
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 23 May 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1156493
 Johannes R. Schmidt1,2†
Johannes R. Schmidt1,2† Janine Haupt1,2†Sina Riemschneider1
Janine Haupt1,2†Sina Riemschneider1 Christoph Kämpf3Dennis Löffler2,3
Christoph Kämpf3Dennis Löffler2,3 Conny Blumert2,3Kristin Reiche3,4
Conny Blumert2,3Kristin Reiche3,4 Ulrike Koehl2,4
Ulrike Koehl2,4 Stefan Kalkhof1,2,5‡
Stefan Kalkhof1,2,5‡ Jörg Lehmann1,2*
Jörg Lehmann1,2*Introduction: The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that regulates a broad range of target genes involved in the xenobiotic response, cell cycle control and circadian rhythm. AhR is constitutively expressed in macrophages (Mϕ), acting as key regulator of cytokine production. While proinflammatory cytokines, i.e., IL-1β, IL-6, IL-12, are suppressed through AhR activation, anti-inflammatory IL-10 is induced. However, the underlying mechanisms of those effects and the importance of the specific ligand structure are not yet completely understood.
Methods: Therefore, we have compared the global gene expression pattern in activated murine bone marrow-derived macrophages (BMMs) subsequently to exposure with either benzo[a]pyrene (BaP) or indole-3-carbinol (I3C), representing high-affinity vs. low-affinity AhR ligands, respectively, by means of mRNA sequencing. AhR dependency of observed effects was proved using BMMs from AhR-knockout (Ahr-/-) mice.
Results and discussion: In total, more than 1,000 differentially expressed genes (DEGs) could be mapped, covering a plethora of AhR-modulated effects on basal cellular processes, i.e., transcription and translation, but also immune functions, i.e., antigen presentation, cytokine production, and phagocytosis. Among DEGs were genes that are already known to be regulated by AhR, i.e., Irf1, Ido2, and Cd84. However, we identified DEGs not yet described to be AhR-regulated in Mϕ so far, i.e., Slpi, Il12rb1, and Il21r. All six genes likely contribute to shifting the Mϕ phenotype from proinflammatory to anti-inflammatory. The majority of DEGs induced through BaP were not affected through I3C exposure, probably due to higher AhR affinity of BaP in comparison to I3C. Mapping of known aryl hydrocarbon response element (AHRE) sequence motifs in identified DEGs revealed more than 200 genes not possessing any AHRE, and therefore being not eligible for canonical regulation. Bioinformatic approaches modeled a central role of type I and type II interferons in the regulation of those genes. Additionally, RT-qPCR and ELISA confirmed a AhR-dependent expressional induction and AhR-dependent secretion of IFN-γ in response to BaP exposure, suggesting an auto- or paracrine activation pathway of Mϕ.
The aryl hydrocarbon receptor (AhR) is a ligand-activated basic helix-loop-helix transcription factor belonging to the Per-Arnt-Sim family (1). Upon ligand binding, the AhR translocates into the nucleus, where it dimerizes with the AhR nuclear translocator and binds to an aryl hydrocarbon response element (AHRE; also known as xenobiotic response element, XRE) in the promoter regions of a broad range of target genes, among them are several genes encoding xenobiotic-metabolizing enzymes, such as cytochrome P450 (CYP) 1A1 and CYP 1B1 (2).
Apart from its role in detoxification of xenobiotics, i.e., 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and benzo[a]pyrene (BaP), through the induction of CYP monooxygenases and circadian rhythm, AhR is of crucial importance in liver homeostasis, cell cycle control and immune regulation (3–8). The constitutive expression in innate immune cells, i.e., macrophages (Mϕ) and dendritic cells (DC), or type-3 innate lymphoid cells, implies a central function of AhR in innate immune regulation (9). This was supported by the observation that AhR expression is increased during inflammation in human DC or murine Mϕ (10, 11). Moreover, exogenous xenobiotic (e.g., BaP) or nutritional (e.g., indole-3-carbinole, I3C) but also endogenous AhR ligands (e.g., kynurenine, a product of tryptophan metabolism) can modulate innate and adaptive immunity, potentially resulting in either increased or decreased susceptibility to infection or cancer and may trigger autoimmune disorders and allergies (12). Since agonistic and antagonistic AhR ligands may cause different, sometimes contrary, cell- and tissue-specific effects in diseases, the AhR is considered as a promising drug target.
