 Yuhao Jiao
Yuhao Jiao Zhiyu Yan
Zhiyu Yan Aiming Yang
Aiming Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 April 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1160035
This article is part of the Research TopicMitochondria in Immune Cells and AutoimmunityView all 9 articles
Autoimmune diseases are characterized by vast alterations in immune responses, but the pathogenesis remains sophisticated and yet to be fully elucidated. Multiple mechanisms regulating cell differentiation, maturation, and death are critical, among which mitochondria-related cellular organelle functions have recently gained accumulating attention. Mitochondria, as a highly preserved organelle in eukaryotes, have crucial roles in the cellular response to both exogenous and endogenous stress beyond their fundamental functions in chemical energy conversion. In this review, we aim to summarize recent findings on the function of mitochondria in the innate immune response and its aberrancy in autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, etc., mainly focusing on its direct impact on cellular metabolism and its machinery on regulating immune response signaling pathways. More importantly, we summarize the status quo of potential therapeutic targets found in the mitochondrial regulation in the setting of autoimmune diseases and wish to shed light on future studies.
Mitochondria are highly preserved double-membrane-bound organelle in eukaryotes with fundamental functions of generating energy via oxidative phosphorylation (OXPHOS) (1). Intriguingly, some theories suggest that mitochondria may have evolved from aprokaryotic organism and resided in eukaryotic cytosol symbiotically (2, 3). The double membrane structure of mitochondria, along with their mitochondrial DNA and a highly independent energy supplication system, supports the exogenous origin hypothesis (2, 3), indicating that mitochondria could possibly be involved in the interplay and regulation of immune reactions. This was first confirmed by findings that pointed out the roles mitochondria play in the downstream signaling pathways of multiple pattern-recognition receptors (PRRs) (4). Investigations into mitochondrial apoptosis further suggest their control over the survival and death of vast types of immune cells (5). However, mounting studies have found even more evidence that mitochondria are involved in numerous signaling pathways beyond what was initially expected and that mitochondrial biology is also affected by various cellular signals and metabolites, and vice versa (6, 7).
In the study of immune disorders, autoimmune diseases provide an example of their core pathogenesis involves the loss of immune tolerance, the generation of autoantibodies, and host tissue damage (8). At the cellular level, exposure to self-antigen has a complex pathogenic process that eventually leads to the loss of self-tolerance and the abnormal activations of autoreactive T cells and/or B-cells (8).
Innate immune cells, as the first-line defense of the immune system, play a pivotal role in early defense against infectious disease and act as the facilitator for the adaptive immune system (9). Compiling studies have demonstrated the importance of antigen-presenting cells, including dendritic cells (DCs) and macrophages, in sensing danger signals and initiating downstream immune responses (10). Neutrophils and other effector cells, including natural killer (NK) cells and the helper-like innate lymphoid cells (ILCs), also contribute to end-stage tissue damage, exacerbate immune responses, and skew immune responses (11). During this process, PRRs, expressed by innate immune cells, are vital players in detecting invading pathogens and host cell damage (12). PRR recognition of pathogen-associated molecular patterns (PAMPs), expressed by microorganisms, and damage-associated molecular patterns (DMAPs), expressed by host cells, is the key driver of initial innate immune reactions (12). More specifically, sensing PAMPs and DMAPs triggers downstream signaling to recruit additional immune cells, augment cytokine production, and activate adaptive immune responses. Mitochondria get involved in this grand banquet through alterations in immune metabolism, participation in immune signaling transduction, and exacerbation of immune reactions.
Among these comprehensive mechanisms involved in the pathogenesis of autoimmune diseases are numer ous intermediates, such as nucleic acids, key transcription factors, and different immune cell subsets, which are under comprehensive study to unveil potential therapeutic targets. At the organelle level, mitochondria have emerged as critical participants in initiating and progressing multiple autoimmune diseases. And the fact that mitochondria interact with the immune system can give a reasonable explanation of how mitochondria are involved in the pathogenesis of autoimmune diseases.
In this review, we summarize current understandings of potential mechanisms by which mitochondria regulate innate immunity and contribute to the pathogenesis of autoimmune diseases, focus on mitochondrial regulation of immune signaling pathways, immunometabolism, and apoptotic pathways.
Innate immune cells recognize pathogenic and damage signals through the expression of PRR family, such as toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), and C-type lectin receptors (CLRs). Recognition of respective PAMPs and DAMPs by these receptors evokes multiple downstream signals to activate and amplify innate immune responses, eventually upregulating inflammatory mediators for participating in anti-microbial reactions and the pathogenesis of autoimmune diseases under certain conditions (13). Mitochondria are involved in this process by releasing host-derived DAMPs, including phospholipid cardiolipin (pCL), reactive oxygen species (ROS), n-formyl peptides (n-fp), and mitochondrial DNA (mtDNA), when activated by both exogenous and endogenous stress signals (14, 15). And the release of DAMPs recognized by PRRs is a way for mitochondria to participate in triggering downstream innate immune responses (14). Additionally, the transduction of certain PRR-related signaling pathways is also controlled by the mitochondrial platform for signaling molecules (15). The dual role of mitochondria in innate immune responses and the pathogenesis of autoimmune diseases may provide great potential in finding therapeutic targets.
The biochemical generation of mitochondrial ROS (mROS) is an enormously complex process linked to multiple immunological and metabolic events. Approximately 90% of the ROS are biochemical byproducts of OXPHOS at the electron transport chain (ETC) in the inner mitochondrial membrane. Theoretically, electrons from nicotinamide adenine dinucleotide hydrogen (NADH) and flavin adenine dinucleotide (FADH2) are transferred through ETC at complex I-IV. However, leakage of electrons in this process results in the formation of superoxide anion radicals, namely, mROS (16, 17). Other biochemical reactions, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) complexes and uncoupled nitric oxide synthase reactions at the meantime also generate certain amounts of non-m ROS, especially in phagocytic cells, including macrophages, DCs, and neutrophils (18–20). Studies have shown that the activation of TLRs in innate immune cells sends signals via tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) and evolutionarily conserved signaling intermediate in toll pathways (ECSIT), leading to the phagocytosis of mitochondria and eventually contributing to mROS production (21). The NLR family, another class of cytoplasmic PRRs, also potentially increases mROS levels upon its activation through downstream regulators, such as NLRP3 inflammasomes and NLRX1 (22).
Notably, mROS is constitutively produced from OXPHOS at the physiological status that supports many signaling pathways and maintains the normal machinery of the innate immune system. Owing to the strong oxidation capability of mROS, it exerts solid activity of anti-microorganisms via direct oxidation of microbial proteins and lipids (23). However, mROS is not only limited to its role in facilitating the antibacterial response but also plays a significant role in immune cell differentiation, maturation, and polarization. During the hematopoiesis of immune cells, metabolic control of cell differentiation and death occurs (24). The accumulation of mitochondrial mass and a decreased demand for glycolysis in the later developmental stages lead to an upregulation of OXPHOS and mROS production, which drives the early differentiation of naïve immune cells (25, 26). A single-cell level transcriptome study has also suggested that the potential relationship between ROS and myeloid cell differentiation via is built by modulating gene expression related to cellular metabolism (27). As to mature innate immune cells, mROS, more specifically, is crucial for the induction and maintenance of M1 macrophage (the proinflammatory macrophage subset) polarization (28). By activating nuclear factor-kappa B (NF-κB) and p38 mitogen-activated protein kinase (MAPK) signaling pathways, mROS can promote the expression of proinflammatory genes in macrophages, synthesizing numerous factors like TNF-α and interleukin (IL)-6 (29). mROS is also a prerequisite for the activation of nucleotide-binding oligomerization domain (NOD)-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome and the subsequent production of IL-1β and IL-18 via initiating caspase-1-dependent cleavage (which is discussed in detail in the following section) (30). Moreover, mROS can directly enhance immune cell function and augment innate immune responses via its downstream upregulation of hypoxia-inducible factor-1α (HIF-1α) and NF-κB, leading to the proinflammatory cytokine production and upregulated cytotoxicity (31, 32).
In recent years, there has been growing interest in the role of mROS in the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE), Sjögren’s syndrome (SjS), and rheumatoid arthritis (RA). While mROS has an essential impact in regulating cellular signaling pathways, an overproduction of mROS in response to cellular or mitochondrial stress, represented by hypoxia and inflammation, could be pathogenic (33). The accumulation of mitochondria mass and mROS production is observed in autoimmune diseases such as SLE, whereas the NOX complexes related ROS in phagocytes is deficient, which may predispose to SLE (34). Studies have shown that the increased mROS level is likely to contribute to NETosis (the formation of neutrophil extracellular traps, NETs), ultimately leading to direct tissue damage, the upregulation of interferons (IFN), and clinical progression of lupus (34, 35). In clinical trials, metformin has been studied in individuals with mild to moderate systemic lupus erythematosus (SLE), and it has been reported to reduce disease relapse, potentially by reducing NET formation and IFNα production by plasmacytoid dendritic cells (36). Moreover, ROS production via NOX complexes is also found to be downregulated or even absent in SLE, SjS, and RA (37–39). The deficiency of ROS released from NOX complexes might be a compensatory reason why mROS production and NETosis are upregulated in autoimmune diseases. When mitochondrial fission factor Drp1 is depleted via Rab4A and mitophagy is diminished, an accumulation of mitochondria mass is observed in SLE patients (40). These findings are consistent with the fact that Rab4A expression is increased in phagocytes in a lupus mouse model, indicating that Drp1 might serve as a potential therapeutic target for SLE (41). mROS is also associated with M2 macrophage (the anti-inflammatory macrophage subset) polarization induced by spermidine (30). Spermidine can markedly increase the level of mitochondrial superoxide and H2O2 that can activate AMP-activated protein kinase (AMPK), ultimately upregulating HIF-1α, which is needed for the expression of anti-inflammatory genes and induction of autophagy (30). And this phenomenon has been proven in a Dextran Sulfate Sodium (DSS)-induced inflammatory bowel disease (IBD) murine model, which could be ameliorated after treatment with spermidine in vitro (30).
