 Shirin Emtenani
Shirin Emtenani Michael Hertl2†
Michael Hertl2† Enno Schmidt
Enno Schmidt Christoph Hudemann
Christoph Hudemann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 27 April 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1169947
This article is part of the Research Topic Pemphigus and pemphigoid diseases: in memoriam Detlef Zillikens View all 38 articles
Autoimmune blistering diseases (AIBD) are paradigms of autoantibody-mediated organ-specific autoimmune disorders that involve skin and/or mucous membranes. Compared to other autoimmune diseases, the pathogenicity of autoantibodies in AIBD is relatively well described. Pemphigus is a potentially lethal autoantibody driven autoimmune disorder with a strong HLA class II association. It is mainly characterized by IgG against the desmosomal adhesion molecules desmoglein 3 (Dsg3) and Dsg1. Several murine pemphigus models were developed subsequently, each allowing the analysis of a characteristic feature, such as pathogenic IgG or Dsg3-specific T or B cells. Thus, the models can be employed to preclinically evaluate potentially novel therapies. We here thoroughly summarize past and recent efforts in developing and utilizing pemphigus mouse models for pathomechanistic investigation and therapeutic interventions.
Pemphigus is a severe autoimmune blistering skin disease characterized by disruption of desmosomes, thereby affecting the epidermis of the skin and surface-close mucosal epithelia (1, 2). Depending on their immunohistological manifestations, three major forms of pemphigus are distinguished. The mucocutaneous pemphigus vulgaris (PV) presents in both mucosal epithelia and the epidermis and is characterized by IgG autoantibodies (auto-abs) directed against desmoglein 1 (Dsg1) and Dsg3 antigens, which were identified over three decades ago (3). Pemphigus foliaceus (PF) is triggered by anti-Dsg1 IgG leading to intraepidermal blistering limited to the skin. Paraneoplastic pemphigus (PNP) is characterized by polymorphic mucocutaneous eruptions and originates from a pool of auto-abs mainly against plakin proteins such as envoplakin, periplakin, desmoplakins, epiplakin, desmocollins, and BP230 as well as against α2 macroglobulin-like 1 and Dsg (4, 5). In addition to Dsg3 and/or Dsg1 auto-abs, IgG auto-abs against several target proteins other than Dsgs such as desmocollin 3 were identified in pemphigus patients, raising speculations about potential synergic effects eventually triggering acantholysis (6, 7).
Autoreactive B cells are key players in the production of pathogenic antigen-specific IgG in specific organs such as the skin (8). Pathogenic auto-abs can be generated after clonal expansion of autoreactive B cells in secondary lymphoid organs, but also in the skin (9). During T-dependent and T-independent responses to Dsg3, Dsg3-specific memory B cells are generated which can remain in immunological niches and become reactivated (10). In remission following immunosuppressive therapy, these cells might potentially lead to reencounter of the clinical symptoms (11). The critical role of auto-abs in pemphigus pathogenesis is supported by the observations that i.) the level of IgG auto-abs frequently correlates with the severity of the disease (12), ii.) pemphigus-associated blister formation can be induced by passive transfer of IgG from pemphigus patients into neonatal mice (13), and iii.) transplacental transmission of pemphigus IgG antibodies from diseased mothers to neonates leading to temporal neonatal pemphigus (14).
Despite the direct pathogenic role of B cells in pemphigus, recent studies shed light on the so far largely neglected autoimmune block, the T cells. While several T-cell subsets with a strong Th2 and Th17 polarization are undoubtedly involved in the pathogenesis of pemphigus (15, 16), identifying immunodominant self-peptides and characterization of autoreactive T cells is still challenging. Indirect characterization via activation markers such as CD154 presents as a reliable method to detect antigen-reactive T cells, while other methods such as ELISPOT or incorporation of 3H-thymidine are unable to distinguish specific cell populations (3H-T, ELISPOT) or focus on a single cytokine secreted by activated T cells (ELISPOT) (17). With the advent of MHC class II–peptide technology a more comprehensive understanding of antigen-specific T cells in the immune pathogenesis of several HLA class II–linked autoimmune disorders is now achievable (18).
Considering the potential individual contributions of loss of tolerance, arising antigen-specific T and B cells as well as antigen-specific IgG as key elements of pemphigus pathogenesis, we next summarize the development of pemphigus-related mouse models focusing on those different aspects of the disease with each of the advantages and disadvantages (Table 1).
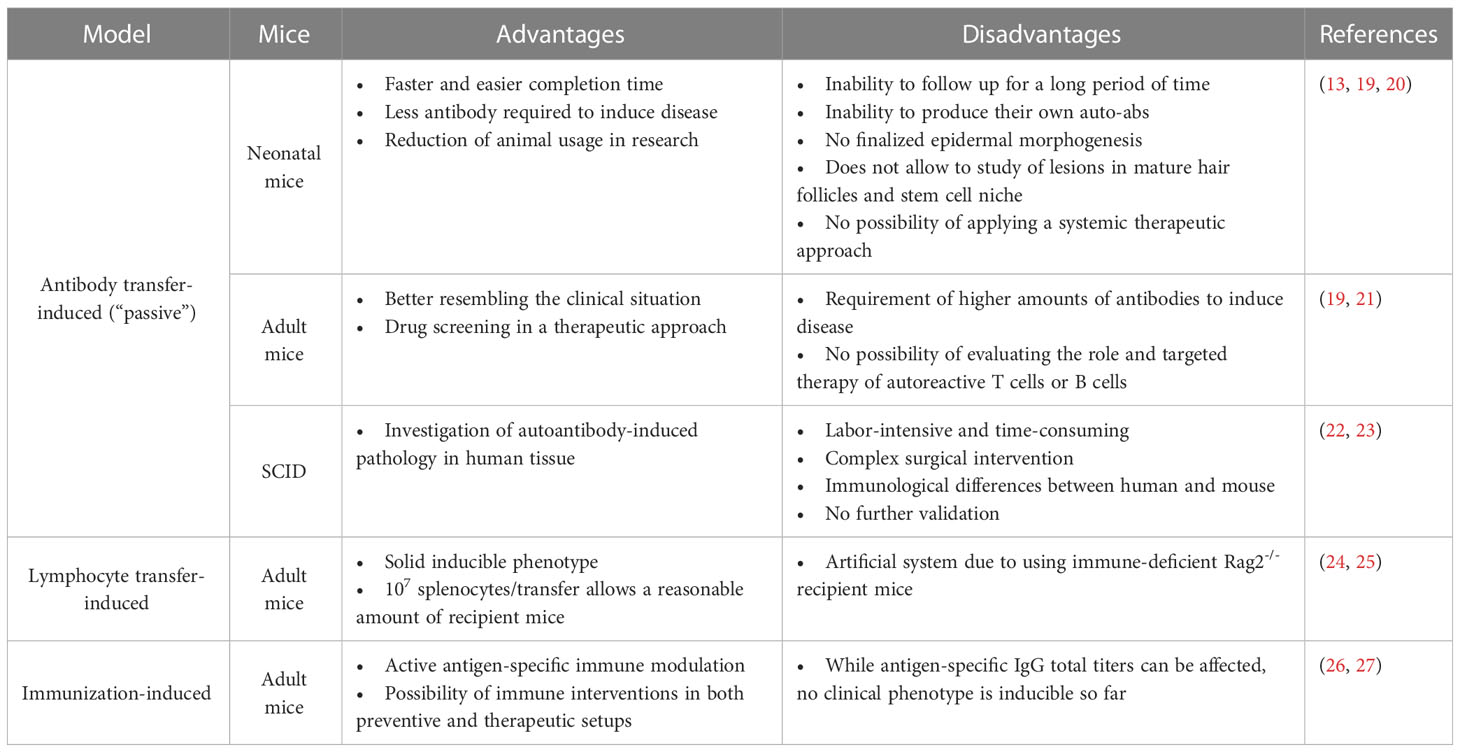
Table 1 Advantages and disadvantages of pemphigus mouse models.