The immunoregulatory function of AhR seems to be realized through both, the canonical and several recently discovered non-canonical AhR signaling pathways (13). The latter are realized at the genomic level through association with other transcription factors, i.e., signal transducer and activator of transcription 1 (STAT1) or nuclear factor-κB (NF-κB) subunits RelA and RelB, causing activation or repression of several target genes such as c-myc or Il6, respectively (14–19). But also at the non-genomic level non-canonical AhR signaling pathways may be implemented by regulating protein ubiquitination, e.g., as a ligand-dependent E3 ubiquitin ligase (i.e., CUL4AhR complex), targeting substrate proteins for proteasomal degradation such as steroid receptors (i.e., estrogen receptor α (ERα), ERβ, and androgen receptor) (20) and phosphorylation, such as the release of the Src kinase from the cytosolic AhR complex in an active form (4, 21, 22).
In terms of murine Mϕ, surface expression of MHC-II, CD64, CD14, and CD86 as well as cytokine secretion could be modulated through AhR activation only upon stimulation via pattern recognition receptors (PRRs) by pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS) or formulations including several PAMPs, such as heat-killed bacteria (10). Moreover, AhR activation interferes with the differentiation of bone marrow-derived myeloid precursors into mature Mϕ (23). Interestingly, AhR activation seems to induce an anti-inflammatory phenotype in PAMPs-activated murine Mϕ, that is characterized by significant upregulation of interleukin (IL)-10 but downregulation of proinflammatory cytokines, such as TNF-α, IL-1β, IL-6, and IL-12 (11). Additionally, AhR activation is linked to interferon (IFN)-α and IFN-γ expression (24, 25). However, it remains unclear so far whether high-affinity vs. low-affinity AhR ligands may activate the same gene expression pattern in Mϕ and other innate immune cells, and thus may have the same impact on innate immune regulation. In this context, also the importance of the source of AhR ligands (xenobiotic vs. nutritional vs. endogenous) is still matter of debate. Therefore, we intended to compare the global gene expression pattern in murine PAMPs-activated bone marrow-derived macrophages (BMMs) following exposure to the AhR ligands BaP (high affinity) or I3C (low affinity) by means of mRNA sequencing.
We identified more than 1,000 differentially expressed AhR-dependent genes that could be related to various functions ranging from basal biological processes to the modulation of innate immunity. Based on computational modeling approaches, the majority of those genes is anticipated to be under canonical control. However, 203 genes do not possess any known AHRE motif, and thus are supposed to be non-canonically or secondarily regulated by AhR. The most interesting cluster of non-canonical signaling concerns to type I and type II interferons. Computational modeling and experimental verification suggest an AhR-mediated induction of IFN-α/β and IFN-γ secretion resulting in autocrine/paracrine Mϕ activation. Of note, those IFN-triggered effects were only observed in BaP- but not I3C-exposed BMMs, suggesting that they were induced ligand-specifically or, more likely, in dependence on the ligand’s affinity and effective concentration.
All chemicals or reagents were obtained from Sigma Aldrich (Taufkirchen, Germany) unless noted otherwise. Cell culture flasks and plates were purchased from Greiner Bio-One (Frickenhausen, Germany). Roswell Park Memorial Institute (RPMI) 1640 culture medium was supplemented with 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) buffer, 2 mM L-glutamine, 10% (v/v) fetal bovine serum (FBS) and 100 U/ml penicillin, 100 µg/ml streptomycin (Biochrom, Berlin, Germany or PAN-Biotech, Aidenbach, Germany), and 50 μM β-mercaptoethanol to be used as complete cell culture medium. BaP and I3C were dissolved in dimethyl sulfoxide (DMSO).
