 Jingjing Ye
Jingjing Ye Xiaodan Hu
Xiaodan Hu Zhiwei Wang3
Zhiwei Wang3 Lebin Gan
Lebin Gan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 July 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1164187
This article is part of the Research Topic Mitochondrial Dysfunction in Inflammation and Autoimmunity View all 5 articles
Systemic inflammatory response syndrome (SIRS) is a non-specific exaggerated defense response caused by infectious or non-infectious stressors such as trauma, burn, surgery, ischemia and reperfusion, and malignancy, which can eventually lead to an uncontrolled inflammatory response. In addition to the early mortality due to the “first hits” after trauma, the trauma-induced SIRS and multiple organ dysfunction syndrome (MODS) are the main reasons for the poor prognosis of trauma patients as “second hits”. Unlike infection-induced SIRS caused by pathogen-associated molecular patterns (PAMPs), trauma-induced SIRS is mainly mediated by damage-associated molecular patterns (DAMPs) including mitochondrial DAMPs (mtDAMPs). MtDAMPs released after trauma-induced mitochondrial injury, including mitochondrial DNA (mtDNA) and mitochondrial formyl peptides (mtFPs), can activate inflammatory response through multiple inflammatory signaling pathways. This review summarizes the role and mechanism of mtDAMPs in the occurrence and development of trauma-induced SIRS.
Systemic inflammatory response syndrome (SIRS) is a non-specific exaggerated defense response caused by infectious or non-infectious stressors such as trauma, burn, surgery, ischemia and reperfusion, and malignancy, which can eventually lead to an uncontrolled inflammatory response (1). In trauma medicine, SIRS can be regarded as an independent predictor of mortality after trauma (2). Moreover, trauma-induced SIRS increases susceptibility to infections, which can lead to multiple organ dysfunction syndrome (MODS), which is often detrimental and leads to poor prognosis in trauma patients (3). Therefore, understanding the role and mechanism of trauma-induced SIRS may provide new perspectives for clinical diagnosis, treatment, and scientific research.
Early studies indicate that trauma-induced tissue damage leads to pathogen invasion and the release of pathogen-associated molecular patterns (PAMPs). The binding of specific pattern recognition receptors (PRRs) by PAMPs activates the innate immune system, a prerequisite step for generating immunogenic signals that ultimately lead to infectious SIRS (4). Gradually, researchers found that SIRS is not caused by pathogens (5) but rather by the endogenous damage-associated molecular patterns (DAMPs) released by tissue injury in many trauma patients. Just like PAMPs, DAMPs can bind to PRRs, activate the immune system, and cause further tissue damage. It is largely accepted that DAMPs can be sensed by several classical PRRs, including Toll-like receptors (TLRs), C-type lectin receptors (CLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), NOD-like receptors (NLRs), and multiple intracellular DNA sensors (6–8). DAMPs are mainly derived from the plasma membrane, nucleus, cytosol, and mitochondria (9). Mitochondrial DAMPs (mtDAMPs) mainly include mitochondrial DNA (mtDNA), mitochondrial formyl peptides (mtFPs), mitochondrial transcription factor A (TFAM), cardiolipin (CL), ATP, cytochrome c, and mitochondrial RNA (mtRNA), which are also related to trauma-induced SIRS (5).
Trauma causes mitochondrial damage and dysfunction, leading to mtDAMP release and the induction of an immune response similar to that against pathogen infection (10). This phenomenon is theoretically supported by the danger model of immune recognition and the endosymbiotic theory. Matzinger’s danger model theory points out that the activation of the body’s immune system occurs through the recognition of danger signals released by damaged tissues rather than by the recognition of non-self-molecules (11). These endogenous danger signals are called DAMPs, which normally avoid contact with the immune system because of the plasma membrane and intracellular compartmentalization (12). When cells and tissues are damaged, DAMPs are released into the extracellular space or circulation (12). According to the endosymbiotic theory, the mitochondria evolved from ancient bacterial endosymbiont; therefore, the endogenous molecules released by the mitochondria are similar to bacterial PAMPs, which mediate inflammatory reactions similar to bacterial infections. This review focuses on the research progress of mtDAMPs in trauma-induced SIRS and summarizes the development and pathophysiology at the cellular and organ levels.
Human mtDNA is a closed-circular double-stranded molecule coding 37 genes. Unlike nuclear DNA, mtDNA can be easily damaged by the lack of repair systems and histone protection (13). Similar to bacterial DNA, mtDNA contains unmethylated CpG repeats that are recognized by the immune system as non-self. Post-traumatic mitochondrial damage causes mtDNA released outside the mitochondria (10, 14, 15). A recent study in a trauma cohort from Briggs GD showed that the size of mtDNA in the circulation post-trauma is a mixture of “larger forms” and low molecular weight mtDNA, and it was the low-molecular-weight version of cell-free mtDNA that is associated with inflammation and poor clinical outcomes post-trauma (16). Free mtDNA mediates inflammation through a variety of signaling pathways (17).
As a PRR, Toll-like receptor 9 (TLR9) can directly bind to CpG DNA. After the stimulation by CpG DNA, TLR9 and its intracellular adaptor protein myeloid differentiation factor 88 (MyD88) localize in the endoplasmic reticulum, rapidly redistribute to the CpG DNA accumulation site, and transfer to the endosomal membrane and lysosomal compartment (18).
MtDNA/TLR9 signaling pathway has been demonstrated in various cell types, such as dendritic cells, neutrophils, macrophages, and natural killer cells (19). Stimulated TLR9 binds to MyD88 to activate the nuclear factor kappa-B (NF-κB) and mitogen-activated protein kinase (MAPK) cascades (6). The activation of NF-κB induces the expression of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and other proinflammatory cytokines (6). The activation of MAPK cascades induces activator protein-1 (AP-1) formation and promotes cytokine expression (6). In plasmacytoid dendritic cells, TLR9/MyD88 activates interferon regulatory factor 7 (IRF7), which transfers to the nucleus, and induces the expression of type I interferon (20). In contrast, in myeloid dendritic cells and macrophages, TLR9/MyD88 activates IRF1 and induces the expression of interferon-β (IFN-β) (21). In addition, mtDNA activates neutrophils through TLR9, causing Ca2+ influx and MAPK phosphorylation, mediating neutrophil degranulation and migration, and inducing inflammation (10).
Cyclic GMP-AMP synthase (cGAS) and interferon gene stimulator (STING) are widely expressed in mammalian cells that mediate the expression of type I interferon and other cytokines in infections and inflammatory diseases (22). cGAS is mainly limited in the cytoplasm to avoid the continuous activation of its DNA in the nucleus. However, recent studies have demonstrated that cGAS also exists in the nucleus (23) and plasma membrane (24).
cGAS binds to mtDNA and catalyzes the generation of 2′-3′ cyclic GMP-AMP (cGAMP) from its substrates GTP and AMP (25). The secondary messenger cGAMP binds to STING in the endoplasmic reticulum (ER) membrane, thereby promoting STING conformational change and forming dimers (26). The activated STING is transferred to the ER–Golgi intermediates and Golgi by vesicle transport (27). In this process, STING triggers inflammatory storms and interferes with autophagy in an interferon-dependent manner (28). There are two main downstream pathways of STING (29, 30). In the first pathway, STING directly binds and phosphorylates TANK-binding kinase 1 (TBK1), mediates the phosphorylation and nuclear transfer of IRF3, and initiates the expression of type I IFN. In the second pathway, STING directly binds and phosphorylates the inhibitor of κB kinase (IKK) complex, activates NF-κB, and promotes the expression of cytokines, such as IL-1β, IL-6, and TNF-α.
Nucleotide oligomerization domain-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome is a macromolecular complex composed of NLRP3, caspase-1, and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) (31). It is usually present in neutrophils, monocytes, dendritic cells, macrophages, and non-hematopoietic cells (32). A study indicated that NLRP3 inflammasomes could be activated through three different signaling pathways: canonical NLRP3 activation, non-canonical NLRP3 activation, and alternative NLRP2 activation (33). The typical NLRP3 activation pathway involves two steps, namely, initiation and activation. The initiation step revolves around TLR recognition of PAMP or DAMP, activation of NF-κB, synthesis of IL-1β and IL-18 precursor, and expression of NLRP3 (34). However, the exact mechanism of the activation step remains unclear. Several NLRP3 agonists trigger the activation of NLRP3 inflammasome; however, these agonists are not chemically or structurally related, and there is little evidence regarding whether NLRP3 directly binds them (33). A possible explanation is that NLRP3 agonist-mediated molecular and cellular events (including K+ outflow, mitochondrial dysfunction, reactive oxygen species (ROS) and mtDNA release, and lysosomal destruction) are upstream signals for inflammasome assembly and activation (35, 36). However, these events do not apply to all NLRP3 agonists, and a consistent NLRP3 activation pathway is lacking (35).
Unlike cGAS, which binds to both oxidized and non-oxidized mtDNAs, NLRP3 prefers oxidized mtDNA (ox-mtDNA) (13, 37). Ox-mtDNA exits the mitochondria through mitochondrial permeability transition pores and voltage-dependent ion channels and acts as a ligand to activate the NLRP3 inflammasome (38). Extracellular mtDNA does not serve as an NLRP3 ligand directly, while it triggers the TLR9/MAPK/NF-κB pathway to induce the initiation and activation of NLRP3 (39). After NLRP3 activation, the activated caspase-1 processes IL-1β and IL-18 precursor into a mature secreted form. Moreover, it cleaves gasdermin D (GSDMD) that is subsequently transferred to the cell membrane to form pores, thereby mediating the release of proinflammatory cytokines and causing pyroptosis (40). In addition, gasdermin D promotes mitochondrial fragmentation and mtDNA release (41).
