 Candice Lee Herd
Candice Lee Herd Juanita Mellet
Juanita Mellet Tsungai Mashingaidze
Tsungai Mashingaidze Chrisna Durandt
Chrisna Durandt Michael Sean Pepper
Michael Sean Pepper
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 July 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1163012
Dysregulation of the bone marrow niche resulting from the direct and indirect effects of HIV infection contributes to haematological abnormalities observed in HIV patients. The bone marrow niche is a complex, multicellular environment which functions primarily in the maintenance of haematopoietic stem/progenitor cells (HSPCs). These adult stem cells are responsible for replacing blood and immune cells over the course of a lifetime. Cells of the bone marrow niche support HSPCs and help to orchestrate the quiescence, self-renewal and differentiation of HSPCs through chemical and molecular signals and cell-cell interactions. This narrative review discusses the HIV-associated dysregulation of the bone marrow niche, as well as the susceptibility of HSPCs to infection by HIV.
The existence of the bone marrow niche was first proposed by Schofield (1) as a specialised microenvironment for the maintenance of haematopoietic stem and progenitor cells (HSPCs), and can be found in the marrow of long bones, vertebrae and iliac crest. The presence of multiple niches within the bone marrow has been proposed due to the presence of distinct subsets of HSPCs in close proximity to non-haematopoietic cell types (2). The bone marrow niche consists of bone matrix and various non-haematopoietic cells, including endothelial cells, stromal cells, neuronal cells and adipocytes (3). Cells of the niche contribute directly to HSPC quiescence, tethering in the bone marrow, homing to niche regions and mobilisation into the circulation, as well as differentiation through intercellular contact and paracrine signalling (3–5).
The bone marrow niche is separated into endosteal (6–8) and perivascular regions (4, 5, 9), each thought to serve a distinct function in the maintenance and mobilisation of HSPCs. Osteolineage cells, perivascular mesenchymal stromal/stem cells (MSCs), CXC chemokine ligand (CXCL)12-abundant reticular (CAR) cells and endothelial cells produce chemoattracting gradients of CXCL12 (also known as stromal-derived factor 1 (SDF-1) and stem cell factor (SCF)) which draw HSPCs to both the endosteal and perivascular regions (4, 5, 7). Non-myelinating Schwann cells and megakaryocytes activate transforming growth factor beta (TGF-β) which has been implicated in maintaining quiescence (5). Megakaryocytes are also thought to contribute to the niche function by releasing CXCL4 and small amounts of thrombopoietin (TPO) which encourage quiescence (4). Coupled with long-range and short-range cytokines regulating haematopoiesis, signalling networks in the bone marrow are extremely complex and have not been fully elucidated. The process of haematopoiesis is well studied and involves many cytokines, chemokines, cell-to-cell interactions and extracellular matrix interactions. However, the in vivo functionality, frequency and longevity of HSPCs in humans has not been fully defined.
The classical model of haematopoiesis is represented by a hierarchical structure with long-term HSPCs at the apex of the hierarchy (10–12). These cells possess self-renewal capabilities and give rise to short-term HSPCs with limited self-renewal capabilities. Short-term HSPCs differentiate to form multipotent progenitors (MPP), which are precursors of common lymphoid and myeloid progenitors (CLPs/CMPs). MPPs are not able to self-renew but are capable of full lineage differentiation (13). Progeny of CLPs differentiate into lymphoid and natural killer (NK) cells, while progeny of CMPs form granulocyte–macrophage progenitors (GMP) or megakaryocyte–erythrocyte progenitors (MEP). These differentiate into granulocytes and macrophages, and erythrocytes and megakaryocytes, respectively (14). Recent studies suggest that haematopoiesis is more complex than the classical model makes provision for, which includes myeloid-restricted progenitors with long-term repopulating potential (15) and HSPCs expressing platelet-biased genes while having the ability to self-renew (16). These examples represent only a fraction of the data demonstrating the non-classical differentiation potential of HSPCs. Technological advances in the past decade have resulted in a revised model depicted in Figure 1.
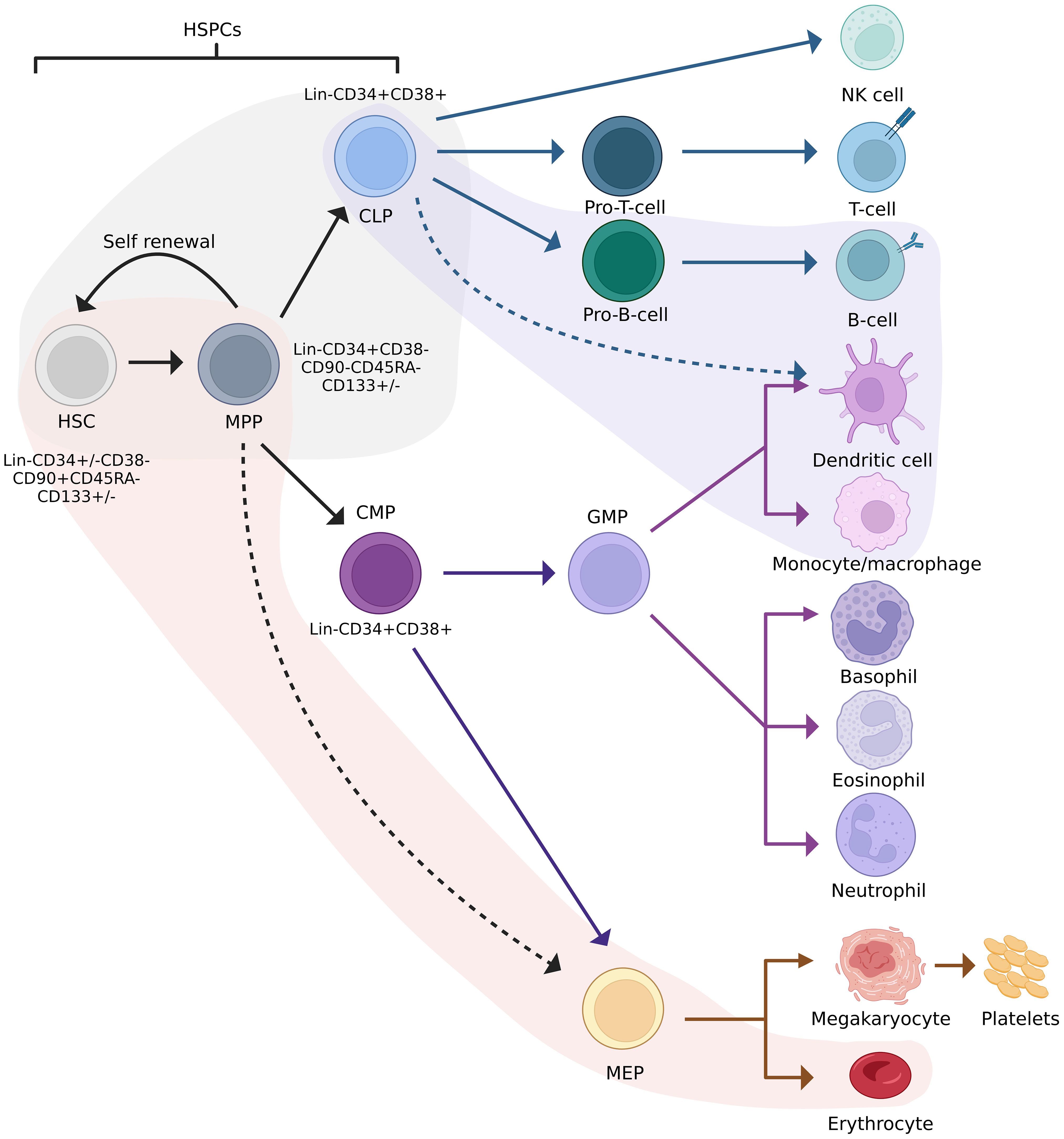
Figure 1 Revised model of haematopoiesis including HSPC phenotypes. Red and purple streams indicate lineage preferences. The grey area indicates cells between which lineage commitment may be reversed. HSC, haematopoietic stem cell; HSPC, haematopoietic stem/progenitor cell; MPP, multipotent progenitor; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GFM, granulocyte-macrophage progenitor; MEP, megakaryocyte-erythrocyte progenitor. Adapted from Velten et al., 2017, Brown et al., 2018, and Liggett & Sankaran, 2020. Figure created in BioRender.com.
Since the discovery of the human immunodeficiency virus (HIV) in the early 1980s (17–20), it has spread globally, infecting more than 38 million people worldwide according to the latest available statistics (21). The ability of the virus to evade the immune system (22) and escape antiretroviral therapy (ART) pressure (23) contributes to its persistence in vivo. Drug escape and immune evasion are achieved through the action of viral proteins (24–26), immune dysregulation as an indirect consequence of infection (27–30), as well as the high mutation rate of the virus conferred by the low-fidelity HIV reverse transcriptase enzyme (31–33). The majority of HIV infections are caused by HIV-1 group M strains (34), with HIV-1 subtype B (HIV-1B) and C (HIV-1C) claiming 11% and 48% of worldwide infections, respectively (35). Most HIV-1 infections in India, southern Brazil (36) and sub-Saharan Africa are due to HIV-1C (37), while HIV-1B is confined to high income regions such as North America, Europe and Australia (37–40). Despite the prevalence of HIV-1C, HIV-1B dominates the research landscape. Variation between subtypes has been documented for phenotypic properties such as co-receptor tropism (41–45), replication rate and disease progression (45–50), transmission mechanics (51–54), and mutation patterns (55–57). Furthermore, reverse transcription (58) and the emergence of drug resistance (59–62) have been reported to vary between subtypes.
HIV primarily infects cells of the immune system that express cluster of differentiation (CD) 4, C-C-motif chemokine receptor type 5 (CCR5) and C-X-C-motif chemokine receptor type 4 (CXCR4), including CD4+ T-cells and monocyte/macrophages (63); in the case of the former, this results in the depletion of CD4+ cells. The viral reservoir is made up of latently-infected cells which harbour proviral DNA but do not produce viral particles (64). Once established, the reservoir is the most challenging barrier to curing HIV. Latency is complex and regulated at several levels, reviewed elsewhere (64–67). Activation of viral production from latently-infected cells contributes to viral persistence throughout the lifetime of an infected individual. This is evidenced through lineage-tracing which has shown the resurgence of sequences that were dominant during early infection, in the later stages of infection (68–74).
HIV has several concomitant effects once an individual becomes infected. Direct infection of cells with HIV is not the only cause of blood cell depletion, referred to as cytopenia, in HIV patients. Multifactorial, indirect effects associated with HIV infection can also cause cytopenia and other haematological abnormalities. A plethora of cytopenias may present in HIV patients including leukopenia, lymphopenia, anaemia, neutropenia, thrombocytopenia, and pancytopenia (10, 75–81). In a large study conducted in Beijing, neutropenia, thrombocytopenia, and anaemia were partially restored in ART-naïve patients following initiation of treatment (76). However, multiple factors influence restoration of cytopenia following induction of ART (82) including concomitant infections, viral load, tropism and drug resistance, and individual response/adherence to treatment. With the exception of lymphopenia, HIV-associated cytopenias cannot be explained by the lytic cycle of HIV infection. It is unclear whether the haematological abnormalities observed in HIV-infected individuals are due to direct or indirect effects of infection on HSPCs. Various studies suggest these cytopenias may be attributed to disruption of the bone marrow niche housing HSPCs, which maintain the continuous production of blood and immune cells throughout life (83–85). This review will discuss both these possibilities in detail.
The bone marrow is considered a primary and secondary lymphoid organ allowing for continuous interactions of immune cells (86). The indirect effects of HIV on HSPCs may stem from infection of bone marrow niche cells (87), the effects of HIV proteins on bone marrow cells, or dysregulation of the cytokine milieu which is instrumental in orchestrating dynamic physiological processes including haematopoiesis. The effects of HIV infection on bone marrow niche cells and the consequences for haematopoiesis are described in detail below and are illustrated in Figure 2.
