
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 22 May 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1163987
This article is part of the Research TopicInsights in Molecular Innate Immunity: 2022View all 13 articles
 Lianna Zhou1†
Lianna Zhou1† Zhaoqi Yan2*†
Zhaoqi Yan2*† Wei Yang3Jessica A. Buckley1Sameer Al Diffalha4Etty N. Benveniste1
Wei Yang3Jessica A. Buckley1Sameer Al Diffalha4Etty N. Benveniste1 Hongwei Qin1*
Hongwei Qin1*Introduction: Myeloid cells play a critical role in the pathogenesis of Inflammatory Bowel Diseases (IBDs), including Ulcerative Colitis (UC) and Crohn’s Disease (CD). Dysregulation of the JAK/STAT pathway is associated with many pathological conditions, including IBD. Suppressors Of Cytokine Signaling (SOCS) are a family of proteins that negatively regulate the JAK/STAT pathway. Our previous studies identified that mice lacking Socs3 in myeloid cells developed a hyper-activated phenotype of macrophages and neutrophils in a pre-clinical model of Multiple Sclerosis.
Methods: To better understand the function of myeloid cell Socs3 in the pathogenesis of colitis, mice with Socs3 deletion in myeloid cells (Socs3ΔLysM) were utilized in a DSS-induced colitis model.
Results: Our results indicate that Socs3 deficiency in myeloid cells leads to more severe colitis induced by DSS, which correlates with increased infiltration of monocytes and neutrophils in the colon and increased numbers of monocytes and neutrophils in the spleen. Furthermore, our results demonstrate that the expression of genes related to the pathogenesis and diagnosis of colitis such as Il1β, Lcn2, S100a8 and S100a9 were specifically enhanced in Socs3-deficient neutrophils localized to the colon and spleen. Conversely, there were no observable differences in gene expression in Ly6C+ monocytes. Depletion of neutrophils using a neutralizing antibody to Ly6G significantly improved the disease severity of DSS-induced colitis in Socs3-deficient mice.
Discussion: Thus, our results suggest that deficiency of Socs3 in myeloid cells exacerbates DSS-induced colitis and that Socs3 prevents overt activation of the immune system in IBD. This study may provide novel therapeutic strategies to IBD patients with hyperactivated neutrophils.
Inflammatory Bowel Diseases (IBDs) are chronic relapsing inflammatory disorders of the gastrointestinal tract, including two major subtypes: Ulcerative Colitis (UC) and Crohn’s Disease (CD) (1, 2). IBD has drawn wide attention with a high rate of prevalence in the Western world and East Asia with connections to the Westernized lifestyle and environmental factors such as smoking, microorganisms, medication, nutrition, and stress (3, 4). Both UC and CD share common symptoms and structural damages, which are associated with multiple pathogenic factors including environmental changes, susceptibility gene variants, and a broadly dysregulated immune response (5, 6). A comprehensive understanding of IBD pathogenesis is still unclear and, consequently, treatment is far from optimal. One of the animal models used to investigate the molecular and cellular mechanisms of IBD is the acute dextran sodium sulfate (DSS) colitis model, which is frequently used to develop and evaluate the efficacy of novel anti-inflammatory drugs (7). The acute inflammatory responses in the DSS colitis model are particularly useful to study the contribution of innate immune mechanisms in intestinal inflammation (8, 9).
The JAK/STAT signaling pathway plays a critical role in activation and regulation of immune responses (10, 11). Dysregulation of the JAK/STAT pathway is associated with many pathological conditions, including Multiple Sclerosis (MS), Rheumatoid Arthritis and IBDs (12, 13). We previously demonstrated that inhibition of the JAK/STAT pathway ameliorates disease severity in a number of pre-clinical models of MS and Parkinson’s disease (14, 15). There is emerging evidence that the JAK/STAT pathway has pathogenic roles in the development of colitis; JAK inhibitors, such as tofacitinib, have been approved for treatment of UC with demonstrated efficacy in early phases of clinical trials (16, 17). As a family of proteins that negatively regulate the JAK/STAT pathway, Suppressors Of Cytokine Signaling 3 (SOCS3) mainly inhibits activation of STAT1 and STAT3 (18, 19). Studies from animal models, including the DSS-induced colitis model, have established that STAT3 activation promotes pathogenesis of UC and carcinogenesis (20–25). However, other results indicate STAT3 deletion in myeloid cells also leads to exacerbation of colitis (26, 27). As such, further study is required to elucidate the role of the STAT3/SOCS3 axis in myeloid cells to understand their role in the pathogenesis of colitis.
The role of myeloid cells in IBD is also inconclusive. Monocytes and macrophages play pivotal roles in the innate immune response to pathogens primarily through phagocytosis and the release of inflammatory mediators such as cytokines and chemokines (28). Previous studies have shown that under inflammatory conditions, monocytes migrate out of the blood utilizing a CCR2-related mechanism and are recruited to the lamina propria, thereby favoring a pro-inflammatory response (29–32). Macrophage- and monocyte-related pro-inflammatory cytokines such as TNF, IL-1β, IL-6 and IL-1 are detected in colonic biopsies from patients with CD and UC and are implicated in the development of IBD in humans (33). Moreover, inhibition of monocyte/macrophage recruitment led to ameliorated murine colitis (34). Neutrophils are the most abundant cell type in human blood and are the first responders of the immune system when the body encounters foreign antigens (35, 36). Neutrophil infiltration into the lamina propria is an important histological activity index in UC (37) and promotes disease progression in animal models of colitis (38). However, other studies demonstrated that bone marrow-derived suppressor cells, which consist of macrophages and neutrophils, suppress overt inflammation, accelerate recovery, and protect the epithelial barrier in the intestine (39–41).
We generated a mouse model, crossing Socs3fl/fl mice with LysMCre mice, to create a conditional knockout mouse possessing Socs3-deficient myeloid cells (Socs3ΔLysM) (42, 43). Previous studies in our lab using Socs3ΔLysM mice demonstrated enhanced basal and stimulus-induced STAT activation, in addition to upregulation of various pro-inflammatory mediators in Socs3-deficient myeloid cells (42, 43). Furthermore, both Socs3-deficient macrophages and neutrophils displayed gene expression patterns reflective of hyper-activated cells with pro-inflammatory properties (43, 44). These pro-inflammatory responses were observed in animal models of MS and LPS-induced sepsis (42, 43). Overall, these studies demonstrate that deletion of Socs3 in myeloid cells contributes to hyper-activation and enhanced inflammation, leading us to postulate that Socs3-deficient myeloid cells may play a potential role in the pathogenesis of DSS-induced colitis.
In this study, we investigated the pathogenic function of myeloid cells in a colitis model induced by DSS in Socs3ΔLysM mice. Our results demonstrate that neutrophils from Socs3ΔLysM mice are critical for disease severity and inflammatory responses in DSS colitis. Furthermore, neutrophil depletion using neutralizing antibody 1A8 significantly reduces inflammation in DSS colitis in Socs3ΔLysM mice. Overall, our findings demonstrate the importance of Socs3 in repressing the hyper-activation of myeloid cells, specifically neutrophils, which contribute to our understanding of the role of neutrophils in IBD/colitis.
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of UAB. C57BL/6 mice were bred in an animal facility at the University of Alabama at Birmingham (UAB). Transgenic mice with the Socs3 locus flanked with flox sequences [Socs3fl/fl; (45)] were the generous gift of Dr. Warren Alexander (Walter and Eliza Hall Institute of Medical Research; Victoria, Australia) and were bred at UAB. Socs3 conditional knockout (Socs3ΔLysM) mice were generated by serial breeding of Socs3fl/fl mice with mice expressing Cre-recombinase under the control of the LysM promoter (42, 44). All mice were used at 8-12 weeks of age.
Colitis was induced in female Socs3fl/fl and Socs3ΔLysM mice with 3% DSS (molecular weight=40 kDa, MP Biomedical. Solon, OH), which was supplied in drinking water as described (8). DSS-induced colitis in male mice shows higher severity and faster progression compared to female mice. Combining both sexes significantly increased variability, therefore only female mice were used in this study. Weight loss of mice was monitored daily, and mice were sacrificed at day 5 for immune cell analysis and at day 7 for histology analysis (8, 46, 47). A scoring system was applied to assess diarrhea and the presence of occult or overt bleeding in the stool. Disease severity scores were calculated at day 7 by weight loss, stool bleeding, and stool consistency, with combined scores ranging from 0 to 12 (Table 1). Disease severity scores were determined by two investigators blinded to the treatment groups.