Treatments targeting mROS and mitochondrial oxidative stress in SLE and RA have great potential. The antioxidants mitoquinone (mitoQ) and [2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-ox-oethyl] triphenylphosphonium chloride monohydrate (mito-TEMPO) have shown promise in ameliorating SLE via inhibition of mROS. Administration with mitoQ can result in the downregulation of mROS releasing, reduced NET formation and NETosis, as well as decreased type I IFN production via inhibition of oligomerization of mitochondrial antiviral signaling protein (MAVS), in both patient and murine models (34, 42, 43). Other novel compounds, such as polydatin, that is extracted from a traditional Chinese herb Polygonum cuspidatum, are also proven to exert therapeutic potential in SLE via by restraining ROS and NETosis-mediated damage (44). In RA, resveratrol could alleviate disease progression via its inhibition of ROS and MAPK signaling, eventually resulting in suppressed angiogenesis (45). Celastrol is also found to inhibit RA via its downregulation of NF-κB-NLRP3 inflammasome axis via ROS (46). These findings verify that mROS and oxidative stress may be promising targets for developing novel therapies for autoimmune diseases, especially for SLE and RA. But further research is also needed to deeply and fully understand how mROS elaborately works in the pathogenesis of these diseases and to develop effective treatments targeting this link upon mechanism.
mtDNA is a circular double-strand molecule composed of 37 genes that encode 13 mitochondrial-specific proteins for OXPHOS, 2 ribosomal RNA and 22 transfer RNA (47). All these molecules are pivotal for cellular metabolism, calcium homeostasis, and programmed cell death (48). However, upon cell death processes such as apoptosis and necrosis or mitochondrial damage, the integrity of mitochondria is ruptured and mtDNA is released into either cytosol or extracellular matrix. In contrast to self-DNA in the nucleus, mtDNA is considered a foreign substance due to its distinct methylation status (49, 50). Therefore, exposure to mtDNA sends a danger signal to innate immune sensors and triggers downstream immune responses, consequently contributing to the pathogenesis of certain autoimmune diseases. For example, mtDNA has been detected in the synovial fluid and serum of RA patients, where it can activate immune cells such as phagocytes and overproduce proinflammatory cytokines, leading to joint inflammation (51). In patients with SLE, abundant mtDNA released from damaged cells emerges in the circulating and can activate the type I IFN pathway via the endosomal TLR9 (52). Moreover, emerging evidence suggests that free circulating mtDNA is present in a range of inflammatory diseases, including autoimmune diseases, infectious diseases, cardiovascular diseases, and trauma, indicating their highly preserved relationship with inflammation (53). In a groundbreaking study, injecting mtDNA into joints in mice has been shown to induce sterile inflammation and arthritis (54). In conclusion, the role of mtDNA in immune responses has significant implications for understanding and treating inflammatory diseases. Meanwhile, with regards to SLE, there is increasing attention on mtDNA instability as a novel antigen whose antibodies have shown a positive correlation with anti-dsDNA, which is also observed in RA, another prototype of autoimmune diseases (55, 56).
As atype of PRR, TLRs can be activated by specific ligands to recognize various targets, covering bacterial and mtDNA, and evoke immune responses through the activation and nuclear localization of NF-κB via p38 and MAPK pathways (57). In humans, 10 types of TLR types have been identified. And the characteristics of ligands for TLR9 are hypomethylated CpG motifs, which are common between bacterial DNA and mtDNA (58, 59). TLRs are widely expressed in various immune cells, including monocytes, macrophages, and plasmacytoid DCs, and are located on the endosomal membrane of these cells. After the formation of the DNA-bound TLR9 complex, myeloid differentiation primary response 88 (MyD88) would be recruited to furtherly induce inflammatory signaling (48). Abnormally released mtDNA into the circulation or mtDNA-associated NETs are capable of instigating drastic immune responses through the above mechanism not only in systemic inflammatory response syndromes (SIRS) but also in hematological malignancies, and neutrophils are thought to play an essential role in this process (60, 61). These innate immune signaling pathways play a crucial role in the development and progression of non-alcoholic steatohepatitis (NASH), mainly involving hepatic stellate cells and macrophage-like Kupffer cells. In a study comparing TLR9 knockout (TLR9-/-) mice and wild-type mice on the same choline-deficient amino acid-defined diet, the wild- type mice were found to bemore prone to develop NASH (62). Meanwhile, the levels of oxidated mtDNA in hepatocytes and plasma of both mice and humans with NASH are immensely elevated, indicating that mitochondrial damage is associated with the development of NASH in a TLR-dependent manner. And strikingly, treatment with a TLR9 antagonist treatment has been found to be effective in treating NASH, suggesting that targeting TLR9 signaling may be a promising therapeutic strategy for this disease (63). In parallel, the pivotal interactions have also contributed to inflammation and autoimmunity that correlate with the pathogenesis of SLE (64). In SLE, neutrophils abundant in oxidized mtDNA could activate TLR7 upon the presence of type I IFN and anti-Sm/ribonucleotide protein (RNP) autoantibodies. TLR7 activation can block the phosphorylation of transcription factor A mitochondrial (TFAM), which leads to inefficient disassembly of oxidized mtDNA from TFAM (65). Consequently, oxidized mtDNA accumulates within mitochondria instead of being directed to lysosomes for degradation and is extruded as endogenous foreign substances, evoking formidable inflammation (65). In RA, while limited research has been conducted on mtDNA-induced innate immune responses through TLRs, research has focused more on the adaptive immune system, particularly T cells (56, 66).
Recent studies have shed light on another mechanism by which cytosolic DNA, including mtDNA, triggers inflammatory responses. Upon recognition of mtDNA, cyclic GMP-AMP synthase (cGAS) undergoes a conformational change that facilitates the conversion of ATP and GTP into 2’3’-cyclic GMP-AMP (cGAMP) (67). Subsequently, cGAMP causes conformational changes in the C-terminal tail of STING, which leads to its translocation from the endoplasmic reticulum to the Golgi apparatus. And this results in the recruitment and activation of Tank binding kinase 1 (TBK1) and IKB kinase (IKK), which phosphorylate and activate the transcription factor interferon regulatory factor 3 (IRF3) and NF-κB, resulting in an evoked inflammatory responses featured by type I IFN (68–70). And this pivotal mechanism has been found to contribute to the occurrence and development of systemic inflammatory diseases by numerous studies. As is well known, RA is one of the classic TNF-driven diseases characterized by an IFN signature in affected tissues (71, 72). It has been found that interactions between mtDNA and cGAS contribute to TNF-induced IFN production, which in turn contributes to the joint pathology of RA. In the presence of TNF, PINK1-mediated mitochondrial mitophagy in macrophages is inhibited, leading to an increase in cytosolic mtDNA levels, subsequently inducing and amplifying the inflammatory responses described above. Arthritis-related indicators, such as inflammatory cell infiltration and joint swelling, are significantly ameliorated in the K/BxN arthritis model with cGAS-/- compared to the wild type (73). Additionally, DNase II, an important catalytic enzyme capable of digesting chromosomal DNA and nuclei from apoptotic cells to maintain host homeostasis, has also been implicated in autoinflammation and arthritis when self-DNA degradation fails due to DNase II deficiency. This has been shown to correlate with increased type I IFN in humans and mice via sustained cGAS-STING stimulation (74, 75). These findings emphasize the crucial role of mtDNA and its recognition in the development of inflammatory responses and associated diseases.
Inflammasomes are multiprotein complexes consisting of three major components: a receptor, an adaptor, and an effector protein. The receptor protein is a member of the NLR family that recognizes PAMPS or DAMPs, which allows the inflammasome activation. The different types of inflammasomes are mainly distinguished by the molecular receptors they contain, with NLRP3 being one of the most well-studied examples (76). The recognition triggers the oligomerization of the receptor protein and then recruits the adaptor protein, defined as an apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). ASC acts as a bridging molecule, with its N-terminal pyrin domain (PYD) binding to the receptor protein and its C-terminal caspase recruitment domain (CARD) interacting with the effector protein. Pro-caspase-1, as the effector molecule, undergoes autoactivation, eventually leading to the cleavage and activation of proinflammatory cytokines, such as IL-1β and IL-18, completing the circular process from cell injury recognition to the induction of inflammatory responses (77–80). Although the precise events during the inflammasome activation process are still unclear, it is now evident that mtDNA may rouse the NLRP3 inflammasome pathway by serving as a danger signal or a DAMP. Upon mitochondrial damage or dysfunction, mtDNA is released where it can directly interact with NLRP3, inducing NLRP3 oligomerization and inflammasome activation (79, 81). Another proposed mechanism is that mtDNA can also contribute to NLRP3 inflammasome activation in an mROS-dependent manner (82). mtDNA can stimulate mROS production by binding to the MAVS and initiating the downstream signaling cascades (83). The mROS then activates the NLRP3 inflammasome by inducing mitochondrial damage, potassium efflux, and lysosomal destabilization (77). Furthermore, recent studies have shown that oxidized mtDNA, which is generated by mtDNA damage caused by mROS or other cellular stressors, has a stronger ability to activate the NLRP3 inflammasome than non-oxidized mtDNA (84, 85).
The pro-inflammatory potential of mtDNA highlights the importance of self-clearance to limit inflammatory priming and storm. Multiple intrinsic mechanisms have been identified for mtDNA clearance, including autophagy, phagocytosis mainly mediate via by macrophages and DNase-associated degradation (86, 87). Dysfunction in mtDNA clearance has been linked to the pathogenesis of various autoimmune diseases. As aforementioned, Upregulated levels of circulating mtDNA are observed in patients with SLE and RA, which is closely related to the proinflammatory cytokine production and disease progression (51, 52). Defective in mtDNA clearance is also found in IBDs, resulting in the accumulation of mtDNA in the cytoplasm of intestinal epithelial cells and inducing inflammasome activation (88–90). In turn, both autophagic elimination of damaged mitochondria and pharmacological inhibition of mtDNA synthesis effectively prevent inflammasome activation and improve conditions (91, 92). Therefore, strategies targeting mtDNA clearance pathways, such as autophagy and DNase, may hold therapeutic potential for treating autoimmune diseases and warrant deeper investigations.
