic melanoma protein B (GPNMB), which also drives fibroblast activation. Tissue injury and fibrosis in the arterial wall may be reflected by circulating levels of TGF-β; hyaluronic acid (HA); matrix metalloproteinases (MMP) 2, 3, and 9; tissue inhibitor of metalloproteinase 1 (TIMP-1); and pro-collagen III amino-terminal pro-peptide (PIIINP). Created with BioRender.com.
Mast cells
Mast cells are involved in tissue repair (22). Le Joncour et al. demonstrated elevated circulating markers of mast cell activation in TAK compared to healthy controls. Furthermore, they stimulated in vitro cultured mast cells with interleukin-33 (IL-33) with or without sera of TAK or healthy controls and treated cultured arterial wall fibroblasts with the supernatant of stimulated mast cells. Platelet-derived growth factor (PDGF) and TGF-β were elevated in the culture supernatant of mast cells incubated with TAK sera than those incubated with sera from healthy controls, unaffected by IL-33 inhibition or IL-6 inhibition. Cultured arterial wall fibroblasts treated with mast cell culture supernatant incubated with TAK sera had increased expression of markers of fibroblast activation, i.e., collagen 1, fibronectin, and alpha-smooth muscle actin (α-SMA) when compared with those treated with sera from TAK alone. Furthermore, greater activation of cultured arterial fibroblasts was observed with mast cell culture supernatant incubated with TAK sera than from healthy controls. These findings suggested a role for mast cells in driving the pathogenesis of vascular wall fibrosis in TAK (23). The role of mast cells in the vascular fibrosis of TAK is summarized in Figure 1.
T lymphocytes
Mammalian target organ of rapamycin complex 1 (mTORC1) is a therapeutic target for fibrosis (24–26). Zhang et al. reported an increased frequency of Th1 and Th17 lymphocytes when naïve CD4+ T lymphocytes from TAK were cultured in vitro when compared with those from healthy controls or with granulomatosis with polyangiitis (GPA, a small vessel vasculitis). Cultured CD4+ T lymphocytes from TAK demonstrated mTORC1 activation when compared with healthy controls or with GPA. In vitro treatment of CD4+ T lymphocytes from TAK with the mTORC1 inhibitor rapamycin or silencing of mTORC1 RNA expression reduced the frequencies of Th1 and Th17 lymphocytes. Circulating CD4+ T lymphocytes expressing mTORC1 moderately correlated with acute phase reactants erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). In a murine model of TAK using human axillary arteries implanted into NSG mice infused with peripheral blood mononuclear cells (PBMCs) from active TAK, treatment with rapamycin or silencing of mTORC1 RNA expression reduced arterial wall inflammation in vivo (27). Jiang et al. further identified Notch-1 upregulation in CD4+ T lymphocytes from TAK compared to healthy controls or GPA. Inhibiting Notch-1 expression using the gamma-secretase inhibitor DAPT or silencing RNA reduced Th1 and Th17 lymphocyte differentiation in cultured CD4+ T lymphocytes from TAK. Furthermore, Notch-1 mediated Th1 and Th17 lymphocyte polarization via mTORC1 activation (28). Maciejewski-Duval et al. further reported mTORC1 activation (identified by phosphorylation of S6 ribosomal protein) in the adventitia of LVV arteries (more in TAK than in GCA), which co-localized with CD3+ and CD4+ lymphocyte infiltration. Greater mTORC1 activation was evident in Th1 than in Th17 lymphocytes from TAK, GCA, and healthy controls. Th17 lymphocytes with mTORC1 activation were more prevalent in TAK than in GCA. Upon culturing PBMCs from TAK in vitro with rapamycin, decreased frequencies of Th1, Th17, and IL-21-expressing CD4+ T lymphocytes were observed than without rapamycin (29).
Programmed cell death 1 (PD1) was previously thought to be a marker of T lymphocyte exhaustion (30). However, a seminal report implicated PD1+CD4+ T lymphocytes and PD1+Th17 lymphocytes as key drivers of fibrosis via TGF-β1 secretion in idiopathic pulmonary fibrosis and sarcoidosis-associated pulmonary fibrosis (31). Elevated Th17 lymphocytes are associated with disease activity in TAK (32–34). Follicular helper T lymphocytes (TFH) that also express PD1 (35) are elevated in TAK (36). Tertiary lymphoid organs (TLOs) were more often identified in the arteries of TAK than of GCA. Increased PD1 expression was also noted in TLOs from the arterial wall of TAK when compared with TLOs from arteries affected by GCA (36). Work from our group identified elevated CD4+ PD1+ T lymphocytes in the peripheral blood of TAK than in healthy controls or sarcoidosis and increased PD1+ Th17 lymphocytes in TAK than in healthy controls. CD4+ PD1+ T lymphocytes and PD1+ Th17 lymphocytes did not significantly differ before or after immunosuppressive therapy in TAK (34). IL-23 helps to maintain the differentiated Th17 cell population (37). Interestingly, a previous genome-wide association study of TAK implicated the A allele in the rs6871626 single-nucleotide polymorphism (SNP) in the IL12B region as a risk allele for TAK (38). Furthermore, those TAK who were homozygous or heterozygous for the A allele of rs6871626 had greater vascular damage as indicated by higher scores on the vasculitis damage index (VDI) and the TAK Damage Score (TADS) (39). These observations suggest that Th17 lymphocytes, particularly the PD1+ Th17 subset, might drive vascular fibrosis in TAK. Inhibiting mTORC1 activation on T lymphocytes through rapamycin, sirolimus, or everolimus might target vascular fibrosis in TAK (13, 40). The role of T lymphocytes in the vascular fibrosis of TAK is summarized in Figure 1.
