 Bi Lian
Bi Lian Xiaosong Chen
Xiaosong Chen Kunwei Shen
Kunwei Shen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 09 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1164514
This article is part of the Research TopicCommunity Series in Post-Translational Modifications of Proteins in Cancer Immunity and Immunotherapy, volume IIView all 12 articles
Breast cancer is one of the common malignancies with poor prognosis worldwide. The treatment of breast cancer patients includes surgery, radiation, hormone therapy, chemotherapy, targeted drug therapy and immunotherapy. In recent years, immunotherapy has potentiated the survival of certain breast cancer patients; however, primary resistance or acquired resistance attenuate the therapeutic outcomes. Histone acetyltransferases induce histone acetylation on lysine residues, which can be reversed by histone deacetylases (HDACs). Dysregulation of HDACs via mutation and abnormal expression contributes to tumorigenesis and tumor progression. Numerous HDAC inhibitors have been developed and exhibited the potent anti-tumor activity in a variety of cancers, including breast cancer. HDAC inhibitors ameliorated immunotherapeutic efficacy in cancer patients. In this review, we discuss the anti-tumor activity of HDAC inhibitors in breast cancer, including dacinostat, belinostat, abexinostat, mocetinotat, panobinostat, romidepsin, entinostat, vorinostat, pracinostat, tubastatin A, trichostatin A, and tucidinostat. Moreover, we uncover the mechanisms of HDAC inhibitors in improving immunotherapy in breast cancer. Furthermore, we highlight that HDAC inhibitors might be potent agents to potentiate immunotherapy in breast cancer.
Breast cancer is one of the common tumors worldwide. Approximately 2.3 million new breast cancer cases were estimated in 2020 in the 185 countries (1). It has been estimated that there are 297,790 new cases of breast cancer and 59,910 deaths due to this deadly disease in the United States (2). Approximately 11%-20% of breast cancer patients are triple negative breast cancer (TNBC) due to lack of expression of HER2, ER and PR (3). TNBC patients often have aggressive behavior, metastasis and poor prognosis (4). For the treatment of local breast cancer, there are surgery and radiation, while the systemic therapies of breast cancer include chemotherapy, hormone therapy, targeted drug therapy and immunotherapy (5, 6). Histone acetyltransferases can lead to histone acetylation on lysine residues, which can be reversed by histone deacetylases (HDACs) (7, 8). It has been known that HDACs function on remodeling of chromatin and modulation of gene expression by specific epigenetic regulation (9). There are 18 HDACs that have been characterized to regulate various biological processes, which are classified into four groups (I-IV). Class I includes HDAC1, HDAC2, HDAC3 and HDAC8, which are related to RPD3 gene. Class II includes HDAC4, HDAC5, HDAC6, HDAC-7, HDAC9 and HDAC10. Class III includes sirtulin 1-7 and class IV includes HDAC11 (10–12). Dysregulation of HDACs via mutation and abnormal expression contributes to oncogenesis and tumor progression (10–12). Therefore, modulation of HDACs could be a potent strategy for cancer treatment.
Immunotherapy has emerged for fighting cancer via using the patient’s own immune system (13). Immunotherapy includes monoclonal antibodies, chimeric antigen receptor (CAR) T-cell therapy, CAR NK cell therapy, tumor infiltrating lymphocyte (TIL) therapy, endogenous T cell (ETC) therapy, immune checkpoint inhibitors (ICIs), cancer vaccines, cytokines and immunomodulators (14–17). It has been known that ICIs block immune checkpoints, which allow immune cells to respond to tumor. Inhibitory immune checkpoint molecules include programmed cell death ligand (PD-1), programmed death ligand (PD-L1), PD-L2, B7-H3 (CD276), B7-H4 (VTCN1), LAG3, TIM-3, and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) (18, 19). Although immunotherapy has improved the survival of certain cancer patients, primary resistance and acquired resistance in immunotherapy attenuate the cancer treatment outcomes (20, 21). Hence, it is pivotal to uncover the mechanism of immunotherapy resistance and to develop the compounds that improve immunotherapy.
Several HDAC inhibitors have been developed and exhibited the potent anti-tumor activity in a various cancer types, including inhibition of tumor growth, metastasis and drug resistance (22–24). For instance, abexinostat, givinostat and mocetinostat decreased the expression of Slug and increased the expression of E-cadherin in mammary tumor cells (25). Breast epithelial cells with E-cadherin depletion were sensitive to several HDAC inhibitors, including entinostat, vorinostat, pracinostat, and mocetinostat, due to inhibition of proliferation and upregulation of cell apoptosis (26). Here, we discuss the function of HDAC inhibitors in tumorigenesis, especially in improving immunotherapy in breast cancer.
Vorinostat, also known as SAHA (suberoylanilide hydroxamic acid), is an oral inhibitor of class I and II of HDACs, which was the first time to approve for clinical application in patients with cutaneous T-cell lymphoma in 2006 (27–29). Vorinostat has been determined by preclinical experiments and clinical trials to decide its therapeutic efficacy in combination with other antitumor drugs in breast cancer (30). Vorinostat plus CDK inhibitor flavopiridol treatments exhibited synergistic lethality in breast cancer cells via suppression of ERK1/2 and AKT pathways and regulation of apoptosis pathways (31). Using breast cancer brain metastatic cells and intracranial xenograft model, radio-sensitivity was increased by vorinostat (32). Vorinostat accelerated radio-sensitivity of breast tumor cells, leading to suppression of lung metastasis via inhibition of MMP-9, DNA repair proteins and modulation of autophagy and endoplasmic reticulum stress (33).
TRAIL-resistant breast cancer cells became more sensitive after vorinostat treatment in BALB/c nude mice because vorinostat inhibited the expression of NF-κB, cyclin D1, Bcl-2, Bcl-xL, VEGF, MMP-2, MMP-9, HIF-1α, IL-6, IL-8, increased the expression of DR4, DR5, p21, PUMA, TIMP-1, TIMP-2, Bax, Bak, Bim and Noxa (34). It has been reported that vorinostat overcame apoptosis-inducing ligand Apo2L/TRAIL resistance via regulation of Bax, DR5, caspase-3, caspase-8, caspase-9 and PARP cleavage in human MDA-MB-231 breast cancer cells (35). Vorinostat increased the sensitivity of olaparib, one PARP inhibitor, in TNBC cells via induction of DNA damage, apoptosis and autophagy (36). Vorinostat restrained brain metastasis and stimulated DNA double-strand breaks and induced the downregulation of Rad52 in a TNBC model (37). Vorinostat promoted taxol-mediated cell death and triggered inhibition of cell growth and induced cell cycle arrest at G2/M phase in breast cancer (38). Vorinostat in combination with Aurora kinase inhibitor (MK-0457) displayed synergistical inhibition of proliferation of breast cancer cells (39). Vorinostat activated the expression of estrogen receptor α (ERα) and sensitized a ligand of the aryl hydrocarbon receptor, aminoflavone, -mediated growth inhibition in mesenchymal-like TNBC cells, such as MDA-MB-231 and Hs578T cells (40). Co-treatment with vorinostat and simvastatin exhibited synergistic functions on cell proliferation and apoptosis via inhibition of Rab7 prenylation in TNBC cells (41). It has been found that tamoxifen sensitivity was enhanced by vorinostat treatment in TNBC cells (42).