Cardiolipin is a unique phospholipid that is a pivotal structural constituent of mitochondria and is also widely found in bacteria, yeast, and plants. It is characterized by a dimeric phospholipid linked by a glycerol moiety (93). In eukaryotes, cardiolipin presents as an unsaturated form and is strictly located in the inner and outer leaflets of the inner mitochondria membrane. Beyond maintaining mitochondrial membrane integrity and regulating mitochondrial dynamics, abundant studies have revealed that cardiolipin has multiple functions in mitochondrial metabolism, apoptosis, and mitophagy via interacting with multiple molecules.
Furthermore, cardiolipin plays a critical role in innate immune responses, particularly under pathological conditions associated with mitochondrial dysfunction, stress, or damage. These abnormal conditions can lead to changes in the level of cardiolipin saturation and oxidation, resulting in the translocation of cardiolipin to the cytosolic side of the outer mitochondrial membrane (94). Once externalized, cardiolipin would act as a DAMP molecule to activate the innate immune system through various mechanisms, and one of them involves the direct activation of TLR4 by cardiolipin. In contrast to the unsaturated form that is unique to physiological conditions, saturated cardiolipin can mimic bacterial lipopolysaccharides and bind to MD2 co-receptor of TLR4, leading to cytokine production, such as TNF-α, IP-10, and IL-1β, and fostering a pro-inflammatory environment (95). Another mechanism involves the activation of the NLRP3 inflammasome. Exposed cardiolipin outside the inner mitochondrial membrane can interact with concomitant pro-caspase-1 and NLRP3 associated with mitochondria to induce inflammasome activation and produce IL-1β and IL-18 (95).
The relationship between cardiolipin and the onset of autoimmune diseases largely relies on the presence of anti-phospholipid autoantibodies, which are considered a hallmark of SLE and anti-phospholipid syndrome (APS). These autoantibodies lead to the formation of immune complexes in multiple tissues and contribute to inflammation and tissue damage. However, the actual role of cardiolipin itself is intensely complicated in the setting of inflammation and autoimmune diseases. Sam50, a mitochondrial outer membrane protein, cooperates with Mic60 to bind to cardiolipin, vital for maintaining mitochondrial membrane integrity. Depletion of Sam50 predisposes cardiolipin to externalization and structural remodeling of the inner and outer mitochondrial membranes, ultimately resulting in liver inflammation in mice models (95). Intriguingly, cardiolipin externalization is also a signal for mtDNA release, which activates the cGAS-STING pathway and induces the downstream responses. And the exogenous expression of Sam50 can reverse it and attenuate liver injury (95). Overall, cardiolipin’s role in autoimmune diseases is complex and multifaceted. Future research is urgently needed to help people fully understand the mechanisms involved and how they may be targeted for therapeutic purposes.
The involvement of mitochondria in innate immunity extends beyond the generation of DAMPs such as mROS, mtDNA, and cardiolipin, as discussed above. Mitochondria also act as a transfer station for TLR, NLR, and RLR-related signaling pathways, serving as a bridge between immunoreactive agents and downstream signaling pathways. Through these pathways, mitochondria play a crucial role in regulating the immune response to infection and tissue damage.
Upon activation, TLR initiates downstream signaling transduction that ultimately leads to the nuclear translocation of NF-kB and upregulation of proinflammatory cytokines, particularly type I IFN (96, 97). Mitochondria play a dual role in TLR signaling, acting as both initiators and enhancers of TLR signaling via through the release of mtDNA and mROS, as well as key intermediates in TLR signaling transduction. Studies have demonstrated that mitochondria and their certain intermediates are closely related to TLR signaling. For example, TRAF6 acts as an intermediate that connects activated TLR with the ubiquitination of ECSIT in mitochondria, which further contributes to an elevated level of mROS, enhancing the anti-microbial effect and potentially favoring the pathogenesis of autoimmune diseases (4, 98). Additionally, TLR activation could also alter the mitochondria function and vice versa. Upregulation of mtDNA is observed upon activation of TLR2 and TLR4, potentially contributing to the induction of local or systemic inflammation (99, 100). Inappropriate activation of TLRs could possibly be linked with autoimmune diseases. Multiple studies have found that the TLR7 and TLR9 signaling pathways could contribute to type I IFN release in SLE (101, 102). In a study of systemic sclerosis, the production of IL-10 by dendritic cells, which induce T helper (Th) 2 immune responses and exacerbate disease progression, is found to be mediated by TLR2 and TLR4 (103).
The NLR family of PRRs is located in the cytoplasm and forms a multi-protein complex known as the inflammasome. Innate immune cells, including dendritic cells, macrophages, monocytes, and neutrophils, are the major source for NLRP3 and NLRX1 inflammasomes to carry out their anti-microbial function (104). However, inappropriate activation of inflammasomes, including NLRP3 and NLRX1, is closely related to multiple autoimmune diseases, such as SLE, RA, and other inflammatory diseases, which is well summarized and discussed in two recently published review articles (105, 106). The priming and activation of NLRP3 and NLRX1 inflammasomes are closely related to mitochondria. Once an immunostimulatory signal transduces through the TLR- MyD88- NF-κB axis, NLRP3 and pro-IL-1β are upregulated for further assembly of NLRP3 inflammasomes (107). The process of NLRP3 activation is also intertwined with several mitochondrial molecules, such as mtDNA and cardiolipin. Oxidized mtDNA can activate NLRP3 and contribute to downstream upregulation of proinflammatory cytokines and activation of immune responses (108). Additionally, cardiolipin can directly bind to the NLRP3 and activate the inflammasome (109). Moreover, if the cardiolipin synthesis pathway is disrupted, it leads to compromised activation of the NLRP3 inflammasome (109). And introducing cardiolipin (excluding other phospholipids) into a damaged cellular system could prompt caspase-1 activation (109). Abnormalities of the NLR family are now recognized as a critical factor invarious inflammatory diseases. Human NLRP1 polymorphisms are correlated with an increased risk of SLE (110), vitiligo (111), and type 1 diabetes (112). In these disease models, a shared pattern of IL-1α, IL-1β, and IL-18 overproduction is observed, which may be the reason for how NLRP1 activation contributes to the pathogenesis of these inflammatory diseases.
The RLR family, comprising of RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) plays a critical role in the innate response to viral infections and the production of type I IFN (113). MAVS is activated upon viral RNA recognition by RIG-I or MDA5 and further recruits multiple molecules including TRAF5 and TRAF6 for downstream activation of NF-κB during a typical response to viral infection (114). MAVS, therefore, acts as a central hub in the mitochondrial antiviral response pathway, mediating the interaction between the RLR family of PRRs and downstream effectors. And mutations or dysregulation of MAVS have been implicated in a variety of diseases, including autoimmune disorders. In SLE patients, an abnormal aggregation of MAVS in a prion-like pattern is observed and could contribute to the release of type I IFN, potentially exacerbating the onset of SLE (115). In non-synonymous single nucleotide polymorphisms (SNPs) screening analysis identifies a loss-of-function variant of MAVS is associated with an uncommon subset of SLE patients and correlates with lowered expression of type I IFN (116). Intriguingly, a recent study has shown that MAVS is critical in promoting B cell activation besides their impact on promoting type I IFN production (117). Meanwhile, aberrant RLR activation has also been proven to be associated with the pathogenesis of various autoimmune or inflammatory diseases such as SLE (118), type 1 diabetes (119), and dermatomyositis (120), due to their ability to induce proinflammatory cytokine production and autoantibody generation (121). MDA5 is the autoantigen that induces the production of the anti-MDA5 autoantibody, which directly contributes to the progression of dermatomyositis and significantly increases the risk of interstitial lung disease in these patients (120). RLR mutations or polymorphisms, such as MDA5, are also observed in SLE and dermatomyositis, leading to an increase in the type I IFN signature (122, 123).
NLRX1 is another critical member of the NLR family that also regulates mitochondrial antiviral signaling and might serve as a link between NLR and RLR pathways. It has been shown to negatively regulate the RLR pathway by inhibiting the MAVS and downstream activation of IRF3 and NF-κB (124). This leads to the suppression of the type I IFN and pro-inflammatory cytokine production, which is important for preventing excessive inflammation and tissue damage during viral infections (22). Additionally, NLRX1 has been implicated in the regulation of mitochondrial homeostasis, including mitochondrial fission and fusion, ROS production, and mitophagy (22). Studies have shown the association between NLRX1 and autoimmune diseases such as SLE, RA, and IBD. In SLE, as aforementioned, the aggregation of MAVS may contribute to the elevation of type I IFN and augment autoimmunity (116). However, although NLRX1 was expressed, its level was not affected by the aggregation of MAVS (116). In IBD, NLRX1 was found to inhibit the development of the disease, and its agonist NX13 could increase OXPHOS levels and prevent the progression of IBD (125, 126). Overall, research on NLRX1 in the field of autoimmunity is rapidly developing, yet its exact mechanisms in autoimmune diseases remain to be fully elucidated.
Chloroquine or hydroxychloroquine, the anti-malarial drug, has long been recognized as critical in the treatment of SLE and RA (127, 128). These drugs work by blocking TLR7, 8, and 9, which are involved in autoimmune disease progression (129). However, clinicians must also manage side effects including myopathy, retinopathy, and gastrointestinal symptoms, which may be due to their off-target effects (130). Hence, specific inhibitors targeting TLRs, NLRs, and RLRs could have even more tremendous potential in developing novel treatments for autoimmune diseases. Small molecule inhibitors and other antibodies, nanoparticles, and oligonucleotides that directly inhibit TLRs, NLRs, RLRs, or intermediates in their signaling pathways are now under comprehensive study, referring to a recently published review (121).
There are six major cellular metabolic pathways that maintain cellular homeostasis: glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway, fatty acid oxidation (FAO) and synthesis, as well as amino acid metabolism. Mitochondria is the site where the TCA cycle and FAO take place. Owing to the diverse functions of different cell types, the metabolic pattern can differ. In various innate immune cells, research has demonstrated abnormalities in mitochondrial metabolism and their association with the development of autoimmune diseases.
Mitochondria are the center for pyruvate oxidation, facilitated by the enzyme pyruvate dehydrogenase (PDH) that converts pyruvate to acetyl-coenzyme A (CoA) in the mitochondrial matrix, enabling carbon flow into the TCA cycle and maximizing energy extraction from glycolysis. PDH can be inactivated through phosphorylation by pyruvate dehydrogenase kinase (PDK), resulting in lower catalytic activity. In contrast, dephosphorylation of PDH via by pyruvate dehydrogenase phosphatase (PDP) activates it. Therefore, PDK and PDP orchestrate the metabolic state within innate immune cells, ultimately affecting the balance between proinflammatory and immunosuppressive functions (131).
