 Yan Liu
Yan Liu Kai Xu
Kai Xu Yin Yao
Yin Yao Zheng Liu
Zheng Liu- 1Department of Otolaryngology-Head and Neck Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Hubei Clinical Research Center for Nasal Inflammatory Diseases, Wuhan, China
- 3Institute of Allergy and Clinical Immunology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Allergic airway diseases are characterized by excessive and prolonged type 2 immune responses to inhaled allergens. Nuclear factor κB (NF-κB) is a master regulator of the immune and inflammatory response, which has been implicated to play a prominent role in the pathogenesis of allergic airway diseases. The potent anti-inflammatory protein A20, termed tumor necrosis factor-α-inducible protein 3 (TNFAIP3), exerts its effects by inhibiting NF-κB signaling. The ubiquitin editing abilities of A20 have attracted much attention, resulting in its identification as a susceptibility gene in various autoimmune and inflammatory disorders. According to the results of genome-wide association studies, several TNFAIP3 gene locus nucleotide polymorphisms have been correlated to allergic airway diseases. In addition, A20 has been found to play a pivotal role in immune regulation in childhood asthma, particularly in the protection against environmentally mediated allergic diseases. The protective effects of A20 against allergy were observed in conditional A20-knockout mice in which A20 was depleted in the lung epithelial cells, dendritic cells, or mast cells. Furthermore, A20 administration significantly decreased inflammatory responses in mouse models of allergic airway diseases. Here, we review emerging findings elucidating the cellular and molecular mechanisms by which A20 regulates inflammatory signaling in allergic airway diseases, as well as discuss its potential as a therapeutic target.
1 Introduction
The incidence rates of allergic airway disorders, including allergic asthma, allergic rhinitis (AR), and chronic rhinosinusitis (CRS), have increased dramatically in recent decades (1–4). Allergies affect the patients’ quality of life and can be life threatening in severe cases, resulting in a heavy financial burden on individuals and the society (5). Allergic airway diseases are characterized by prolonged and exaggerated type 2 immune responses to allergens and immunoglobulin E (IgE)-mediated hypersensitivity (6–8). Under normal physiological conditions, immune responses are tightly controlled at multiple levels to maintain immune homeostasis. Dysfunction of the immune network can lead to a hyperinflammatory state, resulting in the initiation and exacerbation of allergic diseases (8). The transcription factor nuclear factor kappa-B (NF-κB) is involved in both innate and adaptive immunity. Importantly, inappropriate NF-κB activation underlies allergic airway disorder development (8, 9). This phenomenon was confirmed in an allergic-asthma mouse model, where NF-κB translocation blockade led to decreased interleukin (IL)-4 and IL-17 production by type 2 helper T (Th2) and Th17 cells, respectively (10).
A20, encoded by the TNFAIP3 gene, is a cytoplasmic ubiquitin (Ub)-modifying enzyme with dual ubiquitinating and deubiquitinating (DUB) activities (11). It acts as an endogenous negative regulator of NF-κB signaling and has anti-inflammatory and immunomodulatory effects in various inflammatory and autoimmune diseases (12). Single-nucleotide polymorphisms (SNPs) on the TNFAIP3 locus are correlated to several inflammatory disorders, including rheumatoid arthritis (12, 13), systemic lupus erythematosus (14), and inflammatory bowel disease (15). A20 is involved in allergic disease pathogenesis (16). Reduced A20 expression is observed in the epithelium of asthmatic patients (17). Using Tnfaip3 conditional knockout mouse models, its functions in lung in epithelial cells (16), dendritic cells (DCs) (18, 19), T cells (20), and mast cells (21) were explored. Moreover, novel anti-inflammatory functions of A20 have been identified, including those of inhibiting the activation of mitogen-activated protein kinases (MAPKs), modulating the activation of tumor necrosis factor (TNF) receptors and inflammasomes, and limiting the secretion of pro-inflammatory interleukins (22), demonstrating its potential role in anti-inflammation. Notably, intranasal administration of A20 has shown promising effects in alleviating allergic inflammation in animal models of allergic respiratory diseases (23, 24). Herein, we summarize the current data on the mechanism by which A20 regulates allergic diseases, its effects on allergic airway diseases on the basis of experimental evidence from human genetic studies and experimental evidence in animal models, and its potential as a therapeutic target.
2 Regulation of A20 and its relationship to NF-κB signaling in allergic disease
A20 is a highly conserved protein that comprising an amino-terminal ovarian tumor domain (OTU) at its N-terminus, and seven zinc finger (ZnF) domains at its C-terminus (11). The OTU domain is responsible for mediating deubiquitinating (DUB) activity, while ZnF4 and ZnF7 domains are responsible for K63- and M1-linked ubiquitination, respectively, thereby contributing to A20 ubiquitinating activity (12, 22).
Through altering the critical protein ubiquitination status in the toll-like receptor (TLR) and TNF receptor (TNFR) pathways, A20 is well-defined as a potent inhibitor of NF-κB signaling pathway (25, 26). Mechanistically, indicated proteins’ ubiquitination status depends on the OUT-domain’s DUB activity and the corresponding ZnF domains’ ubiquitinating activity of A20 (22). At least four out of seven ZnF domains at the C-terminus of A20 are involved in activating TNF-α-mediated NF-κB by recognizing and degrading receptor-interacting protein 1 (RIP1) (25, 27). Furthermore, A20 is induced by NF-κB activation in the TNFR signaling pathway, forming a negative feedback loop that causes the RIP1 inactivation through ubiquitination (25).
Recently, Das et al. summarized that several proteins regulating the expression and function of A20 proteins at different levels, including transcriptional, post-transcriptional, and post-translational levels (28). However, the upstream initiation and activation of A20 are not well defined. The function and activity of A20 appear to be associated with post-translational alternation, including phosphorylation and protein hydrolysis processes (Figure 1A). At the post-translational level, A20 is generally governed by inhibitory κB (IκB) kinases (IκK). A20 phosphorylation at serine 381 by an IκK leads to NF-κB pathway inhibition (11). In T and B cells, mucosa-associated lymphoid tissue transformation protein 1 (MALT1) mediates A20 cleavage following antigen receptor stimulation (29). The post-translational deubiquitylation modifications of A20 are related to K48- or K63-polyubiquitin chains (22). Multiple A20-binding proteins, including TNFR-associated factor 6 (TRAF6), Tax1-binding protein (TAX1BP1), and A20-binding nuclear inhibitor (ABIN), have been shown to regulate A20 activity (30). In the absence of TAX1BP1, the functions of A20 in regulating RIP1 binding, TRAF6 deubiquitylation, and further NF-κB activation suppression are impaired (30). ABIN-1 physically links A20 to IκK complex, facilitating A20-mediated de-ubiquitination of IκK component to inhibit NF-κB signaling (31).
