 Yanhong Zhang
Yanhong Zhang Jiajun Weng
Jiajun Weng Luyao Huan1,4
Luyao Huan1,4 Song Sheng
Song Sheng Fengqin Xu
Fengqin Xu- 1Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Traditional Chinese Medicine Clinical Medical School (Xiyuan), Peking University, Beijing, China
- 3Department of Integrated Traditional and Western Medicine, Peking University Health Science Center, Beijing, China
- 4Graduate School of Beijing University of Chinese Medicine, Beijing, China
Mitophagy is a type of autophagy that can selectively eliminate damaged and depolarized mitochondria to maintain mitochondrial activity and cellular homeostasis. Several pathways have been found to participate in different steps of mitophagy. Mitophagy plays a significant role in the homeostasis and physiological function of vascular endothelial cells, vascular smooth muscle cells, and macrophages, and is involved in the development of atherosclerosis (AS). At present, many medications and natural chemicals have been shown to alter mitophagy and slow the progression of AS. This review serves as an introduction to the field of mitophagy for researchers interested in targeting this pathway as part of a potential AS management strategy.
1 Introduction
Atherosclerosis (AS) is a systemic disease and an important risk factor for various high-risk cardiovascular and cerebrovascular diseases, such as stroke, myocardial infarction, and even sudden death (1). Vascular endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and macrophages all play important roles in the development and formation of AS. The vascular endothelium is essential to AS, as it can regulate vascular tension, the proliferation of VSMCs, and vascular permeability (2). ECs also exhibit anti-coagulant and fibrinolysis-promoting capabilities, which can reduce platelet aggregation and immune cell adhesion, preventing the development of pathogenic thrombi (3). Oxidized low-density lipoprotein (ox-LDL) damages ECs in the early stages of AS, causing themsecrete a large amount of chemokines and high expression of endothelial adhesion cytokines (2). Furthermore, ECs can attract monocytes into the endangium, where they differentiate into macrophages, resulting in a partial inflammatory response. Subsequently, VSMCs migrate from the mesomembrane to the subintimal space and proliferate massively (4). This is followed by increased extracellular matrix secretion, which finally leads to the formation of a fibrous plaque cap (5). As lipid build-up increases, macrophages die and create a necrotic core (6). The necrotic core is covered by a fibrous cap, which is composed of oxidized lipoproteins, cholesterol crystals, and cellular debris, with varying levels of remodeling and calcification. (7). In addition, macrophages, when activated by ox-LDL, can transform into cholesterol-rich foam cells, producing matrix metalloproteinases capable of hydrolyzing collagen fibers in the fibrous cap (7). This causes a progressive thinning of the fibrous cap around the necrotic core, which eventually transforms into a vulnerable plaque. Therefore, a favorable control of the roles of ECs, VSMCs, and macrophages is crucial for the prevention and treatment of AS.
Mitochondria are organelles found in eukaryotic cells that use oxidative phosphorylation to produce adenosine triphosphate (ATP) and supply energy to the cells (8). Dysfunctional mitochondria, which are a major source of oxidative stress, cause a build-up of excessive reactive oxygen species (ROS) (9, 10). Lemasters et al. (11) developed the idea of “mitophagy” in 2005 and established that mitophagy may selectively remove damaged mitochondria and that this process plays a key role in maintaining mitochondrial homeostasis and lowering ROS generation. The brief process of mitophagy is shown in Figure 1. Under physiological conditions, mitophagy can effectively remove mitochondria with impaired functions, maintain intracellular calcium homeostasis, cell signal transduction, and ATP synthesis, and reduce the level of ROS and oxidative stress damage (12). Under stress conditions (such as nutrient starvation, traumatic injury, or ischemia/hypoxia), deficient mitophagy leads to a build-up of poor-functioning mitochondria, resulting in excessive ROS (13, 14). This further damages mitochondria, causing the release of pro-apoptotic proteins into the cytoplasm, exacerbating partial inflammatory responses, and eventually causing the rupture of unstable AS plaques (15, 16). In actuality, the key cells involved in the formation and development of AS depend on mitophagy to operate normally (17–19). As a result, in this review, we outline the current knowledge on the classical mechanism of mitophagy in AS on the role of these cell types.
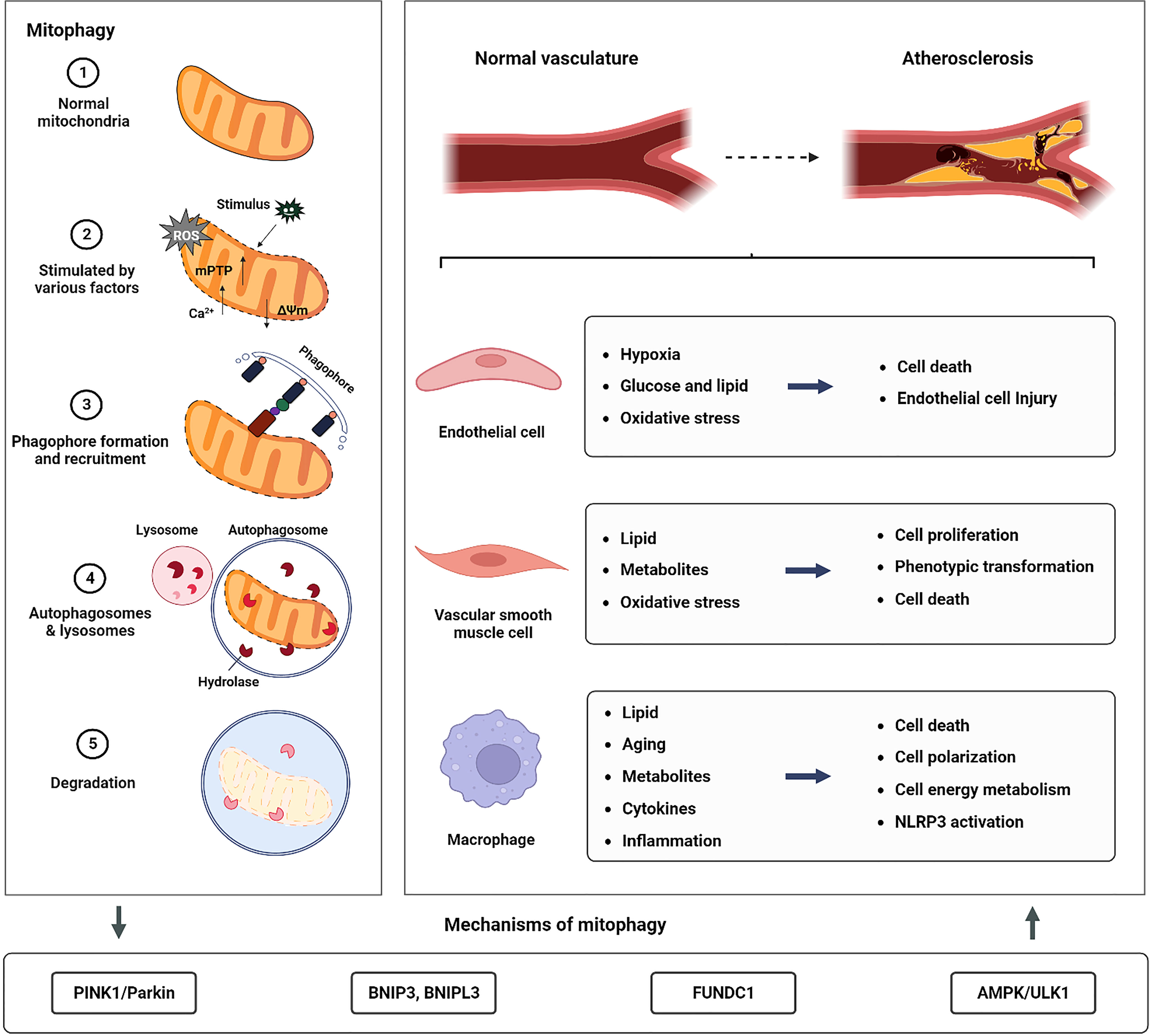
Figure 1 The relationship between mitophagy and AS. The left panel of the image illustrates the macro process of mitophagy. The figure below illustrates the main mitophagy pathway involved in the formation and development of AS. To the right of the figure, the stress conditions and cellular responses reported in current studies on mitophagy in macrophages, ECs, and VSMCs, which are involved in the formation and development of AS are presented.
2 Mechanisms of mitophagy
2.1 PINK1/Parkin-mediated mitophagy
2.1.1 Regulation of PINK1 stability in polarized mitochondria
It was discovered as early as 2006 that in Drosophila, abnormalities in the phosphatase and tensin homolog-long (PTEN)-induced putative kinase protein PINK1 or the Parkin RBR E3 ubiquitin-protein ligase cause mitochondrial dysfunction, muscle degeneration, and decreased longevity (20, 21). PINK1/Parkin-mediated mitophagy may control cell survival or death by eliminating damaged mitochondria (22–24). Under normal conditions, PINK1 enters the mitochondria through the translocase of the outer mitochondrial membrane (TOMM) complex, which is found in the outer mitochondrial membrane (OMM), and the translocase of the inner mitochondrial membrane 23 (TIMM23), and then moves on to the inner mitochondrial membrane (IMM) translocase complex (25). Subsequently, mitochondrial processing peptidase (MPP) in the mitochondrial matrix cleaves the N-terminal mitochondrial-targeting sequence of PINK1, and presenilin-associated rhomboid-like protein (PARL) in the inner mitochondrial matrix cleaves the M-segment of PINK1 (26, 27). The remaining PINK1, with an unstable amino acid at the N-terminus, is released into the cytosol, where it is destroyed by the ubiquitin-proteasome system (UPS) after being targeted by an N-degron2-type E3 ubiquitin ligase (28–30). Due to its degradation, PINK1 remains at very low levels in polarized mitochondria. In addition, Cristina et al. (31) have found that the degradation of PINK1 by the proteasome depends on valosin-containing protein (VCP) and the components of the endoplasmic reticulum-related degradation pathway, such as E3 ligase GP78 and HRD1. However, this does not rule out the possibility that PINK1 serves some sort of physiological purpose.
When mitochondria are damaged, the membrane potential (ΔΨm) of the IMM is depolarized, rendering these mitochondria unable to import PINK1 into the IMM (32), preventing PINK1 from being cleaved by PARL (33). This uncleaved PINK1 is stabilized and accumulates on the OMM, becoming a dimer and attaching to the TOMM complex to limit the import of freshly produced proteins into the mitochondria (29). The TOMM complex supports the autophosphorylation of dimeric PINK1 and aids in its proper localization, both of which are necessary for the recruitment of Parkin and the completion of mitophagy (34). Furthermore, TOMM7 (35), the complex subunit of mitochondrial contact-site and cristae organizing system (36), and the mitochondrial protease OMA1 (35) are involved in controlling PINK1’s stability on the outer membrane.
2.1.2 Recruitment of Parkin and phosphorylation of ubiquitin
PINK1 at the OMM is activated by autophosphorylation at Ser228 (37) and Ser402 (38), and it affects Parkin in two ways. First, PINK1 can phosphorylate ubiquitin (UB) attached to OMM proteins at Ser65 in the mitochondria (39). Due to the high affinity of Parkin for phosphorylated ubiquitin (pSer65-UB), this can drive its translocation from the cytosol to the mitochondria (40, 41). Furthermore, PINK1 directly phosphorylates Parkin at Ser65 in the UB-like domain and attracts Parkin from the cytoplasm to the OMM to recruit autophagy mechanisms (30, 39, 42). In both cases, the activated Parkin conjugates more UB to OMM-associated proteins, which are then phosphorylated by PINK1. This forms a positive feedback loop that amplifies the initial signal, leading to extensive recruitment and ubiquitination of Parkin and the decoration of the OMM with pSer65-UB (43, 44). In addition, the study of crystal structures has provided important insights into the interaction between PINK1 (45, 46), Parkin (47–49), and UB (50). Studies have shown that phosphorylated UB (pUB) can bind two distinct sites on the Parkin structure. The high-affinity site on RING1 controls the Parkin localization, and the low-affinity site on RING0 releases the Parkin autoinhibition (51). The interaction between Parkin’s RING1 and pUB results in the disengagement of the UB-like domain from the core structure and the liberation of Parkin from inhibitory interactions and accumulation on OMM (52, 53). Notably, the UB-like domain of Parkin, when replaced with UB, is more readily activated by PINK1 phosphorylation (51). It can be seen that pUB holds an important role in the feedforward mechanism because it is not only the signal of Parkin recruitment but also the signal of activation.
Parkin is activated by the recruitment and phosphorylation of PINK1, which leads to the ubiquitination of various substrates at the OMM, such as voltage-dependent anion-selective channel protein 1 (VDAC1), mitofusin 1 (MFN1), and mitofusin 2 (MFN2). This results in the general ubiquitination of the mitochondria (54). Indeed, VDAC1 plays a crucial role in regulating mitophagy and apoptosis as a key substrate of Parkin. VDAC1 deficient in polyubiquitination impedes mitophagy, while VDAC1 deficient in monoubiquitination promotes apoptosis by increasing mitochondrial Ca2+ uptake through the mitochondrial calcium uniporter channel (55, 56). Pharmacological inhibition of VDAC1 significantly prevents apoptosis induced by sonodynamic therapy combined with endogenous protoporphyrin IX derived from 5-aminolevulinic acid in human myeloid leukemia-derived (THP-1) monocytes by decreasing Ca2+ levels and oxidative stress (57).
2.1.3 Mitophagy adaptors
The Atg8/LC3 family of autophagy-related proteins is a component of the autophagosome membrane that functions as a bridge between cargo and autophagosomes (58). During autophagy activation, Atg8 family proteins bind to phosphatidyl ethanolamine (PE) through a lipidation process (59). The newly synthesized Atg8 is cleaved by Atg4 family members and then conjugated to PE through a ubiquitination-like reaction cascade (60–62). Once inserted into autophagic membranes, the lipidized LC3 serves as a docking site for various autophagy adaptors and receptors that facilitate the movement of autophagic vesicles and target selection, respectively (63). After the remodeling of the OMM through proteasomal degradation of partially ubiquitinated proteins such as VADC1, MFN2, and MFN1, UB-bound adaptor proteins are recruited to form a substrate poly-UB chain (64, 65). This chain causes the UB-binding protein p62, optineurin (OPTN), nuclear domain 10 protein 52 (NDP52), Tax1-binding protein 1 (TAX1BP1), and the BRCA1 gene 1 protein (NBR1) to aggregate and bind to ubiquitinated proteins (66). Notably, OPTN and NDP52 are the primary, yet redundant, receptors for Parkin-mediated mitophagy, whereas p62 and NBR1 are dispensable (66). Unlike other autophagy receptors for mitochondria, p62 preferentially localizes to the domain between adjacent mitochondria, and aggregates damaged mitochondria using its polymerase basic protein oligomerization domain but does not regulate LC3 recruitment to mitochondria (67, 68). The UB-binding domain of autophagy adaptors recognizes mono- or polyubiquitin chains conjugated to proteins at the surface of the mitochondrion, while the LC3 interaction region (LIR) interacts with Atg8 family proteins. Subsequently, autophagosomes engulfing damaged mitochondria fuse with lysosomes to form “autolysosomes” that degrade the cargo (69, 70). It is worth noting that PINK1, as both a sensor and an effector of mitochondrial damage, can recruit NDP52 and OPTN to damaged mitochondria without the assistance of Parkin (66). Furthermore, tank-binding kinase 1 (TBK1) can phosphorylate some autophagy adaptors, which increases the binding affinity of those proteins to UB chains and Atg8/LC3 proteins (71, 72). TBK1 phosphorylates OPTN (Ser177) (73), NDP52 (Thr137) (74), and p62 (Ser403) (72) to enhance their LC3-binding affinity. In addition, OPTN (Ser473 and Ser513) (73, 75) and p62 (Ser403) (72) are phosphorylated by TBK1, increasing their ability to bind to UB chains, as shown in Figure 2. It is worth noting that PINK1’s phosphorylation of UB is necessary to recruit various autophagy adaptors to mitochondria (66, 76). Mitochondrial damage triggers the phosphorylation of TBK1 (Ser172), which activates its catalytic activity in a PINK1/Parkin-dependent manner (77). This suggests that the assembly of UB chains by Parkin at the OMM may promote a feedforward loop that drives TBK1 phosphorylation to enhance adaptor binding to UB chains.
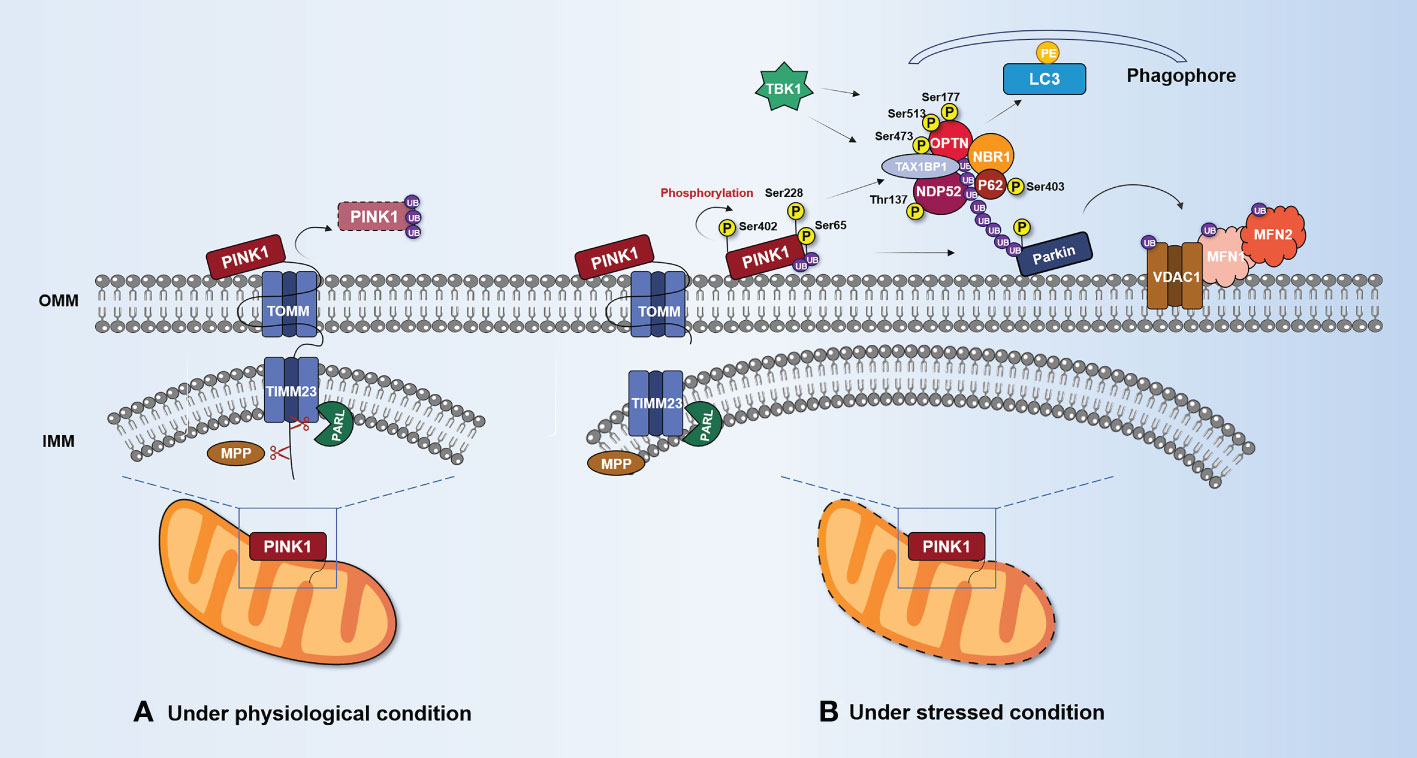
Figure 2 PINK1/Parkin-mediated mitophagy. (A) Under normal circumstances, PINK1 enters the mitochondria via TOMM and TIMM23. MPP and PARL cleave PINK1. The UPS degrades the remaining portion after it is released into the cytosol. (B) Under stress conditions, PINK1 accumulates and stabilizes on the OMM, forming dimers and interacting with TOMM complexes. Accumulated PINK1 (Ser228 and Ser402) is phosphorylated on its own and activated, phosphorylating Ser65 to recruit UB and Parkin. OMM protein substrates (VDAC1, MFN1, and MFN2) are ubiquitinated by activated Parkin. Mitophagy adaptors (p62, OPTN, NDP52, and NBR1) are recruited by the polyubiquitin chain linked to Parkin to start the process of delivering mitochondria to autophagosomes. These mitophagy adaptors work together with LC3 to enclose damaged mitochondria in autophagosomes. TBK1 can promote mitophagy by phosphorylating mitophagy adaptors such as p62 (Ser403), OPTN (Ser177, Ser473, and Ser513), and NDP52 (Thr137). IMM, inner mitochondrial membrane; LC3, microtubule-associated protein 1 light chain 3; MFN1, mitofusin 1; MFN2, mitofusin 2; MPP, mitochondrial processing peptidase; NBR1, neighbor of BRCA1 gene 1; NDP52, nuclear domain 10 protein 52; OMM, outer mitochondrial membrane; OPTN, optineurin; Parkin, E3 ubiquitin-protein ligase Parkin; PARL, presenilin associated rhomboid-like protein; PE, phosphatidyl ethanolamine; PINK1, serine/threonine-protein kinase PINK1; SQSTM1/p62, sequestosome-1/ubiquitin-binding protein p62; TBK1, tank-binding kinase 1; TIMM23, inner mitochondrial membrane 23 translocase; TOMM, outer mitochondrial membrane translocase; UB, ubiquitin; VDAC1, voltage-dependent anion-selective channel protein 1.
2.1.4 Deubiquitinases and PTEN-L as regulators of mitophagy
Recent studies have found that phosphatase and tensin homolog-long (PTEN-L) can effectively prevent the mitochondrial translocation of Parkin, dephosphorylate pSer65-UB, and reduce the level of Parkin phosphorylation (78). This activity keeps Parkin in an autoinhibited state, inhibits its E3 ligase activity, and prevents its recruitment and activation at the OMM. This leads to a blockade of the feedforward mechanism and inhibition of mitophagy (78, 79).