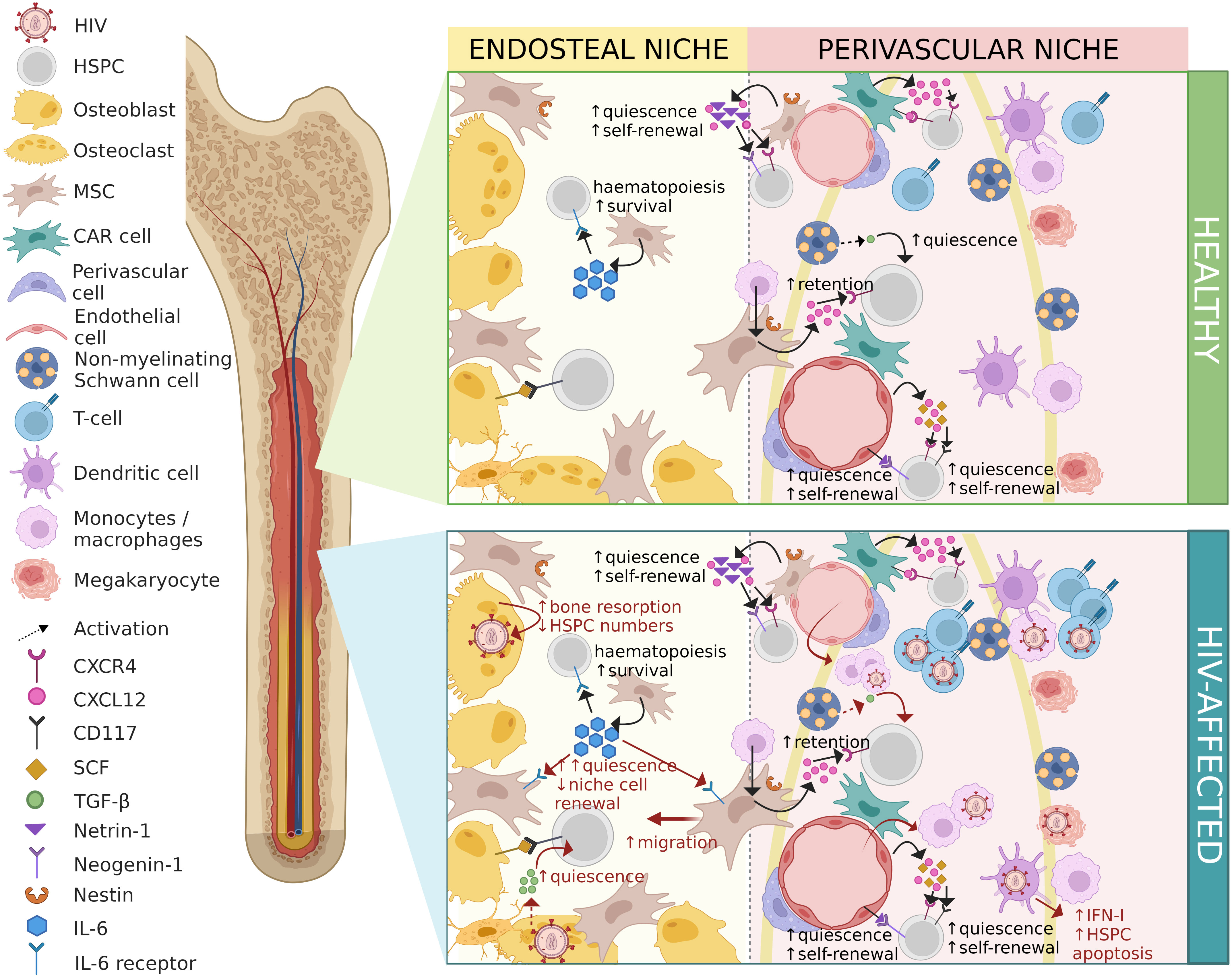
Figure 2 Contrasting healthy and HIV-affected bone marrow niches. HIV-associated changes are shown in red. HIV, human immunodeficiency virus; HSPC, haematopoietic stem/progenitor cell; MSC, mesenchymal stem/stromal cell; CAR cell, CXCL12-abundant reticular cell; CXCR4, C-X-C-motif chemokine receptor type 4; CXCL12, C-X-C-motif chemokine ligand 12; CD117, cluster of differentiation molecule 117; SCF, stem cell factor; TGF-β, transforming growth factor-beta; IL-6, interleukin-6; IFN-I, interferon type-I. Figure created in BioRender.com.
The perivascular region around blood vessels that permeate the bone marrow contains perivascular cells, endothelial cells, CAR cells, and nerve fibres including non-myelinated Schwann cells.
Non-myelinating Schwann cells sheath neuronal axons in the perivascular niche of the bone marrow, and participate in niche regulation (88). These cells have been found to activate latent TGF-β released from the bone marrow extracellular matrix (89), and to facilitate circadian regulation of CXCL12 production in Nestin+ MSCs (88). Together, these functions probably account for the maintenance of a quiescent perivascular HSPC pool very closely associated to neuronal axons. Non-myelinating Schwann cell depletion results in reduced HSPC numbers as early as three days post-depletion (89), although the mechanism is unclear. Investigation into HIV-associated neuropathy revealed that the HIV glycoprotein (gp)120 protein stimulated lysosomal exocytosis in Schwann cells (90), releasing axon-exciting adenosine triphosphate (ATP) into the extracellular environment. Exocytosis of lysosomes increased calcium and induced reactive oxygen species (ROS) generation in neighbouring axons, which in turn activates latent TGF-β (91). This could contribute to impaired haematopoiesis by driving HSPCs toward quiescence. Since infection of Schwann cells has only been documented once by electron microscopy (92), HIV-associated neurotoxicity is likely caused by either viral proteins or neurotoxic cytokines released by activated/infected glial cells. The interaction between gp120 and CXCR4 on Schwann cells results in the release of several chemokines, including CC chemokine ligand (CCL)-5 (also known as RANTES) and CXCL1. Release of CCL5 results in the production of TNF-α by dorsal root ganglion neurons and subsequent autocrine neurotoxicity mediated by TNFR1 (93), whereas the release of CXCL1 results in the recruitment of macrophages in mice (94).
Several subtypes of endothelial cells, the cells which line blood vessels, have been identified in the bone marrow, and the distinct functions of each subtype are still being elucidated. Arteriolar, sinusoidal, and endothelial cells expressing endoglin (CD105) are among these. Netrin-1 is expressed by arteriolar endothelial cells and binds to the receptor Neogenin-1 on HSPCs, which is correlated with quiescence and self-renewal of HSPCs in vivo (95). Furthermore, arteriolar endothelial cells have been found to produce the majority of endothelial cell-derived SCF, in addition to producing CXCL12. Knock-out of SCF in arteriolar endothelial cells results in reduced CD150+CD48-Lin-Sca-1+c-Kit+ primitive HSPCs in mice (96, 97). In bone marrow injury such as irradiation or chemotherapy, an endothelial cell population expressing endoglin (CD105) produces interleukin (IL)-33, which expands umbilical cord blood-derived CD34+ HSPCs in vitro and promotes angiogenesis and osteogenesis for bone marrow regeneration (98). Work on the cytokine profiles produced by different endothelial subtypes is lacking. Previous studies in the 1990s showed that endothelial cells produce messenger RNA (mRNA) for cytokines supporting and inhibiting haematopoiesis (99, 100). Cytokines supporting haematopoiesis include granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1, IL-7, IL-6, TGF-β, IL-8, and IL-11, while thymosin-β4 is a small molecule that inhibits haematopoiesis (100). Endothelial cells also produce mRNA for macrophage inflammatory protein (MIP)-2, platelet-derived growth factor (PDGF), merozoite surface protein (MSP)-1, interferon (IFN)-γ, IL-13 and inhibitin (100). However, the relationship between cytokine mRNA and protein production by endothelial cells for these cytokines is unclear.
In a study comparing bone marrow microvascular endothelial cells from HIV seropositive to those from healthy uninfected donors, these cells have been shown to be permissive to HIV infection in vivo (101). HIV-infected microvascular endothelial cells expressing von Willebrand Factor (vWF) were found to produce the HIV protein p24 in long-term culture. Endothelial cells from HIV seropositive donors showed a significant reduction in IL-6 and GM-CSF production in response to IL-1α stimulation compared to uninfected controls (101). In addition to being susceptible to productive HIV infection in vivo, HIV proteins contribute to endothelial cell activation, apoptosis, and conversely, stimulate angiogenesis, proliferation, and migration of endothelial cells through various mechanisms (102, 103).
The HIV trans-activator of transcription (Tat) protein induces apoptosis in endothelial cells, but also induces the release of IL-6 and the expression of adhesion markers E-selectin, intracellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, and endothelial leukocyte adhesion molecule (ELAM)-1 which recruit monocytes and increase their migration across the endothelial barrier, increasing monocyte tissue pervasion (104–106). The HIV negative factor (Nef) protein induces apoptosis in endothelial cells by increasing ROS production, causing oxidative stress and cell death (107–111) as well as increasing production of monocyte attractant protein (MCP)-1 (109). The HIV matrix protein p17, similar to Tat, promotes angiogenesis (112) and increases monocyte chemoattractant protein (MCP)-1 production in endothelial cells, as does Nef (113, 114). The implications of these contrasting consequences of HIV infection on endothelial cells in vivo remain to be resolved.
CXCL12 is primarily produced by CAR cells (115, 116) and osteoblasts (117) in the bone marrow, and is involved in the homing of cells expressing the cell-surface marker CXCR4 (118, 119), thereby acting as a potent chemoattractant for HSPCs and their progeny in the bone marrow. In addition to chemoattraction, CXCL12-CXCR4 interactions provide physical tethering of CD34+ HSPCs to cell-surface CXCL12 on CAR cells (116, 120). The CXCR4-CXCL12 axis has been found to be critical for the maintenance of the primitive HSPC pool, which is diminished in CXCR4 knock-down mice (116). Neither direct nor indirect effects of HIV infection have been reported for CAR cells to date. There is ongoing debate as to whether a variant in the untranslated region of CXCL12 (designated 3’A) is protective against HIV infection and delays disease progression or whether it is associated with susceptibility to HIV infection and faster progression to AIDS (121).
Dendritic cells and macrophages are both present in the bone marrow stroma in the perivascular niche forming so-called “immune pockets”, where B- and T-lymphocytes are localised. While bone marrow dendritic cells do not play an appreciable role in HSPC maintenance or haematopoiesis, selective ablation of dendritic cells results in increased HSPC mobilisation through an indirect mechanism involving CXCR2 (122). Dendritic cells and macrophages are capable of sustaining HIV replication and could contribute to viral dissemination in the niche (123–125). The perivascular niche is also home to megakaryocytes, which have been found to be susceptible to HIV infection both in vitro (126–128) and in vivo (128, 129). This relates primarily to viral production and release in the bone marrow and could contribute to thrombocytopenia in HIV patients through the loss of megakaryocytes as a consequence of viral replication.
Circulating monocytes are recruited to tissues and differentiate into tissue-resident macrophages, where they fulfil critical functions in tissue homeostasis. Most of the research on bone marrow macrophages to date has involved murine studies, although whether the findings translate to human bone marrow is not clear. Two populations of bone marrow macrophages have been identified in mice, so-called “osteomacs” present in the endosteal niche in close contact to osteoblasts and Nestin+ MSCs (130), and CD169+ macrophages located in the perivascular niche around Nestin+ MSCs (131). Osteomacs, through cell-to-cell contact, increase Nestin+ MSC production of prostaglandin (PG)-E2, which in turn stimulates oncostatin M release by osteomacs, resulting in increased osteoblast mineralisation and differentiation (132). Higher levels of PGE2 correlated with increased release of anti-inflammatory IL-10 (133, 134) by macrophages (135). Depletion of CD169+ bone marrow macrophages severely impaired HSPC retention in the niche, and this was associated with a reduction in Nestin+ MSC expression of CXCL12 and SCF mRNA (136, 137). Bone marrow macrophages have been reported to be permissive to HIV infection, although the cytokine profile did not appear to be altered in vitro following infection with a number of HIV isolates (138). Whether HIV infection occurs predominantly in monocytes prior to tissue specification into macrophages or in tissue-resident macrophages is difficult to establish. Macrophages may be an important tissue reservoir for HIV capable of sustaining HIV infection in vivo (87, 139–142) and their presence would therefore form a large part of the barrier to HIV eradication. Conversely, other researchers have suggested that macrophages play a limited role in HIV replication (143–145). While these cells can be infected by HIV, they may not effectively support viral replication and production. As a result, their contribution to viral spread and long-term persistence in the body may be minimal. Although drawing parallels between murine and human bone marrow is beyond the scope of this review, it is plausible that macrophage depletion resulting from direct HIV infection may result in reduced HSPC retention in the niche and thereby contribute to impaired haematopoiesis in humans.
The endosteal niche is in close proximity to the endosteum of the bone marrow niche which is made up of osteoclasts, osteoblasts, and MSCs. Osteolineage cells (osteoblasts and osteoclasts) were some of the first cells shown to interact with HSPCs and play an important role in HSPC fate. MSCs destined to become osteoblasts occupy the endosteal surface of flat and trabecular bones between the bone and the bone marrow.
Bone marrow MSCs, initially thought to be fibroblasts (146–148), are a heterogenous population of cells forming part of both the perivascular and endosteal stromal cell populations. In addition to replacing osteoblasts and adipocytes as a normal part of cell turnover, bone marrow MSCs play an important role in immunomodulation and HSPC maintenance through cytokine production (149). MSCs constitutively produce IL-6, which is important in haematopoiesis and suppressing the proliferation of MSCs and activated T-cells (149–151). MSCs have also been shown to secrete prostaglandin E2 (PGE2), which is implicated in the expansion of less primitive HSPCs (152) as well as HSPC recovery and repopulation after chemotherapy (153, 154). In the perivascular niche, periarteriolar MSCs produce Netrin-1 (95) similar to arteriolar endothelial cells, thereby contributing to HSPC quiescence and self-renewal. MSC heterogeneity in the bone marrow is much better described in mice than in humans, and this includes single-cell resolution as has been extensively reviewed elsewhere (155, 156). In murine bone marrow, Lepr+ MSCs enriched for adipocyte and osteoblastic precursors secreting SCF (157) and CXCL12 (116) have been described, which are implicated in HSPC self-renewal (158) and quiescence, respectively, have been described. Although LEPR(hi)CD45(low) BM-MSCs were recently identified in human marrow, the function of these cells remains poorly described (159). Nestin+ MSCs are typically periarteriolar and were found to be clustered around nerve fibres where they produce CXCL12 (156). Characterisation of human bone marrow MSC heterogeneity at the single-cell level remains crucial to elucidating the complex niche dynamics supporting HSPCs, haematopoiesis, as well as immunomodulation in the bone marrow.