Vorinostat in combination with chemotherapeutic agent decitabine increased sensitivity of Fas ligand (FasL)-induced apoptosis and CTL immunotherapy via promotion of CD8+ T cells in colon cancer cells (43). Vorinostat increased sensitivity of anti-GD2 monoclonal antibody (mAb) treatment and reduced tumor growth through elevation of macrophage effector cells with high expression of Fc-receptors and reduction of MDSC number in neuroblastoma (44, 45). In pancreatic cancer, vorinostat and sorafenib co-treatment enlarged the efficacy of anti-PD-1 antibody via promotion of CD8+ cells, M1 macrophages and NK cells in mice (46). A combination therapy by vorinostat and anti-PD-L1 to abrogate the immune escape has been reported via induction of cell apoptosis and G1 phase arrest in melanoma (47). In head and neck and salivary cancer patients with vorinostat plus pembrolizumab treatments, NLR, neutrophils, lymphocytes and T helper cells were associated with poor overall survival (48). The MDA-MB-231 breast carcinoma cells and LNCaP prostate cancer cells displayed sensitivity to vorinostat therapy via enhancement of the immune evasion, leading to promotion of T-cell-induced lysis. HDAC1 was further identified to play a pivotal role in tumor immune escape in breast cancer cells (49). Data from ER-positive breast cancer patients after vorinostat, tamoxifen and pembrolizumab treatments revealed that exhausted T cell signature was linked to immunotherapy response (50). Hence, combination of HDAC inhibitors and immunotherapy could obtain synergistic effects in cancer therapy in breast cancer.
Entinostat, a class I HDACs inhibitor, has been uncovered to attenuate cell proliferation and stimulated cell apoptosis in breast cancer (51, 52). Moreover, entinostat was critically involved in reversal of tumor immune escape in breast cancer (51). One study revealed that entinostat promoted lapatinib efficacy via inhibition of AKT phosphorylation, activation of FOXO3 transcription, leading to elevation of Bim1 expression in breast cancer cells with HER2 overexpression (53). Entinostat can attenuate the resistance of trastuzumab/lapatinib-resistant breast cancer cells with HER2 overexpression to the trastuzumab/lapatinib treatment (53). Entinostat plus MEK inhibitor pimasertib retarded cell growth in TNBC cells and inflammatory breast cancer (IBC) cells, and reduced tumor growth in mice via regulation of NOXA-participated MCL1 degradation (54).
One study used microarray analysis and revealed that doxorubicin and entinostat regulated numerous gene expressions related to differentiation, inflammation and proliferation. Entinostat sensitized doxorubicin-mediated cell cycle arrest at G2 phase (55). Doxorubicin and entinostat inhibited the expression of E2F and Myc genes, elevated interferon genes and increased the numbers of tumor-infiltrating lymphocytes. Moreover, entinostat and doxorubicin enhanced the expression of tumor testis antigens, such as IL13RA2, and elevated the expression of ICOSL and GITRL in MDA-MB-231 cells, which were immune checkpoint agonists (55). PD-L1 expression was increased by entinostat and reduced by doxorubicin treatment. Entinostat, all-trans retinoic acid, and doxorubicin together stimulated cell death and differentiation, leading to regression of tumor growth in mice by a xenograft model of TNBC (55). A combination of entinostat, all-trans retinoic acid, and doxorubicin caused tumor regression via targeting tumor-initiating cells in TNBC and modulating the ESE-1 and ELF-3 (56).
Entinostat, a cancer vaccine, and an IL15 agonist N-803 displayed a synergistic effect on tumor growth via upregulation of infiltration of CD8+ T cells, promotion of tumor inflammation-related gene expressions, enhancement of T cell responses to antigens, reduction of VISTA expression in 4T1 TNBC murine carcinoma model and MC38-CEA colon mouse model (57). Combined treatments with vaccine, entinostat, ICIs, and chemotherapy had exhibited a potential efficacy in advanced breast cancer (58). The breast cancer cells and prostate tumor cells exhibited sensitivity to entinostat by T-cell-involved lysis (49). Entinostat altered the tumor-related antigens, including PSA, brachyury, CEA and MUC1, and elevated the expression of several proteins that governed tumor immune recognition and antigen processing (49). Entinostat combined with immunotherapy could be a potential strategy for breast cancer therapy.
Romidepsin (FK228), a class I HDAC inhibitor, has been reported to inhibit the tumor growth in different types of cancers (59, 60). For example, in colon cancer cells, romidepsin attenuated cellular immune functions via upregulation of PD-L1 expression by enhancing the acetylation of histones H3 and H4 and modulation of BRD4 (61). Romidepsin accelerated the number of FOXP3+ regulatory T cells, reduced the number of IFN-γ+ CD8+ T cells, and alleviated Th1/Th2 ratio in TME in subcutaneous model and colitis-related cancer mice. Moreover, Romidepsin-mediated tumor suppression was abrogated by anti-PD-1 antibody treatment in colon cancer cells (61). One case report showed that romidepsin might be safe and effective for treatment of anaplastic large cell lymphoma (ALCL), which did not impair cellular immunity to HTLV-1 (62).
Romidepsin increased paclitaxel sensitivity and blocked tumor metastasis in inflammatory breast cancer (63). Specifically, romidepsin impaired tumor emboli and lymphatic vascular structure, and suppressed the expression of VEGF and HIF-1α in inflammatory breast cancer. Moreover, romidepsin induced the expression of acetylated Histone 3 proteins, triggered cell apoptosis and upregulated p21 expression level (63). Recently, romidepsin treatment upregulated the expression of chemokines, stimulated T-cell infiltration, and promoted T-cell-induced tumor regression. A combination of romidepsin and PD-1 blockade elevated T-cell infiltration and increased the efficacy of anti-PD-1 immunotherapy in lung adenocarcinoma (64). One group reported that a triple combination (gemcitabine, romidepsin, cisplatin) accelerated cell death in MDA-MB-231 and MDA-MB-468 cells (65). Moreover, a triple combination treatment using gemcitabine, romidepsin and cisplatin inhibited cell survival and invasion via targeting EMT in an ROS-dependent way, leading to suppression of tumor development, recurrence, and metastasis in TNBC (66).
It has been known that panobinostat (LBH589), a pan-HDAC inhibitor, performs a tumor suppressive function in various cancer types (67, 68). The function of panobinostat has bene verified in breast carcinogenesis and progression. Panobinostat enhanced the acetylation of GRP78 (glucose-regulated protein 78) and increased endoplasmic reticulum stress via upregulation of p-eIF2α, CHOP and ATF4, and elevation of BIK, BIM, Bax and BAK expression, acceleration of the caspase-7 activity and UPR in breast cancer cells (69). Panobinostat inhibited proliferation of breast cancer cells via modulation of aromatase gene expression, and synergized the anti-tumor function of letrozole in hormone-dependent breast cancer (70). In addition, panobinostat exposure elevated histone acetylation, induced G2/M cell cycle arrest and alleviated cell proliferation in TNBC cells. Panobinostat increased the expression of E-cadherin and changed the cell morphology in MDA-MB-231 cells (71). Another study showed that panobinostat inhibited the expression of ZEB family (ZEB1 and ZEB2) and led to suppression of tumor metastasis in TNBC (72).
The proliferation of breast cancer cells with aromatase inhibitor resistance was mitigated by panobinostat in part via inactivation of NF-κB1 pathway (73). The invasive and migratory ability of breast cancer cells was also repressed by panobinostat via induction of E-cadherin and alteration of Slug, MTA3 and Snail (74). Using a claudin-low TNBC PDX model, one group revealed that panobinostat inhibited the mesenchymal phenotype, such as inhibition of collagen expression (75). Panobinostat accelerated the expression of APCL and blocked Wnt/β-catenin pathway via promotion of β-catenin degradation in breast cancer, resulting in inactivation of β-catenin targets, including c-Myc, CD44, Cyclin D1 and c-Jun, which contributed to inhibition of tumor growth and metastasis (76). Panobinostat plus rapamycin led to increased efficacy against TNBC on inhibition of proliferation, invasion, migration and induction of apoptosis, which could be due to overproduction of ROS ad activation of endoplasmic reticulum stress in breast cancer (77). Panobinostat inhibits tumor growth by induction of autophagy and accelerated secretory autophagy via targeting Vps34/Rab5C pathway in breast cancer (78). Panobinostat has shown the treatment benefits in oncolytic herpes simplex virus in combination with anti-PD-1/PD-L1 therapy in glioma and squamous cell carcinoma (79). The efficacy of panobinostat was spatially correlated with multiple gene expressions, including galectin-3, cleaved caspase-3, PD-L1, neuropilin-1 and calrecticulin in breast cancer, suggesting that panobinostat (80). Without a doubt, the function of panobinostat in altering immunotherapy warrant to further exploration in breast cancer.