Deubiquitinases (DUBs) are members of the cysteine and metalloproteinase families, which cleave UB from protein substrates. Multiple DUBs regulate mitophagy positively or negatively. USP8 has been demonstrated to selectively remove K6-linked UB chains from Parkin and promote Parkin localization to depolarized mitochondria (80, 81). Conversely, USP15, USP30, and USP35 have been shown to remove Parkin-mediated ubiquitination of OMM proteins and attenuate subsequent depolarized mitochondrial clearance (82–84). Among these DUBs, USP30 tends to hydrolyze unanchored and mitochondrially conjugated Lys6- and Lys11-linked UB chains, delaying or antagonizing Parkin-mediated mitophagy by interfering with its recruitment to mitochondria (85–88). Notably, USP14 is one of the major deubiquitinating enzymes that associates with the proteasome, and its primary function is believed to be preventing proteins from being misubiquitinated in vivo by cleaving UB chains on protein substrates, thereby participating in the process of regulated protein degradation (89, 90). Studies have shown (91) that USP14 is highly expressed in human aortic valve tissue, which may be related to its inhibition of the degradative activity of the proteasome. AS is a lipid-inflammatory process. USP14 can activate the non-canonical NF-κB signaling pathway and promote the expression of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-18, which interact to form a positive feedback cycle, promote lipid deposition, and ultimately induce the occurrence of AS (92, 93). Fu et al. (94) have shown that the overexpression of USP14 in ECs can interfere with the activation of NF-κB, and adenovirus injection of USP14 can alleviate AS in ApoE–/– mice. Inhibition of USP14 reduces the formation of foam cells in AS by down-regulating CD36-mediated lipid uptake (95). Furthermore, the knockdown of USP14 prevents the proliferation of VSMCs induced by platelet-derived growth factor (PDGF)-BB, by inhibiting the mTOR complex 1 (mTORC1)/ribosomal protein S6 kinase signal pathway (96). Nonetheless, it remains unclear how OMM-associated poly-UB prevents cleavage by DUBs. One study found that the PINK1-dependent phosphorylation of UB rendered ubiquitinated OMM proteins DUB-resistant (97). Collectively, understanding the function of DUBs and their regulation in cellular mitophagy may provide a novel and effective approach to the treatment of AS and is worthy of further study.
2.2 BNIP3L and BNIP3-mediated mitophagy
2.2.1 BNIP3L/Nix
BCL2-interacting protein 3-like (BNIP3L/Nix) is an OMM protein that belongs to the BCL2 family of BH3-only proteins. Studies have demonstrated (98) that BNIP3L is a mitophagy receptor that may bind LC3 via the LIR motif at its N-terminus. Notably, BNIP3L can inhibit ox-LDL-induced ROS production and nucleotide oligomerization domain (NOD)-like receptor thermoprotein dome-associated protein 3 (NLRP3) inflammasome activation in macrophages through mitophagy (99). In addition, dynamic phosphorylation can regulate BNIP3L-mediated mitophagy. Phosphorylation of Ser17, Ser34, Ser35, and other residues near the LIR domain improves the interaction between the BNIP3L and LC3 homologs, targeting mitochondria into autophagosomes for degradation (100, 101).
Protein kinase PRKA phosphorylates BNIP3L at Ser212, causing it to be exported from mitochondria and sarcoplasmic sites to the cytosol, preventing mitophagy (102). Furthermore, the dimerization of BNIP3L confers greater mitophagy receptor activity than the monomeric form (103). Notably, glycines 204 and 208 in the transmembrane domain of BNIP3L are important for BNIP3L dimerization, whereas phosphorylation of C-terminal BNIP3L disrupts dimerization (103). Unfortunately, it remains unclear how BNIP3L dimers trigger mitophagy and the molecular mechanism underlying BNIP3L dimerization.
2.2.2 BNIP3
BCL2-interacting protein 3 (BNIP3) mRNA is positively correlated with necrotic core size in human AS plaques (104). In its role as a mitophagy receptor, BNIP3 is typically expressed as an inactive monomer in the cytosol. Upon hypoxia, it forms stable homodimers through its C-terminal transmembrane domain and is integrated into the OMM through this domain (103, 105). However, mutations can disrupt homodimerization and cause mitophagy defects (106). Upon autophagy activation, BNIP3 causes mitophagy by triggering mitochondrial depolarization and subsequent sequestration of mitochondria into autophagic bodies (107). Reduced BNIP3 expression in mouse embryonic stem cells can cause the accumulation of abnormal mitochondria, along with decreased ΔΨm, increased ROS production, and decreased ATP production (108). The BNIP3 gene promoter contains a hypoxia-inducible factor (HIF) response element, and HIF-1 promotes the expression of BNIP3 during hypoxia, which limits ROS production via mitophagy (109–111).
Similar to BNIP3L, the interaction between BNIP3 and LC3 is also regulated by phosphorylation (112, 113), as shown in Figure 3A. Zhu et al. (101) have found that phosphorylation at Ser17 and Ser24 on both sides of the LIR domain of BNIP3 enhances BNIP3–LC3 interaction and promotes mitophagy. Further studies have revealed that unc-51-like autophagy-activating kinase 1 (ULK1) is responsible for the phosphorylation of Ser17 in BNIP3 and of Ser35 in BNIP3L, whereas TBK1 may be required for the phosphorylation of Ser24 and Ser34 in BNIP3 and BNIP3L, respectively (114).
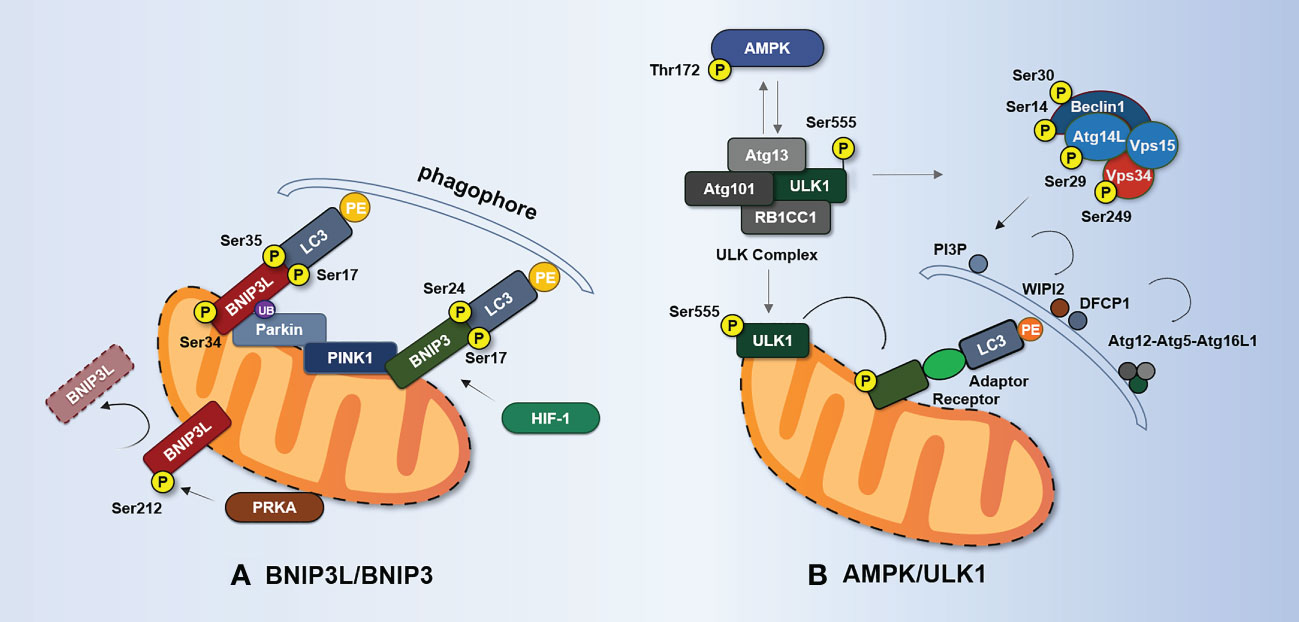
Figure 3 BNIP3L-/BNIP3-mediated and AMPK/ULK1-mediated mechanisms of mitophagy. (A) Phosphorylation of BNIP3L (Ser17, Ser34, and Ser35) enhances the interaction between BNIP3L and LC3 homologs. PRKA phosphorylates BNIP3L at Ser212 and causes BNIP3L to leave the mitochondria, thereby hindering mitophagy. Phosphorylation at Ser17 and Ser24 flanking the LIR domain of BNIP3 enhances the BNIP3–LC3 interaction and promotes mitophagy. Hif-1α enhances mitophagy by promoting the expression of BNIP3 during hypoxia. BNIP3L can be regulated by Parkin ubiquitination. BNIP3 interacts with PINK1 and promotes PINK1 accumulation on OMM. (B) When activated by AMPK phosphorylation, ULK1 recruits ULK complexes consisting of ULK1, Atg13, RB1CC1, and Atg101. The activated ULK complex recruits the Beclin1–Atg14L–Vps34–Vps15 complex. This complex is responsible for the production of Ptdlns3P and the formation of omegasomes at autophagosome formation sites and the recruitment of WIPI2 and DFCP1. WIPI2 and DFCP1 recruit and bind the complex of Atg16L1–Atg5–Atg12 and mediate covalent modification of PE and Atg8/LC3. ULK1 directly phosphorylates Beclin1 at Ser14 and Ser30, Atg14L at Ser29, and Vps34 at Ser249. Under stress, AMPK phosphorylates Ser555 of ULK1, and phosphorylated ULK1 in turn reduces the phosphorylation of Thr172 of the AMPK α-subunit. Phosphorylated ULK1 also phosphorylates the receptor regulating mitophagy. AMPK, adenosine 5′-monophosphate-activated protein kinase; Atg, autophagy-related protein; BNIP3, BCL2-interacting protein 3; BNIP3L/Nix, BCL2-interacting protein 3-like; DFCP1, double FYVE-containing protein 1; RB1CC1, RB1-inducible coiled-coil protein 1; HIF-1, hypoxia-inducible factor 1; PI3P, phosphatidylinositol 3-phosphate; PRKA, protein kinase A; ULK1, unc-51-like autophagy-activating kinase 1; Vps, vacuolar protein sorting-associated protein; WIPI2, WD repeat domain phosphoinositide-interacting protein 2.
2.2.3 Relationship between BNIP3/BNIP3L and PINK1/Parkin-dependent mitophagy
BNIP3 and BNIP3L may be involved in PINK1/Parkin-mediated mitophagy. According to Onishi et al. (115, 116), Parkin may ubiquitinate BNIP3L to attract NBR1, a specific autophagic adaptor of LC3, to mitochondria, causing autophagosomes to form around damaged mitochondria. This study demonstrated that BNIP3L acts as a substrate for Parkin and that mitochondrial clearance induced by BNIP3L depends on the presence of Parkin. In addition, Zhang et al. (117) have found that BNIP3 interacts with PINK1 and promotes PINK1 accumulation at the OMM, leading to Parkin translocation to mitochondria. In conclusion, BNIP3 and BNIP3L can trigger mitophagy through various signaling pathways and cytokines (103). Loss of mitophagy speeds up cell death, but its relationship with AS is poorly understood, necessitating further research on the kinases and phosphatases of BNIP3 and BNIP3L in this context.
2.3 FUNDC1-mediated mitophagy
2.3.1 Phosphorylation and dephosphorylation of FUNDC1
FUN14 domain-containing protein 1 (FUNDC1) is a mitophagy receptor in mammals. Phosphorylation and dephosphorylation of Ser17, Ser13, and Tyr18 in FUNDC1’s LIR domain can have a direct impact on the FUNDC1–LC3 interaction mechanism (118, 119). Casein kinase 2 (CK2) protein kinase, SRC proto-oncogene, non-receptor tyrosine kinase (SRC), and ULK1 are the main regulators of FUNDC1 phosphorylation, while mitochondrial phosphatase PGAM family member 5 (PGAM5) and Bcl-2 like protein 1 (BCL2L1) are considered key regulators of FUNDC1 dephosphorylation (120–122), as shown in Figure 4. FUNDC1 can exist in OMM under normal physiological conditions without mediating mitophagy (123). Furthermore, FUNDC1 is phosphorylated and has little mitophagy activity. FUNDC1 is phosphorylated by CK2 kinase at Ser13 and SRC kinase at Tyr18, which reduces the binding of FUNDC1 and LC3, thereby preventing mitophagy (119, 124). However, under hypoxia, the dephosphorylation of FUNDC1 at Tyr18 and the dephosphorylation of Ser13 by PGAM5 phosphatase both enhance the interaction between FUNDC1 and LC3, which further leads to the selective removal of dysfunctional mitochondria (123). On the other hand, dephosphorylation of FUNDC1 at Ser17 inactivates FUNDC1, but it is phosphorylated by ULK1 kinase under hypoxia or mitochondrial uncoupled stimulation, which increases the binding to LC3 and promotes mitophagy (125).
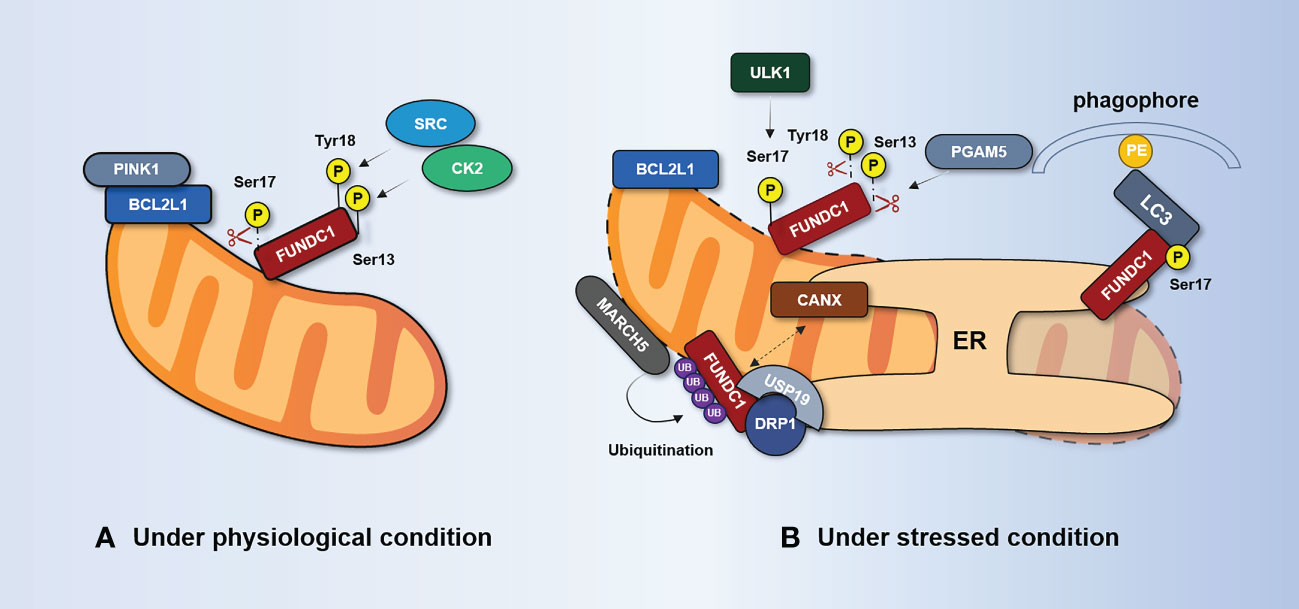
Figure 4 FUNDC1-mediated regulation of mitophagy. (A) Under physiological conditions, FUNDC1 is phosphorylated at Ser13 by CK2, phosphorylated at Tyr18 by SRC, and dephosphorylated at Ser17. It suppresses the interaction of FUNDC1 with LC3. BCL2L1 inhibits PGAM5 activity through the BH3 domain and prevents dephosphorylation at Ser13. (B) Under stress conditions, PGAM5 dissociates from BCL2L1 and dephosphorylates Ser13, thereby promoting FUNDC1-mediated mitophagy. The kinase ULK1 translocates to the mitochondria and phosphorylates FUNDC1 at Ser17. The separation of CANX from FUNDC1 and the incorporation of deubiquitinated FUNDC1 by the deubiquitinating enzyme USP19 at the MAM junction enhances the interaction between DRP1 and FUNDC1. BCL2L1, Bcl-2 like protein 1; CANX, calnexin; CK2, casein kinase 2; DRP1, dynamic-related protein 1; ER, endoplasmic reticulum; FUNDC1, FUN14 domain-containing protein 1; MARCH5, mitochondrial E3 ubiquitin ligase MARCH5; PGAM5, PGAM family member 5; SRC, SRC proto-oncogene; non-receptor tyrosine kinase; USP, ubiquitin-specific protease.
It is worth noting that the regulation of FUNDC1-mediated mitophagy is also influenced by the dephosphorylation and phosphorylation of other proteins. In normoxic conditions, BCL2L1 can interact with PGAM5 through the BH3 domain and inhibit PGAM5 activity, preventing the dephosphorylation of Ser13 (126). During hypoxia, BCL2L1 is degraded, releasing PGAM5 to promote Ser13 dephosphorylation, thereby initiating or enhancing FUNDC1-mediated mitophagy (120, 127). Meanwhile, the kinase ULK1 is recruited to mitochondria and phosphorylates FUNDC1 at Ser17, enhancing its interaction with LC3 to promote mitophagy (125). However, the regulatory mechanism of dephosphorylation of FUNDC1 at Tyr18 remains unclear. These findings highlight the complex and intricate nature of FUNDC1-mediated mitophagy, and further research is needed to fully understand its regulation and signaling pathways.
2.3.2 Phosphorylation-independent mechanisms of FUNDC1 regulation
Recent research has demonstrated that FUNDC1’s negative regulation of phosphorylation-independent activity plays a significant role in mitophagy. Mitochondria-associated endoplasmic reticulum (MAM) is a dynamic membrane-coupling region formed by the coupling of the OMM and the ER and is involved in mitochondrial dynamics, mitophagy, Ca2+ exchange, and other processes (128). FUNDC1 mediates the formation of the MAM and accumulates at the MAM, promoting mitochondrial activity through indirect binding to calnexin (CANX) on the ER (129). A high expression of FUNDC1 promotes MAM formation, leads to increased cytosol Ca2+ levels, promotes phosphorylation of serum response factor, and enhances its binding to the vascular endothelial growth factor receptor 2 (VEGFR2) promoter (130). As a result, angiogenesis is encouraged and VEGFR2 expression is elevated.
Wu et al. (129) found that the association between FUNDC1 and CANX disappears in the late stage of hypoxia, and FUNDC1 mainly interacts with dynamic-related protein 1 (DRP1), which causes DRP1 to initiate mitochondrial fission and mitophagy at MAMs. Interestingly, the deubiquitinating enzyme USP19 incorporates deubiquitinated FUNDC1 at the MAM junction, which enhances the interaction between DRP1 and FUNDC1 to promote mitophagy and mitochondrial fission (131). This suggests that FUNDC1 might serve as a bridging link between mitophagy and mitochondrial dynamics. Furthermore, Chen et al. (132) have discovered that ubiquitination of FUNDC1 at K119, mediated by the mitochondrial E3 ubiquitin ligase MARCH5, could inhibit hypoxia-induced mitophagy via the proteasomal degradation of FUNDC1 during hypoxia.
2.3.3 FUNDC1 and angiogenesis
In the context of AS, neoangiogenesis may increase the flow of local nutrients and oxygen, promoting the progress and remodeling of plaques (133, 134). However, due to the vulnerability of the neovasculature, this may lead to plaque instability, rupture, and thromboembolism (135, 136). Wang et al. (130) have found that FUNDC1-specific deletion in ECs disrupts ECs’ ability to form MAMs and lowers the expression of VEGFR2, which inhibits the growth of functional blood vessels both in vitro and in vivo. However, Wang et al. (137) have discovered that FUNDC1 silencing impairs mitophagy flux and worsens angiotensin II (Ang II)-induced VSMC proliferation and migration. Overexpression of FUNDC1 inhibits Ang II-induced proliferation and migration of VSMCs, blocking Ang II-induced expression of cell adhesion cytokines and matrix metalloproteinase-9, increasing ΔΨm, and reducing intracellular ROS production (137). These findings suggest that FUNDC1 has different effects on participation in angiogenesis in different cell types and more research is needed to fully understand its role in AS.
2.4 AMPK/ULK1-mediated mitophagy
2.4.1 Role of ULK1 in autophagosome formation
As previously mentioned, ULK complexes and Atg9 are required for the formation of mitochondrial autophagosomes (138). ULK1, Atg13, RB1-inducible coiled-coil 1 (RB1CC1), and Atg101, which can be activated by adenosine 5′-monophosphate-activated protein kinase (AMPK) phosphorylation, are the components of the ULK complex (139). The activated ULK complex translocates to the ER tubule vesicle region labeled by Atg9 vesicles and recruits the Beclin1–Atg14L–vacuolar protein sorting-associated protein (Vps) 34–Vps15 complex (140). This complex is responsible for the production of phosphatidylinositol 3-phosphate (PI3P) at the autophagosome formation site and the formation of omegasomes (141–143). Omegasomes amplify local PI3P signaling to attract members of the WIPI protein family, such as WD repeat domain, phosphoinositide interacting 2 (WIPI2), and double FYVE-containing protein 1 (DFCP1) (144, 145). These proteins then attract and bind the Atg16L1–Atg5–Atg12 complex, which is responsible for the covalent modification of PE and Atg8/LC3 (146). This is a prerequisite for the formation of autophagosomes. Notably, ULK1 directly phosphorylates Beclin1 at Ser14 and Ser30 (147, 148), Atg14L at Ser29 (149), and Vps34 itself at Ser249 (148), thereby enhancing Vps34 activity, PI3P production, and initiation of autophagy.
2.4.2 The AMPL/ULK1 axis signaling cascade
AMPK is a metabolic and redox state sensor that is widely expressed in various tissues and organs. It consists of one catalytic subunit (α-subunit) and two regulatory subunits (a β-subunit and a γ-subunit). AMPK senses the energy available in cells by binding directly to adenine nucleotides, and is activated in response to changes in the ratio of ATP to ADP or ATP to AMP in cellular energy (150–152). AMPK plays a crucial role in regulating cellular metabolism by shifting metabolism toward increased catabolism and decreased anabolism through phosphorylation of key proteins in various pathways, such as mTORC1 (153), lipid homeostasis (154, 155), and glycolysis (156, 157). In addition, AMPK is involved in various physiological processes, such as metabolism, cytoskeleton assembly, transcriptional control, apoptosis, and autophagy (158, 159). Zhang et al. (160) have shown that AMPKα1 deficiency impairs autophagy-mediated macromonocyte differentiation and decreases macrophage survival, attenuating AS in ApoE–/– mice. Furthermore, Chen et al. (161) believe that chronic exercise training may improve aortic endothelial and mitochondrial function by increasing the expression of AMPKα2, decreasing the expression of BNIP3L and LC3B, and increasing aortic mitochondrial content.