Aortic adventitial fibroblasts as drivers of fibrosis in TAK
IL-6 drives both vascular inflammation and vascular fibrosis in TAK. Kong et al. reported the co-localization of IL-6 and IL-6 receptor (IL-6R) with alpha-smooth muscle actin (α-SMA) in the arterial wall adventitia of TAK. Increased proliferation of human aortic adventitial fibroblasts (AAFs) was observed upon treatment in vitro with a combination of IL-6 and IL-6R, coupled with increased production of α-SMA, collagen 1, collagen 3, fibronectin, and TGF-β1, mediated through JAK2 acting downstream mainly through STAT3 and also via AKT (41). Chen et al. reported that IL-6 co-localized with Atg3 (a marker of autophagy) and α-SMA in the TAK aortic wall adventitia. Thereafter, they observed in vitro induction of autophagy in AAF upon treatment with IL-6 and IL-6R. The fibrotic phenotype of AAF induced by treatment with IL-6 and IL-6R could be reversed with the late-phase autophagy inhibitor bafilomycin A1, with decreased collagen-1 and fibronectin in the culture supernatant. Addition of JAK1 inhibitors tofacitinib and itacitinib to IL-6 and IL-6R in vitro reduced LC3-II expression (a marker of autophagy), autophagosome formation, and production of collagen 1 and fibronectin in the culture supernatant of AAF, mediated through STAT3 (42). These experiments identify potential mechanisms for the observed effectiveness of tocilizumab (12) and tofacitinib (11) in TAK.
Th17 lymphocytes, particularly the PD1+ Th17 population, had been associated with TAK (34). Ma et al. further dissected the potential role of IL-17 in the vascular fibrosis of TAK. They reported increased cysteine-rich protein 61 (CYR61) expression in the adventitia of arteries from TAK. Upon stimulating the AAF with CYR61, increased production of fibronectin, collagen 1, collagen 3, and TGF-β1 were observed, mediated by αVβ1 receptor via ERK1 and ERK2. Treatment of AAF with recombinant human IL-17 stimulated CYR61 secretion. Co-culture of AAF with IL-17 and CYR-61 markedly enhanced the pro-fibrotic phenotype when compared with CYR61 alone (43).
A GWAS study identified the A allele in the rs2069837 SNP in the IL6 region as a risk allele for TAK (44). This SNP repressed glycoprotein non-metastatic melanoma protein B (GPNMB), which has anti-inflammatory effects (44, 45). Interestingly, M2 macrophages that are more prevalent in fibrotic arteries than in inflamed arteries of TAK secrete GPNMB (46). Dai et al. identified GPNMB co-expression with collagen 1, fibronectin, TGF-β, matrix metalloproteinase 2 (MMP2), and MMP9 in the adventitial layer of arteries from TAK than from non-inflammatory controls. In the TAK arterial wall, GPNMB co-localized with macrophages and fibroblasts. The culture supernatant of THP-1 macrophages overexpressing GPNMB induced the expression of collagen 1, fibronectin, TGF-β, MMP2, and MMP9 in AAF when compared with the culture supernatant from THP-1 macrophages with a knock-down of GPNMB. After treating AAF with soluble GPNMB in the culture media, a similar overexpression of collagen 1, fibronectin, TGF-β, MMP2, and MMP9 with increased fibroblast proliferation and migration was observed. Opposite effects were observed after knocking down GPNMB in the AAF. The effect of GPNMB on AAF was mediated via the αVβ1 receptor acting downstream on AKT and ERK-1/2 pathways. In vitro treatment of AAF with GPNMB along with leflunomide, tofacitinib, or baricitinib reduced collagen 1, fibronectin, TGF-β, MMP2, and MMP9 expression when compared with GPNMB alone. Leflunomide also suppressed GPNMB production from THP-1 macrophages in vitro. However, in 22 patients with TAK treated with leflunomide and corticosteroids, inconsistent changes in circulating GBNMB were observed (47). The role of adventitial fibroblasts in the vascular fibrosis of TAK is summarized in Figure 1.
Circulating proteins as biomarkers of fibrosis in TAK
Kong et al. identified, among numerous serum chemokines, an increase in CCL22 (produced by M2 macrophages) (48) following immunosuppressive therapy, whereas the levels of IL-16 did not change following immunosuppressive therapy (49). CCL22 has been implicated previously in lung fibrosis (50) and IL-16 in cardiac fibrosis (51). The persistent elevation of these chemokines in TAK despite immunosuppressive therapy might also contribute towards vascular fibrosis while inflammation resolves.
Another recent study explored the enhanced liver fibrosis (ELF) score, a validated circulating biomarker of liver fibrosis derived from serum levels of hyaluronic acid (HA), tissue inhibitor of metalloproteinase 1 (TIMP-1), and pro-collagen III amino-terminal pro-peptide (PIIINP), in 24 patients with TAK. The ELF score moderately correlated with clinical damage indices VDI and TADS and strongly correlated with angiographically assessed vascular damage using the Combined Arteritis Damage Score (52).
MMP2, MMP3, and MMP9 are secreted by fibroblasts during tissue remodeling, including in the inflamed arterial wall. Some studies (15) but not others have associated circulating levels of MMP2, MMP3 (53), and MMP9 with TAK disease activity (54). Whether MMP2, MMP3, and MMP9 are associated with vascular fibrosis in TAK remains to be evaluated. Supplementary Table S1 summarizes potential biomarkers for further evaluation of their association with vascular fibrosis in TAK.