Mocetinostat, a class I/IV HDAC inhibitor, has been identified to suppress the tumorigenesis and tumor development in a various types of human cancers (81). Mocetinostat increased PD-L1 expression and elevated the expression of antigen presentation genes in NSCLC (82). Mocetinostat interacted with the promoters of a class I HDAC and increased active histone marks, and enhanced IFN-γ activity in governing class II transactivator. In mice, mocetinostat reduced the number of Tregs and MDSCs, but elevated the number of CD8+ population in tumors. Mocetinostat and PD-L1 antibody displayed a synergistic function in mouse lung tumor models (82). Mocetinostat plus the BET inhibitor JQ1 reduced viability of breast cancer cells via modulation of cell cycle-associated gene expressions. Mocetinostat and JQ1 cotreatment upregulated the expression of USP17 family members in breast cancer cells, resulting in inactivation of Ras/MAPK pathway and attenuation of cell viability (83).
Fyn-related kinase (FRK) has been known to be repressed in cancer cells due to its promoter CpG methylation (84). Cell migration and invasion were reduced by FRK overexpression via inactivation of MAPK, AKT and JAK/STAT pathways and blockade of EMT in breast cancer cells, including inhibition of slug, vimentin, fibronectin, and upregulation of E-cadherin (85). Mocetinostat and entinostat can induce re-expression of FRK at mRNA and protein levels in basal B breast cancer cells, contributing to tumor regression (86). Similarly, mocetinostat exhibited anti-cancer functions in basal-like breast cancer cells with HDAC2 overexpression (87). Moreover, mocetinostat plus azacytidine increased chemotherapeutic sensitivity in mammary mesenchymal tumors via targeting EMT process (25). One group used TCGA database and found that mocetinostat and vorinostat exhibited the functional similarity with the FDA-approved drugs for the treatment of HER2-postive breast cancer (88). Mocetinostat combined with capecitabine showed a synergistic effect on suppression of proliferation and induction of apoptosis in 4T1 breast cancer cells via targeting Bax, Bcl-2, PI3K/AKT, c-Myc, PTEN, p53, caspase-7, -9, and cleaved PARP (89). It is required to further dissect the function of mocetinostat in improving immunotherapy in breast cancer.
Abexinostat (PCI-24781, CRA-024781) is a Pan-HDACs mainly targeting HDAC1. It has been reported that abexinostat increased tumor radio-sensitivity in NSCLC (90). PCI-24781 was developed to decrease cell proliferation, differentiation and metastasis via influencing calcium influx by activation of RGS2 in breast cancer (91). Abexinostat triggered the differentiation of cancer stem cells in breast cancer with low level of lncRNA Xist expression (92). Moreover, low expression of lncRNA Xist could indicate abexinostat response in breast tumor PDXs and linked to an inhibition of cancer stem cells in breast cancer (92). Interestingly, administration of abexinostat did not change the expression of ESR1, ERα, and ESR1-associated genes in xenograft models (93). This study indicated that it is doubtable to use a combination of abexinostat and hormone therapy for the management of breast cancer patients. Due to unclear role of abexinostat in immune response, it is pivotal to define the function of abexinostat in regulation of immunotherapy of breast cancer patients.
Belinostat (Beleodaq, PXD101) is a HDACi with antineoplastic function in part via targeting HDAC6. One study showed that TNBC cells and HER2-enriched breast cancer cells were remarkably sensitive to belinostat and panobinostat treatment. Moreover, belinostat and panobinostat increased doxorubicin sensitivity in TNBC cells (94). Belinostat and SAHA sensitized TNBC cells to the PARP inhibitor olaparib treatment, showing the synergistic inhibition of proliferation of TNBC cells and induction of cell apoptosis (95). Belinostat plus Hsp90 inhibitor 17-AAG displayed a synergistic effect on suppression of invasion and cell growth in TNBC cells via inhibiting the expression of TEAD family proteins and elevating YY1AP1 phosphorylation and MLC1 (modulator of VRAC current 1) (96). Chemotherapeutic drugs led to cancer stem cell (ALDH+/CD44+) abundance in breast cancer, which was abrogated by belinostat exposure (97). One group has demonstrated that belinostat stimulated the expression of CXCL1 in TBNC cells, suggesting that CXCL1 clone evolution could be an indicator for TNBC prognosis (98).
Dacinostat (LAQ-824) has been observed to tackle cancer chemoresistance in multiple myeloma ad acute myeloid leukemia (99). One study demonstrated that dacinostat and givinostat can restore the activity of cytotoxic T lymphocytes in in pancreatic cancer cells (100). NVP-LAQ824 attenuated tumor growth and angiogenesis and enhanced VEGFR inhibitor PTK787/ZK222584-mediated inhibition of angiogenesis via upregulation of p21 and downregulation of angioprotein-2, Tie-2, VEGF, HIF-1α, and survivin (101). Using an orthotopic breast tumor model, NVP-LAQ824 plus PTK787/ZK222584 induced a greater suppression of tumor growth (101). LAQ824 can regulate the expression of miRNAs in SKBR-3 breast cancer cells (102). It has been known that noncoding RNAs, including microRNAs, lncRNAs and circRNAs, are critical in carcinogenesis in a variety of human cancers (103–105). LAQ824 increased 22 miRNA expressions and decreased 5 miRNA expressions in breast cancer cells (102). LAQ824 in combination with 5-Aza-2’-deoxycytidine, known as decitabine, displayed a greater antineoplastic effect on breast cancer cells (106). LAQ824 reduced the expression of ERα, PRβ, c-Myc, cyclin D1 and HDAC6 in breast cancer cells, leading to suppression of cellular proliferation (107). LAQ-824 sensitized drug sensitivity, including taxotere, epothilone B, trastuzumab and gemcitabine, via downregulation of HER-2 expression in breast cancer cells (108). LAQ824 was found to work as a sensitizer to immunotherapy with adoptive T-cell transfer in melanoma (109). Further exploration is pivotal to determine the LAQ824-enhanced immunotherapy in cancer patients via improving the anticancer function of tumor antigen-specific lymphocytes.
Pracinostat (SB939) attenuated tumor growth and metastasis via blocking the IL6/STAT3 pathway in breast cancer (110). YF479, a HDACi, exhibited antitumor functions in breast cancer, including suppression of growth, metastasis and recurrence (111). NK-HDAC-1 was designed and synthesized for fighting breast cancer, which induced apoptosis and cell cycle arrest via upregulation of p21 and inhibition of Cyclin D1 (112). Givinostat (ITF2357) increased cell death and reduced cell proliferation in urothelial carcinoma cells and acute lymphocytic leukemia (113, 114). Givinostat enhanced CTL sensitivity in pancreatic cancer cells (100). In addition, givinostat reduced cancer stemness and reversed transformed phenotype in glioblastoma (115, 116). The function of givinostat is breast tumorigenesis is unclear, which should be explored in the future. Tubastatin A and alisertib reduced the number of pulmonary metastases via suppression of HDAC6 and AURKA in breast tumor xenograft models (117). Tubastatin A in combination with palladium nanoparticles triggered cell apoptosis in breast cancer cells (118). MPT0G211, a HDAC6 inhibitor, exhibited an inhibition of tumor metastasis via attenuation of HDAC6 activity in breast cancer cells (119).