Recent studies have shown that the AMPK/ULK1 axis is critical for selective mitochondrial clearance (162, 163). Cells deficient in ULK1 or AMPK show an increased accumulation of damaged mitochondria (164–166). Hypoxia significantly induces AMPKα1 phosphorylation at Thr172 and phosphorylation of ULK1 at Ser555 and Ser317, whereas phosphorylation of ULK1 at Ser757 is decreased (163). This may be due to the enhanced combination of AMPKα1 and ULK1, while inhibiting or silencing AMPKα leads to the loss of ULK1 phosphorylation at Ser555 and interferes with the translocation of ULK1 to mitochondria (163). Furthermore, under glucose starvation, AMPK promotes autophagy by directly phosphorylating ULK1 at Ser317 and Ser777 (167). Under nutrient-sufficient conditions, high activity of mTOR prevents ULK1 activation by phosphorylating Ser757 of ULK1 and inhibiting the interaction between ULK1 and AMPK (167).
Notably, ULK1 regulates AMPK activity by directly phosphorylating all three AMPK subunits (168). This suggests that phosphorylation of ULK1 in turn acts in a feedback loop, as shown in Figure 3B. Although there is currently insufficient evidence showing that AMPK/ULK1-mediated mitophagy is relevant to AS, it has been found that translocation of the apoptosis-triggering factor AIF from mitochondria to the nucleus, which induces macrophage cell death, is observed in ULK1-deficient THP-1 cells upon lipopolysaccharide (LPS) plus nigericin stimulation (169). Additionally, a high-protein diet has been shown to induce macrophage apoptosis through increased mTORC1 targets, phosphorylation of ULK1, and concomitant inhibition of autophagosome formation, ultimately promoting the progression of AS plaque (170).
2.4.3 Series of AMPK/ULK1 axis and PINK1/Parkin axis
As mentioned above, the AMPK/ULK1 axis is linked to the FUNDC1, BNIP3, and BNIP3L-mediated mitophagy axes. In fact, the AMPK/ULK1 axis also interacts with the PINK1/Parkin axis. Under hypoxia or glucose or nutrient starvation, AMPK is activated in the cytoplasm and subsequently phosphorylates Ser555 and Ser757 of ULK1 (163). ULK1 activation results in the phosphorylation of Parkin at Ser108 (171). Inhibition of AMPKα1 activation or silencing of AMPKα1 causes loss of ULK1 phosphorylation and interferes with ULK1 translocation to mitochondria. AMPKα1 deficiency impairs autophagy-mediated macromonocyte differentiation and decreases macrophage survival, thereby attenuating AS in ApoE–/– mice (160). Notably, PINK1-mediated phosphorylation of Parkin (Ser65) requires ULK1-dependent phosphorylation of Parkin at Ser108 (171). This highlights the critical and early role played by the AMPK/ULK1 axis in mitophagy. The interaction between the AMPK/ULK1 axis and the PINK1/Parkin axis deserves further investigation.
In addition, the effects of AMPK and ULK1 on the PINK1/Parkin axis may be independent. Overexpression of AMPKα2 can interact with Ser495 - the phosphorylation site of PINK1 - to enhance mitophagy mediated by it (172). Lee et al. (173) have reported that Parkin is an AMPK substrate and can be activated by AMPK phosphorylation at Ser9. As a result, cell death during necrotizing apoptosis is decreased, and receptor-interacting protein kinase 3 (RIPK3) is polyubiquitinated. Likewise, according to Hung et al. (171), ULK1 phosphorylates Parkin at Ser108 and facilitates Parkin translocation to the mitochondria. Inhibition of ULK1 or mutation of this phosphorylation site results in delayed activation of Parkin and defective mitophagy. Thus, as an early mechanism of mitophagy, the AMPK/ULK1 axis is located upstream of many classical mitophagy pathways. In addition, independent of AMPK, the mitophagy adaptors NDP52 and OPTN can also recruit ULK1 (174). In fact, it is well known that the AMPK/ULK1 axis plays an important role in the prevention and treatment of AS. It can prevent oxidative stress and inflammation, control energy metabolism, prevent apoptosis, play anti-proliferation and anti-migration roles in VSMCs, and inhibit vascular calcification, to protect ECs (160, 175, 176). However, further research is necessary to determine whether AMPK/ULK1 can control mitophagy to prevent and treat AS.
2.5 Other receptors for mitophagy
In addition to the above pathways, it has been found that some receptors are located in the OMM, such as BECN1-regulated activation molecule in autophagy protein 1 (AMBRA1) (177), Bcl-2-like protein 13 (BCL2L13) (178), and FKBP prolyl isomerase FKBP8 (179). These receptors share a common LIR motif in their N-terminal domain that can interact with LC3B. According to studies, AMBRA1 has the ability to start mitophagy in both Parkin-dependent and Parkin-independent ways. AMBRA1 phosphorylation at Ser1014 in LIR regulates AMBRA1–LC3 interaction and mitophagy activity (180). This motif has the capacity both to enhance Parkin-mediated mitochondrial clearance and to control Parkin-independent mitophagy (181). As a result of damaged mitochondria, choline dehydrogenase also builds up at the OMM and interacts with p62 to trigger the recruitment of LC3 and autophagosomes (182). A recently identified receptor that controls mitophagy is the IMM-localized protein prohibitin 2 (PHB2), which differs from the OMM-localized mitophagy receptor (183, 184). PHB2 depletion can prevent the stabilization of PINK1 and recruitment of Parkin, UB, and OPTN to mitochondria via the PARL–PGAM5 axis, resulting in the inhibition of mitophagy upon mitochondrial membrane depolarization or aggregation of misfolded proteins (185). Overexpression of PHB2 directly induces Parkin recruitment to mitochondria. In fact, Parkin can directly bind to PHB2 through its RING1 domain and promote the ubiquitination of PHB2, thereby enhancing the interaction between PHB2 and LC3 (186). In addition, PHB2 may play an important role in maintaining the contractile phenotype of VSMCs by interacting with cartilage oligomeric matrix protein (COMP) and regulating mitochondrial oxidative phosphorylation (187). However, it is not yet known whether the mitophagy receptors on the OMM and IMM act synergistically to promote mitophagy.
MAMs play important roles in Ca2+ exchange, mitochondrial dynamics, and lipid metabolism. Numerous proteins in MAMs have been implicated in mitophagy (188). For example, Phosphofurin acidic cluster sorting protein-2 (PACS-2) plays a critical role in cellular functions by regulating the formation and transfer of cargo proteins at MAMs also in addition to regulating mitochondrial dynamics, endoplasmic reticulum stress, Ca2+ transport, autophagy, and apoptosis (189). Studies have shown that ox-LDL induces clustering of PACS-2 at mitochondria–endoplasmic reticulum contact sites, thereby increasing MAM contacts to promote the formation of phagosomal membranes (190, 191). The knockdown of PACS-2 interferes with mitochondrial formation and mitophagy, thereby increasing apoptosis of VSMCs, which may contribute to the formation of unstable AS plaques.
Recently, the disruption of lipid homeostasis in AS has received increased attention. Cardiolipin (CL) is a phospholipid molecule that exists in the IMM and translocates to the OMM upon mitochondrial damage. Externalized CL can interact with the dynamin-related protein Drp1 and promote its oligomerization, which is crucial for mitochondrial fission (192, 193). Furthermore, externalized CL induces a proinflammatory response. The proinflammatory response is associated with the activation of NLRP3 inflammasome (194, 195) and the activation of inflammatory signal transduction upstream from NF-κB (196). In addition, CL is particularly susceptible to lipid peroxidation. Oxidized CL increases the concentration of Ca2+ in monocytes/macrophages and neutrophils, and elevates the levels of the adhesion cytokines ICAM-1 and VCAM-1 in ECs (197). The hydrolysis of CL may lead to endothelial barrier dysfunction and necrosis (198). The accumulation of oxidized CL in OMM, leading to mPTP opening, and the release of cytochrome c from the mitochondria to the cytosol, induces apoptosis and programmed cell death (199, 200). Notably, externalized CL interacts with LC3 at the OMM, identifying damaged and dysfunctional mitochondria, and initiating mitophagy (201). Further research has found that CL accumulates at MAMs after mitophagy stimulation, interacting not only with MFN2 and CANX, but also with the multiple molecular complexes (AMBRA1/BECN1/WIPI1) involved in autophagosome formation (202). This suggests that CL plays an important role in both the early and late stages of mitophagy. Inhibition or knockdown of the convertase catalyzing the synthesis of CL with a high peroxidation index promotes mitophagy by up-regulating the expression of PINK1 and promoting the recruitment of Parkin to dysfunctional mitochondria, thereby alleviating oxidative stress, insulin resistance, and mitochondrial dysfunction (203–205). Although there no studies have yet shown a relationship between CL-related mitophagy and AS, it has been found that antibodies to oxidized CL correlate with the extent of AS progression, which warrants further investigation (206).
Ceramides are a class of bioactive lipids with structural and regulatory functions. Functionally, ceramide inhibits respiratory chain activity, induces ROS production and oxidative stress, disrupts ΔΨm, and may even induce apoptosis (207, 208). There is a large amount of evidence that ceramides are involved in the pathophysiological process of AS. Studies have shown that a high ceramide risk score is significantly associated with a higher risk of all-cause death in patients with coronary artery disease (209, 210). From the cellular level, ceramide may affect the activity of ECs by directly controlling NO levels (211, 212), promoting the transformation of macrophages into foam cells, mediating the apoptosis of foam cells (213), and inducing osteogenic differentiation of VSMCs (214). Recently, Vos M. et al. (215) showed that the accumulation of mitochondrial ceramide is sufficient to damage the mitochondrial electron transport chain, increase ROS production, and promote mitophagy. The ceramide in the mitochondrial membrane interacts directly with LC3-II on the autophagosomal membrane, thereby selectively targeting mitochondria for autophagy (216). Unfortunately, there has been no study on the relationship between ceramide and AS in the context of mitophagy.
3 Mechanisms and therapeutic advances of mitophagy in cells
3.1 ECs
3.1.1 Mitophagy, ROS, and oxidative stress in ECs
Under normal conditions, ECs lining the inner surface of blood vessels maintain vascular homeostasis by regulating antithrombotic and anti-inflammatory processes, vascular tone, adhesion, and proliferation of VSMCs (217). In the development of AS, various factors cause endothelial dysfunction, resulting in vascular insufficiency, permeability, and proliferation of VSMCs, among other effects (218). However, a growing number of studies have shown that mitophagy plays an important role in endothelial dysfunction in AS. Mitochondria produce the vast majority of ROS, promoting poor activation/function of ECs, which leads to increased intravascular thrombosis, leakage, and inflammation (219–221). Therefore, regulating ROS-triggered oxidative damage in ECs may be an important way to prevent AS exacerbation.
Copper oxide nanoparticles can directly trigger oxidative stress and inflammation, leading to significant genotoxicity and cytotoxicity. Zhang et al. (222) have shown that copper oxide nanoparticles are ubiquitously deposited in lysosomes, leading to lysosomal dysfunction, impairment of autophagic flux, and accumulation of undegraded autophagosomes. They also induce EC death through a caspase-independent pathway, resulting in endothelial lesions. The mitophagy inhibitor Mdivi-1 specifically inhibits mitophagy, significantly exacerbating copper oxide nanoparticle-induced death of ECs (223). Activation of PINK1-mediated mitophagy contributes to the removal of damaged mitochondria in copper oxide nanoparticle-induced vascular ECs (224). This indicates that mitophagy has a protective effect on the copper oxide nanoparticle-induced death of ECs.
Some substances, such as melatonin and hydrogen sulfide, have been shown to exert a protective effect on ECs by alleviating oxidative stress damage through the regulation of mitophagy-related pathways. Tert-butyl hydroperoxide (TBHP) is a potent oxidizing agent that is commonly used to induce oxidative stress in ECs (225, 226). Oxidative stress is a key contributor to the development of AS. Melatonin reverses the damage in ECs caused by TBHP by enhancing the phosphorylation of AMPKα, promoting transcription factor EB (TFEB) nuclear translocation, up-regulating the protein levels of LC3, Parkin, and PINK1, and decreasing p62 (227). This indicates that melatonin may protect ECs against oxidative stress injury via Parkin and AMPK-TFEB mediated-mitophagy. In addition, hydrogen sulfide is a direct scavenger of ROS and peroxynitrite. It can protect cells from oxidative stress by reducing mitochondrial ROS production, increasing ATP synthesis, and preventing mitochondrial membrane depolarization at the cardiovascular and cellular levels (228). Cysteine β-synthase is the predominant hydrogen sulfide-producing enzyme in ECs. Silencing cysteine β-synthase in ECs significantly increases ROS production and ultimately reduces the lifespan and resilience of cells by affecting the mechanisms involved in MFN2 down-regulation, uncoupling of endoplasmic reticulum mitochondrial contact, increasing mitochondrial fission, and enhancing BNIP3-induced mitophagy (229). However, exogenous hydrogen sulfide may protect aortic ECs from high glucose and palmitate by inhibiting apoptosis and oxidative stress and up-regulating mitophagy through the PINK1/Parkin signaling pathway (230).
Notably, p66Shc acts as an oxidoreductase that can generate ROS by oxidizing cytochrome c. Piao et al. (231) have shown that the knockdown of p66shc inhibits mitochondrial ROS increase, membrane potential depolarization, and mitochondrial fusion triggered by the deletion of the CR6-interacting factor CRIF1. It also inhibits CRIF1 deficiency-induced mitophagy by reducing the levels of LC3-II/I, PINK1, and Parkin. Indeed, autophagy initiation requires processes such as autophagosome formation, fusion of autophagosomes with lysosomes, and degradation of autophagy substrates in lysosomes. LC3 is mainly involved in the formation of autophagosomes. In this study, only the expressions of LC3-II/I, PINK1, and Parkin, and the co-localization of LC3 with the nucleus, are observed, which cannot prove that the subsequent procedures of autophagy would take place.
3.1.2 Mitophagy, glucose, and lipids in ECs
Exposure of ECs to high levels of glucose and lipids induces excessive production of ROS and reduces mitophagy, thereby accelerating the accumulation of dysfunctional mitochondria. This, in turn, results in poor EC function and apoptosis, and ultimately contribute to the development and progression of AS (232–235). Similarly, mitophagy deficiency in ECs accelerates the accumulation of dysfunctional mitochondria in ECs and triggers oxidative stress, thereby accelerating the aging of vascular ECs (236). Specifically, the most common type of mitophagy in ECs under high-glucose and -lipid conditions is PINK1-/Parkin-induced mitophagy. High levels of glucose and lipids decrease the expression of PINK1, Parkin, LC3II, Beclin1, and other mitophagy-related proteins in ECs, whereas the expression of p62 is up-regulated (8, 230, 236). Knockout of PINK1 or Parkin may inhibit mitophagy, resulting in the accumulation of mitochondrial fragments and leading to mitochondrial dysfunction, ROS overproduction, and apoptosis in ECs (237). Interestingly, the advanced glycation end products and their main precursor (methylglyoxal) may play an important role in the progression of AS and plaque rupture. Glycosylation leads to structural modifications and functional changes in the fibrinogen molecule, forming a cleavage-resistant, dense, and less porous fibrin network that leads to the progression of AS in diabetic patients (238, 239). The oxidative stress associated with the diabetic metabolite methylglyoxal may cause mitochondrial damage and the activation of Parkin-mediated mitophagy, resulting in the dysregulation of junction proteins and impaired permeabilization of ECs (240). Furthermore, Li et al. (233) have suggested that ox-LDL-induced endothelial injuries may be associated with PTEN overexpression. Inhibition of PTEN expression protects ECs by activating the AMPK/cAMP-response element-binding protein CREB/MFN2/mitophagy signaling pathway (233). These studies suggest that high glucose and lipid levels inhibit mitophagy in ECs by reducing the expression levels of ubiquitin-mediated mitophagy proteins.
Several drugs have shown properties that modulate mitophagy function (as shown in Tables 1, 2). The combination of rivaroxaban and aspirin may increase the expressions of PINK1 and Parkin, restore ΔΨm, and decrease the level of ROS in high-glucose-induced ECs (263). Scutellarin, a plant extract, up-regulates mitophagy through the PINK1/Parkin signal pathway, thereby resisting hyperglycemia-induced endothelial injury (244). Moreover, resveratrol also reduces hyperlipidemia-induced endothelial injuries by enhancing BNIP3-associated mitophagy (241). This implies that promoting mitophagy protects mitochondrial integrity and prevents EC damage induced by high levels of glucose and lipids.
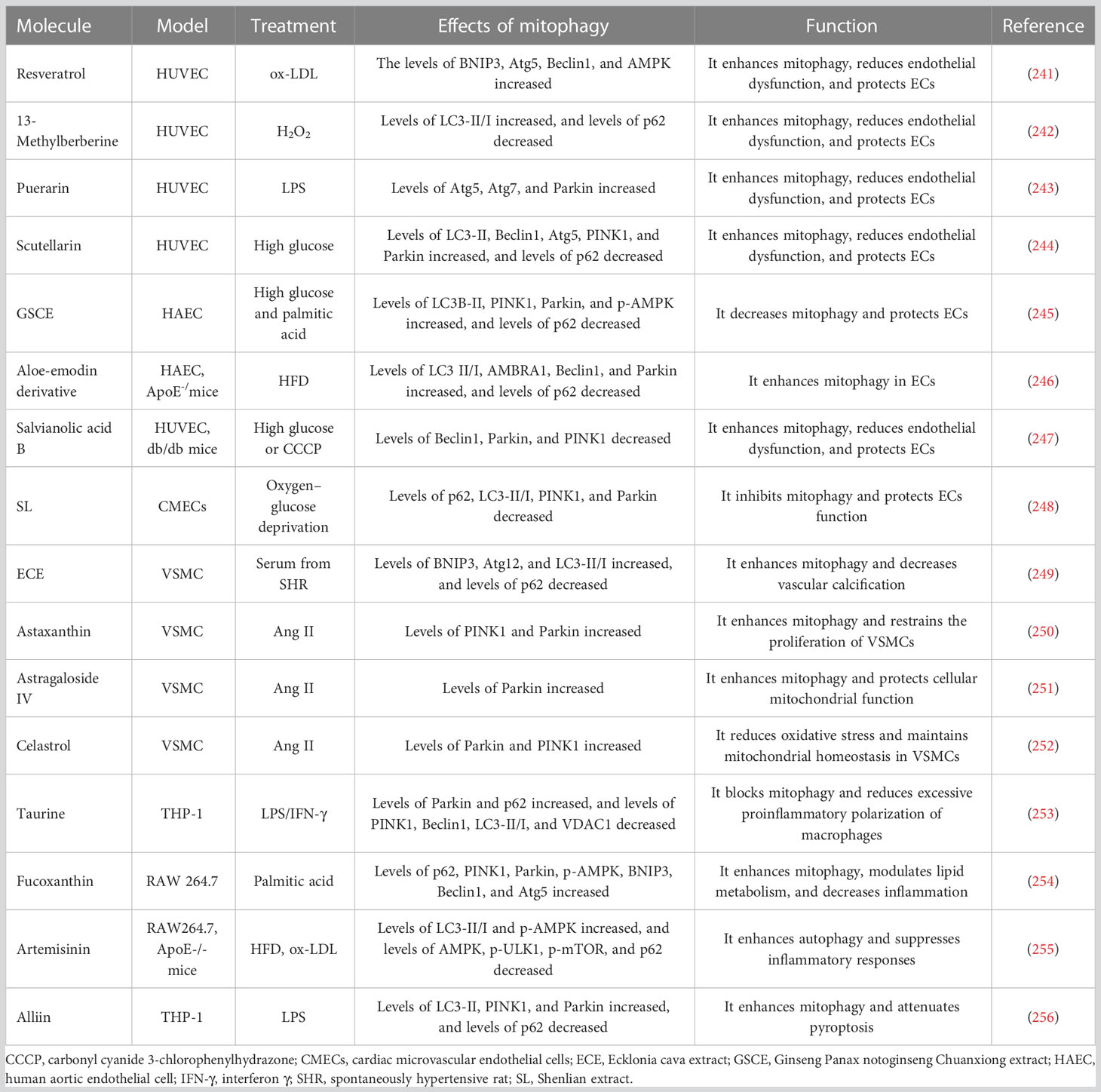
Table 1 Progress of natural compounds in regulating mitophagy in AS.
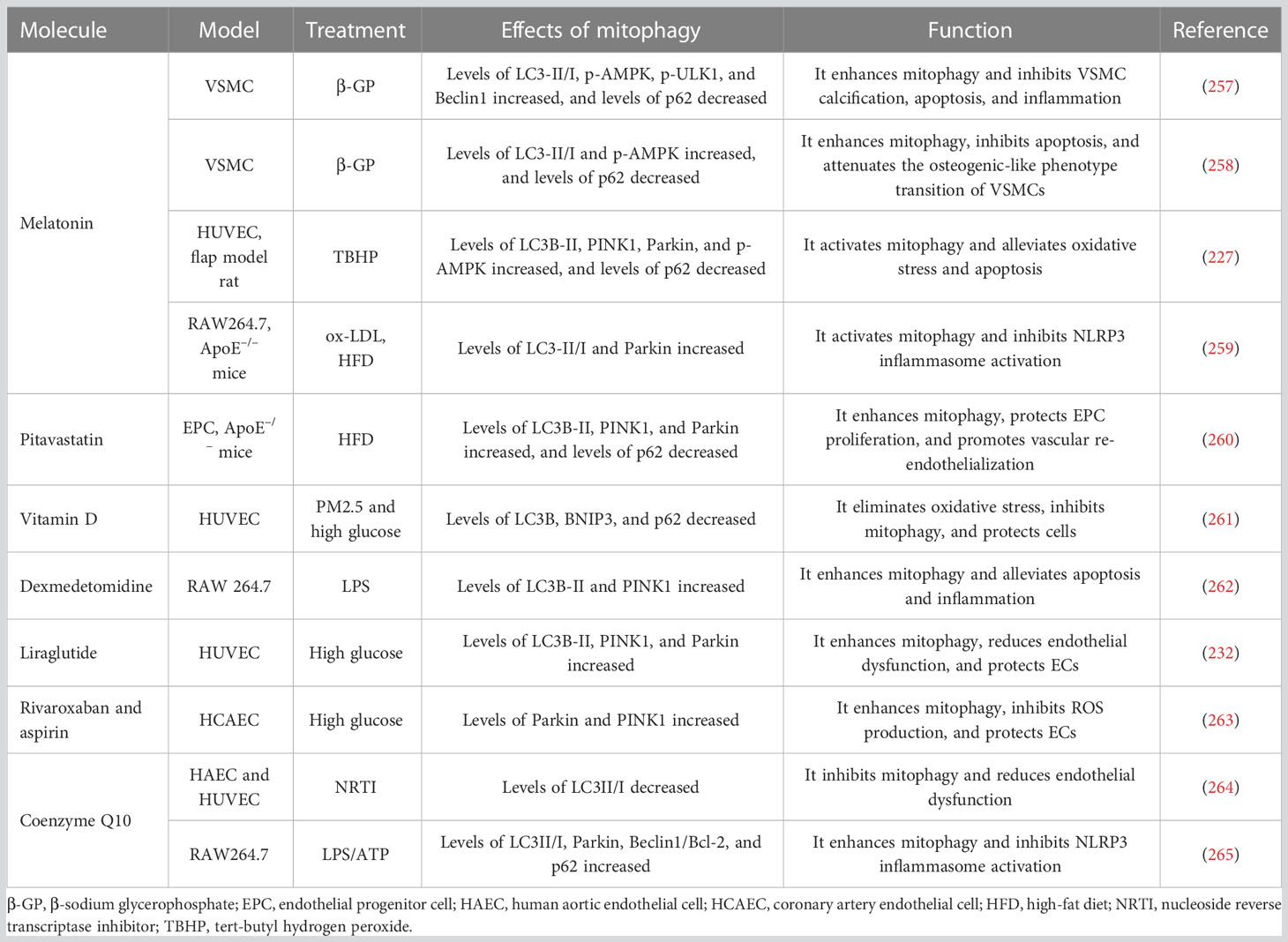
Table 2 Progress of drug regulation of mitophagy in AS.