Macrophage polarization towards the M1 type is a critical factor in the pathogenesis of several autoimmune diseases. Pyruvate oxidation is associated with enhanced inflammation status in certain situations and PDH activity is directly linked to the polarization of macrophages towards M1 type. Studies using mouse models have verified that inhibiting PDK2 and PDK4, either through genetic deletion or pharmacological inhibition, can entirely prevent this polarization (132). This occurs through enhancing mitochondrial respiration and metabolite rewiring from glycolysis and the TCA cycle. Interestingly, transplantation of PDK2/4-deficient bone marrow into irradiated wild-type mice can alleviate inflammation and reduce obesity-associated insulin resistance. PDK inhibitors, such as sodium dichloroacetate (DCA), have the potential to treat inflammatory diseases, including colitis and experimental autoimmune encephalomyelitis (EAE) (133).
In turn, M1 macrophages exhibit a reduced α-ketoglutarate/succinate ratio, leading to pseudo-hypoxia gloss and an increased stability of HIF-1α, which further PDK1 expression and favors PDH phosphorylated. As a result, the carbon flux into the TCA cycle is blocked at pyruvate oxidation, while the concomitant, represented by lactate, is increased (134–136). In addition, the activity of PDH is also downregulated by nitro oxide (NO) by directly affecting its PDH-E3 via S-nitrosation, which is independent of HIF-1α (137). Moreover, inducible nitric oxide synthase (iNOS or Nos2) continuously produces abundant NO to support this process in M1 macrophages (138). Similarly, DCs exhibit increased expression of Nos2 and NO production, indicating their glycolytic commitment to Warburg metabolism (139).
Inhibition of PDH via pharmacological strategies including using melatonin was shown to be able to suppress the proinflammatory polarization and associated with relieved clinical outcomes of chronic inflammatory diseases, represented by multiple sclerosis (133, 140).
In the mitochondrial matrix, pyruvate or fatty acid-derived acetyl-CoA combines with oxaloacetate, a four-carbon compound, to form citrate and release CoA. Through a series of eight-step reactions, the TCA cycle regenerates oxaloacetate to generate a complete loop, producing energy and small molecules for anabolism NADH and FADH2 are representative molecules that can yield ATP via the ETC, consisting of five multiprotein complexes on the inner mitochondrial membrane. Citrate can also act as a carrier for acetyl-CoA, crossing the mitochondrial membrane and being hydrolyzed by ATP-citrate lyase to generate cytosolic acetyl-CoA, which participates in epigenetic modification (141, 142). The TCA cycle plays a crucial role in maintaining normal cellular functions, including those of innate immune cells. Studies have suggested potential roles for the TCA cycle in regulating immune functions and disease pathogenesis.
From the perspective of energy supply, the TCA cycle is the primary source of long-term activation and energetic supplies for most quiescent or non-proliferative cells, such as M2 macrophages, due to its highly efficient modes of ATP generation. Nevertheless, the metabolic requirements of M1 macrophages undergoing rapid activation necessitate a shift toward glycolysis, even in the absence of hypoxia (143). And glycolysis in this scenario is essential for generating ATP and NADH, as well as providing biosynthetic intermediates for nucleotides, amino acids, and others (143). It is noteworthy that M2 macrophages exhibit an increased mitochondrial oxygen-consumption rate and spare respiratory capacity, in contrast to M1 macrophages.
Although both activated DCs and M1 macrophages primarily rely on glycolysis as their metabolic pathway, their objectives differ. While M1 macrophages use glycolysis to generate additional fuel, DCs prioritize the de novo synthesis of fatty acids from glycolysis metabolites to support the synthesis, transport, and secretion of proteins necessary for DCs activation. This is because DCs are non-proliferative cells (144). Additionally, glucose uptake increases at the early stages of DC activation, but as time progresses, OXPHOS becomes the dominant energy source required (145).
After being activated by esterification with CoA in the cytosol, fatty acids are transported into the mitochondrion through conjugation to carnitine via carnitine palmitoyl-transferase (CPT) 1 located at the outer mitochondrial membrane, which is also the rate-limiting step in the pathway. Once inside the mitochondrion, fatty acids are converted back to the activated form by CPT2 and undergo cyclic β-oxidation, resulting in the continuous degradation of fatty acyl-CoA thioesters and yielding numerous acetyl-CoA, NADH, and FADH2. Interestingly, this metabolic pathway has been found to regulate immune functions and support the cellular longevity of macrophages (143).
In contrast to M1 macrophages, where glycolytic metabolism is the predominant way to satisfy their needs in energy and materials, FAO plays an essential role in M2 macrophages, especially for optimal activation, under the drive by signal transducer and activator of transcription 6 (STAT6) and peroxisome proliferator-activated receptor (PPAR) γ coactivator-1β (PGC-1β) (146, 147). Additionally, this pathway is also important to fuel mitochondrial OXPHOS.
Impaired FAO in M1 macrophages can result in the aberrant degeneration and accumulation of fatty acids and their derivative lipoproteins, leading to the production of proinflammatory cytokines, including IL-1α, which can contribute to the occurrence and development of inflammatory pathologies (148). In contrast, the main role of FAO in M2 macrophages is to maintain sustained induction and prolonged survival of the anti-inflammatory subtype at later stages, rather than during early differentiation (149). Furthermore, inducing increased FAO with didymin, which upregulates Hadhb expression, can even convert polarized M1 macrophage into the M2 type, attenuating acute and chronic colitis in mice models (150).
Similarly, macrophages expressing the permanently active mutant form of CPT1A, CPT1AM, exhibit enhanced levels of FAO, leading to lower triglyceride content and reduced production of proinflammatory cytokines, represented by TNF-α and IL-1β, while the expression of anti-inflammatory cytokines remains un affected (151). Overexpression of PGC-1β can also weaken M1 macrophage activation in response to stimuli, such as saturated fatty acids (152). Conversely, inhibition of CPT1A with etomoxir suppresses the FAO commitment and spare respiratory capacity of M2 macrophages which in turn limits their activation in response to IL-4 (153).
The immune functions of DCs are also regulated by the status of FAO. Solid evidence has demonstrated that the expression of the CPT1A gene, whose translation product is the rate-limiting enzyme for β-oxidation, is increased in tolerogenic DCs, leading to the active state of OXPHOS. Inhibition of FAO by etomoxir significantly reduces the secretory levels of tolerogenic cytokines IL-10 and IL-27 whereas proinflammatory cytokines IL-12 and IL-6 remain stable. Moreover, compromised tolerogenic DCs under treatment with dual KC7F2, a selective HIF-1α translation inhibitor, and etomoxir perturb the polarization of naïve CD4+ Th cells towards Th1 (145). Additionally, in DCs, upregulation of PPARα induced by tumor-derived exosomes promotes FAO activity and shifts the predominant pattern from glycolysis toward OXPHOS, resulting in a dysfunctional state. Importantly, the PPARα inhibitor, GW6471, can abrogate this suppressive effect and restore the function of DCs, enhancing IFN-γ producing ability of T cells to promote inflammation (154).
Apolipoprotein C-III (Apoc3) is a component of triglyceride-rich lipoproteins that is known to stimulate monocytes in an NLRP3 inflammasome-dependent manner, resulting in pro-inflammatory activity (155). In addition, a study on Apoc3-transgenic mice, which are characterized by high levels of plasma triglycerides and free fatty acids, showed an imbalance in the energy supply of NK cells, with increased FAO and decreased glycolysis (155). This led to abnormal activation of the PPAR-γ signaling pathway and downstream signaling through PTEN-AKT-mTOR/FOXO1. The changes also severely impaired the number and function of NK cells in multiple dimensions, including decreased capacity for proliferation, IFN-γ production and secretion, and degranulation against pathogenic targets. However, the impaired state can be partially reversed by administering PPAR-γ inhibitors such as GW9662 or CTP1α inhibitors like etomoxir (155).
Mitochondrial metabolites produced by innate immune cells undergoing metabolic reprogramming also own powerful capacity to precisely influence the direction of immune responses and modulate actual functions for adopting the surrounding environment in a macroscopic perspective rather than leading to nuanced or specific responses to stimuli (131). Perturbations to metabolites can have significant impacts on inflammatory signaling, epigenomic regulation, and other cellular functions.
Macrophage subtypes exhibit distinct differences in their TCA cycle patterns. M2 macrophages have an intact TCA cycle that is closely connected to the ETC, providing a steady energy supply while avoiding excessive mROS production. The TCA cycle generates Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) intermediates essential for N-glycosylation, including for the lectin/mannose receptor, which is critical for the normal immune function of M2 macrophages. Inhibitors of N-glycosylation significantly decreased the expression of CD206 and CD301, notable M2 markers, indicating that M2 activation and polarization were prevented (156). The transcription factor c-Maf plays a crucial role in this process (157). In contrast, in M1 macrophages, the TCA cycle is “broken” at two sites where citrate and succinate are converted to isocitrate and fumarate, respectively (15). Fumarate forms a variant of a pathway called the aspartate-arginosuccinate shunt. These changes not only alter the energy supply pattern of M1 macrophages but also lead to the accumulation of by-products that maintain a high-intensity pro-inflammatory status, such as malates (156).
In the context of metabolic reprogramming, citrate, an important TCA cycle metabolite, is exported from the mitochondria via the mitochondrial citrate carrier and utilized for producing mediators with proinflammatory potentials (15). Citrate-derived lipogenesis supports membrane biogenesis, which is essential for cell differentiation and proliferation but also can be a source of arachidonic acid needed for prostaglandin synthesis. And this phenomenon has been observed in activated DCs, where citrate is used to enhance antigen presentation efficiency. Additionally, citrate can be shunted into synthetic pathways of nitric oxide through two steps, NADPH is first produced via malic enzyme and pyruvate and ultimately converted to NO by iNOS. At the same time, NAPDH can also be furtherly oxidated to produce ROS (143). In M2 macrophages, however, there is limited NO due to increased expression of arginase, leading to a low concentration of arginine (158). In contrast, M1 macrophages utilize immune response gene 1 protein to decarboxylate citrate-derived cis-aconitate to itaconate. This metabolic product possesses direct antimicrobial effects and inhibits succinate dehydrogenase, resulting in massive accumulation of succinate due to failed conversion to fumarate and regulating ROS production (159–161).