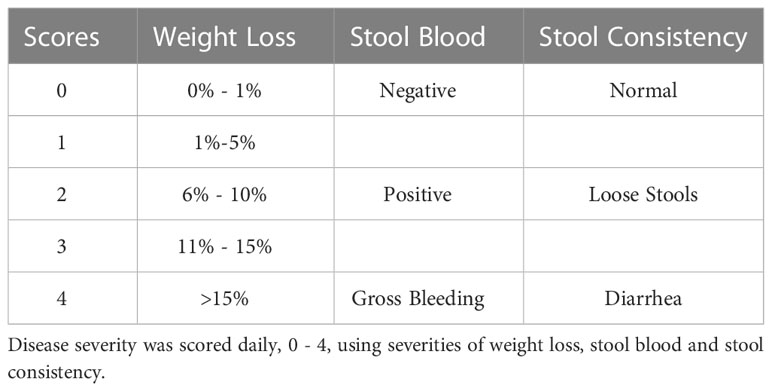
Table 1 DSS disease severity scoring criteria.
Colon lamina propria cells were prepared using the Lamina Propria Dissociation Kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions (48). Lamina propria immune cells were from the intestines as previously described (49). Tissues were rinsed, sliced into small pieces, and incubated at 37°C for 40 min. The tissues were then digested for 50 min at 37°C by continuous stirring with magnetic micro stir bars. The liberated cells were collected by passage through a 100 μm cell strainer (BD Falcon). Mononuclear cells were isolated from the lamina propria using a 40%/75% Percoll (GE Healthcare, 17089101) gradient. The isolated cells were resuspended in R10 medium (RPMI 1640 with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 50 μM β-mercaptoethanol). The cell yield was typically 2-3 x 106 immune cells per mouse with 90% cell viability.
Single-cell suspensions of spleen were prepared as previously described (50, 51) and resuspended in R10 medium.
Swiss rolls of colons were prepared as previously described (52). Tissues were fixed in 10% formalin in water overnight prior to paraffin-embedding. Paraffin-embedded samples were cut into 5 μm sections followed by hematoxylin and eosin (H&E) staining, and histopathology was evaluated in a double-blinded manner by a GI pathologist. Epithelium damage and inflammation severity were scored separately, and a total score was given accordingly. These scores were comprised of the following factors: severity of lesion, degree of hyperplasia, degree of ulceration, and percent of area involved. Severity of lesion referred to the overall size, quantity, and spatial dispersion of lesions. Degree of hyperplasia referred to crypt and goblet cell morphology, hyperchromasia, and mitotic index of goblet cells. Degree of ulceration referred to number of ulcers and the number of crypts involved. Epithelial damage was evaluated in all sections by hyperplasia, goblet cell loss, degeneration and necrosis, ulceration, and dysplasia. Inflammation severity was evaluated by crypt exudate, lamina propria and submucosal inflammatory cell accumulation intensity, submucosal edema distribution and transmural inflammation. Severity of lesion was graded as follows: 0 = normal, 1 = mild, 2 = moderate, 3 = severe (7, 48, 53).
For flow cytometry experiments, antibodies (Abs) directed against murine CD11b (M1/70), CD45 (30-F11), Ly6C (HK1.4), Ly6G (1A8), CD3 (17A2), CD4 (GK1.5), and CD19 (6D5) were from BioLegend. Cell phenotypes were determined based on surface staining patterns analyzed by flow cytometry, as previously described (14, 42, 44, 48). All flow cytometry data were analyzed using FlowJo software (BD), as previously described (44). For neutrophil depletion experiments, Ly6G was detected after by intracellular staining of Ly6G after fixation and permeablization with eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Invitrogen, 00-5523-00) (44).
Total RNA was purified from FACS-sorted neutrophils and Ly6C+ monocytes isolated by flow cytometry using TRIzol reagent extraction. For qRT-PCR analysis, 500-1000 ng of RNA was used as a template for cDNA synthesis (Promega). qRT-PCR was performed using TaqMan primers purchased from Thermo Fisher Scientific. The resulting data were analyzed using the comparative cycle threshold method to calculate relative RNA quantities (44, 50).
ELISA Analysis. Fecal samples were collected from day 4 post-DSS administration. LCN2 levels were measured by ELISA using the Lipocalin-2 (LCN2) Mouse ELISA Kit (ThermoFisher) (8, 54).
Significant differences between two groups were analyzed by the nonparametric Mann-Whitney test. One-way ANOVA was used to compare differences between more than two variables. P-values less than 0.05 were considered statistically significant. All error bars represent standard error of the mean (SEM).
To assess the function of Socs3 in myeloid cells in the pathogenesis of colitis, Socs3fl/fl mice crossed with LysM-Cre mice (Socs3ΔLysM), which leads to deletion of Socs3 in myeloid cells, including macrophages, neutrophils and partially in dendritic cells, were used (43, 44). Acute colitis was induced in mice by administering water containing 3% DSS polymers for 7 days (Figure 1A), which induces a colitis characterized by diarrhea, bloody feces, and weight loss as seen in human IBD. Results demonstrate that Socs3ΔLysM mice have more severe colitis compared to Socs3fl/fl mice at days 6 and 7 after DSS feeding. Socs3ΔLysM mice exhibited reduced body weight, increased disease severity scores, and reduced colon lengths compared to Socs3fl/fl mice (Figures 1B–E). Colon length was recorded as a proxy for degree of longitudinal contraction as a result of inflammation, diarrhea, and hypertrophy of the muscularis mucosae (55). Consistent with these findings, histopathological analysis showed more severe inflammation in the colon of Socs3ΔLysM mice compared to Socs3fl/fl mice (Figures 1F, G). Fecal lipocalin 2 (LCN2), measured by ELISA, is a non-invasive and well-characterized biomarker of intestinal inflammation in the DSS colitis model (8, 54). ELISA was performed on fecal samples collected from day 4 post-DSS administration due to difficulties in sample collection on later days. LCN2 levels were significantly higher in the fecal samples from Socs3ΔLysM mice compared to Socs3fl/fl mice at day 4 (Figure 1H). Colon length, colon histology, spleen weight, and distribution of immune cell populations in the colon and spleen from naïve Socs3fl/fl and Socs3ΔLysM mice were comparable (Supplementary Figure 1). These results indicate that Socs3 expression in myeloid cells is required for suppressing the development of DSS-induced colitis.
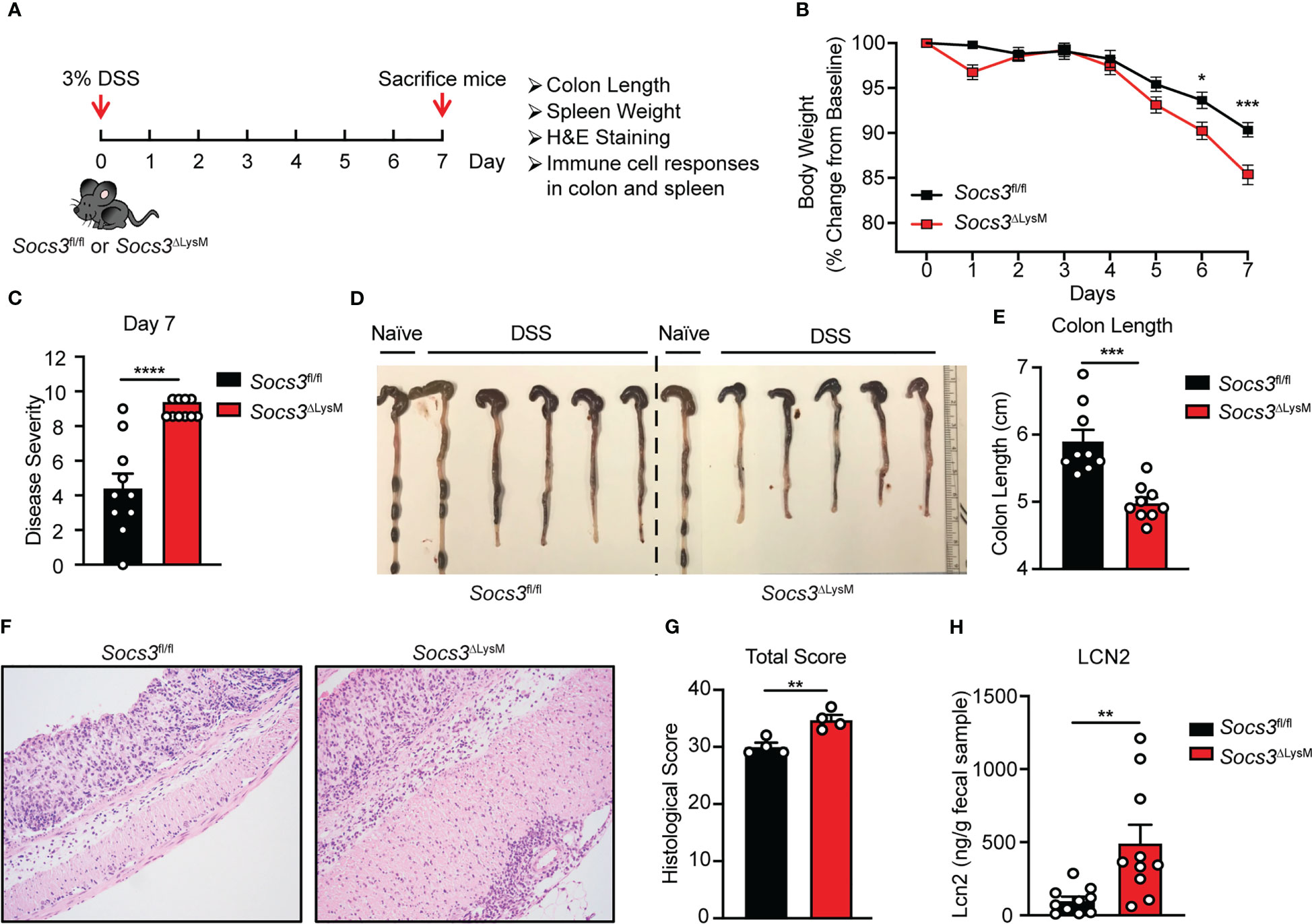
Figure 1 Socs3 deficiency in myeloid cells leads to more severe colitis induced by DSS. (A) Female Socs3fl/fl (n = 10) or Socs3ΔLysM (n = 10) mice were fed with 3% DSS for 7 days. Mice were weighed daily, and weight loss was determined by comparing to weight on Day 0. Mice were euthanized on day 7. Colon and spleen were collected for colon length, spleen weight, colon histology and immune cell responses. (B) Mice were weighed daily, and weight loss was determined by comparing to weight on Day 0. (C) Disease severity was calculated on day 7. (D) Representative pictures of the colon. (E) Statistics of colon length (n = 9). (F) Haemotoxylin and Eosin (H&E) staining of the distal colon. Pictures are representation of 4 mice. (G) Total pathological scores of colons are shown using severity of lesion, degree of hyperplasia, degree of ulceration and percent of area involved (n = 4) in Socs3fl/fl or Socs3ΔLysM mice. (H) Fecal samples were collected on day 4 post-DSS administration. LCN2 levels were determined by ELISA (n = 10). *p < 0.05, **p<0.01, ***p < 0.001, ****p < 0.0001.