In contrast, another study has found that ox-LDL can up-regulate the expression of NR4A1, a member of nuclear receptor subfamily 4A, and CaMKII, a calcium/calmodulin-dependent protein kinase, by post-translational modification, thereby activating Parkin-mediated mitophagy (266). The lack of NR4A1 mitophagy protects excess aortic ECs from ox-LDL-induced apoptosis. There are two possible reasons for this phenomenon. First, excessive mitophagy may consume mitochondrial mass, leading to an energy shortage and poor mitochondrial function. Second, acute lipid challenge up-regulates cellular autophagy in response to injury, but long-term lipid stimulation decreases cellular autophagy by inhibiting autophagosome–lysosome fusion (267). In chronic lipid loading, decreased macrolipophagy may promote cellular lipid accumulation, forming a vicious cycle of excessive lipid accumulation. In addition, human umbilical vein endothelial cells (HUVECs) exposed to particulate matter exhibit a significant increase in ROS production, inflammatory response, mitophagy, and apoptosis under high-glucose conditions (261). Vitamin D effectively attenuates oxidative stress, mitophagy, and inflammation in mice induced by streptozotocin and particulate matter (261). Moreover, in the thoracic aorta of diabetic mice with high-glucose- and carbonyl cyanide 3-chlorophenylhydrazone (CCCP)-induced ECs, salvianolic acid B significantly increases the expression of the apoptosis regulator Bcl-2, decreases the expression of the apoptosis regulators Bax, Beclin1, Parkin, and PINK1, inhibits mitophagy, and protects ECs from apoptosis (247). By comparing the findings of these studies, we speculate that acute exposure to hyperglycemia is an acute stress condition. Cells repair mitochondrial dysfunction by activating mitophagy. However, long-term exposure to hyperglycemia causes decreased glycolysis and massive generation of ROS, which inhibits ATP production and thus leads to suppressed mitophagy (268).
3.1.2 Mitophagy and hypoxia in ECs
Recent studies have demonstrated that the imbalance between oxygen supply and demand in the arterial vessel wall is a key contributor to AS and plaque instability (269). Indeed, hypoxia can cause ROS overproduction and poor mitochondrial function, leading to enhanced local inflammatory responses and EC injury and death and ultimately promoting the formation and progression of AS plaques (270–273). The beneficial effects of mitophagy activation in protecting cells after hypoxia are controversial. Some scholars believe that mitophagy activation can reduce hypoxia-induced impairment of endothelial function by improving mitochondrial quality control, reducing ROS production, and increasing cell viability and proliferation (274–276). Conversely, Wang et al. (277, 278) believe that inhibiting hypoxia-induced autophagy/mitophagy can reduce the inflammatory response, injury, and apoptosis in ECs. Li et al. (248) have found that Shenlian extract may inhibit mitophagy through the PINK/Parkin pathway and regulate mitochondrial function to prevent hypoxia-induced EC dysfunction.
In fact, there may be multiple reasons for the activation of autophagy in ECs under hypoxia. Autophagy can be considered a cell protection/stress adaptation mechanism that ECs activate in response to energy deprivation (hypoxia, ischemia, nutrient deprivation, etc.) to protect themselves and maintain their own functions. From the perspective of energy metabolism, hypoxia is often accompanied by the consumption of nutrients. This activates AMPK, which phosphorylates and activates ULK1 to initiate autophagy (279). Regarding autophagy procedures, autophagy requires the formation of autophagosomes, the fusion of autophagosomes with lysosomes, and the degradation of autophagy substrates in lysosomes. Although some experiments have observed autophagosome formation and upstream regulatory mechanisms, this does not necessarily mean that mitophagy can successfully occur at the end or reverse the impairment due to insufficient mitophagy.
In the aforementioned studies, hypoxia resulted in the appearance of a large number of autophagosomes in tissues and cells, visible under transmission electron microscopy, and increases in the level of lysosomes co-localized with autophagosomes. However, the investigators concluded that hypoxia-induced hyperactivation of mitophagy, the opening of swollen mitochondria, fragmentation of mitochondrial ridges, and even fragmentation of the cytoplasm continued, although the ROS-scavenging ability of autophagosomes was reduced. (248). Existing evidence suggests that excessive accumulation of autophagosomes can interfere with cell function by damaging organelles and disrupting cell transport, ultimately leading to cell death (280). Moreover, under long-term stress conditions such as nutrient deprivation, hypoxia/ischemia, and oxidative damage, excessive activation of autophagy leads to excessive self-consumption and bioenergetic failure (281). Nevertheless, the detailed mechanism by which cell death occurs is unclear.
Collectively, mitophagy in ECs is induced by high blood glucose and lipid levels, ROS, hypoxia, and other vascular risk factors. Basal mitophagy is required to maintain EC function. However, insufficiently activated mitophagy to eliminate damaged mitochondria impairs vascular endothelial function, thereby promoting the development of AS. Under long-term stress conditions such as nutrient deprivation, hypoxia/ischemia, or oxidative stress, mitophagy may lead to cell death in a time-dependent manner.
3.2 VSMCs
3.2.1 Mitophagy and ox-LDL in VSMCs
VSMCs are a significant cellular component of the blood vessel wall, which regulates blood pressure and blood flow through vasoconstriction and vasodilation (282). Under physiological conditions, differentiated VSMCs are located in the arterial intima and have a contractile phenotype. It is characterized by less proliferation, less extracellular matrix secretion, and the expression of specific contractile proteins such as smooth muscle myosin heavy chain, smooth muscle α-actin, and calmodulin (283–285). However, vascular damage results in the loss of VSMC contractility and a transition to a proliferative phenotype, migrating to the intimal layer of the vessel wall, which accelerates the formation of AS plaques (286). With the development of AS plaques, the apoptosis and extracellular matrix degradation of VSMCs increase, rendering them more susceptible to rupture (287). Therefore, the regulation of VSMC phenotype, proliferation, and death is the key to stabilizing AS plaques.
In VSMCs, ox-LDL may induce mitochondrial DNA damage in AS plaques, leading to decreased aerobic respiration in the cap and core regions (288) and promoting VSMC phenotype conversion (289) and calcification (290), ultimately resulting in VSMC apoptosis (291). Apoptosis of VSMCs in the plaque promotes the formation of unstable plaques (292). Recent studies have shown that mitophagy regulates the VSMC phenotype and proliferation, and can prevent apoptosis caused by low levels of ox-LDL (293) or autophagy (294). First discovered by Swiader et al. (293), knockdown of PINK1 or Parkin impairs mitophagy flux and increases ox-LDL-induced apoptosis in VSMCs. However, overexpression of PINK1 and Parkin can activate granulocyte autophagy and protect VSMCs. It may also suppress NLRP3 inflammasome hyperactivation to reduce inflammation in AS plaques (295) and promote a compensatory glycolytic program linked to AMPK and Hex2 in response to mitochondrial dysfunction (296).
3.2.2 Mitophagy, ROS, and oxidative stress in VSMCs
The production of ROS and oxidative stress are important factors contributing to VSMC proliferation. Several studies have demonstrated the role of ROS in VSMC proliferation. For example, hydrogen peroxide (H2O2), a type of ROS, was found to induce the proliferation of VSMCs in vitro (297, 298). Similarly, angiotensin II (Ang II) is a peptide hormone that regulates blood pressure and fluid balance. Studies have shown that it can cause mitochondrial fragmentation and dysfunction, leading to increased production of ROS and oxidative stress in VSMCs (250). This can promote the proliferation and migration of VSMCs (297, 299). Geng et al. (252) demonstrated that nuclear receptor NR4A1 controls mitophagy and mitochondrial fission in VSMCs to maintain mitochondrial homeostasis and protect VSMCs from Ang II-induced oxidative stress damage. Celastrol restores mitochondrial autophagy flux in VSMCs through up-regulating nuclear receptor NR4A1 expression. Meanwhile, Wang et al. (300) suggested that corticostatin can inhibit mitophagy through its receptor somatostatin receptor types 3 and 5, thereby reducing the Ang II-induced production of ROS and proliferation of VSMCs.
Vascular calcification is a marker of vascular stiffness in AS. It is caused by advanced glycation end products and excess ROS (301–303). Excessive ROS production can lead to oxidative stress, which has a destructive effect on lysosomal membrane integrity, resulting in the release of lysosomal hydrolases and insufficient mitophagy after prolonged exposure (301, 304, 305). This is primarily connected to the onset and development of vascular calcification. The change in VSMC phenotype is a crucial factor in vascular calcification, and the transition from a contractile to an osteogenesis phenotype promotes vascular calcification (306). There have been groundbreaking advances in the regulation of VSMC phenotype conversion with certain drugs. Melatonin protects VSMCs from calcification by promoting mitochondrial fusion and mitophagy through the AMPK/mTOR/ULK1 pathway, thus reducing the risk of AS (307). In this process, the dwarf-related protein Runx2 and cleaved caspase-3, which regulate osteogenic phenotype and transcription factors, are down-regulated. This indicates that mitophagy and mitochondrial fusion play vital roles in regulating the phenotype of VSMCs and the development of vascular calcification, which is a breakthrough in reducing the risk of AS. In addition, metformin has also been proven to affect mitochondrial biogenesis by increasing mitophagy, thereby preventing β-glycerophosphate-induced osteogenic phenotypic transformation of VSMCs (258).
Aging is a well-known risk factor for AS. Oxidative stress and ROS may be key to the link between aging and vascular calcification (308). Tyrrell et al. (309, 310) have shown that aging in the presence of normal lipids can lead to mitochondrial dysfunction, the activation of mitophagy, and an increase in the levels of the proinflammatory cytokine IL-6 in the aorta. Meanwhile, enhancing mitophagy by adding spermidine to drinking water during hyperlipidemia inhibits aortic mitochondrial dysfunction, IL-6 levels, and the development of AS with age (311). Excessive accumulation of lipids is an important factor that leads to vascular aging (312). Uchikado et al. (313) have found that ox-LDL induces mitochondrial division via the combination of lectin-like ox-LDL scavenger receptors and angiotensin type II1 receptors, and the rapidly accelerated fibrosarcom (RAF) proto-oncogene serine/threonine-protein kinase CRAF/mitogen-activated protein kinase MEK/extracellular signal-regulated kinase ERK pathway, causes massive production of ROS, leading to mitochondrial dysfunction and aging. However, inhibition of the angiotensin type II1 receptor attenuates senescence by blocking this cascade and inducing the replacement of autophagy by mitophagy through the CRAF/MEK axis and mitochondrial quality control processes in human VSMCs, which are dependent on Ras-related protein (313). Additionally, astragaloside IV alleviates the structural and biochemical abnormalities caused by Ang II in VSMCs by eliminating mtROS levels and enhancing both mitophagy and mitochondrial biosynthesis (251).
3.2.3 Mitophagy and metabolic reprogramming of VSMCs
Recent studies have shown that increased expression of pyruvate dehydrogenase kinase isotype PDK4 is associated with calcified vessels in patients with AS (314). Pyruvate produced during glycolysis is transported to the mitochondria under normoxic conditions, where it is converted to acetyl-CoA by pyruvate dehydrogenase complexes and then enters the tricarboxylic acid cycle (315, 316). However, under hypoxic conditions, pyruvate dehydrogenase complexes are inhibited, leading to reduced mitochondrial tricarboxylic acid cycling activity and increased conversion of pyruvate to lactate in the cytoplasm, known as the Warburg effect (317, 318). This metabolic shift is observed in calcified vessels in patients with AS (314). Recent research has shown that the metabolic reprogramming of VSMCs toward high-rate glycolysis and lactate production in the cytosol is driven by PDK4 (319). However, PDK4 inhibition promotes lysosomal activity and mitophagy, reducing VSMC calcification, and alleviating AS (319). In addition, lactic acid inhibits BNIP3-mediated mitophagy, leading to VSMC calcification in vitro (320). Further research has revealed that lactate may inhibit mitophagy by inducing apoptosis and accelerating the phenotypic transition of osteoblasts and calcium deposition in VSMCs through activation of the NR4A1/DNA-dependent protein kinase catalytic subunit/cell tumor antigen p53 pathway (321). Notably, metabolic reprogramming and mitophagy seem to interact (322). Mitophagy controls metabolic reprogramming, which has an impact on how cells develop and differentiate (323, 324). Therefore, targeting mitophagy and metabolic reprogramming in VSMCs may hold promise as novel treatments for AS.
3.2.4 Mitophagy in VSMCs may not always be beneficial
The migration of VSMCs from the vascular media to the intima is a key factor in the pathogenesis of AS. Microtubules, an important part of the cytoskeleton, play an important role in VSMCs’ migration. A balance between dynamically unstable and stable microtubules is required for cell migration, and the microtubule stability is regulated by multiple microtubule-associated proteins and post-translational modifications (325). KAT2a is a histone acetyltransferase that increases the acetylation of tubulin-α (326). Acetylation of tubulin-α increases the polymerization and stability of microtubules, ultimately inhibiting the directional migration of cells (327, 328). However, activation of autophagy selectively degrades KAT2a, disrupts the microtubule stability, and promotes the directional migration of VSMCs (329). Further research has revealed that specific deletion of ULK1 in VSMCs can inhibit the autophagic degradation of KAT2A, which increases the protein level of acetylated tubulin-α and inhibits the directional migration of VSMCs and the formation of new intima (330). Thus, inhibiting autophagy in VSMCs may be a crucial therapeutic strategy for AS. The relationship between autophagy and microtubule stability remains controversial. Choi et al. (331) have found that glucocorticoids induce microtubule dysfunction and inhibit autophagy by activating mTOR, increasing the protein level of SCG10, a microtubule destabilizing protein, and finally inducing microtubule instability. Autophagy increases microtubule stability by degrading SCG10 and promotes axonal regeneration after injury (332). Kirchenwitz et al. (333) have found that SMER28, an inducer of autophagy, significantly alters microtubule dynamics in cells, promotes acetylation of microtubules, and increases resistance to excitotoxin-induced axonal degeneration.
Apelin is an endogenous ligand of the G protein-coupled receptor APJ. Both apelin and APJ receptors are expressed in VSMCs and exert a variety of effects on the cardiovascular system, namely, angiogenesis, vasodilation, and cardiac contractility (334). He et al. (335) have shown that in the AS lesions of ApoE-/- mice and VSMCs induced by apelin-13, the expression of several mitophagy-related proteins, such as PINK1, Parkin, p-AMPKα, and VADC1, is significantly up-regulated. They believe that PINK1/Parkin-mediated mitophagy promotes apelin-13-induced human aortic VSMC proliferation by activating p-AMPKα and exacerbates the progression of AS lesions in vivo. Furthermore, Chen et al. (336) have confirmed this view further and found that mitochondrial calcium uniporter uptake-dependent mitochondrial calcium-induced mitophagy is involved in the proliferation of VSMCs induced by apelin-13. In fact, some scholars believe that apelin and APJ receptors are protective against AS. Kostopoulos et al. (337) have found that apelin expression is negatively correlated with AS by analyzing human aorta and coronary arteries using immunohistochemical staining, concluding that apelin and its APJ receptor have anti-AS effects in human arteries. Chun et al. (338) have found that apelin produces an anti-AS effect by promoting nitric oxide production and inhibiting the cell signaling of Ang II. Additionally, apelin-13 treatment improves blood lipid levels and reduces the vulnerability of AS plaque (339). Apelin inhibits the increased proliferation of cells and blocks the progression of the cell cycle in VSMCs in response to hypoxia. This may be due to apelin's inhibition of autophagy levels in VSMCs. Zhang et al. (340) have shown that apelin protects VSMCs from apoptosis and inhibits the migration of VSMCs under hypoxic conditions. Another study found that apelin can inhibit the osteogenic differentiation of VSMCs and has a protective effect on the calcification of arteries (341). Therefore, the role of the apelin/APJ system in VSMCs needs further investigation.
In summary, most scholars believe that autophagy deficiency in VSMCs promotes an unstable plaque phenotype in AS (342). As a regulatory mechanism of mitochondrial homeostasis, mitophagy may protect VSMCs by regulating ROS generation, oxidative stress, and metabolic levels and by preventing VSMCs from phenotype change, proliferation, and death. This provides possibilities for the prevention and treatment of atherosclerotic, unstable plaques. Although current studies believe that inhibition of autophagy can prevent abnormal proliferation and migration of VSMCs, the degree and duration of autophagy are essential for cell health, and different stress conditions may also be significant reasons for the divergence of research conclusions. In conclusion, new therapies for AS that focus on mitophagy in VSMCs may show great prospects and need further investigation.
3.3 Macrophages
3.3.1 Mitophagy and lipids in macrophages
Macrophages have the ability to actively engulf ox-LDL, leading to the expression of high levels of proinflammatory cytokines, such as TNF, IL-6, and IL-1β, and releasing matrix metalloproteinases during the development of AS (6). This weakens the stability of vulnerable AS plaques by hydrolyzing the collagen fibers in the fibrous cap. However, excessive lipid accumulation impairs mitophagy, which may be associated with excessive ROS generation and exacerbated inflammatory responses (343). Ultimately, this leads to macrophage dysfunction, secondary necrosis, and increased inflammation, which can manifest AS plaque erosion or rupture of vulnerable AS plaques. Mice deficient in FUNDC1 and stimulated by a high-fat diet show increased macrophage infiltration, enhanced M1 macrophage polarization, impaired mitophagy, and develop more severe obesity and insulin resistance (344). Onat et al. (345) have shown that lipid-activated eukaryotic initiation factor EIF2α signaling blocks Parkin-mediated mitophagy in macrophages, leading to greater mitochondrial oxidative stress, inflammasome activation, and IL-1β secretion. Gupta et al. (346) have shown that palmitate induces acetylation of the forkhead box protein FOXO3a, which inhibits the LPS-induced binding of FOXO3a to the PINK1 promoter. This inhibition may be related to LPS-induced mtROS production, mtDNA release, apoptosis-associated speck-like protein (ASC) oligomerization, and caspase-1 activation of macrophages under palmitate conditions. As a result, the activation of the NLRP3 inflammasome increases while PINK1-mediated mitophagy is reduced. The up-regulation of autophagy not only reduces the accumulation of intracellular lipid droplets but also inhibits cell apoptosis and the progression of AS by removing dysfunctional mitochondria and reducing intracellular ROS levels (347). Fucoxanthin treatment increases the expression of PINK1, Parkin, BNIP3, and p-AMPK and significantly increases p62 accumulation, and attenuates inflammation in raw 264.7 induced by palmitic acid (254). Notably, in addition to the high-fat diet, abrupt high-protein diet intake increases amino acid levels in the blood and AS plaques, promotes macrophage mTOR signaling to suppress mitophagy, exacerbates the build-up of dysfunctional mitochondria, and induces AS lipid-induced macrophage apoptosis (170).
3.3.2 Mitophagy and NLRP3 inflammasome activation in macrophages
NLRP3 inflammasome activation and mitophagy must coexist in a balanced manner to maintain cellular and mitochondrial homeostasis, but the exact nature of this relationship is still unknown. Following oligomerization, NLRP3 binds to ASC, which contains the C-terminal caspase-recruitment domain, through its pyrin domain (348). The pro-cysteinyl aspartate-specific proteinase pro-caspase-1 is then recruited to the C-terminal caspase recruitment domain on ASC, resulting in the autocatalytic cleavage of pro-caspase-1 to its active form, caspase-1 (349, 350). However, inhibiting caspase-1 can increase mitophagy, exocytosis, and M2 polarization of macrophages, prevent foam cell formation, and inhibit NLRP3 inflammasome assembly, thereby reducing vascular inflammation and AS (351).
NF-κB is a well-known key activator of inflammation that induces NLRP3 inflammasome activation by up-regulating pro-IL-1β and NLRP3 expression (352–354). Zhong et al. (355) have found that NF-κB can prevent excessive inflammation and inhibit NLRP3 inflammasome activation by inducing delayed accumulation of autophagy receptor p62 in macrophages. It is beneficial for self-limited host response to tissue repair. This suggests that NF-κB has both proinflammatory and anti-inflammatory effects in macrophages, but the complex mechanism remains to be elucidated.
Endogenous lipidoids can activate the NLRP3 inflammasome, potentially driving metabolic inflammation relevant to the pathogenesis of AS (356). Ma et al. (259) have shown that melatonin can reduce the production of mitochondrial ROS by activating the silent information regulator sir2-like protein 3/FOXO3/Parkin-mediated mitophagy, thereby weakening the inflammatory activation of NLRP3 in macrophages induced by ox-LDL. In contrast, dietary polyunsaturated fatty acids may slow the development of AS by triggering macrophage autophagy and reducing NLRP3 inflammasome activation, reducing the number of dysfunctional mitochondria (357).
Recent research has demonstrated that the relationship between macrophages and heme is crucial to homeostasis and inflammation (358). Btb-and-cnc homolog 1 (BACH1) is a key regulator of the cell cycle, cell differentiation, oxidative stress response, and heme homeostasis, which are closely related to the occurrence and development of AS (359, 360). Pradhan et al. (361) have found that mitochondrial energy metabolism in BACH1–/– macrophages shift toward increased glycolysis and reduced oxidative phosphorylation, resulting in increased ΔΨm and mitochondrial ROS production and reduced mitophagy. These changes ultimately trigger the activation of the NLRP3 inflammasome. This enhances the correlation between metabolic reprogramming and mitophagy.
Overall, understanding the relationship between the NLRP3 inflammasome and mitophagy is essential for maintaining macrophage and mitochondrial homeostasis and preventing the development of AS. Further research is needed to fully elucidate the complex mechanisms involved in these processes.