Female wild-type (Ahr+/+) C57BL/6JRj mice were originally purchased from Janvier Labs (St. Berthevin Cedex, France). C57BL/6 AhR knockout (Ahr-/-) mice (26) were originally purchased from Jackson Laboratory (Bar Harbor, USA) and back crossed in-house to Ahr+/+ C57BL/6JRj mice for several times, while the Ahr deletion was confirmed by genotyping by means of PCR. Animals were used at 8-12 weeks of age for the experiments. Mice were housed as five or six animals per cage in the animal care facility of the Fraunhofer Institute for Cell Therapy and Immunology (Leipzig, Germany) in a temperature- and humidity-controlled room (23 °C, 50 % humidity) under specific pathogen-free conditions with 12 h/12 h of light/dark cycle and free access to pelleted standard rodent chow and water ad libitum. All experiments involving laboratory animals had been conducted according to the European Communities Council Directive (86/609/EEC) and were approved by local authorities (registration no. T 10/17, Landesdirektion Sachsen, Leipzig, Germany). All animals were sacrificed using flow-controlled carbon dioxide (1 L/min). All efforts were made to minimize suffering of the animals.
Bone marrow cells were isolated from femur and tibia of each four Ahr+/+ and Ahr-/- mice by flushing with phosphate-buffered saline and harvested by centrifugation (260 x g, 10 min, room temperature, RT). For differentiation, the cells were plated at a density of 4 x 105/ml into tissue culture dishes in RPMI 1640 medium without phenol red (20% FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/ml penicillin, 100 µg/ml streptomycin, 50 μM β-mercaptoethanol) supplemented with 30 % macrophage colony-stimulating factor-enriched medium (culture supernatant from the fibroblastic cell line L929) prepared in-house as previously described (11). Fresh medium containing the same ingredients was added after 3 days. At day 6 of the myeloid differentiation process adherent cells were scraped off and tested for the surface expression of CD11b (Miltenyi Biotec, Bergisch-Gladbach, Germany) and F4/80 (Miltenyi Biotec) by flow cytometry (CytoFLEX, Beckman Coulter, Krefeld, Germany) in order to verify the differentiation to BMMs. Cells were harvested for further experiments if they revealed more than 80 % expression of CD11b and F4/80. For RNA sequencing experiments, 1 x 106 BMMs/ml were cultured in complete RPMI 1640 medium without phenol red in the presence of 1 µM BaP, 10 µM I3C or 0.01 % (v/v) DMSO (vehicle control) for 6 h. Subsequently, the BMMs were activated for 3 h or 20 h by adding 1 x 107 heat-killed Salmonella enterica Serovar Enteritidis (hk S.E.)/ml to each culture. Hk S.E. that represent an excellent source of several PAMPs for broad-spectrum PRR activation were prepared from a S.E. vaccine strain that was kindly provided from IDT Biologika (Dessau-Roßlau, Germany) as previously described (27). For quantitative RT-PCR (Il10, Il1b, Il12rb1, Slpi, Ido2, Gsta3, Spn and Igf1r), BMMs were exposed to increasing doses of BaP and I3C in the range of 0.5–100 µM and for Ifng expression analyses, BMMs were exposed to 8 nM or 800 nM BaP.
Total RNA was isolated with TriReagent® according to manufacturer’s instructions (Sigma Aldrich). After quantifying by absorbance at 260 nm/280 nm DNA contamination was degraded by DNaseI (Fermentas, St. Leon Rot, France). The single strand cDNA synthesis was performed using the Transcriptor First Strand cDNA Synthesis kit (Roche Diagnostics, Mannheim, Germany). Optimal primer design and the Universal ProbeLibrary® (UPL; Roche Life Science, Penzberg, Germany) probe selection was performed using the web-based software tool ProbeFinder (Roche) (Table 1). Designed primers were purchased from TibMolBiol (Berlin, Germany) and the real-time RT-PCR was performed using the LightCycler® 480 instrument (Roche). PCR assays were prepared using LightCycler® Probes Master kit (Roche) and the appropriate UPL probe with optimal primers.

Table 1 Forward and reverse primer sequences for real-time PCR.