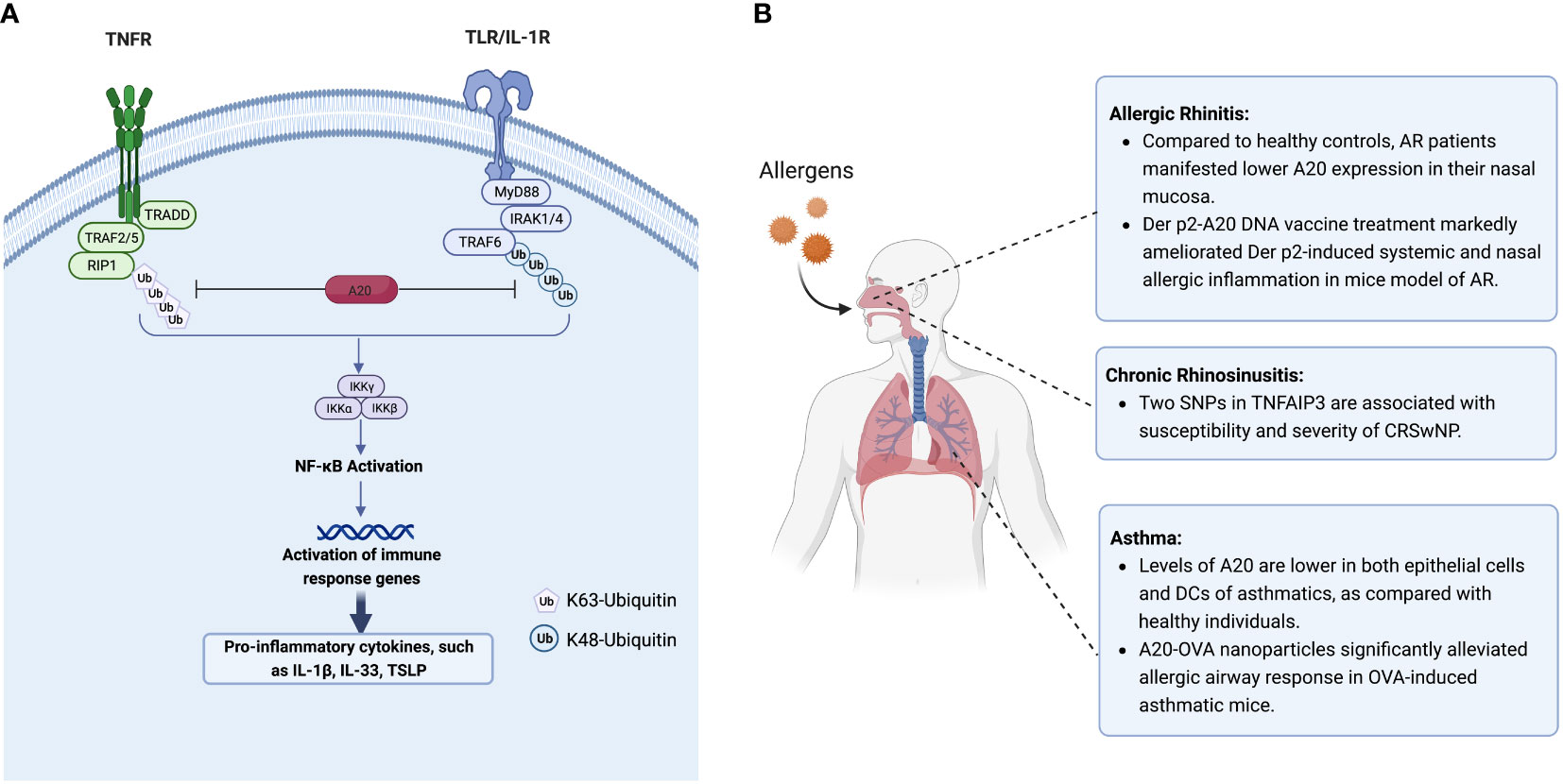
Figure 1 (A) A20 inhibits NF-κB signaling by targeting and degradation of key proteins in TNFR and TLR pathway. The canonical NF-κB pathway is triggered by signals from TNFR and TLR, leading to the activation of NF-κB, and further increased the transcription of cytokines IL-1β, IL-33 and TSLP. A20 inhibits the activation of IKK complex by degradation of ubquitinated RIP1 and TRAF6 in TNFR and TLR/IL-1R pathway, respectively. (B) The expression and regulation of A20 in allergic respiratory diseases. Figure panels were created with BioRender.com. NF-κB, Nuclear factor κB; TNFR, tumor necrosis factor receptor; TLR, toll-like receptor; TSLP, thymic stromal lymphopoietin; IL-1β/IL-33, interleukin 1β/33; IL-1R, interleukin-1 receptor; IKK, IκB kinase.
3 A20 in allergic airway diseases
3.1 A20 in asthma
Allergic asthma is a common asthma type triggered by inhaled allergens and is defined by airway hyperresponsiveness, mucus hyperproduction, and eosinophilic inflammation (1). Inhaled allergens activate epithelial cells and DCs by pattern recognition receptors, leading to Th2 polarization and IgE production, ultimately resulting in the initiation of allergic inflammation (32). In this section, we summarize the phenotypes of existing asthma mouse models that specifically entail depletion of the Tnfaip3 gene in different cell types as well as the critical TNFAIP3 SNPs in asthmatic patients.
A20 protein is hardly detectable in most cell types under resting conditions except specific immune cells (peripheral T cells, DCs) and epithelial cells (18, 28). Studies have shown that A20 levels in both epithelial cells and DCs are significantly lower in asthmatic patients than those in healthy individuals (17). Roles of A20 in allergic asthma have been demonstrated by the observation that administration of A20 significantly attenuated allergic inflammation in asthmatic mice (23, 33). Using an adenovirus containing A20 cDNA, Kang et al. found that A20 significantly diminished inflammatory cell infiltration in the lungs and decreased IL-5 and IL-13 levels in the bronchoalveolar lavage fluid in ovalbumin (OVA)-induced asthmatic mice lungs, accompanied by compromised activation of RIP1 and NF-κB (33). In another study, Luo et al. encapsulated both A20 and OVA into poly(lactic-co-glycolic) acid (PLGA) to create a nano vaccine (23). They found that vaccinated mice showed significantly reduced serum OVA-specific IgE levels, alleviated local inflammatory cell infiltration, and increased regulatory T (Treg) cells numbers in the lungs (23), indicating that A20 might be a potential treatment target for allergic asthma (Figure 1B).