3.3.3 Macrophage mitophagy and macrophage polarization
It is well known that several phenotypically different macrophages play different roles. M2 macrophages are associated with anti-inflammatory effects, tissue repair, and wound healing, whereas M1 macrophages are linked to proinflammatory effects and plaque rupture (362). The polarization classification of macrophages affects the progression and regression of AS. Choi et al. (343) have shown that apolipoprotein A-I binding protein (AIBP) is a novel regulator of macrophage autophagy. In AS, mitochondrial AIBP is involved in mitophagy and mitochondrial quality control, which reduces ROS production and prevents cell death (343). Duan et al. (363) suggest that mitochondrial AIBP may play an anti-AS role by regulating PINK1-dependent mitophagy and M1/M2 polarization.
Notably, reprogramming of macrophages from M1 to M2 can be achieved by targeting metabolic events. Taurine, one of the byproducts of S-adenosylmethionine’s metabolism downstream to the sulfur pathway, helps to maintain the homeostasis of the anti-inflammatory process and cellular energy metabolism (364). Meng et al. (253) have discovered that taurine inhibits methylation of the S-adenosylmethionine-dependent protein phosphatase 2 catalytic subunit, which blocks PINK1-mediated mitophagy flux. This maintains high mitochondrial density and eventually prevents the glycolytic conversion of energy metabolism to M1.
In addition, the cytokines IL-25 (365, 366) and IL-33 (367–369) are considered to have the potential to regulate the progression of AS. Lin et al. (370) have found that IL-33 stimulates the generation of ROS production and subsequently up-regulates the expression of PINK1, Parkin, and LC3 through the AMPK signaling pathway. Furthermore, IL-33 significantly decreases the production of the M1 macrophage-related cytokines CXCL-10 and TNF-α, while increasing the production of the M2 macrophage-related cytokine CCL-22. Tsai et al. (371) have demonstrated that IL-25 induces ROS production and significantly activates the expression of p-AMPK and PINK1, p-Parkin, and LC3 in a dose-dependent manner, promoting mitophagy and M2 macrophage polarization in THP-1-derived macrophages.
3.3.4 Mitophagy and macrophage aging
Aging is an important factor affecting mitophagy in macrophages. The simulator of interference response protein STING is considered a cell solid DNA sensor that mediates sterile inflammation closely associated with atherosclerosis and aging (372, 373). Zhong et al. reported a decrease in PINK1/Parkin-mediated mitochondrial polyubiquitination in aging macrophages (370). In addition, aging changes caused impairment of lysosomal biogenesis and function in macrophages via regulation of the mTOR/TFEB signaling pathway. Overexpression of PINK1 did not promote mitochondrial lysosome formation in aged macrophages, but it did reverse the inhibitory effect of mitochondrial ubiquitination. Combining PINK1 overexpression and treatment with the mTOR inhibitor Torin-1 restored mitophagy flux in aged macrophages and attenuated STING activation. In addition, the guanine nucleotide-binding protein (Gbp1) plays an important role in regulating macrophage polarization, metabolic reprogramming, and cellular senescence (374). Qiu et al. (375) have shown that Gbp1 participates in the removal of damaged or unhealthy mitochondria brought on by proinflammatory cytokine stimulation during mitophagy. Specifically, Gbp1 plays a protective role against inflammation-induced macrophage inflammatory response and metabolic dysfunction by maintaining mitochondrial function and preventing mitochondrial dysfunction-associated senescence through the promotion of mitophagy. However, down-regulation of Gbp1 reduces mitophagy activity, leading to mitochondrial dysfunction, oxidative stress, inflammation, and aging.
In conclusion, dysregulation of lipid metabolism, inflammation, and aging are important factors leading to impaired mitophagy in macrophages. Mitophagy can prevent the formation and development of AS by removing dysfunctional mitochondria, reducing intracellular ROS levels, inhibiting NLRP3 activation, and regulating macrophage energy metabolism, among other pathways.
4 Conclusions
Recent research has demonstrated that the occurrence, development, and pathological mechanisms of AS are all closely related to mitophagy. Mitophagy dysfunction has been linked to several factors, such as ROS, glucose and lipid metabolism disorders, and hypoxia. This dysfunction may cause damage to ECs and result in the proliferation and phenotypic switching of VSMCs. Additionally, it can induce altered polarization of macrophages and metabolic dysfunction, and potentially lead to cell death. However, there are still many limitations and problems in the current research. There is a dearth of reports on animal or clinical studies, with most studies being limited to the cellular level. The correlation between these studies and AS requires additional verification. Is there a common or specific mitophagy pathway present in different cells or tissues within the study model? Does mitophagy in the same cells and tissues have different feedback in different stress modes, to varying degrees and over time? Can some stress conditions closely relate to AS, such as lipid accumulation, abnormal glucose metabolism, inflammation, and oxidative stress, only activate specific mitophagy pathways? It should be noted that further investigations are necessary to determine whether mitophagy is the preferred stress or compensatory pathway of cells under these stress conditions.
In terms of research mechanisms, the classical PINK1/Parkin pathway is currently receiving more attention, while research on other pathways is relatively scarce. In addition, there is a crosstalk between different mitochondrial autophagy pathways and classical AS mechanisms. The same protein can participate in various mitophagy pathways and respond to various stresses showing different expression patterns. It is worth noting that mitophagy can protect cells, but it may also lead to cell damage and cell death. This is an important issue that needs to be clarified in the future. In drug research, a variety of natural and auxiliary drugs that can directly or indirectly regulate mitochondrial quality control have been further explored. However, there is still a lack of safe and effective mitochondrial-targeted therapies. In conclusion, mitophagy, as an important mechanism for regulating AS, needs to be further explored.
Author contributions
YZ and JW conceived the article and wrote the original draft. LH gathered materials and revised the manuscript. SS designed and drew pictures of the mechanism and reviewed the manuscript. FX reviewed the manuscript and critically revised important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Major Key Projects of the Science and Technology Innovation Project of the China Academy of Chinese Medical Sciences (Project no. CI2021A01406) and the National Natural Science Foundation of China (General Program and Young Scholars, project nos. 81973679 and 82204923).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
AIBP: A-I binding protein
Ang II: angiotensin II
AMBRA1: activating molecule in Beclin 1-regulated autophagy protein 1
AMPK: adenosine 5′-monophosphate-activated protein kinase
AS: atherosclerosis
ASC: apoptosis-associated speck-like protein
Atg: autophagy-related protein
ATP: adenosine triphosphate
BACH1: Btb-And-Cnc homolog 1
BCL2L1: Bcl-2-like protein 1
BCL2L13: Bcl-2-like protein 13
BNIP3: BCL2-interacting protein 3
BNIP3L/Nix: BCL2 interacting protein 3-like
CANX: calnexin
CCCP: carbonyl cyanide 3- chlorophenylhydrazone
CK2: casein kinase 2
CL: cardiolipin
COMP: cartilage oligomeric matrix protein
DFCP1: double FYVE-containing protein 1
DRP1: dynamic-related protein 1
EC: vascular endothelial cell
EPC: endothelial progenitor cell
ER: endoplasmic reticulum
RB1CC1: RB1-inducible coiled-coil protein 1
FUNDC1: FUN14 domain-containing protein 1
H2O2: hydrogen peroxide
HAEC: human aortic endothelial cell
HFD: high-fat diet
HIF: hypoxia-inducible factor
HUVEC: human umbilical vein endothelial cell
Hcy: hyperhomocysteine
IL: interleukin
IMM: inner mitochondrial membrane
LC3: microtubule-associated protein 1 light chain 3
LIR: LC3- interaction region
LPS: lipopolysaccharide
MAM: mitochondria-associated endoplasmic reticulum
MARCH5: mitochondrial E3 ubiquitin ligase MARCH5
MFN1: mitofusin 1
MFN2: mitofusin 2
MPP: mitochondrial processing peptidase
mPTP: mitochondrial permeability transition pore
mTORC1: mTOR complex 1
NBR1: next to BRCA1 gene 1 protein
NDP52: nuclear domain 10 protein 52
NF-κB: nuclear factor-kappa B
NLRP3: NOD-like receptor thermoprotein dome-associated protein 3
OMM: outer mitochondrial membrane
NOD: nucleotide oligomerization domain
OPTN: optineurin
ox-LDL: oxidized low-density lipoprotein
PACS-2: phosphofurin acidic cluster sorting protein 2
PARL: presenilin associated rhomboid-like protein
PE: phosphatidyl ethanolamine
PGAM5: PGAM family member 5
PHB2: prohibitin 2
PI3P: phosphatidylinositol 3-phosphate
PINK1: PTEN-induced putative kinase protein 1
PTEN: phosphatase and tensin homolog
PTEN-L: phosphatase and tensin homolog-long
pUB: phosphorylated UB
Rab: Ras-related protein
ROS: reactive oxygen species
SQSTM1/p62: sequestosome-1/ubiquitin-binding protein p62
SRC: SRC proto-oncogene, non-receptor tyrosine kinase
TBHP: tert-butyl hydroperoxide
TBK1: tank-binding kinase 1
TIMM23: inner mitochondrial membrane 23 translocase
THP-1: human myeloid leukemia-derived
TNF: tumor necrosis factor
TOMM: translocase of the outer mitochondrial membrane
UB: ubiquitin
ULK1: unc-51-like autophagy activating kinase 1
UPS: ubiquitin-proteasome system
USP: ubiquitin-specific protease
VDAC1: voltage-dependent anion-selective channel protein 1
VEGFR2: vascular endothelial growth factor receptor 2
Vps: vacuolar protein sorting-associated protein
VSMC: vascular smooth muscle cell
WIPI2: WD repeat domain phosphoinositide-interacting protein 2
ΔΨm: mitochondrial membrane potential.
References
1. Bjorkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell (2022) 185(10):1630–45. doi: 10.1016/j.cell.2022.04.004
2. Pi X, Xie L, Patterson C. Emerging roles of vascular endothelium in metabolic homeostasis. Circ Res (2018) 123(4):477–94. doi: 10.1161/CIRCRESAHA.118.313237
3. van Hinsbergh VW. Endothelium–role in regulation of coagulation and inflammation. Semin Immunopathol (2012) 34(1):93–106. doi: 10.1007/s00281-011-0285-5
4. Li J, Fu X, Yang R, Zhang W. Atherosclerosis vascular endothelial secretion dysfunction and smooth muscle cell proliferation. J Healthc Eng. (2022) 2022:9271879. doi: 10.1155/2022/9271879
5. Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis: time for a re-assessment. Cardiovasc Res (2021) 117(11):2326–39. doi: 10.1093/cvr/cvab046
6. Back M, Yurdagul A Jr., Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol (2019) 16(7):389–406. doi: 10.1038/s41569-019-0169-2
7. Jing L, Shu-Xu D, Yong-Xin R. A review: pathological and molecular biological study on atherosclerosis. Clin Chim Acta (2022) 531:217–22. doi: 10.1016/j.cca.2022.04.012
8. Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol (2022) 23(2):141–61. doi: 10.1038/s41580-021-00415-0
9. Zhou J, Li XY, Liu YJ, Feng J, Wu Y, Shen HM, et al. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy (2022) 18(6):1240–55. doi: 10.1080/15548627.2021.1984656
10. Suarez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Alvarez-Cordoba M, Villalon-Garcia I, Talaveron-Rey M, et al. From mitochondria to atherosclerosis: the inflammation path. Biomedicines (2021) 9(3):258. doi: 10.3390/biomedicines9030258
11. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res (2005) 8(1):3–5. doi: 10.1089/rej.2005.8.3
12. Wu Y, Jiang T, Hua J, Xiong Z, Dai K, Chen H, et al. PINK1/Parkin-mediated mitophagy in cardiovascular disease: from pathogenesis to novel therapy. Int J Cardiol (2022) 361:61–9. doi: 10.1016/j.ijcard.2022.05.025
13. Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol-Heart C (2015) 309(9):H1453–H67. doi: 10.1152/ajpheart.00554.2015
14. Chakrabarti S, Jahandideh F, Wu JP. Food-derived bioactive peptides on inflammation and oxidative stress. BioMed Res Int (2014) 2014:608979. doi: 10.1155/2014/608979
15. Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation (2013) 128(7):702–12. doi: 10.1161/CIRCULATIONAHA.113.002271
16. Zhu H, Wang Z, Dong Z, Wang C, Cao Q, Fan F, et al. Aldehyde dehydrogenase 2 deficiency promotes atherosclerotic plaque instability through accelerating mitochondrial ROS-mediated vascular smooth muscle cell senescence. Bba-Mol Basis Dis (2019) 1865(7):1782–92. doi: 10.1016/j.bbadis.2018.09.033
17. Qu K, Yan F, Qin X, Zhang K, He W, Dong MQ, et al. Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis. Front Physiol (2022) 13. doi: 10.3389/fphys.2022.1084604
18. Ajoolabady A, Bi YG, McClements DJ, Lip GYH, Richardson D, Reiter RJ, et al. Melatonin-based therapeutics for atherosclerotic lesions and beyond: focusing on macrophage mitophagy. Pharmacol Res (2022) 176:106072. doi: 10.1016/j.phrs.2022.106072
19. Huynh DTN, Heo KS. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res (2021) 44(12):1051–61. doi: 10.1007/s12272-021-01360-4
20. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature (2006) 441(7097):1162–6. doi: 10.1038/nature04779
21. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in drosophila PINK1 mutants is complemented by parkin. Nature (2006) 441(7097):1157–61. doi: 10.1038/nature04788
22. Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, et al. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ (2013) 20(7):920–30. doi: 10.1038/cdd.2013.19
23. Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet (2003) 12(5):517–26. doi: 10.1093/hmg/ddg044
24. Li W, Jiang WS, Su YR, Tu KW, Zou L, Liao CR, et al. PINK1/Parkin-mediated mitophagy inhibits osteoblast apoptosis induced by advanced oxidation protein products. Cell Death Dis (2023) 14(2):88. doi: 10.1038/s41419-023-05595-5
25. Rasool S, Veyron S, Soya N, Eldeeb MA, Lukacs GL, Fon EA, et al. Mechanism of PINK1 activation by autophosphorylation and insights into assembly on the TOM complex. Mol Cell (2022) 82(1):44–59.e6. doi: 10.1016/j.molcel.2021.11.012
26. Jin SM, Lazarou M, Wang CX, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol (2010) 191(5):933–42. doi: 10.1083/jcb.201008084
27. Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and parkin recruitment. EMBO Rep (2012) 13(4):378–85. doi: 10.1038/embor.2012.14
28. Liu Y, Guardia-Laguarta C, Yin J, Erdjument-Bromage H, Martin B, James M, et al. The ubiquitination of PINK1 is restricted to its mature 52-kDa form. Cell Rep (2017) 20(1):30–9. doi: 10.1016/j.celrep.2017.06.022
29. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase parkin. Dev Cell (2012) 22(2):320–33. doi: 10.1016/j.devcel.2011.12.014
30. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PloS Biol (2010) 8(1):e1000298. doi: 10.1371/journal.pbio.1000298
31. Guardia-Laguarta C, Liu YH, Lauritzen KH, Erdjument-Bromage H, Martin B, Swayne TC, et al. PINK1 content in mitochondria is regulated by ER-associated degradation. J Neurosci (2019) 39(36):7074–85. doi: 10.1523/JNEUROSCI.1691-18.2019
32. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183(5):795–803. doi: 10.1083/jcb.200809125
33. Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem (2011) 117(5):856–67. doi: 10.1111/j.1471-4159.2011.07253.x
34. Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, et al. A dimeric PINK1-containing complex on depolarized mitochondria stimulates parkin recruitment. J Biol Chem (2013) 288(51):36372–84. doi: 10.1074/jbc.M113.509653
35. Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, Youle RJ. Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol Cell (2019) 73(5):1028–43 e5. doi: 10.1016/j.molcel.2019.01.002
36. Akabane S, Uno M, Tani N, Shimazaki S, Ebara N, Kato H, et al. PKA regulates PINK1 stability and parkin recruitment to damaged mitochondria through phosphorylation of MIC60. Mol Cell (2016) 62(3):371–84. doi: 10.1016/j.molcel.2016.03.037
37. Kakade P, Ojha H, Raimi OG, Shaw A, Waddell AD, Ault JR, et al. Mapping of a n-terminal alpha-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells. Open Biol (2022) 12(1):210264. doi: 10.1098/rsob.210264
38. Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for parkin recruitment to damaged mitochondria. Nat Commun (2012) 3:1016. doi: 10.1038/ncomms2016
39. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates parkin E3 ligase activity by phosphorylating serine 65. Open Biol (2012) 2:120080. doi: 10.1098/rsob.120080
40. Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, et al. Phosphorylated ubiquitin chain is the genuine parkin receptor. J Cell Biol (2015) 209(1):111–28. doi: 10.1083/jcb.201410050
41. Wauer T, Simicek M, Schubert A, Komander D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature (2015) 526(7575):728–. doi: 10.1038/nature15531
42. Ramirez A, Old W, Selwood DL, Liu X. Cannabidiol activates PINK1-parkin-dependent mitophagy and mitochondrial-derived vesicles. Eur J Cell Biol (2022) 101(1):151185. doi: 10.1016/j.ejcb.2021.151185
43. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Bio. (2018) 19(2):93–108. doi: 10.1038/nrm.2017.129
44. Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell (2014) 56(3):360–75. doi: 10.1016/j.molcel.2014.09.007
45. Gan ZY, Callegari S, Cobbold SA, Cotton TR, Mlodzianoski MJ, Schubert AF, et al. Activation mechanism of PINK1. Nature (2022) 602(7896):328–35. doi: 10.1038/s41586-021-04340-2
46. Kumar A, Tamjar J, Waddell AD, Woodroof HI, Raimi OG, Shaw AM, et al. Structure of PINK1 and mechanisms of parkinson's disease-associated mutations. Elife (2017) 6:e29985. doi: 10.7554/eLife.29985
47. Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science (2013) 340(6139):1451–5. doi: 10.1126/science.1237908
48. Fakih R, Sauve V, Gehring K. Structure of the second phosphoubiquitin-binding site in parkin. J Biol Chem (2022) 298(7):102114. doi: 10.1016/j.jbc.2022.102114
49. Wauer T, Komander D. Structure of the human parkin ligase domain in an autoinhibited state. EMBO J (2013) 32(15):2099–112. doi: 10.1038/emboj.2013.125
50. Schubert AF, Gladkova C, Pardon E, Wagstaff JL, Freund SMV, Steyaert J, et al. Structure of PINK1 in complex with its substrate ubiquitin. Nature (2017) 552(7683):51–+. doi: 10.1038/nature24645
51. Sauve V, Sung G, MacDougall EJ, Kozlov G, Saran A, Fakih R, et al. Structural basis for feedforward control in the PINK1/Parkin pathway. EMBO J (2022) 41(12):e109460. doi: 10.15252/embj.2021109460
52. Ham SJ, Lee SY, Song S, Chung JR, Choi S, Chung J. Interaction between RING1 (R1) and the ubiquitin-like (UBL) domains is critical for the regulation of parkin activity. J Biol Chem (2016) 291(4):1803–16. doi: 10.1074/jbc.M115.687319
53. Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, et al. A ubl/ubiquitin switch in the activation of parkin. EMBO J (2015) 34(20):2492–505. doi: 10.15252/embj.201592237
54. Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature (2013) 496(7445):372–+. doi: 10.1038/nature12043
55. Ham SJ, Lee D, Yoo H, Jun K, Shin H, Chung J. Decision between mitophagy and apoptosis by parkin via VDAC1 ubiquitination. P Natl Acad Sci USA (2020) 117(8):4281–91. doi: 10.1073/pnas.1909814117
56. Liao YJ, Hao YM, Chen H, He Q, Yuan ZQ, Cheng JB. Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell (2015) 6(6):434–42. doi: 10.1007/s13238-015-0144-6
57. Chen HB, Gao WW, Yang Y, Guo SY, Wang H, Wang W, et al. Inhibition of VDAC1 prevents Ca2+-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis (2014) 19(12):1712–26. doi: 10.1007/s10495-014-1045-5
58. Huang X, Yao J, Liu L, Luo Y, Yang A. Atg8-PE protein-based in vitro biochemical approaches to autophagy studies. Autophagy (2022) 18(9):2020–35. doi: 10.1080/15548627.2022.2025572
59. Subramani S, Farre JC. A ubiquitin-like protein involved in membrane fusion. Cell (2007) 130(1):18–20. doi: 10.1016/j.cell.2007.06.041
60. Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou SS, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy (2012) 8(6):883–92. doi: 10.4161/auto.19652
61. Agrotis A, Pengo N, Burden JJ, Ketteler R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy (2019) 15(6):976–97. doi: 10.1080/15548627.2019.1569925
62. Zhang SD, Yazaki E, Sakamoto H, Yamamoto H, Mizushima N. Evolutionary diversification of the autophagy-related ubiquitin-like conjugation systems. Autophagy (2022) 18(12):2969–84. doi: 10.1080/15548627.2022.2059168
63. Chang C, Shi X, Jensen LE, Yokom AL, Fracchiolla D, Martens S, et al. Reconstitution of cargo-induced LC3 lipidation in mammalian selective autophagy. Sci Adv (2021) 7(17):eabg4922. doi: 10.1126/sciadv.abg4922
64. Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol (2011) 23(4):476–82. doi: 10.1016/j.ceb.2011.05.007
65. Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem (2011) 286(22):19630–40. doi: 10.1074/jbc.M110.209338
66. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang CX, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524(7565):309–+. doi: 10.1038/nature14893
67. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Mol Biol Cell (2014) 25:E4439–48. doi: 10.1073/pnas.1405752111
68. Narendra DP, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy (2010) 6(8):1090–106. doi: 10.4161/auto.6.8.13426
69. Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, et al. Mitophagy in human diseases. Int J Mol Sci (2021) 22(8):3903. doi: 10.3390/ijms22083903
70. Popelka H, Klionsky DJ. Multiple structural rearrangements mediated by high-plasticity regions in Atg3 are key for efficient conjugation of Atg8 to PE during autophagy. Autophagy (2021) 17(8):1805–8. doi: 10.1080/15548627.2021.1954457
71. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell (2015) 60(1):7–20. doi: 10.1016/j.molcel.2015.08.016
72. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet (2015) 24(15):4429–42. doi: 10.1093/hmg/ddv179
73. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. (2016) 113(15):4039–44. doi: 10.1073/pnas.1523926113
74. Fu T, Zhang M, Zhou Z, Wu P, Peng C, Wang Y, et al. Structural and biochemical advances on the recruitment of the autophagy-initiating ULK and TBK1 complexes by autophagy receptor NDP52. Sci Adv (2021) 7(33):eabi6582. doi: 10.1126/sciadv.abi6582
75. Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, et al. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy (2018) 14(1):66–79. doi: 10.1080/15548627.2017.1391970
76. Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy (2013) 9(11):1750–7. doi: 10.4161/auto.26122