Depending on the desired complexity of parameter induction, pemphigus-associated blister research brought forth different mouse models of pemphigus during the last three decades. In vivo models of pemphigus are classified as follows (Figure 1) (19, 28, 29):
1. Antibody transfer-induced (“passive”) models based on the injection of pemphigus auto-abs into mice to reproduce a transient disease phenotype
2. Lymphocyte transfer-induced disease models based on the transfer of autoreactive lymphocytes into mice, using transgenic models that induce a complex auto-ab-driven clinical phenotype
3. Antigen-specific immunization of mice to induce B and T cell-specific autoimmune responses including induction of pathogenic auto-abs
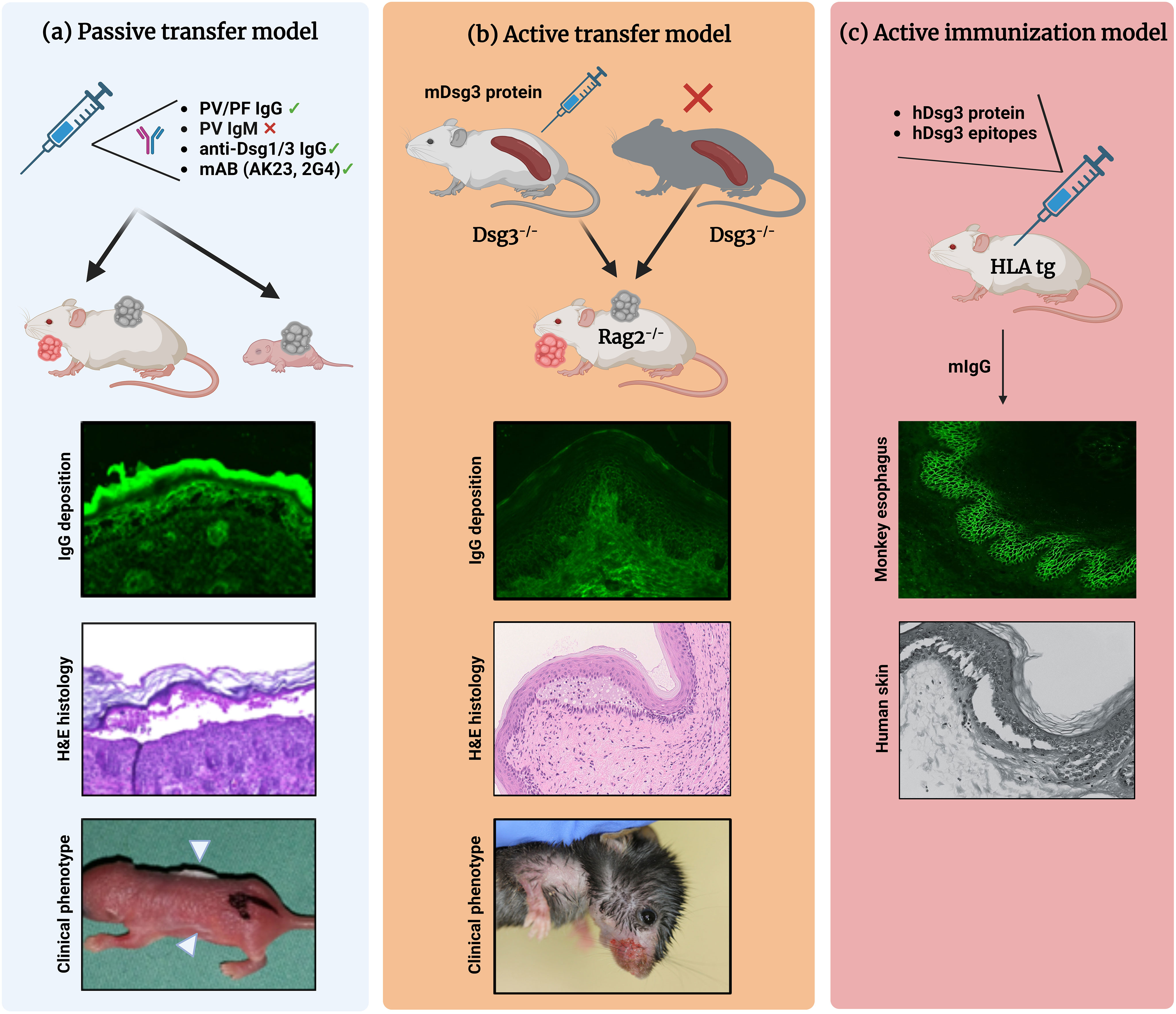
Figure 1 Different mouse models of pemphigus. (A) Antibody transfer-induced (“passive”) models. Total pemphigus vulgaris (PV) or pemphigus foliaceus (PF) IgG fractions, anti-desmoglein (Dsg) 1/3 IgG isolated from patients′ sera, or monoclonal anti-Dsg3 antibodies (e.g., AK23, 2G4) are injected into adult or neonatal mice leading to blister formation. Direct immunofluorescence (IF) microscopy, hematoxylin and eosin (H&E)-stained histology, and clinical presentation (white arrowheads indicate the blister sites) of a neonatal mouse injected with purified IgG from a patient with PV. (B) Lymphocyte transfer-induced models. Following adoptive transfer of naive lymphocytes from Dsg3-/- mice or splenocytes from Dsg3-/- mice immunized with recombinant Dsg3 protein, Rag2-/- immunodeficient mice produce anti-Dsg3 IgG antibodies and display the PV phenotype, allowing us to study the loss of tolerance. (C) Immunization-induced models. Immunization of humanized HLA-DBR1*04:02 transgenic mice with human Dsg3 recapitulates the effector phase of T and B cells followed by the formation of pathogenic Dsg3-specific IgG formation. Binding of the murine IgG from immunized mice to the keratinocyte cell surface was confirmed by indirect IF staining on monkey esophagus. Injection into human skin with murine IgG resulted in intraepidermal acantholysis and typical pathology. Figure was created with BioRender.com.
The direct pathogenicity of pemphigus auto-abs has been confirmed by their passive transfer into mice. The first experimental model for pemphigus was the passive transfer model described by Anhalt and coworkers in 1982 (13). Intraperitoneal (i.p.) administration of IgG from sera of PV patients at doses of 1.5 to 16 mg/g/day into neonatal BALB/c mice resulted in cutaneous blisters and erosions with clinical, histological, ultrastructural, and immunological features of human pathology. In the course of 18-72 h following IgG administration, mice developed skin lesions in a dose- and titer-dependent manner. Continuous IgG administration led to the development of new erosions and crusts; however, skin lesions healed when the IgG injections were discontinued. This model further allowed mechanistical investigations of of auto-ab-induced acantholysis in pemphigus. In a time-course study, the ultrastructural immunological changes in the epidermis of mice injected with PV IgG were examined by electron microscopy (30). Early detachment of the epidermis was observed 1 h post IgG injection as a widening of the interdesmosomal spaces, followed by splitting of the desmosomes and complete cell detachment in the suprabasal epidermal layers within 6 h. Of note, basal cells remained unaffected in a tombstone-like pattern. Anti-Dsg3 auto-abs purified from PNP sera were also pathogenic and caused blistering in neonatal mice (31).
The principle of disease induction by passive transfer of pathogenic PV IgG into neonatal mice has also been successfully applied to other pemphigus variants. Roscoe et al., in 1985 published the first in vivo study on the pathogenic role of anti-Dsg1 antibodies (32). When injected into neonatal mice, PF IgG (at 10 mg/g/day for a total of four injections) induced superficial skin blisters reproducing the clinical and immunopathological findings of human disease. Similar to the PV model (13), there was a close correlation between disease severity and the amount of injected IgG. In contrast to PV IgG, PF IgG did not cause suprabasal acantholysis in the epidermis but rather a superficial loss of epidermal adhesion, suggesting that PV and PF are mediated by different auto-abs specificities. Further studies showed that the pathogenic auto-abs in pemphigus are predominantly of the IgG4 subclass (33, 34). In contrast, some PF patients only had IgG1 auto-abs, which caused acantholysis in mice (35).
Unlike the pemphigoids, IgG-induced acantholysis in pemphigus is independent of complement activation or Fc-effector function. In an experimental passive transfer model, i.p. injection of intact pathogenic IgG and F(ab′)2 from PV sera led to the same extensive blistering, while the latter failed to induce C3 deposition. In addition, blistering was observed in C5-deficient or cobra venom factor (a structural analog of complement component C3) pretreated neonatal mice following administration of PV IgG (36). Hence, it was speculated that crosslinking of PV antigen on the surface of keratinocytes by bivalent PV auto-abs may be a necessary step for acantholysis.