The airway epithelium is the first line of defense against microorganisms and allergens due to their interface location between the host and the environment. In the lung epithelium, A20 mediates protection against farm dust and endotoxin in patients with asthma (16). Compared with Tnfaip3 wild type (Tnfaip3EC-WT) controls, a lung epithelium Tnfaip3-knockout (Tnfaip3EC-KO) mouse model produced less granulocyte-macrophage colony-stimulating factor and less eosinophil inflammation and Th2 response after house dust mites (HDMs) stimulation (16). Related phenomena were confirmed in bronchial epithelium air-liquid interface cultures from asthmatic patients (16). Lipopolysaccharide (LPS) low-dose exposure increased A20 expression in lung epithelial cells, significantly suppressed airway eosinophilia and bronchial hyperreactivity, and decreased allergen-specific IgE and IgG1 levels induced by HDMs in wild-type mice (16). These changes were not observed in the lung epithelial cells in the mice lacking A20 (16). Indeed, asthmatic patients had lower A20 levels in the lung epithelium than healthy subjects (16). Besides suppressing NF-κB activation, A20 also affected epithelial cells in other ways by facilitating staphylococcal enterotoxin B degradation in nasal epithelium via promoting endosomes and lysosome tethering (34), suggesting the central role of A20 in maintaining epithelial cell homeostasis.
DCs are antigen-presenting cells that regulate T-cell differentiation in the lung immune system. However, DCs activation depends on NF-κB activation. A20, an upstream regulator of NF-κB signaling pathway, is typically considered an anti-inflammatory mediator with therapeutic potential in certain allergic airway diseases (33). The expression of A20 in the DCs of the lungs has been correlated with Th2/Th17 cell differentiation in eosinophilic or neutrophilic asthma (18). Specific Tnfaip3 deletion in mouse DCs leads to an increase in the levels of cytokines IL-6 and IL-23 correlated to the Th17 response, resulting in severe neutrophil inflammation (18). Conventional type 1 DCs (cDC1s) are a specific subset of DCs associated with antiviral and anti-tumor immune responses. Vroman et al. found that A20 knockout in cDC1s led to enhanced IL-12 production by cDC1s and increased PD-L1 expression in all pulmonary DC subsets (19). Mice with specific knockout A20 in the cDC1s displayed increased interferon-gamma (IFN-γ)-expressing CD8+ T cell numbers, and absence of Th2-driven eosinophilic airway inflammation in the lungs upon exposure to HDMs (19). These data indicate the importance of NF-κB signaling activation in the DCs for the differentiation of IFN-γ-expressing CD8+ T cells and Th17 cells. However, lower A20 expression in DCs in asthmatic patients than those in healthy controls was observed under unstimulated conditions (17). Therefore, careful consideration of animal models is required when assessing the role of A20 in DCs in asthma.
Increased Th2 cells are a hallmark of allergic asthma. A20, a negative regulator of NF-κB signaling, has been shown to inhibit the development of Th2-driven airway inflammation in OVA-challenged mice (33). Using mice with specific depletion of A20 in T cells (Cd4CreTnfaip3fl/fl), Yokoyama et al. found that infiltration of eosinophils in the lungs, airway hyperresponsiveness, and levels of IL-5 and IL-13 in the lungs were significantly increased in the Cd4CreTnfaip3fl/fl mice compared to those in the wild type mice upon HDMs stimulation (20) (Figure 1). In in vitro studies, depletion of A20 in CD4+ T cells significantly enhanced IL-5 and IL-13 production under Th2 conditions. Mechanistically, the induction of GATA binding protein 3 (GATA3) was faster in CD4+ T cells from Cd4CreTnfaip3fl/fl mice than those from wild type mice, implying that A20 may act as a stabilizer of GATA3 levels during Th2 cell differentiation (20). Consistently, administration of an adenovirus containing A20 cDNA markedly reduced inflammatory cell infiltration in the lungs, inhibited inflammatory cytokines production in bronchoalveolar fluid, and prevented the development of airway hyperresponsiveness (33).
Using acute and chronic HDMs-driven asthma models, Vroman et al. recently found that conditional deletion of Tnfaip3 gene in mouse myeloid cells promotes the secretion of Th17-inducing cytokines IL-1β, IL-6, and IL-23, thereby increasing Th17 cell numbers and exacerbating neutrophilic inflammation (18). In contrast, increased IL-6 and IL-12 robustly inhibited the differentiation of HDMs-specific Th2 cells (18).
Mast cell involvement in early- and late-phase asthma responses has been recognized. Using an A20 knockout mouse asthma model, Heger et al. reported that A20 does not govern the instant mast cell degranulation but acts as a negative feedback inhibitor of NF-κB pathway (21). They also found that loss of A20 in mast cells led to enhanced pro-inflammatory responses downstream of the mast cell antigen receptor module, TLRs, and IL-33R (21). Although mast cell-specific A20 ablation did not trigger any spontaneous pathology phenotype, it markedly elevated IL-33 production and aggravated allergic lung inflammation upon HDMs stimulation (21). A20 depletion, specifically in connective tissue-type mast cells (Mcpt5CreTnfaip3fl/fl), prolonged the survival and enhanced the proliferation activity of LPS- and IL-33-activated mast cells, further promoting inflammation in an asthma model (21). To better illustrate the roles of A20 in asthma, we summarize the phenotypes associated with specific knockout of Tnfaip3 in distinct cell types in mouse asthma models in Figure 2.