77. Sarraf SA, Sideris DP, Giagtzoglou N, Ni LN, Kankel MW, Sen A, et al. PINK1/Parkin influences cell cycle by sequestering TBK1 at damaged mitochondria, inhibiting mitosis. Cell Rep (2019) 29(1):225–+. doi: 10.1016/j.celrep.2019.08.085
78. Wang L, Cho YL, Tang Y, Wang J, Park JE, Wu Y, et al. PTEN-l is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-parkin-mediated mitophagy. Cell Res (2018) 28(8):787–802. doi: 10.1038/s41422-018-0056-0
79. Wang L, Wang J, Tang Y, Shen HM. PTEN-l puts a brake on mitophagy. Autophagy (2018) 14(11):2023–5. doi: 10.1080/15548627.2018.1502565
80. Faesen AC, Luna-Vargas MPA, Geurink PP, Clerici M, Merkx R, van Dijk WJ, et al. The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem Biol (2011) 18(12):1550–61. doi: 10.1016/j.chembiol.2011.10.017
81. Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J (2014) 33(21):2473–91. doi: 10.15252/embj.201489729
82. Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature (2014) 510(7505):370–5. doi: 10.1038/nature13418
83. Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, et al. The deubiquitinase USP15 antagonizes parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet (2014) 23(19):5227–42. doi: 10.1093/hmg/ddu244
84. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol (2018) 20(9):1013–22. doi: 10.1038/s41556-018-0176-2
85. Sato Y, Okatsu K, Saeki Y, Yamano K, Matsuda N, Kaiho A, et al. Structural basis for specific cleavage of Lys6-linked polyubiquitin chains by USP30. Nat Struct Mol Biol (2017) 24(11):911–+. doi: 10.1038/nsmb.3469
86. Gersch M, Gladkova C, Schubert AF, Michel MA, Maslen S, Komander D. Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat Struct Mol Biol (2017) 24(11):920–+. doi: 10.1038/nsmb.3475
87. Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT, et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy (2015) 11(4):595–606. doi: 10.1080/15548627.2015.1034408
88. Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, et al. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol (2015) 17(2):160–9. doi: 10.1038/ncb3097
89. Viguerie N, Tahirijouti N, Esteve JP, Clerc P, Logsdon C, Svoboda M, et al. Functional somatostatin receptors on a rat pancreatic acinar cell-line. Am J Physiol (1988) 255(1):G113–G20. doi: 10.1152/ajpgi.1988.255.1.G113
90. Wang F, Ning S, Yu BM, Wang YF. USP14: structure, function, and target inhibition. Front Pharmacol (2022) 12. doi: 10.1055/s-0041-1742095
91. Yang Y, Wang LQ, Yao BC, Guo ZG. Ubiquitin-specific protease as the underlying gene biomarker for aortic stenosis. Lipids Health Dis (2020) 19(1):115. doi: 10.1186/s12944-020-01299-3
92. Li HR, Quan JZ, Zhao XB, Ling J, Chen WL. USP14 negatively regulates RIG-i-mediated IL-6 and TNF-alpha production by inhibiting NF-kappa b activation. Mol Immunol (2021) 130:69–76. doi: 10.1016/j.molimm.2020.12.022
93. Mialki RK, Zhao J, Wei JX, Mallampalli DF, Zhao YT. Overexpression of USP14 protease reduces I-kappa b protein levels and increases cytokine release in lung epithelial cells. J Biol Chem (2013) 288(22):15437–41. doi: 10.1074/jbc.C112.446682
94. Fu Y, Qiu J, Wu J, Zhang L, Wei F, Lu L, et al. USP14-mediated NLRC5 upregulation inhibits endothelial cell activation and inflammation in atherosclerosis. Biochim Biophys Acta Mol Cell Biol Lipids. (2022) 1868(5):159258. doi: 10.1016/j.bbalip.2022.159258
95. Zhang FC, Xia XH, Chai RJ, Xu RQ, Xu Q, Liu MK, et al. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J Cell Mol Med (2020) 24(6):3292–302. doi: 10.1111/jcmm.15002
96. Liu HB, Li XL, Yan GB, Lun RH. Knockdown of USP14 inhibits PDGF-BB-induced vascular smooth muscle cell dedifferentiation via inhibiting mTOR/P70S6K signaling pathway. Rsc Adv (2019) 9(63):36649–57. doi: 10.1039/C9RA04726C
97. Huguenin-Dezot N, De Cesare V, Peltier J, Knebel A, Kristaryianto YA, Rogerson DT, et al. Synthesis of isomeric phosphoubiquitin chains reveals that phosphorylation controls deubiquitinase activity and specificity. Cell Rep (2016) 16(4):1180–93. doi: 10.1016/j.celrep.2016.06.064
98. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11(1):45–51. doi: 10.1038/embor.2009.256
99. Peng X, Chen H, Li Y, Huang D, Huang B, Sun D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol Int (2020) 44(7):1481–90. doi: 10.1002/cbin.11343
100. Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor nix enhances its interaction with LC3 proteins. Sci Rep-Uk (2017) 7:1131. doi: 10.1038/s41598-017-01258-6
101. Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem (2013) 288(2):1099–113. doi: 10.1074/jbc.M112.399345
102. da Silva Rosa SC, Martens MD, Field JT, Nguyen L, Kereliuk SM, Hai Y, et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy (2021) 17(9):2257–72. doi: 10.1080/15548627.2020.1821548
103. Marinkovic M, Sprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy (2021) 17(5):1232–43. doi: 10.1080/15548627.2020.1755120
104. van Kuijk K, Demandt JAF, Perales-Patón J, Theelen TL, Kuppe C, Marsch E, et al. Deficiency of myeloid PHD proteins aggravates atherogenesis via macrophage apoptosis and paracrine fibrotic signalling. Cardiovasc Res (2022) 118(5):1232–46. doi: 10.1093/cvr/cvab152
105. Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol (2000) 296(3):911–9. doi: 10.1006/jmbi.1999.3489
106. Sulistijo ES, MacKenzie KR. Sequence dependence of BNIP3 transmembrane domain dimerization implicates side-chain hydrogen bonding and a tandem GxxxG motif in specific helix-helix interactions. J Mol Biol (2006) 364(5):974–90. doi: 10.1016/j.jmb.2006.09.065
107. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem (2008) 283(16):10892–903. doi: 10.1074/jbc.M800102200
108. Liu K, Zhao Q, Sun H, Liu L, Wang C, Li Z, et al. BNIP3 (BCL2 interacting protein 3) regulates pluripotency by modulating mitochondrial homeostasis via mitophagy. Cell Death Dis (2022) 13(4):334. doi: 10.1038/s41419-022-04795-9
109. Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. (2000) 97(16):9082–7. doi: 10.1073/pnas.97.16.9082
110. Zhang L, Li L, Liu H, Prabhakaran K, Zhang X, Borowitz JL, et al. HIF-1alpha activation by a redox-sensitive pathway mediates cyanide-induced BNIP3 upregulation and mitochondrial-dependent cell death. Free Radic Biol Med (2007) 43(1):117–27. doi: 10.1016/j.freeradbiomed.2007.04.005
111. Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol (2020) 36:101671. doi: 10.1016/j.redox.2020.101671
112. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem (2012) 287(23):19094–104. doi: 10.1074/jbc.M111.322933
113. Field JT, Gordon JW. BNIP3 and nix: atypical regulators of cell fate. Biochim Biophys Acta Mol Cell Res (2022) 1869(10):119325. doi: 10.1016/j.bbamcr.2022.119325
114. Poole LP, Bock-Hughes A, Berardi DE, Macleod KF. ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci Rep (2021) 11(1):20526. doi: 10.1038/s41598-021-00170-4
115. Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, et al. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet (2015) 24(9):2528–38. doi: 10.1093/hmg/ddv017
116. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J (2021) 40(3):e104705. doi: 10.15252/embj.2020104705
117. Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, et al. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem (2016) 291(41):21616–29. doi: 10.1074/jbc.M116.733410
118. Zhang W, Siraj S, Zhang R, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy (2017) 13(6):1080–1. doi: 10.1080/15548627.2017.1300224
119. Kuang Y, Ma K, Zhou C, Ding P, Zhu Y, Chen Q, et al. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy (2016) 12(12):2363–73. doi: 10.1080/15548627.2016.1238552
120. Ma K, Zhang Z, Chang R, Cheng H, Mu C, Zhao T, et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ (2020) 27(3):1036–51. doi: 10.1038/s41418-019-0396-4
121. Zheng T, Wang HY, Chen Y, Chen X, Wu ZL, Hu QY, et al. Src activation aggravates podocyte injury in diabetic nephropathy via suppression of FUNDC1-mediated mitophagy. Front Pharmacol (2022) 13:897046. doi: 10.3389/fphar.2022.897046
122. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell (2014) 54(3):362–77. doi: 10.1016/j.molcel.2014.02.034
123. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol (2012) 14(2):177–85. doi: 10.1038/ncb2422
124. Lv M, Wang C, Li F, Peng J, Wen B, Gong Q, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell (2017) 8(1):25–38. doi: 10.1007/s13238-016-0328-8
125. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep (2014) 15(5):566–75. doi: 10.1002/embr.201438501
126. Cheng M, Lin N, Dong D, Ma J, Su J, Sun L. PGAM5: a crucial role in mitochondrial dynamics and programmed cell death. Eur J Cell Biol (2021) 100(1):151144. doi: 10.1016/j.ejcb.2020.151144
127. Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy (2014) 10(10):1712–25. doi: 10.4161/auto.29568
128. Mao H, Chen W, Chen L, Li L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem Pharmacol (2022) 199:115011. doi: 10.1016/j.bcp.2022.115011
129. Wu W, Li W, Chen H, Jiang L, Zhu R, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy (2016) 12(9):1675–6. doi: 10.1080/15548627.2016.1193656
130. Wang C, Dai X, Wu S, Xu W, Song P, Huang K. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun (2021) 12(1):2616. doi: 10.1038/s41467-021-22771-3
131. Chai P, Cheng Y, Hou C, Yin L, Zhang D, Hu Y, et al. USP19 promotes hypoxia-induced mitochondrial division via FUNDC1 at ER-mitochondria contact sites. J Cell Biol (2021) 220(7):e202010006. doi: 10.1083/jcb.202010006
132. Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep (2017) 18(3):495–509. doi: 10.15252/embr.201643309
133. Poellinger L, Johnson RS. HIF-1 and hypoxic response: the plot thickens. Curr Opin Genet Dev (2004) 14(1):81–5. doi: 10.1016/j.gde.2003.12.006
134. Sedding DG, Boyle EC, Demandt JAF, Sluimer JC, Dutzmann J, Haverich A, et al. Vasa vasorum angiogenesis: key player in the initiation and progression of atherosclerosis and potential target for the treatment of cardiovascular disease. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00706
135. Parma L, Baganha F, Quax PHA, de Vries MR. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur J Pharmacol (2017) 816:107–15. doi: 10.1016/j.ejphar.2017.04.028
136. Michel JB, Martin-Ventura JL, Nicoletti A, Ho-Tin-Noe B. Pathology of human plaque vulnerability: mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis (2014) 234(2):311–9. doi: 10.1016/j.atherosclerosis.2014.03.020
137. Wang X, Han X, Liu N. FUNDC1 mediated mitophagy alleviates rat aortic smooth muscle cell proliferation, migration induced by angiotensin II. Atherosclerosis (2017) 263:e132. doi: 10.1016/j.atherosclerosis.2017.06.420
138. Mercer TJ, Gubas A, Tooze SA. A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem (2018) 293(15):5386–95. doi: 10.1074/jbc.R117.810366
139. Zachari M, Gudmundsson SR, Li Z, Manifava M, Cugliandolo F, Shah R, et al. Selective autophagy of mitochondria on a ubiquitin-Endoplasmic-Reticulum platform. Dev Cell (2019) 50(5):627–43 e5. doi: 10.1016/j.devcel.2019.06.016
140. Kim BW, Jin Y, Kim J, Kim JH, Jung J, Kang S, et al. The c-terminal region of ATG101 bridges ULK1 and PtdIns3K complex in autophagy initiation. Autophagy (2018) 14(12):2104–16. doi: 10.1080/15548627.2018.1504716
141. Walker S, Chandra P, Manifava M, Axe E, Ktistakis NT. Making autophagosomes: localized synthesis of phosphatidylinositol 3-phosphate holds the clue. Autophagy (2008) 4(8):1093–6. doi: 10.4161/auto.7141
142. Nascimbeni AC, Giordano F, Dupont N, Grasso D, Vaccaro MI, Codogno P, et al. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J (2017) 36(14):2018–33. doi: 10.15252/embj.201797006
143. Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol (2010) 190(4):511–21. doi: 10.1083/jcb.200911141
144. Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy (2010) 6(4):506–22. doi: 10.4161/auto.6.4.11863
145. Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell (2012) 23(10):1860–73. doi: 10.1091/mbc.e11-09-0746
146. Dooley HC, Wilson MI, Tooze SA. WIPI2B links PtdIns3P to LC3 lipidation through binding ATG16L1. Autophagy (2015) 11(1):190–1. doi: 10.1080/15548627.2014.996029
147. Park JM, Seo M, Jung CH, Grunwald D, Stone M, Otto NM, et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy (2018) 14(4):584–97. doi: 10.1080/15548627.2017.1422851
148. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol (2013) 15(7):741–50. doi: 10.1038/ncb2757
149. Pyo KE, Kim CR, Lee M, Kim JS, Kim KI, Baek SH. ULK1 O-GlcNAcylation is crucial for activating VPS34 via ATG14L during autophagy initiation. Cell Rep (2018) 25(10):2878–90 e4. doi: 10.1016/j.celrep.2018.11.042
150. Crozet P, Margalha L, Confraria A, Rodrigues A, Martinho C, Adamo M, et al. Mechanisms of regulation of SNF1/AMPK/SnRK1 protein kinases. Front Plant Sci (2014) 5:190. doi: 10.3389/fpls.2014.00190
151. Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature (2007) 449(7161):496–U14. doi: 10.1038/nature06161
152. Rafaeloff-Phail R, Ding LY, Conner L, Yeh WK, McClure D, Guo HH, et al. Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH. J Biol Chem (2004) 279(51):52934–9. doi: 10.1074/jbc.M409574200
153. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
154. Lee MKS, Cooney OD, Lin X, Nadarajah S, Dragoljevic D, Huynh K, et al. Defective AMPK regulation of cholesterol metabolism accelerates atherosclerosis by promoting HSPC mobilization and myelopoiesis. Mol Metab (2022) 61:101514. doi: 10.1016/j.molmet.2022.101514
155. Desjardins EM, Smith BK, Day EA, Ducommun S, Sanders MJ, Nederveen JP, et al. The phosphorylation of AMPKβ1 is critical for increasing autophagy and maintaining mitochondrial homeostasis in response to fatty acids. P Natl Acad Sci USA (2022) 119(48):e2119824119. doi: 10.1073/pnas.2119824119
156. Salvati KA, Ritger ML, Davoudian PA, O'Dell F, Wyskiel DR, Souza GMPR, et al. AMPK-mediated potentiation of GABAergic signalling drives hypoglycaemia-provoked spike-wave seizures. Brain (2022) 145(7):2332–46. doi: 10.1093/brain/awac037
157. Haythorne E, Lloyd M, Walsby-Tickle J, Tarasov AI, Sandbrink J, Portillo I, et al. Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells. Nat Commun (2022) 13(1):6754. doi: 10.1038/s41467-022-34095-x
158. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med (2016) 48(7):e245. doi: 10.1038/emm.2016.81
159. Ou H, Liu C, Feng W, Xiao X, Tang S, Mo Z. Role of AMPK in atherosclerosis via autophagy regulation. Sci China Life Sci (2018) 61(10):1212–21. doi: 10.1007/s11427-017-9240-2
160. Zhang M, Zhu H, Ding Y, Liu Z, Cai Z, Zou M-H. AMP-activated protein kinase α1 promotes atherogenesis by increasing monocyte-to-macrophage differentiation. J Biol Chem (2017) 292(19):7888–903. doi: 10.1074/jbc.M117.779447
161. Chen XH, An XB, Chen DR, Ye MQ, Shen WL, Han WQ, et al. Chronic exercise training improved aortic endothelial and mitochondrial function via an AMPK alpha 2-dependent manner. Front Physiol (2016) 7. doi: 10.3389/fphys.2016.00631
162. Liang J, Xu ZX, Ding Z, Lu Y, Yu Q, Werle KD, et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun (2015) 6:7926. doi: 10.1038/ncomms8926
163. Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett (2015) 589(15):1847–54. doi: 10.1016/j.febslet.2015.05.020
164. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331(6016):456–61. doi: 10.1126/science.1196371
165. Watanabe K, Tsubata T. Autophagy connects antigen receptor signaling to costimulatory signaling in b lymphocytes. Autophagy (2009) 5(1):108–10. doi: 10.4161/auto.5.1.7278
166. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood (2008) 112(4):1493–502. doi: 10.1182/blood-2008-02-137398
167. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–U71. doi: 10.1038/ncb2152
168. Loffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy (2011) 7(7):696–706. doi: 10.4161/auto.7.7.15451
169. Shen Y, Liu WW, Zhang X, Shi JG, Jiang S, Zheng LS, et al. TRAF3 promotes ROS production and pyroptosis by targeting ULK1 ubiquitination in macrophages. FASEB J (2020) 34(5):7144–59. doi: 10.1096/fj.201903073R
170. Zhang X, Sergin I, Evans TD, Jeong SJ, Rodriguez-Velez A, Kapoor D, et al. High-protein diets increase cardiovascular risk by activating macrophage mTOR to suppress mitophagy. Nat Metab (2020) 2(1):110–25. doi: 10.1038/s42255-019-0162-4
171. Hung CM, Lombardo PS, Malik N, Brun SN, Hellberg K, Van Nostrand JL, et al. AMPK/ULK1-mediated phosphorylation of parkin ACT domain mediates an early step in mitophagy. Sci Adv (2021) 7(15):eabg4544. doi: 10.1126/sciadv.abg4544
172. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res (2018) 122(5):712–29. doi: 10.1161/CIRCRESAHA.117.312317
173. Lee SB, Kim JJ, Han SA, Fan Y, Guo LS, Aziz K, et al. The AMPK-parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat Cell Biol (2019) 21(8):940–51. doi: 10.1038/s41556-019-0356-8
174. Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell (2019) 74(2):347–62 e6. doi: 10.1016/j.molcel.2019.02.010
175. Sun LQ, Zhao MM, Liu AH, Lv M, Zhang JB, Li YX, et al. Shear stress induces phenotypic modulation of vascular smooth muscle cells via AMPK/mTOR/ULK1-mediated autophagy. Cell Mol Neurobiol (2018) 38(2):541–8. doi: 10.1007/s10571-017-0505-1
176. Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep (2017) 21(1):1–9. doi: 10.1016/j.celrep.2017.09.026
177. Van Humbeeck C, Cornelissen T, Vandenberghe W. Ambra1: a parkin-binding protein involved in mitophagy. Autophagy (2011) 7(12):1555–6. doi: 10.4161/auto.7.12.17893
178. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun (2015) 6:7527. doi: 10.1038/ncomms8527
179. Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, et al. FKBP8 recruits LC3A to mediate parkin-independent mitophagy. EMBO Rep (2017) 18(6):947–61. doi: 10.15252/embr.201643147
180. Di Rita A, Peschiaroli A, Acunzo D P, Strobbe D, Hu Z, Gruber J, et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat Commun (2018) 9(1):3755. doi: 10.1038/s41467-018-05722-3
181. Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ (2015) 22(3):419–32. doi: 10.1038/cdd.2014.139
182. Park S, Choi SG, Yoo SM, Son JH, Jung YK. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy (2014) 10(11):1906–20. doi: 10.4161/auto.32177
183. Lahiri V, Klionsky DJ. PHB2/prohibitin 2: an inner membrane mitophagy receptor. Cell Res (2017) 27(3):311–2. doi: 10.1038/cr.2017.23
184. Wei Y, Chiang WC, Sumpter R Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell (2017) 168(1-2):224–38 e10. doi: 10.1016/j.cell.2016.11.042
185. Yan C, Gong L, Chen L, Xu M, Abou-Hamdan H, Tang M, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy (2020) 16(3):419–34. doi: 10.1080/15548627.2019.1628520
186. Sun S, Hou HY, Ma GQ, Ma QL, Li NN, Zhang L, et al. The interaction between E3 ubiquitin ligase parkin and mitophagy receptor PHB2 links inner mitochondrial membrane ubiquitination to efficient mitophagy. J Biol Chem (2022) 298(12):102704. doi: 10.1016/j.jbc.2022.102704
187. Jia YT, Wang ML, Mao CF, Yu F, Wang YB, Xiao R, et al. COMP-prohibitin 2 interaction maintains mitochondrial homeostasis and controls smooth muscle cell identity. Cell Death Dis (2018) 9:676. doi: 10.1038/s41419-018-0703-x
188. Yang M, Li CR, Yang SK, Xiao Y, Xiong XF, Chen W, et al. Mitochondria-associated ER membranes - the origin site of autophagy. Front Cell Dev Biol (2020) 8. doi: 10.3389/fcell.2020.00595
189. Li CR, Li L, Yang M, Zeng LF, Sun L. PACS-2: a key regulator of mitochondria-associated membranes (MAMs). Pharmacol Res (2020) 160:105080. doi: 10.1016/j.phrs.2020.105080
190. Moulis M, Grousset E, Faccini J, Richetin K, Thomas G, Vindis C. The multifunctional sorting protein PACS-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells (2019) 8(6):638. doi: 10.3390/cells8060638
191. Wang X, Luo D, Wu S. Molecular dysfunctions of mitochondria-associated endoplasmic reticulum contacts in atherosclerosis. Oxid Med Cell Longev (2021) 2021:2424509. doi: 10.1155/2021/2424509
192. Mahajan M, Bharambe N, Shang YT, Lu B, Mandal A, Mohan PM, et al. NMR identification of a conserved Drp1 cardiolipin-binding motif essential for stress-induced mitochondrial fission. P Natl Acad Sci USA (2021) 118(29):e2023079118. doi: 10.1073/pnas.2023079118
193. Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basanez G, Terrones O, Martinou JC. Specific interaction with cardiolipin triggers functional activation of dynamin-related protein 1. PloS One (2014) 9(7):e102738. doi: 10.1371/journal.pone.0102738
194. O'Neill LAJ. Cardiolipin and the Nlrp3 inflammasome. Cell Metab (2013) 18(5):610–2. doi: 10.1016/j.cmet.2013.10.013
195. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
196. Reynolds MB, Hong HS, Michmerhuizen BC, Lawrence ALE, Zhang L, Knight JS, et al. Cardiolipin coordinates inflammatory metabolic reprogramming through regulation of complex II disassembly and degradation. Sci Adv (2023) 9(5):eade8701. doi: 10.1126/sciadv.ade8701
197. Wan M, Hua X, Su J, Thiagarajan D, Frostegård AG, Haeggström JZ, et al. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by annexin A5. Atherosclerosis (2014) 235(2):592–8. doi: 10.1016/j.atherosclerosis.2014.05.913
198. Buland JR, Wasserloos KJ, Tyurin VA, Tyurina YY, Amoscato AA, Mallampalli RK, et al. Biosynthesis of oxidized lipid mediators via lipoprotein-associated phospholipase A2 hydrolysis of extracellular cardiolipin induces endothelial toxicity. Am J Physiol Lung Cell Mol Physiol (2016) 311(2):L303–L16. doi: 10.1152/ajplung.00038.2016
199. Zhao K, Zhao G-M, Wu D, Soong Y, Birk AV, Schiller PW, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem (2004) 279(33):34682–90. doi: 10.1074/jbc.M402999200
200. Birk AV, Chao WM, Liu SY, Soong Y, Szeto HH. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Bba-Bioenergetics (2015) 1847(10):1075–84. doi: 10.1016/j.bbabio.2015.06.006
201. Anton Z, Landajuela A, Hervas JH, Montes LR, Hernandez-Tiedra S, Velasco G, et al. Human Atg8-cardiolipin interactions in mitophagy: specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy (2016) 12(12):2386–403. doi: 10.1080/15548627.2016.1240856
202. Manganelli V, Capozzi A, Recalchi S, Riitano G, Mattei V, Longo A, et al. The role of cardiolipin as a scaffold mitochondrial phospholipid in autophagosome formation: In Vitro evidence. Biomolecules (2021) 11(2). doi: 10.3390/biom11020222
203. Song CJ, Zhang J, Qi SS, Liu Z, Zhang XY, Zheng Y, et al. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to parkinson's diseases. Aging Cell (2019) 18(3):e12941. doi: 10.1111/acel.12941
204. Liu XL, Ye BL, Miller S, Yuan HJ, Zhang HX, Tian L, et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol Cell Biol (2012) 32(21):4493–504. doi: 10.1128/MCB.01092-12
205. Li J, Romestaing C, Han XL, Li YA, Hao XB, Wu YY, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab (2010) 12(2):154–65. doi: 10.1016/j.cmet.2010.07.003