Zhong et al. confirmed that the newly synthesized mtDNA is indispensable for NLRP3 inflammasome activation (42). TLR activation triggers the IRF1-dependent transcription of CMPK2, which acts as a rate-limiting enzyme to catalyze mtDNA synthesis after exposure to NLRP3 agonists (42). The newly synthesized mtDNA is oxidized by mitochondrial ROS to generate ox-mtDNA, which subsequently triggers NLRP3 inflammasome activation (42). Xian et al. stimulated mice with myeloid-specific knockout of cytidine/uridine monophosphate kinase (CMPK2) with lipopolysaccharide (LPS) and observed decreased NLRP3 inflammasome activation and blocked IL-1β secretion in mouse alveolar macrophages, which is consistent with the results of Zhong et al. (42, 43). However, in the study on severe fever caused by thrombocytopenia syndrome virus infection, ox-mtDNA activated NLRP3 inflammasome, but no significant new mtDNA synthesis was observed (44). Therefore, whether newly synthesized mtDNA is necessary for NLRP3 inflammasome activation remains unclear.
MtFPs are proteins synthesized from mtDNA, and like in bacteria, N-formyl methionine is the first amino acid during translation initiation. Severe trauma causes mitochondrial damage and the release of mtFPs into the blood circulation, where they bind and activate formyl peptide receptor (FPR), and recruit immune cells to mediate inflammatory response (45). FPR1, FPR2, and FPR3 are three human FPRs (5) expressed in multiple cell types. The highest expression levels of FPR1 and FPR2 have been detected in neutrophils, and those of FPR3 have been detected in mononuclear macrophages (46). Among FPR ligands, mtFPs are the only ligand common to all human FPRs (46).
Downstream signaling pathways after FPR activation remain unclear. Hazeldine et al. demonstrated that mtFPs activate neutrophils through extracellular signal-related kinase 1/2 (ERK1/2) and MAPK signaling pathways (47). MtFP-stimulated FPR1 induces chemotaxis, degranulation, and Ca2+ outflow of the polymorphonuclear neutrophils (PMNs); promotes ROS and proinflammatory cytokine production; and enhances cytoskeletal rearrangement and network formation (5, 48). Liu et al. discovered that FPR2 can directly interact with transforming growth factor-β-activated kinase 1 (TAK1), thereby enhancing inflammation and oxidative stress related to Nrf2 activation (49). Lee et al. confirmed the presence of FPR3 in neutrophils; moreover, the use of FPR3 agonists could activate neutrophils, inhibit inflammatory cytokines generation, and kill bacteria through ROS generation (50). Studies have discovered that the binding of mtFPs to PMN stimulating FPR1 leads to the desensitization and inline of multiple PMN chemokine receptors, thereby reducing the number of PMNs that can migrate to secondary infection sites (51, 52). Therefore, the blockage of FPR1 not only protects the receptors from desensitization but also preserves the immune response at the injection site (triggered by the stimulation of monocytes with PAMP), which improves overall anti-pathogen efficacy and diminishes SIRS (5).
TFAM is abundant in the mitochondria and plays a key role in stabilizing the mtDNA structure and protecting mitochondrial function (13). TFAM damage has a dual role in the inflammatory response. On the one hand, TFAM damage disrupts mtDNA stability, leading to mtDNA escape and inflammatory response through multiple signaling pathways (53). On the other hand, TFAM, as a DAMP, enhances the secretion of proinflammatory cytokines (54).
mtRNA, synthesized through mtDNA transcription, can also lead to inflammation because of abnormal accumulation and release. Mitochondrial ssRNA can activate TLR8/MyD88 signaling pathway (55). After recognition by RIG-I, dsRNA activates NF-κB and IRF3/7 through MAVS to induce type I IFN expression (56). However, dsRNA cannot activate the type I IFN response in all cells, such as human islet β-cells (57).
CL is a crucial phospholipid in the bacterial membrane and mitochondrial inner membrane and plays a pivotal role in maintaining the electron transport chain, mitophagy, and apoptosis (58). Chen et al. discovered that the loss of Sam50 (which connects the mitochondrial inner and outer membrane) led to CL externalization. This causes mitochondrial membrane remodeling, mtDNA aggregation, and release through Bax/Bak mitochondrial pore, mtDNA/cGAS/STING signaling pathway activation, and hepatitis damage (59).
Extracellular ATP (eATP) induces the generation and release of IL-1β, IL-6, and TNF-α by activating phospholipase A2/D, MAPK, NF-κB, and other pathways (60). In the mouse model of acute pancreatitis, the levels of eATP increased and promoted proinflammatory cytokine production and induced SIRS by activating MAPK and NF-κB signaling pathways (61).
Cytochrome c is released from damaged cells into the extracellular space and acts as DAMP to trigger the inflammatory response. Wenzel et al. discovered that cytochrome c induced the inflammatory activation of microglia through the TLR4 signaling pathway (62). Moreover, Pullerits et al. discovered that extracellular cytochrome c triggered neutrophil-mediated and monocyte-mediated inflammation through the NF-κB signaling pathway (63).
Multiple signaling pathways of mtDAMPs that mediate inflammatory response interact with each other.
MtDAMPs mediate proinflammatory cytokine release and form positive feedback, leading to further release of mtDAMPs from the mitochondria and exacerbation of the inflammatory response. Aarreberg et al. demonstrated that exogenous IL-1β promotes mitochondrial aggregation in bystander cells, such as fibroblasts and epithelial cells, lowers mitochondrial membrane potential, and induces mtDNA release, thereby activating the cGAS/STING-dependent type I IFN response (64). Willemsen et al. discovered that long-term TNF stimulation also triggers the release of mtDNA and induces type I IFN response through cGAS/STING (65). In diABZIP (a kind of STING agonist)-administered mice, STING activation-induced cell death and mtDNA release, thereby activating the cGAS/STING signaling pathway (66). The excessive activation of STING amplifies the inflammatory cycle (67).
Post-traumatic tissue damage leads to mtDAMP release from the mitochondria, which promotes inflammatory cytokine release through a variety of signaling pathways and mediates inflammatory response. Clinical studies have demonstrated that the plasma mtDNA levels of trauma patients were significantly higher than those of healthy individuals (10, 67); the levels continued to increase 24 h after injury (10), indicating that plasma mtDNA has a moderate discriminative power in predicting the risk of SIRS after trauma (68). In addition, the plasma mtFP levels of trauma patients with SIRS and sepsis were higher than those of controls (69). Hu et al. claimed that plasma mtDNA concentration was remarkably high in patients with intraperitoneal infection and MODS; moreover, the baseline plasma mtDNA concentration at admission could effectively predict their prognosis (70). Martinez-Quinones et al. evaluated critical patients who had undergone open laparotomy and discovered that peritoneal lavage reduced the level of mtDAMPs in ascites (71). They proposed that increasing the frequency of peritoneal lavage may decrease systemic absorption of mtDAMPs, thereby reducing the risk of aseptic SIRS (71).
One of the features of the SIRS response is a widespread inflammatory response, defined in part by immune cell activation and the production of proinflammatory cytokines. In-depth studies have demonstrated that mtDAMPs including mtDNA/mtFPs can activate innate immune cells such as antigen-presenting cells, macrophages, neutrophils, and dendritic cells play crucial roles in recognizing, phagocytosing, and releasing inflammatory mediators (8). For example, via PRRs activating antigen-presenting cells and neutrophils, the production of local ROS, cytokines, chemokines, MMPs, and NETs increased (8, 72–74). Moreover, injection of DAMPs into rodents has been shown to be associated with organ damage, while patients who have high levels of mtDNA in their circulation when sampled 2 h post-injury are at higher risk of developing MODS (75). Therefore, scavenging DAMPs may help alleviate the proinflammatory response triggered by DAMPs. For example, Lee et al. have demonstrated that nucleic acid scavenging microfiber meshes represent an effective strategy to inhibit trauma-induced inflammation and thrombosis in vitro and in vivo (76). Moreover, Aswani et al. have shown in vitro that the use of nucleic acid scavenging polymers, for example, hexadimethrine bromide (HDMBr), can reduce circulating mtDAMP levels and reduce the severity of organ injury in rat hemorrhagic shock models (75).
SIRS often involves multiple organ injuries and inflammation such as acute lung injury (ALI), acute respiratory distress syndrome (ARDS), acute kidney injury (AKI), and traumatic brain injury (TBI) (8). The following describes the latest research on mtDAMPs in trauma-induced SIRS at the organ level. The mechanisms and therapeutic targets of mtDAMPs in various organs of trauma-induced SIRS are also summarized in Tables 1, 2.