Trichostatin A (TSA) inhibited the expression of DNMT1 (DNA methyltransferase 1) via reduction of DNMT1 mRNA stability in Jurkat T leukemia cells (120). TSA decreased the transcript and protein levels of aromatase CYP19 and phospholipase C gamma-1 (PLC-γ1) in MCF-7 breast cancer cells (121, 122). SK-7041, a HDACi via a hybrid of TSA and MS-275, induced cell apoptosis and G2/M arrest in breast cancer cells (123). MAGE-C1 (melanoma-associated antigen-C1) and MAGE-C2 expressions were linked to advanced tumor grade and poor survival in breast cancer patients. TSA treatment increased 5-aza-CdR-induced MAGE-C2 transcription in breast cancer cells, indicating that MAGE-C2 could be a target for cancer immunotherapy (124). Tucidinostat, an inhibitor of HDAC1, HDAC2, HDAC3 and HDAC10, has shown a remarkable anticancer activity and a synergistic ability with immunotherapy (125). Tucidinostat combined with selinexor, an exportin 1 inhibitor, showed a greater antitumor effect on TP53 wild-type breast cancer (126). Breast cancer patients with HR+/HER2- received CDK4/6 inhibitor treatment and then obtained tucidinostat-based therapy, which displayed better clinical outcomes (127). DNMT inhibitor 5-zazcytidine and HDACi butyrate ameliorated the tumorigenicity of CSCs and retarded breast tumor growth (128). We believe more HDAC inhibitors will be developed for potentiating immunotherapy in the future.
In conclusion, HDAC inhibitors improve immunotherapy via targeting HDACs and their downstream targets in breast cancer (Figure 1). Although HDAC inhibitors might be useful to enhance tumor immunotherapy, several concerns should be mentioned. So far, only five HDAC inhibitors have been approved by FDA for cancer therapy, including vorinostat, belinostat, panobinostat, pracinostat and romidepsin (129). These HDAC inhibitors exhibited clinical advantage in hematological malignancies. It is required to measure the efficacy of HDAC inhibitors in solid tumors (130). Sirtuins inhibitors, such as nicotinamide, sirtinol and splitomicin, have shown their activities in regulation of metabolism, DNA repair, proliferation, drug resistance and immunotherapy (131). Due to limited space, we do not discuss the role of sirtuins inhibitors in modulation of breast cancer immunotherapy. Among dozens of HDAC inhibitors, which one is the best choice for enhancement of immunotherapy in breast cancer? The development of inhibitors based on the differential expression of HDAC isoforms is pivotal to rationally develop selective and effective inhibitors for personalized-medicine treatment (132, 133). Notably, HDAC inhibitors also have adverse side effects and cause drug resistance, which should be overcome. The resistant reasons of HDAC inhibitors are still incomplete. This might be due to cancer cell types, tumor-specific mutations, tumor microenvironmental conditions, upregulation of efflux pumps (P-glycoprotein), overexpression of HDAC enzymes. Lastly, triple combination of HDACi, immunotherapy and other inhibitors could be a promising approach for the treatment of breast cancer.
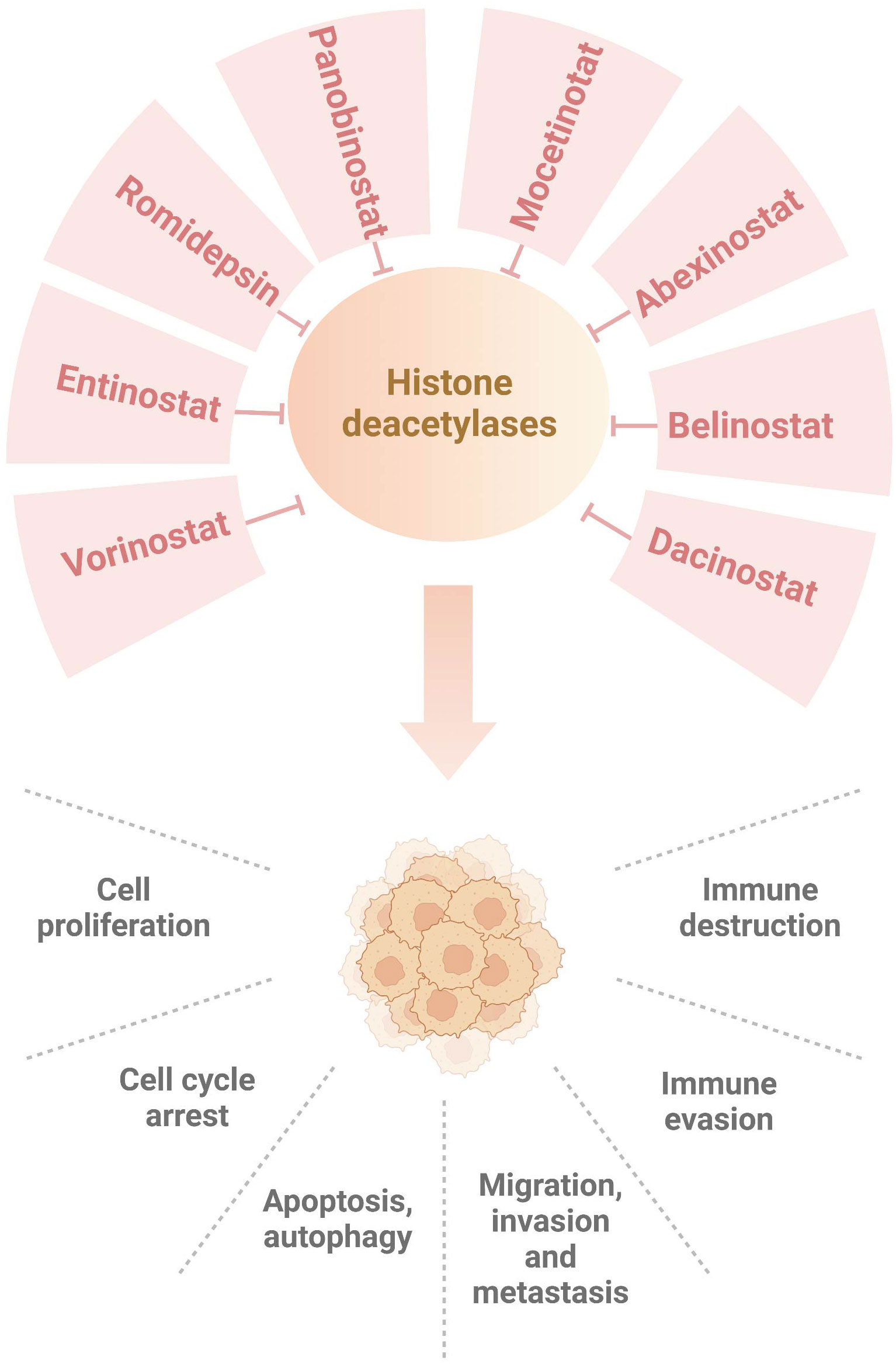
Figure 1 Numerous HDAC inhibitors suppress histone deacetylases in breast cancer. Dacinostat, belinostat, abexinostat, mocetinotat, panobinostat, romidepsin, entinostat and vorinostat, inhibit histone deacetylases and regulate breast tumorigenesis, progression and immunotherapy.