To elucidate the primary cell type(s) responsible for the inflammation observed in this model, we used multicolor flow cytometry analysis to observe changes in cell populations. We characterized immune cell infiltration when weight loss was apparent and signs of inflammation were macroscopically visible, specifically day 5 post-DSS administration (Figure 2B). Immune cell populations were determined and assessed using the gating strategy represented in Figure 2A. As a percentage of total CD45+ cells, Socs3ΔLysM mice displayed a significant increase in CD45+CD11b+ myeloid cells and a significant decrease in the CD45+CD11b- lymphocyte population (Figure 2C). As a measure of total cell counts, Socs3ΔLysM mice displayed significantly increased CD45+CD11b+Ly6G+ neutrophil (Neu), Ly6G-Ly6C+ monocyte (Mono Ly6C+) and Ly6G-Ly6C- monocyte (Mono Ly6C-) populations (Figure 2D). Lymphocytes were also further characterized into CD4+ and CD4- T-cell subsets and B-cells, with no differences observed in any population in the colon (Supplementary Figure 2A). These results suggest that myeloid cells, specifically neutrophils, Ly6C+ monocytes and Ly6C- monocytes, may have a potential role in the hyper-inflammation observed in DSS-induced colitis in Socs3ΔLysM mice.
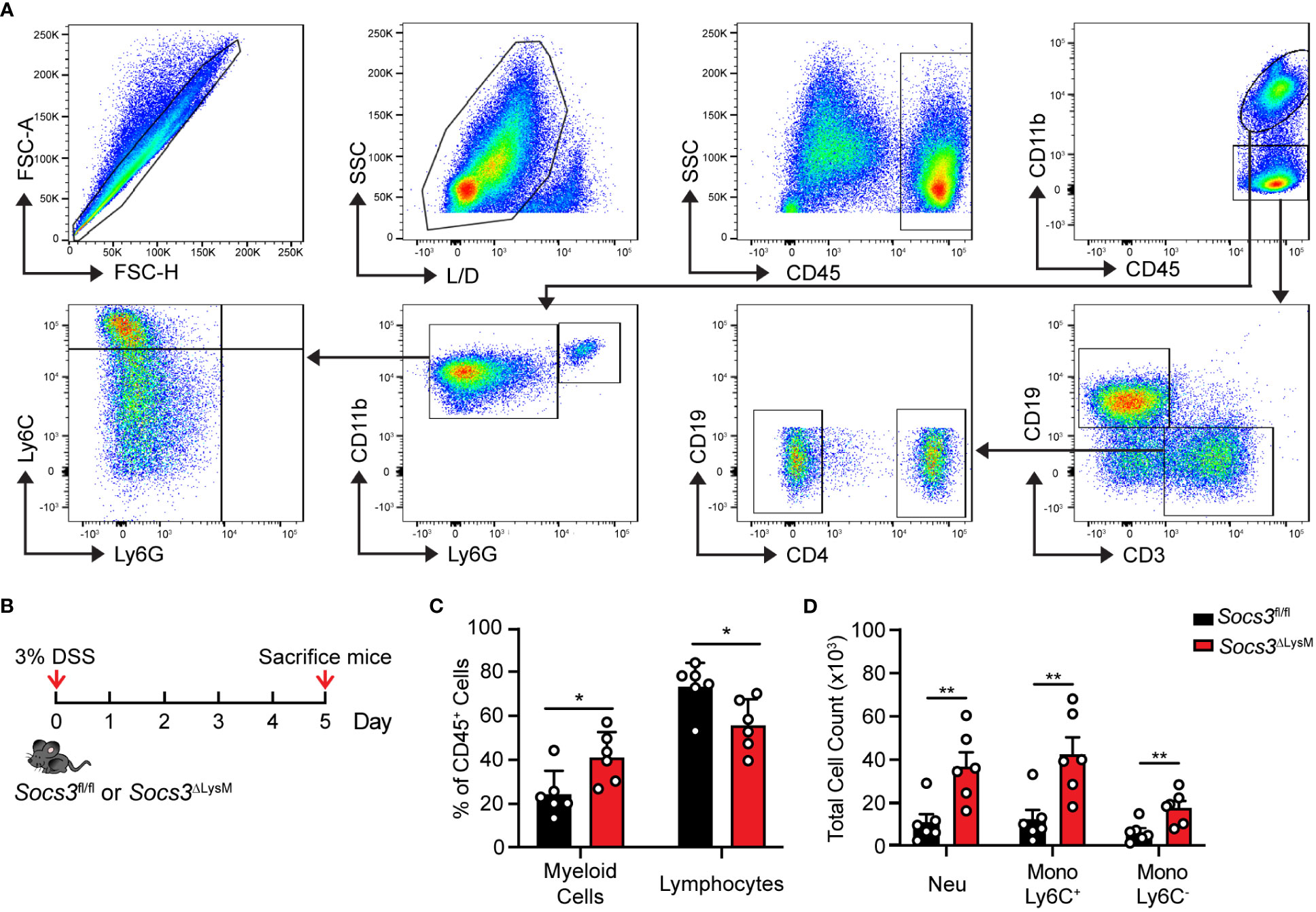
Figure 2 Socs3 deficiency increases colonic myeloid cell infiltration induced by DSS. (A) Singlets were gated for live CD45+ immune cells, CD45+CD11b+ myeloid cells and CD45+CD11b- lymphocytes. Ly6G+ neutrophils and Ly6G- monocytes were gated from the myeloid population; CD19-CD4+ T-cells, CD19-CD4- T-cells and CD3-CD19+ B-cells were gated from lymphocytes. Ly6C+ monocytes and Ly6C- monocytes were gated from the Ly6G- population. (B) Socs3fl/fl (n = 6) or Socs3ΔLysM (n = 6) mice were fed with 3% DSS. Mice were euthanized on day 5 and colonic infiltrating cells were isolated and stained for multicolor flow cytometry analysis. Total number of infiltrating myeloid cells and lymphocytes (C) and neutrophils and different monocyte subsets (D) in the colon from Socs3fl/fl or Socs3ΔLysM mice were calculated and compared. not significant (ns) p ≥ 0.05, *p < 0.05, **p < 0.01 and ***p<0.001.