Succinate significantly stabilizes the transcription factor HIF-1α, which is vital for the sustained production of IL-1β, under normoxia by inhibiting the activity of prolyl hydroxylases, resulting in enhancing the inflammation responses (134). In addition, succinate can also inhibit PDH to limit acetyl-CoA and restrict acetyl-CoA from entering the TCA cycle, forming a vicious cycle together with the aforementioned commitments (162). However, the HIF-1α expression in the tolerogenic DCs is more stabilized and coordinated by increased AKT and mTORC1, responsible for the upregulation of several glycolytic enzymes to promote this metabolism. KC7F2, a HIF-1α inhibitor, significantly reduces the percentage of HIF-1α positive tolerogenic DCs and suppresses the secretion of tolerogenic cytokines, without affecting the production of proinflammatory cytokines (145).
Glutamine is a critical source of α‐ketoglutarate, which serves as a vital intermediate in the TCA cycle and plays a role in controlling innate immune responses (131). Glutamine is converted to glutamate via glutaminase, which is further dehydrogenated into α‐ketoglutarate via glutamate dehydrogenases (131). Studies have found an increased glutamine consumption and upregulated glutaminase activity in immune cells upon their stimulation, indicating the role of glutamine in immune cell proliferation (163). In macrophages, α‐ketoglutarate could drive the polarization of macrophages via modulating marker genes of M1 and M2 types to exert their anti-inflammatory effect (164). α‐ketoglutarate could inhibit the production of proinflammatory cytokines such as IL-6 and IL-12 and also the expression of IL-1β, IL-6, and TNF-α in macrophages possible via the induction of Arg1 and Mrc1 (165). Supplement with dimethyl-α‐ketoglutarate, a cell membrane-permeable analog of α‐ketoglutarate, could attenuate inflammation via driving M2 polarization and inhibition of proinflammatory cytokine production (165, 166). Treatment with melatonin, which could induce α‐ketoglutarate production, could promote M2 polarization and potentially prevent inflammatory diseases (167).
Mitochondria, as a critical organelle in a cell for energy supply, is now recognized to have comprehensive and sophisticated functions in maintaining homeostasis and also innate immunological responses. The immunological function of mitochondria is of great importance in deciphering the pathogenesis of immune disorders. And compiling studies show the close relationship between mitochondria abnormalities and the pathogenesis of autoimmune diseases (Figure 1). In response to immunological stimuli, the stress signal activates respective signaling pathways including TLRs, NLRs, and RLRs, eventually leading to the production of proinflammatory cytokines and augmentation of immune responses. As one of the major sources of host- derived DAMPs, mitochondria generate multiple types of alarmins including mROS, mtDNA, and cardiolipins. These molecules play a pivotal role in initiation and activation of certain immune responses or mediating direct tissue damage. Abnormalities of DAMPs generation of signal transduction of TLRs, NLRs, and RLRs are found in multiple autoimmune diseases. By directly targeting DAMPs or signaling pathways of TLRs, NLRs, and RLRs, people could discover novel treatments for autoimmune diseases. And the attempts to develop therapeutic strategies for SLE and RA by targeting mitochondrial molecules and their respective signaling pathways are summarized in Table 1. Mitochondria also controls the metabolism of innate immune cells, which further interferes with their function in innate immunity. The dysregulation of metabolic pathways and metabolites has been linked to several autoimmune diseases. Although several mitochondrial pathways involved in the pathogenesis of autoimmune diseases have been targeted for therapeutic strategies (as shown in Figure 1), many remain unexplored. Targeting these unattended pathways with novel agents that modulate metabolic pathways and metabolites could offer potential therapeutic options for autoimmune diseases and should be further investigated.
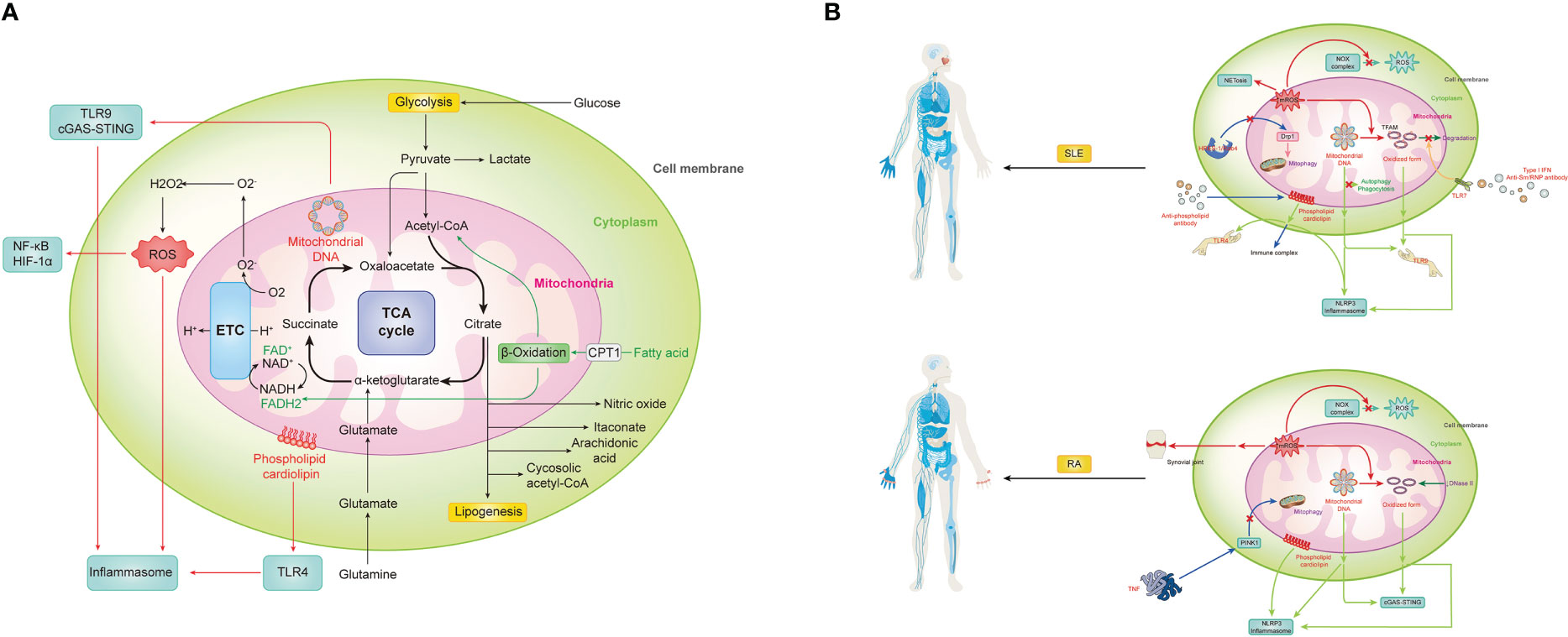
Figure 1 Mitochondrial in innate immunity and aberrant status for autoimmune diseases, represented by two prototypes, SLE and RA. (A) Mitochondria can trigger vigoroso inflammation by activating inflammasomes in multiple ways, including reactive oxygen species (ROS), mitochondrial DNA (mtDNA) and cardiolipin. Meanwhile, mtROS-related activation of NF-kB and HIF-1a, mtDNA-induced cGAS-STING pathway, as well as cardiolipin-driven TLRs also contribute a lot during the immune response. Notably, as the center of metabolism, various metabolic pathways and metabolites within mitochondria must also not be ignored in the process. (B) SLE is promoted by mitochondrial signaling through numerous mechanisms. 1) mROS not only directly causes severe inflammation and tissue damage by itself but also contributes to NETosis and oxidize mtDNA into oxidized form with enhanced inflammatory capability. However, with the accumulation of mROS, NOX complex-related ROS is inhibited. 2) Both prototype and oxidized form mtDNA can be recognized by TLR9 or NLRP3 to evoke downstream immune signaling. Meanwhile, there is a brunch of pathways for their accumulation. With the activation of TLR7 by anti-Sm/RNP antibody under type I IFN environment, the phosphorylation of TAFM is blocked and persistently covered in the oxidized mtDNA to protect it. And other self-clearance ways are also weakened. The HRES-1/Rab4 enzyme can digest Drp1 to prevent physiological mitophagy, resulting in increased mitochondria mass for indirect mtDNA accumulation. 3) Similarity to mtDNA, cardiolipin can be recognized by TLR4 or NLRP3 to evoke inflammation, but it can also be recognized and bound by anti-phospholipid antibodies, one type of classical autoantibodies of SLE, to form the immune complex and induce subsequent effects. (C) RA is promoted by mitochondrial signaling in the following mechanisms. 1) Except for systematically accumulating mROS and correspondingly decreased NOX complex-related ROS and increased oxidized mtDNA, it can directly gather in the synovial joint to cause immensity local damage. 2) mtDNA and its oxidized form can activate cGAS-STING or NLRP3 for inflammation. Meanwhile, failed self-DNA degradation by DNase II and attenuation of PINK1-mediated mitophagy caused by TNF promote this process. 3) Cardiolipin can trigger NLRP3 inflammasome for inflammation.