Differences in the pathogenesis of TAK and GCA—are differences in Th17 subtypes a key feature?
Vascular fibrosis is prominent in affected arteries of TAK but less so in GCA (2, 4–6). Th17 lymphocytes were initially implicated in GCA (55). Subsequent reports identified a role for Th17 lymphocytes in TAK (32–34), including an association with active TAK (32, 34). Th17 lymphocytes attract neutrophils to the arterial wall (56). Recently, IL-17 has been implicated in granuloma formation (57, 58), a pathological feature of both TAK and GCA (2). Th17.1 lymphocytes implicated in TAK (34) also secrete interferon-gamma (classically secreted by Th1 lymphocytes) (59), which also drives granuloma formation (60).
The Th17 population in GCA is corticosteroid-responsive (55). However, a reduction in Th17 lymphocytes following immunosuppressive therapy was not observed in TAK (32, 33). The poor responsiveness of Th17 lymphocytes in TAK could be explained by the elevated Th17.1 population known to express the drug efflux protein p-glycoprotein (thereby conferring corticosteroid resistance) (34, 61). Th17.1 lymphocytes have not yet been described in GCA (61). Despite elevated tumor necrosis factor-alpha (TNF-α) levels in both TAK (62) and GCA (63), TNF-α inhibitors (TNFi) are effective in TAK (10) but not in GCA (64). Blocking TNF-α inhibits p-glycoprotein expression (65), which might be one of the mechanisms driving the effectiveness of TNFi in TAK.
PD1+ Th17 lymphocytes secrete TGF-β, which drives arterial wall fibrosis in TAK (34) along with IL-17 secreted by Th17 lymphocytes. PD1+ Th17 lymphocytes have not yet been described in GCA. Such differences in Th17 sub-populations could explain distinct vascular pathology encountered in TAK or GCA (Figure 2).
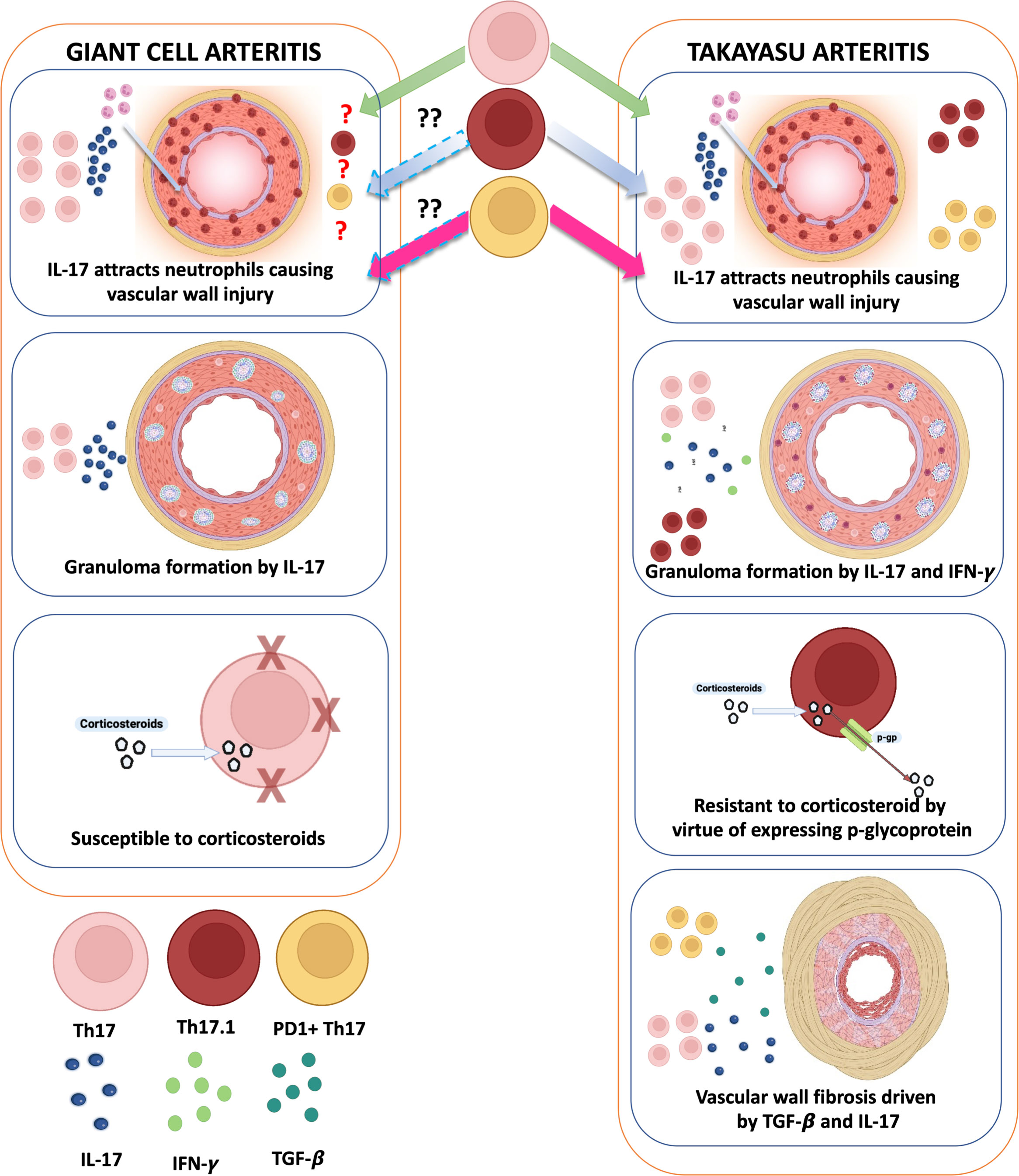
Figure 2 Distinct roles of Th17 lymphocytes in giant cell arteritis and Takayasu arteritis: do they underlie mechanistic differences in vascular fibrosis? In giant cell arteritis (GCA), Th17 lymphocytes, which are sensitive to corticosteroids, predominantly secrete IL-17, which attracts neutrophils causing inflammation of the vascular wall. IL-17 also contributes to granuloma formation in the vascular wall. In Takayasu arteritis (TAK), apart from these roles, Th17.1 lymphocytes also secrete IFN-γ (contributing towards granuloma formation) and express p-glycoprotein (contributing towards corticosteroid resistance). Th17 lymphocytes expressing programmed cell death 1 (PD1) also secrete TGF-β1, which, along with IL-17, contributes towards vascular fibrosis. Created with BioRender.com.