In accordance with our findings of immune cells derived from the colon, we wanted to confirm whether these were mirrored in systemic inflammation as represented by splenic size and weight. In correlation with colon lengths depicted in Figures 1D, E, we first report splenic weight after 7 days of DSS administration and found that regardless of mouse phenotype, both groups of mice developed splenomegaly as compared to spleens taken from healthy controls (Figure 3A). However, mice lacking Socs3 displayed an exacerbated effect, having significantly larger spleens than those with intact Socs3 (Figure 3B). Because the overall increase in size and weight suggested an increase in immune cell infiltration, we further characterized isolated splenocytes at our designated early disease time point, day 5, using multicolor flow cytometry. Using the same gating strategy from Figure 2, we found results comparable to those from isolated colonic cells. Socs3ΔLysM mice not only displayed a significant increase in overall myeloid cells, but this increase was observed in the Neu and Mono Ly6C+ populations but not the Mono Ly6C- population (Figures 3C, D). While there was no difference between the overall lymphocyte population (Figure 3E), a significant increase in the CD4+ T-cell subpopulation in the spleen was observed, but not in the CD4- T-cell subpopulation nor the B-cell population (Supplementary Figure 2B). Overall, these data indicate that Socs3 deficiency leads to increased inflammatory responses in the colon and spleen of these mice.
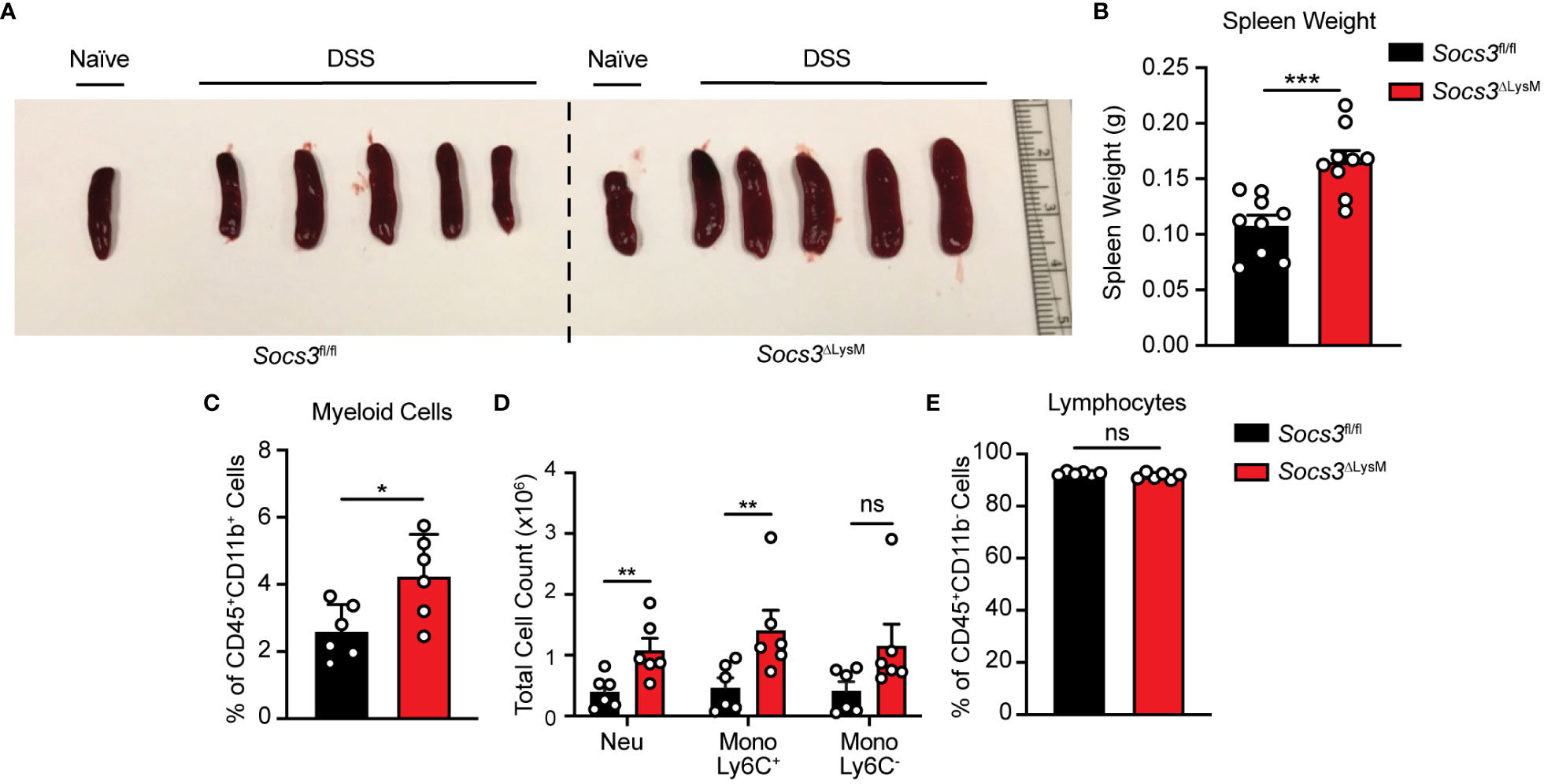
Figure 3 Socs3 deficiency increases splenic myeloid cell numbers in DSS-induced colitis. Representative spleen picture (A) and statistics of spleen weight (B) at day 7 (n = 9). Splenocytes were isolated and stained at day 5 as described in Figures 2A, B. Percentage of CD45+CD11b+ myeloid cells (C) and cell numbers of neutrophils and different monocyte subsets (D) from Socs3fl/fl (n = 6) or Socs3ΔLysM (n = 6) mice fed with 3% DSS on day 5. Percentage of CD45+CD11b- lymphocytes (n = 6) (E) in the spleen from Socs3fl/fl or Socs3ΔLysM mice. *p < 0.05, ***p < 0.001.
Considering that Socs3ΔLysM mice displayed an increase in both monocyte and neutrophil populations, we compared the expression of proinflammatory mediators at the RNA level through qRT-PCR using sorted Ly6C+ monocytes and neutrophils from colonic tissue. Previous studies have suggested that Ly6Chi monocytes are precursors to macrophages during inflammatory conditions while Ly6Clo monocytes are precursors to steady-state tissue macrophages (56–58). Therefore, these experiments were performed using Ly6C+ monocytes instead of Ly6C- monocytes. Our results indicated that colonic neutrophils from Socs3ΔLysM mice exhibited higher expression levels of Il1β compared to those of Socs3fl/fl mice (Figure 4A). Additionally, we examined changes in expression of genes pertinent to the pathogenesis of colitis. We found that neutrophils from Socs3ΔLysM mice displayed higher levels of expression of Lcn2, S100a8, and S100a9 (Figures 4B–D), with the latter two having emerging roles as pre-clinical biomarkers for neutrophilic inflammation such as that observed in IBDs (39, 53, 54). Interestingly, there were no observable differences between Socs3fl/fl and Socs3ΔLysM Ly6C+ monocyte populations for any of these genes (Figures 4A–D). Additional genes reportedly expressed by monocytes/macrophages, including Cd74, Tnfα, Il6, Il12α, Il12β, Ccl2, Cxcl1, Cxcl10 and iNOS, were determined by qRT-PCR (Supplementary Figure 3) (42, 43, 59). Interestingly, the only significant difference we found was that Socs3ΔLysM neutrophils displayed significantly higher levels of Il12β expression compared to Socs3fl/fl neutrophils. These findings suggest that monocytes/macrophages may not be the culprit of the heightened colonic inflammation in Socs3ΔLysM mice. Rather, Socs3-deficient neutrophils are critical for the exacerbation of DSS-induced colitis in Socs3ΔLysM mice.