While the susceptibility of bone marrow MSCs to HIV infection has not yet been conclusively determined (160), more studies suggest low levels of productive infection - referring to the production of new virus particles (161–163) than studies which demonstrate resistance to infection (138). However, exposure to HIV proteins has varying effects on the differentiation of MSCs. HIV Tat and Nef proteins reduce MSC proliferation and differentiation, and encourage senescence, corresponding to increased oxidative stress and mitochondrial dysfunction (164). Tat increases nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) activity and inflammatory cytokine secretion, while Nef reduces autophagy, and it was found that the effects of Tat and Nef are cumulative (164). NF-κB expression also drives IL-6 production in MSCs (149), resulting in increased MSC senescence which could explain dysregulation of both bone and fat metabolism in HIV patients. Regulator of expression of virion (Rev) and p55-gag protein expression results in temporal and quantitative changes in key osteo- and adipogenic signals, hampering differentiation (165). Expression of both Rev and p55-gag increase alkaline phosphatase activity and decrease lipid levels, where Rev increases calcium deposition in non-differentiating MSCs. Rev also increases potent peroxisome proliferator-activated receptor gamma (PPAR-γ) expression which drives adipogenic differentiation, and Runt-related transcription factor (RUNX)2 which drives osteogenic differentiation in non-differentiating MSCs (165). In a study assessing the effects of HIV proteins on human MSCs and osteoblast cell lines, HIV proteins p55 and gp120 reduced calcium deposition, alkaline phosphatase activity, and key bone remodelling proteins (166). In contrast, the HIV Rev protein augments MSC osteogenesis (166). HIV gp120 improves adipogenic differentiation, impairs endothelial differentiation, and induces apoptosis of vessel wall-derived MSCs (161). These findings support a role for MSCs in haematopoietic abnormalities resulting from HIV-associated bone marrow niche dysregulation.
Osteoclasts are large, multinucleated monocyte/macrophage-derived cells responsible for resorption of the bone matrix produced by osteoblasts in the continuous, dynamic process of bone remodelling (167, 168). Osteoblasts are smaller osteolineage cells derived from MSCs and produce macrophage colony-stimulating factor (M-CSF) for osteoclastogenesis, osteopontin for continuous formation of bone matrix, as well as a variety of bone marrow niche regulatory cytokines (169). These include IL-6, MIP-1α, SCF, CXCL12, granulocyte colony-stimulating factor (G-CSF), TPO, angiotensin-1, and annexin 2 (169). The bone matrix maintained by osteoclasts and osteoblasts results in a protected environment for long-term HSPCs. Bone remodelling results in the release and activation of TGF-β stored in the bone matrix (168), and a calcium gradient (170), both of which contribute to the quiescence of HSPCs in the endosteal niche. Immature osteoblasts release CXCL12, a potent chemoattractant for cells such as HSPCs which express CXCR4 (169), drawing them towards the endosteum where they bind to SCF on mature osteoblast cell surfaces through the CD117 receptor (169). Angiotensin-1 (171) and TPO (172) produced by osteoblasts also assist in maintaining HSPC quiescence in the endosteal niche.
In HIV infection, bone resorption is increased due to the stimulation of osteoclastogenesis by HIV proteins and direct infection of osteoclasts (173–175). This increased osteoclast activity has been associated with reduced HSPC numbers in the bone marrow (176). In addition to propagating virus through replication, osteoclasts have been implicated in cell-to-cell transmission of HIV-1 between cells of the bone marrow niche (177). Osteoblasts are reportedly not susceptible to HIV infection in vitro (178), but respond deleteriously to the presence of HIV proteins. Alkaline phosphatase activity, receptor activator of nuclear factor kappa-B ligand (RANKL) secretion, and calcium (Ca2+) deposition by osteoblasts have been reported to be impaired in the presence of HIV p55-gag and gp120 proteins (179). Studies on osteoclasts have produced conflicting findings regarding the induction of apoptosis versus proliferation following exposure to HIV-1 gp120 (180, 181). Degradation of the bone matrix during bone resorption releases and activates an excess of TGF-β in the endosteal niche (182). In addition to stimulating and recruiting MSCs from the perivascular niche to the endosteal niche, high levels of active TGF-β induce quiescence in primitive HSPCs (91), promote proliferation and differentiation of myeloid-primed HSPCs, and hinder lymphoid-primed HSPCs (183). Bone resorption, reduced osteoblast activity in the presence of HIV proteins, and impaired differentiation of MSCs into osteoblasts all contribute to loss of bone density and overall osteopenia observed in HIV patients (184, 185). Consequently, reductions in IL-6, MIP-1α, SCF, CXCL12, G-CSF, and TPO usually produced by osteoblasts are expected in HIV patients. Bone disease in HIV-infected individuals suggests that some of the in vitro findings may be transferable (186), although anti-retroviral therapy (ART) has also been implicated (185–187).
Productive infection of bone marrow niche cells would result in the release of HIV proteins which consequently would have an adverse effect on the infected cell and surrounding cells which in turn would negatively affect HSPCs.
The bone marrow is highly vascularised, allowing the trafficking of cells and chemical signals to and from the bone marrow through the circulation. In addition to localised effects of HIV on bone marrow cells, altered cytokine profiles produced by trafficked cells affect the niche microenvironment. Monocytes originating from the bone marrow enter the circulation and differentiate into macrophages or dendritic cells in tissues, where they may become infected with HIV (188). Upon infection and pathogen-associated molecular pattern (PAMP)/toll-like receptor (TLR)-initiated migration to secondary lymphoid tissues, dendritic cells are involved in cell-to-cell transfer of HIV to T-cells (189–191). In addition to perpetuating viral transmission, activation of PAMP triggers type I IFN production by dendritic cells (192–195) and the cascade towards chronic immune activation observed in HIV patients (196, 197). Acute type I IFN exposure has been shown to induce proliferation of c-Kit+ HSPCs in mice, whereas chronic type I IFN treatment led to HSPC apoptosis due to proapoptotic induction by IFN exposure irrespective of duration (198). The presence of HIV-infected dendritic cells in the bone marrow therefore contributes to haematopoietic dysfunction by directly affecting HSPCs (199).
While monocyte-derived tissue-resident macrophages do not return to the bone marrow, macrophages support viral replication and are important members of the viral reservoir (200) in combination with circulating CD4+ T-cells (201). Circulating CD4+ T-cells are present at higher rates in the bone marrow of HIV positive individuals compared to HIV negative individuals (202), exacerbating the effects of aberrant T-cell cytokine production on the bone marrow. During HIV infection, increased production of IL-4 by T-helper 2 (Th2) CD4+ T-cells was observed (203), which was reported to impair megakaryocyte production in leukaemia (204) and could reasonably be expected to contribute to thrombocytopenia.
It has been suggested that a population of resident memory T-cells in the bone marrow niche may play a role in long-lived immunity against systemic pathogens (205–208), although this is not yet fully understood (209, 210). Resting CD4+ memory and T-cells are well-described as an important latent reservoir for HIV, extensively reviewed elsewhere (206, 211). Bone marrow CD4+ memory T-cells were found to harbour similar levels of virus to circulating CD4+ T-cells in simian immunodeficiency virus (SIV)-infected rhesus macaques (212) and HIV-infected individuals (211). The susceptibility of these cells to HIV infection and their resident status in the bone marrow presents a source of HIV and HIV proteins in the bone marrow outside of circulating infected cells. Activation of this latent reservoir in the bone marrow could result in an increase in HIV load in the bone marrow, which may infect surrounding cells and cause dysregulation of bone marrow niche cells as a consequence of the presence of HIV proteins.
Interactions between cells in the bone marrow are critical for normal haematopoiesis as well as HSPC maintenance and regulation. Bone marrow homeostasis is disrupted during HIV infection as a consequence of direct infection of niche cells and/or the effects of HIV proteins. As a consequence of HIV infection or the effects of HIV proteins on perivascular niche cells, perivascular niche HSPCs are driven towards quiescence and mobilisation, thereby impairing haematopoiesis. Endosteal niche HSPCs are driven to mobilisation due to the breakdown of normal bone remodelling and MSC senescence resulting from HIV infection. The consequences of the indirect effects of HIV infection on HSPCs are therefore deleterious, ultimately reducing the number of HSPCs in the bone marrow and the creation of a quiescence-supporting environment for the remaining HSPCs.
The indirect effects of HIV infection on HSPCs are complex and cumulative, contributing to the impairment of HSPC function. The direct effects of HIV on HSPCs encompass both direct infection and the effect of HIV proteins on HSPC function. However, the literature presents opposing conclusions regarding the susceptibility of HSPCs to HIV infection.
HSPCs in the bone marrow are directed to remain quiescent or divide and differentiate, forming blood and immune cells in response to the cytokine milieu. At key points, differentiating HSPCs will become lineage restricted and only form the cell types dictated by the cytokine milieu. This means that one infected HSPC would produce a limited number of haematopoietic cell types harbouring HIV. Nixon et al., 2013 demonstrated that HSPC progeny generated through colony-forming assays of CD34+ cells from HIV-infected humanised mice, harboured HIV. Clonally infected cells resulting from HSPC division and differentiation would therefore be restricted to a single or limited number of haematopoietic cell types, depending on the differentiation potential of the infected HSPC and on external stimulus directing haematopoiesis. Transcriptional activation during differentiation could activate HIV replication from integrated or episomal provirus, possibly resulting in cell death due to the lytic nature of HIV replication. Carter et al., 2010 (213) showed that CD34+ cells expressing HIV gene products were markedly depleted in culture compared to a transduced control, which could indicate some other mechanism of cell death in infected HSPCs. In vivo, host cell lysis or death might contribute to the absence of terminally differentiated HIV-infected cells of all haematopoietic lineages harbouring clonal virus initially of HSPC-origin. HIV infection may skew haematopoiesis towards or away from certain lineages (214, 215), which may contribute to cytopenia and the lack of clonal infection of certain haematopoietic cell types. This is not well described in literature as bone marrow research is limited to static snapshots of a highly dynamic environment.
The controversy in literature dates back to the early 1990s with Stanley and colleagues detecting HIV in CD34+ HSPCs from seropositive patients (216) and Neal and colleagues presenting alternate data showing that CD34+ HSPCs were rarely infected with HIV in asymptomatic patients (217). This was followed by a number of studies with different conclusions and one paper suggesting that HIV-1 subtypes may differ in their ability to infect HSPCs (218). Several studies found HSPC subsets to be resistant to HIV infection (217–230), the suggested mechanism being through a p21-mediated pre-integration block (231). Given the inducible expression of HIV proteins in the presence of a pre-integration block, the findings may suggest transient transcription from episomal proviral DNA in HSPCs (213). In contrast, a number of studies have detected HIV in HSPCs (213, 214, 216, 218, 222, 226, 229, 230, 232–236). Carter and colleagues showed latent infection of Lin-CD34+CD133+CD38- primitive HSPC subsets in vitro and corroborated these findings with bone marrow CD34+ HSPCs from HIV infected individuals with high viral load (213). However, their findings also suggest that HIV-infected cells actively expressing HIV proteins were short-lived compared to their latently-infected counterparts (213). Several follow-up studies reported similar findings (214, 230, 235, 236), which are presented in Table 1. A recent study reported that a small subset of the heterogenous CD34+ HSPC population expresses low levels of CD4, and that this subset was found to harbour HIV genomes in vivo (237). While detection of HIV in this subset is not necessarily surprising, the fact that both R5- and X4-tropic HIV genomes were detected was notable as CXCR4 is usually expressed in a greater fraction of CD34+ HSPCs than is CCR5 (213). While HSPC susceptibility to HIV infection hinges largely on the expression of CD4, CXCR4, and CCR5, CD4-independent infection mechanisms have been described (238–241) and should not be discounted for infection of HSPCs. Most recently, Renelt and colleagues made a strong argument for HIV infection of CD133+ and CD34+CD133- HSPC subpopulations in some donors, and their contribution to viremia using proviral sequence tracing (222).
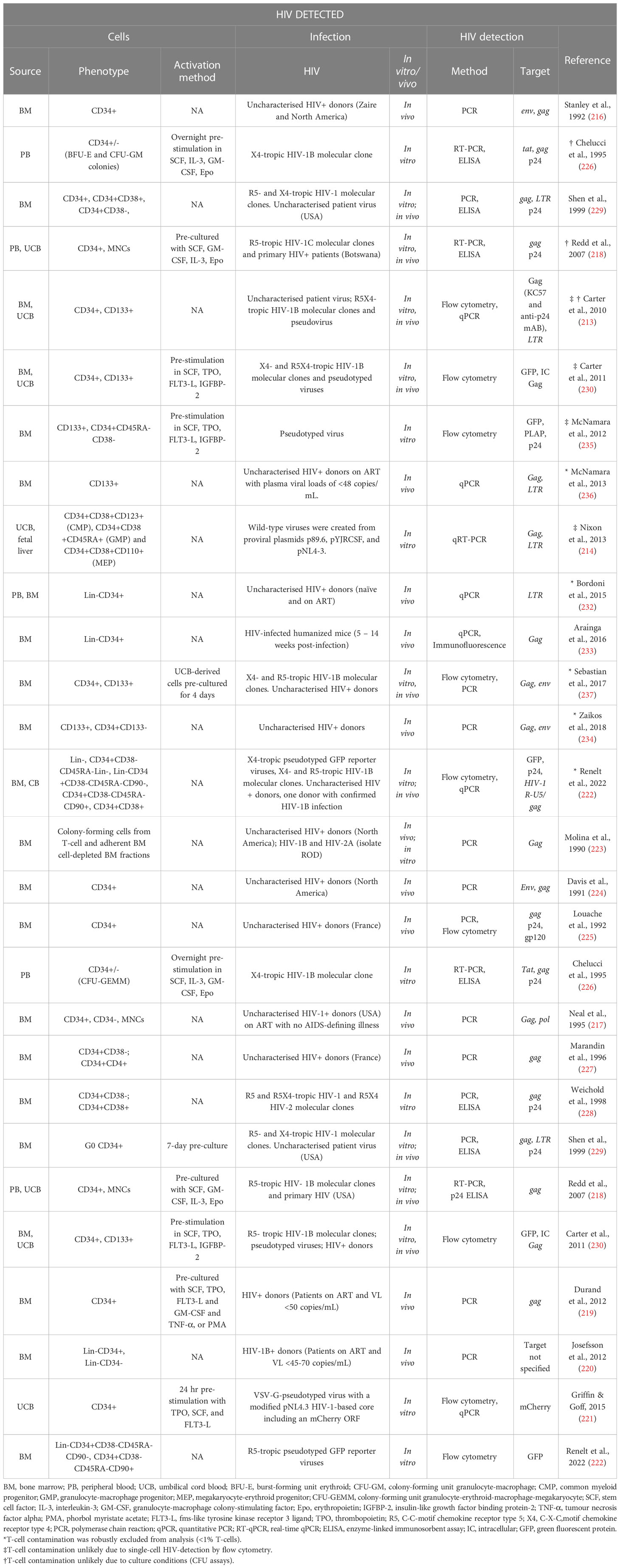
Table 1 Comparison of literature on the susceptibility of HSPCs to HIV infection.