Detection of fibrosis in vivo in Takayasu arteritis
Computed tomography (CTA), magnetic resonance (MRA), or conventional angiography helps delineate the arterial tree anatomy in TAK and other LVV. CTA and MRA also provide information about arterial wall characteristics, more easily appreciated when intravenous contrast is administered during angiography (15). Late gadolinium enhancement of the arterial wall on MRA may indicate either inflammation or fibrosis (66, 67).
18-Fluorodeoxyglucose (18F-FDG) positron emission tomography (PET), combined with computed tomography (CT) or more recently magnetic resonance imaging (MRI) for anatomic localization, is increasingly being used to visualize metabolic activity in the arterial wall in TAK (15). A recent paper proposed that concomitant arterial wall 18F-FDG uptake and wall thickening evident on MRA might indicate ongoing inflammation, whereas wall thickening without 18F-FDG uptake might indicate vascular fibrosis (68).
68-Gadolinium (68-Ga)-tagged fibroblast activation protein inhibitor (FAPI)-PET identifies in vivo fibroblast activity in solid organ tumors or lungs in the context of interstitial lung diseases (69). A case report described a young female with clinically active TAK without 18-FDG-PET uptake but with extensive arterial uptake using 68-Ga-FAPI-PET (70). It remains to be explored whether 68-Ga-FAPI-PET can enable the identification of areas of ongoing arterial fibrosis in TAK.
Prospects to therapeutically target arterial fibrosis in Takayasu arteritis
mTORC1 activation drives the differentiation to Th1, Th17, and IL-21+CD4+ T lymphocytes in TAK (27–29). Elevated PD1+ Th17 lymphocytes in TAK serve as a source of TGF-β (34). mTORC1 activation has also been noted in the endothelial cells of TAK but not in GCA. Purified immunoglobulin G1 (IgG1) from TAK induced endothelial cell proliferation in vitro through the PI3K/AKT pathway, inhibited by the mTORC1 inhibitor sirolimus or through a direct inhibitor of PI3K (71). Sirolimus and everolimus are mTORC1 inhibitors commonly used in clinical practice (40). mTORC1 inhibition has antifibrotic effects in kidneys affected with antiphospholipid antibody syndrome (25). mTORC1 inhibition has also been proposed as a therapeutic modality for pulmonary fibrosis (24). Therefore, mTORC1 inhibition might ameliorate both inflammation and fibrosis in TAK. Sirolimus has been anecdotally used in TAK (72). Leflunomide suppresses M2 macrophages and aortic adventitial fibroblasts in vitro (21, 47). The JAK inhibitors tofacitinib and baricitinib also suppressed aortic adventitial fibroblasts in vitro (47).
Sirolimus-coated stents inhibit endothelial proliferation and reduce re-stenosis after endovascular stenting. A case report described a patient with TAK with repeated coronary artery stenosis despite a sirolimus-eluting stent where the addition of systemic corticosteroids prevented restenosis from occurring with a sirolimus-eluting stent alone (73). This suggests the potential benefits of combining immunosuppressive therapy with antifibrotic drugs in TAK.
To explore this concept further, we treated PBMCs from TAK cultured in vitro with tacrolimus (a calcineurin inhibitor) and tadalafil (a phosphodiesterase 5 inhibitor that increases intracellular levels of cyclic guanosine monophosphate, thereby suppressing canonical and non-canonical signaling pathways downstream to TGF-β). Upon stimulation of the PBMCs with anti-CD3/CD28, treatment with tacrolimus and tadalafil significantly reduced the levels of IL-6, IL-17A, IL-1β, and IL-10 in the culture supernatant than tacrolimus alone (34). IL-6 (41, 42) and IL-17 (43) activate aortic adventitial fibroblasts in TAK. IL-10 (74) and IL-1β (75) have been associated with organ fibrosis in clinical and pre-clinical models. The synergistic effect of tacrolimus and tadalafil on various cytokines involved in fibrosis in vitro on cultured PBMCs from TAK (34) suggests the potential to explore a combination of immunosuppressive and antifibrotic therapies in TAK. Given that no clinical trial of an immunosuppressive agent in TAK has met its primary endpoint, future trials should consider combining DMARDs with antifibrotic drugs.