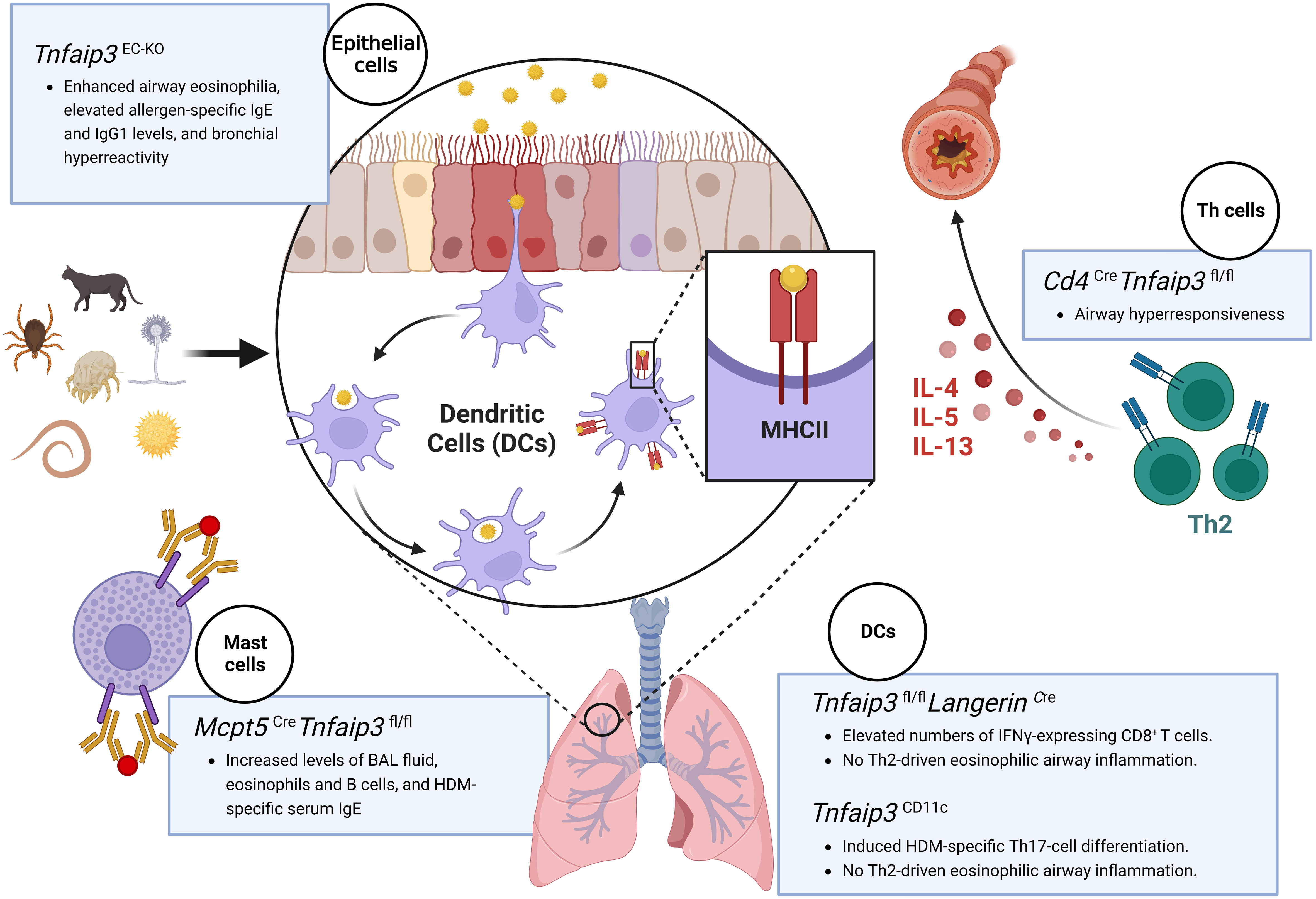
Figure 2 Schematic illustration of various cell-specific knockout mouse models used to study the role of the A20 protein in asthma. Asthma mouse models with specific knockout of the Tnfaip3 gene in the epithelial cells (Tnfaip3EC-KO) exhibit enhanced airway eosinophilia, elevated allergen-specific IgE and IgG1 levels, and bronchial hyperreactivity; mice with Tnfaip3 gene knockout in the dendritic cells (Tnfaip3fl/fl LangerinCre or Tnfaip3CD11c) exhibit the absence of Th2-driven eosinophilic airway inflammation, accomplished with elevated numbers of IFN-γ-expressing CD8+ T cells or increased levels of Th17 cell differentiation; mice with Tnfaip3 gene knockout in T cells (Cd4Cre Tnfaip3fl/fl) display airway hyperresponsiveness; and mice with Tnfaip3 gene knockout in the mast cells (Mcpt5CreTnfaip3fl/fl) display increased BAL fluid levels, serum IgE levels, and eosinophil and B-cell counts. Figure was created with BioRender.com. IgE/IgG1, immunoglobulin E/G1; IFN-γ, interferon-gamma; BAL, bronchoalveolar lavage.
Genetic and environmental factors are involved in asthma pathogenesis, as illustrated by the significant difference in its prevalence among children living in urban versus rural areas (32). Genome-wide association studies of large patient cohorts indicated that the TNFAIP3 gene should be considered as an asthma susceptibility locus (Table 1). In the GABRIELA study population, children growing up on farms had a higher risk of developing asthma than those growing up in urban areas, and this risk was associated with SNP rs2230926, a Phe127 to Cys127 mutation in exon 3 of TNFAIP3 (16). TNFAIP3 interacting protein 1 (TNIP1) interacts with A20 and suppresses the TNF-α-induced NF-κB activation. Li et al. found that several SNPs of the TNIP1 gene, including rs1422673 and rs10036748, were associated with asthma risk (35). Taken together, these findings indicate that genetic factors of A20 play important roles in the development of asthma.
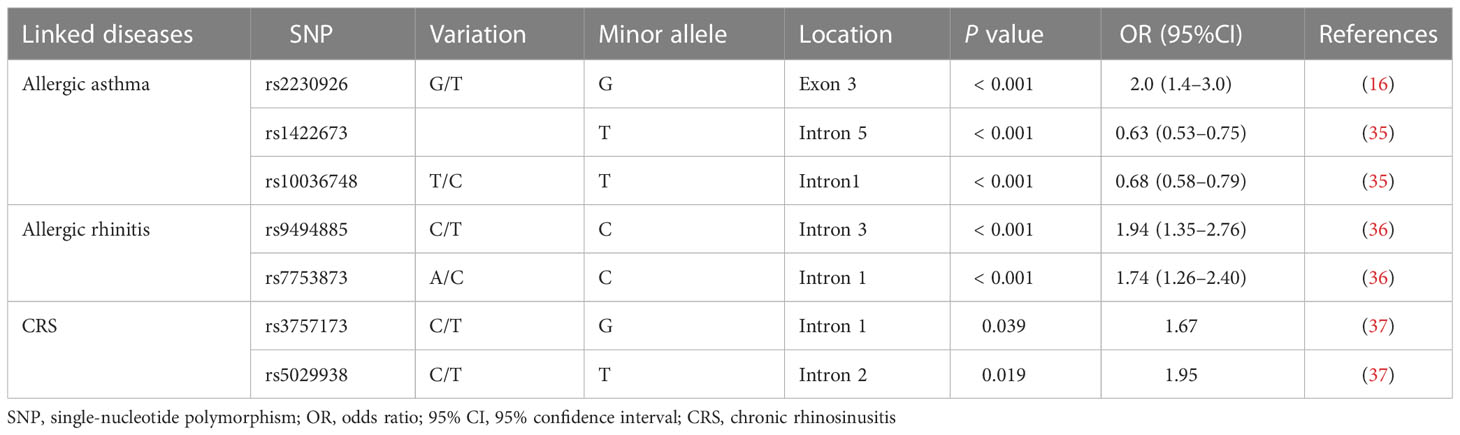
Table 1 Allergic airway diseases associated with TNFAIP3 single-nucleotide polymorphisms.