A closer inspection of studies exploring the susceptibility of HSPCs to HIV infection outlined in Table 1 reveals that different experimental approaches may in part explain the lack of consensus between studies to some degree. Variations in culturing, the use of growth factors, HIV moieties, infection strategy, and HIV detection method could contribute to variation between results. These factors and how each could affect the outcome of the study are discussed in more detail below.
HSPCs are rare cells that often require in vitro expansion so that enough cells are obtained to optimally perform experiments. Numerous studies have cultured or expanded HSPCs for several days before in vitro infection with HIV. Expression of CXCR4 is upregulated on murine HSPCs after overnight incubation (242), which may artefactually increase susceptibility to CXCR4-tropic HIV. Similarly, HIV integration and replication is dependent on the activation state of target cells (243) with dividing cells being more susceptible to productive HIV infection. The majority of HSPCs (>90%) are in a quiescent state in vivo (244) in the bone marrow niche; expansion prior to infection would therefore create an ex vivo artefactual state. Expansion and culturing of HSPCs in vitro is often performed in the presence of haematopoietic cytokines which promote expansion and HSPC survival in culture. However, there is not currently a standardised cytokine cocktail for HSPC expansion and maintenance. Studies investigating the susceptibility of HSPCs to HIV infection have been performed with (218, 226, 230, 235) and without (229, 231, 245) cytokines. The duration of culture and the supplementation of medium with cytokines could therefore result in increased susceptibility of HSPCs to HIV infection that is not inherent but rather an artefact of culturing.
The use of HIV propagated in vitro in the form of laboratory-generated HIV molecular clones, pseudo- or pseudo-typed virus, or cultured primary virus has several aspects where outcome-critical variation between studies could occur. The most glaring differences between studies are (i) multiplicity of infection (MOI), (ii) infection strategy, and (iii) nature of virus used. Unrealistic bombardment of target cells with extremely high MOIs, referring to the number of infectious units per target cell, could result in artefactual infection in vitro, which is unlikely in vivo. The infection strategy is similarly crucial to a translatable experimental outcome. Infection in small volumes or using centrifugal force (termed “spinoculation”) to create close contact between cells are two methods commonly used to increase the potential for infection. Spinoculation is not a physiological condition and could therefore also result in artefactual infection. The nature of the virus used in experiments is constrained by several factors including but not limited to biosafety, availability of comparable research tools, and the effect of HIV proteins on target cells which can affect results. As previously mentioned, HIV-1B (being the most-studied subtype) epitomises what is known about HIV infection. However, distinct characteristics including coreceptor usage during early and late infection (246) and reduced cytopathic effects (58) have been documented for HIV-1C, which could affect research outcomes in terms of latency and host cell susceptibility. Variation from HIV-1B has been reported for non-B subtypes in several aspects related to viral fitness and disease progression (45–48, 50, 51, 55, 58, 59, 61, 218, 247–250) which are outside the scope of this review but are important when comparing research findings.
The method used to detect HIV is another critical factor to be considered when comparing studies. In addition to the increased sensitivity that comes with improvements in detection technologies over time, studies have varied with the technology used to detect the presence of HIV in target cells. Proviral DNA, viral transcripts, or viral proteins can be targeted, and each detection method comes with a limit of detection and considerations for use. This is particularly important where, as with HSPCs, it is reasonable to expect low to very low proportions of infected cells. This is illustrated in a study by Izopet et al. (251) who were able to detect four proviral genomes per million cells and found that the frequency of infection of highly susceptible CD4+ T-cells in vivo can be lower than 1%, as reported in other studies (252–254). The sensitivity of the HIV detection method is often not reported, and the improvements of technologies over time are difficult to categorise, but these are equally important to consider when comparing older and more recent studies.
Disruption of HSPC function can also be caused by the presence of HIV proteins. The HIV receptor protein gp120 has been shown to impair the clonogenic potential of HSPCs and induce apoptosis through Fas-dependent endogenous TGF-β upregulation (255). Suppression of HSPC colony formation is caused by HIV-1 p24 (256). Exposure to HIV Tat protein stimulates TGF-β production in macrophages resulting in myelosuppression in vitro (255), and viral protein R (Vpr) has been shown to induce phagocytosis of bone marrow cells by mononuclear phagocytes (257). Blocking TGF-β in purified CD34+ HSPCs exposed to HIV reportedly improved growth and survival (255), and this is supported by the simultaneous downregulation of a proliferation-inducing ligand (APRIL) with TGF-β upregulation induced by exposure to gp120 (258). Furthermore, Nef has been shown to act as a PPARγ agonist with deleterious effects on early haematopoiesis in macaques (259). Cumulatively, the effects of HIV proteins in the bone marrow are deleterious to HSPC and niche cell function and survival, and ultimately contribute to haematological abnormalities present in HIV patients independently of direct HSPC infection.
The conditions under which HSPCs may become susceptible to HIV infection in the bone marrow are not clear based on current information from the literature; what is clear however is that HIV proteins have a direct suppressive effect on HSPC function.
Haematopoietic dysfunction in HIV patients is well-documented and results from the combined direct and indirect effects of HIV on HSPCs. The bone marrow niche is a uniquely complex environment which is yet to be fully understood. Healthy human bone marrow is therefore not completely represented in literature, which makes it difficult to fully model the marrow under conditions of HIV infection. The limited understanding of in vivo susceptibility of bone marrow cells to HIV and the fact that bone marrow cell types have largely been studied in vitro or in animal models contribute to the paucity of literature on the HIV-infected marrow. Moreover, the evolution of HIV detection methods over time, and the understanding that detection of HIV proteins or partial DNA does not necessarily indicate productive infection, compound this challenge. Although it is undeniable that HIV affects haematopoiesis, the susceptibility of HSPCs to HIV has long been debated. Studies investigating HIV infection in HSPCs differ critically in methodology and HSPC subpopulations used. This review has aimed to highlight what is currently known about the consequences of HIV infection on the bone marrow niche, and to summarise the studies to date which have attempted to determine the susceptibility of HSPCs to HIV infection. This is particularly relevant to the fields of stem cell transplantation and HIV pathogenesis, and potentially to the treatment of HIV-associated haematological malignancies.
CH and JM wrote, compiled, and edited the manuscript. CH made the figures. TM contributed to the manuscript. MP and CD were involved in manuscript conception. MP edited the manuscript. All authors contributed to the article and approved the submitted version.
CH is funded by the NRF-DAAD program and the University of Pretoria. TM is funded by the National Research Foundation and the University of Pretoria. JM receives funding from the Bill and Melinda Gates Foundation (INV-022216). The group is funded by the South African Medical Research Council, and the University of Pretoria (through the ICMM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells (1978) 4(1–2):7–25.
2. Beerman I, Luis TC, Singbrant S, Lo Celso C, Méndez-Ferrer S. The evolving view of the hematopoietic stem cell niche. Exp Hematol (2017) 50:22–6. doi: 10.1016/j.exphem.2017.01.008
3. Szade K, Gulati GS, Chan CKF, Kao KS, Miyanishi M, Marjon KD, et al. Where hematopoietic stem cells live: the bone marrow niche. Antioxid Redox Signal (2018) 29(2):191–204. doi: 10.1089/ars.2017.7419
4. Asada N, Takeishi S, Frenette PS. Complexity of bone marrow hematopoietic stem cell niche. Int J Hematol (2017) 106(1):45–54. doi: 10.1007/s12185-017-2262-9
5. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. (2014) 505(7483):327–34. doi: 10.1038/nature12984
6. Cordeiro-Spinetti E, Taichman RS, Balduino A. The bone marrow endosteal niche: how far from the surface? J Cell Biochem (2015) 116(1):6–11. doi: 10.1002/jcb.24952
7. Lévesque JP, Helwani FM, Winkler IG. The endosteal osteoblastic niche and its role in hematopoietic stem cell homing and mobilization. Leukemia. (2010) 24(12):1979–92. doi: 10.1038/leu.2010.214
8. Tamma R, Ribatti D. Bone niches, hematopoietic stem cells, and vessel formation. Int J Mol Sci (2017) 18(1):151. doi: 10.3390/ijms18010151
9. Yu VWC, Scadden DT. Chapter two - hematopoietic stem cell and its Bone marrow niche. In: Bresnick EH, editor. Hematopoiesis. Academic Press (2016). p. 21–44. doi: 10.1016/bs.ctdb.2016.01.009
10. Durandt C, Potgieter JC, Mellet J, Herd C, Khoosal R, Nel JG, et al. HIV And haematopoiesis. S Afr Med J (2019) 109(8):40–5. doi: 10.7196/SAMJ.2019.v109i8b.13829
11. Liggett LA, Sankaran VG. Unraveling hematopoiesis through the lens of genomics. Cell. (2020) 182(6):1384–400. doi: 10.1016/j.cell.2020.08.030
12. Brown G, Tsapogas P, Ceredig R. The changing face of hematopoiesis: a spectrum of options is available to stem cells. Immunol Cell Biol (2018) 96(9):898–911. doi: 10.1111/imcb.12055
13. Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol (2017) 19(4):271–81. doi: 10.1038/ncb3493
14. Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev (2010) 238(1):37–46. doi: 10.1111/j.1600-065X.2010.00963.x
15. Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. (2013) 154(5):1112–26. doi: 10.1016/j.cell.2013.08.007
16. Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. (2013) 502(7470):232–6. doi: 10.1038/nature12495
17. Montagnier L, Chermann JC, Barre-Sinoussi F, Klatzmann D, Wain-Hobson S, Alizon M, et al. Lymphadenopathy associated virus and its etiological role in AIDS. Princess Takamatsu Symp (1984) 15:319–31.
18. Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Sci (80-). (1983) 220(4599):868–71. doi: 10.1126/science.6189183
19. Durack DT. Opportunistic infections and kaposi’s sarcoma in homosexual men. N Engl J Med (1981) 305(24):1465–7. doi: 10.1056/NEJM198112103052408
20. Masur H, Michelis MA, Greene JB, Onorato I, Vande Stouwe RA, Holzman RS, et al. An outbreak of community-acquired pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction. N Engl J Med (1981) 305(24):1431–8. doi: 10.1056/NEJM198112103052402
21. UNAIDS. Global HIV & AIDS statistics [[/amp]]mdash; fact sheet. In: Global HIV & AIDS statistics [[/amp]]mdash; fact sheet (2022). Available at: https://www.unaids.org/en/resources/fact-sheet.