Another study showed that subcutaneous (s.c.) injection of F(ab′)2 and Fab fragments caused blistering in mice (37). Pemphigus-like lesions have also been induced in neonatal mice by passive transfer of F(ab′)2 and Fab′ fragments purified from PF IgG (33).
These early studies relied predominantly on polyclonal IgG or its cleavage products. Although patient’s IgG is certainly an excellent tool for investigation of pemphigus pathomechanisms, its limited availability and heterogeneity over the course of the disease and between individuals may complicate systematic studies (38, 39). Monoclonal antibodies (mAbs), developed by immunization of mice with the recombinant mouse Dsg3 ectodomain, such as AK23, or isolated by phage display from active pemphigus patients, are now being extensively used to model PV pathogenesis (40–44). Payne et al. cloned anti-Dsg3 mAbs as single-chain variable fragments (scFvs) from a mucocutaneous PV patient using antibody phage display, which induced typical PV lesions in neonatal mice (40). Using the same technique, Ishii et al., isolated anti-Dsg1 scFv mAbs from a PF patient that were pathogenic in mouse and human skin (44). While most pathogenic Dsg3-related mAbs bind to the membrane distal EC-1/2 regions of the Dsg3 ectodomain, a novel Dsg3-EC5 binding mAb 2G4 allows the characterization of membrane proximal pathogenic binding (45). Additionally, experimental pemphigus can also be induced by Dsg3-hybridoma cell lines. Passive transfer of AK23-IgG hybridoma derived from splenocytes of the PV model caused blistering in neonatal mice (41). The practical advantage of hybridoma technology is that once stable somatic cell lines are established, they can be used to produce sensitive and specific mAbs in unlimited quantities. Furthermore, this technique preserves the native pairing of antibody variable and constant regions (46). Despite these advantages, hybridoma cells are limited by long generation times and the difficulty of controlling the epitopes against which antibodies are produced. Immunoadsorption of pathologic auto-abs from PV sera using the entire EC domains of Dsg1 and Dsg3 abolished the blister-inducing ability of IgG fractions in mice, suggesting that anti-Dsg1/3 IgG alone is pathogenic; other factors such as additional non-desmoglein PV auto-abs alone or in combination are not required (47, 48). In line with these observations, adjuvant immunoadsorption has been successfully applied in patients with severe PV and PF to efficiently reduce circulating pathogenic auto-abs (49–51).
The possible involvement of IgM in the pathogenesis of PV has also been investigated. In experimental PV, in contrast to AK23 IgG, administration of its AK23-IgM hybridoma cells isotype did not induce clinically overt blisters (52). As monovalent (Fab), bivalent (F(ab)’)2), or scFv fragments of anti-Dsg auto-abs induce keratinocyte dissociation in vitro and in vivo, the Fc portion of pathogenic pemphigus auto-abs is probably of minor significance (53).
Taken together, these results demonstrate that Fc-mediated mechanisms are not required for blister formation in PV and PF. Accordingly, research in pemphigus has focused on characterizing anti-Dsg B cells and antibodies as well as signaling pathways that modulate the pathogenic effects of auto-abs.
Dsg1 or Dsg3 can, at least in part, compensate for the adhesive functions of each other (1, 54). The desmoglein compensation model has been supported by several experimental studies. Passive transfer of PF IgG to neonatal mice caused loss of adhesion in the superficial epidermis (55). Consistent with this finding, exfoliative toxins produced by Staphylococcus aureus specifically cleave Dsg1, resulting in blistering just below the stratum corneum of neonatal mice (56, 57). On the other hand, forced superficial expression of Dsg3 in transgenic mice protected them from PF antibody-induced blistering. Another proof comes from the passive transfer of pemphigus IgG to normal and neonatal Dsg3null mice. Basically, Dsg3null neonatal mice do not present skin blisters because Dsg1 is present throughout the epidermis to compensate for the genetic loss of Dsg3. However, challenging these mice with anti-Dsg1 IgG led to severe PV-like blistering similar to cutaneous PV patients (54, 58). Additionally, the telogen hair sheds early in Dsg3−/− mice leading to an alopecia areata-like phenotype, whereas transgenic Dsg1 over-expression in the telogen club leads to a decreased or delayed balding phenotype (59).
Beyond steric hindrance, signaling pathways induced by binding of pemphigus IgG have been proposed to indirectly trigger acantholysis (60–62). Pharmacological modulation of signaling molecules blocked blister formation in the passive transfer mouse models for pemphigus. Consequently, some of the therapies derived from treating these experimental models might be translatable to human disease. For instance, 38 mitogen-activated protein kinase (p38MAPK) was shown to be activated by polyclonal PV (63, 64) and PF IgG (65) in vivo. Furthermore, pretreatment with p38MAPK inhibitors (i.e., SB202190 and SB203580) blocked PV and PF IgG-induced blistering in mice, suggesting a pivotal role for p38MAPK signaling in acantholysis (64–66). On the other hand, blisters induced by mAbs isolated from PV patients were not affected by p38 or MAPK-activated protein kinase 2 (MK2) inhibition, pointing to the significance of steric hindrance mechanisms (42, 67). Application of a Dsg3-specific tandem peptide stabilized adhesion inhibited PV IgG-mediated activation of p38MAPK and skin blistering (68). Unfortunately, clinical trials with a p38MAPK inhibitor (NCT00606749) in PV patients were terminated due to its dose-limiting hepatotoxicity (69). Downstream of p38MAPK, epidermal growth factor receptor (EGFR) signaling was shown to be activated in PV IgG-treated keratinocytes. In line, treatment with AG1478, an inhibitor of EGFR signaling, blocked blister formation in mice (70). Moreover, other clinically approved EGFR (Erlotinib) and dual EGFR/ErbB2 (Lapatinib) inhibitors prevented blistering in a neonatal PV mouse model (71). ADAM10 together with Src regulates signaling downstream of Dsg3 which may also include EGFR signaling. It was shown that PV IgG increased ADAM10 activation in a Src-dependent manner. Thus, inhibition of ADAM10 prevented histological acantholysis and skin blistering in mice injected with PV IgG (72). In addition, in neonatal mice injected with PV IgG, enhanced phosphorylation of mammalian target of rapamycin (m-TOR) and Src as well as increased blister formation and apoptosis were observed, all of which were eliminated or reduced by focal adhesion kinase (FAK) inhibition (73). In another experimental setting, modulation of mTOR with sirolimus (or rapamycin) and Src with PP1 abolished acantholysis (74). Additionally, Src blockade was protective against AK23-induced skin blistering (75). In a neonatal PV model, pharmacological inhibition of cyclin-dependent kinase 2 (Cdk2) by roscovitine prevented blister formation (76). Pretreatment of mice with inhibitors of tyrosine-kinase (TK) completely prevented the clinical and histological findings of PV (77). PV IgG was demonstrated to induce protein kinase C (PKC) activation as well. In line with these findings, PKC inhibition reduced PV IgG-induced skin blistering in vivo (78, 79). Finally, elevated levels of cyclic adenosine monophosphate (cAMP) have been shown to interfere with signaling pathways and prevent blister formation in vivo (80). A recent study showed that the phosphodiesterase 4 (PDE4) inhibitor apremilast inhibited blister formation in a passive transfer PV model via protective cAMP signaling in keratinocytes. Hence, apremilast offers a promising approach to target loss of desmosomal adhesion in pemphigus patients (81). PV IgG not only induces direct signaling effects but also changes gene expression patterns, which may contribute to pemphigus pathogenesis. In contrast, corticosteroids antagonistically upregulate DSG3 transcription through inhibition of the transcription factor Stat3 (82, 83). Additionally, it has been demonstrated that methylprednisolone reduces the extent of PV IgG-induced acantholysis in the skin of neonatal mice.