Conclusion
Excessive vascular fibrosis characterizes TAK. Th17 lymphocyte populations, M2 macrophages, and mast cells drive aortic adventitial fibroblast activation leading to arterial wall fibrosis. In vitro experiments suggest the potential to target arterial wall fibrosis in TAK with leflunomide, tofacitinib, baricitinib, or mTORC1 inhibitors. Since arterial wall inflammation begets fibrosis, a strategy of combining immunosuppressive agents with antifibrotic drugs merits exploration in future clinical trials of TAK.
Author contributions
Conception and design of the study: DM, AS, and VA. Acquisition of data, analysis, and interpretation of data: DM and KS. Drafting the article: DM and KS. Critically revising the article for important intellectual content: AS and VA. Final approval of the version to be submitted: DM, KS, AS, and VA. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: DM, KS, AS, and VA. All authors contributed to the article and approved the submitted version.
Funding
DM acknowledges support from the Indian Council of Medical Research (grant number 5/4/ 1-2/2019-NCD-II) for some of the work on which this paper is based. KS is supported by a Senior Research Fellowship from the Indian Council of Medical Research (grant number No. 3/1/1(20)/2022-NCD-I).
Acknowledgments
DM would like to acknowledge the support of his late father Dr. Sasanka Sekhar Misra for his academic endeavors.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1174249/full#supplementary-material
References
1. Pugh D, Karabayas M, Basu N, Cid MC, Goel R, Goodyear CS, et al. Large-Vessel vasculitis. Nat Rev Dis Primers (2022) 7(1):93. doi: 10.1038/s41572-021-00327-5
2. Bajocchi G, Cavazza A. Histopathology of vasculitis. Reumatismo (2018) 70(3):155–64. doi: 10.4081/reumatismo.2018.1096
3. Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z. Pathogenesis of takayasu's arteritis: a 2011 update. Autoimmun Rev (2011) 11(1):61–7. doi: 10.1016/j.autrev.2011.08.001
4. Gornik HL, Creager MA. Aortitis. Circulation (2008) 117(23):3039–51. doi: 10.1161/CIRCULATIONAHA.107.760686
5. Harky A, Fok M, Balmforth D, Bashir M. Pathogenesis of Large vessel vasculitis: implications for disease classification and future therapies. Vasc Med (2018) 24(1):79–88. doi: 10.1177/1358863X18802989
6. Tombetti E, Mason J. Large Vessel vasculitis: the search for response biomarkers. Expert Rev Clin Immunol (2016) 12(10):1011–3. doi: 10.1080/1744666X.2016.1224180
7. Misra DP, Wakhlu A, Agarwal V, Danda D. Recent advances in the management of takayasu arteritis. Int J Rheum Dis (2019) 22 Suppl 1:60–8. doi: 10.1111/1756-185x.13285
8. Misra DP, Rathore U, Mishra P, Singh K, Thakare DR, Behera MR, et al. Comparison of presentation and prognosis of takayasu arteritis with or without stroke or transient ischemic attack-a retrospective cohort study. Life (Basel) (2022) 12(11):1904. doi: 10.3390/life12111904
9. Misra DP, Rathore U, Patro P, Agarwal V, Sharma A. Corticosteroid monotherapy for the management of takayasu arteritis-a systematic review and meta-analysis. Rheumatol Int (2021) 41(10):1729–42. doi: 10.1007/s00296-021-04958-5
10. Misra DP, Rathore U, Patro P, Agarwal V, Sharma A. Disease-modifying anti-rheumatic drugs for the management of takayasu arteritis–a systematic review and meta-analysis. Clin Rheumatol (2021) 40(11):4391–416. doi: 10.1007/s10067-021-05743-2
11. Rathore U, Thakare DR, Patro P, Agarwal V, Sharma A, Misra DP. A systematic review of clinical and preclinical evidences for janus kinase inhibitors in Large vessel vasculitis. Clin Rheumatol (2022) 41(1):33–44. doi: 10.1007/s10067-021-05973-4
12. Misra DP, Singh K, Rathore U, Patro P, Tomelleri A, Campochiaro C, et al. The effectiveness of tocilizumab and its comparison with tumor necrosis factor alpha inhibitors for takayasu arteritis: a systematic review and meta-analysis. Autoimmun Rev (2023) 22(3):103275. doi: 10.1016/j.autrev.2023.103275
13. Misra DP, Sharma A, Kadhiravan T, Negi VS. A scoping review of the use of non-biologic disease modifying anti-rheumatic drugs in the management of Large vessel vasculitis. Autoimmun Rev (2017) 16(2):179–91. doi: 10.1016/j.autrev.2016.12.009
14. Ferfar Y, Mirault T, Desbois AC, Comarmond C, Messas E, Savey L, et al. Biotherapies in Large vessel vasculitis. Autoimmun Rev (2016) 15(6):544–51. doi: 10.1016/j.autrev.2016.02.012
15. Misra DP, Jain N, Ora M, Singh K, Agarwal V, Sharma A. Outcome measures and biomarkers for disease assessment in takayasu arteritis. Diagnostics (Basel) (2022) 12(10):2565. doi: 10.3390/diagnostics12102565
16. Tombetti E, Godi C, Ambrosi A, Doyle F, Jacobs A, Kiprianos AP, et al. Novel angiographic scores for evaluation of Large vessel vasculitis. Sci Rep (2018) 8(1):15979. doi: 10.1038/s41598-018-34395-7
17. Direskeneli H, Aydin SZ, Merkel PA. Assessment of disease activity and progression in takayasu's arteritis. Clin Exp Rheumatol (2011) 29(1 Suppl 64):S86–91.