22. Guha D, Ayyavoo V. Innate immune evasion strategies by human immunodeficiency virus type 1. Isrn Aids. (2013) 2013:1–10. doi: 10.1155/2013/954806
23. Günthard HF, Calvez V, Paredes R, Pillay D, Shafer RW, Wensing AM, et al. Human immunodeficiency virus drug resistance: 2018 recommendations of the international antiviral society-USA panel. Clin Infect Dis (2019) 68(2):177–87. doi: 10.1093/cid/ciy463
24. Frankel AD, Young JAT. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem (1998) 67:1–25. doi: 10.1146/annurev.biochem.67.1.1
25. Rose KM, Marin M, Kozak SL, Kabat D. The viral infectivity factor (Vif) of HIV-1 unveiled. Trends Mol Med (2004) 10(6):291–7. doi: 10.1016/j.molmed.2004.04.008
26. Pereira EA, daSilva LLP. HIV-1 nef: taking control of protein trafficking. Traffic. (2016) 17(9):976–96. doi: 10.1111/tra.12412
27. Boasso A, Shearer GM, Chougnet C. Immune dysregulation in human immunodeficiency virus infection: know it, fix it, prevent it? J Intern Med (2009) 265(1):78–96. doi: 10.1111/j.1365-2796.2008.02043.x
28. Vishnu P, Aboulafia DM. Haematological manifestations of human immune deficiency virus infection. Br J Haematol (2015) 171(5):695–709. doi: 10.1111/bjh.13783
29. Kirchhoff F, Silvestri G. Is nef the elusive cause of HIV-associated hematopoietic dysfunction? J Clin Invest (2008) 118(5):1622–5. doi: 10.1172/JCI35487
30. Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol (2013) 119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3
31. Ji J, Loeb LA. Fidelity of HIV-1 reverse transcriptase copying a hypervariable region of the HIV-1 env gene. Virology. (1994) 199(2):323–30. doi: 10.1006/viro.1994.1130
32. Cuevas JM, Geller R, Garijo R, López-Aldeguer J, Sanjuán R. Extremely high mutation rate of HIV-1 In vivo. PloS Biol (2015) 13(9):e1002251. doi: 10.1371/journal.pbio.1002251
33. Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1. Sci (80-). (1988) 242(4882):1171–3. doi: 10.1126/science.2460925
34. Castro-Nallar E, Pérez-Losada M, Burton GF, Crandall KA. The evolution of HIV: inferences using phylogenetics. Mol Phylogenet Evol (2012) 62(2):777–92. doi: 10.1016/j.ympev.2011.11.019
35. Abecasis AB, Wensing AMJ, Paraskevis D, Vercauteren J, Theys K, Van de Vijver DAMC, et al. HIV-1 subtype distribution and its demographic determinants in newly diagnosed patients in Europe suggest highly compartmentalized epidemics. Retrovirology. (2013) 10(1):7. doi: 10.1186/1742-4690-10-7
36. Souto B, Triunfante V, Santos-Pereira A, Martins J, Araújo PMM, Osório NS. Evolutionary dynamics of HIV-1 subtype c in Brazil. Sci Rep (2021) 11(1):23060. doi: 10.1038/s41598-021-02428-3
37. Geretti AM. HIV-1 subtypes: epidemiology and significance for HIV management. Curr Opin Infect Dis (2006) 19(1):1–7. doi: 10.1097/01.qco.0000200293.45532.68
38. Siddappa NB, Dash PK, Mahadevan A, Desai A, Jayasuryan N, Ravi V, et al. Identification of unique B/C recombinant strains of HIV-1 in the southern state of karnataka, India. Aids. (2005) 19(13):1426–9. doi: 10.1097/01.aids.0000180795.49016.89
39. Soares MA, De Oliveira T, Brindeiro RM, Diaz RS, Sabino EC, Brigido L, et al. A specific subtype c of human immunodeficiency virus type 1 circulates in Brazil. AIDS. (2003) 17(1):11–21. doi: 10.1097/00002030-200301030-00004
40. Bbosa N, Kaleebu P, Ssemwanga D. HIV Subtype diversity worldwide. Curr Opin HIV AIDS. (2019) 14(3):153–60. doi: 10.1097/COH.0000000000000534
41. Riemenschneider M, Cashin KY, Budeus B, Sierra S, Shirvani-Dastgerdi E, Bayanolhagh S, et al. Genotypic prediction of Co-receptor tropism of HIV-1 subtypes a and c. Sci Rep (2016) 6(1):24883. doi: 10.1038/srep24883
42. Esbjörnsson J, Månsson F, Martínez-Arias W, Vincic E, Biague AJ, da Silva ZJ, et al. Frequent CXCR4 tropism of HIV-1 subtype a and CRF02_AG during late-stage disease - indication of an evolving epidemic in West Africa. Retrovirology. (2010) 7:23. doi: 10.1186/1742-4690-7-23
43. Ketseoglou I, Lukhwareni A, Steegen K, Carmona S, Stevens WS, Papathanasopoulos MA. Viral tropism and antiretroviral drug resistance in HIV-1 subtype c-infected patients failing highly active antiretroviral therapy in Johannesburg, south Africa. AIDS Res Hum Retroviruses (2014) 30(3):289–93. doi: 10.1089/aid.2013.0267
44. Cashin K, Paukovics G, Jakobsen MR, Østergaard L, Churchill MJ, Gorry PR, et al. Differences in coreceptor specificity contribute to alternative tropism of HIV-1 subtype c for CD4+ T-cell subsets, including stem cell memory T-cells. Retrovirology. (2014) 11(1):97. doi: 10.1186/s12977-014-0097-5
45. Ball SC, Abraha A, Collins KR, Marozsan AJ, Baird H, Quiñones-Mateu ME, et al. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes b and c. J Virol (2003) 77(2):1021–38. doi: 10.1128/JVI.77.2.1021-1038.2003
46. Venner CM, Nankya I, Kyeyune F, Demers K, Kwok C, Chen P-LL, et al. Infecting HIV-1 subtype predicts disease progression in women of Sub-Saharan Africa. EBioMedicine. (2016) 13:305–14. doi: 10.1016/j.ebiom.2016.10.014
47. de Mendoza C, Garrido C. Different disease progression rate according to HIV-1 subtype. Futur HIV Ther (2008) 2(4):319–22. doi: 10.2217/17469600.2.4.319
48. Kanki PJ, Hamel DJ, Sankalé JL, cheng HC, Thior I, Barin F, et al. Human immunodeficiency virus type 1 subtypes differ in disease progression. J Infect Dis (1999) 179(1):68–73. doi: 10.1086/314557
49. Marozsan AJ, Moore DM, Lobritz MA, Fraundorf E, Abraha A, Reeves JD, et al. Differences in the fitness of two diverse wild-type human immunodeficiency virus type 1 isolates are related to the efficiency of cell binding and entry. J Virol (2005) 79(11):7121–34. doi: 10.1128/JVI.79.11.7121-7134.2005
50. Kaleebu P, French N, Mahe C, Yirrell D, Watera C, Lyagoba F, et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes a and d on disease progression in a large cohort of HIV-1-positive persons in Uganda. J Infect Dis (2002) 185(9):1244–50. doi: 10.1086/340130
51. Lynch RM, Shen T, Gnanakaran S, Derdeyn CA. Appreciating HIV type 1 diversity: subtype differences in env. AIDS Res Hum Retroviruses (2009) 25(3):237–48. doi: 10.1089/aid.2008.0219
52. Gnanakaran S, Lang D, Daniels M, Bhattacharya T, Derdeyn CA, Korber B. Clade-specific differences between human immunodeficiency virus type 1 clades b and c: diversity and correlations in C3-V4 regions of gp120. J Virol (2007) 81(9):4886–91. doi: 10.1128/JVI.01954-06
53. Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Sci (80-). (2004) 303(5666):2019–22. doi: 10.1126/science.1093137
54. Frost SDW, Liu Y, Pond SLK, Chappey C, Wrin T, Petropoulos CJ, et al. Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype b. J Virol (2005) 79(10):6523–7. doi: 10.1128/JVI.79.10.6523-6527.2005
55. Choisy M, Woelk CH, Guégan J-F, Robertson DL. Comparative study of adaptive molecular evolution in different human immunodeficiency virus groups and subtypes. J Virol (2004) 78(4):1962–70. doi: 10.1128/JVI.78.4.1962-1970.2004
56. Patel MB, Hoffman NG, Swanstrom R. Subtype-specific conformational differences within the V3 region of subtype b and subtype c human immunodeficiency virus type 1 env proteins. J Virol (2008) 82(2):903 LP – 916. doi: 10.1128/JVI.01444-07
57. Travers SAA, Connell MJ, McCormack GP, McInerney JO. Evidence for heterogeneous selective pressures in the evolution of the env gene in different human immunodeficiency virus type 1 subtypes. J Virol (2005) 79(3):1836 LP – 1841. doi: 10.1128/JVI.79.3.1836-1841.2005
58. Iordanskiy S, Waltke M, Feng Y, Wood C. Subtype-associated differences in HIV-1 reverse transcription affect the viral replication. Retrovirology. (2010) 7(1):85. doi: 10.1186/1742-4690-7-85
59. Han YS, Mesplède T, Wainberg MA. Differences among HIV-1 subtypes in drug resistance against integrase inhibitors. Infect Genet Evol (2016) 46:286–91. doi: 10.1016/j.meegid.2016.06.047
60. Koning FA, Castro H, Dunn D, Tilston P, Cane PA, Mbisa JL. Subtype-specific differences in the development of accessory mutations associated with high-level resistance to hiv-1 nucleoside reverse transcriptase inhibitors. J Antimicrob Chemother (2013) 68(6):1220–36. doi: 10.1093/jac/dkt012
61. Spira S. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J Antimicrob Chemother (2003) 51(2):229–40. doi: 10.1093/jac/dkg079
62. Zazzi M, Hu H, Prosperi M. The global burden of HIV-1 drug resistance in the past 20 years. PeerJ. (2018) 6:e4848–8. doi: 10.7717/peerj.4848
63. Woodham AW, Skeate JG, Sanna AM, Taylor JR, Da Silva DM, Cannon PM, et al. Human immunodeficiency virus immune cell receptors, coreceptors, and cofactors: implications for prevention and treatment. AIDS Patient Care STDS. (2016) 30(7):291–306. doi: 10.1089/apc.2016.0100
64. Siliciano JD, Siliciano RF. Latency and viral persistence in HIV-1 infection. J Clin Invest. (2000) 106(7):823–5. doi: 10.1172/JCI11246
65. Ta TM, Malik S, Anderson EM, Jones AD, Perchik J, Freylikh M, et al. Insights into persistent HIV-1 infection and functional cure: novel capabilities and strategies. Front Microbiol (2022) 13. doi: 10.3389/fmicb.2022.862270
66. Siliciano RF, Greene WC. HIV Latency. Cold Spring Harb Perspect Med (2011) 1(1):a007096. doi: 10.1101/cshperspect.a007096
67. Dufour C, Gantner P, Fromentin R, Chomont N. The multifaceted nature of HIV latency. J Clin Invest. (2020) 130(7):3381–90. doi: 10.1172/JCI136227
68. Brooks K, Jones BR, Dilernia DA, Wilkins DJ, Claiborne DT, McInally S, et al. HIV-1 variants are archived throughout infection and persist in the reservoir. PloS Pathog (2020) 16(6):e1008378. doi: 10.1371/journal.ppat.1008378
69. Jones BR, Kinloch NN, Horacsek J, Ganase B, Harris M, Harrigan PR, et al. Phylogenetic approach to recover integration dates of latent HIV sequences within-host. Proc Natl Acad Sci U S A. (2018) 115(38):E8958–67. doi: 10.1073/pnas.1802028115
70. Buzon MJ, Martin-Gayo E, Pereyra F, Ouyang Z, Sun H, Li JZ, et al. Long-term antiretroviral treatment initiated at primary HIV-1 infection affects the size, composition, and decay kinetics of the reservoir of HIV-1-infected CD4 T cells. J Virol (2014) 88(17):10056–65. doi: 10.1128/JVI.01046-14
71. Ruff CT, Ray SC, Kwon P, Zinn R, Pendleton A, Hutton N, et al. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J Virol (2002) 76(18):9481–92. doi: 10.1128/JVI.76.18.9481-9492.2002
72. Verhofstede C, Noë A, Demecheleer E, De Cabooter N, Van Wanzeele F, van der Gucht B, et al. Drug-resistant variants that evolve during nonsuppressive therapy persist in HIV-1-infected peripheral blood mononuclear cells after long-term highly active antiretroviral therapy. J Acquir Immune Defic Syndr (2004) 35(5):473–83. doi: 10.1097/00126334-200404150-00005
73. Kieffer TL, Finucane MM, Nettles RE, Quinn TC, Broman KW, Ray SC, et al. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J Infect Dis (2004) 189(8):1452–65. doi: 10.1086/382488
74. Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol (2006) 80(13):6441–57. doi: 10.1128/JVI.00591-06
75. Tsukamoto T. Hematopoietic Stem/Progenitor cells and the pathogenesis of HIV/AIDS. Front Cell Infect Microbiol (2020) 10. doi: 10.3389/fcimb.2020.00060
76. Fan L, Li C, Zhao H. Prevalence and risk factors of cytopenia in HIV-infected patients before and after the initiation of HAART. BioMed Res Int (2020) 2020:1–10. doi: 10.1155/2020/3132589
77. Gebreweld A, Fiseha T, Girma N, Haileslasie H, Gebretsadik D. Prevalence of cytopenia and its associated factors among HIV infected adults on highly active antiretroviral therapy at mehal meda hospital, north shewa zone, Ethiopia. PloS One (2020) 15(9):e0239215. doi: 10.1371/journal.pone.0239215
78. Kyeyune R, Saathoff E, Ezeamama AE, Löscher T, Fawzi W, Guwatudde D. Prevalence and correlates of cytopenias in HIV-infected adults initiating highly active antiretroviral therapy in Uganda. BMC Infect Dis (2014) 14(1):496. doi: 10.1186/1471-2334-14-496
79. Fiseha T, Ebrahim H. Prevalence and predictors of cytopenias in HIV-infected adults at initiation of antiretroviral therapy in mehal meda hospital, central Ethiopia. J Blood Med (2022) 13:201–11. doi: 10.2147/JBM.S355966
80. Kaner J, Thibaud S, Sridharan A, Assal A, Polineni R, Zingman B, et al. HIV Is associated with a high rate of unexplained multilineage cytopenias and portends a poor prognosis in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Blood. (2016) 128(22):4345–5. doi: 10.1182/blood.V128.22.4345.4345
81. Sharma S, Sachdeva RK, Sachdeva MUS, Sreedharanunni S, Naseem S, Sharma P, et al. Bone marrow examination of HIV-infected children in HAART era reveals a spectrum of abnormalities: a study from single tertiary care center of north India. J Hematop. (2021) 14(4):283–90. doi: 10.1007/s12308-021-00471-7
82. Gunda DW, Godfrey KG, Kilonzo SB, Mpondo BC. Cytopenias among ART-naive patients with advanced HIV disease on enrolment to care and treatment services at a tertiary hospital in Tanzania: a crosssectional study. Malawi Med J (2017) 29(1):43. doi: 10.4314/mmj.v29i1.9
83. Banda NK, Simon GR, Sipple JD, Terrell KL, Archer P, Shpall EJ, et al. Depletion of CD34+CD4+ cells in bone marrow from HIV-1-infected individuals. Biol Blood Marrow Transplant. (1999) 5(3):162–72. doi: 10.1053/bbmt.1999.v5.pm10392962
84. Shah I, Murthy A. Bone marrow abnormalities in HIV infected children, report of three cases and review of the literature. J Res Med Sci (2014) 19(2):181–3.
85. Koka P, Reddy S. Cytopenias in HIV infection: mechanisms and alleviation of hematopoietic inhibition. Curr HIV Res (2004) 2(3):275–82. doi: 10.2174/1570162043351282
86. Alexaki A, Wigdahl B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PloS Pathog (2008) 4(12):e1000215. doi: 10.1371/journal.ppat.1000215
87. Wong ME, Jaworowski A, Hearps AC. The HIV reservoir in monocytes and macrophages. Front Immunol (2019) 10(JUN). doi: 10.3389/fimmu.2019.01435
88. Maryanovich M, Takeishi S, Frenette PS. Neural regulation of bone and bone marrow. Cold Spring Harb Perspect Med (2018) 8(9):a031344. doi: 10.1101/cshperspect.a031344
89. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. (2011) 147(5):1146–58. doi: 10.1016/j.cell.2011.09.053
90. Datta G, Miller NM, Afghah Z, Geiger JD, Chen X. HIV-1 gp120 promotes lysosomal exocytosis in human schwann cells. Front Cell Neurosci (2019) 13. doi: 10.3389/fncel.2019.00329
91. Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, et al. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res (2018) 6(1):2. doi: 10.1038/s41413-017-0005-4
92. Mahadevan A, Gayathri N, Taly AB, Santosh V, Yasha TC, Shankar SK. Vasculitic neuropathy in HIV infection: a clinicopathological study. Neurol India. (2001) 49(3):277–83.