Several lines of evidence indicate that certain mediators involved in apoptosis contribute to the pathological mechanisms of pemphigus, while apoptosis itself is not apparent in pemphigus lesions nor required for acantholysis in vitro (84–86). In particular, cleaved caspase-8 and -3 were detected in pemphigus lesions, and upregulation of Fas ligand (FasL) was observed in keratinocytes co-cultured with PV IgG (87–89). Accordingly, a study by Lotti et al., showed that an anti-FasL antibody blocked blister formation in a neonatal mouse model of PV. Following PV IgG injection, sFasL-/- mice (lacking secreted soluble form of FasL) revealed reduced acantholytic area, whereas mFasL-/- mice (lacking membrane-bound form of FasL) developed blisters. These observations indicate that sFasL, which is upregulated and released from keratinocytes, plays a critical role in pemphigus pathogenesis (90). In a neonatal PF model, the administration of caspase inhibitors blocked intraepidermal blistering (91). In line, a novel anti-sFasL human mAb (PC111) has been tested for pemphigus therapy due to low its potential for immunogenicity, favorable chemical and physical stability, and high binding affinity of the compound (92).
Several studies have shown a pivotal role for FcRn in pemphigus. Li et al., demonstrated that FcRn-deficient mice were resistant to experimental PV and PF, in which circulating auto-ab levels were significantly reduced as compared to wild type mice. Besides, the therapeutic efficacy of high-dose intravenous immunoglobulins (IVIg) might be attributed to FcRn saturation and increased catabolism of pathogenic IgG (93). Of note, IVIg protected target cells from apoptosis by interfering with signaling pathways and increasing the sensitivity to corticosteroids (89). In an auto-ab-induced model of PV, IVIg selectively inhibited anti-Dsg3 antibodies, decreased the number of circulating auto-abs, and reduced blister formation (94). Additionally, disease-specific IVIg was shown to be more effective in neutralizing pathogenic antibodies and preventing blister formation in vivo than commercial IVIg (95). Clinical trials are currently evaluating the safety and efficacy of several FcRn‐targeting compounds (e.g., efgartigimod (ARGX-113) and SYNT001 (ALXN1830)) in pemphigus (96–98). Showing unexpected effects for efgartigimod in vitro, a recent study additionally suggested that efgartigimod-induced blockade of FcRn may have functions on B cell homeostasis beyond IgG recycling. Improvement of keratinocyte adhesion by FcRn blockade may provide a novel treatment option for pemphigus (39). However, the antibody-induced model is not suitable to study the role and specific targeting of autoreactive T cells or B cells.
Due to incomplete epidermal development in neonates, the principle of autoantibody transfer has been further adapted to adult mice. Experimental PV was induced in adult C57Bl/6J or Rag2-/- mice by passive transfer of a murine pathogenic Dsg3 mAb, AK23 (21). This model has proved useful in exploring non-apoptotic aspects of pemphigus related signaling. For instance, EGFR was shown to be activated in response to AK23, followed by an increase in c-myc levels. Adult passive transfer model of pemphigus was also applied in fully humanized Dsg3 (hDsg3) mice, showing that the administration of anti-Dsg3 serum IgG from patients with mucosal PV (mPV) caused IgG deposits on the surface of epidermal keratinocytes and suprabasilar acantholysis in mucosal tissues of hDsg3 mice, but not in WT mice. This finding confirms the in vivo pathogenicity of mPV anti-hDsg3 IgG (99).
An alternative approach for studying the pathogenesis of human auto-abs in mice is to passively transfer pemphigus serum into athymic nude mice grafted with human oral mucosa. This model combines a mouse model with human skin allowing in vivo studies with pathogenic anti-human Dsg3 auto-abs (22, 100). A low degree of epithelial cell detachment was observed in this mucosal graft model, whereas human IgG was detected in the skin of all mice, and nearly two-thirds of the transplants showed basal epithelial edema (23). When full-thickness human skin was grafted onto the backs of severe combined immunodeficient (SCID) mice followed by injection of PF and PV IgG, subcorneal and suprabasal loss of adhesion as well as intercellular IgG deposits in the upper and lower layers of the epidermis, respectively, was seen (22). Because the xenograft model in pemphigus is complex and adds little to a classical passive model, few experiences have been gained with these models.
Considering the half-life of serum IgG, passive transfer models can only be used and analyzed for a limited amount of time. The main obstacle to an active immunization model is immunological self-tolerance. The usage of pemphigus mouse models was transformed by the development of Dsg3-/- knockout mice. These mice show a loss of keratinocyte cell adhesion mainly in oral mucous membranes, a phenotype that resembles that of patients with PV (24). These erosions, also occasionally found in the skin with suprabasal acantholysis demonstrated the importance of Dsg3 in vivo, however, no immunological inherent components were involved. Immunization with murine Dsg3 induced a more severe phenotype in terms of affected skin area and eosinophilic spongiosis (25). To further pinpoint Dsg3-specific immune modulation, immunization of Dsg3-/- mice with recombinant murine Dsg3 was followed by the intravenous transfer of the splenocyte pool into immunodeficient Rag2-/- Dsg3+/+ recipient mice (25). Newly formed antigen-specific T and B cells then induced a Dsg3-specific autoimmune response presenting with suprabasal acantholysis, rows of tombstone basal keratinocytes, and half-desmosomes (101). Interestingly, antigen-specific T cell infiltration into Dsg3-expressing tissues led to interface dermatitis, a distinct form of T cell-mediated autoimmunity causing acantholysis and can be found in autoimmune skin diseases such as PNP (102). Establishing a retroviral transduction system, Takahashi et al., generated a C57BL/6J-Tg (Dsg3TCR140) mouse line which enabled them to show that while tolerized Dsg3H1 T cells could induce interface dermatitis, but not PV, non-tolerized Dsg3H1 T cells induced both anti-Dsg3 IgG production and interface dermatitis. These results demonstrated that induced anti-Dsg3 auto-abs potentially interfere with cell-cell adhesion of keratinocytes in the PV model. Based on those studies, a series of human, murine, and bispecific T cell clones were generated. Further characterization indicated that although an individual anti-Dsg3 IgG is not sufficient to cause acantholysis in adult mice, several clones together can induce a pemphigus phenotype (43). An array of various immunosuppressive agents frequently used in PV patient treatment was evaluated by means of ELISA, weight, and PV severity score (103). While cyclophosphamide displayed the strongest immunosuppressive properties, other agents such as azathioprine were shown to be less effective or not suppressive at all such as methylprednisolone and dexamethasone. Those findings are partially in contrast to the experience in humans (104), showing that observations in preclinical mouse models cannot be directly transferred to the human situation. Unfortunately, no follow-up studies applying novel treatment modalities in this model have been published so far.
Another modification of the pemphigus mouse model involved the transfer of naїve splenocytes from Dsg3-/- mice into Rag2-/- Dsg3+/+ recipients (105). Here, Dsg3-specific naїve lymphocytes in Dsg3-/- mice can be primed and activated by endogenous Dsg3 in recipient mice to ultimately induce pathogenic anti-Dsg3 IgG without active immunization. While the time and overall levels of IgG and phenotype induction were delayed by two weeks, once the phenotype developed, no apparent differences in disease severity between Rag2-/- recipients of naïve or immunized splenocytes were found (105). Immunosuppressive drugs such as cyclophosphamide successfully inhibited disease development in a preventive setting, and mice were free of symptoms 35 days after discontinuing the treatment (106). To further pinpoint crucial CD4+ subpopulations for anti-Dsg3 IgG formation, Kim et al., developed a modified murine transfer protocol based on Rag1-/- recipient mice and provided compelling data that Dsg3-specific ICOS+ T follicular helper (Tfh) cells play a pivotal role in pathogenic humoral immunity in PV (107). Since alterations in the Tfh compartment have previously been found in PV (108), targeting ICOS represents a valid therapeutic option for the treatment of PV.
PV is an oligoparametric disease, arising from a combination of different environmental factors in predisposed individuals carrying individual susceptibility genes. While genome-wide association studies are lacking to complete the picture of protective, susceptible, or neutral alleles (109), two highly associated class II alleles have been found in PV patients (DQB1*0503 and DRB1*0402) (110). PV patients carrying these alleles showed Dsg3-specific auto-aggressive Th2 cells, while healthy carriers preferentially displayed an autoreactive Th1 response (111). This prompted us to develop an active mouse model using a C57B/6J transgenic mouse line with the respective alleles HLA-DQB1*04:02 in a linkage disequilibrium with HLA-DQB1*03:02 that additionally expresses the human CD4 co-receptor while lacking a functional murine major histocompatibility class II molecule (I-Aβ−/−) (26). Antigen presentation after Dsg3 immunization is therefore restricted to human alleles which are presented in association with these human HLA class II alleles to CD4+ T cells. Intraperitoneal immunization with human Dsg3 protein or with a set of immunodominant Dsg3-peptides (50 µg/mouse day 0 and day 14), which share a positively charged anchor motif for HLA-DRB1*04:02, led to the formation of Dsg3-reactive CD4+ T cells followed by a profound and lasting induction of anti-Dsg3 IgG (26). This model was used to show that T and B cell interaction is crucial for pemphigus pathology. Blockade of this interaction by anti-CD40L mab completely prevented anti-Dsg3 IgG formation. Furthermore, the induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 also significantly reduced anti-Dsg3 IgG induction (112).