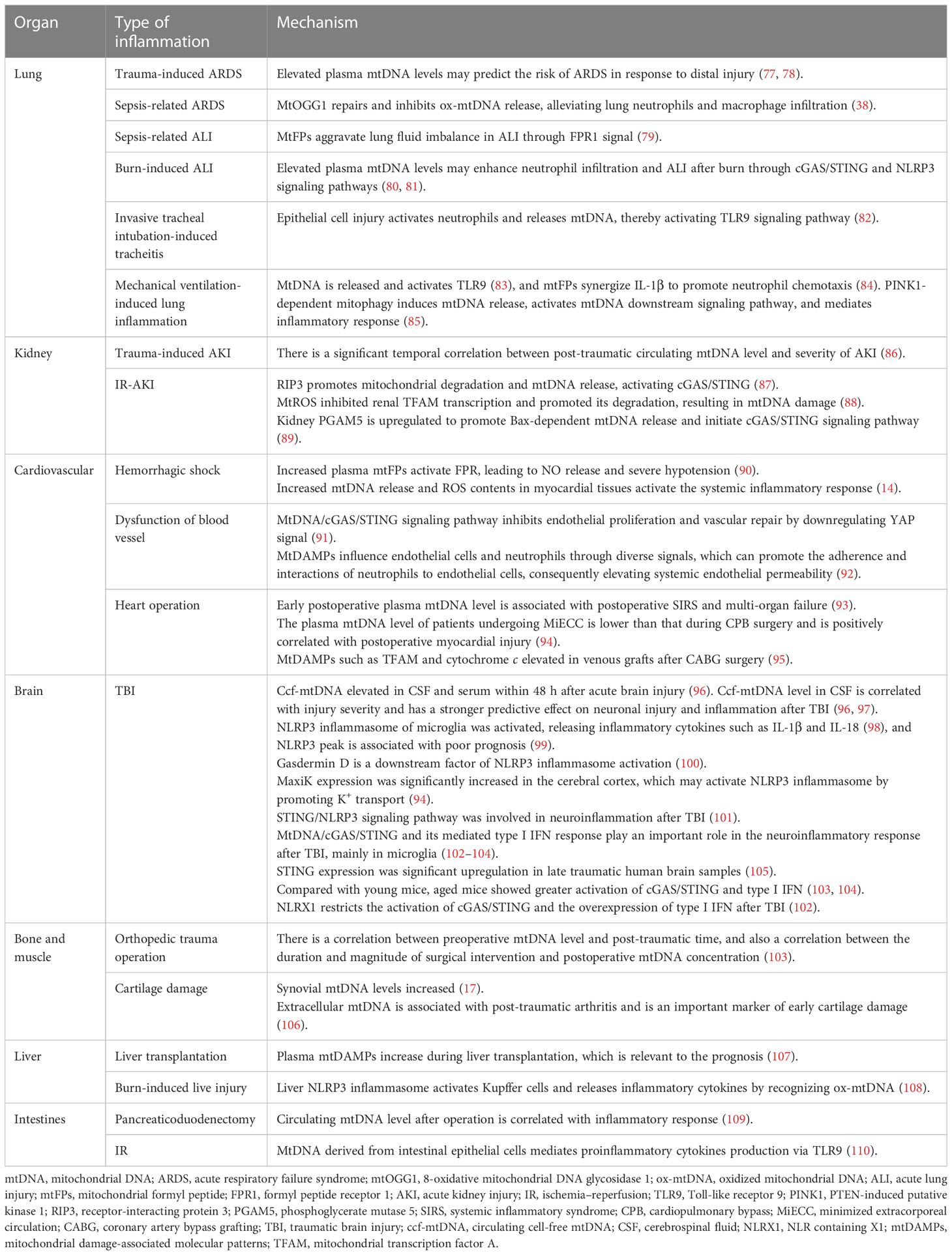
Table 1 Mechanisms of post-trauma organ inflammation induced by mtDAMPs.
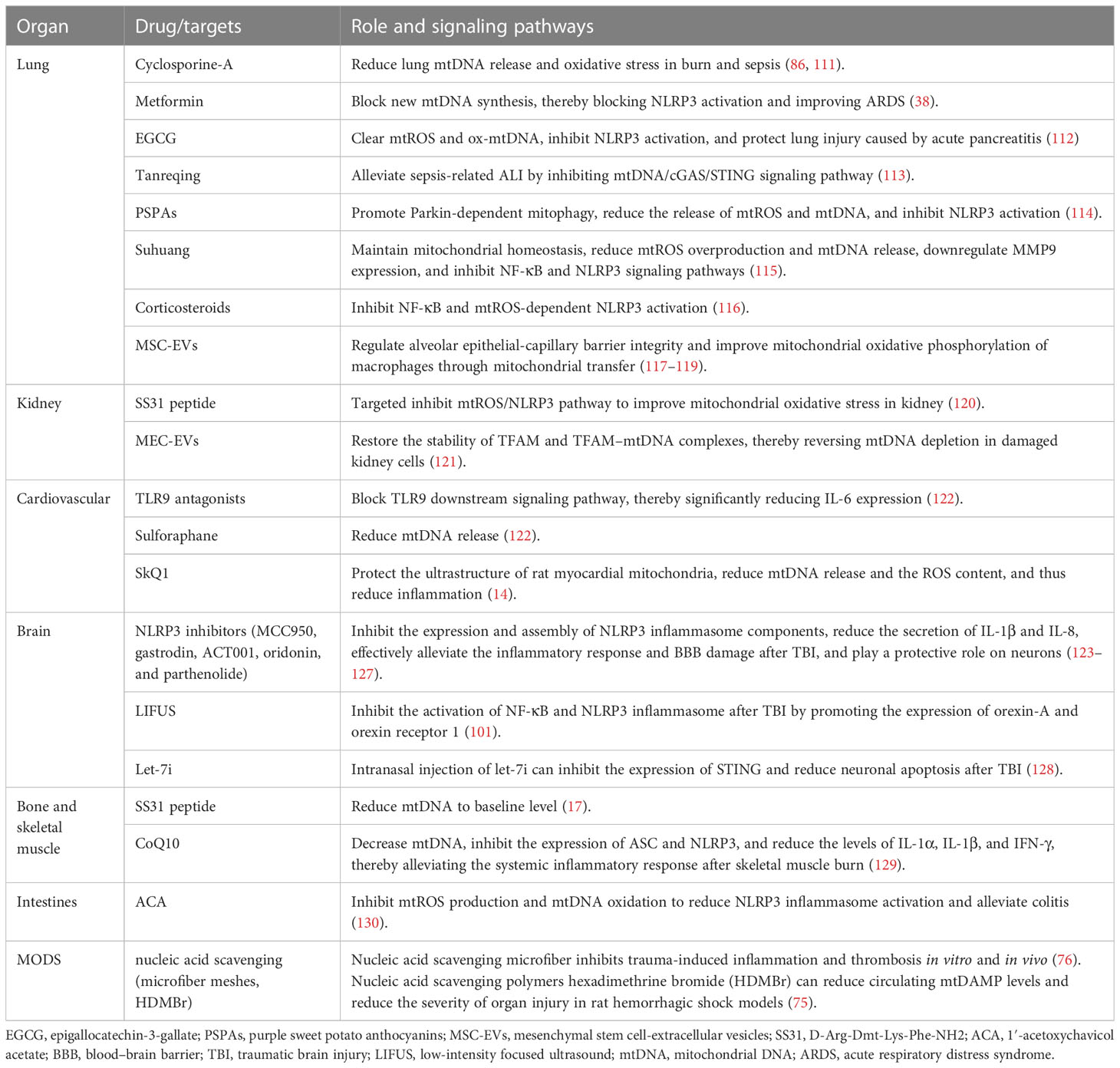
Table 2 Therapeutic measures targeting mtDAMPs and the signaling pathways.
ARDS is an acute inflammatory lung injury. Clinical studies have discovered that plasma mtDNA levels correlate with ARDS severity in trauma patients and can also predict the risk of ARDS during distal injury (77, 78).
MtFPs aggravate lung fluid imbalance through the FPR1 signaling pathway (79). In vivo experiments of burn-induced ALI mice revealed that elevated plasma mtDNA levels may enhance neutrophil infiltration and post-burn ALI through cGAS/STING and NLRP3 signaling pathways (80, 81).
Acute tracheitis is often secondary to invasive endotracheal intubation because of epithelial cell damage. This activates neutrophils and mtDNA release, which mediate proinflammatory cytokine secretion through the mtDNA/TLR9/NF-κB signaling pathway (82). In addition, mechanical ventilation can lead to excessive lung traction and mechanical damage, resulting in the activation of the TLR9/MyD88/NF-κB signaling pathway (83). MtFPs cooperate with IL-1β to promote neutrophil chemotaxis (84), thereby aggravating inflammation and lung injury. In the cyclic stretching cell culture model constructed by Jing et al., the overexpression of PTEN-induced putative kinase 1 (PINK1) in lung epithelial cells exacerbated stretch-induced inflammatory response (85) through PINK1-dependent mitophagy to induce mtDNA release (85). Therefore, the inhibition of mitophagy and mtDNA-mediated TLR9/MyD88/NF-κB signaling pathway may be a potential therapeutic approach for lung injury caused by mechanical ventilation (85). Xian et al. discovered that 8-oxoguanine DNA glycosylase 1 (mtOGG1), an mtDNA base excision repair enzyme, repaired ox-mtDNA and inhibited its release, thereby alleviating pulmonary vascular endothelial injury and infiltration of neutrophil and macrophage; this provided resistance to LPS-induced ARDS in mtOGG1 transgenic mice (38).
The inhibiting of mtDAMP synthesis and release, blockage of downstream signaling pathways of mtDAMPs, and maintenance of mitochondrial homeostasis may control pulmonary inflammation. Cyclosporine-A attenuates oxidative stress and mtDNA release in lung tissue in a dose-dependent manner, thereby exerting a protective effect on both burn-induced ALI and LPS-induced ALI (86, 111). Xian et al. discovered that metformin ameliorated ARDS by inhibiting mtDNA synthesis and blocking NLRP3 inflammasome activation (43). Epigallocatechin-3-gallate (EGCG) may inhibit NLRP3 inflammasome activation by clearing mtROS and ox-mtDNA, thereby protecting against lung injury caused by acute pancreatitis (112). In a study, Tanreqing significantly alleviated LPS-induced ALI by inhibiting the mtDNA/cGAS/STING signaling pathway (113). Dong et al. reported that purple sweet potato anthocyanins (PSPAs) inhibited NLRP3 inflammasome activation by promoting parkin-dependent mitophagy and reducing the release of mtROS and mtDNA; this resulted in reduced lung inflammation and mortality of Klebsiella pneumoniae-infected mice (114). Suhuang exhibited a positive effect on mitochondrial homeostasis in ALI mice by reducing mtROS overproduction and mtDNA release, downregulating MMP9 expression, and inhibiting NF-κB and NLRP3 inflammasome activation (115). Corticosteroids protect against inflammation response and ALI by inhibiting the NF-κB signaling pathway and mtROS-dependent NLRP3 inflammasome activation (116). Mesenchymal stem cell-extracellular vesicles (MSC-EVs) regulate alveolar epithelial–capillary barrier integrity through mitochondrial transfer; this restores metabolic and immune homeostasis of airway macrophages and thereby reduces the release of alveolar mtDNA, which effectively alleviates lung inflammation and improves organ function (117–119).
In critically injured patients, the continuous monitoring of circulating mtDNA levels within 48 h after trauma has a significant temporal correlation with AKI (131). AKI is frequent among patients with severe burns and is associated with high mortality (132). Although the incidence of AKI is low, late AKI is severe and is a poor prognostic factor for severe burns (132).