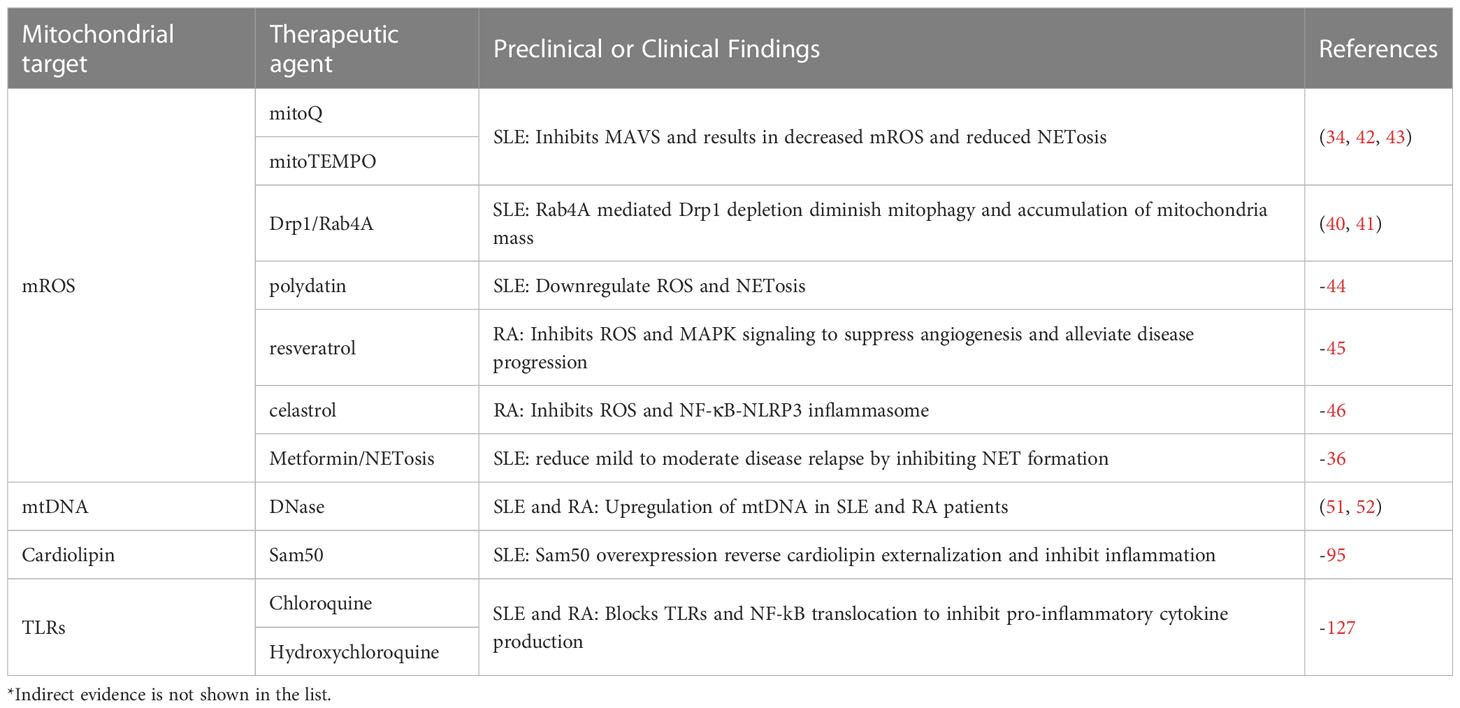
Table 1 Potential therapeutic agents targeting mitochondrial molecules in SLE and RA.
In general, the actual mechanisms that induce autoimmune disease pathogenesis are complicated and might involve far more molecules and signaling pathways. In the study into such disease, mitochondria with their related molecules, signaling pathways, and metabolic pathways could possibly be a potential key to the question.
YJ and ZY did the literature review and drafted the manuscript. AY supervised on finishing this review. All authors read and approved the final manuscript for publication.
This article is under the support of the National Key R&D Program of China (Grant No. 2022YFC3602103) to AY; CAMS Initiative for Innovative Medicine (Grant No. 2021-I2M-1-013) to AY; National High Level Hospital Clinical Research Funding (2022-PUMCH-B-024) to AY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Emelyanov VV. Mitochondrial connection to the origin of the eukaryotic cell. Eur J Biochem (2003) 270(8):1599–618. doi: 10.1046/j.1432-1033.2003.03499.x
3. Martin W, Hoffmeister M, Rotte C, Henze K. An overview of endosymbiotic models for the origins of eukaryotes, their ATP-producing organelles (mitochondria and hydrogenosomes), and their heterotrophic lifestyle. Biol Chem (2001) 381(11):1521–39. doi: 10.1515/BC.2001.187
4. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472(7344):476–80. doi: 10.1038/nature09973
5. Banjara S, Suraweera CD, Hinds MG, Kvansakul M. The bcl-2 family: ancient origins, conserved structures, and divergent mechanisms. Biomolecules (2020) 10(1):128. doi: 10.3390/biom10010128
6. Soubannier V, McBride HM. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta (BBA)-Molecular Cell Res (2009) 1793(1):154–70. doi: 10.1016/j.bbamcr.2008.07.008
7. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci (2010) 35(9):505–13. doi: 10.1016/j.tibs.2010.04.002
8. Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell (2007) 130(1):25–35. doi: 10.1016/j.cell.2007.06.033
9. Lang KS, Burow A, Kurrer M, Lang PA, Recher M. The role of the innate immune response in autoimmune disease. J Autoimmunity (2007) 29(4):206–12. doi: 10.1016/j.jaut.2007.07.018
10. Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest (2004) 113(7):990–7. doi: 10.1172/JCI19388
11. Jiao Y, Wu L, Huntington ND, Zhang X. Crosstalk between gut microbiota and innate immunity and its implication in autoimmune diseases. Front Immunol (2020) 11:282. doi: 10.3389/fimmu.2020.00282
12. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
13. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
14. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol (2022) 23(3):159–73. doi: 10.1038/s41577-022-00760-x
15. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18(5):488–98. doi: 10.1038/ni.3704
16. Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA. Mitochondrial ROS metabolism: 10 years later. Biochem (Mosc) (2015) 80(5):517–31. doi: 10.1134/S0006297915050028
17. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell (2005) 120(4):483–95. doi: 10.1016/j.cell.2005.02.001
18. Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol (2004) 122:277–91. doi: 10.1007/s00418-004-0679-8
19. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33(7):829–37. doi: 10.1093/eurheartj/ehr304
20. Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol (2013) 9(11):674–86. doi: 10.1038/nrrheum.2013.147
21. Chaitanya NS, Tammineni P, Nagaraju GP, Reddy AB. Pleiotropic roles of evolutionarily conserved signaling intermediate in toll pathway (ECSIT) in pathophysiology. J Cell Physiol (2022) 237(9):3496–504. doi: 10.1002/jcp.30832
22. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Trans Res (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
23. Tiku V, Tan M-W, Dikic I. Mitochondrial functions in infection and immunity. Trends Cell Biol (2020) 30(4):263–75. doi: 10.1016/j.tcb.2020.01.006
24. Huang N, Perl A. Metabolism as a target for modulation in autoimmune diseases. Trends Immunol (2018) 39(7):562–76. doi: 10.1016/j.it.2018.04.006
25. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature (2017) 543(7644):205–10. doi: 10.1038/nature21388
26. Ushio-Fukai M, Rehman J. Redox and metabolic regulation of stem/progenitor cells and their niche. Antioxidants Redox Signaling (2014) 21(11):1587–90. doi: 10.1089/ars.2014.5931
27. Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood J Am Soc Hematology. (2016) 128(8):e20–31. doi: 10.1182/blood-2016-05-716480
28. Yuan Y, Chen Y, Peng T, Li L, Zhu W, Liu F, et al. Mitochondrial ROS-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin Sci (2019) 133(15):1759–77. doi: 10.1042/CS20190672
29. Tan H-Y, Wang N, Li S, Hong M, Wang X, Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longevity (2016). doi: 10.1155/2016/2795090
30. Liu R, Li X, Ma H, Yang Q, Shang Q, Song L, et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, hif-1α upregulation and autophagy. Free Radical Biol Med (2020) 161:339–50. doi: 10.1016/j.freeradbiomed.2020.10.029
31. Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab (2005) 1(6):409–14. doi: 10.1016/j.cmet.2005.05.002
32. Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J Immunol (2000) 165(2):1013–21. doi: 10.4049/jimmunol.165.2.1013
33. Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol (2018) 19(2):77–92. doi: 10.1038/nrm.2017.66
34. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi: 10.1038/nm.4027
35. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Trans Med (2011) 3(73):73ra19–9. doi: 10.1126/scitranslmed.3001180
36. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol (2015) 67(12):3190–200. doi: 10.1002/art.39296
37. Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang S-Y, et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet (2017) 49(3):433–7. doi: 10.1038/ng.3782
38. Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci (2012) 109(2):E59–67. doi: 10.1073/pnas.1113251108
39. Olsson LM, Johansson ÅC, Gullstrand B, Jönsen A, Saevarsdottir S, Rönnblom L, et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann rheumatic diseases. (2017) 76(9):1607–13. doi: 10.1136/annrheumdis-2017-211287
40. Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol (2016) 68(11):2728–39. doi: 10.1002/art.39791
41. Caza TN, Fernandez DR, Talaber G, Oaks Z, Haas M, Madaio MP, et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis (2014) 73(10):1888–97. doi: 10.1136/annrheumdis-2013-203794
42. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signaling (2016) 9(456):ra115–ra. doi: 10.1126/scisignal.aaf1933
43. Fortner KA, Blanco LP, Buskiewicz I, Huang N, Gibson PC, Cook DL, et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci Med (2020) 7(1):e000387. doi: 10.1136/lupus-2020-000387
44. Liao P, He Y, Yang F, Luo G, Zhuang J, Zhai Z, et al. Polydatin effectively attenuates disease activity in lupus-prone mouse models by blocking ROS-mediated NET formation. Arthritis Res Ther (2018) 20(1):1–11. doi: 10.1186/s13075-018-1749-y
45. Yang G, Chang C-C, Yang Y, Yuan L, Xu L, Ho C-T, et al. Resveratrol alleviates rheumatoid arthritis via reducing ROS and inflammation, inhibiting MAPK signaling pathways, and suppressing angiogenesis. J Agric Food Chem (2018) 66(49):12953–60. doi: 10.1021/acs.jafc.8b05047
46. Jing M, Yang J, Zhang L, Liu J, Xu S, Wang M, et al. Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis. Int Immunopharmacol (2021) 98:107879. doi: 10.1016/j.intimp.2021.107879
47. Lee SR, Han J. Mitochondrial mutations in cardiac disorders. In: Santulli G, editor. Mitochondrial dynamics in cardiovascular medicine. Cham: Springer International Publishing (2017). p. 81–111.