3.2 A20 in allergic rhinitis
AR, like asthma, is characterized by Th2-driven eosinophilic airway inflammation. Patients with AR had lower A20 expression levels in their nasal mucosa as compared with healthy controls (38). However, higher A20 expression was observed in nasal mucosa of OVA-induced AR mice compared to controls (38). Since A20 is an endogenous NF-κB inhibitor, its expression is strictly regulated (39). In an AR mouse model, antigen invasions, including OVA or pathogens, triggered pathogen recognition receptors, including TLRs, inducing A20 expression at both mRNA and protein levels (24). Subsequently, A20 plays a role in a negative feedback loop by inhibiting key pro-inflammatory signaling pathways, including those controlling NF-κB signaling (11, 22). Hu et al. reported that intranasal administration of an over-expression vector which could cause simultaneous overexpression of a dust mite antigen, dermatophagoides pteronyssinus (Der p) 2, along with A20 protein in a mouse AR model, led to significantly enhanced infiltration of mononuclear cells in the nasal mucosa, and attenuated the levels of Der p2−specific IgE, IL−4, and IL−13 in serum (24). CD4+CD25+Foxp3+ Treg populations were elevated substantially in both serum and spleen by upregulating A20 in Derp 2-induced AR mouse model (Figure 1B). It should be noted that there is a gap between experimental allergens animal models induced by certain allergens and allergic patients as the consequence of environmental exposures. The potential application of A20 for relieving the symptoms of AR patients warrants further exploration. The SNPs of TNFAIP3 were also identified as susceptibility factors for AR. Ke et al. found that two SNPs of TNFAIP3, rs9494885 and rs7753873, were linked to AR’s susceptibility in the Chinese Han population (36) (Table 1).
3.3 A20 in CRS
CRS involves chronic inflammation of the mucous membranes of the nasal cavity and sinuses. Several studies have found associations between the TNFAIP3 gene and the occurrence of CRS (37) (Table 1). Significantly, two SNPs (rs3757173 and rs5029938) in TNFAIP3 are associated with the susceptibility and severity of CRS and nasal polyposis (37).
Activation of NF-κB signaling is associated with the production of various pro-inflammatory cytokines, including IL-1, IL-33 and thymic stromal lymphopoietin (TSLP), in human nasal epithelial cells (40). Glucocorticoid treatment, which is considered the first-line treatment of CRS (3), has been found to hinder the recurrence of CRS by inhibiting NF-κB signaling (41). Enhanced NF-κB pathway activation and increased IL-6 and IL-8 levels are observed in patients with CRS (42), implying the involvement of A20 in CRS pathogenesis and development. Nevertheless, neither expression levels nor the role of A20 in CRS has been investigated. Whether A20 contributes to the pathogenesis of CRS or nasal polyposis awaits to be explored. The application of an A20 overexpression vector in primary human nasal epithelial cells or conducting A20 depletion in cell-type specific CRS mice models would help illustrate the exact role of A20 in CRS, especially Th2-type CRS.
4 A20 as a potential therapeutic target in allergic airway diseases
A20 downregulation may be used as a biomarker to predict the development of childhood asthma (17). Moreover, induction of A20 expression after allergen-induced NF-κB activation is required to resolve inflammation (32, 43). Therefore, the pharmacological induction of A20 may provide resolutions for the inappropriate inflammatory immune response in allergic airway diseases. Farm dust and pathogen exposure, along with induction of A20 expression, reduced the childhood asthma risk (16). Using publicly available gene expression data and a statistically significant connections’ map, Malcomson et al. found that two medications, ikarugamycin and quercetin, significantly reduced A20 expression in both primary nasal and bronchial epithelium lines (44). Intranasally administration of IRL201104, a peptide derived from M. tuberculosis chaperonin 60.1, led to a significant increase in A20 expression in the lung of OVA-sensitized mice and a long-lasting anti-allergic effect, which at least partially explained the protective effects of tuberculosis against allergic diseases (45). A diterpenoid generated from plants called gibberellic acid has potent anti-inflammatory properties. Reihill et al. found that pre-treatment with gibberellic acid considerably increased A20 expression in LPS-induced bronchial epithelium and markedly decreased IL-6 and IL-8 production (46). Vitamin E in γ-tocotrienol natural forms upregulated A20 and inhibited NF-κB activation and its upstream regulator transforming growth factor β-activated kinase 1 in murine macrophages (47). Hand et al. observed that adiponectin, a pleiotropic adipokine, suppressed primary macrophage responses to LPS and pro-inflammatory fatty acids through the induction of A20 in adipose tissue (48). Chrysin is a flavone capable of exerting anti-neuroinflammatory effects in vegetables, fruits, plants, and honey, and it significantly enhanced A20 expression and inhibited LPS-induced NF-κB pathway activation and TRAF6 expression in primary microglial cells and cell lines (49).
Glucocorticoid administration is considered the most effective anti-inflammatory treatment for allergic airway diseases. A20 mRNA and protein expression levels were regulated by dexamethasone, a commonly used glucocorticoid, in human bronchial epithelial cell lines (50). Inflammatory cytokines also governed A20 expression (51). TNF-α and IL-1 promote A20 expression in various cell types. IL-4 and IL-13 significantly downregulate A20 expression in cultured sinonasal epithelial cells (51). Patients suffering from allergic illnesses, including asthma, CRS, and atopic dermatitis, have been demonstrated to benefit significantly from biologics that target IL-4 and IL-13, such as Dupilumab (52–59). Exploration of whether these biologics affect A20 expression in patients with allergic disorders is of great interest. Notably, A20 is expressed in various cell and tissue types. Mouse model studies have shown the effects of cell type-specific regulation of A20 expression, elucidating the A20 function in specific contexts. Therefore, pharmaceuticals targeting A20 to normalize allergic airway responses could be good candidates for future research on this topic.