Dsg3-specific immunization of the HLA tg mice allows cellular characterization and therapeutic intervention studies in a preclinical setting (i.e., during the formation of antigen-specific T and B cells and subsequent auto-Ab formation) or a therapeutic setting (i.e., after the onset of immunization with an ongoing auto-Ab formation as in human PV). Using this model, CD4+ T cell-specific tolerance could be induced and analyzed based on the application of a set of immunodominant Dsg3 CD4+ T cell epitopes linked to nanoparticles (113). For identification and characterization of low-frequent Dsg3-specific CD4+ T cells in lymphatic tissues, a novel detection based on DRB1*04:02 HLA class II specific dextramers loaded with the aforementioned immunodominant Dsg3-T cell epitopes was developed. CD4+ T cells were primarily detected in lymphoid tissue after initial immunization (4% Dsg3+ CD4+ CD19+ cells) which steadily decreased thereafter. Additionally, applying IVIg as an established treatment for pemphigus was shown to modulate antigen-specific T and B cell formation (114). A significant reduction of Dsg3-specific serum IgG correlated with an upregulation of regulatory T cells.
Recent endeavors to break tolerance against Dsg3 in various strains of Dsg3-expressing mice employing different immunization protocols with recombinant human and murine Dsg3/Dsg1 forms did not elicit a clinical phenotype while non-pathogenic anti-Dsg3/Dsg1 IgG was induced (115). However, unpublished work currently focuses on the establishment of a Dsg3-transgenic mouse model displaying the formation of HLA-dependent antigen-specific T and B cells as well as a solid antigen-specific IgG as a basis of a lasting clinical phenotype. This model will be suitable for in-depth analysis of autoreactive B and T cells and IgG formation as well as a preclinical model for testing novel specific immune interventions in pemphigus.
Even though our understanding of T- and B-cell-related induction of antigen-specific IgG in AIBD PV and PF has dramatically improved over the last decades, we still lack a proper mouse model that reflects the “grande picture” of patient-relevant characteristics. The interplay between blister formation based on steric hindrance by auto-abs against Dsg3 and prominent induced signaling pathways such as p38MAPK or ERK is still under debate. The identification of novel PV-related antigens allows the further distinction of novel clinical subtypes with specific clinical outcomes. Different preclinical models reflect distinct hallmarks and pathways of PV immune pathogenesis, i.e., induction of autoreactive T and B cells and auto-ab-induced acantholysis. They are constantly improved and reflect, so far, only a few PV-related parameters. Novel therapeutic interventions in PV address these parameters and be further characterized in vivo. Nonetheless, a mouse model reproducing PV pathology based on HLA-dependent T- and B-cell mediated antibody formation leading to mucosal acantholysis would be a desired gold standard.
All authors contributed to the writing and review of this manuscript. All authors approved the final version of this manuscript. All authors contributed to the article and approved the submitted version.
This work received support through the Deutsche Forschungsgemeinschaft (DFG) FOR 2497/TP01 to CH and TP08 to MH (HE 1602/16-2), the Collaborative Research Center CRC 1526 (Project-ID 454193335 to ES), the Schleswig-Holstein Excellence Cluster Precision Medicine in Chronic Inflammation (DFG EXC 2167/1 to ES), and the research grant from the University of Lübeck (J13-2022 to SE). Open Access funding is provided to CH by the Open Access Publication Fund of Philipps-Universität Marburg with support of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation).
We are highly grateful to Dr. Masayuki Amagai and Dr. Hayato Takahashi (Keio University School of Medicine, Tokyo, Japan) for providing the murine images of the active transfer model, and Dr. Rüdiger Eming (Philipps-Universität Marburg, Marburg, Germany) for the human skin histology after transfer of pathogenic murine IgG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet (2019) 394(10201):882–94. doi: 10.1016/S0140-6736(19)31778-7
2. Beek NV, Zillikens D, Schmidt E. Bullous autoimmune dermatoses. Dtsch Arztebl Int (2021) 118(24):413–20. doi: 10.3238/arztebl.m2021.0136
3. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell (1991) 67(5):869–77. doi: 10.1016/0092-8674(91)90360-B
4. Didona D, Fania L, Didona B, Eming R, Hertl M, Di Zenzo G. Paraneoplastic dermatoses: a brief general review and an extensive analysis of paraneoplastic pemphigus and paraneoplastic dermatomyositis. Int J Mol Sci (2020) 21(6):2178. doi: 10.3390/ijms21062178
5. Kim JH, Kim SC. Paraneoplastic pemphigus: paraneoplastic autoimmune disease of the skin and mucosa. Front Immunol (2019) 10:1259. doi: 10.3389/fimmu.2019.01259
6. Sajda T, Hazelton J, Patel M, Seiffert-Sinha K, Steinman L, Robinson W, et al. Multiplexed autoantigen microarrays identify HLA as a key driver of anti-desmoglein and -non-desmoglein reactivities in pemphigus. Proc Natl Acad Sci U.S.A. (2016) 113(7):1859–64. doi: 10.1073/pnas.1525448113
7. Mindorf S, Dettmann IM, Krüger S, Fuhrmann T, Rentzsch K, Karl I, et al. Routine detection of serum antidesmocollin autoantibodies is only useful in patients with atypical pemphigus. Exp Dermatol (2017) 26(12):1267–70. doi: 10.1111/exd.13409
8. Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol (2011) 36(7):703–7. doi: 10.1111/j.1365-2230.2011.04092.x
9. Fetter T, Niebel D, Braegelmann C, Wenzel J. Skin-associated b cells in the pathogenesis of cutaneous autoimmune diseases-implications for therapeutic approaches. Cells (2020) 9(12):2627. doi: 10.3390/cells9122627
10. Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, Payne AS, et al. Persistence of anti-desmoglein 3 IgG(+) b-cell clones in pemphigus patients over years. J Invest Dermatol (2015) 135(3):742–9. doi: 10.1038/jid.2014.291
11. Nishifuji K, Amagai M, Kuwana M, Iwasaki T, Nishikawa T. Detection of antigen-specific b cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol (2000) 114(1):88–94. doi: 10.1046/j.1523-1747.2000.00840.x
12. Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, Gamou S, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol (1997) 159(4):2010–7. doi: 10.4049/jimmunol.159.4.2010
13. Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med (1982) 306(20):1189–96. doi: 10.1056/NEJM198205203062001
14. Wasserstrum N, Laros RK Jr. Transplacental transmission of pemphigus. JAMA (1983) 249(11):1480–2. doi: 10.1001/jama.1983.03330350056029
15. Satyam A, Khandpur S, Sharma VK, Sharma A. Involvement of T(H)1/T(H)2 cytokines in the pathogenesis of autoimmune skin disease-pemphigus vulgaris. Immunol Invest (2009) 38(6):498–509. doi: 10.1080/08820130902943097
16. Holstein J, Solimani F, Baum C, Meier K, Pollmann R, Didona D, et al. Immunophenotyping in pemphigus reveals a T(H)17/T(FH)17 cell-dominated immune response promoting desmoglein1/3-specific autoantibody production. J Allergy Clin Immunol (2021) 147(6):2358–69. doi: 10.1016/j.jaci.2020.11.008