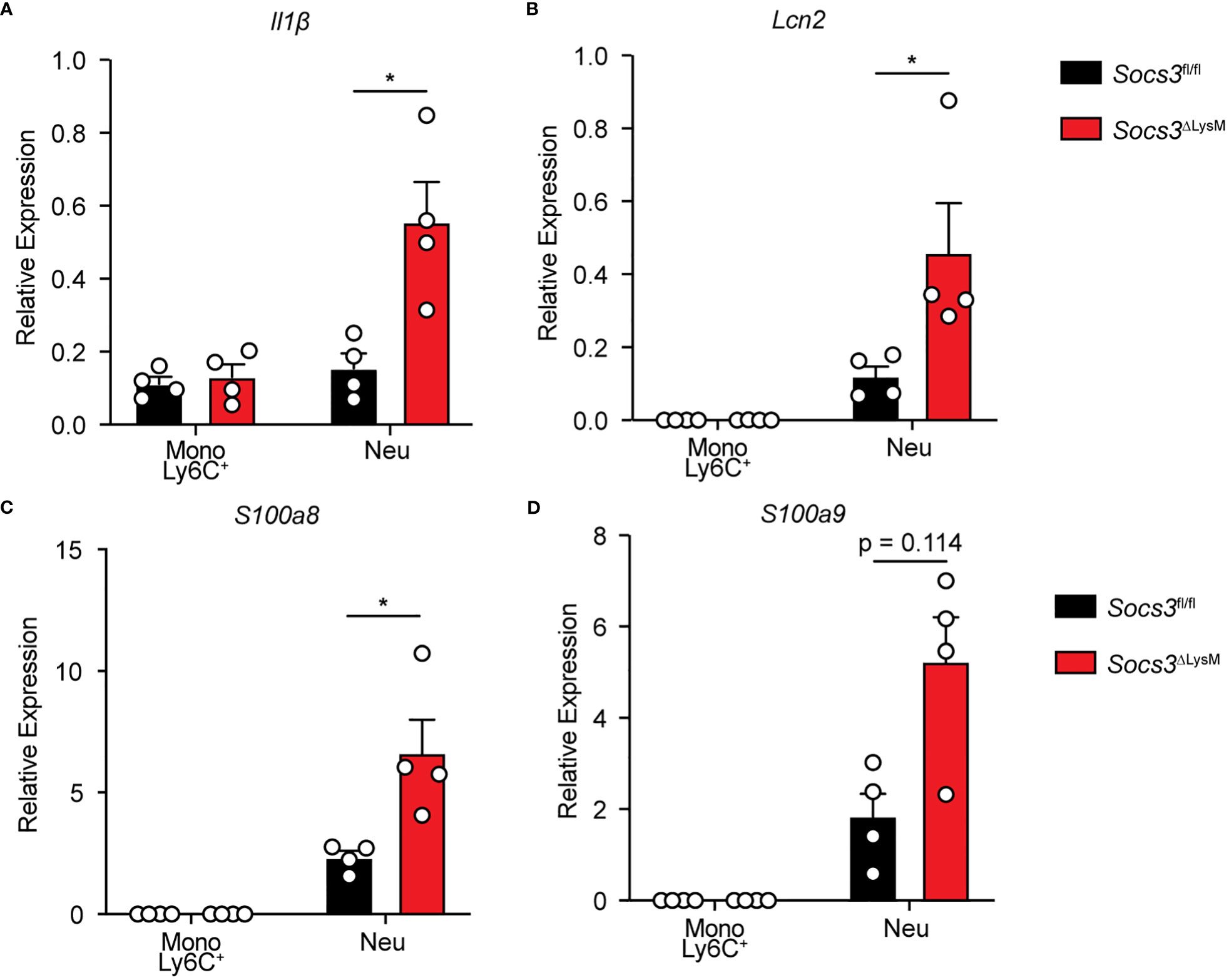
Figure 4 Socs3-deficient neutrophils produce increased proinflammatory mediators in DSS-induced colitis. Socs3fl/fl (n = 8) or Socs3ΔLysM (n = 8) mice were fed with 3% DSS. Mice were euthanized on day 5, and immune cells were sorted from colon tissue by flow cytometry. Gene expression in Ly6C+ monocytes and neutrophils from Socs3fl/fl or Socs3ΔLysM mice was examined. Gene expression of Il1β (A), Lcn2 (B), S100a8 (C), and S100a9 (D) were determined by qRT-PCR, using 18s rRNA as an internal control. Two mice were combined for each experiment with 4 experiments in each group. *p < 0.05.
Thus far, our results have suggested a strong correlation between Socs3 deletion, augmented neutrophil infiltration, increased expression of pro-inflammatory mediators by neutrophils, and exacerbated colitis. We, therefore, hypothesized that depletion of neutrophils would ameliorate intestinal inflammation and mitigate the effects of DSS colitis in Socs3ΔLysM mice. To determine the role of neutrophils in the exacerbation of DSS-induced colitis in Socs3ΔLysM mice, we developed an acute neutrophil depletion regimen by injection of anti-Ly6G antibody. Our method results in a significantly lower frequency of circulating neutrophils for seven days in the anti-Ly6G group compared to the isotype control Ab group (Supplementary Figure 4), confirming successful and conserved neutrophil depletion. We depleted neutrophils two days prior to DSS administration, followed by one dose on the initial day of DSS administration, and a final dose two days thereafter (Figure 5A). After seven days of DSS administration, mice were euthanized for spleen and colon tissue collection. Importantly, Socs3ΔLysM mice that received the neutrophil depletion antibody exhibited less severe forms of DSS-induced colitis as indicated by reduced body weight loss and lower disease severity scores (Figures 5B, C). To confirm our findings at the histopathological level, we stained the colonic tissue using H&E staining for imaging to assess and quantify the degree of intestinal damage. As shown in the representative images, we found that neutrophil depletion led to partial conservation of tissue structure, which was supported by significantly lower total histological scores (Figures 5D, E). Mice treated with the anti-Ly6G antibody displayed significantly lower severity of lesion scores compared to those that received the isotype control (Figure 5F).
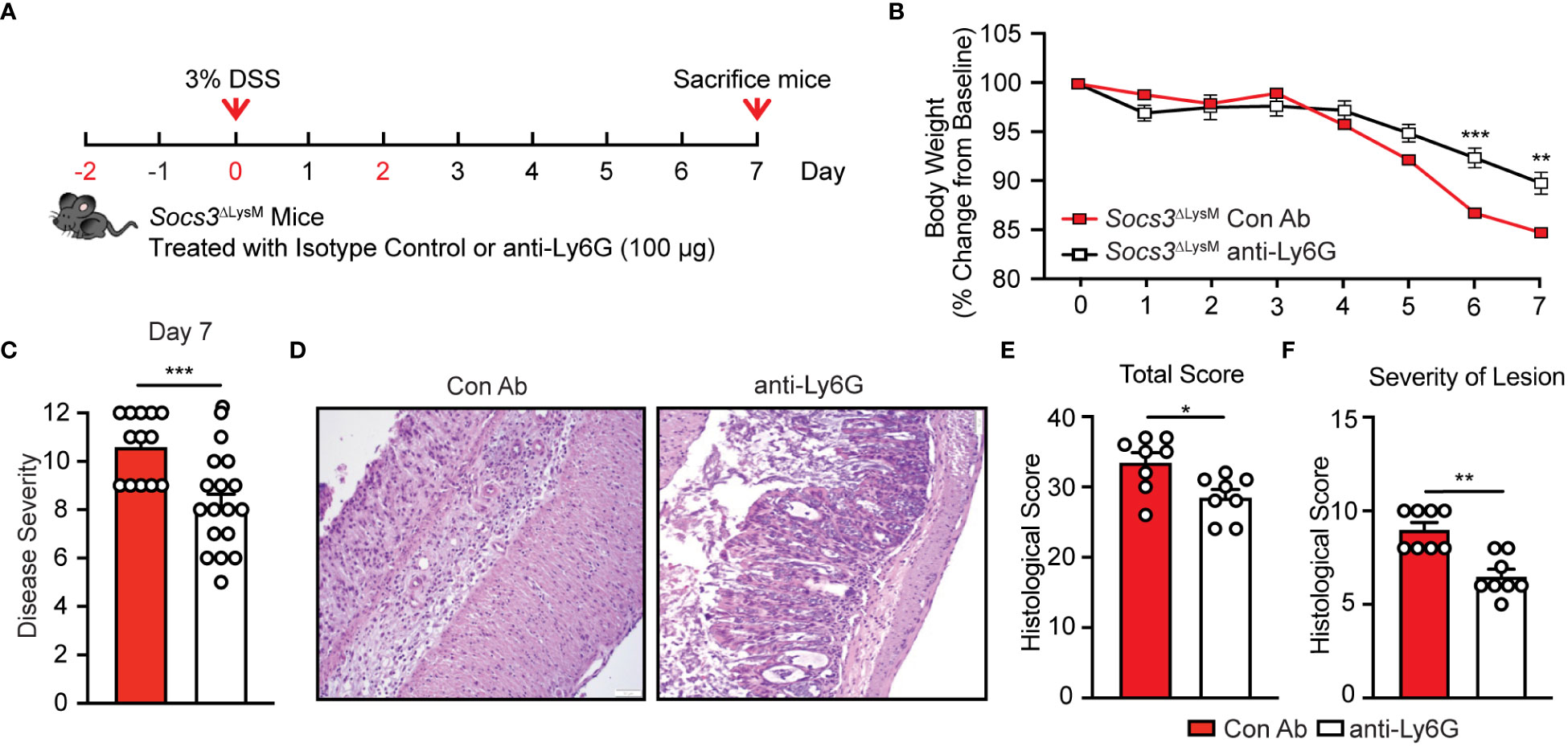
Figure 5 Neutrophil depletion significantly improves DSS-induced colitis in Socs3ΔLysM mice. (A) Female Socs3ΔLysM mice were injected with either anti-Ly6G (anti-Ly6G; n = 17) or Isotype Control (Con Ab; n = 13) Abs (100 µg/dose) intraperitoneally at days -2, 0 and 2 (shown in red). Mice were fed with 3% DSS for 7 days. (B) Anti-Ly6G (n = 17) and Con Ab (n = 13) mice were weighed daily, and weight loss was determined by comparing to weight daily from day 0 to day 7. (C) Disease severity on day 7. (D) H&E staining of the distal colon. Pictures are representation of 8 mice. (E) Total pathological scores of colons are shown using severity of lesion, degree of hyperplasia, degree of ulceration and percent of area involved (n = 8). (F) Severity of lesion was calculated in Socs3ΔLysM mice administered anti-Ly6G or isotype control abs. *p < 0.05, **p < 0.01 and ***p<0.001.