According to Feng et al., receptor-interacting protein 3 (RIP3) promotes mitochondrial degradation and mtDNA release, which activates the cGAS/STING signaling pathway and exacerbates inflammation and kidney injury after renal IR (87). Zhao et al. noted renal TFAM deficiency and mtDNA damage in patients with IR-AKI; in the mouse model, mtROS disrupted TFAM and mtDNA homeostasis by inhibiting renal TFAM transcription and promoting its degradation, thereby driving mtDNA release and renal inflammatory response (88). Li et al. discovered that phosphoglycerate mutase 5 (PGAM5) was upregulated in the kidneys of AKI human biopsy samples and mouse models; the upregulation promoted Bax-dependent mtDNA release and initiated the mtDNA/cGAS/STING signaling pathway (89).
In a cisplatin-induced AKI mouse model, mitochondrial-targeted therapy with SS31 peptide (D-ARG-DMT-lys-Ph-NH2, a mitochondrial targeting antioxidant) improved renal oxidative stress by inhibiting the mtROS/NLRP3 signaling pathway (120). In addition to lung inflammation alleviation, MSC-EVs play an active role in kidney inflammation. Zhao et al. reported that the application of MSC-EVs restored the stability of TFAM and TFAM–mtDNA complexes, thereby reversing damage caused by mitochondrial oxidative phosphorylation in renal tubular cells and alleviating kidney inflammation (121). However, intravenous injection of MSC-EVs into mice with weakened TFAM expression had poor efficacy, and TFAM overexpression had better efficacy (121). These results suggest that MSC-EVs are a promising nanotherapy for diseases with mitochondrial damage; the TFAM signaling pathway is essential for maintaining mitochondrial regenerative capacity (121).
AKI can damage distal organs such as the lung. Hepokoski et al. discovered that mitochondrial dysfunction occurred in the lungs and systemic circulation of IR-AKI mice, leading to an increase in extracellular mtDNA and TFAM levels and enhanced infiltration of pulmonary neutrophils (133). Intraperitoneal injection of renal-derived mtDAMPs resulted in metabolomic changes caused by lung mitochondrial dysfunction in vivo (133). Therefore, mitochondrial function and mtDAMPs may be potential therapeutic targets for preventing AKI-related lung injury (133).
MtFPs mediate inflammation through FPR activation, also dilate resistant arteries, and induce vascular endothelial cell dysfunction in a blood-dependent manner (90). Specifically, the collapse of blood vessels is the primary pathophysiology feature of the sepsis-like syndrome. F-MIT does not affect the arteries’ relaxation induced by acetylcholine. However, arteries incubated in blood containing F-MIT or blood from rats treated with F-MIT show reduced relaxation compared to their respective control groups. These findings indicate that F-MIT induces blood-dependent endothelial dysfunction (90). In a rat model of hemorrhagic shock, mtFPs lead to NO release and severe hypotension through FPR, and elevated plasma mtFP levels were associated with aseptic trauma-induced ALI (90). Moreover, the activation of the mtDNA/cGAS/STING signaling pathway inhibits endothelial proliferation and vascular repair by downregulation of the YAP signaling pathway (91). Therefore, mtFPs may serve as a bridge between trauma, SIRS, and cardiovascular failure (90). Moreover, the release of mtDAMPs into the bloodstream can occur through various mechanisms, causing cellular damage and resulting in increased pathological endothelial permeability. Sun et al. demonstrated that the mitochondria contain numerous DAMP motifs capable of influencing endothelial cells and neutrophils through diverse signals, which can promote the adherence and interactions of neutrophils to endothelial cells, consequently elevating systemic endothelial permeability (92). Recent research by our team has also identified myocardial mitochondrial structure disruption in the rat model of hemorrhagic shock (HS), resulting in increased mtDNA release and ROS contents, which in turn activates the systemic inflammatory response (131). More importantly, we found the mitochondrial antioxidant SkQ1 can protect myocardial mitochondria, improve the ultrastructure of rat myocardial mitochondria, reduce mtDNA release and the ROS content, and thus reduce inflammation (14). Cardiopulmonary bypass, surgical trauma, and ischemia–reperfusion injury stimulate a systemic inflammatory response in cardiac surgery. Early postoperative plasma mtDNA level is a predictive marker for SIRS and multi-organ failure in patients undergoing cardiac surgery (93). The elevated plasma mtDNA level during cardiopulmonary bypass (CPB) surgery may be involved in SIRS pathogenesis and the related postoperative inflammatory events (such as postoperative atrial fibrillation and infection) (67). Compared with traditional CPB, minimally invasive extracorporeal circulation (MiECC) results in a lower plasma mtDNA level, positively correlated with CPB duration and postoperative myocardial injury (94). Naase et al. further confirmed that mtDNA-mediated inflammation through TLR9 and TLR9 antagonist administration significantly reduced IL-6 expression (122). The antioxidant sulforaphane reduces mtDNA release and may have a potential therapeutic role in stimulating the systemic inflammatory response in cardiac surgery (122).
TBI can damage the blood–brain barrier (BBB), which can lead to neuroinflammation. This inflammation can be caused by mtDAMPs, which trigger the release of proinflammatory cytokines and polarize microglia/macrophages toward an M1-like phenotype (134). A series of previous studies have proved that the levels of IL-1β, IL-8, TNF-α, and other cytokines in serum and cerebrospinal fluid (CSF) of TBI patients increased, which were correlated with the degree of tissue damage; IL-1β might especially be an independent prognostic factor after TBI (135, 136). The increased expression of these cytokines is related to TBI-mediated mtDNA release (121). Within 48 h after TBI, circulating cell-free mtDNA (ccf-mtDNA) levels in both CSF and serum are elevated (96). Moreover, compared with serum, ccf-mtDNA level in CSF is correlated with injury severity and inflammatory cytokine response and has a more robust predictive effect on neuronal injury and inflammation after TBI (96, 97).
The circulating mtDNA was first identified as the mtDAMP activated by NLRP3 (10) and a crucial component of the NLRP3 activation pathway (37, 42). Moreover, mitochondrial dysfunction-induced mtDNA and mtROS release after TBI could serve as stimuli regulating the NLRP3 inflammasome downstream by transcriptional or post-translational modifications (137). Therefore, we further elucidate the role of the NLRP3 inflammasome in TBI. NLRP3 inflammasome of microglia is involved in the development of neuroinflammation after TBI. Liu et al. showed for the first time that TBI induced upregulation of NLRP3-related genes and proteins in the mouse cerebral cortex, activating NLRP3 inflammasome assembly and releasing inflammatory cytokines such as IL-1β and IL-18 (98). The concentration of NLRP3 in the CSF of infants with TBI changes significantly with time after trauma, and the NLRP3 peak is associated with poor prognosis (99). It is worth noting that NLRP3 knockdown does not lead to changes in IL-1β production, but some markers of microglia and astrocytes will be overexpressed, increasing cytokine levels (138). As a downstream factor of NLRP3, the expression of GSDMD is also blocked (100). According to RNA sequencing, both GSDMD knockdown and NLRP3 knockdown reversed the expression of genes related to neuroinflammation after TBI (100). MaxiK channel, also known as large-conductance Ca2+-activated K+ channels, big K+ (BK) channels, is important for K+ transport. MaxiK channels are important in a variety of physiological functions, including regulation of neuronal firing, endocrine cell secretion, smooth muscle tone, and cellular proliferation and migration (139, 140). In explosion-induced TBI, MaxiK significantly increased in the cerebral cortex, which may activate NLRP3 inflammasome by promoting K+ transport. Blocking this channel can effectively inhibit NLRP3 activation and neuroinflammatory response (139). The STING/NLRP3 signaling pathway has also been shown to be involved in neuroinflammation, and the NLRP3-mediated inflammatory response can be partially inhibited by blocking the expression of STING (101, 136).
NLRP3 inhibitors can restrain the expression and assembly of NLRP3 inflammasome components and reduce the secretion of caspase-1-activated IL-1β and IL-8, thereby effectively alleviating the inflammatory response and BBB damage after TBI, playing a protective role on neurons (123, 126, 127). As a specific inhibitor of NLRP3, the neuroprotective effect of MCC950 depends on the presence of microglia and is limited to the first 6 h after TBI (123). MCC950 combined with rapamycin treatment further enhances neuroprotection after TBI through rapamycin-mediated mitochondrial phagocytosis (141). In addition to inhibiting the expression of NLRP3, oridonin extracted from Chinese herbal medicine can also improve mitochondrial function by enhancing the activation of the Nrf2 signal and reducing the number of degenerated neurons and the volume of cortical lesions (124). Parthenolide treatment simultaneously suppresses STAT3/NF-κB and NLRP3 inflammasomes, thereby inhibiting microglial activation, alleviating neurological deficits, and improving memory and learning in TBI mice (125). In addition to drug therapy, low-intensity focused ultrasound (LIFUS), as a novel treatment for neurological diseases, significantly inhibited the activation of NF-κB and NLRP3 inflammasome after TBI by promoting the expression of orexin-A and orexin receptor 1 (142).
Previous studies have demonstrated that STING mRNA is significantly upregulated in post-mortem human TBI brain samples (105). In recent years, many experimental studies have shown that mtDNA/cGAS/STING and its mediated type I IFN response play an important role in the neuroinflammatory response after TBI, and microglia are the main cell type expressing cGAS and STING in the brain (102–104). STING activation is also age-related. Compared with young mice, aged mice showed greater activation of cGAS/STING and significantly upregulated type I IFN response (103, 104). Fritsch et al. found that NLR containing X1 (NLRX1) could limit the activation of cGAS/STING after brain injury and inhibit the overexpression of type I IFN (102). Let-7i is the upstream signal of STING, whose expression level decreases in TBI mice (128). Intranasal injection of let-7i helps to inhibit the expression of STING, reduce neuronal apoptosis, and improve the cognitive function of mice (128).