17. Polakova A, Kauter L, Ismagambetova A, Didona D, Solimani F, Ghoreschi K, et al. Detection of rare autoreactive T cell subsets in patients with pemphigus vulgaris. Front Immunol (2022) 13:979277. doi: 10.3389/fimmu.2022.979277
18. Wieber K, Zimmer CL, Hertl M. Detection of autoreactive CD4+ T cells by MHC class II multimers in HLA-linked human autoimmune diseases. J Clin Invest (2021) 131(9):e148674. doi: 10.1172/JCI148674
19. Bieber K, Sun S, Ishii N, Kasperkiewicz M, Schmidt E, Hirose M, et al. Animal models for autoimmune bullous dermatoses. Exp Dermatol (2010) 19(1):2–11. doi: 10.1111/j.1600-0625.2009.00948.x
20. McClain RM, Keller D, Casciano D, Fu P, MacDonald J, Popp J. Neonatal mouse model: review of methods and results. Toxicol Pathol (2001) 29 Suppl:128–37. doi: 10.1080/019262301753178537
21. Schulze K, Galichet A, Sayar BS, Scothern A, Howald D, Zymann H, et al. An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J Invest Dermatol (2012) 132(2):346–55. doi: 10.1038/jid.2011.299
22. Zillikens D, Schmidt E, Reimer S, Chimanovitch I, Hardt-Weinelt K, Rose C, et al. Antibodies to desmogleins 1 and 3, but not to BP180, induce blisters in human skin grafted onto SCID mice. J Pathol (2001) 193(1):117–24. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH742>3.0.CO;2-W
23. Buschard K, Dabelsteen E, Bretlau P. A model for the study of autoimmune diseases applied to pemphigus: transplants of human oral mucosa to athymic nude mice binds pemphigus antibodies in vivo. J Invest Dermatol (1981) 76(3):171–3. doi: 10.1111/1523-1747.ep12525667
24. Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol (1997) 137(5):1091–102. doi: 10.1083/jcb.137.5.1091
25. Ohyama M, Amagai M, Tsunoda K, Ota T, Koyasu S, Hata J, et al. Immunologic and histopathologic characterization of an active disease mouse model for pemphigus vulgaris. J Invest Dermatol (2002) 118(1):199–204. doi: 10.1046/j.0022-202x.2001.01643.x
26. Eming R, Hennerici T, Bäcklund J, Feliciani C, Visconti KC, Willenborg S, et al. Pathogenic IgG antibodies against desmoglein 3 in pemphigus vulgaris are regulated by HLA-DRB1*04:02-restricted T cells. J Immunol (2014) 193(9):4391–9. doi: 10.4049/jimmunol.1401081
27. Hudemann C, Maglie R, Llamazares-Prada M, Beckert B, Didona D, Tikkanen R, et al. Human desmocollin 3-Specific IgG antibodies are pathogenic in a humanized HLA class II transgenic mouse model of pemphigus. J Invest Dermatol (2022) 142(3 Pt B):915–923 e3. doi: 10.1016/j.jid.2021.06.017
28. Egu DT, Schmitt T, Waschke J. Mechanisms causing acantholysis in pemphigus-lessons from human skin. Front Immunol (2022) 13:884067. doi: 10.3389/fimmu.2022.884067
29. Pollmann R, Eming R. Research techniques made simple: mouse models of autoimmune blistering diseases. J Invest Dermatol (2017) 137(1):e1–6. doi: 10.1016/j.jid.2016.11.003
30. Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ. Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol (1985) 84(1):41–6. doi: 10.1111/1523-1747.ep12274679
31. Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T. Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest (1998) 102(4):775–82. doi: 10.1172/JCI3647
32. Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, et al. Brazilian Pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol (1985) 85(6):538–41. doi: 10.1111/1523-1747.ep12277362
33. Rock B, Labib RS, Diaz LA. Monovalent fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal balb/c mice. J Clin Invest (1990) 85(1):296–9. doi: 10.1172/JCI114426
34. Labib RS, Rock B, Martins CR, Diaz LA. Pemphigus foliaceus antigen: characterization of an immunoreactive tryptic fragment from BALB/c mouse epidermis recognized by all patients’ sera and major autoantibody subclasses. Clin Immunol Immunopathol (1990) 57(2):317–29. doi: 10.1016/0090-1229(90)90045-R
35. Hacker-Foegen MK, Janson M, Amagai M, Fairley JA, Lin MS. Pathogenicity and epitope characteristics of anti-desmoglein-1 from pemphigus foliaceus patients expressing only IgG1 autoantibodies. J Invest Dermatol (2003) 121(6):1373–8. doi: 10.1111/j.1523-1747.2003.12608.x
36. Anhalt GJ, Till GO, Diaz LA, Labib RS, Patel HP, Eaglstein N. Defining the role of complement in experimental pemphigus vulgaris in mice. J Immunol (1986) 137(9):2835–40. doi: 10.4049/jimmunol.137.9.2835
37. Mascaro JM Jr., España A, Liu Z, Ding X, Swartz SJ, Fairley JA, et al. Mechanisms of acantholysis in pemphigus vulgaris: role of IgG valence. Clin Immunol Immunopathol (1997) 85(1):90–6. doi: 10.1006/clin.1997.4408
38. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PloS One (2012) 7(12):e50696. doi: 10.1371/journal.pone.0050696
39. Zakrzewicz A, Würth C, Beckert B, Feldhoff S, Vanderheyden K, Foss S, et al. Correction: zakrzewicz et al. stabilization of keratinocyte monolayer integrity in the presence of anti-Desmoglein-3 antibodies through FcRn blockade with efgartigimod: novel treatment paradigm for pemphigus? cells 2022, 11, 942. Cells (2022) 11(10):1700. doi: 10.3390/cells11101700
40. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest (2005) 115(4):888–99. doi: 10.1172/JCI24185
41. Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol (2003) 170(4):2170–8. doi: 10.4049/jimmunol.170.4.2170
42. Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem (2011) 286(2):1283–91. doi: 10.1074/jbc.M110.172874
43. Kawasaki H, Tsunoda K, Hata T, Ishii K, Yamada T, Amagai M. Synergistic pathogenic effects of combined mouse monoclonal anti-desmoglein 3 IgG antibodies on pemphigus vulgaris blister formation. J Invest Dermatol (2006) 126(12):2621–30. doi: 10.1038/sj.jid.5700450
44. Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol (2008) 128(4):939–48. doi: 10.1038/sj.jid.5701132
45. Hudemann C, Exner Y, Pollmann R, Schneider K, Zakrzewicz A, Feldhoff S, et al. IgG against the membrane-proximal portion of the desmoglein 3 ectodomain induces loss of keratinocyte adhesion, a hallmark in pemphigus vulgaris. J Invest Dermatol (2023) 143(2):254–263 e3. doi: 10.1016/j.jid.2022.07.030
46. Moraes JZ, Hamaguchi B, Braggion C, Speciale ER, Cesar FBV, Soares GFDS, et al. Hybridoma technology: is it still useful? Curr Res Immunol (2021) 2:32–40. doi: 10.1016/j.crimmu.2021.03.002
47. Hofrichter M, Dworschak J, Emtenani S, Langenhan J, Weiß F, Komorowski L, et al. Immunoadsorption of desmoglein-3-Specific IgG abolishes the blister-inducing capacity of pemphigus vulgaris IgG in neonatal mice. Front Immunol (2018) 9:1935. doi: 10.3389/fimmu.2018.01935
48. Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest (1994) 94(1):59–67. doi: 10.1172/JCI117349
49. Kasperkiewicz M, Shimanovich I, Meier M, Schumacher N, Westermann L, Kramer J, et al. Treatment of severe pemphigus with a combination of immunoadsorption, rituximab, pulsed dexamethasone and azathioprine/mycophenolate mofetil: a pilot study of 23 patients. Br J Dermatol (2012) 166(1):154–60. doi: 10.1111/j.1365-2133.2011.10585.x
50. Meyersburg D, et al. Immunoadsorption in dermatology. Ther Apher Dial (2012) 16(4):311–20. doi: 10.1111/j.1744-9987.2012.01075.x