To determine whether these therapeutic effects could be recapitulated in Socs3fl/fl mice, these mice were subjected to the same neutrophil-depleting treatment as the Socs3ΔLysM mice. Neutrophil depletion had no effect on DSS-induced colitis in Socs3fl/fl mice. As shown in Supplementary Figure 5, weight loss, disease severity scores, and histological analysis at day 7 were comparable between the anti-Ly6G treated mice and the isotype control Ab treated mice. Taken together, these data suggest the detrimental role of hyperactivated neutrophils in the pathogenesis of DSS-induced colitis in Socs3ΔLysM mice.
In the present study, we report the detrimental role of myeloid cell Socs3 deletion in DSS-induced colitis. We performed unbiased FACS analysis and discovered that Socs3 deficiency in myeloid cells leads to profound infiltration of macrophages and neutrophils in the lamina propria. Interestingly, only neutrophils from Socs3ΔLysM mice show increased pro-inflammatory properties, compared to Socs3fl/fl controls. Furthermore, depleting Socs3-deficient neutrophils using a monoclonal neutralizing antibody significantly improved the pathology of colitis in mice. Our findings support the critical role of SOCS3, a negative regulator of the JAK/STAT pathway, in neutrophils for mitigating intestinal inflammation in a DSS-induced model of IBD.
To date, there is no evidence indicating that Socs3 is a risk factor for IBD. However, recent studies have shown that Socs3 mRNA expression is increased in the mucosa tissue of UC patients, compared to inactive control tissues (60). The same study also reported reduced Socs3 in IBD patients to be correlated with more frequent remission (60). Another study supported these findings by identifying high levels of SOCS3 protein in UC patients (61). Conversely, other groups found lower Socs3 expression to be associated with IBD-associated dysplasia (62) as well as an increased susceptibility to IBD (63). These findings demonstrate the complex role of Socs3 in IBD, suggesting context-dependent functionality, either beneficial or detrimental.
While the original objective of this study was to focus on myeloid cells, our data unveiled a detrimental role of neutrophils in an animal model of IBD. Although this is in line with some reports (38), other studies suggest that neutrophil depletion exacerbates intestinal inflammation (39, 64, 65). This discrepancy in findings can be explained by the ever-growing importance of neutrophil heterogeneity as well as neutrophil subsets. Historically, neutrophils were thought to be a homogeneous population of terminally differentiated cells. However, recent studies have shown that neutrophils display phenotypic and transcriptional plasticity and diversity in terms of functionality (66, 67). The occurrence of neutrophil heterogeneity has recently been confirmed, and phenotypic clusters have been characterized by scRNA-Seq (68–70). More importantly, these neutrophil subsets play opposite roles during intestinal inflammation. Recent studies using scRNA-Seq revealed complex heterogeneity of neutrophils in homeostasis and inflammation (71, 72) and demonstrated dual roles of neutrophils during inflammation. Future studies will use scRNA-Seq to examine the composition of neutrophils in animal models of IBD and determine whether hyperactive JAK/STAT signaling led by Socs3 deficiency skews neutrophil subsets towards a pro-inflammatory status.
Our current study revealed significant up-regulation of antimicrobial peptides in Socs3-deficient neutrophils during inflammation, namely LCN2, S100A8, and S100A9. The role of antimicrobial peptides is also complex. LCN2 promotes neutrophil-mediated anti-bacterial activity (73), and the serum level of LCN2 is an ideal biomarker of active UC (74). However, Lcn2 deficiency leads to severe intestinal inflammation in Il10-/- mice (75), indicating a complex role for Lcn2 in IBDs. S100A8/S100A9 are also sensitive biomarkers for IBD (76) and work as endogenous activators of Toll-like receptor 4 (TLR4) (77). On the other hand, S100A8/S100A9 are also critical to contain inflammation at the local level, as S100A9-/- mice result in TNF-α-driven fatal inflammation (78). Future studies will determine the role of increased anti-microbial peptides in Socs3-deficient neutrophils in colitis.
In addition to antimicrobial peptides, neutrophils have been shown to increase production of pro-inflammatory cytokines and chemokines during intestinal inflammation (79). Cytokines such as IL-1β, IL-6, IL-8, TNF-α, GM-CSF and G-CSF are critical for secondary neutrophil recruitment and infiltration into the intestinal mucosa, and chemokines such as CXCL-1, -8, and -10 are necessary for recruitment of other immune cells (80). Our study revealed an increase in Il1β expression by Socs3-deficient neutrophils at the RNA level during DSS-induced colitis. This result is congruent with clinical findings of higher IL-1β concentrations present in the intestines of IBD patients (81). The same study described crosstalk between IL-1β and IL-23 to sustain both innate and adaptive inflammatory responses during intestinal inflammation (81). A recent study also demonstrated that a subset of IBD patients, non-responsive to anti-TNF, corticosteroid, and anti-integrin therapy, showed significant IL-1β-driven neutrophil interaction with intestinal stromal cells (82). Future studies will perform comprehensive profiling to observe changes in a broader selection of pro-inflammatory mediators, including cytokines (e.g., IL-1β, G-CSF), chemokines (e.g., CXCL1, CXCL10) and oxidative stress-related free radicals, as a result of Socs3 deletion.
Our data also suggest that inhibiting neutrophil activity attenuates experimental colitis as neutrophil depletion significantly improved disease in the DSS colitis model in Socs3ΔLysM mice. However, because these findings were not observed in Socs3fl/fl mice, further studies on the potential protective mechanisms of Socs3 in human IBD development are needed. Additionally, the exact source of neutrophils’ deleterious effects is unclear. Some studies report impairment of neutrophil recruitment and trafficking significantly improving the pathology of colitis (83, 84), while others have demonstrated efficacy in degrading neutrophil extracellular traps (85). Intestinal oxidative damage is prominent in both forms of IBD (86–88), and we have previously shown that deletion of Socs3 in myeloid cells leads to elevated neutrophil activation and increased production of reactive oxygen species (44). Therefore, further investigation will be conducted to elucidate the mechanisms behind neutrophil-specific inflammatory responses in this model.
In summary, our study using a clinically relevant experimental colitis model provides further insights into the role of neutrophils in IBDs. Not only do these results unveil the potential for exploitation of neutrophil function, but they also demonstrate how targeting neutrophil activity holds potential clinical significance. As such, this may pave the way for therapies aimed at mitigating the effects of pro-inflammatory neutrophils.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by University of Alabama at Birmingham.
HQ contributes to experimental designs, results and data analysis and manuscript writing. ZY contributes to experimental designs, experiment performance, results and data analysis and manuscript writing. LZ contributes to experimental designs, experiment performance, results and data analysis and manuscript writing. WY contributes to experimental designs and experiment performance. JB contributes to experiment performance. SD contributes to Histology data analysis. EB contributes to experimental designs and manuscript writing.
This work was supported by National Institutes of Health Grants R01NS057563 and R01CA194414 (to EB), and National Multiple Sclerosis Society Grant RG-1606-24794 (to HQ).