18. Tombetti E, Manfredi A, Sabbadini MG, Baldissera E. Management options for takayasu arteritis. Expert Opin Orphan Drugs (2013) 1(9):685–93. doi: 10.1517/21678707.2013.827570
19. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
20. Kong X, Xu M, Cui X, Ma L, Cheng H, Hou J, et al. Potential role of macrophage phenotypes and Ccl2 in the pathogenesis of takayasu arteritis. Front Immunol (2021) 12:646516. doi: 10.3389/fimmu.2021.646516
21. Cui X, Kong X, Chen R, Ma L, Jiang L. The potential role of leflunomide in inhibiting vascular fibrosis by down-regulating type-ii macrophages in takayasu's arteritis. Clin Exp Rheumatol (2020) 38 Suppl 124(2):69–78.
22. Ng MF. The role of mast cells in wound healing. Int Wound J (2010) 7(1):55–61. doi: 10.1111/j.1742-481X.2009.00651.x
23. Le Joncour A, Desbois AC, Leroyer AS, Tellier E, Régnier P, Maciejewski-Duval A, et al. Mast cells drive pathologic vascular lesions in takayasu arteritis. J Allergy Clin Immunol (2022) 149(1):292–301.e3. doi: 10.1016/j.jaci.2021.05.003
24. Platé M, Guillotin D, Chambers RC. The promise of mtor as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur Respir Rev (2020) 29(157):200269. doi: 10.1183/16000617.0269-2020
25. Canaud G, Bienaimé F, Tabarin F, Bataillon G, Seilhean D, Noël LH, et al. Inhibition of the mtorc pathway in the antiphospholipid syndrome. N Engl J Med (2014) 371(4):303–12. doi: 10.1056/NEJMoa1312890
26. Woodcock HV, Eley JD, Guillotin D, Platé M, Nanthakumar CB, Martufi M, et al. The Mtorc1/4e-Bp1 axis represents a critical signaling node during fibrogenesis. Nat Commun (2019) 10(1):6. doi: 10.1038/s41467-018-07858-8
27. Zhang J, Zhao L, Wang J, Cheng Z, Sun M, Zhao J, et al. Targeting mechanistic target of rapamycin complex 1 restricts proinflammatory T cell differentiation and ameliorates takayasu arteritis. Arthritis Rheumatol (2020) 72(2):303–15. doi: 10.1002/art.41084
28. Jiang W, Sun M, Wang Y, Zheng M, Yuan Z, Mai S, et al. Critical role of notch-1 in mechanistic target of rapamycin hyperactivity and vascular inflammation in patients with takayasu arteritis. Arthritis Rheumatol (2022) 74(7):1235–44. doi: 10.1002/art.42103
29. Maciejewski-Duval A, Comarmond C, Leroyer A, Zaidan M, Le Joncour A, Desbois AC, et al. Mtor pathway activation in Large vessel vasculitis. J Autoimmun (2018) 94:99–109. doi: 10.1016/j.jaut.2018.07.013
30. Lee J, Ahn E, Kissick HT, Ahmed R. Reinvigorating exhausted T cells by blockade of the pd-1 pathway. For Immunopathol Dis Therap (2015) 6(1-2):7–17. doi: 10.1615/ForumImmunDisTher.2015014188
31. Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT, et al. Pd-1 up-regulation on Cd4(+) T cells promotes pulmonary fibrosis through Stat3-mediated il-17a and tgf-Β1 production. Sci Transl Med (2018) 10(460):eaar8356. doi: 10.1126/scitranslmed.aar8356
32. Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, et al. Th1 and Th17 cytokines drive inflammation in takayasu arteritis. Arthritis Rheumatol (2015) 67(5):1353–60. doi: 10.1002/art.39037
33. Misra DP, Chaurasia S, Misra R. Increased circulating Th17 cells, serum il-17a, and il-23 in takayasu arteritis. Autoimmune Dis (2016) 2016:7841718. doi: 10.1155/2016/7841718
34. Singh K, Rathore U, Rai MK, Behera MR, Jain N, Ora M, et al. Novel Th17 lymphocyte populations, Th17.1 and Pd1+Th17, are increased in takayasu arteritis, andboth Th17 and Th17.1 Sub-populations associate with active disease. J Inflamm Res (2022) 15:1521–41. doi: 10.2147/JIR.S355881
35. Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. Pd-1 controls follicular T helper cell positioning and function. Immunity (2018) 49(2):264–74.e4. doi: 10.1016/j.immuni.2018.06.012
36. Desbois AC, Régnier P, Quiniou V, Lejoncour A, Maciejewski-Duval A, Comarmond C, et al. Specific follicular helper T cell signature in takayasu arteritis. Arthritis Rheumatol (2021) 73(7):1233–43. doi: 10.1002/art.41672
37. Stritesky GL, Yeh N, Kaplan MH. Il-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol (2008) 181(9):5948–55. doi: 10.4049/jimmunol.181.9.5948
38. Terao C, Yoshifuji H, Kimura A, Matsumura T, Ohmura K, Takahashi M, et al. Two susceptibility loci to takayasu arteritis reveal a synergistic role of the Il12b and hla-b regions in a Japanese population. Am J Hum Genet (2013) 93(2):289–97. doi: 10.1016/j.ajhg.2013.05.024
39. Kadoba K, Watanabe R, Iwasaki T, Nakajima T, Kitagori K, Akizuki S, et al. A susceptibility locus in the Il12b but not Lilra3 region is associated with vascular damage in takayasu arteritis. Sci Rep (2021) 11(1):13667. doi: 10.1038/s41598-021-93213-9
40. Xie J, Wang X, Proud CG. Mtor inhibitors in cancer therapy. F1000Res (2016) 5:F1000 Faculty Rev-2078. doi: 10.12688/f1000research.9207.1
41. Kong X, Ma L, Ji Z, Dong Z, Zhang Z, Hou J, et al. Pro-fibrotic effect of il-6 Via aortic adventitial fibroblasts indicates il-6 as a treatment target in takayasu arteritis. Clin Exp Rheumatol (2018) 36(1):62–72.