51. Schmidt E, Sticherling M, Sárdy M, Eming R, Goebeler M, Hertl M, et al. S2k guidelines for the treatment of pemphigus vulgaris/foliaceus and bullous pemphigoid: 2019 update. J Dtsch Dermatol Ges (2020) 18(5):516–26. doi: 10.1111/ddg.14097
52. Tsunoda K, Ota T, Saito M, Hata T, Shimizu A, Ishiko A, et al. Pathogenic relevance of IgG and IgM antibodies against desmoglein 3 in blister formation in pemphigus vulgaris. Am J Pathol (2011) 179(2):795–806. doi: 10.1016/j.ajpath.2011.04.015
53. Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a comprehensive review on pathogenesis, clinical presentation and novel therapeutic approaches. Clin Rev Allergy Immunol (2018) 54(1):1–25. doi: 10.1007/s12016-017-8662-z
54. Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest (1999) 103(4):461–8. doi: 10.1172/JCI5252
55. Wu H, Wang ZH, Yan A, Lyle S, Fakharzadeh S, Wahl JK, et al. Protection against pemphigus foliaceus by desmoglein 3 in neonates. N Engl J Med (2000) 343(1):31–5. doi: 10.1056/NEJM200007063430105
56. Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR. Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat Med (2000) 6(11):1275–7. doi: 10.1038/81385
57. Amagai M, Yamaguchi T, Hanakawa Y, Nishifuji K, Sugai M, Stanley JR, et al. Staphylococcal exfoliative toxin b specifically cleaves desmoglein 1. J Invest Dermatol (2002) 118(5):845–50. doi: 10.1046/j.1523-1747.2002.01751.x
58. Vu TN, Lee TX, Ndoye A, Shultz LD, Pittelkow MR, Dahl MV, et al. The pathophysiological significance of nondesmoglein targets of pemphigus autoimmunity. development of antibodies against keratinocyte cholinergic receptors in patients with pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol (1998) 134(8):971–80. doi: 10.1001/archderm.134.8.971
59. Hanakawa Y, Matsuyoshi N, Stanley JR. Expression of desmoglein 1 compensates for genetic loss of desmoglein 3 in keratinocyte adhesion. J Invest Dermatol (2002) 119(1):27–31. doi: 10.1046/j.1523-1747.2002.01780.x
60. Brescacin A, Baig Z, Bhinder J, Lin S, Brar L, Cirillo N. What protein kinases are crucial for acantholysis and blister formation in pemphigus vulgaris? a systematic review. J Cell Physiol (2022) 237(7):2825–37. doi: 10.1002/jcp.30784
61. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol (2017) 8:603. doi: 10.3389/fimmu.2017.00603
62. Schmitt T, Waschke J. Autoantibody-specific signalling in pemphigus. Front Med (Lausanne) (2021) 8:701809. doi: 10.3389/fmed.2021.701809
63. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem (2005) 280(25):23778–84. doi: 10.1074/jbc.M501365200
64. Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U.S.A. (2006) 103(34):12855–60. doi: 10.1073/pnas.0602973103
65. Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol (2008) 173(6):1628–36. doi: 10.2353/ajpath.2008.080391
66. Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP, Rubenstein DS. Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis. J Biol Chem (2009) 284(18):12524–32. doi: 10.1074/jbc.M808204200
67. Mao X, Li H, Sano Y, Gaestel M, Mo Park J, Payne AS. MAPKAP kinase 2 (MK2)-dependent and -independent models of blister formation in pemphigus vulgaris. J Invest Dermatol (2014) 134(1):68–76. doi: 10.1038/jid.2013.224
68. Spindler V, Rötzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest (2013) 123(2):800–11. doi: 10.1172/JCI60139
69. Ellebrecht CT, Maseda D, Payne AS. Pemphigus and pemphigoid: from disease mechanisms to druggable pathways. J Invest Dermatol (2022) 142(3 Pt B):907–14. doi: 10.1016/j.jid.2021.04.040
70. Bektas M, olly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem (2013) 288(13):9447–56. doi: 10.1074/jbc.M112.438010
71. Sayar BS, Rüegg S, Schmidt E, Sibilia M, Siffert M, Suter MM, et al. EGFR inhibitors erlotinib and lapatinib ameliorate epidermal blistering in pemphigus vulgaris in a non-linear, V-shaped relationship. Exp Dermatol (2014) 23(1):33–8. doi: 10.1111/exd.12290
72. Ivars M, España A, Alzuguren P, Pelacho B, Lasarte JJ, López-Zabalza MJ. The involvement of ADAM10 in acantholysis in mucocutaneous pemphigus vulgaris depends on the autoantibody profile of each patient. Br J Dermatol (2020) 182(5):1194–204. doi: 10.1111/bjd.18382
73. Gil MP, Modol T, España A, López-Zabalza MJ. Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp Dermatol (2012) 21(4):254–9. doi: 10.1111/j.1600-0625.2012.01441.x
74. Pretel M, España A, Marquina M, Pelacho B, López-Picazo JM, López-Zabalza MJ, et al. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp Dermatol (2009) 18(9):771–80. doi: 10.1111/j.1600-0625.2009.00893.x
75. Kugelmann D, Rötzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of src and cortactin in pemphigus skin blistering. Front Immunol (2019) 10:626. doi: 10.3389/fimmu.2019.00626
76. Lanza A, Cirillo N, Rossiello R, Rienzo M, Cutillo L, Casamassimi A, et al. Evidence of key role of Cdk2 overexpression in pemphigus vulgaris. J Biol Chem (2008) 283(13):8736–45. doi: 10.1074/jbc.M702186200
77. Marquina M, España A, Fernández-Galar M, López-Zabalza MJ. The role of nitric oxide synthases in pemphigus vulgaris in a mouse model. Br J Dermatol (2008) 159(1):68–76. doi: 10.1111/j.1365-2133.2008.08582.x
78. Sanchez-Carpintero I, España A, Pelacho B, López Moratalla N, Rubenstein DS, Diaz LA, et al. In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol (2004) 151(3):565–70. doi: 10.1111/j.1365-2133.2004.06147.x
79. Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting? Am J Pathol (2011) 179(4):1905–16. doi: 10.1096/fasebj.25.1_supplement.242.1
80. Spindler V, Vielmuth F, Schmidt E, Rubenstein DS, Waschke J. Protective endogenous cyclic adenosine 5’-monophosphate signaling triggered by pemphigus autoantibodies. J Immunol (2010) 185(11):6831–8. doi: 10.4049/jimmunol.1002675
81. Sigmund AM, Winkler M, Engelmayer S, Kugelmann D, Egu DT, Steinert LS, et al. Apremilast prevents blistering in human epidermis and stabilizes keratinocyte adhesion in pemphigus. Nat Commun (2023) 14(1):116. doi: 10.1038/s41467-022-35741-0
82. Mao X, Cho MJT, Ellebrecht CT, Mukherjee EM, Payne AS. Stat3 regulates desmoglein 3 transcription in epithelial keratinocytes. JCI Insight (2017) 2(9):e92253. doi: 10.1172/jci.insight.92253
83. Nguyen VT, Arredondo J, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA. Pemphigus vulgaris IgG and methylprednisolone exhibit reciprocal effects on keratinocytes. J Biol Chem (2004) 279(3):2135–46. doi: 10.1074/jbc.M309000200
84. Grando SA, Bystryn JC, Chernyavsky AI, Frusić-Zlotkin M, Gniadecki R, Lotti R, et al. Apoptolysis: a novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp Dermatol (2009) 18(9):764–70. doi: 10.1111/j.1600-0625.2009.00934.x
85. Schmidt E, Gutberlet J, Siegmund D, Berg D, Wajant H, Waschke J. Apoptosis is not required for acantholysis in pemphigus vulgaris. m J Physiol Cell Physiol (2009) 296(1):C162–72. doi: 10.1152/ajpcell.00161.2008
86. Schmidt E, Waschke J. Apoptosis in pemphigus. Autoimmun Rev (2010) 10(2):84–9. doi: 10.1016/j.autrev.2009.01.011
87. Wang X, Brégégère F, Frusić-Zlotkin M, Feinmesser M, Michel B, Milner Y. Possible apoptotic mechanism in epidermal cell acantholysis induced by pemphigus vulgaris autoimmunoglobulins. Apoptosis (2004) 9(2):131–43. doi: 10.1023/B:APPT.0000018795.05766.1f