5 Discussion
Allergic airway inflammation is usually characterized by airway hyperreactivity, increased mucus secretion, and eosinophil aggregation. Cytokines, including IL-25, IL-33, and TSLP are involved in activating the immune responses (1, 8). Accumulating evidence implies that A20 plays a protective role in allergic airway diseases. Findings from animal models reveal the A20 involvement in eosinophilic inflammation development; its role has been observed in the epithelium, T cells, DCs, and mast cells (16, 18–20). Conditional deletion of Tnfaip3 gene in above mentioned cell types in mouse models of airway diseases resulted in an enhanced respiratory inflammatory response and increased local inflammatory factor release, thus facilitating our understanding of A20’s immunological and molecular mechanisms underlying allergic airway disorders. Multiple SNPs of A20/TNFAIP3 are correlated to patients with allergic asthma, AR, and CRS (16, 35–37), supporting the notion that the genetic aspect of A20 is critical in allergic airway disorders. Despite the knowledge gained regarding the role of A20 in allergic airway diseases, several key questions still remain unsolved. Firstly, studies have demonstrated that A20 also protects cells from death, such as pyroptosis and apoptosis, independent of NF-κB signaling (11, 60). Thus, the additional roles of A20 in allergic diseases beyond orchestrating NF-κB signaling require further study. Secondly, other cell types, including follicular helper T cells, B cells, and group 2 innate lymphoid cells, are also crucial for the initiation and development of allergic diseases. Future cell-specific gene targeting studies related to the above-mentioned cell types may provide novel information on A20 function in allergic airway disorders. Furthermore, considering most of current researches are at preclinical level, clinical trials with large sample size are needed to evaluate the efficacy and safety of A20-targeted therapies in allergic respiratory diseases. Meticulously designed randomized cohort studies would help to select the patients who are likely to benefit from A20-targeted therapies. Lastly, the observation windows of current allergic airway disease mice models, either with OVA or HDMs stimulation, are usually shorter than two months, making it impossible fully manipulate the complex pathogenesis of allergic airway diseases in humans. Whether boosting A20 expression and/or its function is a promising strategy for resolving allergic airway disorders is yet to be elucidated.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (NSFC) 81630024 and 81920108011 (ZL), 82101198 (YY), and 81702687 (KX), Knowledge Innovation Program of Wuhan-Shuguang Project 2022020801020455 (YY), the National Key Research and Development Program of China 2022YFE0131200 (ZL), and the Key Research and Development Program of Hubei Province 2021BCA119 (ZL).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Porsbjerg C, Melen E, Lehtimaki L and Shaw D. Asthma. Lancet (2023) 401(10379):858–73. doi: 10.1016/S0140-6736(22)02125-0
2. Bousquet J, Anto JM, Bachert C, Baiardini I, Bosnic-Anticevich S, Walter Canonica G, et al. Allergic rhinitis. Nat Rev Dis Primers (2020) 6(1):95. doi: 10.1038/s41572-020-00227-0
3. Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, Reitsma S, et al. European Position paper on rhinosinusitis and nasal polyps 2020. Rhinology (2020) 58(Suppl S29):1–464. doi: 10.4193/Rhin20.600
4. Huang K, Yang T, Xu J, Yang L, Zhao J, Zhang X, et al. Prevalence, risk factors, and management of asthma in China: a national cross-sectional study. Lancet (2019) 394(10196):407–18. doi: 10.1016/S0140-6736(19)31147-X
5. Pawankar R. Allergic diseases and asthma: a global public health concern and a call to action. World Allergy Organ J (2014) 7(1):12. doi: 10.1186/1939-4551-7-12
6. Han X, Krempski JW and Nadeau K. Advances and novel developments in mechanisms of allergic inflammation. Allergy (2020) 75(12):3100–11. doi: 10.1111/all.14632
7. Yao Y, Chen CL, Yu D and Liu Z. Roles of follicular helper and regulatory T cells in allergic diseases and allergen immunotherapy. Allergy (2021) 76(2):456–70. doi: 10.1111/all.14639
8. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell (2021) 184(6):1469–85. doi: 10.1016/j.cell.2021.02.016
9. Hurrell BP, Galle-Treger L, Jahani PS, Howard E, Helou DG, Banie H, et al. TNFR2 signaling enhances ILC2 survival, function, and induction of airway hyperreactivity. Cell Rep (2019) 29(13):4509–24. doi: 10.1016/j.celrep.2019.11.102
10. Gregorczyk I, Jasiecka-Mikolajczyk A, Maslanka T. Blockade of NF-kappaB translocation and of RANKL/RANK interaction decreases the frequency of Th2 and Th17 cells capable of IL-4 and IL-17 production, respectively, in a mousemodel of allergic asthma. Molecules (2021) 26(11):3117. doi: 10.3390/molecules26113117
11. Bai W, Huo S, Li J and Shao J. Advances in the study of the ubiquitin-editing enzyme A20. Front Pharmacol (2022) 13:845262. doi: 10.3389/fphar.2022.845262
12. Wu Y, He X, Huang N, Yu J and Shao B. A20: a master regulator of arthritis. Arthritis Res Ther (2020) 22(1):220. doi: 10.1186/s13075-020-02281-1