A study of heterogeneous orthopedic trauma patients emphasizes the sustained presence of predominantly mtDNA in the plasma of trauma patients following surgical intervention. The correlation between the degree and timing of surgery and mtDNA concentration suggests that mtDNA may serve as a potential marker for a postoperative secondary strike and post-traumatic complications (143). Seewald et al. demonstrated for the first time the relationship between extracellular mtDNA and post-traumatic osteoarthritis (PTOA) (106). In another study in horses, mechanically induced cartilage injury resulted in increased synovial mtDNA concentration either through the selective release of mtDNA from living cells or through cell death or rupture; however, treatment with SS31 peptide effectively reduced mtDNA levels to the baseline level (17). These results suggest that synovial mtDNA concentration is a non-invasive method to detect cartilage dysfunction and acute cartilage injury; moreover, mitochondrial protective drugs may be a novel PTOA prevention and treatment strategy (17).
In smooth muscle, Thankam et al. found that TFAM, cytochrome c, and other mtDAMPs were elevated in venous grafts after coronary artery bypass grafting (CABG), which was related to the increase of ROS content in hypoxic smooth muscle and the damage of membrane integrity, leading to graft failure (95). In mice with skeletal muscle burn, CoQ10 treatment decreased mtDNA level, inhibited the expression of ASC and NLRP3, and reduced the levels of IL-1α, IL-1β, and IFN-γ, thereby alleviating the systemic inflammatory response after burn (129).
In a retrospective study on patients undergoing a liver transplant, Nagakawa et al. discovered that the measurement of plasma mtDAMPs might predict the post-transplantation recovery of the patients (107). In burn and delayed resuscitation experiments in rats, liver NLRP3 inflammasomes activated Kupffer cells by recognizing ox-mtDNA, releasing inflammatory cytokines, and causing liver injury (108).
The circulating mtDNA level after pancreaticoduodenectomy is associated with inflammatory responses and may be used as an early marker of the postoperative disease course (109). During intestinal IR, mtDNA derived from intestinal epithelial cells mediates proinflammatory cytokine generation through the TLR9 signaling pathway, which exacerbates acute inflammatory response (110). Sok et al. constructed mouse models of peritonitis and colitis and demonstrated that 1′-acetoxychavicol acetate (ACA; a natural compound in the rhizome of tropical ginger) inhibited mtROS production and mtDNA oxidation to reduce NLRP3 inflammasome activation, thereby alleviating the colitis in mice (130).
Post-trauma tissue damage releases mtDAMPs from the mitochondria into the cytoplasm or the extracellular space, leading to proinflammatory cytokine release and immune cell activation through a series of signaling pathways. MtDAMP-induced inflammatory reaction protects the body; however, excessive inflammation damages organ function and causes SIRS and MODS, which are poor prognostic factors for trauma patients. Figure 1 summarizes the role of mtDAMPs in the trauma-induced systemic inflammatory response syndrome. For the optimal management of trauma-induced SIRS, further clinical studies on mtDAMPs are required such as the quantification of mtDAMPs levels in different trauma patients, the correlation of mtDAMPs with trauma injury severity or clinical outcomes, the potential intervention strategies targeting mtDAMPs to reduce traumatic SIRS, and the effectiveness and safety assessments. Such studies may lead to the usage of mtDAMPs, especially mtDNA, as biomarkers for predicting the course and prognosis of SIRS in trauma patients. Drugs targeting mtDAMPs and maintaining mitochondrial homeostasis are promising therapeutic strategies.
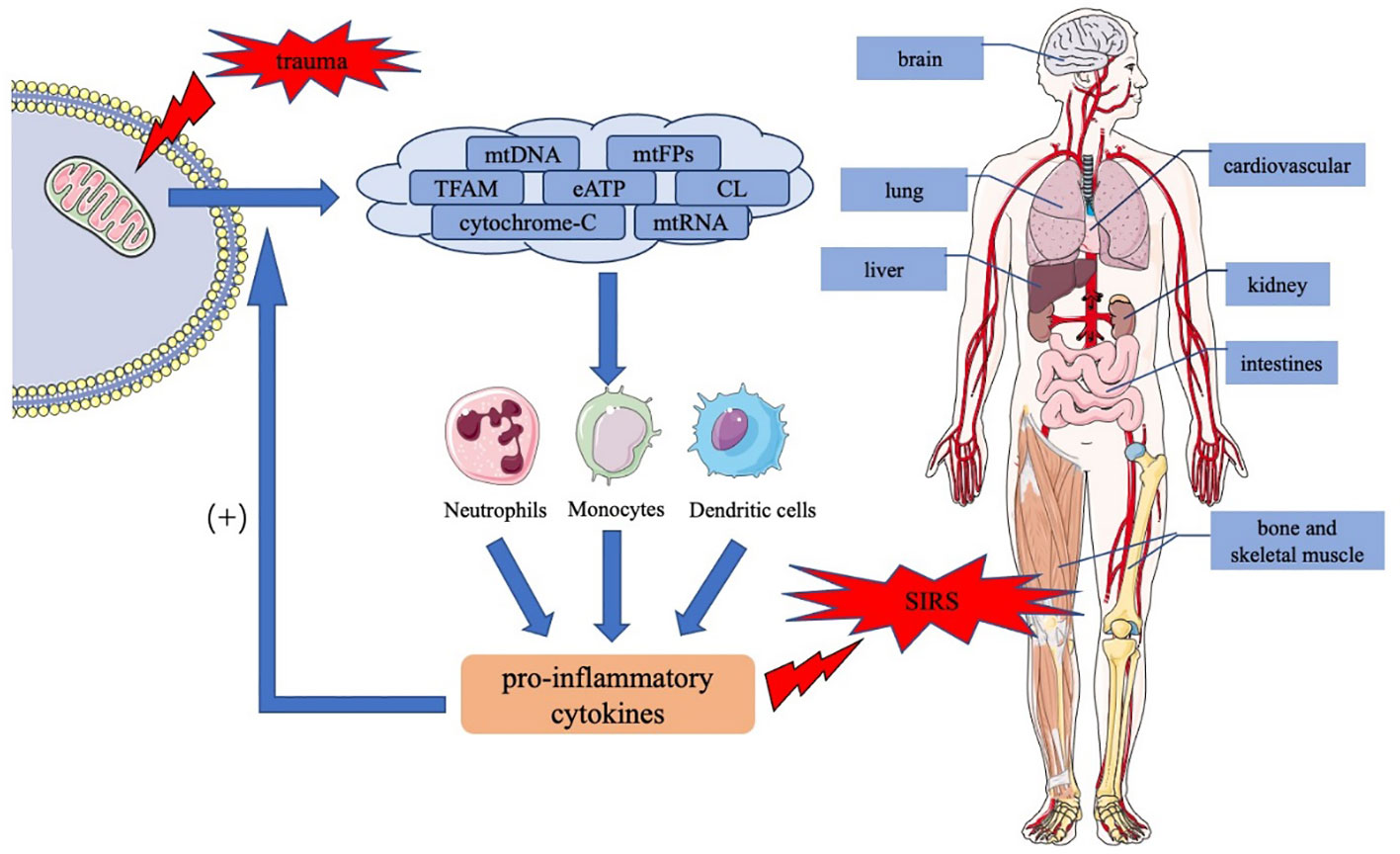
Figure 1 The role of mtDAMPs in the trauma-induced systemic inflammatory response syndrome. Trauma causes tissue damage, resulting in the release of mtDAMPs including mtDNA, mtFPs, TFAM, eATP, CL, cytochrome c, and mtRNA, in the injured mitochondria. MtDAMPs activate downstream signaling pathways in immune cells, such as neutrophils, monocytes, and dendritic cells, thereby inducing the production and release of proinflammatory cytokines. In addition, proinflammatory cytokines act positively on normal cells, increasing mtDAMP release and exacerbating inflammatory response. An uncontrolled inflammatory response causes SIRS and the dysfunction of multiple organs throughout the body. mtDAMPs, mitochondrial damage-associated molecular patterns; mtDNA, mitochondrial DNA; mtFPs, mitochondrial formyl peptides; TFAM, mitochondrial transcription factor A; CL, cardiolipin; eATP, extracellular ATP; mtRNA, mitochondrial RNA; SIRS, systemic inflammatory response syndrome.
TW and JY designed the project; JY and XH wrote the paper; ZW, RL, LG and MZ performed literature search and revision; and all authors participated in the data analysis and interpretation. All authors contributed to the article and approved the submitted version.
Natural Science Foundation of Beijing (7222193), National Natural Science Foundation of China (82000348), Research and Development Fund of Peking University People’s Hospital (RDY2020-05, RDX2021-09), UMHS-PUHSC Joint Institute Grant (BMU 2020JI007), and Medical Science and Technology joint project of Henan Province (LHGJ20220356).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chakraborty RK, Burns B. Systemic inflammatory response syndrome. Treasure Island (FL): StatPearls Publishing (2022).
2. Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg (2012) 99(Suppl 1):12–20. doi: 10.1002/bjs.7717
3. Fehrenbach E, Schneider ME. Trauma-induced systemic inflammatory response versus exercise-induced immunomodulatory effects. Sports Med (2006) 36(5):373–84. doi: 10.2165/00007256-200636050-00001
4. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
5. Itagaki K, Riça I, Konecna B, Kim HI, Park J, Kaczmarek E, et al. Role of mitochondria-derived danger signals released after injury in systemic inflammation and sepsis. Antioxid Redox Signal (2021) 35(15):1273–90. doi: 10.1089/ars.2021.0052
6. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
7. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
8. Vourc'h M, Roquilly A, Asehnoune K. Trauma-induced damage-associated molecular patterns-mediated remote organ injury and immunosuppression in the acutely Ill patient. Front Immunol (2018) 9:1330. doi: 10.3389/fimmu.2018.01330
9. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol (2011) 32(4):157–64. doi: 10.1016/j.it.2011.01.005
10. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi: 10.1038/nature08780
11. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
12. Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC, et al. Mitochondrial damage-associated molecular patterns and vascular function. Eur Heart J (2014) 35(18):1172–7. doi: 10.1093/eurheartj/ehu047
13. Wu Z, Sainz AG, Shadel GS. Mitochondrial DNA: cellular genotoxic stress sentinel. Trends Biochem Sci (2021) 46(10):812–21. doi: 10.1016/j.tibs.2021.05.004
14. Jia B, Ye J, Gan L, Li R, Zhang M, Sun D, et al. Mitochondrial antioxidant SkQ1 decreases inflammation following hemorrhagic shock by protecting myocardial mitochondria. Front Physiol (2022) 13:1047909. doi: 10.3389/fphys.2022.1047909
15. Thurairajah K, Briggs GD, Balogh ZJ. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur J Trauma Emerg Surg (2018) 44(3):325–34. doi: 10.1007/s00068-018-0954-3
16. Briggs GD, Gelzinnis S, Meakes S, King KL, Balogh ZJ. NOT ALL CELL-FREE MITOCHONDRIAL DNA IS EQUAL IN TRAUMA PATIENTS. Shock (2022) 58(3):231–5. doi: 10.1097/SHK.0000000000001969
17. Seewald LA, Keller LE, Thomas M, Casey J, Delco ML. Mitoprotection prevents increased synovial fluid mitochondrial DNA concentrations after articular injury. Osteoarthritis Cartilage (2020) 28:S96. doi: 10.1016/j.joca.2020.02.148
18. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol (2004) 5(2):190–8. doi: 10.1038/ni1028
19. Zhang X, Wu X, Hu Q, Wu J, Wang G, Hong Z, et al. Lab For trauma and surgical infections. mitochondrial DNA in liver inflammation and oxidative stress. Life Sci (2019) 236:116464. doi: 10.1016/j.lfs.2019.05.020
20. Hayashi K, Taura M, Iwasaki A. The interaction between IKKα and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci Signal (2018) 11(528):eaan4144. doi: 10.1126/scisignal.aan4144
21. Schmitz F, Heit A, Guggemoos S, Krug A, Mages J, Schiemann M, et al. Interferon-regulatory-factor 1 controls toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur J Immunol (2007) 37(2):315–27. doi: 10.1002/eji.200636767
22. Bryant JD, Lei Y, VanPortfliet JJ, Winters AD, West AP. Assessing mitochondrial DNA release into the cytosol and subsequent activation of innate immune-related pathways in mammalian cells. Curr Protoc (2022) 2(2):e372. doi: 10.1002/cpz1.372
23. Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The n-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep (2019) 26(9):2377–93.e13. doi: 10.1016/j.celrep.2019.01.105
24. Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell (2019) 176(6):1432–46.e11. doi: 10.1016/j.cell.2019.01.049
25. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498(7454):380–4. doi: 10.1038/nature12306
26. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell (2013) 51(2):226–35. doi: 10.1016/j.molcel.2013.05.022
27. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461(7265):788–92. doi: 10.1038/nature08476
28. Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, et al. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis (2021) 12(7):673. doi: 10.1038/s41419-021-03961-9
29. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal (2012) 5(214):ra20. doi: 10.1126/scisignal.2002521
30. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol (2014) 88(10):5328–41. doi: 10.1128/JVI.00037-14
31. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
32. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
33. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
34. Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev (2011) 243(1):136–51. doi: 10.1111/j.1600-065X.2011.01046.x
35. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18(9):2114–27. doi: 10.1038/s41423-021-00740-6
36. Wu XY, Lv JY, Zhang SQ, Yi X, Xu ZW, Zhi YX, et al. ML365 inhibits TWIK2 channel to block ATP-induced NLRP3 inflammasome. Acta Pharmacol Sin (2022) 43(4):992–1000. doi: 10.1038/s41401-021-00739-9
37. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
38. Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity (2022) 55(8):1370–85.e8. doi: 10.1016/j.immuni.2022.06.007
39. Wu G, Zhu Q, Zeng J, Gu X, Miao Y, Xu W, et al. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-κB. J Thorac Dis (2019) 11(11):4816–28. doi: 10.21037/jtd.2019.10.26
40. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
41. de Torre-Minguela C, Gómez AI, Couillin I, Pelegrín P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J (2021) 35(8):e21757. doi: 10.1096/fj.202100085R
42. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
43. Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity (2021) 54(7):1463–77.e11. doi: 10.1016/j.immuni.2021.05.004
44. Li S, Li H, Zhang YL, Xin QL, Guan ZQ, Chen X, et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep (2020) 30(13):4370–85. doi: 10.1016/j.celrep.2020.02.105
45. Kong C, Song W, Fu T. Systemic inflammatory response syndrome is triggered by mitochondrial damage (Review). Mol Med Rep (2022) 25(4):147. doi: 10.3892/mmr.2022.12663
46. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev (2009) 61(2):119–61. doi: 10.1124/pr.109.001578
47. Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM. N-formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury (2015) 46(6):975–84. doi: 10.1016/j.injury.2015.03.028
48. Dorward DA, Lucas CD, Doherty MK, Chapman GB, Scholefield EJ, Conway Morris A, et al. Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax (2017) 72(10):928–36. doi: 10.1136/thoraxjnl-2017-210030
49. Liu H, Lin Z, Ma Y. Suppression of Fpr2 expression protects against endotoxin-induced acute lung injury by interacting with Nrf2-regulated TAK1 activation. BioMed Pharmacother (2020) 125:109943. doi: 10.1016/j.biopha.2020.109943
50. Lee HY, Kim HS, Jeong YS, Kim JC, Bae YS, Jo YH, et al. A membrane-tethering pepducin derived from formyl peptide receptor 3 shows strong therapeutic effects against sepsis. Biochem Biophys Res Commun (2020) 524(1):156–62. doi: 10.1016/j.bbrc.2020.01.058
51. Zhang Q, Kwon WY, Vlková B, Riça I, Kaczmarek E, Park J, et al. Direct airway instillation of neutrophils overcomes chemotactic deficits induced by injury. Shock (2021) 56(1):119–24. doi: 10.1097/SHK.0000000000001691
52. Itagaki K, Kaczmarek E, Kwon WY, Chen L, Vlková B, Zhang Q, et al. Formyl peptide receptor-1 blockade prevents receptor regulation by mitochondrial danger-associated molecular patterns and preserves neutrophil function after trauma. Crit Care Med (2020) 48(2):e123–32. doi: 10.1097/CCM.0000000000004094
53. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520(7548):553–7. doi: 10.1038/nature14156
54. Schindler SM, Frank MG, Annis JL, Maier SF, Klegeris A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor a (TFAM). Mol Cell Neurosci (2018) 89:71–9. doi: 10.1016/j.mcn.2018.04.005
55. Krüger A, Oldenburg M, Chebrolu C, Beisser D, Kolter J, Sigmund AM, et al. Human TLR8 senses UR/URR motifs in bacterial and mitochondrial RNA. EMBO Rep (2015) 16(12):1656–63. doi: 10.15252/embr.201540861
56. Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol (2019) 10:325. doi: 10.3389/fimmu.2019.00325
57. Coomans de Brachène A, Castela A, Musuaya AE, Marselli L, Marchetti P, Eizirik DL. Endogenous mitochondrial double-stranded RNA is not an activator of the type I interferon response in human pancreatic beta cells. Auto Immun Highlights (2021) 12(1):6. doi: 10.1186/s13317-021-00148-2
58. Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol (2017) 5:90. doi: 10.3389/fcell.2017.00090
59. Chen L, Dong J, Liao S, Wang S, Wu Z, Zuo M, et al. Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology (2022) 76(5):1389–408. doi: 10.1002/hep.32471
60. Li S, Hu Q, Huang J, Wu X, Ren J. Mitochondria-derived damage-associated molecular patterns in sepsis: from bench to bedside. Oxid Med Cell Longev (2019) 2019:6914849. doi: 10.1155/2019/6914849
61. Dixit A, Cheema H, George J, Iyer S, Dudeja V, Dawra R, et al. Extracellular release of ATP promotes systemic inflammation during acute pancreatitis. Am J Physiol Gastrointest Liver Physiol (2019) 317(4):G463–75. doi: 10.1152/ajpgi.00395.2018
62. Wenzel TJ, Bajwa E, Klegeris A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim Biophys Acta Gen Subj (2019) 1863(11):129400. doi: 10.1016/j.bbagen.2019.07.009