The Comprehensive Flow Cytometry Core at the University of Alabama at Birmingham is supported by National Institutes of Health Grants P30 AR048311 and P30 AI27667. Tissue Biorepository at the University of Alabama at Birmingham/Tissue Procurement Shared Resource (UAB-TBR/TPRO).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1163987/full#supplementary-material
1. Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol (2003) 3:521–33. doi: 10.1038/nri1132
2. Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol (2008) 8:458–66. doi: 10.1038/nri2340
3. Plichta DR, Graham DB, Subramanian S, Xavier RJ. Therapeutic opportunities in inflammatory bowel disease: mechanistic dissection of host-microbiome relationships. Cell (2019) 178:1041–56. doi: 10.1016/j.cell.2019.07.045
4. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
5. Valatas V, Bamias G, Kolios G. Experimental colitis models: insights into the pathogenesis of inflammatory bowel disease and translational issues. Eur J Pharmacol (2015) 759:253–64. doi: 10.1016/j.ejphar.2015.03.017
6. Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med (2011) 365:1713–25. doi: 10.1056/NEJMra1102942
7. Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol (1998) 274:G544–551. doi: 10.1152/ajpgi.1998.274.3.G544
8. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol (2014) 104(15):25. doi: 10.1002/0471142735.im1525s104
9. Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J BioMed Biotechnol (2012) 2012:718617. doi: 10.1155/2012/718617
10. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of jak-STAT signaling in the immune system. Nat Immunol (2017) 18:374–84. doi: 10.1038/ni.3691
11. Philips RL, Wang Y, Cheon H, Kanno Y, Gadina M, Sartorelli V, et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell (2022) 185:3857–76. doi: 10.1016/j.cell.2022.09.023
12. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
13. Yan Z, Gibson SA, Buckley JA, Qin H, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol (2016) 189:4–13. doi: 10.1016/j.clim.2016.09.014
14. Liu Y, Holdbrooks AT, De Sarno P, Rowse AL, Yanagisawa LL, McFarland BC, et al. Therapeutic efficacy of suppressing the JAK/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J Immunol (2014) 192:59–72. doi: 10.4049/jimmunol.1301513
15. Qin H, Buckley JA, Li X, Liu Y, Fox TH, Meares GP, et al. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci (2016) 36:5144–59. doi: 10.1523/JNEUROSCI.4658-15.2016
16. Salas A, Hernandez-Rocha C, Duijvestein M, Faubion W, McGovern D, Vermeire S, et al. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol (2020) 17:323–37. doi: 10.1038/s41575-020-0273-0
17. Coskun M, Salem M, Pedersen J, Nielsen OH. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol Res (2013) 76:1–8. doi: 10.1016/j.phrs.2013.06.007
18. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol (2008) 19:414–22. doi: 10.1016/j.semcdb.2008.07.010
19. Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity (2004) 20:153–65. doi: 10.1016/S1074-7613(04)00022-6
20. Periasamy S, Wu WH, Chien SP, Liu CT, Liu MY. Dietary ziziphus jujuba fruit attenuates colitis-associated tumorigenesis: a pivotal role of the NF-kappaB/IL-6/JAK1/STAT3 pathway. Nutr Cancer (2020) 72:120–32. doi: 10.1080/01635581.2019.1615515
21. Lee HN, Choi YS, Kim SH, Zhong X, Kim W, Park JS, et al. Resolvin D1 suppresses inflammation-associated tumorigenesis in the colon by inhibiting IL-6-induced mitotic spindle abnormality. FASEB J (2021) 35:e21432. doi: 10.1096/fj.202002392R
22. Lin H, Feng L, Cui KS, Zeng LW, Gao D, Zhang LX, et al. The membrane-associated E3 ubiquitin ligase MARCH3 downregulates the IL-6 receptor and suppresses colitis-associated carcinogenesis. Cell Mol Immunol (2021) 18:2648–59. doi: 10.1038/s41423-021-00799-1
23. Dabritz J, Judd LM, Chalinor HV, Menheniott TR, Giraud AS. Altered gp130 signalling ameliorates experimental colitis via myeloid cell-specific STAT3 activation and myeloid-derived suppressor cells. Sci Rep (2016) 6:20584. doi: 10.1038/srep20584
24. Pathria P, Gotthardt D, Prchal-Murphy M, Putz EM, Holcmann M, Schlederer M, et al. Myeloid STAT3 promotes formation of colitis-associated colorectal cancer in mice. Oncoimmunology (2015) 4:e998529. doi: 10.1080/2162402X.2014.998529
25. Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med (2001) 193:471–81. doi: 10.1084/jem.193.4.471
26. Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, et al. STAT3 deletion during hematopoiesis causes crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA (2003) 100:1879–84. doi: 10.1073/pnas.0237137100
27. Reindl W, Weiss S, Lehr H-A, Förster I. Essential crosstalk between myeloid and lymphoid cells for development of chronic colitis in myeloid-specific signal transducer and activator of transcription 3-deficient mice. Immunology (2007) 120:19–27. doi: 10.1111/j.1365-2567.2006.02473.x
28. Gren ST, Grip O. Role of monocytes and intestinal macrophages in crohn's disease and ulcerative colitis. Inflammation Bowel Dis (2016) 22:1992–8. doi: 10.1097/MIB.0000000000000824
29. Grimm MC, Pullman WE, Bennett GM, Sullivan PJ, Pavli P, Doe WF. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J Gastroenterol Hepatol (1995) 10:387–95. doi: 10.1111/j.1440-1746.1995.tb01589.x
30. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118:2269–80. doi: 10.1172/JCI34610
31. Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J Immunol (2010) 184:6843–54. doi: 10.4049/jimmunol.0903987
32. Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol (2013) 6:498–510. doi: 10.1038/mi.2012.89
33. Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol (2008) 14:4280–8. doi: 10.3748/wjg.14.4280
34. Liu Y, Xu J, Ren X, Zhang Y, Ke Z, Zhou J, et al. Cholecystectomy-induced secondary bile acids accumulation ameliorates colitis through inhibiting monocyte/macrophage recruitment. Gut Microbes (2022) 14:2107387. doi: 10.1080/19490976.2022.2107387
35. Nicolas-Avila JA, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity (2017) 46:15–28. doi: 10.1016/j.immuni.2016.12.012
36. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science (2017) 358:111–6. doi: 10.1126/science.aam9690
37. Bressenot A, Salleron J, Bastien C, Danese S, Boulagnon-Rombi C, Peyrin-Biroulet L. Comparing histological activity indexes in UC. Gut (2015) 64:1412–8. doi: 10.1136/gutjnl-2014-307477
38. Natsui M, Kawasaki K, Takizawa H, Hayashi S-I, Matsuda Y, Sugimura K, et al. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J Gastroenterol Hepatol (1997) 12:801–8. doi: 10.1111/j.1440-1746.1997.tb00375.x
39. Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci United States America (2013) 110:12768–73. doi: 10.1073/pnas.1300318110
40. Zhang R, Ito S, Nishio N, Cheng Z, Suzuki H, Isobe KI. Dextran sulphate sodium increases splenic Gr1(+)CD11b(+) cells which accelerate recovery from colitis following intravenous transplantation. Clin Exp Immunol (2011) 164:417–27. doi: 10.1111/j.1365-2249.2011.04374.x
41. Bai R, Sun D, Chen M, Shi X, Luo L, Yao Z, et al. Myeloid cells protect intestinal epithelial barrier integrity through the angiogenin/plexin-B2 axis. EMBO J (2020) 39:e103325. doi: 10.15252/embj.2019103325
42. Qin H, Yeh W-I, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci USA (2012) 109:5004–9. doi: 10.1073/pnas.1117218109
43. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol (2012) 189:3439–48. doi: 10.4049/jimmunol.1201168
44. Yan Z, Yang W, Parkitny L, Gibson SA, Lee KS, Collins F, et al. Deficiency of Socs3 leads via enhanced neutrophil activation and ROS production. JCI Insight (2019) 5:e126520. doi: 10.1172/jci.insight.126520
45. Croker BA, Krebs DL, Zhang J-G, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol (2003) 4:540–5. doi: 10.1038/ni931
46. Urushima H, Nishimura J, Mizushima T, Hayashi N, Maeda K, Ito T. Perilla frutescens extract ameliorates DSS-induced colitis by suppressing proinflammatory cytokines and inducing anti-inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol (2015) 308:G32–41. doi: 10.1152/ajpgi.00294.2014
47. Jin BR, Chung KS, Cheon SY, Lee M, Hwang S, Noh Hwang S, et al. Rosmarinic acid suppresses colonic inflammation in dextran sulphate sodium (DSS)-induced mice via dual inhibition of NF-kappaB and STAT3 activation. Sci Rep (2017) 7:46252. doi: 10.1038/srep46252