BL wrote the manuscript and made the figure. XC and KS edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin (2023) 73(1):17–48. doi: 10.3322/caac.21763
3. Agostinetto E, Gligorov J, Piccart M. Systemic therapy for early-stage breast cancer: learning from the past to build the future. Nat Rev Clin Oncol (2022) 19(12):763–74. doi: 10.1038/s41571-022-00687-1
4. Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat Rev Clin Oncol (2022) 19(2):91–113. doi: 10.1038/s41571-021-00565-2
5. Loizides S, Constantinidou A. Triple negative breast cancer: Immunogenicity, tumor microenvironment, and immunotherapy. Front Genet (2022) 13:1095839. doi: 10.3389/fgene.2022.1095839
6. Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: Advances and future directions. Nat Rev Drug Discov (2023) 22(2):101–26. doi: 10.1038/s41573-022-00579-0
7. Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer (2001) 1(3):194–202. doi: 10.1038/35106079
8. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov (2014) 13(9):673–91. doi: 10.1038/nrd4360
9. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
10. Johnstone RW. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat Rev Drug Discov (2002) 1(4):287–99. doi: 10.1038/nrd772
11. Kelly WK, Marks PA. Drug insight: Histone deacetylase inhibitors–development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol (2005) 2(3):150–7. doi: 10.1038/ncponc0106
12. Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov (2013) 12(12):917–30. doi: 10.1038/nrd4154
13. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12(4):237–51. doi: 10.1038/nrc3237
14. Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer (2022) 3(8):911–26. doi: 10.1038/s43018-022-00418-6
15. Laskowski TJ, Biederstadt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer (2022) 22(10):557–75. doi: 10.1038/s41568-022-00491-0
16. Wolf NK, Kissiov DU, Raulet DH. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol (2023) 23(2):90–105. doi: 10.1038/s41577-022-00732-1
17. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol (2017) 17(9):559–72. doi: 10.1038/nri.2017.49
18. Liu J, Chen T, Li S, Liu W, Wang P, Shang G. Targeting matrix metalloproteinases by E3 ubiquitin ligases as a way to regulate the tumor microenvironment for cancer therapy. Semin Cancer Biol (2022) 86(Pt 2):259–68. doi: 10.1016/j.semcancer.2022.06.004
19. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
20. Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of cancer resistance to immunotherapy. Front Oncol (2020) 10:1290. doi: 10.3389/fonc.2020.01290
21. Hou B, Chen T, Zhang H, Li J, Wang P, Shang G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front Immunol (2023) 14:1123244. doi: 10.3389/fimmu.2023.1123244
22. Fedele P, Orlando L, Cinieri S. Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert Opin Investig Drugs (2017) 26(11):1199–206. doi: 10.1080/13543784.2017.1386172
23. De Souza C, Chatterji BP. HDAC inhibitors as novel anti-cancer therapeutics. Recent Pat Anticancer Drug Discov (2015) 10(2):145–62. doi: 10.2174/1574892810666150317144511
24. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5(9):769–84. doi: 10.1038/nrd2133
25. Zhao N, Powell RT, Yuan X, Bae G, Roarty KP, Stossi F, et al. Morphological screening of mesenchymal mammary tumor organoids to identify drugs that reverse epithelial-mesenchymal transition. Nat Commun (2021) 12(1):4262. doi: 10.1038/s41467-021-24545-3
26. Decourtye-Espiard L, Bougen-Zhukov N, Godwin T, Brew T, Schulpen E, Black MA, et al. E-Cadherin-Deficient epithelial cells are sensitive to HDAC inhibitors. Cancers (2021) 14(1). doi: 10.3390/cancers14010175
27. Patra S, Praharaj PP, Klionsky DJ, Bhutia SK. Vorinostat in autophagic cell death: A critical insight into autophagy-mediated, -associated and -dependent cell death for cancer prevention. Drug Discov Today (2022) 27(1):269–79. doi: 10.1016/j.drudis.2021.08.004
28. Zagni C, Floresta G, Monciino G, Rescifina A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after vorinostat. Med Res Rev (2017) 37(6):1373–428. doi: 10.1002/med.21437
29. Duvic M, Vu J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs (2007) 16(7):1111–20. doi: 10.1517/13543784.16.7.1111
30. Wawruszak A, Borkiewicz L, Okon E, Kukula-Koch W, Afshan S, Halasa M. Vorinostat (SAHA) and breast cancer: An overview. Cancers (2021) 13(18). doi: 10.3390/cancers13184700
31. Mitchell C, Park MA, Zhang G, Yacoub A, Curiel DT, Fisher PB, et al. Extrinsic pathway- and cathepsin-dependent induction of mitochondrial dysfunction are essential for synergistic flavopiridol and vorinostat lethality in breast cancer cells. Mol Cancer Ther (2007) 6(12 Pt 1):3101–12. doi: 10.1158/1535-7163.MCT-07-0561
32. Baschnagel A, Russo A, Burgan WE, Carter D, Beam K, Palmieri D, et al. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther (2009) 8(6):1589–95. doi: 10.1158/1535-7163.MCT-09-0038
33. Chiu HW, Yeh YL, Wang YC, Huang WJ, Chen YA, Chiou YS, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PloS One (2013) 8(10):e76340. doi: 10.1371/journal.pone.0076340
34. Shankar S, Davis R, Singh KP, Kurzrock R, Ross DD, Srivastava RK. Suberoylanilide hydroxamic acid (Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells orthotopically implanted in BALB/c nude mice. Mol Cancer Ther (2009) 8(6):1596–605. doi: 10.1158/1535-7163.MCT-08-1004
35. Butler LM, Liapis V, Bouralexis S, Welldon K, Hay S, Thai le M, et al. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAIL. Int J Cancer (2006) 119(4):944–54. doi: 10.1002/ijc.21939
36. Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res (2015) 17. doi: 10.1186/s13058-015-0534-y
37. Palmieri D, Lockman PR, Thomas FC, Hua E, Herring J, Hargrave E, et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin Cancer Res (2009) 15(19):6148–57. doi: 10.1158/1078-0432.CCR-09-1039
38. Shi YK, Li ZH, Han XQ, Yi JH, Wang ZH, Hou JL, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances taxol-induced cell death in breast cancer. Cancer Chemother Pharmacol (2010) 66(6):1131–40. doi: 10.1007/s00280-010-1455-1
39. Fiskus W, Hembruff SL, Rao R, Sharma P, Balusu R, Venkannagari S, et al. Co-Treatment with vorinostat synergistically enhances activity of aurora kinase inhibitor against human breast cancer cells. Breast Cancer Res Treat (2012) 135(2):433–44. doi: 10.1007/s10549-012-2171-9
40. Stark K, Burger A, Wu J, Shelton P, Polin L, Li J. Reactivation of estrogen receptor alpha by vorinostat sensitizes mesenchymal-like triple-negative breast cancer to aminoflavone, a ligand of the aryl hydrocarbon receptor. PloS One (2013) 8(9):e74525. doi: 10.1371/journal.pone.0074525
41. Kou X, Yang Y, Jiang X, Liu H, Sun F, Wang X, et al. Vorinostat and simvastatin have synergistic effects on triple-negative breast cancer cells via abrogating Rab7 prenylation. Eur J Pharmacol (2017) 813:161–71. doi: 10.1016/j.ejphar.2017.08.022
42. Ma W, Sun J, Xu J, Luo Z, Diao D, Zhang Z, et al. Sensitizing triple negative breast cancer to tamoxifen chemotherapy via a redox-responsive vorinostat-containing polymeric prodrug nanocarrier. Theranostics (2020) 10(6):2463–78. doi: 10.7150/thno.38973
43. Yang D, Torres CM, Bardhan K, Zimmerman M, McGaha TL, Liu K. Decitabine and vorinostat cooperate to sensitize colon carcinoma cells to fas ligand-induced apoptosis in vitro and tumor suppression in vivo. J Immunol (2012) 188(9):4441–9. doi: 10.4049/jimmunol.1103035