13. Martens A, Hertens P, Priem D, Rinotas V, Meletakos T, Gennadi M, et al. A20 controls RANK-dependent osteoclast formation and bone physiology. EMBO Rep (2022) 23(12):e55233. doi: 10.15252/embr.202255233
14. Pasula S, Gopalakrishnan J, Fu Y, Tessneer KL, Wiley MM, Pelikan RC, et al. Systemic lupus erythematosus variants modulate the function of an enhancer upstream of TNFAIP3. Front Genet (2022) 13:1011965. doi: 10.3389/fgene.2022.1011965
15. Zheng C, Huang Y, Ye Z, Wang Y, Tang Z, Lu J, et al. Infantile onset intractable inflammatory bowel disease due to novel heterozygous mutations in TNFAIP3 (A20). Inflammation Bowel Dis (2018) 24(12):2613–20. doi: 10.1093/ibd/izy165
16. Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science (2015) 349(6252):1106–010. doi: 10.1126/science.aac6623
17. Krusche J, Twardziok M, Rehbach K, Bock A, Tsang MS, Schroder PC, et al. TNF-alpha-induced protein 3 is a key player in childhood asthma development and environment-mediated protection. J Allergy Clin Immunol (2019) 144(6):1684–96. doi: 10.1016/j.jaci.2019.07.029
18. Vroman H, Bergen IM, van Hulst JAC, van Nimwegen M, van Uden D, Schuijs MJ, et al. TNF-alpha-induced protein 3 levels in lung dendritic cells instruct T(H)2 or T(H)17 cell differentiation in eosinophilic or neutrophilic asthma. J Allergy Clin Immunol (2018) 141(5):1620–33. doi: 10.1016/j.jaci.2017.08.012
19. Vroman H, van Uden D, Bergen IM, van Hulst JAC, Lukkes M, van Loo G, et al. Tnfaip3 expression in pulmonary conventional type 1 langerin-expressing dendritic cells regulates T helper 2-mediated airway inflammation in mice. Allergy (2020) 75(10):2587–98. doi: 10.1111/all.14334
20. Yokoyama Y, Tamachi T, Iwata A, Maezawa Y, Meguro K, Yokota M, et al. A20 (Tnfaip3) expressed in CD4(+) T cells suppresses Th2 cell-mediated allergic airway inflammation in mice. Biochem Biophys Res Commun (2022) 629:47–53. doi: 10.1016/j.bbrc.2022.08.097
21. Heger K, Fierens K, Vahl JC, Aszodi A, Peschke K, Schenten D, et al. A20-deficient mast cells exacerbate inflammatory responses in vivo. PLoS Biol (2014) 12(1):e1001762. doi: 10.1371/journal.pbio.1001762
22. Martens A, van Loo G. A20 at the crossroads of cell death, inflammation, and autoimmunity. Cold Spring Harb Perspect Biol (2020) 12(1):a036418. doi: 10.1101/cshperspect.a036418
23. Luo XQ, Zhong JW, Qiu SY, Zhi M, Yang LQ, Zhou YL, et al. A20-OVA nanoparticles inhibit allergic asthma in a murine model. Inflammation (2020) 43(3):953–61. doi: 10.1007/s10753-020-01181-5
24. Hu W, Ma L, Yang G, Zeng X, Liu J, Cheng B, et al. Der p2−A20 DNA vaccine attenuates allergic inflammation in mice with allergic rhinitis. Mol Med Rep (2019) 20(6):4925–32. doi: 10.3892/mmr.2019.10760
25. Shembade N, Ma A and Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science (2010) 327(5969):1135–9. doi: 10.1126/science.1182364
26. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430(7000):694–9. doi: 10.1038/nature02794
27. Heaton SM, Borg NA and Dixit VM. Ubiquitin in the activation and attenuation of innate antiviral immunity. J Exp Med (2016) 213(1):1–13. doi: 10.1084/jem.20151531
28. Das T, Chen Z, Hendriks RW and Kool M. A20/Tumor necrosis factor alpha-induced protein 3 in immune cells controls development of autoinflammation and autoimmunity: lessons from mouse models. Front Immunol (2018) 9104:104. doi: 10.3389/fimmu.2018.00104
29. Yin H, Karayel O, Chao YY, Seeholzer T, Hamp I, Plettenburg O, et al. A20 and ABIN-1 cooperate in balancing CBM complex-triggered NF-kappaB signaling in activated T cells. Cell Mol Life Sci (2022) 79(2):112. doi: 10.1007/s00018-022-04154-z
30. Shembade N, Harhaj NS, Liebl DJ and Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J (2007) 26(17):3910–22. doi: 10.1038/sj.emboj.7601823
31. Yuan S, Dong X, Tao X, Xu L, Ruan J, Peng J, et al. Emergence of the A20/ABIN-mediated inhibition of NF-kappaB signaling via modifying the ubiquitinated proteins in a basal chordate. Proc Natl Acad Sci U S A (2014) 111(18):6720–5. doi: 10.1073/pnas.1321187111
32. Holt PG, Sly PD. Environmental microbial exposure and protection against asthma. N Engl J Med (2015) 373(26):2576–8. doi: 10.1056/NEJMcibr1511291
33. Kang NI, Yoon HY, Lee YR, Won M, Chung MJ, Park JW, et al. A20 attenuates allergic airway inflammation in mice. J Immunol (2009) 183(2):1488–95. doi: 10.4049/jimmunol.0900163
34. An YF, Li TL, Geng XR, Yang G, Zhao CQ and Yang PC. Ubiquitin E3 ligase A20 facilitates processing microbial product in nasal epithelial cells. J Biol Chem (2012) 287(42):35318–23. doi: 10.1074/jbc.M112.392639
35. Li X, Ampleford EJ, Howard TD, Moore WC, Torgerson DG, Li H, et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J Allergy Clin Immunol (2012) 130(4):861–8. doi: 10.1016/j.jaci.2012.04.041
36. Ke X, Yang Y, Shen Y, Wang X and Hong S. Association between TNFAIP3 gene polymorphisms and risk of allergic rhinitis in a Chinese han population. Iran J Allergy Asthma Immunol (2016) 15(1):46–52.
37. Cormier C, Bosse Y, Mfuna L, Hudson TJ and Desrosiers M. Polymorphisms in the tumour necrosis factor alpha-induced protein 3 (TNFAIP3) gene are associated with chronic rhinosinusitis. J Otolaryngol Head Neck Surg (2009) 38(1):133–41.