63. Pullerits R, Bokarewa M, Jonsson IM, Verdrengh M, Tarkowski A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatol (Oxford) (2005) 44(1):32–9. doi: 10.1093/rheumatology/keh406
64. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1β induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell (2019) 74(4):801–15.e6. doi: 10.1016/j.molcel.2019.02.038
65. Willemsen J, Neuhoff MT, Hoyler T, Noir E, Tessier C, Sarret S, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep (2021) 37(6):109977. doi: 10.1016/j.celrep.2021.109977
66. Messaoud-Nacer Y, Culerier E, Rose S, Maillet I, Rouxel N, Briault S, et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis (2022) 13(3):269. doi: 10.1038/s41419-022-04664-5
67. Sandler N, Kaczmarek E, Itagaki K, Zheng Y, Otterbein L, Khabbaz K, et al. Mitochondrial DAMPs are released during cardiopulmonary bypass surgery and are associated with postoperative atrial fibrillation. Heart Lung Circ (2018) 27(1):122–9. doi: 10.1016/j.hlc.2017.02.014
68. Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PloS One (2013) 8(8):e72834. doi: 10.1371/journal.pone.0072834
69. Wenceslau CF, Szasz T, McCarthy CG, Baban B, NeSmith E, Webb RC. Mitochondrial n-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm Pharmacol Ther (2016) 37:49–56. doi: 10.1016/j.pupt.2016.02.005
70. Hu Q, Ren J, Wu J, Li G, Wu X, Liu S, et al. Elevated levels of plasma mitochondrial DNA are associated with clinical outcome in intra-abdominal infections caused by severe trauma. Surg Infect (Larchmt) (2017) 18(5):610–8. doi: 10.1089/sur.2016.276
71. Martinez-Quinones PA, McCarthy CG, Mentzer CJ, Wenceslau CF, Holsten SB, Webb RC, et al. Peritoneal cavity lavage reduces the presence of mitochondrial damage associated molecular patterns in open abdomen patients. J Trauma Acute Care Surg (2017) 83(6):1062–5. doi: 10.1097/TA.0000000000001676
72. Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma (2010) 24:534–8. doi: 10.1097/BOT.0b013e3181ec4991
73. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62:600–14. doi: 10.1002/hep.27841
74. Hadass O, Tomlinson BN, Gooyit M, Chen S, Purdy JJ, Walker JM, et al. Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury. PloS One (2013) 8:e76904. doi: 10.1371/journal.pone.0076904
75. Aswani A, Manson J, Itagaki K, Chiazza F, Collino M, Wupeng WL, et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front Immunol (2018) 9:891. doi: 10.3389/fimmu.2018.00891
76. Lee J, Jackman JG, Kwun J, Manook M, Moreno A, Elster EA, et al. Nucleic acid scavenging microfiber mesh inhibits trauma-induced inflammation and thrombosis. Biomaterials (2017) 120:94–102. doi: 10.1016/j.biomaterials.2016.12.024
77. Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, et al. Plasma mitochondrial DNA levels are associated with ARDS in trauma and sepsis patients. Chest (2020) 157(1):67–76. doi: 10.1016/j.chest.2019.09.028
78. Huang L, Chang W, Huang Y, Xu X, Yang Y, Qiu H. Prognostic value of plasma mitochondrial DNA in acute respiratory distress syndrome (ARDS): a single-center observational study. J Thorac Dis (2020) 12(4):1320–8. doi: 10.21037/jtd.2020.02.49
79. Yuan ZC, Zeng N, Liu L, Wang T, Dai LQ, Wang H, et al. Mitochondrial damage-associated molecular patterns exacerbate lung fluid imbalance Via the formyl peptide receptor-1 signaling pathway in acute lung injury. Crit Care Med (2021) 49(1):e53–62. doi: 10.1097/CCM.0000000000004732
80. Comish PB, Liu MM, Huebinger R, Carlson D, Kang R, Tang D. The cGAS-STING pathway connects mitochondrial damage to inflammation in burn-induced acute lung injury in rat. Burns (2022) 48(1):168–75. doi: 10.1016/j.burns.2021.04.007
81. Xiao MJ, Zou XF, Li B, Li BL, Wu SJ, Zhang B. Simulated aeromedical evacuation exacerbates burn induced lung injury: targeting mitochondrial DNA for reversal. Mil Med Res (2021) 8(1):30. doi: 10.1186/s40779-021-00320-9
82. Puyo CA, Earhart A, Staten N, Prince OA, Haug C, Kollef M, et al. Endotracheal intubation results in acute tracheal damage induced by mtDNA/TLR9/NF-κB activity. J Leukoc Biol (2019) 105(3):577–87. doi: 10.1002/JLB.5A0718-254RR
83. Lin JY, Jing R, Lin F, Ge WY, Dai HJ, Pan L. High tidal volume induces mitochondria damage and releases mitochondrial DNA to aggravate the ventilator-induced lung injury. Front Immunol (2018) 9:1477. doi: 10.3389/fimmu.2018.01477
84. Grazioli S, Dunn-Siegrist I, Pauchard LA, Blot M, Charles PE, Pugin J. Mitochondrial alarmins are tissue mediators of ventilator-induced lung injury and ARDS. PLoS One (2019) 14(11):e0225468. doi: 10.1371/journal.pone.0225468
85. Jing R, Hu ZK, Lin F, He S, Zhang SS, Ge WY, et al. Mitophagy-mediated mtDNA release aggravates stretching-induced inflammation and lung epithelial cell injury via the TLR9/MyD88/NF-κB pathway. Front Cell Dev Biol (2020) 8:819. doi: 10.3389/fcell.2020.00819
86. Liu R, Xu F, Bi S, Zhao X, Jia B, Cen Y. Mitochondrial DNA-induced inflammatory responses and lung injury in thermal injury murine model: protective effect of cyclosporine-a. J Burn Care Res (2019) 40(3):355–60. doi: 10.1093/jbcr/irz029
87. Feng Y, Imam Aliagan A, Tombo N, Draeger D, Bopassa JC. RIP3 translocation into mitochondria promotes mitofilin degradation to increase inflammation and kidney injury after renal ischemia-reperfusion. Cells (2022) 11(12):1894. doi: 10.3390/cells11121894
88. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics (2021) 11(4):1845–63. doi: 10.7150/thno.50905
89. Li J, Sun X, Yang N, Ni J, Xie H, Guo H, et al. Phosphoglycerate mutase 5 initiates inflammation in acute kidney injury by triggering mitochondrial DNA release by dephosphorylating the pro-apoptotic protein bax. Kidney Int (2023) 103(1):115–33. doi: 10.1016/j.kint.2022.08.022
90. Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial n-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol (2015) 308(7):H768–777. doi: 10.1152/ajpheart.00779.2014
91. Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity (2020) 52(3):475–86.e5. doi: 10.1016/j.immuni.2020.02.002
92. Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PloS One (2013) 8(3):e59989. doi: 10.1371/journal.pone.0059989
93. Zabelina TS, Grebenchikov OA, Skripkin YV, Yavorovsky AG, Lihvantsev VV. Plasma mitochondrial DNA level as an early marker of systemic inflammatory response syndrome and organ failure in patients undergoing cardiac surgery. Patologiya krovoobrashcheniya i kardiokhirurgiya (2019) 23(1):33. doi: 10.21688/1681-3472-2019-1-33-41
94. Zajonz T, Koch C, Schwiddessen J, Markmann M, Hecker M, Edinger F, et al. Minimized extracorporeal circulation is associated with reduced plasma levels of free-circulating mitochondrial DNA compared to conventional cardiopulmonary bypass: a secondary analysis of an exploratory, prospective, interventional study. J Clin Med (2022) 11(11):2994. doi: 10.3390/jcm11112994
95. Thankam FG, Ayoub JG, Ahmed MMR, Siddique A, Sanchez TC, Peralta RA, et al. Association of hypoxia and mitochondrial damage associated molecular patterns in the pathogenesis of vein graft failure: a pilot study. Transl Res (2021) 229:38–52. doi: 10.1016/j.trsl.2020.08.010
96. Kayhanian S, Glynos A, Mair R, Lakatos A, Hutchinson PJA, Helmy AE, et al. Cell-free mitochondrial DNA in acute brain injury. Neurotrauma Rep (2022) 3(1):415–20. doi: 10.1089/neur.2022.0032
97. Walko TD 3rd, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, et al. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock (2014) 41(6):499–503. doi: 10.1097/SHK.0000000000000160
98. Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res (2013) 38(10):2072–83. doi: 10.1007/s11064-013-1115-z
99. Wallisch JS, Simon DW, Bayır H, Bell MJ, Kochanek PM, Clark RSB. Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care (2017) 27(1):44–50. doi: 10.1007/s12028-017-0378-7
100. Du H, Li CH, Gao RB, Cen XQ, Li P. Ablation of GSDMD attenuates neurological deficits and neuropathological alterations after traumatic brain injury. Front Cell Neurosci (2022) 16:915969. doi: 10.3389/fncel.2022.915969
101. Zhang LM, Xin Y, Wu ZY, Song RX, Miao HT, Zheng WC, et al. STING mediates neuroinflammatory response by activating NLRP3-related pyroptosis in severe traumatic brain injury. J Neurochem (2022) 162(5):444–62. doi: 10.1111/jnc.15678
102. Fritsch LE, Ju J, Gudenschwager Basso EK, Soliman E, Paul S, Chen J, et al. Type I interferon response is mediated by NLRX1-cGAS-STING signaling in brain injury. Front Mol Neurosci (2022) 15:852243. doi: 10.3389/fnmol.2022.852243
103. Barrett JP, Knoblach SM, Bhattacharya S, Gordish-Dressman H, Stoica BA, Loane DJ. Traumatic brain injury induces cGAS activation and type I interferon signaling in aged mice. Front Immunol (2021) 12:710608. doi: 10.3389/fimmu.2021.710608
104. Wangler LM, Bray CE, Packer JM, Tapp ZM, Davis AC, O'Neil SM, et al. Amplified gliosis and interferon-associated inflammation in the aging brain following diffuse traumatic brain injury. J Neurosci (2022) 42(48):9082–96. doi: 10.1523/JNEUROSCI.1377-22.2022
105. Abdullah A, Zhang M, Frugier T, Bedoui S, Taylor JM, Crack PJ. STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflammation (2018) 15(1):323. doi: 10.1186/s12974-018-1354-7
106. Seewald LA, Sabino IG, Montney KL, Delco ML. Synovial fluid mitochondrial DNA concentration reflects the degree of cartilage damage after naturally occurring articular injury. Osteoarthritis Cartilage (2023) S1063-4584(23)00735-5. doi: 10.1101/2021.08.26.457571