206. Sherer Y, Shemesh J, Tenenbaum A, Praprotnik S, Harats D, Fisman EZ, et al. Coronary calcium and anti-cardiolipin antibody are elevated in patients with typical chest pain. Am J Cardiol (2000) 86(12):1306–11. doi: 10.1016/S0002-9149(00)01232-7
207. Kogot-Levin A, Saada A. Ceramide and the mitochondrial respiratory chain. Biochimie (2014) 100:88–94. doi: 10.1016/j.biochi.2013.07.027
208. Siskind LJ, Colombini M. The lipids C2- and C16-ceramide form large stable channels. implications for apoptosis. J Biol Chem (2000) 275(49):38640–4. doi: 10.1074/jbc.C000587200
209. Li Q, Wang X, Pang J, Zhang Y, Zhang HY, Xu ZL, et al. Associations between plasma ceramides and mortality in patients with coronary artery disease. Atherosclerosis (2020) 314:77–83. doi: 10.1016/j.atherosclerosis.2020.09.004
210. Akawi N, Checa A, Antonopoulos AS, Akoumianakis I, Daskalaki E, Kotanidis CP, et al. Fat-secreted ceramides regulate vascular redox state and influence outcomes in patients with cardiovascular disease. J Am Coll Cardiol (2021) 77(20):2494–513. doi: 10.1016/j.jacc.2021.03.314
211. Xu XX, Tabas I. Sphingomyelinase enhances low-Density-Lipoprotein uptake and ability to induce cholesteryl ester accumulation in macrophages. J Biol Chem (1991) 266(36):24849–58. doi: 10.1016/S0021-9258(18)54306-4
212. Cantalupo A, Sasset L, Gargiulo A, Rubinelli L, Del Gaudio I, Benvenuto D, et al. Endothelial sphingolipid De Novo synthesis controls blood pressure by regulating signal transduction and NO via ceramide. Hypertension (2020) 75(5):1279–88. doi: 10.1161/HYPERTENSIONAHA.119.14507
213. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res (2009) 50:S382–S7. doi: 10.1194/jlr.R800032-JLR200
214. Liao LZ, Zhou Q, Song Y, Wu WK, Yu HM, Wang S, et al. Ceramide mediates ox-LDL-Induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PloS One (2013) 8(12):e82379. doi: 10.1371/journal.pone.0082379
215. Vos M, Dulovic-Mahlow M, Mandik F, Frese L, Kanana Y, Diaw SH, et al. Ceramide accumulation induces mitophagy and impairs beta-oxidation in PINK1 deficiency. P Natl Acad Sci USA (2021) 118(43):e2025347118. doi: 10.1073/pnas.2025347118
216. Sentelle RD, Senkal CE, Jiang WH, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol (2012) 8(10):831–8. doi: 10.1038/nchembio.1059
217. Cong X, Kong W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell Signal (2020) 66:109485. doi: 10.1016/j.cellsig.2019.109485
218. Duan H, Zhang Q, Liu J, Li R, Wang D, Peng W, et al. Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol Res (2021) 168:105599. doi: 10.1016/j.phrs.2021.105599
219. Zheng DD, Liu J, Piao HL, Zhu ZC, Wei R, Liu KX. ROS-triggered endothelial cell death mechanisms: focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.1039241
220. Chang MC, Chang HH, Chan CP, Yeung SY, Hsien HC, Lin BR, et al. P-cresol affects reactive oxygen species generation, cell cycle arrest, cytotoxicity and Inflammation/Atherosclerosis-related modulators production in endothelial cells and mononuclear cells. PloS One (2014) 9(12):e114446. doi: 10.1371/journal.pone.0114446
221. Liu X, Sun JA. Endothelial cells dysfunction induced by silica nanoparticles through oxidative stress via JNK/P53 and NF-kappa b pathways. Biomaterials (2010) 31(32):8198–209. doi: 10.1016/j.biomaterials.2010.07.069
222. Zhang J, Wang B, Wang H, He H, Wu Q, Qin X, et al. Disruption of the superoxide anions-mitophagy regulation axis mediates copper oxide nanoparticles-induced vascular endothelial cell death. Free Radical Bio Med (2018) 129:268–78. doi: 10.1016/j.freeradbiomed.2018.09.032
223. Zhang J, Zou Z, Wang B, Xu G, Wu Q, Zhang YC, et al. Lysosomal deposition of copper oxide nanoparticles triggers HUVEC cells death. Biomaterials (2018) 161:228–39. doi: 10.1016/j.biomaterials.2018.01.048
224. Fan YZ, Cheng ZL, Mao LJ, Xu G, Li N, Zhang ML, et al. PINK1/TAX1BP1-directed mitophagy attenuates vascular endothelial injury induced by copper oxide nanoparticles. J Nanobiotechnol (2022) 20(1):149. doi: 10.1186/s12951-022-01338-4
225. Huber J, Vales A, Mitulovic G, Blumer M, Schmid R, Witztum JL, et al. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler Thromb Vasc Biol (2002) 22(1):101–7. doi: 10.1161/hq0102.101525
226. Sestili P, Martinelli C, Ricci D, Fraternale D, Bucchini A, Giamperi L, et al. Cytoprotective effect of preparations from various parts of punica granatum l. fruits in oxidatively injured mammalian cells in comparison with their antioxidant capacity in cell free systems. Pharmacol Res (2007) 56(1):18–26. doi: 10.1016/j.phrs.2007.02.003
227. Chen ZT, Wu HQ, Yang JX, Li BL, Ding J, Cheng S, et al. Activating parkin-dependent mitophagy alleviates oxidative stress, apoptosis, and promotes random-pattern skin flaps survival. Commun Biol (2022) 5(1):616. doi: 10.1038/s42003-022-03556-w
228. Wen YD, Wang H, Kho SH, Rinkiko S, Sheng X, Shen HM, et al. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PloS One (2013) 8(2):e53147. doi: 10.1371/journal.pone.0053147
229. Rao GT, Murphy B, Dey A, Dwivedi SKD, Zhang YS, Roy RV, et al. Cystathionine beta synthase regulates mitochondrial dynamics and function in endothelial cells. FASEB J (2020) 34(7):9372–92. doi: 10.1096/fj.202000173R
230. Liu N, Wu JC, Zhang LX, Gao ZP, Sun Y, Yu M, et al. Hydrogen sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J Cell Mol Med (2017) 21(12):3190–203. doi: 10.1111/jcmm.13223
231. Piao S, Nagar H, Kim S, Lee I, Choi SJ, Kim T, et al. CRIF1 deficiency induced mitophagy via p66shc-regulated ROS in endothelial cells. Biochem Biophys Res Commun (2020) 522(4):869–75. doi: 10.1016/j.bbrc.2019.11.109
232. Zhang Y, Wang S, Chen X, Wang Z, Wang X, Zhou Q, et al. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol Cell Endocrinol (2022) 545:111560. doi: 10.1016/j.mce.2022.111560
233. Li P, Wang J, Zhao X, Ru J, Tian T, An Y, et al. PTEN inhibition attenuates endothelial cell apoptosis in coronary heart disease via modulating the AMPK-CREB-Mfn2-mitophagy signaling pathway. J Cell Physiol (2020) 235(5):4878–89. doi: 10.1002/jcp.29366
234. Cho YE, Basu A, Dai AZ, Heldak M, Makino A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am J Physiol-Cell Ph (2013) 305(10):C1033–C40. doi: 10.1152/ajpcell.00234.2013
235. Matsunaga T, Iguchi K, Nakajima T, Koyama I, Miyazaki T, Inoue I, et al. Glycated high-density lipoprotein induces apoptosis of endothelial cells via a mitochondrial dysfunction. Biochem Bioph Res Co. (2001) 287(3):714–20. doi: 10.1006/bbrc.2001.5625
236. Zhu WZ, Yuan YJ, Liao GN, Li L, Liu JP, Chen YN, et al. Mesenchymal stem cells ameliorate hyperglycemia-induced endothelial injury through modulation of mitophagy. Cell Death Dis (2018) 9:837. doi: 10.1038/s41419-018-0861-x
237. Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, et al. PINK1-Parkin-Mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PloS One (2015) 10(7):e0132499. doi: 10.1371/journal.pone.0132499
238. Dunn EJ, Ariens RAS, Grant PJ. The influence of type 2 diabetes on fibrin structure and function. Diabetologia (2005) 48(6):1198–206. doi: 10.1007/s00125-005-1742-2
239. Dunn EJ, Ariens RAS. Fibrinogen and fibrin clot structure in diabetes. Herz (2004) 29(5):470–9. doi: 10.1007/s00059-004-2607-z
240. Kim D, Kim KA, Kim JH, Kim EH, Bae ON. Methylglyoxal-induced dysfunction in brain endothelial cells via the suppression of Akt/HIF-1 alpha pathway and activation of mitophagy associated with increased reactive oxygen species. Antioxidants-Basel (2020) 9(9):820. doi: 10.3390/antiox9090820
241. Li C, Tan Y, Wu J, Ma Q, Bai S, Xia Z, et al. Resveratrol improves Bnip3-related mitophagy and attenuates high-Fat-Induced endothelial dysfunction. Front Cell Dev Biol (2020) 8:796. doi: 10.3389/fcell.2020.00796
242. Peng Z, Zhan H, Shao Y, Xiong Y, Zeng L, Zhang C, et al. 13-methylberberine improves endothelial dysfunction by inhibiting NLRP3 inflammasome activation via autophagy induction in human umbilical vein endothelial cells. Chin Med (2020) 15:8. doi: 10.1186/s13020-020-0286-1
243. Chang X, Zhang T, Liu D, Meng Q, Yan P, Luo D, et al. Puerarin attenuates LPS-induced inflammatory responses and oxidative stress injury in human umbilical vein endothelial cells through mitochondrial quality control. Oxid Med Cell Longev (2021) 2021:6659240. doi: 10.1155/2021/6659240
244. Xi J, Rong Y, Zhao Z, Huang Y, Wang P, Luan H, et al. Scutellarin ameliorates high glucose-induced vascular endothelial cells injury by activating PINK1/Parkin-mediated mitophagy. J Ethnopharmacol (2021) 271:113855. doi: 10.1016/j.jep.2021.113855
245. Wang X, Zhang JQ, Xiu CK, Yang J, Fang JY, Lei Y. Ginseng-Sanqi-Chuanxiong (GSC) extracts ameliorate diabetes-induced endothelial cell senescence through regulating mitophagy via the AMPK pathway. Oxid Med Cell Longev (2020) 2020:7151946. doi: 10.1155/2020/7151946
246. Tang XQ, Zhang Y, Liu X, Li XH, Zhao HR, Cui H, et al. Aloe-emodin derivative produces anti-atherosclerosis effect by reinforcing AMBRA1-mediated endothelial autophagy. Eur J Pharmacol (2022) 916:174641. doi: 10.1016/j.ejphar.2021.174641
247. Xiang J, Zhang C, Di T, Chen L, Zhao W, Wei L, et al. Salvianolic acid b alleviates diabetic endothelial and mitochondrial dysfunction by down-regulating apoptosis and mitophagy of endothelial cells. Bioengineered (2022) 13(2):3486–502. doi: 10.1080/21655979.2022.2026552
248. Li JJ, Wang YJ, Wang CM, Li YJ, Yang Q, Cai WY, et al. Shenlian extract decreases mitochondrial autophagy to regulate mitochondrial function in microvascular to alleviate coronary artery no-reflow. Phytother Res (2023). doi: 10.1002/ptr.7703
249. Byun KA, Oh S, Yang JY, Lee SY, Son KH, Byun K. Ecklonia cava extracts decrease hypertension-related vascular calcification by modulating PGC-1alpha and SOD2. BioMed Pharmacother. (2022) 153:113283. doi: 10.1016/j.biopha.2022.113283
250. Chen Y, Li S, Guo Y, Yu H, Bao Y, Xin X, et al. Astaxanthin attenuates hypertensive vascular remodeling by protecting vascular smooth muscle cells from oxidative stress-induced mitochondrial dysfunction. Oxid Med Cell Longev (2020) 2020:4629189. doi: 10.1155/2020/4629189
251. Lu Y, Li S, Wu HF, Bian ZP, Xu JD, Gu CR, et al. Beneficial effects of astragaloside IV against angiotensin II-induced mitochondrial dysfunction in rat vascular smooth muscle cells. Int J Mol Med (2015) 36(5):1223–32. doi: 10.3892/ijmm.2015.2345
252. Geng N, Chen T, Chen L, Zhang H, Sun L, Lyu Y, et al. Nuclear receptor Nur77 protects against oxidative stress by maintaining mitochondrial homeostasis via regulating mitochondrial fission and mitophagy in smooth muscle cell. J Mol Cell Cardiol (2022) 170:22–33. doi: 10.1016/j.yjmcc.2022.05.007
253. Meng L, Lu C, Wu B, Lan C, Mo L, Chen C, et al. Taurine antagonizes macrophages M1 polarization by mitophagy-glycolysis switch blockage via dragging SAM-PP2Ac transmethylation. Front Immunol (2021) 12:648913. doi: 10.3389/fimmu.2021.648913
254. Li S, Ren X, Wang Y, Hu J, Wu H, Song S, et al. Fucoxanthin alleviates palmitate-induced inflammation in RAW 264.7 cells through improving lipid metabolism and attenuating mitochondrial dysfunction. Food Funct (2020) 11(4):3361–70. doi: 10.1039/d0fo00442a
255. Cao Q, Du HJ, Fu X, Duan N, Liu CH, Li XD. Artemisinin attenuated atherosclerosis in high-fat diet-fed ApoE(-/-) mice by promoting macrophage autophagy through the AMPK/mTOR/ULK1 pathway. J Cardiovasc Pharm (2020) 75(4):321–32. doi: 10.1097/FJC.0000000000000794
256. Liu M, Lu J, Yang S, Chen Y, Yu J, Guan S. Alliin alleviates LPS-induced pyroptosis via promoting mitophagy in THP-1 macrophages and mice. Food Chem Toxicol (2022) 160:112811. doi: 10.1016/j.fct.2022.112811
257. Chen WR, Zhou YJ, Yang JQ, Liu F, Wu XP, Sha Y. Melatonin attenuates calcium deposition from vascular smooth muscle cells by activating mitochondrial fusion and mitophagy via an AMPK/OPA1 signaling pathway. Oxid Med Cell Longev (2020) 2020:5298483. doi: 10.1155/2020/5298483
258. Ma W-Q, Sun X-J, Wang Y, Zhu Y, Han X-Q, Liu N-F. Restoring mitochondrial biogenesis with metformin attenuates β-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol Cell Endocrinology. (2019) 479:39–53. doi: 10.1016/j.mce.2018.08.012
259. Ma S, Chen JW, Feng J, Zhang R, Fan MM, Han D, et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid Med Cell Longevity (2018) 2018:9286458. doi: 10.1155/2018/9286458
260. Yang J, Sun M, Cheng R, Tan H, Liu C, Chen R, et al. Pitavastatin activates mitophagy to protect EPC proliferation through a calcium-dependent CAMK1-PINK1 pathway in atherosclerotic mice. Commun Biol (2022) 5(1):124. doi: 10.1038/s42003-022-03081-w
261. Lai TC, Chen YC, Cheng HH, Lee TL, Tsai JS, Lee IT, et al. Combined exposure to fine particulate matter and high glucose aggravates endothelial damage by increasing inflammation and mitophagy: the involvement of vitamin d. Part Fibre Toxicol (2022) 19(1):25. doi: 10.1186/s12989-022-00462-1
262. Wang Y, Mao X, Chen H, Feng J, Yan M, Wang Y, et al. Dexmedetomidine alleviates LPS-induced apoptosis and inflammation in macrophages by eliminating damaged mitochondria via PINK1 mediated mitophagy. Int Immunopharmacol. (2019) 73:471–81. doi: 10.1016/j.intimp.2019.05.027
263. Zekri-Nechar K, Zamorano-León JJ, Cortina-Gredilla M, López-de-Andrés A, Jiménez-García R, Navarro-Cuellar C, et al. Mitochondrial mitophagy protection combining rivaroxaban and aspirin in high glucose-exposed human coronary artery endothelial cell. an in vitro study. Diabetes Vasc Dis Res (2022) 19(5):14791641221129877. doi: 10.1177/14791641221129877
264. Xue SY, Hebert VY, Hayes DM, Robinson CN, Glover M, Dugas TR. Nucleoside reverse transcriptase inhibitors induce a mitophagy-associated endothelial cytotoxicity that is reversed by coenzyme Q10 cotreatment. Toxicol Sci (2013) 134(2):323–34. doi: 10.1093/toxsci/kft105
265. Hseu YC, Tseng YF, Pandey S, Shrestha S, Lin KY, Lin CW, et al. Coenzyme Q0 inhibits NLRP3 inflammasome activation through mitophagy induction in LPS/ATP-stimulated macrophages. Oxid Med Cell Longev (2022) 2022:4266214. doi: 10.1155/2022/4266214
266. Li P, Bai Y, Zhao X, Tian T, Tang L, Ru J, et al. NR4A1 contributes to high-fat associated endothelial dysfunction by promoting CaMKII-parkin-mitophagy pathways. Cell Stress Chaperones. (2018) 23(4):749–61. doi: 10.1007/s12192-018-0886-1
267. Szeto HH, Liu SY, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int (2016) 90(5):997–1011. doi: 10.1016/j.kint.2016.06.013
268. Sun DN, Wang J, Toan S, Muid D, Li RB, Chang X, et al. Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: focus on mitochondrial quality surveillance. Angiogenesis (2022) 25(3):307–29. doi: 10.1007/s10456-022-09835-8
269. Jain T, Nikolopoulou EA, Xu QB, Qu AJ. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol Therapeut. (2018) 183:22–33. doi: 10.1016/j.pharmthera.2017.09.003
270. Wu JR, Hsu JH, Dai ZK, Wu BN, Chen IJ, Liou SF, et al. Activation of endothelial NO synthase by a xanthine derivative ameliorates hypoxia-induced apoptosis in endothelial progenitor cells. J Pharm Pharmacol (2016) 68(6):810–8. doi: 10.1111/jphp.12555
271. Tuleta I, Franca CN, Wenzel D, Fleischmann B, Nickenig G, Werner N, et al. Intermittent hypoxia impairs endothelial function in early preatherosclerosis. Adv Exp Med Biol (2015) 858:1–7. doi: 10.1007/5584_2015_114
272. Song DM, Fang GQ, Mao SZ, Ye XB, Liu G, Miller EJ, et al. Selective inhibition of endothelial NF-kappa b signaling attenuates chronic intermittent hypoxia-induced atherosclerosis in mice. Atherosclerosis (2018) 270:68–75. doi: 10.1016/j.atherosclerosis.2018.01.027
273. Li LY, Yang YY, Zhang HA, Du YH, Jiao XL, Yu HH, et al. Salidroside ameliorated intermittent hypoxia-aggravated endothelial barrier disruption and atherosclerosis via the cAMP/PKA/RhoA signaling pathway. Front Pharmacol (2021) 12. doi: 10.3389/fphar.2021.723922
274. Wu D, Ji HZ, Du WJ, Ren LN, Qian G. Mitophagy alleviates ischemia/reperfusion-induced microvascular damage through improving mitochondrial quality control. Bioengineered (2022) 13(2):3596–607. doi: 10.1080/21655979.2022.2027065
275. Sun YM, Wen F, Yan C, Su LS, Luo JW, Chi W, et al. Mitophagy protects the retina against anti-vascular endothelial growth factor therapy-driven hypoxia via hypoxia-inducible factor-1 alpha signaling. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.727822
276. Sun LJ, Hao YW, An R, Li HX, Xi C, Shen GH. Overexpression of Rcan1-1L inhibits hypoxia-induced cell apoptosis through induction of mitophagy. Mol Cells (2014) 37(11):785–94. doi: 10.14348/molcells.2014.0103
277. Peng ZL, Ji DF, Qiao LK, Chen YD, Huang HJ. Autophagy inhibition by ATG3 knockdown remits oxygen-glucose Deprivation/Reoxygenation-induced injury and inflammation in brain microvascular endothelial cells. Neurochem Res (2021) 46(12):3200–12. doi: 10.1007/s11064-021-03423-w
278. Tang Q, Cao YP, Xiong W, Ke XX, Zhang J, Xia Y, et al. Glycyrrhizic acid exerts protective effects against hypoxia/reoxygenation-induced human coronary artery endothelial cell damage by regulating mitochondria. Exp Ther Med (2020) 20(1):335–42. doi: 10.3892/etm.2020.8668
279. Chun Y, Kim J. AMPK-mTOR signaling and cellular adaptations in hypoxia. Int J Mol Sci (2021) 22(18):9765. doi: 10.3390/ijms22189765
280. Nah J, Zhai PY, Huang CY, Fernandez AF, Mareedu S, Levine B, et al. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest. (2020) 130(6):2978–91. doi: 10.1172/JCI132366
281. Chen G, Zhang W, Li YP, Ren JG, Xu N, Liu H, et al. Hypoxia-induced autophagy in endothelial cells: a double-edged sword in the progression of infantile haemangioma? Cardiovasc Res (2013) 98(3):437–48. doi: 10.1093/cvr/cvt035
282. Phadwal K, Vrahnas C, Ganley IG, MacRae VE. Mitochondrial dysfunction: cause or consequence of vascular calcification? Front Cell Dev Biol (2021) 9:611922. doi: 10.3389/fcell.2021.611922
283. Sazonova OV, Isenberg BC, Herrmann J, Lee KL, Purwada A, Valentine AD, et al. Extracellular matrix presentation modulates vascular smooth muscle cell mechanotransduction. Matrix Biol (2015) 41:36–43. doi: 10.1016/j.matbio.2014.11.001
284. Shanahan CM, Weissberg PL, Metcalfe JC. Isolation of gene markers of differentiated and proliferating vascular smooth-muscle cells. Circ Res (1993) 73(1):193–204. doi: 10.1161/01.RES.73.1.193
285. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol (2019) 16(12):727–44. doi: 10.1038/s41569-019-0227-9
286. Aherrahrou R, Guo L, Nagraj VP, Aguhob A, Hinkle J, Chen L, et al. Genetic regulation of atherosclerosis-relevant phenotypes in human vascular smooth muscle cells. Circ Res (2020) 127(12):1552–65. doi: 10.1161/CIRCRESAHA.120.317415
287. Miano JM, Fisher EA, Majesky MW. Fate and state of vascular smooth muscle cells in atherosclerosis. Circulation (2021) 143(21):2110–6. doi: 10.1161/CIRCULATIONAHA.120.049922
288. Yu EPK, Reinhold J, Yu H, Starks L, Uryga AK, Foote K, et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler Thromb Vasc Biol (2017) 37(12):2322–32. doi: 10.1161/ATVBAHA.117.310042
289. Burger F, Baptista D, Roth A, da Silva RF, Montecucco F, Mach F, et al. NLRP3 inflammasome activation controls vascular smooth muscle cells phenotypic switch in atherosclerosis. Int J Mol Sci (2022) 23(1):340. doi: 10.3390/ijms23010340
290. Goettsch C, Rauner M, Hamann C, Sinningen K, Hempel U, Bornstein SR, et al. Nuclear factor of activated T cells mediates oxidised LDL-induced calcification of vascular smooth muscle cells. Diabetologia (2011) 54(10):2690–701. doi: 10.1007/s00125-011-2219-0
291. He X, Bai QQ, Zhang XS, Zhang LM. MgCl2 attenuates ox-LDL-Induced vascular smooth muscle-derived foam cells pyroptosis by downregulating the TLR4/NF-kappa b signaling pathway. Biol Trace Elem Res (2023). doi: 10.1007/s12011-023-03585-4
292. Clarke MCH, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med (2006) 12(9):1075–80. doi: 10.1038/nm1459
293. Swiader A, Nahapetyan H, Faccini J, D'Angelo R, Mucher E, Elbaz M, et al. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget (2016) 7(20):28821–35. doi: 10.18632/oncotarget.8936
294. Nahapetyan H, Moulis M, Grousset E, Faccini J, Grazide MH, Mucher E, et al. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death Dis (2019) 10(2):119. doi: 10.1038/s41419-019-1400-0
295. Mishra SR, Mahapatra KK, Behera BP, Patra S, Bhol CS, Panigrahi DP, et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int J Biochem Cell Biol (2021) 136:106013. doi: 10.1016/j.biocel.2021.106013
296. Docherty CK, Carswell A, Friel E, Mercer JR. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: a potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis (2018) 268:1–11. doi: 10.1016/j.atherosclerosis.2017.11.009
297. Shi R, Hu C, Yuan Q, Yang T, Peng J, Li Y, et al. Involvement of vascular peroxidase 1 in angiotensin II-induced vascular smooth muscle cell proliferation. Cardiovasc Res (2011) 91(1):27–36. doi: 10.1093/cvr/cvr042
298. Li PF, Dietz R, von Harsdorf R. Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells. Circulation (1997) 96(10):3602–9. doi: 10.1161/01.CIR.96.10.3602
299. Lv P, Miao S-B, Shu Y-N, Dong L-H, Liu G, Xie X-L, et al. Phosphorylation of smooth muscle 22α facilitates angiotensin II-induced ROS production via activation of the PKCδ-P47phox axis through release of PKCδ and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ Res (2012) 111(6):697–707. doi: 10.1161/CIRCRESAHA.112.272013
300. Wang Y, Zhang X, Chen W, Gao L, Li J, Song T, et al. Cortistatin ameliorates ang II-induced proliferation of vascular smooth muscle cells by inhibiting autophagy through SSTR3 and SSTR5. Life Sci (2020) 253:117726. doi: 10.1016/j.lfs.2020.117726
301. Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H, et al. Reactive oxygen-forming Nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res (2020) 127(7):911–27. doi: 10.1161/CIRCRESAHA.119.316159
302. Liu YY, Li J, Han YT, Chen YY, Liu LX, Lang JL, et al. Advanced glycation end-products suppress autophagy by AMPK/mTOR signaling pathway to promote vascular calcification. Mol Cell Biochem (2020) 471(1-2):91–100. doi: 10.1007/s11010-020-03769-9
303. Brodeur MR, Bouvet C, Bouchard S, Moreau S, Leblond J, deBlois D, et al. Reduction of advanced-glycation end products levels and inhibition of RAGE signaling decreases rat vascular calcification induced by diabetes. PloS One (2014) 9(1):e85922. doi: 10.1371/journal.pone.0085922
304. Hu CT, Shao YD, Liu YZ, Xiao X, Cheng ZB, Qu SL, et al. Oxidative stress in vascular calcification. Clin Chim Acta (2021) 519:101–10. doi: 10.1016/j.cca.2021.04.012
305. Liberman M, Bassi E, Martinatti MK, Bortolotto LA, Laurindo FRM. Oxidative stress mediates expansive vascular remodeling in a rabbit model of atherosclerosis and vascular calcification. Free Radical Bio Med (2006) 41:S39–S.