48. De Gaetano A, Solodka K, Zanini G, Selleri V, Mattioli AV, Nasi M, et al. Molecular mechanisms of mtDNA-mediated inflammation. Cells (2021) 10(11):2898. doi: 10.3390/cells10112898
49. Mechta M, Ingerslev LR, Fabre O, Picard M, Barrès R. Evidence suggesting absence of mitochondrial DNA methylation. Front Genet (2017) 8:166. doi: 10.3389/fgene.2017.00166
50. Nass MM. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells in vivo and in vitro methylation. J Mol Biol (1973) 80(1):155–75. doi: 10.1016/0022-2836(73)90239-8
51. Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther (2003) 5:1–7. doi: 10.1186/ar787
52. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Trans Med (2011) 3(73):73ra20–0. doi: 10.1126/scitranslmed.3001201
53. Boyapati RK, Tamborska A, Dorward DA, Ho G-T. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Research (2017) 6:169. doi: 10.12688/f1000research.10397.1
54. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukocyte Biol (2004) 75(6):995–1000. doi: 10.1189/jlb.0703328
55. Becker Y, Marcoux G, Allaeys I, Julien A-S, Loignon R-C, Benk-Fortin H, et al. Autoantibodies in systemic lupus erythematosus target mitochondrial RNA. Front Immunol (2019) 10:1026. doi: 10.3389/fimmu.2019.01026
56. Qiu J, Wu B, Goodman SB, Berry GJ, Goronzy JJ, Weyand CM. Metabolic control of autoimmunity and tissue inflammation in rheumatoid arthritis. Front Immunol (2021) 12:652771. doi: 10.3389/fimmu.2021.652771
57. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduction Targeted Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
58. Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, et al. Cutting edge: Role of toll-like receptor 9 in CpG DNA-induced activation of human Cells1. J Immunol (2001) 167(7):3555–8. doi: 10.4049/jimmunol.167.7.3555
59. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi: 10.1038/nature08780
60. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock (2010) 34(1):55–9. doi: 10.1097/SHK.0b013e3181cd8c08
61. Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, et al. Neutrophil extracellular traps induced by IL8 promote diffuse Large b-cell lymphoma progression via the TLR9 SignalingNETs promote DLBCL progression. Clin Cancer Res (2019) 25(6):1867–79. doi: 10.1158/1078-0432.CCR-18-1226
62. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology (2010) 139(1):323–34.e7. doi: 10.1053/j.gastro.2010.03.052
63. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest (2016) 126(3):859–64. doi: 10.1172/JCI83885
64. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy (2020) 16(1):3–17. doi: 10.1080/15548627.2019.1603547
65. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med (2016) 213(5):697–713. doi: 10.1084/jem.20151876
66. Barberá A, Lorenzo N, Garrido G, Mazola Y, Falcón V, Torres AM, et al. APL-1, an altered peptide ligand derived from human heat-shock protein 60, selectively induces apoptosis in activated CD4+ CD25+ T cells from peripheral blood of rheumatoid arthritis patients. Int Immunopharmacol (2013) 17(4):1075–83. doi: 10.1016/j.intimp.2013.10.010
67. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep (2014) 6(3):421–30. doi: 10.1016/j.celrep.2014.01.003
68. Zhang C, Shang G, Gui X, Zhang X, Bai X-C, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature (2019) 567(7748):394–8. doi: 10.1038/s41586-019-1000-2
69. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci (2009) 106(49):20842–6. doi: 10.1073/pnas.0911267106
70. Rongvaux A, Jackson R, Harman Christian CD, Li T, West AP, de Zoete Marcel R, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell (2014) 159(7):1563–77. doi: 10.1016/j.cell.2014.11.037
71. Lewis MJ, Barnes MR, Blighe K, Goldmann K, Rana S, Hackney JA, et al. Molecular portraits of early rheumatoid arthritis identify clinical and treatment response phenotypes. Cell Rep (2019) 28(9):2455–70.e5. doi: 10.1016/j.celrep.2019.07.091
72. Muskardin TW, Vashisht P, Dorschner JM, Jensen MA, Chrabot BS, Kern M, et al. Increased pretreatment serum IFN-β/α ratio predicts non-response to tumour necrosis factor α inhibition in rheumatoid arthritis. Ann Rheum Dis (2016) 75(10):1757–62. doi: 10.1136/annrheumdis-2015-208001
73. Willemsen J, Neuhoff M-T, Hoyler T, Noir E, Tessier C, Sarret S, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep (2021) 37(6):109977. doi: 10.1016/j.celrep.2021.109977
74. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun (2017) 8(1):2176. doi: 10.1038/s41467-017-01932-3
75. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature (2006) 443(7114):998–1002. doi: 10.1038/nature05245
76. Bai H, Zhang Q. Activation of NLRP3 inflammasome and onset of alzheimer’s disease. Front Immunol (2021) 12:701282. doi: 10.3389/fimmu.2021.701282
77. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
78. Schroder K, Tschopp J. The inflammasomes. cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
79. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, X-j L, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
80. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep (2020) 21(4):e49799. doi: 10.15252/embr.201949799
81. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
82. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
83. Kuriakose T, Man SM, Subbarao Malireddi R, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol (2016) 1(2):aag2045–aag. doi: 10.1126/sciimmunol.aag2045
84. Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity (2022) 55(8):1370–85.e8. doi: 10.1016/j.immuni.2022.06.007
85. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520(7548):553–7. doi: 10.1038/nature14156
86. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature (2012) 485(7397):251–5. doi: 10.1038/nature10992
87. Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor–independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med (2005) 202(10):1333–9. doi: 10.1084/jem.20051654
88. Novak EA, Mollen KP. Mitochondrial dysfunction in inflammatory bowel disease. Front Cell Dev Biol (2015) 3:62. doi: 10.3389/fcell.2015.00062
89. Aguilera M, Darby T, Melgar S. The complex role of inflammasomes in the pathogenesis of inflammatory bowel diseases–lessons learned from experimental models. Cytokine Growth Factor Rev (2014) 25(6):715–30. doi: 10.1016/j.cytogfr.2014.04.003
90. Boyapati R, Satsangi J, Ho G. Pathogenesis of crohn’s disease. F1000Prime Rep (2015) 2(7):44. doi: 10.12703/P7-44
91. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol (2016) 34(4 Suppl 98):12–6.
92. Stacey KJ, Sweet MJ, Hume DA. Macrophages ingest and are activated by bacterial DNA. J Immunol (Baltimore Md: 1950). (1996) 157(5):2116–22. doi: 10.4049/jimmunol.157.5.2116
93. Oemer G, Lackner K, Muigg K, Krumschnabel G, Watschinger K, Sailer S, et al. Molecular structural diversity of mitochondrial cardiolipins. Proc Natl Acad Sci (2018) 115(16):4158–63. doi: 10.1073/pnas.1719407115
94. Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol (2020) 30(11):892–903. doi: 10.1016/j.tcb.2020.09.004
95. Pizzuto M, Lonez C, Baroja-Mazo A, Martínez-Banaclocha H, Tourlomousis P, Gangloff M, et al. Saturation of acyl chains converts cardiolipin from an antagonist to an activator of toll-like receptor-4. Cell Mol Life Sci (2019) 76:3667–78. doi: 10.1007/s00018-019-03113-5
96. Xiao C, Ghosh S. NF-κB, an evolutionarily conserved mediator of immune and inflammatory responses. Mech Lymphocyte Activation Immune Regul X: Innate Immun (2005) 560:41–5. doi: 10.1007/0-387-24180-9_5
97. Muzio M, Polentarutti N, Bosisio D, Kumar PM, Mantovani A. Toll-like receptor family and signalling pathway. Biochem Soc Trans (2000) 28(5):563–6. doi: 10.1042/bst0280563
98. Carneiro FR, Lepelley A, Seeley JJ, Hayden MS, Ghosh S. An essential role for ECSIT in mitochondrial complex I assembly and mitophagy in macrophages. Cell Rep (2018) 22(10):2654–66. doi: 10.1016/j.celrep.2018.02.051
99. Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated e. coli. FASEB J (2005) 19(11):1531–3. doi: 10.1096/fj.04-3500fje
100. Sweeney TE, Suliman HB, Hollingsworth JW, Piantadosi CA. Differential regulation of the PGC family of genes in a mouse model of staphylococcus aureus sepsis. PloS One (2010) 5(7):e11606. doi: 10.1371/journal.pone.0011606
101. Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol (2012) 24(5):499–505. doi: 10.1097/BOR.0b013e3283562c3e
102. Hamerman JA, Pottle J, Ni M, He Y, Zhang ZY, Buckner JH. Negative regulation of TLR signaling in myeloid cells–implications for autoimmune diseases. Immunol Rev (2016) 269(1):212–27. doi: 10.1111/imr.12381
103. Van Bon L, Popa C, Huijbens R, Vonk M, York M, Simms R, et al. Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Ann Rheum Dis (2010) 69(8):1539–47. doi: 10.1136/ard.2009.128207
104. Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol (2011) 186(4):2529–34. doi: 10.4049/jimmunol.1002720
105. Li Z, Guo J, Bi L. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed Pharmacotherapy (2020) 130:110542. doi: 10.1016/j.biopha.2020.110542
106. Liu M, Liu K, Cheng D, Zheng B, Li S, Mo Z. The regulatory role of NLRX1 in innate immunity and human disease. Cytokine (2022) 160:156055. doi: 10.1016/j.cyto.2022.156055
107. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
108. Nakahira K, Haspel JA, Rathinam VA, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
109. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
110. Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity (2012) 45(4):271–8. doi: 10.3109/08916934.2011.637532
111. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med (2007) 356(12):1216–25. doi: 10.1056/NEJMoa061592
112. Magitta N, Bøe Wolff A, Johansson S, Skinningsrud B, Lie B, Myhr K, et al. A coding polymorphism in NALP1 confers risk for autoimmune addison’s disease and type 1 diabetes. Genes Immunity (2009) 10(2):120–4. doi: 10.1038/gene.2008.85