107. Nagakawa K, Soyama A, Hidaka M, Adachi T, Ono S, Hara T, et al. Elevated plasma levels of mitochondria-derived damage-associated molecular patterns during liver transplantation: predictors for postoperative multi-organ dysfunction syndrome. Tohoku J Exp Med (2020) 250(2):87–93. doi: 10.1620/tjem.250.87
108. Wu Y, Hao C, Liu X, Han G, Yin J, Zou Z, et al. MitoQ protects against liver injury induced by severe burn plus delayed resuscitation by suppressing the mtDNA-NLRP3 axis. Int Immunopharmacol (2020) 80:106189. doi: 10.1016/j.intimp.2020.106189
109. Pencovich N, Nevo N, Weiser R, Bonder E, Bogoch Y, Nachmany I. Postoperative rise of circulating mitochondrial DNA is associated with inflammatory response in patients following pancreaticoduodenectomy. Eur Surg Res (2021) 62(1):18–24. doi: 10.1159/000514661
110. Hu Q, Ren H, Ren J, Liu Q, Wu J, Wu X, et al. Released mitochondrial DNA following intestinal ischemia reperfusion induces the inflammatory response and gut barrier dysfunction. Sci Rep (2018) 8(1):7350. doi: 10.1038/s41598-018-25387-8
111. Xiao Z, Jia B, Zhao X, Bi S, Meng W. Attenuation of lipopolysaccharide-induced acute lung injury by cyclosporine-a via suppression of mitochondrial DNA. Med Sci Monit (2018) 24:7682–8. doi: 10.12659/MSM.909909
112. Luo ZL, Sun HY, Wu XB, Cheng L, Ren JD. Epigallocatechin-3-gallate attenuates acute pancreatitis induced lung injury by targeting mitochondrial reactive oxygen species triggered NLRP3 inflammasome activation. Food Funct (2021) 12(12):5658–67. doi: 10.1039/D1FO01154E
113. He YQ, Zhou CC, Deng JL, Wang L, Chen WS. Tanreqing inhibits LPS-induced acute lung injury In vivo and In vitro through downregulating STING signaling pathway. Front Pharmacol (2021) 12:746964. doi: 10.3389/fphar.2021.746964
114. Dong G, Xu N, Wang M, Zhao Y, Jiang F, Bu H, et al. Anthocyanin extract from purple sweet potato exacerbate mitophagy to ameliorate pyroptosis in klebsiella pneumoniae infection. Int J Mol Sci (2021) 22(21):11422. doi: 10.3390/ijms222111422
115. Qin W, Tong X, Liang R, Tang K, Wu X, Jia Y, et al. Preservation of mitochondrial homeostasis is responsible for the ameliorative effects of suhuang antitussive capsule on non-resolving inflammation via inhibition of NF-κB signaling and NLRP3 inflammasome activation. J Ethnopharmacol (2021) 271:113827. doi: 10.1016/j.jep.2021.113827
116. Yang JW, Mao B, Tao RJ, Fan LC, Lu HW, Ge BX, et al. Corticosteroids alleviate lipopolysaccharide-induced inflammation and lung injury via inhibiting NLRP3-inflammasome activation. J Cell Mol Med (2020) 24(21):12716–25. doi: 10.1111/jcmm.15849
117. Dutra Silva J, Su Y, Calfee CS, Delucchi KL, Weiss D, McAuley DF, et al. Mesenchymal stromal cell extracellular vesicles rescue mitochondrial dysfunction and improve barrier integrity in clinically relevant models of ARDS. Eur Respir J (2021) 58(1):2002978. doi: 10.1183/13993003.02978-2020
118. Xia L, Zhang C, Lv N, Liang Z, Ma T, Cheng H, et al. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics (2022) 12(6):2928–47. doi: 10.7150/thno.69533
119. Blot M, Jacquier M, Pauchard LA, Rebaud C, Marlin C, Hamelle C, et al. Adverse mechanical ventilation and pneumococcal pneumonia induce immune and mitochondrial dysfunctions mitigated by mesenchymal stem cells in rabbits. Anesthesiology (2022) 136(2):293–313. doi: 10.1097/ALN.0000000000004083
120. Zhu Y, Luo M, Bai X, Li J, Nie P, Li B, et al. SS-31, a mitochondria-targeting peptide, ameliorates kidney disease. Oxid Med Cell Longev (2022) 2022:1295509. doi: 10.1155/2022/1295509
121. Zhao M, Liu S, Wang C, Wang Y, Wan M, Liu F, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate mitochondrial damage and inflammation by stabilizing mitochondrial DNA. ACS Nano (2021) 15(1):1519–38. doi: 10.1021/acsnano.0c08947
122. Naase H, Harling L, Kidher E, Sepehripour A, Nguyen B, Kapelouzou A, et al. Toll-like receptor 9 and the inflammatory response to surgical trauma and cardiopulmonary bypass. J Cardiothorac Surg (2020) 15(1):137. doi: 10.1186/s13019-020-01179-y
123. Xu X, Yin D, Ren H, Gao W, Li F, Sun D, et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis (2018) 117:15–27. doi: 10.1016/j.nbd.2018.05.016
124. Zhao XJ, Zhu HY, Wang XL, Lu XW, Pan CL, Xu L, et al. Oridonin ameliorates traumatic brain injury-induced neurological damage by improving mitochondrial function and antioxidant capacity and suppressing neuroinflammation through the Nrf2 pathway. J Neurotrauma (2022) 39(7-8):530–43. doi: 10.1089/neu.2021.0466
125. Ding W, Cai C, Zhu X, Wang J, Jiang Q. Parthenolide ameliorates neurological deficits and neuroinflammation in mice with traumatic brain injury by suppressing STAT3/NF-κB and inflammasome activation. Int Immunopharmacol (2022) 108:108913. doi: 10.1016/j.intimp.2022.108913
126. Yang F, Li G, Lin B, Zhang K. Gastrodin suppresses pyroptosis and exerts neuroprotective effect in traumatic brain injury model by inhibiting NLRP3 inflammasome signaling pathway. J Integr Neurosci (2022) 21(2):72. doi: 10.31083/j.jin2102072
127. Cai L, Gong Q, Qi L, Xu T, Suo Q, Li X, et al. ACT001 attenuates microglia-mediated neuroinflammation after traumatic brain injury via inhibiting AKT/NFκB/NLRP3 pathway. Cell Commun Signal (2022) 20(1):56. doi: 10.1186/s12964-022-00862-y
128. He XC, Wang J, Du HZ, Liu CM, Teng ZQ. Intranasal administration of agomir-let-7i improves cognitive function in mice with traumatic brain injury. Cells (2022) 11(8):1348. doi: 10.3390/cells11081348
129. Nakazawa H, Ikeda K, Shinozaki S, Yasuhara S, Yu YM, Martyn JAJ, et al. Coenzyme Q10 protects against burn-induced mitochondrial dysfunction and impaired insulin signaling in mouse skeletal muscle. FEBS Open Bio (2019) 9(2):348–63. doi: 10.1002/2211-5463.12580
130. Sok SPM, Ori D, Wada A, Okude H, Kawasaki T, Momota M, et al. 1'-acetoxychavicol acetate inhibits NLRP3-dependent inflammasome activation via mitochondrial ROS suppression. Int Immunol (2021) 33(7):373–86. doi: 10.1093/intimm/dxab016
131. Faust HE, Oniyide O, Wang Y, Forker CM, Dunn T, Yang W, et al. Early plasma nuclear DNA, mitochondrial DNA, and nucleosome concentrations are associated with acute kidney injury in critically ill trauma patients. Crit Care Explor (2022) 4(4):e0663. doi: 10.1097/CCE.0000000000000663
132. You B, Yang Z, Zhang Y, Chen Y, Gong Y, Chen Y, et al. Late-onset acute kidney injury is a poor prognostic sign for severe burn patients. Front Surg (2022) 9:842999. doi: 10.3389/fsurg.2022.842999
133. Hepokoski M, Wang J, Li K, Li Y, Gupta P, Mai T, et al. Altered lung metabolism and mitochondrial DAMPs in lung injury due to acute kidney injury. Am J Physiol Lung Cell Mol Physiol (2021) 320(5):L821–31. doi: 10.1152/ajplung.00578.2020
134. Simon DW, McGeachy MJ, Bayır H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol (2017) 13(3):171–91. doi: 10.1038/nrneurol.2017.13
135. Doğanyiğit Z, Erbakan K, Akyuz E, Polat AK, Arulsamy A, Shaikh MF. The role of neuroinflammatory mediators in the pathogenesis of traumatic brain injury: a narrative review. ACS Chem Neurosci (2022) 13(13):1835–48. doi: 10.1021/acschemneuro.2c00196
136. Pérez-Bárcena J, Rodríguez Pilar J, Salazar O, Crespí C, Frontera G, Novo MA, et al. Serum caspase-1 as an independent prognostic factor in traumatic brain injured patients. Neurocrit Care (2022) 36(2):527–35. doi: 10.1007/s12028-021-01340-y
137. O'Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation (2020) 17(1):104. doi: 10.1186/s12974-020-01778-5
138. Lopez-Rodriguez AB, Decouty-Perez C, Farré-Alins V, Palomino-Antolín A, Narros-Fernández P, Egea J. Activation of NLRP3 is required for a functional and beneficial microglia response after brain trauma. Pharmaceutics (2022) 14(8):1550. doi: 10.3390/pharmaceutics14081550
139. Ma J, Wang J, Deng K, Gao Y, Xiao W, Hou J, et al. The effect of MaxiK channel on regulating the activation of NLRP3 inflammasome in rats of blast-induced traumatic brain injury. Neuroscience (2022) 482:132–42. doi: 10.1016/j.neuroscience.2021.12.019
140. Wang Y, Wang M, Ning F, Ren D, Tao J, Xie W, et al. A novel role of BK potassium channel activity in preventing the development of kidney fibrosis[J]. Kidney Int (2022) 101(5):945–62. doi: 10.1016/j.kint.2021.11.033
141. Chen Y, Meng J, Xu Q, Long T, Bi F, Chang C, et al. Rapamycin improves the neuroprotection effect of inhibition of NLRP3 inflammasome activation after TBI. Brain Res (2019) 1710:163–72. doi: 10.1016/j.brainres.2019.01.005
142. Huang L, Kang J, Chen G, Ye W, Meng X, Du Q, et al. Low-intensity focused ultrasound attenuates early traumatic brain injury by OX-A/NF-κB/NLRP3 signaling pathway. Aging (Albany NY) (2022) 14(18):7455–69. doi: 10.18632/aging.204290
Keywords: mtDAMPs, trauma-induced SIRS, mtDNA, mtFPs, trauma
Citation: Ye J, Hu X, Wang Z, Li R, Gan L, Zhang M and Wang T (2023) The role of mtDAMPs in the trauma-induced systemic inflammatory response syndrome. Front. Immunol. 14:1164187. doi: 10.3389/fimmu.2023.1164187
Received: 12 February 2023; Accepted: 26 June 2023;
Published: 18 July 2023.
Edited by:
Luz Pamela Blanco, National Institutes of Health (NIH), United StatesReviewed by:
Julien Pottecher, Hôpitaux Universitaires de Strasbourg, FranceCopyright © 2023 Ye, Hu, Wang, Li, Gan, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianbing Wang, d2FuZ3RpYW5iaW5nQHBrdXBoLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.