93. Keswani SC, Polley M, Pardo CA, Griffin JW, McArthur JC, Hoke A. Schwann cell chemokine receptors mediate HIV-1 gp120 toxicity to sensory neurons. Ann Neurol (2003) 54(3):287–96. doi: 10.1002/ana.10645
94. Ntogwa M, Imai S, Hiraiwa R, Koyanagi M, Matsumoto M, Ogihara T, et al. Schwann cell-derived CXCL1 contributes to human immunodeficiency virus type 1 gp120-induced neuropathic pain by modulating macrophage infiltration in mice. Brain Behav Immun (2020) 88:325–39. doi: 10.1016/j.bbi.2020.03.027
95. Renders S, Svendsen AF, Panten J, Rama N, Maryanovich M, Sommerkamp P, et al. Niche derived netrin-1 regulates hematopoietic stem cell dormancy via its receptor neogenin-1. Nat Commun (2021) 12(1):608. doi: 10.1038/s41467-020-20801-0
96. Xu C, Gao X, Wei Q, Nakahara F, Zimmerman SE, Mar J, et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun (2018) 9(1):2449. doi: 10.1038/s41467-018-04726-3
97. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. (2012) 481(7382):457–62. doi: 10.1038/nature10783
98. Kenswil KJG, Jaramillo AC, Ping Z, Chen S, Hoogenboezem RM, Mylona MA, et al. Characterization of endothelial cells associated with hematopoietic niche formation in humans identifies IL-33 as an anabolic factor. Cell Rep (2018) 22(3):666–78. doi: 10.1016/j.celrep.2017.12.070
99. Almeida-Porada G, Ascensāo JL. Isolation, characterization, and biologic features of bone marrow endothelial cells. J Lab Clin Med (1996) 128(4):399–407. doi: 10.1016/S0022-2143(96)80012-6
100. Li WM, Huang WQ, Huang YH, De Jiang Z, Wang QR. Positive and negative haematopoietic cytokines produced by bone marrow endothelial cells. Cytokine. (2000) 12(7):1017–23. doi: 10.1006/cyto.1999.0678
101. Moses AV, Williams S, Heneveld ML, Strussenberg J, Rarick M, Loveless M, et al. Human immunodeficiency virus infection of bone marrow endothelium reduces induction of stromal hematopoietic growth factors. Blood. (1996) 87(3):919–25. doi: 10.1182/blood.V87.3.919.bloodjournal873919
102. Herrmann CH, Rice AP. Specific interaction of the human immunodeficiency virus tat proteins with a cellular protein kinase. Virology. (1993) 197(2):601–8. doi: 10.1006/viro.1993.1634
103. Zidovetzki R, Wang JL, Chen P, Jeyaseelan R, Hofman F. Human immunodeficiency virus tat protein induces interleukin 6 mRNA expression in human brain endothelial cells via protein kinase c- and cAMP- dependent protein kinaes pathways. AIDS Res Hum Retroviruses (1998) 14(10):825–33. doi: 10.1089/aid.1998.14.825
104. Kamtchum-Tatuene J, Mwandumba H, Al-Bayati Z, Flatley J, Griffiths M, Solomon T, et al. HIV Is associated with endothelial activation despite ART, in a sub-Saharan African setting. Neurol - Neuroimmunol Neuroinflammation. (2019) 6(2):e531. doi: 10.1212/NXI.0000000000000531
105. Dhawan S, Puri RK, Kumar A, Duplan H, Masson JM, Aggarwal BB. Human immunodeficiency virus-1-tat protein induces the cell surface expression of endothelial leukocyte adhesion molecule-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 in human endothelial cells. Blood. (1997) 90(4):1535–44. doi: 10.1182/blood.V90.4.1535
106. Hofman FM, Wright AD, Dohadwala MM, Wong-Staal F, Walker SM. Exogenous tat protein activates human endothelial cells. Blood. (1993) 82(9):2774–80. doi: 10.1182/blood.V82.9.2774.2774
107. Acheampong EA, Parveen Z, Muthoga LW, Kalayeh M, Mukhtar M, Pomerantz RJ. Human immunodeficiency virus type 1 nef potently induces apoptosis in primary human brain microvascular endothelial cells via the activation of caspases. J Virol (2005) 79(7):4257–69. doi: 10.1128/JVI.79.7.4257-4269.2005
108. Qi M, Aiken C. Nef enhances HIV-1 infectivity via association with the virus assembly complex. Virology. (2008) 373(2):287–97. doi: 10.1016/j.virol.2007.12.001
109. Wang T, Green LA, Gupta SK, Kim C, Wang L, Almodovar S, et al. Transfer of intracellular HIV nef to endothelium causes endothelial dysfunction. PloS One (2014) 9(3):e91063. doi: 10.1371/journal.pone.0091063
110. Vilhardt F, Plastre O, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, et al. The HIV-1 nef protein and phagocyte NADPH oxidase activation. J Biol Chem (2002) 277(44):42136–43. doi: 10.1074/jbc.M200862200
111. Duffy P, Wang X, Lin PH, Yao Q, Chen C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J Surg Res (2009) 156(2):257–64. doi: 10.1016/j.jss.2009.02.005
112. Caccuri F, Giagulli C, Bugatti A, Benetti A, Alessandri G, Ribatti D, et al. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc Natl Acad Sci (2012) 109(36):14580–5. doi: 10.1073/pnas.1206605109
113. Marini E, Tiberio L, Caracciolo S, Tosti G, Guzman CA, Schiaffonati L, et al. HIV-1 matrix protein p17 binds to monocytes and selectively stimulates MCP-1 secretion: role of transcriptional factor AP-1. Cell Microbiol (2008) 10(3):655–66. doi: 10.1111/j.1462-5822.2007.01073.x
114. Mazzuca P, Caruso A, Caccuri F. Endothelial cell dysfunction in HIV-1 Infection. In: Endothelial dysfunction - old concepts and new challenges. (Rijeka: InTech) (2018). doi: 10.5772/intechopen.73023
115. Aoki K, Kurashige M, Ichii M, Higaki K, Sugiyama T, Kaito T, et al. Identification of CXCL12-abundant reticular cells in human adult bone marrow. Br J Haematol (2021) 193(3):659–68. doi: 10.1111/bjh.17396
116. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. (2006) 25(6):977–88. doi: 10.1016/j.immuni.2006.10.016
117. Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. (2005) 106(9):3020–7. doi: 10.1182/blood-2004-01-0272
118. Möhle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. (1998) 91(12):4523–30. doi: 10.1182/blood.V91.12.4523.412k04_4523_4530
119. Schajnovitz A, Itkin T, D’Uva G, Kalinkovich A, Golan K, Ludin A, et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat Immunol (2011) 12(5):391–8. doi: 10.1038/ni.2017
120. Abe-Suzuki S, Kurata M, Abe S, Onishi I, Kirimura S, Nashimoto M, et al. CXCL12+ stromal cells as bone marrow niche for CD34+ hematopoietic cells and their association with disease progression in myelodysplastic syndromes. Lab Investig (2014) 94(11):1212–23. doi: 10.1038/labinvest.2014.110
121. Ding J, Zhao J, Zhou J, Li X, Wu Y, Ge M, et al. Association of gene polymorphism of SDF1(CXCR12) with susceptibility to HIV-1 infection and AIDS disease progression: a meta-analysis. PloS One (2018) 13(2):e0191930. doi: 10.1371/journal.pone.0191930
122. Zhang J, Supakorndej T, Krambs JR, Rao M, Abou-Ezzi G, Ye RY, et al. Bone marrow dendritic cells regulate hematopoietic stem/progenitor cell trafficking. J Clin Invest. (2019) 129(7):2920–31. doi: 10.1172/JCI124829
123. Clayton KL, Garcia JV, Clements JE, Walker BD. Hiv infection of macrophages: implications for pathogenesis and cure. Pathog Immun (2017) 2(2):179–92. doi: 10.20411/pai.v2i2.204
124. Hendricks CM, Cordeiro T, Gomes AP, Stevenson M. The interplay of HIV-1 and macrophages in viral persistence. Front Microbiol (2021) 12. doi: 10.3389/fmicb.2021.646447
125. Veenhuis RT, Abreu CM, Costa PAG, Ferreira EA, Ratliff J, Pohlenz L, et al. Monocyte-derived macrophages contain persistent latent HIV reservoirs. Nat Microbiol (2023) 8(5):833–44. doi: 10.1038/s41564-023-01349-3
126. Sakaguchi M, Sato T, Groopman JE. Human immunodeficiency virus infection of megakaryocytic cells. Blood. (1991) 77(3):481–5. doi: 10.1182/blood.V77.3.481.481
127. Sato T, Sekine H, Kakuda H, Miura N, Sunohara M, Fuse A. HIV Infection of megakaryocytic cell lines. Leuk Lymphoma. (2000) 36(3–4):397–404. doi: 10.3109/10428190009148861
128. Voulgaropoulou F, Pontow SE, Ratner L. Productive infection of CD34+-Cell-Derived megakaryocytes by X4 and R5 HIV-1 isolates. Virology. (2000) 269(1):78–85. doi: 10.1006/viro.2000.0193
129. Zucker-Franklin D, Cao YZ. Megakaryocytes of human immunodeficiency virus-infected individuals express viral RNA. Proc Natl Acad Sci U S A. (1989) 86(14):5595–9. doi: 10.1073/pnas.86.14.5595
130. Chen K, Jiao Y, Liu L, Huang M, He C, He W, et al. Communications between bone marrow macrophages and bone cells in bone remodeling. Front Cell Dev Biol (2020) 8. doi: 10.3389/fcell.2020.598263
131. Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med (2011) 208(3):421–8. doi: 10.1084/jem.20110132
132. Sims NA, Quinn JMW. Osteoimmunology: oncostatin m as a pleiotropic regulator of bone formation and resorption in health and disease. Bonekey Rep (2014) 3. doi: 10.1038/bonekey.2014.22
133. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00119
134. Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol (2012) 32(1):23–63. doi: 10.1615/CritRevImmunol.v32.i1.30
135. Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzón IM, Nepomnaschy I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. Neyrolles O editor. PloS One (2010) 5(2):e9252. doi: 10.1371/journal.pone.0009252
136. Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. (2010) 116(23):4815–28. doi: 10.1182/blood-2009-11-253534
137. Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med (2011) 208(2):261–71. doi: 10.1084/jem.20101688
138. Canque B, Marandin A, Rosenzwajg M, Louache F, Vainchenker W, Gluckman JC. Susceptibility of human bone marrow stromal cells to human immunodeficiency virus (HIV). Virology. (1995) 208(2):779–83. doi: 10.1006/viro.1995.1211
139. Campbell JH, Hearps AC, Martin GE, Williams KC, Crowe SM. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. AIDS. (2014) 28(15):2175–87. doi: 10.1097/QAD.0000000000000408
140. Araínga M, Edagwa B, Mosley RL, Poluektova LY, Gorantla S, Gendelman HE. A mature macrophage is a principal HIV-1 cellular reservoir in humanized mice after treatment with long acting antiretroviral therapy. Retrovirology. (2017) 14(1):17. doi: 10.1186/s12977-017-0344-7
141. Ladinsky MS, Khamaikawin W, Jung Y, Lin S, Lam J, An DS, et al. Mechanisms of virus dissemination in bone marrow of HIV-1–infected humanized BLT mice. Elife (2019) 8. doi: 10.7554/eLife.46916
142. Honeycutt JB, Wahl A, Baker C, Spagnuolo RA, Foster J, Zakharova O, et al. Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest. (2016) 126(4):1353–66. doi: 10.1172/JCI84456
143. Guo L, Xu X-Q, Zhou L, Zhou R-H, Wang X, Li J-L, et al. Human intestinal epithelial cells release antiviral factors that inhibit HIV infection of macrophages. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00247
144. Xu X-Q, Guo L, Wang X, Liu Y, Liu H, Zhou R-H, et al. Human cervical epithelial cells release antiviral factors and inhibit HIV replication in macrophages. J Innate Immun (2019) 11(1):29–40. doi: 10.1159/000490586
145. Zhang B, Liu J, Zhou L, Wang X, Khan S, Hu W, et al. Cytosolic DNA sensor activation inhibits HIV infection of macrophages. J Med Virol (2023) 95(1). doi: 10.1002/jmv.28253
146. Denu RA, Nemcek S, Bloom DD, Goodrich AD, Kim J, Mosher DF, et al. Fibroblasts and mesenchymal Stromal/Stem cells are phenotypically indistinguishable. Acta Haematol (2016) 136(2):85–97. doi: 10.1159/000445096
147. Haniffa MA, Collin MP, Buckley CD, Dazzi F. Mesenchymal stem cells: the fibroblasts’ new clothes? Haematologica (2009) 94(2):258–63. doi: 10.3324/haematol.13699
148. Soundararajan M, Kannan S. Fibroblasts and mesenchymal stem cells: two sides of the same coin? J Cell Physiol (2018) 233(12):9099–109. doi: 10.1002/jcp.26860
149. Dorronsoro A, Lang V, Ferrin I, Fernández-Rueda J, Zabaleta L, Pérez-Ruiz E, et al. Intracellular role of IL-6 in mesenchymal stromal cell immunosuppression and proliferation. Sci Rep (2020) 10(1):21853. doi: 10.1038/s41598-020-78864-4
150. Pricola KL, Kuhn NZ, Haleem-Smith H, Song Y, Tuan RS. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J Cell Biochem (2009) 108(3):577–88. doi: 10.1002/jcb.22289