38. Long X, Wang N and Zhang X. Roles of Clara cell 10-kD protein and type 2 innate lymphoid cells in allergic rhinitis. Cell Cycle (2021) 20(18):1923–34. doi: 10.1080/15384101.2021.1966961
39. Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC and Beyaert R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem Pharmacol (2010) 80(12):2009–20. doi: 10.1016/j.bcp.2010.06.044
40. Liu R, Du J, Zhou J, Zhong B, Ba L, Zhang J, et al. Elevated microRNA-21 is a brake of inflammation involved in the development of nasal polyps. Front Immunol (2021) 12530488. doi: 10.3389/fimmu.2021.530488
41. Du J, Lv H, Dou X and Cao Z. Nuclear factor kappaB/microRNA-155 upregulates the expression pattern of cytokines in regulating the relapse of chronic sinusitis with nasal polyps and the underlying mechanism of glucocorticoid. Med Sci Monit (2020) 13:e923618. doi: 10.12659/MSM.923618
42. Xu R, Xu G, Shi J and Wen W. A correlative study of NF-kappaB activity and cytokines expression in human chronic nasal sinusitis. J Laryngol Otol (2007) 121(7):644–9. doi: 10.1017/S0022215106001824
43. Singh PB, Pua HH, Happ HC, Schneider C, von Moltke J, Locksley RM, et al. MicroRNA regulation of type 2 innate lymphoid cell homeostasis and function in allergic inflammation. J Exp Med (2017) 214(12):3627–43. doi: 10.1084/jem.20170545
44. Malcomson B, Wilson H, Veglia E, Thillaiyampalam G, Barsden R, Donegan S, et al. Connectivity mapping (ssCMap) to predict A20-inducing drugs and their antiinflammatory action in cystic fibrosis. Proc Natl Acad Sci U S A (2016) 113(26):E3725–3734. doi: 10.1073/pnas.1520289113
45. Riffo-Vasquez Y, Kanabar V, Keir SD, RR EL, Man F, Jackson DJ, et al. Modulation of allergic inflammation in the lung by a peptide derived from mycobacteria tuberculosis chaperonin 60.1. Clin Exp Allergy (2020) 50(4):508–19. doi: 10.1111/cea.13550
46. Reihill JA, Malcomson B, Bertelsen A, Cheung S, Czerwiec A, Barsden R, et al. Induction of the inflammatory regulator A20 by gibberellic acid in airway epithelial cells. Br J Pharmacol (2016) 173(4):778–89. doi: 10.1111/bph.13200
47. Wang Y, Park NY, Jang Y, Ma A and Jiang Q. Vitamin e gamma-tocotrienol inhibits cytokine-stimulated NF-kappaB activation by induction of anti-inflammatory A20 via stress adaptive response due to modulation of sphingolipids. J Immunol (2015) 195(1):126–33. doi: 10.4049/jimmunol.1403149
48. Hand LE, Usan P, Cooper GJ, Xu LY, Ammori B, Cunningham PS, et al. Adiponectin induces A20 expression in adipose tissue to confer metabolic benefit. Diabetes (2015) 64(1):128–36. doi: 10.2337/db13-1835
49. Li Z, Chu S, He W, Zhang Z, Liu J, Cui L, et al. A20 as a novel target for the anti-neuroinflammatory effect of chrysin via inhibition of NF-kappaB signaling pathway. Brain Behav Immun (2019) 79:228–235. doi: 10.1016/j.bbi.2019.02.005
50. Altonsy MO, Sasse SK, Phang TL and Gerber AN. Context-dependent cooperation between nuclear factor kappaB (NF-kappaB) and the glucocorticoid receptor at a TNFAIP3 intronic enhancer: a mechanism to maintain negative feedback control of inflammation. J Biol Chem (2014) 289(12):8231–9. doi: 10.1074/jbc.M113.545178
51. Li P, Wang Y and Turner JH. Proinflammatory mediators alter expression of nuclear factor kappa b-regulating deubiquitinases in sinonasal epithelial cells. Int Forum Allergy Rhinol (2015) 5(7):583–9. doi: 10.1002/alr.21538
52. Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med (2018) 378(26):2486–96. doi: 10.1056/NEJMoa1804092
53. Rabe KF, Nair P, Brusselle G, Maspero JF, Castro M, Sher L, et al. Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N Engl J Med (2018) 378(26):2475–85. doi: 10.1056/NEJMoa1804093
54. Wechsler ME, Ford LB, Maspero JF, Pavord ID, Papi A, Bourdin A, et al. Long-term safety and efficacy of dupilumab in patients with moderate-to-severe asthma (TRAVERSE): an open-label extension study. Lancet Respir Med (2022) 10(1):11–25. doi: 10.1016/S2213-2600(21)00322-2
55. Bacharier LB, Maspero JF, Katelaris CH, Fiocchi AG, Gagnon R, de Mir I, et al. Dupilumab in children with uncontrolled moderate-to-severe asthma. N Engl J Med (2021) 385(24):2230–40. doi: 10.1056/NEJMoa2106567
56. Bachert C, Han JK, Desrosiers M, Hellings PW, Amin N, Lee SE, et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet (2019) 394(10209):1638–50. doi: 10.1016/S0140-6736(19)31881-1
57. Laidlaw TM, Bachert C, Amin N, Desrosiers M, Hellings PW, Mullol J, et al. Dupilumab improves upper and lower airway disease control in chronic rhinosinusitis with nasal polyps and asthma. Ann Allergy Asthma Immunol (2021) 126(5):584–92. doi: 10.1016/j.anai.2021.01.012
58. Akenroye A, Lassiter G, Jackson JW, Keet C, Segal J, Alexander GC, et al. Comparative efficacy of mepolizumab, benralizumab, and dupilumab in eosinophilic asthma: A Bayesian network meta-analysis. J Allergy Clin Immunol (2022) 150(5):1097–105. doi: 10.1016/j.jaci.2022.05.024
59. Boguniewicz M, Beck LA, Sher L, Guttman-Yassky E, Thaci D, Blauvelt A, et al. Dupilumab improves asthma and sinonasal outcomes in adults with moderate to severe atopic dermatitis. J Allergy Clin Immunol Pract (2021) 9(3):1212–23.e6. doi: 10.1016/j.jaip.2020.12.059
Keywords: A20, allergic airway diseases, nuclear factor κB, protection, inflammation
Citation: Liu Y, Xu K, Yao Y and Liu Z (2023) Current research into A20 mediation of allergic respiratory diseases and its potential usefulness as a therapeutic target. Front. Immunol. 14:1166928. doi: 10.3389/fimmu.2023.1166928
Received: 15 February 2023; Accepted: 16 March 2023;
Published: 28 March 2023.
Edited by:
Ruoxi Yuan, Hospital for Special Surgery, United StatesReviewed by:
Yichao Shen, Baylor College of Medicine, United StatesKaimeng Huang, Dana–Farber Cancer Institute and Harvard Medical School, United States
Cuncai Guo, Washington University in St. Louis, United States
Copyright © 2023 Liu, Xu, Yao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng Liu, emhlbmdsaXVlbnRAaG90bWFpbC5jb20=; Yin Yao, ZHIueWFveWluQGhvdG1haWwuY29t; Kai Xu, a3h1dG9uZ2ppQGhvdG1haWwuY29t