44. Kroesen M, Bull C, Gielen PR, Brok IC, Armandari I, Wassink M, et al. Anti-GD2 mAb and vorinostat synergize in the treatment of neuroblastoma. Oncoimmunology (2016) 5(6):e1164919. doi: 10.1080/2162402X.2016.1164919
45. van den Bijgaart RJE, Kroesen M, Brok IC, Reijnen D, Wassink M, Boon L, et al. Anti-GD2 antibody and vorinostat immunocombination therapy is highly effective in an aggressive orthotopic neuroblastoma model. Oncoimmunology (2020) 9(1):1817653. doi: 10.1080/2162402X.2020.1817653
46. Booth L, Roberts JL, Poklepovic A, Dent P. Prior exposure of pancreatic tumors to [sorafenib + vorinostat] enhances the efficacy of an anti-PD-1 antibody. Cancer Biol Ther (2019) 20(1):109–21. doi: 10.1080/15384047.2018.1507258
47. Lu F, Hou L, Wang S, Yu Y, Zhang Y, Sun L, et al. Lysosome activable polymeric vorinostat encapsulating PD-L1KD for a combination of HDACi and immunotherapy. Drug Deliv (2021) 28(1):963–72. doi: 10.1080/10717544.2021.1927246
48. Pan C, Wu QV, Voutsinas J, Houlton JJ, Barber B, Futran N, et al. Neutrophil to lymphocyte ratio and peripheral blood biomarkers correlate with survival outcomes but not response among head and neck and salivary cancer treated with pembrolizumab and vorinostat. Head Neck (2023) 45(2):391–7. doi: 10.1002/hed.27252
49. Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget (2016) 7(7):7390–402. doi: 10.18632/oncotarget.7180
50. Terranova-Barberio M, Pawlowska N, Dhawan M, Moasser M, Chien AJ, Melisko ME, et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat Commun (2020) 11(1):3584. doi: 10.1038/s41467-020-17414-y
51. Trapani D, Esposito A, Criscitiello C, Mazzarella L, Locatelli M, Minchella I, et al. Entinostat for the treatment of breast cancer. Expert Opin Investig Drugs (2017) 26(8):965–71. doi: 10.1080/13543784.2017.1353077
52. Connolly RM, Rudek MA, Piekarz R. Entinostat: a promising treatment option for patients with advanced breast cancer. Future Oncol (2017) 13(13):1137–48. doi: 10.2217/fon-2016-0526
53. Lee J, Bartholomeusz C, Mansour O, Humphries J, Hortobagyi GN, Ordentlich P, et al. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res Treat (2014) 146(2):259–72. doi: 10.1007/s10549-014-3014-7
54. Torres-Adorno AM, Lee J, Kogawa T, Ordentlich P, Tripathy D, Lim B, et al. Histone deacetylase inhibitor enhances the efficacy of MEK inhibitor through NOXA-mediated MCL1 degradation in triple-negative and inflammatory breast cancer. Clin Cancer Res (2017) 23(16):4780–92. doi: 10.1158/1078-0432.CCR-16-2622
55. Merino VF, Cho S, Nguyen N, Sadik H, Narayan A, Talbot C Jr., et al. Induction of cell cycle arrest and inflammatory genes by combined treatment with epigenetic, differentiating, and chemotherapeutic agents in triple-negative breast cancer. Breast Cancer Res (2018) 20(1):145. doi: 10.1186/s13058-018-1068-x
56. Merino VF, Nguyen N, Jin K, Sadik H, Cho S, Korangath P, et al. Combined treatment with epigenetic, differentiating, and chemotherapeutic agents cooperatively targets tumor-initiating cells in triple-negative breast cancer. Cancer Res (2016) 76(7):2013–24. doi: 10.1158/0008-5472.CAN-15-1619
57. Hicks KC, Knudson KM, Lee KL, Hamilton DH, Hodge JW, Figg WD, et al. Cooperative immune-mediated mechanisms of the HDAC inhibitor entinostat, an IL15 superagonist, and a cancer vaccine effectively synergize as a novel cancer therapy. Clin Cancer Res (2020) 26(3):704–16. doi: 10.1158/1078-0432.CCR-19-0727
58. Gatti-Mays ME, Gameiro SR, Ozawa Y, Knudson KM, Hicks KC, Palena C, et al. Improving the odds in advanced breast cancer with combination immunotherapy: Stepwise addition of vaccine, immune checkpoint inhibitor, chemotherapy, and HDAC inhibitor in advanced stage breast cancer. Front Oncol (2020) 10:581801. doi: 10.3389/fonc.2020.581801
59. Petrich A, Nabhan C. Use of class I histone deacetylase inhibitor romidepsin in combination regimens. Leuk Lymphoma (2016) 57(8):1755–65. doi: 10.3109/10428194.2016.1160082
60. Bertino EM, Otterson GA. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin Investig Drugs (2011) 20(8):1151–8. doi: 10.1517/13543784.2011.594437
61. Shi Y, Fu Y, Zhang X, Zhao G, Yao Y, Guo Y, et al. Romidepsin (FK228) regulates the expression of the immune checkpoint ligand PD-L1 and suppresses cellular immune functions in colon cancer. Cancer Immunol Immunother (2021) 70(1):61–73. doi: 10.1007/s00262-020-02653-1
62. Jo T, Sakai T, Matsuzaka K, Shioya H, Tominaga H, Kaneko Y, et al. Successful treatment of a patient with brentuximab vedotin-refractory ALK-negative anaplastic Large cell lymphoma with romidepsin. Case Rep Oncol (2020) 13(3):1402–9. doi: 10.1159/000511111
63. Robertson FM, Chu K, Boley KM, Ye Z, Liu H, Wright MC, et al. The class I HDAC inhibitor romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J Exp Ther Oncol (2013) 10(3):219–33.
64. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res (2016) 22(16):4119–32. doi: 10.1158/1078-0432.CCR-15-2584
65. Pattarawat P, Wallace S, Pfisterer B, Odoi A, Wang HR. Formulation of a triple combination gemcitabine plus romidepsin + cisplatin regimen to efficaciously and safely control triple-negative breast cancer tumor development. Cancer Chemother Pharmacol (2020) 85(1):141–52. doi: 10.1007/s00280-019-04013-y
66. Pattarawat P, Hunt JT, Poloway J, Archibald CJ, Wang HR. A triple combination gemcitabine + romidepsin + cisplatin to effectively control triple-negative breast cancer tumor development, recurrence, and metastasis. Cancer Chemother Pharmacol (2021) 88(3):415–25. doi: 10.1007/s00280-021-04298-y
67. Li X, Zhang J, Xie Y, Jiang Y, Yingjie Z, Xu W. Progress of HDAC inhibitor panobinostat in the treatment of cancer. Curr Drug Targets (2014) 15(6):622–34. doi: 10.2174/1389450115666140306152642
68. Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett (2009) 280(2):233–41. doi: 10.1016/j.canlet.2009.02.019
69. Rao R, Nalluri S, Kolhe R, Yang Y, Fiskus W, Chen J, et al. Treatment with panobinostat induces glucose-regulated protein 78 acetylation and endoplasmic reticulum stress in breast cancer cells. Mol Cancer Ther (2010) 9(4):942–52. doi: 10.1158/1535-7163.MCT-09-0988
70. Chen S, Ye J, Kijima I, Evans D. The HDAC inhibitor LBH589 (panobinostat) is an inhibitory modulator of aromatase gene expression. Proc Natl Acad Sci U.S.A. (2010) 107(24):11032–7. doi: 10.1073/pnas.1000917107
71. Tate CR, Rhodes LV, Segar HC, Driver JL, Pounder FN, Burow ME, et al. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res (2012) 14(3):R79. doi: 10.1186/bcr3192
72. Rhodes LV, Tate CR, Segar HC, Burks HE, Phamduy TB, Hoang V, et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat (2014) 145(3):593–604. doi: 10.1007/s10549-014-2979-6
73. Kubo M, Kanaya N, Petrossian K, Ye J, Warden C, Liu Z, et al. Inhibition of the proliferation of acquired aromatase inhibitor-resistant breast cancer cells by histone deacetylase inhibitor LBH589 (panobinostat). Breast Cancer Res Treat (2013) 137(1):93–107. doi: 10.1007/s10549-012-2332-x
74. Fortunati N, Marano F, Bandino A, Frairia R, Catalano MG, Boccuzzi G. The pan-histone deacetylase inhibitor LBH589 (panobinostat) alters the invasive breast cancer cell phenotype. Int J Oncol (2014) 44(3):700–8. doi: 10.3892/ijo.2013.2218