151. Starc N, Ingo D, Conforti A, Rossella V, Tomao L, Pitisci A, et al. Biological and functional characterization of bone marrow-derived mesenchymal stromal cells from patients affected by primary immunodeficiency. Sci Rep (2017) 7(1):8153. doi: 10.1038/s41598-017-08550-5
152. Frisch BJ, Porter RL, Gigliotti BJ, Olm-Shipman AJ, Weber JM, O’Keefe RJ, et al. In vivo prostaglandin E2 treatment alters the bone marrow microenvironment and preferentially expands short-term hematopoietic stem cells. Blood. (2009) 114(19):4054–63. doi: 10.1182/blood-2009-03-205823
153. Porter RL, Georger MA, Bromberg O, McGrath KE, Frisch BJ, Becker MW, et al. Prostaglandin E2 increases hematopoietic stem cell survival and accelerates hematopoietic recovery after radiation injury. Stem Cells (2013) 31(2):372–83. doi: 10.1002/stem.1286
154. Porter RL, Calvi LM. Prostaglandin E2 is rapidly produced in response to bone marrow injury and improves survival of primitive hematopoietic cells. Blood. (2010) 116(21):407–7. doi: 10.1182/blood.V116.21.407.407
155. Dolgalev I, Tikhonova AN. Connecting the dots: resolving the bone marrow niche heterogeneity. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.622519
156. Mabuchi Y, Okawara C, Méndez-Ferrer S, Akazawa C. Cellular heterogeneity of mesenchymal Stem/Stromal cells in the bone marrow. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.689366
157. Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol (2017) 19(8):891–903. doi: 10.1038/ncb3570
158. Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Sci (80-). (2007) 318(5854):1296–9. doi: 10.1126/science.1149726
159. Wang Z, Li X, Yang J, Gong Y, Zhang H, Qiu X, et al. Single-cell RNA sequencing deconvolutes the in vivo heterogeneity of human bone marrow-derived mesenchymal stem cells. Int J Biol Sci (2021) 17(15):4192–206. doi: 10.7150/ijbs.61950
160. Kallmeyer K, Ryder MA, Pepper MS. Mesenchymal stromal cells: a possible reservoir for HIV-1? Stem Cell Rev Rep (2022) 18(4):1253–80. doi: 10.1007/s12015-021-10298-5
161. Gibellini D, Alviano F, Miserocchi A, Tazzari PL, Ricci F, Clò A, et al. HIV-1 and recombinant gp120 affect the survival and differentiation of human vessel wall-derived mesenchymal stem cells. Retrovirology. (2011) 8(1):40. doi: 10.1186/1742-4690-8-40
162. Scadden DT, Zeira M, Woon A, Wang Z, Schieve L, Ikeuchi K, et al. Human immunodeficiency virus infection of human bone marrow stromal fibroblasts. Blood. (1990) 76(2):317–22. doi: 10.1182/blood.V76.2.317.317
163. Wang L, Mondal D, La Russa VF, Agrawal KC. Suppression of clonogenic potential of human bone marrow mesenchymal stem cells by HIV type 1: putative role of HIV type 1 tat protein and inflammatory cytokines. AIDS Res Hum Retroviruses (2002) 18(13):917–31. doi: 10.1089/088922202760265597
164. Beaupere C, Garcia M, Larghero J, Fève B, Capeau J, Lagathu C. The HIV proteins tat and nef promote human bone marrow mesenchymal stem cell senescence and alter osteoblastic differentiation. Aging Cell (2015) 14(4):534–46. doi: 10.1111/acel.12308
165. Cotter EJ, Ip HSM, Powderly WG, Doran PP. Mechanism of HIV protein induced modulation of mesenchymal stem cell osteogenic differentiation. BMC Musculoskelet Disord (2008) 9(1):33. doi: 10.1186/1471-2474-9-33
166. Cotter EJ, Malizia AP, Chew N, Powderly WG, Doran PP. HIV Proteins regulate bone marker secretion and transcription factor activity in cultured human osteoblasts with consequent potential implications for osteoblast function and development. AIDS Res Hum Retroviruses (2007) 23(12):1521–9. doi: 10.1089/aid.2007.0112
167. Weivoda MM, Chew CK, Monroe DG, Farr JN, Atkinson EJ, Geske JR, et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat Commun (2020) 11(1):87. doi: 10.1038/s41467-019-14003-6
168. Blank U, Karlsson S. TGF-β signaling in the control of hematopoietic stem cells. Blood. (2015) 125(23):3542–50. doi: 10.1182/blood-2014-12-618090
169. Galán-Díez M, Kousteni S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr Mol Biol Rep (2017) 3(2):53–62. doi: 10.1007/s40610-017-0055-9
170. Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. (2006) 439(7076):599–603. doi: 10.1038/nature04247
171. Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. (2004) 118(2):149–61. doi: 10.1016/j.cell.2004.07.004
172. Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell (2007) 1(6):685–97. doi: 10.1016/j.stem.2007.10.020
173. Gohda J, Ma Y, Huang Y, Zhang Y, Gu L, Han Y, et al. HIV-1 replicates in human osteoclasts and enhances their differentiation in vitro. Retrovirology (2015) 12(1):12. doi: 10.1186/s12977-015-0139-7
174. Raynaud-Messina B, Bracq L, Dupont M, Souriant S, Usmani SM, Proag A, et al. Bone degradation machinery of osteoclasts: an HIV-1 target that contributes to bone loss. Proc Natl Acad Sci U S A. (2018) 115(11):E2556–65. doi: 10.1073/pnas.1713370115
175. Delpino MV, Quarleri J. Influence of HIV infection and antiretroviral therapy on bone homeostasis. Front Endocrinol (Lausanne). (2020) 11:11. doi: 10.3389/fendo.2020.00502
176. Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SEW, Wakkach A, Blin-Wakkach C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med (2012) 209(3):537–49. doi: 10.1084/jem.20110994
177. Xie M, Leroy H, Mascarau R, Woottum M, Dupont M, Ciccone C, et al. Cell-to-Cell spreading of HIV-1 in myeloid target cells escapes SAMHD1 restriction. MBio. (2019) 10(6):e02457–19. doi: 10.1128/mBio.02457-19
178. Nacher M, Serrano S, González A, Hernández A, Mariñoso ML, Vilella R, et al. Osteoblasts in HIV-infected patients: HIV-1 infection and cell function. Aids. (2001) 15(17):2239–43. doi: 10.1097/00002030-200111230-00004
179. Venhoff N, Walker UA. Pathogenesis of bone disorders in HIV infection. Int J Clin Rheumtol. (2009) 4(2):147–59. doi: 10.2217/ijr.09.10
180. Cummins NW, Klicpera A, Sainski AM, Bren GD, Khosla S, Westendorf JJ, et al. Human immunodeficiency virus envelope protein Gp120 induces proliferation but not apoptosis in osteoblasts at physiologic concentrations. PloS One (2011) 6(9):e24876–6. doi: 10.1371/journal.pone.0024876
181. Gibellini D, De Crignis E, Ponti C, Cimatti L, Borderi M, Tschon M, et al. HIV-1 triggers apoptosis in primary osteoblasts and HOBIT cells through TNFα activation. J Med Virol (2008) 80(9):1507–14. doi: 10.1002/jmv.21266
182. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med (2009) 15(7):757–65. doi: 10.1038/nm.1979
183. Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-β1. Cell Stem Cell (2010) 6(3):265–78. doi: 10.1016/j.stem.2010.02.002
184. Lima A. Osteopenia and osteoporosis in people living with HIV: multiprofessional approach. HIV/AIDS - Res Palliat Care (2011) 117:117–24. doi: 10.2147/HIV.S6617
185. Kruger MJ, Nell TA. Bone mineral density in people living with HIV: a narrative review of the literature. AIDS Res Ther (2017) 14(1):35. doi: 10.1186/s12981-017-0162-y
186. McComsey GA, Tebas P, Shane E, Yin MT, Overton ET, Huang JS, et al. Bone disease in HIV infection: a practical review and recommendations for HIV care providers. Clin Infect Dis (2010) 51(8):937–46. doi: 10.1086/656412
187. Wakabayashi Y, Yoshino Y, Seo K, Koga I, Kitazawa T, Ota Y. Inhibition of osteoblast differentiation by ritonavir. BioMed Rep (2018) 9(6):491–6. doi: 10.3892/br.2018.1154
188. Bergamaschi A, Pancino G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology. (2010) 7(1):31. doi: 10.1186/1742-4690-7-31
189. Izquierdo-Useros N, Lorizate M, McLaren PJ, Telenti A, Kräusslich H-G, Martinez-Picado J, et al. HIV-1 capture and transmission by dendritic cells: the role of viral glycolipids and the cellular receptor siglec-1. PloS Pathog (2014) 10(7):e1004146. doi: 10.1371/journal.ppat.1004146
190. Ahmed Z, Kawamura T, Shimada S, Piguet V. The role of human dendritic cells in HIV-1 infection. J Invest Dermatol (2015) 135(5):1225–33. doi: 10.1038/jid.2014.490
191. Manches O, Frleta D, Bhardwaj N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol (2014) 35(3):114–22. doi: 10.1016/j.it.2013.10.003
192. Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-Infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology. (2005) 343(2):256–66. doi: 10.1016/j.virol.2005.09.059
193. Lepelley A, Louis S, Sourisseau M, Law HKW, Pothlichet J, Schilte C, et al. Innate sensing of HIV-infected cells. Emerman M editor. PloS Pathog (2011) 7(2):e1001284. doi: 10.1371/journal.ppat.1001284
194. Martín-Moreno A, Muñoz-Fernández MA. Dendritic cells, the double agent in the war against HIV-1. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02485
195. Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol (2006) 6(11):859–68. doi: 10.1038/nri1960
196. Ziegler SM, Altfeld M. Human immunodeficiency virus 1 and type I interferons–where sex makes a difference. Front Immunol (2017) 8:8. doi: 10.3389/fimmu.2017.01224
197. Murira A, Lamarre A. Type-I interferon responses: from friend to foe in the battle against chronic viral infection. Front Immunol (2016) 7. doi: 10.3389/fimmu.2016.00609
198. Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211(2):245–62. doi: 10.1084/jem.20131043
199. Demerdash Y, Kain B, Essers MAG, King KY. Yin and yang: the dual effects of interferons on hematopoiesis. Exp Hematol (2021) 96:1–12. doi: 10.1016/j.exphem.2021.02.002
200. Huot N, Bosinger SE, Paiardini M, Reeves RK, Müller-Trutwin M. Lymph node cellular and viral dynamics in natural hosts and impact for HIV cure strategies. Front Immunol (2018) 9:780. doi: 10.3389/fimmu.2018.00780
201. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JAM, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. (1997) 94(24):13193–7. doi: 10.1073/pnas.94.24.13193
202. Chen JJ-Y, Huang JC, Shirtliff M, Briscoe E, Ali S, Cesani F, et al. CD4 lymphocytes in the blood of HIV(+) individuals migrate rapidly to lymph nodes and bone marrow: support for homing theory of CD4 cell depletion. J Leukoc Biol (2002) 72(2):271–8. doi: 10.1189/jlb.72.2.271
203. Reuter MA, Pombo C, Betts MR. Cytokine production and dysregulation in HIV pathogenesis: lessons for development of therapeutics and vaccines. Cytokine Growth Factor Rev (2012) 23(4–5):181–91. doi: 10.1016/j.cytogfr.2012.05.005
204. Gao A, Gong Y, Zhu C, Yang W, Li Q, Zhao M, et al. Bone marrow endothelial cell-derived interleukin-4 contributes to thrombocytopenia in acute myeloid leukemia. Haematologica. (2019) 104(10):1950–61. doi: 10.3324/haematol.2018.214593
205. Okhrimenko A, Grün JR, Westendorf K, Fang Z, Reinke S, von Roth P, et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc Natl Acad Sci (2014) 111(25):9229–34. doi: 10.1073/pnas.1318731111
206. Chahroudi A, Silvestri G, Lichterfeld M. T Memory stem cells and HIV: a long-term relationship. Curr HIV/AIDS Rep (2015) 12(1):33–40. doi: 10.1007/s11904-014-0246-4
207. Di Rosa F, Gebhardt T. Bone marrow T cells and the integrated functions of recirculating and tissue-resident memory T cells. Front Immunol (2016) 7. doi: 10.3389/fimmu.2016.00051
208. Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol (2005) 26(7):360–6. doi: 10.1016/j.it.2005.04.011
209. Di Rosa F. Maintenance of memory T cells in the bone marrow: survival or homeostatic proliferation? Nat Rev Immunol (2016) 16(4):271–1. doi: 10.1038/nri.2016.31
210. Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, et al. Bone marrow and the control of immunity. Cell Mol Immunol (2012) 9(1):11–9. doi: 10.1038/cmi.2011.47
211. Sebastian NT, Collins KL. Targeting HIV latency: resting memory T cells, hematopoietic progenitor cells and future directions. Expert Rev Anti Infect Ther (2014) 12(10):1187–201. doi: 10.1586/14787210.2014.956094
212. Hoang TN, Harper JL, Pino M, Wang H, Micci L, King CT, et al. Bone marrow-derived CD4 + T cells are depleted in simian immunodeficiency virus-infected macaques and contribute to the size of the replication-competent reservoir. J Virol (2019) 93(1). doi: 10.1128/JVI.01344-18
213. Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J, Bixby D, Savona MR, et al. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med (2010) 16(4):446–51. doi: 10.1038/nm.2109
214. Nixon CC, Vatakis DN, Reichelderfer SN, Dixit D, Kim SG, Uittenbogaart CH, et al. HIV-1 infection of hematopoietic progenitor cells in vivo in humanized mice. Blood. (2013) 122(13):2195–204. doi: 10.1182/blood-2013-04-496950
215. Zou W, Xing J, Wang F, Chen X, Liu Q, Wang J, et al. HIV-1LAI nef blocks the development of hematopoietic stem/progenitor cells into T lymphoid cells. AIDS. (2021) 35(6):851–60. doi: 10.1097/QAD.0000000000002837
216. Stanley SK, Kessler SW, Justement JS, Schnittman SM, Greenhouse JJ, Brown CC, et al. CD34+ bone marrow cells are infected with HIV in a subset of seropositive individuals. J Immunol (1992) 149(2):689–97. doi: 10.4049/jimmunol.149.2.689
217. Neal TF, Holland HK, Baum CM, Villinger F, Ansari AA, Saral R, et al. CD34+ progenitor cells from asymptomatic patients are not a major reservoir for human immunodeficiency virus-1. Blood. (1995) 86(5):1749–56. doi: 10.1182/blood.V86.5.1749.bloodjournal8651749
218. Redd AD, Avalos A, Essex M. Infection of hematopoietic progenitor cells by HIV-1 subtype c, and its association with anemia in southern Africa. Blood. (2007) 110(9):3143–9. doi: 10.1182/blood-2007-04-086314
219. Durand CM, Ghiaur G, Siliciano JD, Rabi SA, Eisele EE, Salgado M, et al. HIV-1 DNA is detected in bone marrow populations containing CD4 + T cells but is not found in purified CD34 + hematopoietic progenitor cells in most patients on antiretroviral therapy. J Infect Dis (2012) 205(6):1014–8. doi: 10.1093/infdis/jir884
220. Josefsson L, Eriksson S, Sinclair E, Ho T, Killian M, Epling L, et al. Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J Infect Dis (2012) 206(1):28–34. doi: 10.1093/infdis/jis301
221. Griffin DO, Goff SP. HIV-1 is restricted prior to integration of viral DNA in primary cord-derived human CD34 + cells. J Virol (2015) 89(15):8096–100. doi: 10.1128/JVI.01044-15
222. Renelt S, Schult-Dietrich P, Baldauf H-M, Stein S, Kann G, Bickel M, et al. HIV-1 infection of long-lived hematopoietic precursors in vitro and In vivo. Cells (2022) 11(19):2968. doi: 10.3390/cells11192968
223. Molina J, Scadden D, Sakaguchi M, Fuller B, Woon A, Groopman J. Lack of evidence for infection of or effect on growth of hematopoietic progenitor cells after in vivo or in vitro exposure to human immunodeficiency virus. Blood. (1990) 76(12):2476–82. doi: 10.1182/blood.V76.12.2476.2476
224. Davis BR, Schwartz DH, Marx JC, Johnson CE, Berry JM, Lyding J, et al. Absent or rare human immunodeficiency virus infection of bone marrow stem/progenitor cells in vivo. J Virol (1991) 65(4):1985–90. doi: 10.1128/jvi.65.4.1985-1990.1991
225. Louache F, Henri A, Bettaieb A, Oksenhendler E, Raguin G, Tulliez M, et al. Role of human immunodeficiency virus replication in defective in vitro growth of hematopoietic progenitors. Blood. (1992) 80(12):2991–9. doi: 10.1182/blood.V80.12.2991.2991
226. Chelucci C, Hassan HJ, Locardi C, Bulgarini D, Pelosi E, Mariani G, et al. In vitro human immunodeficiency virus-1 infection of purified hematopoietic progenitors in single-cell culture. Blood. (1995) 85(5):1181–7. doi: 10.1182/blood.V85.5.1181.bloodjournal8551181
227. Marandin A, Katz A, Oksenhendler E, Tulliez M, Picard F, Vainchenker W, et al. Loss of primitive hematopoietic progenitors in patients with human immunodeficiency virus infection. Blood. (1996) 88(12):4568–78. doi: 10.1182/blood.V88.12.4568.bloodjournal88124568
228. Weichold FF, Zella D, Barabitskaja O, Maciejewski JP, Dunn DE, Sloand EM, et al. Neither human immunodeficiency virus-1 (HIV-1) nor HIV-2 infects most-primitive human hematopoietic stem cells as assessed in long-term bone marrow cultures. Blood. (1998) 91(3):907–15. doi: 10.1182/blood.V91.3.907
229. Shen H, Cheng T, Preffer FI, Dombkowski D, Tomasson MH, Golan DE, et al. Intrinsic human immunodeficiency virus type 1 resistance of hematopoietic stem cells despite coreceptor expression. J Virol (1999) 73(1):728–37. doi: 10.1128/JVI.73.1.728-737.1999
230. Carter CC, McNamara LA, Onafuwa-Nuga A, Shackleton M, Riddell IVJ, Bixby D, et al. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe (2011) 9(3):223–34. doi: 10.1016/j.chom.2011.02.005
231. Zhang J, Scadden DT, Crumpacker CS. Primitive hematopoietic cells resist HIV-1 infection via p21 Waf1/Cip1/Sdi1. J Clin Invest. (2007) 117(2):473–81. doi: 10.1172/JCI28971
232. Bordoni V, Bibas M, Abbate I, Viola D, Rozera G, Agrati C, et al. Bone marrow CD34+ progenitor cells may harbour HIV-DNA even in successfully treated patients. Clin Microbiol Infect (2015) 21(3):290.e5–8. doi: 10.1016/j.cmi.2014.11.003
233. Araínga M, Su H, Poluektova LY, Gorantla S, Gendelman HE. HIV-1 cellular and tissue replication patterns in infected humanized mice. Sci Rep (2016) 6:23513. doi: 10.1038/srep23513
234. Zaikos TD, Terry VH, Sebastian Kettinger NT, Lubow J, Painter MM, Virgilio MC, et al. Hematopoietic stem and progenitor cells are a distinct HIV reservoir that contributes to persistent viremia in suppressed patients. Cell Rep (2018) 25(13):3759–3773.e9. doi: 10.1016/j.celrep.2018.11.104
235. McNamara LA, Ganesh JA, Collins KL. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-kappaB activation. J Virol (2012) 86(17):9337–50. doi: 10.1128/JVI.00895-12
236. McNamara LA, Onafuwa-Nuga A, Sebastian NT, Riddell J4, Bixby D, Collins KL. CD133+ hematopoietic progenitor cells harbor HIV genomes in a subset of optimally treated people with long-term viral suppression. J Infect Dis (2013) 207(12):1807–16. doi: 10.1093/infdis/jit118
237. Sebastian NT, Zaikos TD, Terry V, Taschuk F, McNamara LA, Onafuwa-Nuga A, et al. CD4 is expressed on a heterogeneous subset of hematopoietic progenitors, which persistently harbor CXCR4 and CCR5-tropic HIV proviral genomes in vivo. PloS Pathog (2017) 13(7):e1006509. doi: 10.1371/journal.ppat.1006509
238. Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol (2004) 78(8):4120–33. doi: 10.1128/JVI.78.8.4120-4133.2004
239. Saha K, Zhang J, Gupta A, Dave R, Yimen M, Zerhouni B. Isolation of primary HIV-1 that target CD8+ T lymphocytes using CD8 as a receptor. Nat Med (2001) 7(1):65–72. doi: 10.1038/83365
240. Xiao P, Usami O, Suzuki Y, Ling H, Shimizu N, Hoshino H, et al. Characterization of a CD4-independent clinical HIV-1 that can efficiently infect human hepatocytes through chemokine (C-X-C motif) receptor 4. Aids. (2008) 22(14):1749–57. doi: 10.1097/QAD.0b013e328308937c
241. Yoshii H, Kamiyama H, Goto K, Oishi K, Katunuma N, Tanaka Y, et al. CD4-independent human immunodeficiency virus infection involves participation of endocytosis and cathepsin b. PloS One (2011) 6(4):e19352. doi: 10.1371/journal.pone.0019352
242. Skinner AM, O’Neill SL, Grompe M, Kurre P. CXCR4 induction in hematopoietic progenitor cells from fanca–/–, -c–/–, and -d2–/– mice. Exp Hematol (2008) 36(3):273–82. doi: 10.1016/j.exphem.2007.11.006
243. Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J (1990) 9(5):1551–60. doi: 10.1002/j.1460-2075.1990.tb08274.x
244. Sumide K, Matsuoka Y, Kawamura H, Nakatsuka R, Fujioka T, Asano H, et al. A revised road map for the commitment of human cord blood CD34-negative hematopoietic stem cells. Nat Commun (2018) 9(1):2202. doi: 10.1038/s41467-018-04441-z
245. Griffin DO, Goff SP. Restriction of HIV-1-based lentiviral vectors in adult primary marrow-derived and peripheral mobilized human CD34+ hematopoietic stem and progenitor cells occurs prior to viral DNA integration. Retrovirology. (2016) 13(1):14. doi: 10.1186/s12977-016-0246-0
246. Cilliers T, Nhlapo J, Coetzer M, Orlovic D, Ketas T, Olson WC, et al. The CCR5 and CXCR4 coreceptors are both used by human immunodeficiency virus type 1 primary isolates from subtype c. J Virol (2003) 77(7):4449–56. doi: 10.1128/JVI.77.7.4449-4456.2003
247. Baeten JM, Chohan B, Lavreys L, Chohan V, McClelland RS, Certain L, et al. HIV-1 subtype d infection is associated with faster disease progression than subtype a in spite of similar plasma HIV-1 loads. J Infect Dis (2007) 195(8):1177–80. doi: 10.1086/512682
248. Desfosses Y, Solis M, Sun Q, Grandvaux N, Van Lint C, Burny A, et al. Regulation of human immunodeficiency virus type 1 gene expression by clade-specific tat proteins. J Virol (2005) 79(14):9180–91. doi: 10.1128/JVI.79.14.9180-9191.2005
249. Rogers L, Obasa AE, Jacobs GB, Sarafianos SG, Sönnerborg A, Neogi U, et al. Structural implications of genotypic variations in HIV-1 integrase from diverse subtypes. Front Microbiol (2018) 9:1754. doi: 10.3389/fmicb.2018.01754
250. Roof P, Ricci M, Genin P, Montano MA, Essex M, Wainberg MA, et al. Differential regulation of HIV-1 clade-specific b, c, and e long terminal repeats by NF-kappaB and the tat transactivator. Virology. (2002) 296(1):77–83. doi: 10.1006/viro.2001.1397
251. Izopet J, Tamalet C, Pasquier C, Sandres K, Marchou B, Massip P, et al. Quantification of HIV-1 proviral DNA by a standardized colorimetric PCR-based assay. J Med Virol (1998) 54(1):54–9. doi: 10.1002/(SICI)1096-9071(199801)54:1<54::AID-JMV8>3.0.CO;2-O
252. Brinchmann JE, Albert J, Vartdal F. Few infected CD4+ T cells but a high proportion of replication-competent provirus copies in asymptomatic human immunodeficiency virus type 1 infection. J Virol (1991) 65(4):2019–23. doi: 10.1128/jvi.65.4.2019-2023.1991
253. Josefsson L, King MS, Makitalo B, Brännström J, Shao W, Maldarelli F, et al. Majority of CD4 + T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc Natl Acad Sci U S A. (2011) 108(27):11199–204. doi: 10.1073/pnas.1107729108
254. Psallidopoulos MC, Schnittman SM, Thompson 3LM, Baseler M, Fauci AS, HC L, et al. Integrated proviral human immunodeficiency virus type 1 is present in CD4+ peripheral blood lymphocytes in healthy seropositive individuals. J Virol (1989) 63(11):4626–31. doi: 10.1128/jvi.63.11.4626-4631.1989
255. Zauli G, Vitale M, Gibellini D, Capitani S. Inhibition of purified CD34+ hematopoietic progenitor cells by human immunodeficiency virus 1 or gp120 mediated by endogenous transforming growth factor beta 1. J Exp Med (1996) 183(1):99–108. doi: 10.1084/jem.183.1.99
256. Rameshwar P, Denny TN, Gascón P. Enhanced HIV-1 activity in bone marrow can lead to myelopoietic suppression partially contributed by gag p24. J Immunol (1996) 157(9):4244–50. doi: 10.4049/jimmunol.157.9.4244
257. Kulkosky J, Laptev A, Shetty S, Srinivasan A, BouHamdan M, Prockop DJ, et al. Human immunodeficiency virus type 1 vpr alters bone marrow cell function. Blood. (1999) 93(6):1906–15. doi: 10.1182/blood.V93.6.1906.406k11_1906_1915
258. Gibellini D, Vitone F, Buzzi M, Schiavone P, De Crignis E, Cicola R, et al. HIV-1 negatively affects the survival/maturation of cord blood CD34+ hematopoietic progenitor cells differentiated towards megakaryocytic lineage by HIV-1 gp120/CD4 membrane interaction. J Cell Physiol (2007) 210(2):315–24. doi: 10.1002/jcp.20815
Keywords: bone marrow niche, HIV, haematopoiesis, haematopoietic stem/progenitor cell, dysregulation
Citation: Herd CL, Mellet J, Mashingaidze T, Durandt C and Pepper MS (2023) Consequences of HIV infection in the bone marrow niche. Front. Immunol. 14:1163012. doi: 10.3389/fimmu.2023.1163012
Received: 17 February 2023; Accepted: 21 June 2023;
Published: 11 July 2023.
Edited by:
Florence Buseyne, Institut Pasteur, FranceReviewed by:
Celina Monteiro Abreu, Johns Hopkins Medicine, United StatesCopyright © 2023 Herd, Mellet, Mashingaidze, Durandt and Pepper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Sean Pepper, bWljaGFlbC5wZXBwZXJAdXAuYWMuemE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.