113. Brennan K, Bowie AG. Activation of host pattern recognition receptors by viruses. Curr Opin Microbiol (2010) 13(4):503–7. doi: 10.1016/j.mib.2010.05.007
114. Seth RB, Sun L, Ea C-K, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell (2005) 122(5):669–82. doi: 10.1016/j.cell.2005.08.012
115. Shao W-H, Shu DH, Zhen Y, Hilliard B, Priest SO, Cesaroni M, et al. Prion-like aggregation of mitochondrial antiviral signaling protein in lupus patients is associated with increased levels of type I interferon. Arthritis Rheumatol (2016) 68(11):2697–707. doi: 10.1002/art.39733
116. Pothlichet J, Niewold TB, Vitour D, Solhonne B, Crow MK, Si-Tahar M. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol Med (2011) 3(3):142–52. doi: 10.1002/emmm.201000120
117. Sun W, Wang H, Qi CF, Wu J, Scott B, Bolland S. Antiviral adaptor MAVS promotes murine lupus with a B cell autonomous role. Front Immunol (2019) 10:2452. doi: 10.3389/fimmu.2019.02452
118. Munroe ME, Pezant N, Brown MA, Fife DA, Guthridge JM, Kelly JA, et al. Association of IFIH1 and pro-inflammatory mediators: Potential new clues in SLE-associated pathogenesis. PloS One (2017) 12(2):e0171193. doi: 10.1371/journal.pone.0171193
119. Blum SI, Tse HM. Innate viral sensor MDA5 and coxsackievirus interplay in type 1 diabetes development. Microorganisms (2020) 8(7):993. doi: 10.3390/microorganisms8070993
120. Wu W, Guo L, Fu Y, Wang K, Zhang D, Xu W, et al. Interstitial lung disease in anti-MDA5 positive dermatomyositis. Clin Rev Allergy Immunol (2021) 60:293–304. doi: 10.1007/s12016-020-08822-5
121. Liu J, Zhang H, Su Y, Zhang B. Application and prospect of targeting innate immune sensors in the treatment of autoimmune diseases. Cell Bioscience. (2022) 12(1):68. doi: 10.1186/s13578-022-00810-w
122. Guo L, Zhang X, Pu W, Zhao J, Wang K, Zhang D, et al. WDFY4 polymorphisms in Chinese patients with anti-MDA5 dermatomyositis is associated with rapid progressive interstitial lung disease. Rheumatology (2023). doi: 10.1093/rheumatology/kead006
123. Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J Immunol (2011) 187(3):1298–303. doi: 10.4049/jimmunol.1100857
124. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature (2008) 451(7178):573–7. doi: 10.1038/nature06501
125. Leber A, Hontecillas R, Zoccoli-Rodriguez V, Ehrich M, Chauhan J, Bassaganya-Riera J. Exploratory studies with NX-13: oral toxicity and pharmacokinetics in rodents of an orally active, gut-restricted first-in-class therapeutic for IBD that targets NLRX1. Drug Chem toxicology. (2022) 45(1):209–14. doi: 10.1080/01480545.2019.1679828
126. Leber A, Hontecillas R, Zoccoli-Rodriguez V, Bienert C, Chauhan J, Bassaganya-Riera J. Activation of NLRX1 by NX-13 alleviates inflammatory bowel disease through immunometabolic mechanisms in CD4+ T cells. J Immunol (2019) 203(12):3407–15. doi: 10.4049/jimmunol.1900364
127. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheumatic Dis (2019) 78(6):736–45. doi: 10.1136/annrheumdis-2019-215089
128. O’Dell JR, Blakely KW, Mallek JA, Eckhoff PJ, Leff RD, Wees SJ, et al. Treatment of early seropositive rheumatoid arthritis: a two-year, double-blind comparison of minocycline and hydroxychloroquine. Arthritis Rheumatism (2001) 44(10):2235–41. doi: 10.1002/1529-0131(200110)44:10<2235::AID-ART385>3.0.CO;2-A
129. Chen J-Q, Szodoray P, Zeher M. Toll-like receptor pathways in autoimmune diseases. Clin Rev Allergy Immunol (2016) 50:1–17. doi: 10.1007/s12016-015-8473-z
130. Ponticelli C, Moroni G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin Drug Safety (2017) 16(3):411–9. doi: 10.1080/14740338.2017.1269168
131. Wang T, Jiao Y, Zhang X. Immunometabolic pathways and its therapeutic implication in autoimmune diseases. Clin Rev Allergy Immunol (2021) 60(1):55–67. doi: 10.1007/s12016-020-08821-6
132. Min B-K, Park S, Kang H-J, Kim DW, Ham HJ, Ha C-M, et al. Pyruvate dehydrogenase kinase is a metabolic checkpoint for polarization of macrophages to the M1 phenotype. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00944
133. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi: 10.1172/JCI76012
134. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
135. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2015) 213(1):15–23. doi: 10.1084/jem.20151570
136. MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, et al. Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol (2007) 27(9):3282–9. doi: 10.1128/MCB.01927-06
137. Palmieri EM, Gonzalez-Cotto M, Baseler WA, Davies LC, Ghesquière B, Maio N, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun (2020) 11(1):698. doi: 10.1038/s41467-020-14433-7
138. MacMicking J, Xie Q-w, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15(1):323–50. doi: 10.1146/annurev.immunol.15.1.323
139. Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood (2012) 120(7):1422–31. doi: 10.1182/blood-2012-03-419747
140. Ghareghani M, Scavo L, Jand Y, Farhadi N, Sadeghi H, Ghanbari A, et al. Melatonin therapy modulates cerebral metabolism and enhances remyelination by increasing PDK4 in a mouse model of multiple sclerosis. Front Pharmacol (2019) 10:147. doi: 10.3389/fphar.2019.00147
141. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
142. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324(5930):1076–80. doi: 10.1126/science.1164097
143. O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
144. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi: 10.1038/ni.2833
145. Sen K, Pati R, Jha A, Mishra GP, Prusty S, Chaudhary S, et al. NCoR1 controls immune tolerance in conventional dendritic cells by fine-tuning glycolysis and fatty acid oxidation. Redox Biol (2023) 59:102575. doi: 10.1016/j.redox.2022.102575
146. Huang SC-C, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15(9):846–55. doi: 10.1038/ni.2956
147. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab (2006) 4(1):13–24. doi: 10.1016/j.cmet.2006.05.011
148. Freigang S, Ampenberger F, Weiss A, Kanneganti T-D, Iwakura Y, Hersberger M, et al. Fatty acid–induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol (2013) 14(10):1045–53. doi: 10.1038/ni.2704
149. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol (2020) 10. doi: 10.3389/fimmu.2019.02993
150. Lv Q, Xing Y, Liu Y, Chen Q, Xu J, Hu L, et al. Didymin switches M1-like toward M2-like macrophage to ameliorate ulcerative colitis via fatty acid oxidation. Pharmacol Res (2021) 169:105613. doi: 10.1016/j.phrs.2021.105613
151. Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiology-Endocrinology Metab (2015) 308(9):E756–E69. doi: 10.1152/ajpendo.00362.2014
152. Chen H, Liu Y, Li D, Song J, Xia M. PGC-1β suppresses saturated fatty acid-induced macrophage inflammation by inhibiting TAK1 activation. IUBMB Life (2016) 68(2):145–55. doi: 10.1002/iub.1470
153. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity (2015) 42(3):406–17. doi: 10.1016/j.immuni.2015.02.002
154. Yin X, Zeng W, Wu B, Wang L, Wang Z, Tian H, et al. PPARα inhibition overcomes tumor-derived exosomal lipid-induced dendritic cell dysfunction. Cell Rep (2020) 33(3):108278. doi: 10.1016/j.celrep.2020.108278
155. Hu X, Jia X, Xu C, Wei Y, Wang Z, Liu G, et al. Downregulation of NK cell activities in apolipoprotein c-III-induced hyperlipidemia resulting from lipid-induced metabolic reprogramming and crosstalk with lipid-laden dendritic cells. Metabolism (2021) 120:154800. doi: 10.1016/j.metabol.2021.154800
156. O’Neill LA. A broken krebs cycle in macrophages. Immunity (2015) 42(3):393–4. doi: 10.1016/j.immuni.2015.02.017
157. Liu M, Tong Z, Ding C, Luo F, Wu S, Wu C, et al. Transcription factor c-maf is a checkpoint that programs macrophages in lung cancer. J Clin Invest (2020) 130(4):2081–96. doi: 10.1172/JCI131335
158. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front Immunol (2014) 5. doi: 10.3389/fimmu.2014.00532
159. Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Society (2011) 133(41):16386–9. doi: 10.1021/ja2070889
160. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab (2016) 24(1):158–66. doi: 10.1016/j.cmet.2016.06.004
161. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem (2016) 291(27):14274–84. doi: 10.1074/jbc.M115.685792
162. Meiser J, Krämer L, Sapcariu SC, Battello N, Ghelfi J, D’Herouel AF, et al. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J Biol Chem (2016) 291(8):3932–46. doi: 10.1074/jbc.M115.676817
163. Ardawi MSM, Newsholme EA. Glutamine metabolism in lymphocytes of the rat. Biochem J (1983) 212(3):835–42. doi: 10.1042/bj2120835
164. Liu S, Yang J, Wu Z. The regulatory role of α-ketoglutarate metabolism in macrophages. Mediators Inflammation. (2021) 2021:1–7. doi: 10.1155/2021/5577577
165. Cheng M-X, Cao D, Chen Y, Li J-Z, Tu B, Gong J-P. α-ketoglutarate attenuates ischemia-reperfusion injury of liver graft in rats. Biomed Pharmacotherapy (2019) 111:1141–6. doi: 10.1016/j.biopha.2018.12.149
166. Yu S, Ding L, Liang D, Luo L. Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing α-ketoglutarate production in mice. Mol Oral Microbiol (2018) 33(5):388–95. doi: 10.1111/omi.12241
Keywords: mitochondria, innate immunity, immune metabolism, autoimmune disease, therapeutic targets
Citation: Jiao Y, Yan Z and Yang A (2023) Mitochondria in innate immunity signaling and its therapeutic implications in autoimmune diseases. Front. Immunol. 14:1160035. doi: 10.3389/fimmu.2023.1160035
Received: 06 February 2023; Accepted: 28 March 2023;
Published: 12 April 2023.
Edited by:
Tuantuan Zhao, Mayo Clinic, United StatesReviewed by:
Charles E. McCall, Wake Forest Baptist Medical Center, United StatesCopyright © 2023 Jiao, Yan and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiming Yang, eWFuZ2FpbWluZ0BtZWRtYWlsLmNvbS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.