75. Matossian MD, Burks HE, Elliott S, Hoang VT, Bowles AC, Sabol RA, et al. Panobinostat suppresses the mesenchymal phenotype in a novel claudin-low triple negative patient-derived breast cancer model. Oncoscience (2018) 5(3-4):99–108. doi: 10.18632/oncoscience.412
76. Qin G, Li Y, Xu X, Wang X, Zhang K, Tang Y, et al. Panobinostat (LBH589) inhibits wnt/beta-catenin signaling pathway via upregulating APCL expression in breast cancer. Cell Signal (2019) 59:62–75. doi: 10.1016/j.cellsig.2019.03.014
77. Wu K, Zhang H, Zhou L, Chen L, Mo C, Xu S, et al. Histone deacetylase inhibitor panobinostat in combination with rapamycin confers enhanced efficacy against triple-negative breast cancer. Exp Cell Res (2022) 421(1):113362. doi: 10.1016/j.yexcr.2022.113362
78. Wang X, Yin X. Panobinostat inhibits breast cancer progression via Vps34-mediated exosomal pathway. Hum Cell (2023) 36(1):366–76. doi: 10.1007/s13577-022-00812-3
79. Wu Y, Chen X, Wang L, Zhou X, Liu Y, Ji D, et al. Histone deacetylase inhibitor panobinostat benefits the therapeutic efficacy of oncolytic herpes simplex virus combined with PD-1/PD-L1 blocking in glioma and squamous cell carcinoma models. Viruses (2022) 14(12). doi: 10.3390/v14122796
80. Tatarova Z, Blumberg DC, Bensen A, Mills GB, Jonas O. Panobinostat induced spatial In situ biomarkers predictive of anti-PD-1 efficacy in mouse mammary carcinoma. Cells (2023) 12(2). doi: 10.3390/cells12020308
81. Boumber Y, Younes A, Garcia-Manero G. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin Investig Drugs (2011) 20(6):823–9. doi: 10.1517/13543784.2011.577737
82. Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother (2018) 67(3):381–92. doi: 10.1007/s00262-017-2091-y
83. Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget (2015) 6(32):33623–35. doi: 10.18632/oncotarget.5601
84. Goel RK, Lukong KE. Understanding the cellular roles of fyn-related kinase (FRK): implications in cancer biology. Cancer Metastasis Rev (2016) 35(2):179–99. doi: 10.1007/s10555-016-9623-3
85. Ogunbolude Y, Dai C, Bagu ET, Goel RK, Miah S, MacAusland-Berg J, et al. FRK inhibits breast cancer cell migration and invasion by suppressing epithelial-mesenchymal transition. Oncotarget (2017) 8(68):113034–65. doi: 10.18632/oncotarget.22958
86. Bagu ET, Miah S, Dai C, Spriggs T, Ogunbolude Y, Beaton E, et al. Repression of fyn-related kinase in breast cancer cells is associated with promoter site-specific CpG methylation. Oncotarget (2017) 8(7):11442–59. doi: 10.18632/oncotarget.14546
87. Shan W, Jiang Y, Yu H, Huang Q, Liu L, Guo X, et al. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am J Cancer Res (2017) 7(5):1213–26.
88. Khanjani F, Jafari L, Azadiyan S, Roozbehi S, Moradian C, Zahiri J, et al. Drug repositioning based on gene expression data for human HER2-positive breast cancer. Arch Biochem Biophys (2021) 712:109043. doi: 10.1016/j.abb.2021.109043
89. Kaya Cakir H, Eroglu O. In vitro anti-proliferative effect of capecitabine (Xeloda) combined with mocetinostat (MGCD0103) in 4T1 breast cancer cell line by immunoblotting. Iran J Basic Med Sci (2021) 24(11):1515–22. doi: 10.22038/IJBMS.2021.58393.12971
90. Rivera S, Leteur C, Megnin F, Law F, Martins I, Kloos I, et al. Time dependent modulation of tumor radiosensitivity by a pan HDAC inhibitor: abexinostat. Oncotarget (2017) 8(34):56210–27. doi: 10.18632/oncotarget.14813
91. Yang T, Wang P, Yin X, Zhang J, Huo M, Gao J, et al. The histone deacetylase inhibitor PCI-24781 impairs calcium influx and inhibits proliferation and metastasis in breast cancer. Theranostics (2021) 11(5):2058–76. doi: 10.7150/thno.48314
92. Salvador MA, Wicinski J, Cabaud O, Toiron Y, Finetti P, Josselin E, et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low xist expression. Clin Cancer Res (2013) 19(23):6520–31. doi: 10.1158/1078-0432.CCR-13-0877
93. de Cremoux P, Dalvai M, N’Doye O, Moutahir F, Rolland G, Chouchane-Mlik O, et al. HDAC inhibition does not induce estrogen receptor in human triple-negative breast cancer cell lines and patient-derived xenografts. Breast Cancer Res Treat (2015) 149(1):81–9. doi: 10.1007/s10549-014-3233-y
94. Hsu KW, Huang CY, Tam KW, Lin CY, Huang LC, Lin CL, et al. The application of non-invasive apoptosis detection sensor (NIADS) on histone deacetylation inhibitor (HDACi)-induced breast cancer cell death. Int J Mol Sci (2018) 19(2). doi: 10.3390/ijms19020452
95. Marijon H, Lee DH, Ding L, Sun H, Gery S, de Gramont A, et al. Co-Targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. BioMed Pharmacother (2018) 99:543–51. doi: 10.1016/j.biopha.2018.01.045
96. Zuo Y, Xu H, Chen Z, Xiong F, Zhang B, Chen K, et al. 17−AAG synergizes with belinostat to exhibit a negative effect on the proliferation and invasion of MDA−MB−231 breast cancer cells. Oncol Rep (2020) 43(6):1928–44. doi: 10.3892/or.2020.7563
97. Chi F, Liu J, Brady SW, Cosgrove PA, Nath A, McQuerry JA, et al. A;one-two punch’ therapy strategy to target chemoresistance in estrogen receptor positive breast cancer. Transl Oncol (2021) 14(1):100946. doi: 10.1016/j.tranon.2020.100946
98. Han XL, Du J, Zheng YD, Dai JJ, Lin SW, Zhang BY, et al. CXCL1 clone evolution induced by the HDAC inhibitor belinostat might be a favorable prognostic indicator in triple-negative breast cancer. BioMed Res Int (2021) 2021:5089371. doi: 10.1155/2021/5089371
99. Ganai SA. Strategy for enhancing the therapeutic efficacy of histone deacetylase inhibitor dacinostat: the novel paradigm to tackle monotonous cancer chemoresistance. Arch Pharm Res (2015). doi: 10.1007/s12272-015-0673-9
100. Looi CK, Gan LL, Sim W, Hii LW, Chung FF, Leong CO, et al. Histone deacetylase inhibitors restore cancer cell sensitivity towards T lymphocytes mediated cytotoxicity in pancreatic cancer. Cancers (2022) 14(15). doi: 10.3390/cancers14153709
101. Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res (2004) 64(18):6626–34. doi: 10.1158/0008-5472.CAN-04-0540
102. Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res (2006) 66(3):1277–81. doi: 10.1158/0008-5472.CAN-05-3632
103. Liu J, Shang G. The roles of noncoding RNAs in the development of osteosarcoma stem cells and potential therapeutic targets. Front Cell Dev Biol (2022) 10:773038. Cited in: Pubmed. doi: 10.3389/fcell.2022.773038
104. Chen T, Liu J, Zhang H, Li J, Shang G. Long intergenic noncoding RNA00265 enhances cell viability and metastasis via targeting miR-485-5p/USP22 axis in osteosarcoma. Front Oncol (2022) 12:907472. Cited in: Pubmed. doi: 10.3389/fonc.2022.907472
105. Su J, Deng L, Wang YD. Roles and mechanisms of long non-coding RNAs in breast cancer. Int J Mol Sci (2022) 24(1). doi: 10.3390/ijms24010089
106. Hurtubise A, Momparler RL. Effect of histone deacetylase inhibitor LAQ824 on antineoplastic action of 5-Aza-2’-deoxycytidine (decitabine) on human breast carcinoma cells. Cancer Chemother Pharmacol (2006) 58(5):618–25. doi: 10.1007/s00280-006-0225-6
107. Fiskus W, Ren Y, Mohapatra A, Bali P, Mandawat A, Rao R, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: A result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res (2007) 13(16):4882–90. doi: 10.1158/1078-0432.CCR-06-3093
108. Fuino L, Bali P, Wittmann S, Donapaty S, Guo F, Yamaguchi H, et al. Histone deacetylase inhibitor LAQ824 down-regulates her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone b. Mol Cancer Ther (2003) 2(10):971–84.
109. Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res (2009) 69(22):8693–9. doi: 10.1158/0008-5472.CAN-09-1456
110. Chen J, Li N, Liu B, Ling J, Yang W, Pang X, et al. Pracinostat (SB939), a histone deacetylase inhibitor, suppresses breast cancer metastasis and growth by inactivating the IL-6/STAT3 signalling pathways. Life Sci (2020) 248:117469. doi: 10.1016/j.lfs.2020.117469
111. Zhang T, Chen Y, Li J, Yang F, Wu H, Dai F, et al. Antitumor action of a novel histone deacetylase inhibitor, YF479, in breast cancer. Neoplasia (2014) 16(8):665–77. doi: 10.1016/j.neo.2014.07.009
112. Li ZH, Zhang XB, Han XQ, Feng CR, Wang FS, Wang PG, et al. Antitumor effects of a novel histone deacetylase inhibitor NK-HDAC-1 on breast cancer. Oncol Rep (2013) 30(1):499–505. doi: 10.3892/or.2013.2434
113. Pinkerneil M, Hoffmann MJ, Deenen R, Kohrer K, Arent T, Schulz WA, et al. Inhibition of class I histone deacetylases 1 and 2 promotes urothelial carcinoma cell death by various mechanisms. Mol Cancer Ther (2016) 15(2):299–312. doi: 10.1158/1535-7163.MCT-15-0618
114. Savino AM, Sarno J, Trentin L, Vieri M, Fazio G, Bardini M, et al. The histone deacetylase inhibitor givinostat (ITF2357) exhibits potent anti-tumor activity against CRLF2-rearranged BCP-ALL. Leukemia (2017) 31(11):2365–75. doi: 10.1038/leu.2017.93
115. Marampon F, Leoni F, Mancini A, Pietrantoni I, Codenotti S, Ferella L, et al. Histone deacetylase inhibitor ITF2357 (givinostat) reverts transformed phenotype and counteracts stemness in in vitro and in vivo models of human glioblastoma. J Cancer Res Clin Oncol (2019) 145(2):393–409. doi: 10.1007/s00432-018-2800-8
116. Angeletti F, Fossati G, Pattarozzi A, Wurth R, Solari A, Daga A, et al. Inhibition of the autophagy pathway synergistically potentiates the cytotoxic activity of givinostat (ITF2357) on human glioblastoma cancer stem cells. Front Mol Neurosci (2016) 9:107. doi: 10.3389/fnmol.2016.00107
117. Kozyreva VK, McLaughlin SL, Livengood RH, Calkins RA, Kelley LC, Rajulapati A, et al. NEDD9 regulates actin dynamics through cortactin deacetylation in an AURKA/HDAC6-dependent manner. Mol Cancer Res (2014) 12(5):681–93. doi: 10.1158/1541-7786.MCR-13-0654
118. Yuan YG, Peng QL, Gurunathan S. Combination of palladium nanoparticles and tubastatin-a potentiates apoptosis in human breast cancer cells: a novel therapeutic approach for cancer. Int J Nanomed (2017) 12:6503–20. doi: 10.2147/IJN.S136142
119. Hsieh YL, Tu HJ, Pan SL, Liou JP, Yang CR. Anti-metastatic activity of MPT0G211, a novel HDAC6 inhibitor, in human breast cancer cells in vitro and in vivo. Biochim Biophys Acta Mol Cell Res (2019) 1866(6):992–1003. doi: 10.1016/j.bbamcr.2019.03.003
120. Januchowski R, Dabrowski M, Ofori H, Jagodzinski PP. Trichostatin a down-regulate DNA methyltransferase 1 in jurkat T cells. Cancer Lett (2007) 246(1-2):313–7. doi: 10.1016/j.canlet.2006.03.010
121. Luczak MW, Jagodzinski PP. Trichostatin a down-regulates CYP19 transcript and protein levels in MCF-7 breast cancer cells. BioMed Pharmacother (2009) 63(4):262–6. doi: 10.1016/j.biopha.2008.05.002
122. Drzewiecka H, Jagodzinski PP. Trichostatin a reduced phospholipase c gamma-1 transcript and protein contents in MCF-7 breast cancer cells. BioMed Pharmacother (2012) 66(1):1–5. doi: 10.1016/j.biopha.2011.09.005
123. Lee KW, Kim JH, Park JH, Kim HP, Song SH, Kim SG, et al. Antitumor activity of SK-7041, a novel histone deacetylase inhibitor, in human lung and breast cancer cells. Anticancer Res (2006) 26(5A):3429–38.
124. Hou S, Sang M, Zhao L, Hou R, Shan B. The expression of MAGE-C1 and MAGE-C2 in breast cancer and their clinical significance. Am J Surg (2016) 211(1):142–51. doi: 10.1016/j.amjsurg.2015.05.028
125. Sun Y, Hong JH, Ning Z, Pan D, Fu X, Lu X, et al. Therapeutic potential of tucidinostat, a subtype-selective HDAC inhibitor, in cancer treatment. Front Pharmacol (2022) 13:932914. doi: 10.3389/fphar.2022.932914
126. Shi Y, Xu S, Li S. Selinexor improves the anti-cancer effect of tucidinostat on TP53 wild-type breast cancer. Mol Cell Endocrinol (2022) 545:111558. doi: 10.1016/j.mce.2022.111558
127. Zhou J, Wu X, Zhang H, Wang X, Yuan Y, Zhang S, et al. Clinical outcomes of tucidinostat-based therapy after prior CDK4/6 inhibitor progression in hormone receptor-positive heavily pretreated metastatic breast cancer. Breast (2022) 66:255–61. doi: 10.1016/j.breast.2022.10.018
128. Pathania R, Ramachandran S, Mariappan G, Thakur P, Shi H, Choi JH, et al. Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res (2016) 76(11):3224–35. doi: 10.1158/0008-5472.CAN-15-2249
129. Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schioth HB. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br J Clin Pharmacol (2021) 87(12):4577–97. doi: 10.1111/bcp.14889
130. Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anticancer Drugs (2014) 25(2):140–9. doi: 10.1097/CAD.0000000000000040
131. Wu QJ, Zhang TN, Chen HH, Yu XF, Lv JL, Liu YY, et al. The sirtuin family in health and disease. Signal Transduct Target Ther (2022) 7(1):402. doi: 10.1038/s41392-022-01257-8
132. Ganai SA. Novel approaches towards designing of isoform-selective inhibitors against class II histone deacetylases: The acute requirement for targetted anticancer therapy. Curr Top Med Chem (2016) 16(22):2441–52. doi: 10.2174/1568026616666160212122609
Keywords: HDAC, inhibitors, breast cancer, immunotherapy, targets
Citation: Lian B, Chen X and Shen K (2023) Inhibition of histone deacetylases attenuates tumor progression and improves immunotherapy in breast cancer. Front. Immunol. 14:1164514. doi: 10.3389/fimmu.2023.1164514
Received: 12 February 2023; Accepted: 27 February 2023;
Published: 09 March 2023.
Edited by:
Xiangpeng Dai, Jilin University, ChinaReviewed by:
Xueqiong Zhu, Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, ChinaCopyright © 2023 Lian, Chen and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaosong Chen, Y2hlbnhpYW9zb25nMDE1NkBob3RtYWlsLmNvbQ==; Kunwei Shen, a3dzaGVuQG1lZG1haWwuY29tLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.