88. Frusic-Zlotkin M, Pergamentz R, Michel B, David M, Mimouni D, Brégégère F, et al. The interaction of pemphigus autoimmunoglobulins with epidermal cells: activation of the fas apoptotic pathway and the use of caspase activity for pathogenicity tests of pemphigus patients. Ann N Y Acad Sci (2005) 1050:371–9. doi: 10.1196/annals.1313.040
89. Arredondo J, Chernyavsky AI, Karaouni A, Grando SA. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. Am J Pathol (2005) 167(6):1531–44. doi: 10.1016/S0002-9440(10)61239-4
90. Lotti R, Shu E, Petrachi T, Marconi A, Palazzo E, Quadri M, et al. Soluble fas ligand is essential for blister formation in pemphigus. Front Immunol (2018) 9:370. doi: 10.3389/fimmu.2018.00370
91. Li N, Zhao M, Wang J, Liu Z, Diaz LA. Involvement of the apoptotic mechanism in pemphigus foliaceus autoimmune injury of the skin. J Immunol (2009) 182(1):711–7. doi: 10.4049/jimmunol.182.1.711
92. Kridin K, Ahn C, Huang WC, Ansari A, Sami N. Treatment update of autoimmune blistering diseases. Dermatol Clin (2019) 37(2):215–28. doi: 10.1016/j.det.2018.12.003
93. Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, et al. Complete FcRn dependence for intravenous ig therapy in autoimmune skin blistering diseases. J Clin Invest (2005) 115(12):3440–50. doi: 10.1172/JCI24394
94. Mimouni D, Blank M, Ashkenazi L, Milner Y, Frusic-Zlotkin M, Anhalt GJ, et al. Protective effect of intravenous immunoglobulin (IVIG) in an experimental model of pemphigus vulgaris. Clin Exp Immunol (2005) 142(3):426–32. doi: 10.1111/j.1365-2249.2005.02947.x
95. Mimouni D, Blank M, Payne AS, Anhalt GJ, Avivi C, Barshack I, et al. Efficacy of intravenous immunoglobulin (IVIG) affinity-purified anti-desmoglein anti-idiotypic antibodies in the treatment of an experimental model of pemphigus vulgaris. Clin Exp Immunol (2010) 162(3):543–9. doi: 10.1111/j.1365-2249.2010.04265.x
96. Bilgic Temel A, Murrell DF. Pharmacological advances in pemphigus. Curr Opin Pharmacol (2019) 46:44–9. doi: 10.1016/j.coph.2019.01.001
97. Goebeler M, Bata-Csörgő Z, De Simone C, Didona B, Remenyik E, Reznichenko N, et al. Treatment of pemphigus vulgaris and foliaceus with efgartigimod, a neonatal fc receptor inhibitor: a phase II multicentre, open-label feasibility trial. Br J Dermatol (2022) 186(3):429–39. doi: 10.1111/bjd.20782
98. Werth VP, Culton DA, Concha JSS, Graydon JS, Blumberg LJ, Okawa J, et al. Safety, tolerability, and activity of ALXN1830 targeting the neonatal fc receptor in chronic pemphigus. J Invest Dermatol (2021) 141(12):2858–2865 e4. doi: 10.1016/j.jid.2021.04.031
99. Culton DA, McCray SK, Park M, Roberts JC, Li N, Zedek DC, et al. Mucosal pemphigus vulgaris anti-Dsg3 IgG is pathogenic to the oral mucosa of humanized Dsg3 mice. J Invest Dermatol (2015) 135(6):1590–7. doi: 10.1038/jid.2015.54
100. Garcia M, Escamez MJ, Carretero M, Mirones I, Martinez-Santamaria L, Navarro M, et al. Modeling normal and pathological processes through skin tissue engineering. Mol Carcinog (2007) 46(8):741–5. doi: 10.1002/mc.20327
101. Shimizu A, Ishiko A, Ota T, Tsunoda K, Koyasu S, Amagai M, et al. Ultrastructural changes in mice actively producing antibodies to desmoglein 3 parallel those in patients with pemphigus vulgaris. Arch Dermatol Res (2002) 294(7):318–23. doi: 10.1007/s00403-002-0341-z
102. Takahashi H, Kouno M, Nagao K, Wada N, Hata T, Nishimoto S, et al. Desmoglein 3-specific CD4+ T cells induce pemphigus vulgaris and interface dermatitis in mice. J Clin Invest (2011) 121(9):3677–88. doi: 10.1172/JCI57379
103. Takae Y, Nishikawa T, Amagai M. Pemphigus mouse model as a tool to evaluate various immunosuppressive therapies. Exp Dermatol (2009) 18(3):252–60. doi: 10.1111/j.1600-0625.2008.00776.x
104. Joly P, et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the european academy of dermatology and venereology (EADV). J Eur Acad Dermatol Venereol (2020) 34(9):1900–13. doi: 10.1111/jdv.16752
105. Aoki-Ota M, Tsunoda K, Ota T, Iwasaki T, Koyasu S, Amagai M, et al. A mouse model of pemphigus vulgaris by adoptive transfer of naive splenocytes from desmoglein 3 knockout mice. Br J Dermatol (2004) 151(2):346–54. doi: 10.1111/j.1365-2133.2004.06056.x
106. Holm TL, Markholst H. Confirmation of a disease model of pemphigus vulgaris: characterization and correlation between disease parameters in 90 mice. Exp Dermatol (2010) 19(8):e158–65. doi: 10.1111/j.1600-0625.2009.01033.x
107. Kim AR, Han D, Choi JY, Seok J, Kim SE, Seo SH, et al. Targeting inducible costimulator expressed on CXCR5(+)PD-1(+) T(H) cells suppresses the progression of pemphigus vulgaris. J Allergy Clin Immunol (2020) 146(5):1070–1079 e8. doi: 10.1016/j.jaci.2020.03.036
108. Hennerici T, Pollmann R, Schmidt T, Seipelt M, Tackenberg B, Möbs C, et al. Increased frequency of T follicular helper cells and elevated interleukin-27 plasma levels in patients with pemphigus. PloS One (2016) 11(2):e0148919. doi: 10.1371/journal.pone.0148919
109. Petzl-Erler ML. Beyond the HLA polymorphism: a complex pattern of genetic susceptibility to pemphigus. Genet Mol Biol (2020) 43(3):e20190369. doi: 10.1590/1678-4685-gmb-2019-0369
110. Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Front Med (Lausanne) (2018) 5:226. doi: 10.3389/fmed.2018.00226
111. Hertl M, Amagai M, Sundaram H, Stanley J, Ishii K, Katz SI. Recognition of desmoglein 3 by autoreactive T cells in pemphigus vulgaris patients and normals. J Invest Dermatol (1998) 110(1):62–6. doi: 10.1046/j.1523-1747.1998.00086.x
112. Schmidt T, Willenborg S, Hünig T, Deeg CA, Sonderstrup G, Hertl M, et al. Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris. Exp Dermatol (2016) 25(4):293–8. doi: 10.1111/exd.12919
113. Hudemann C, Pollmann R, Hertl M, Fleischer S, Digigow R, Eming R. Development of a mouse model of pemphigus vulgaris as a tool to evaluate nanoparticle induced antigen-specific tolerance. J Invest Dermatol (2019) 139(9):S220–0. doi: 10.1016/j.jid.2019.07.034
114. 47th annual meeting of the arbeitsgemeinschaft dermatologische forschung (ADF): virtual, march 04-06, 2021. Exp Dermatol (2021) 30(3):e1–e135. doi: 10.1111/exd.14263
Keywords: pemphigus, desmoglein, autoantibody, mouse model, autoimme diseases
Citation: Emtenani S, Hertl M, Schmidt E and Hudemann C (2023) Mouse models of pemphigus: valuable tools to investigate pathomechanisms and novel therapeutic interventions. Front. Immunol. 14:1169947. doi: 10.3389/fimmu.2023.1169947
Received: 20 February 2023; Accepted: 10 April 2023;
Published: 27 April 2023.
Edited by:
Pietro Ghezzi, University of Urbino Carlo Bo, ItalyReviewed by:
Jong Hoon Kim, Yonsei University, Republic of KoreaCopyright © 2023 Emtenani, Hertl, Schmidt and Hudemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Hudemann, Y2hyaXN0b3BoLmh1ZGVtYW5uQHN0YWZmLnVuaS1tYXJidXJnLmRl
†ORCID: Shirin Emtenani, orcid.org/0000-0002-7965-8045
Michael Hertl, orcid.org/0000-0002-5494-2461
Enno Schmidt, orcid.org/0000-0002-1206-8913
Christoph Hudemann, orcid.org/0000-0001-5807-6882
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.