306. Sun M, Chang Q, Xin M, Wang Q, Li H, Qian J. Endogenous bone morphogenetic protein 2 plays a role in vascular smooth muscle cell calcification induced by interleukin 6 in vitro. Int J Immunopathol Pharmacol (2017) 30(3):227–37. doi: 10.1177/0394632016689571
307. Chen WR, Yang JQ, Liu F, Shen XQ, Zhou YJ. Melatonin attenuates vascular calcification by activating autophagy via an AMPK/mTOR/ULK1 signaling pathway. Exp Cell Res (2020) 389(1):111883. doi: 10.1016/j.yexcr.2020.111883
308. Arefin S, Buchanan S, Hobson S, Steinmetz J, Alsalhi S, Shiels PG, et al. Nrf2 in early vascular ageing: calcification, senescence and therapy. Clin Chim Acta (2020) 505:108–18. doi: 10.1016/j.cca.2020.02.026
309. Tyrrell DJ, Blin MG, Song J, Wood SC, Goldstein DR. Aging impairs mitochondrial function and mitophagy and elevates interleukin 6 within the cerebral vasculature. J Am Heart Assoc (2020) 9(23):e017820. doi: 10.1161/JAHA.120.017820
310. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol (2021) 18(1):58–68. doi: 10.1038/s41569-020-0431-7
311. Tyrrell DJ, Blin MG, Song J, Wood SC, Zhang M, Beard DA, et al. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ Res (2020) 126(3):298–314. doi: 10.1161/CIRCRESAHA.119.315644
312. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res (2018) 114(4):622–34. doi: 10.1093/cvr/cvy007
313. Uchikado Y, Ikeda Y, Sasaki Y, Iwabayashi M, Akasaki Y, Ohishi M. Association of lectin-like oxidized low-density lipoprotein receptor-1 with angiotensin II type 1 receptor impacts mitochondrial quality control, offering promise for the treatment of vascular senescence. Front Cardiovasc Med (2021) 8:788655. doi: 10.3389/fcvm.2021.788655
314. Lee SJ, Jeong JY, Oh CJ, Park S, Kim JY, Kim HJ, et al. Pyruvate dehydrogenase kinase 4 promotes vascular calcification via SMAD1/5/8 phosphorylation. Sci Rep (2015) 5:16577. doi: 10.1038/srep16577
315. Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase Complex/Pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst (2017) 109(11):djx071. doi: 10.1093/jnci/djx071
316. Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, et al. Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell (2018) 175(2):502–13 e13. doi: 10.1016/j.cell.2018.08.040
317. Feron O. Pyruvate into lactate and back: from the warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol (2009) 92(3):329–33. doi: 10.1016/j.radonc.2009.06.025
318. Vaupel P, Schmidberger H, Mayer A. The warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol (2019) 95(7):912–9. doi: 10.1080/09553002.2019.1589653
319. Ma WQ, Sun XJ, Zhu Y, Liu NF. PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming. Cell Death Dis (2020) 11(11):991. doi: 10.1038/s41419-020-03162-w
320. Zhu Y, Ji JJ, Yang R, Han XQ, Sun XJ, Ma WQ, et al. Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal (2019) 58:53–64. doi: 10.1016/j.cellsig.2019.03.006
321. Zhu Y, Han XQ, Sun XJ, Yang R, Ma WQ, Liu NF. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis (2020) 25(5-6):321–40. doi: 10.1007/s10495-020-01592-7
322. Esteban-Martinez L, Boya P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy (2018) 14(5):915–7. doi: 10.1080/15548627.2017.1332567
323. Ferro F, Servais S, Besson P, Roger S, Dumas JF, Brisson L. Autophagy and mitophagy in cancer metabolic remodelling. Semin Cell Dev Biol (2020) 98:129–38. doi: 10.1016/j.semcdb.2019.05.029
324. Naik PP, Birbrair A, Bhutia SK. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell Mol Life Sci (2019) 76(1):27–43. doi: 10.1007/s00018-018-2922-9
325. Song YY, Brady ST. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol (2015) 25(3):125–36. doi: 10.1016/j.tcb.2014.10.004
326. Wang YG, Guo YR, Liu K, Yin Z, Liu R, Xia Y, et al. KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature (2017) 552(7684):273–+. doi: 10.1038/nature25003
327. Eshun-Wilson L, Zhang R, Portran D, Nachury MV, Toso DB, Lohr T, et al. Effects of alpha-tubulin acetylation on microtubule structure and stability. P Natl Acad Sci USA (2019) 116(21):10366–71. doi: 10.1073/pnas.1900441116
328. Seetharaman S, Vianay B, Roca V, Farrugia AJ, De Pascalis C, Boeda B, et al. Microtubules tune mechanosensitive cell responses. Nat Mater (2022) 21(3):366–+. doi: 10.1038/s41563-021-01108-x
329. Ouyang CH, Mu J, Lu QL, Li J, Zhu HP, Wang QL, et al. Autophagic degradation of KAT2A/GCN5 promotes directional migration of vascular smooth muscle cells by reducing TUBA/alpha-tubulin acetylation. Autophagy (2020) 16(10):1753–70. doi: 10.1080/15548627.2019.1707488
330. Ouyang CH, Li J, Zheng XX, Mu J, Torres G, Wang QL, et al. Deletion of Ulk1 inhibits neointima formation by enhancing KAT2A/GCN5-mediated acetylation of TUBA/alpha-tubulin in vivo. Autophagy (2021) 17(12):4305–22. doi: 10.1080/15548627.2021.1911018
331. Choi GE, Oh JY, Lee HJ, Chae CW, Kim JS, Jung YH, et al. Glucocorticoid-mediated ER-mitochondria contacts reduce AMPA receptor and mitochondria trafficking into cell terminus via microtubule destabilization. Cell Death Dis (2018) 9:1137. doi: 10.1038/s41419-018-1172-y
332. He M, Ding Y, Chu C, Tang J, Xiao Q, Luo ZG. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. P Natl Acad Sci USA (2016) 113(40):11324–9. doi: 10.1073/pnas.1611282113
333. Kirchenwitz M, Stahnke S, Grunau K, Melcher L, van Ham M, Rottner K, et al. The autophagy inducer SMER28 attenuates microtubule dynamics mediating neuroprotection. Sci Rep-Uk (2022) 12(1):17805. doi: 10.1038/s41598-022-20563-3
334. Liu W, Yan JL, Pan WN, Tang MJ. Apelin/Elabela-APJ: a novel therapeutic target in the cardiovascular system. Ann Transl Med (2020) 8(5):243. doi: 10.21037/atm.2020.02.07
335. He L, Zhou QL, Huang Z, Xu J, Zhou H, Lv DG, et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPK alpha and exacerbates atherosclerotic lesions. J Cell Physiol (2019) 234(6):8668–82. doi: 10.1002/jcp.27527
336. Chen Z, Zhou QL, Chen J, Yang YY, Chen W, Mao H, et al. MCU-dependent mitochondrial calcium uptake-induced mitophagy contributes to apelin-13-stimulated VSMCs proliferation. Vasc Pharmacol (2022) 144:106979. doi: 10.1016/j.vph.2022.106979
337. Kostopoulos CG, Spiroglou SG, Varakis JN, Apostolakis E, Papadaki HH. Adiponectin/T-cadherin and apelin/APJ expression in human arteries and periadventitial fat: implication of local adipokine signaling in atherosclerosis? Cardiovasc Pathol (2014) 23(3):131–8. doi: 10.1016/j.carpath.2014.02.003
338. Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, et al. Apelin signaling antagonizes ang II effects in mouse models of atherosclerosis. J Clin Invest. (2008) 118(10):3343–54. doi: 10.1172/JCI34871
339. Fraga-Silva RA, Seeman H, Montecucco F, da Silva AR, Burger F, Costa-Fraga FP, et al. Apelin-13 treatment enhances the stability of atherosclerotic plaques. Eur J Clin Invest (2018) 48(3):e12891. doi: 10.1111/eci.12891
340. Zhang HY, Gong YS, Wang ZG, Jiang LP, Chen R, Fan XF, et al. Apelin inhibits the proliferation and migration of rat PASMCs via the activation of PI3K/Akt/mTOR signal and the inhibition of autophagy under hypoxia. J Cell Mol Med (2014) 18(3):542–53. doi: 10.1111/jcmm.12208
341. Shan PF, Lu Y, Cui RR, Jiang Y, Yuan LQ, Liao EY. Apelin attenuates the osteoblastic differentiation of vascular smooth muscle cells. PloS One (2011) 6(3):e17938. doi: 10.1371/journal.pone.0017938
342. Nahapetyan H, Faccini J, Mucher E, Grazide MH, Elbaz M, Martinet W, et al. Defective autophagy in vascular smooth muscle cells promotes an unstable atherosclerotic plaque phenotype and increased expression of mitophagy markers in apo e-/- mice. Atherosclerosis (2017) 263:e5–e. doi: 10.1016/j.atherosclerosis.2017.06.043
343. Choi SH, Agatisa-Boyle C, Gonen A, Kim A, Kim J, Alekseeva E, et al. Intracellular AIBP (Apolipoprotein a-I binding protein) regulates oxidized LDL (Low-density lipoprotein)-induced mitophagy in macrophages. Arterioscler Thromb Vasc Biol (2021) 41(2):e82–96. doi: 10.1161/ATVBAHA.120.315485
344. Wu H, Wang Y, Li WH, Chen H, Du L, Liu D, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy (2019) 15(11):1882–98. doi: 10.1080/15548627.2019.1596482
345. Onat UI, Yildirim AD, Tufanli O, Cimen I, Kocaturk B, Veli Z, et al. Intercepting the lipid-induced integrated stress response reduces atherosclerosis. J Am Coll Cardiol (2019) 73(10):1149–69. doi: 10.1016/j.jacc.2018.12.055
346. Gupta P, Sharma G, Lahiri A, Barthwal MK. FOXO3a acetylation regulates PINK1, mitophagy, inflammasome activation in murine palmitate-conditioned and diabetic macrophages. J Leukoc Biol (2022) 111(3):611–27. doi: 10.1002/JLB.3A0620-348RR
347. Liu X, Tang Y, Cui Y, Zhang H, Zhang D. Autophagy is associated with cell fate in the process of macrophage-derived foam cells formation and progress. J BioMed Sci (2016) 23(1):57. doi: 10.1186/s12929-016-0274-z
348. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
349. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18(9):2114–27. doi: 10.1038/s41423-021-00740-6
350. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol (2021) 22(5):550–9. doi: 10.1038/s41590-021-00886-5
351. Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y, et al. Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis (2022) 13(5):512. doi: 10.1038/s41419-022-04966-8
352. Goto M, Katayama KI, Shirakawa F, Tanaka I. Involvement of NF-kappaB p50/p65 heterodimer in activation of the human pro-interleukin-1beta gene at two subregions of the upstream enhancer element. Cytokine (1999) 11(1):16–28. doi: 10.1006/cyto.1998.0390
353. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
354. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol (2017) 18(8):861–9. doi: 10.1038/ni.3772
355. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164(5):896–910. doi: 10.1016/j.cell.2015.12.057
356. Liang JJ, Fraser LDC, Bryant CE. Lipid regulation of NLRP3 inflammasome activity through organelle stress. Trends Immunol (2021) 42(9):807–23. doi: 10.1016/j.it.2021.07.005
357. Shen L, Yang Y, Ou T, Key CC, Tong SH, Sequeira RC, et al. Dietary PUFAs attenuate NLRP3 inflammasome activation via enhancing macrophage autophagy. J Lipid Res (2017) 58(9):1808–21. doi: 10.1194/jlr.M075879
358. Pradhan P, Vijayan V, Gueler F, Immenschuh S. Interplay of heme with macrophages in homeostasis and inflammation. Int J Mol Sci (2020) 21(3):740. doi: 10.3390/ijms21030740
359. Jiang L, Jia M, Wei X, Guo J, Hao S, Mei A, et al. Bach1-induced suppression of angiogenesis is dependent on the BTB domain. EBioMedicine (2020) 51:102617. doi: 10.1016/j.ebiom.2019.102617
360. Jia M, Li Q, Guo J, Shi W, Zhu L, Huang Y, et al. Deletion of BACH1 attenuates atherosclerosis by reducing endothelial inflammation. Circ Res (2022) 130(7):1038–55. doi: 10.1161/CIRCRESAHA.121.319540
361. Pradhan P, Vijayan V, Cirksena K, Buettner FFR, Igarashi K, Motterlini R, et al. Genetic BACH1 deficiency alters mitochondrial function and increases NLRP3 inflammasome activation in mouse macrophages. Redox Biol (2022) 51:102265. doi: 10.1016/j.redox.2022.102265
362. Kadomoto S, Izumi K, Mizokami A. Macrophage polarity and disease control. Int J Mol Sci (2021) 23(1):144. doi: 10.3390/ijms23010144
363. Duan M, Chen H, Yin L, Zhu X, Novak P, Lv Y, et al. Mitochondrial apolipoprotein a-I binding protein alleviates atherosclerosis by regulating mitophagy and macrophage polarization. Cell Commun Signal (2022) 20(1):60. doi: 10.1186/s12964-022-00858-8
364. Jong CJ, Sandal P, Schaffer SW. The role of taurine in mitochondria health: more than just an antioxidant. Molecules (2021) 26(16):4913. doi: 10.3390/molecules26164913
365. Mantani PT, Duner P, Bengtsson E, Ljungcrantz I, Sundius L, To F, et al. Interleukin-25 (IL-25) has a protective role in atherosclerosis development in the aortic arch in mice. J Biol Chem (2018) 293(18):6791–801. doi: 10.1074/jbc.RA117.000292
366. Mantani PT, Duner P, Bengtsson E, Alm R, Ljungcrantz I, Soderberg I, et al. IL-25 inhibits atherosclerosis development in apolipoprotein e deficient mice. PloS One (2015) 10(1):e0117255. doi: 10.1371/journal.pone.0117255
367. Zhang HF, Lin YQ, Liu WH, Wang SH, Xu CZ, Chen YX, et al. IL-33 promotes IL-10 productions: mechanism of IL-33 reducing macrophages foam cell formation. J Am Coll Cardiol (2017) 70(16):C38–C9. doi: 10.1016/j.jacc.2017.07.132
368. McLaren JE, Michael DR, Salter RC, Ashlin TG, Calder CJ, Miller AM, et al. IL-33 reduces macrophage foam cell formation. J Immunol (2010) 185(2):1222–9. doi: 10.4049/jimmunol.1000520
369. Miller A, Xu D, Asquith D, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. Ann Rheum Dis (2008) 67:A16–A. doi: 10.1084/jem.20071868
370. Zhong W, Rao Z, Xu J, Sun Y, Hu H, Wang P, et al. Defective mitophagy in aged macrophages promotes mitochondrial DNA cytosolic leakage to activate STING signaling during liver sterile inflammation. Aging Cell (2022) 21(6):e13622. doi: 10.1111/acel.13622
371. Tsai ML, Tsai YG, Lin YC, Hsu YL, Chen YT, Tsai MK, et al. IL-25 induced ROS-mediated M2 macrophage polarization via AMPK-associated mitophagy. Int J Mol Sci (2022) 23(1):3. doi: 10.3390/ijms23010003
372. Pham PT, Fukuda D, Nishimoto S, Kim-Kaneyama JR, Lei XF, Takahashi Y, et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur Heart J (2021) 42(42):4336–48. doi: 10.1093/eurheartj/ehab249
373. Foley JF. Taking the STING out of aging? Sci Signal (2022) 15(741):eadd7524. doi: 10.1126/scisignal.add7524
374. Feng S, Man SM. Captain GBP1: inflammasomes assemble, pyroptotic endgame. Nat Immunol (2020) 21(8):829–30. doi: 10.1038/s41590-020-0727-0
Keywords: atherosclerosis, mitophagy, different cell types, mechanism progression, drug and natural product progression
Citation: Zhang Y, Weng J, Huan L, Sheng S and Xu F (2023) Mitophagy in atherosclerosis: from mechanism to therapy. Front. Immunol. 14:1165507. doi: 10.3389/fimmu.2023.1165507
Received: 14 February 2023; Accepted: 12 April 2023;
Published: 16 May 2023.
Edited by:
XuQiang Nie, Zunyi Medical University, ChinaReviewed by:
Valeria Manganelli, Sapienza University of Rome, ItalyDavid Edward Nelson, Middle Tennessee State University, United States
Copyright © 2023 Zhang, Weng, Huan, Sheng and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengqin Xu, 2111210640@stu.pku.edu.cn
†These authors have contributed equally to this work and share first authorship