48. Yang W, Gibson SA, Yan Z, Wei H, Tao J, Sha B, et al. Protein kinase 2 (CK2) controls CD4(+) T cell effector function in the pathogenesis of colitis. Mucosal Immunol (2020) 13:988–798. doi: 10.1038/s41385-020-0258-x
49. Feng T, Qin H, Wang L, Benveniste EN, Elson CO, Cong Y. Th17 cells induce colitis and promote Th1 cell responses through IL-17 induction of innate IL-12 and IL-23 production. J Immunol (2011) 186:6313–8. doi: 10.4049/jimmunol.1001454
50. Gibson SA, Yang W, Yan Z, Liu Y, Rowse AL, Weinmann AS, et al. Protein kinase CK2 controls the fate between Th17 cell and regulatory T cell differentiation. J Immunol (2017) 198:4244–54. doi: 10.4049/jimmunol.1601912
51. Gibson SA, Yang W, Yan Z, Qin H, Benveniste EN. CK2 controls Th17 and regulatory T cell differentiation through inhibition of FoxO1. J Immunol (2018) 201:383–92. doi: 10.4049/jimmunol.1701592
52. Bialkowska AB, Ghaleb AM, Nandan MO, Yang VW. Improved Swiss-rolling technique for intestinal tissue preparation for immunohistochemical and immunofluorescent analyses. J Vis Exp (2016) 13:54161. doi: 10.3791/54161
53. Bleich A, Mahler M, Most C, Leiter EH, Liebler-Tenorio E, Elson CO, et al. Refined histopathologic scoring system improves power to detect colitis QTL in mice. Mamm Genome (2004) 15:865–71. doi: 10.1007/s00335-004-2392-2
54. Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PloS One (2012) 7:e44328. doi: 10.1371/journal.pone.0044328
55. Gore RM. Colonic contour changes in chronic ulcerative colitis: reappraisal of some old concepts. AJR Am J Roentgenol (1992) 158:59–61. doi: 10.2214/ajr.158.1.1727359
56. Ma H, Wan S, Xia CQ. Immunosuppressive CD11b+Ly6Chi monocytes in pristane-induced lupus mouse model. J Leukoc Biol (2016) 99:1121–9. doi: 10.1189/jlb.3A0415-158R
57. Neal LM, Knoll LJ. Toxoplasma gondii profilin promotes recruitment of Ly6Chi CCR2+ inflammatory monocytes that can confer resistance to bacterial infection. PloS Pathog (2014) 10:e1004203. doi: 10.1371/journal.ppat.1004203
58. Helk E, Bernin H, Ernst T, Ittrich H, Jacobs T, Heeren J, et al. TNFalpha-mediated liver destruction by kupffer cells and Ly6Chi monocytes during entamoeba histolytica infection. PloS Pathog (2013) 9:e1003096. doi: 10.1371/journal.ppat.1003096
59. Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev (2010) 24:1236–41. doi: 10.1101/gad.1920310
60. Miyanaka Y, Ueno Y, Tanaka S, Yoshioka K, Hatakeyama T, Shimamoto M, et al. Clinical significance of mucosal suppressors of cytokine signaling 3 expression in ulcerative colitis. World J Gastroenterol (2007) 13:2939–44. doi: 10.3748/wjg.v13.i21.2939
61. Li Y, de Haar C, Chen M, Deuring J, Gerrits MM, Smits R, et al. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut (2010) 59:227–35. doi: 10.1136/gut.2009.184176
62. Gui X, Iacucci M, Ghosh S. Dysregulation of IL6/IL6R-STAT3-SOCS3 signaling pathway in IBD-associated colorectal dysplastic lesions as compared to sporadic colorectal adenomas in non-IBD patients. Pathol Res Pract (2020) 216:153211. doi: 10.1016/j.prp.2020.153211
63. Sanati G, Jafari D, Noruzinia M, Ebrahimi Daryani N, Ahmadvand M, Teimourian S, et al. Association of aberrant promoter methylation changes in the suppressor of cytokine signaling 3 (SOCS3) gene with susceptibility to crohn's disease. Avicenna J Med Biotechnol (2022) 14:165–9. doi: 10.18502/ajmb.v14i2.8887
64. Kuhl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology (2007) 133:1882–92. doi: 10.1053/j.gastro.2007.08.073
65. Zhang R, Ito S, Nishio N, Cheng Z, Suzuki H, Isobe K. Up-regulation of Gr1+CD11b+ population in spleen of dextran sulfate sodium administered mice works to repair colitis. Inflammation Allergy Drug Targets (2011) 10:39–46. doi: 10.2174/187152811794352114
66. Silvestre-Roig C, Fridlender ZG, Glogauer M, Scapini P. Neutrophil diversity in health and disease. Trends Immunol (2019) 40:565–83. doi: 10.1016/j.it.2019.04.012
67. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
68. Grieshaber-Bouyer R, Nigrovic PA. Neutrophil heterogeneity as therapeutic opportunity in immune-mediated disease. Front Immunol (2019) 10:346. doi: 10.3389/fimmu.2019.00346
69. Rossi B, Constantin G, Zenaro E. The emerging role of neutrophils in neurodegeneration. Immunobiol (2020) 225:151865. doi: 10.1016/j.imbio.2019.10.014
70. Woodberry T, Bouffler SE, Wilson AS, Buckland RL, Brustle A. The emerging role of neutrophil granulocytes in multiple sclerosis. J Clin Med (2018) 7:511. doi: 10.3390/jcm7120511
71. Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol (2020) 21:1119–33. doi: 10.1038/s41590-020-0736-z
72. Sas AR, Carbajal KS, Jerome AD, Menon R, Yoon C, Kalinski AL, et al. A new neutrophil subset promotes CNS neuron survival and axon regeneration. Nat Immunol (2020) 21:1496–1505. doi: 10.1038/s41590-020-00813-0
73. Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci USA (2006) 103:1834–9. doi: 10.1073/pnas.0510847103
74. Stallhofer J, Friedrich M, Konrad-Zerna A, Wetzke M, Lohse P, Glas J, et al. Lipocalin-2 is a disease activity marker in inflammatory bowel disease regulated by IL-17A, IL-22, and TNF-alpha and modulated by IL23R genotype status. Inflammation Bowel Dis (2015) 21:2327–40. doi: 10.1097/MIB.0000000000000515
75. Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ, et al. Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host Microbe (2016) 19:455–69. doi: 10.1016/j.chom.2016.03.007
76. Okada K, Okabe M, Kimura Y, Itoh H, Ikemoto M. Serum S100A8/A9 as a potentially sensitive biomarker for inflammatory bowel disease. Lab Med (2019) 50:370–80. doi: 10.1093/labmed/lmz003
77. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13:1042–9. doi: 10.1038/nm1638
78. Vogl T, Stratis A, Wixler V, Voller T, Thurainayagam S, Jorch SK, et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J Clin Invest (2018) 128:1852–66. doi: 10.1172/JCI89867
79. Nikolaus S, Bauditz J, Gionchetti P, Witt C, Lochs H, Schreiber S. Increased secretion of pro-inflammatory cytokines by circulating polymorphonuclear neutrophils and regulation by interleukin 10 during intestinal inflammation. Gut (1998) 42:470–6. doi: 10.1136/gut.42.4.470
80. Chen H, Wu X, Xu C, Lin J, Liu Z. Dichotomous roles of neutrophils in modulating pathogenic and repair processes of inflammatory bowel diseases. Precis Clin Med (2021) 4:246–57. doi: 10.1093/pcmedi/pbab025
81. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med (2012) 209:1595–609. doi: 10.1084/jem.20111453
82. Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med (2021) 27:1970–81. doi: 10.1038/s41591-021-01520-5
83. Pavlidis P, Tsakmaki A, Pantazi E, Li K, Cozzetto D, Digby-Bell J, et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat Commun (2022) 13:5820. doi: 10.1038/s41467-022-33331-8
84. Manzanares LD, David J, Ren X, Yalom LK, Piccolo EB, Dehghan Y, et al. Atovaquone attenuates experimental colitis by reducing neutrophil infiltration of colonic mucosa. Front Pharmacol (2022) 13:1011115. doi: 10.3389/fphar.2022.1011115
85. Dong W, Liu D, Zhang T, You Q, Huang F, Wu J. Oral delivery of staphylococcal nuclease ameliorates DSS induced ulcerative colitis in mice via degrading intestinal neutrophil extracellular traps. Ecotoxicol Environ Saf (2021) 215:112161. doi: 10.1016/j.ecoenv.2021.112161
86. Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci (1996) 41:2078–86. doi: 10.1007/BF02093613
87. Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol (2003) 201:28–36. doi: 10.1002/path.1409
Keywords: inflammatory bowel diseases (IBDs), ulcerative colitis (uc), suppressors of cytokine signaling (SOCS), myeloid cells, neutrophils
Citation: Zhou L, Yan Z, Yang W, Buckley JA, Al Diffalha S, Benveniste EN and Qin H (2023) Socs3 expression in myeloid cells modulates the pathogenesis of dextran sulfate sodium (DSS)-induced colitis. Front. Immunol. 14:1163987. doi: 10.3389/fimmu.2023.1163987
Received: 11 February 2023; Accepted: 05 May 2023;
Published: 22 May 2023.
Edited by:
Francesca Granucci, University of Milano-Bicocca, Milan, ItalyReviewed by:
Francesco Strati, University of Milano-Bicocca, ItalyCopyright © 2023 Zhou, Yan, Yang, Buckley, Al Diffalha, Benveniste and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwei Qin, aHFpbkB1YWIuZWR1; Zhaoqi Yan, emhhb3FpLnlhbkBnbGFkc3RvbmUudWNzZi5lZHU=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.