42. Chen R, Sun Y, Cui X, Ji Z, Kong X, Wu S, et al. Autophagy promotes aortic adventitial fibrosis Via the il-6/Jak1 signaling pathway in takayasu's arteritis. J Autoimmun (2019) 99:39–47. doi: 10.1016/j.jaut.2019.01.010
43. Ma L, Kong X, Cui X, Wu S, Wang Y, Dai X, et al. Cyr61/Tgf-Β axis promotes adventitial fibrosis of takayasu's arteritis in the il-17 mediated inflammatory microenvironment. Clin Exp Rheumatol (2020) 38(6):1102–11.
44. Kong X, Sawalha AH. Takayasu arteritis risk locus in Il6 represses the anti-inflammatory gene gpnmb through chromatin looping and recruiting Mef2-hdac complex. Ann Rheum Dis (2019) 78(10):1388–97. doi: 10.1136/annrheumdis-2019-215567
45. Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, et al. Identification of susceptibility loci in Il6, Rps9/Lilrb3, and an intergenic locus on chromosome 21q22 in takayasu arteritis in a genome-wide association study. Arthritis Rheumatol (2015) 67(5):1361–8. doi: 10.1002/art.39035
46. Zhang H, Zhang S, Dang X, Lin L, Ren L, Song R. Gpnmb plays an active role in the M1/M2 balance. Tissue Cell (2022) 74:101683. doi: 10.1016/j.tice.2021.101683
47. Dai X, Sun Y, Ma L, Hou J, Wang L, Gong Y, et al. A novel molecular mechanism of vascular fibrosis in takayasu arteritis: macrophage-derived gpnmb promoting adventitial fibroblast extracellular matrix production in the aorta. Transl Res (2023) 255:128–139. doi: 10.1016/j.trsl.2022.12.004
48. Soldano S, Pizzorni C, Paolino S, Trombetta AC, Montagna P, Brizzolara R, et al. Alternatively activated (M2) macrophage phenotype is inducible by endothelin-1 in cultured human macrophages. PloS One (2016) 11(11):e0166433. doi: 10.1371/journal.pone.0166433
49. Kong X, Wu S, Dai X, Yu W, Wang J, Sun Y, et al. A comprehensive profile of chemokines in the peripheral blood and vascular tissue of patients with takayasu arteritis. Arthritis Res Ther (2022) 24(1):49. doi: 10.1186/s13075-022-02740-x
50. Yogo Y, Fujishima S, Inoue T, Saito F, Shiomi T, Yamaguchi K, et al. Macrophage derived chemokine (Ccl22), thymus and activation-regulated chemokine (Ccl17), and Ccr4 in idiopathic pulmonary fibrosis. Respir Res (2009) 10(1):80. doi: 10.1186/1465-9921-10-80
51. Tamaki S, Mano T, Sakata Y, Ohtani T, Takeda Y, Kamimura D, et al. Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PloS One (2013) 8(7):e68893. doi: 10.1371/journal.pone.0068893
52. Stojanovic M, Raskovic S, Milivojevic V, Miskovic R, Soldatovic I, Stankovic S, et al. Enhanced liver fibrosis score as a biomarker for vascular damage assessment in patients with takayasu arteritis-a pilot study. J Cardiovasc Dev Dis (2021) 8(12):187. doi: 10.3390/jcdd8120187
53. Ishihara T, Haraguchi G, Kamiishi T, Tezuka D, Inagaki H, Isobe M. Sensitive assessment of activity of takayasu's arteritis by Pentraxin3, a new biomarker. J Am Coll Cardiol (2011) 57(16):1712–3. doi: 10.1016/j.jacc.2010.10.058
54. Arraes AE, de Souza AW, Mariz HA, Silva NP, Torres IC, Pinto PN, et al. (18)F-fluorodeoxyglucose positron emission tomography and serum cytokines and matrix metalloproteinases in the assessment of disease activity in takayasu's arteritis. Rev Bras Reumatol Engl Ed (2016) 56(4):299–308. doi: 10.1016/j.rbre.2015.08.007
55. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation (2010) 121(7):906–15. doi: 10.1161/circulationaha.109.872903
56. Xu S, Cao X. Interleukin-17 and its expanding biological functions. Cell Mol Immunol (2010) 7(3):164–74. doi: 10.1038/cmi.2010.21
57. Tristão FSM, Rocha FA, Carlos D, Ketelut-Carneiro N, Souza COS, Milanezi CM, et al. Th17-inducing cytokines il-6 and il-23 are crucial for granuloma formation during experimental paracoccidioidomycosis. Front Immunol (2017) 8:949. doi: 10.3389/fimmu.2017.00949
58. Shen H, Chen ZW. The crucial roles of Th17-related Cytokines/Signal pathways in m. Tuberculosis Infection Cell Mol Immunol (2018) 15(3):216–25. doi: 10.1038/cmi.2017.128
59. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-inflammatory human Th17 cells selectively express p-glycoprotein and are refractory to glucocorticoids. J Exp Med (2014) 211(1):89–104. doi: 10.1084/jem.20130301
60. Kak G, Raza M, Tiwari BK. Interferon-gamma (Ifn-Γ): exploring its implications in infectious diseases. Biomol Concepts (2018) 9(1):64–79. doi: 10.1515/bmc-2018-0007
61. Misra DP, Agarwal V. Th17.1 lymphocytes: emerging players in the orchestra of immune-mediated inflammatory diseases. Clin Rheumatol (2022) 41(8):2297–308. doi: 10.1007/s10067-022-06202-2
62. Tripathy NK, Gupta PC, Nityanand S. High tnf-alpha and low il-2 producing T cells characterize active disease in takayasu's arteritis. Clin Immunol (2006) 118(2-3):154–8. doi: 10.1016/j.clim.2005.09.010
63. Lozano E, Segarra M, García-Martínez A, Espígol-Frigolé G, Hernández-Rodríguez J, Cid MC. New therapeutic targets in giant-cell arteritis. considerations based on the current pathogenic model and the availability of new therapeutic agents. Clin Exp Rheumatol (2008) 26(3 Suppl 49):S141–50.
64. Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. 2018 Update of the eular recommendations for the management of Large vessel vasculitis. Ann Rheum Dis (2020) 79(1):19–30. doi: 10.1136/annrheumdis-2019-215672
65. Berguetti T, Quintaes LSP, Hancio T, Robaina MC, Cruz ALS, Maia RC, et al. Tnf-Α modulates p-glycoprotein expression and contributes to cellular proliferation Via extracellular vesicles. Cells (2019) 8(5):500. doi: 10.3390/cells8050500
66. Desai MY, Stone JH, Foo TK, Hellmann DB, Lima JA, Bluemke DA. Delayed contrast-enhanced mri of the aortic wall in takayasu's arteritis: initial experience. AJR Am J Roentgenol (2005) 184(5):1427–31. doi: 10.2214/ajr.184.5.01841427
67. Schneeweis C, Schnackenburg B, Stuber M, Berger A, Schneider U, Yu J, et al. Delayed contrast-enhanced mri of the coronary artery wall in takayasu arteritis. PloS One (2012) 7(12):e50655. doi: 10.1371/journal.pone.0050655
68. Laurent C, Ricard L, Fain O, Buvat I, Adedjouma A, Soussan M, et al. Pet/Mri in Large-vessel vasculitis: clinical value for diagnosis and assessment of disease activity. Sci Rep (2019) 9(1):12388. doi: 10.1038/s41598-019-48709-w
69. Röhrich M, Leitz D, Glatting FM, Wefers AK, Weinheimer O, Flechsig P, et al. Fibroblast activation protein-specific Pet/Ct imaging in fibrotic interstitial lung diseases and lung cancer: a translational exploratory study. J Nucl Med (2022) 63(1):127–33. doi: 10.2967/jnumed.121.261925
70. Wu S, Pang Y, Zhao L, Zhao L, Chen H. 68ga-fapi Pet/Ct versus 18f-fdg Pet/Ct for the evaluation of disease activity in takayasu arteritis. Clin Nucl Med (2021) 46(10):847–9. doi: 10.1097/rlu.0000000000003692
71. Hadjadj J, Canaud G, Mirault T, Samson M, Bruneval P, Régent A, et al. Mtor pathway is activated in endothelial cells from patients with takayasu arteritis and is modulated by serum immunoglobulin G. Rheumatol (Oxford) (2018) 57(6):1011–20. doi: 10.1093/rheumatology/key017
72. Misra DP, Tomelleri A, Rathore U, Benanti G, Singh K, Behera MR, et al. Impact of geographic location on diagnosis and initial management of takayasu arteritis: a tale of two cohorts from Italy and India. Diagnostics (Basel) (2022) 12(12):3102. doi: 10.3390/diagnostics12123102
73. Yokota K, Shimpo M, Iwata T, Hirose M, Ikemoto T, Ohya K, et al. A case of takayasu arteritis with repeated coronary artery restenosis after drug-eluting stent implantation successfully treated with a combination of steroids. Intern Med (2012) 51(7):739–43. doi: 10.2169/internalmedicine.51.6344
74. Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for tgf-beta and il-10. Eur Respir J (2003) 22(1):69–76. doi: 10.1183/09031936.03.00014703
Keywords: Takayasu arteritis, giant cell arteritis, fibrosis, T lymphocyte, fibroblast, mechanistic target of rapamycin complex 1
Citation: Misra DP, Singh K, Sharma A and Agarwal V (2023) Arterial wall fibrosis in Takayasu arteritis and its potential for therapeutic modulation. Front. Immunol. 14:1174249. doi: 10.3389/fimmu.2023.1174249
Received: 26 February 2023; Accepted: 20 April 2023;
Published: 15 May 2023.
Edited by:
Ryu Watanabe, Osaka Metropolitan University, JapanReviewed by:
Enrico Tombetti, University of Milan, ItalyCopyright © 2023 Misra, Singh, Sharma and Agarwal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Durga Prasanna Misra, ZHVyZ2FwbWlzcmFAZ21haWwuY29t; ZHBtaXNyYUBzZ3BnaS5hYy5pbg==
†These authors share first authorship
‡ORCID: Durga Prasanna Misra, orcid.org/0000-0002-5035-7396
Kritika Singh, orcid.org/0000-0002-3814-4179
Aman Sharma, orcid.org/0000-0003-0813-1243
Vikas Agarwal, orcid.org/0000-0002-4508-1233