Total RNA was isolated with TriReagent® according to manufacturer’s instructions (Sigma Aldrich). To eliminate all traces of genomic DNA DNAse-digestion (TURBO DNA free Kit, Invitrogen, Thermo Fisher Scientific, Darmstadt, Germany) was performed twice in each sample. Extracted RNA was quantified using a Qubit RNA-Kit and the DeNovix instrument (Biozym, Oldendorf, Germany). Quality of RNA was analyzed by means of a Bioanalyzer 2100 instrument (Agilent Technologies, Waldbronn, Germany).
For subsequent RNA sequencing analyses 500 ng total RNA per sample was used. Library preparation was conducted using TruSeq-Stranded mRNA Sample Prep kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Fragmentation step was performed for 8 min, following Illumina’s recommendation for high quality input RNA. Quality and quantity of each prepared library was analyzed with the DeNovix instrument (Qubit DNA-Kit) and the Bioanalyzer 2100 instrument (Agilent Technologies). Molarity of each library was calculated and equal amounts were pooled and used for subsequent sequencing (12 pM). Sequencing was performed with 2 x 126-bp paired-end reads using SBS V4 chemistry on a HiSeq 2500 instrument (Illumina). One flow-cell containing 8 lanes was sequenced with 64 pooled libraries.
Reads were demultiplexed by Illumina’s bcl2fastq (v2.19.0.316). Adapter sequences were removed from reads using adapter removal (v2.2.1a) using parameters –trimns, –trimqualities, –minquality ‘20’, and –minlength ‘30’). HISAT2 (v2.1.0) with parameters –fr and rna-strandness: RF was used to align reads against the mouse genome mm10 (GENCODE release M22) (28, 29). Number of reads per gene were counted by htseq-count (v0.9.1) using parameters –mode intersection-strict, –stranded reverse and –type exon (30). These steps were orchestrated by the workflow-manager uap (31).
Gene set enrichment analysis (GSEA) of whole expression profiles was performed using the GSEA software (v4.1.0, Broad Institute, Cambridge, MA, USA) and the implemented tool “GSEAPreranked” (32). Genes were ranked by log2-fold change and false discovery rate (FDR). Gene symbols were not collapsed and default parameters were applied. MSigDB gene set databases “c2.cp.reactome”, “c5.go.bp” were used and complemented with “WP_ARYL_HYDROCARBON_RECEPTOR_ PATHWAY_WP2873” from “c2.cp.wikipathways” (all v7.5.1) (33). Gene sets with FDR < 0.25 and normalized enrichment score (NES) > 1.5 in at least one ligand/time combination were considered for further analysis.
Genome-wide screening for known aryl hydrocarbon response elements (AHRE) in gene promoter regions (3,000 nt upstream of transcriptional start site on both strands) was performed using genome-scale dna-pattern matching function of the Regulatory Sequence Analysis Tools program (RSAT) (34). Known AHRE sequences (AHRE I: 5’-GCGTG-3’, AHRE II: 5’ CATGnnnnnnC(A/T)TG-3’ and RelB AHRE: 5’-GGGTGCAT-3’ were searched against the full length C57BL/6 mouse genome (GRCm38, mm10). Only perfect matches were considered and hits were mapped to next generation sequencing results (Supplementary Table 1).
For upstream regulator analysis, RNA sequencing profiles were imported to Ingenuity Pathway Analysis (IPA) software (35) and filtered for AhR-dependent differentially expressed genes (DEGs). Gained upstream regulators were filtered by activation or inhibition (absolute z-score > 2) and targeting AhR-dependent DEGs not possessing any AHRE motif. Functional network analysis was performed using the IPA software. Therefore, all filtered upstream regulators were connected interferon-γ (IFN-γ)-centrically based on IPA knowledge base. Elements, that were computed upstream of direct IFN-γ interactions were subsequently connected by Path Explorer.
IFN-γ secretion of BMM was assessed by determination of its concentration in cell culture supernatants by enzyme-linked immunosorbent assay (Mouse IFNg ‘Femto-HS’ High Sensitivity Uncoated ELISA, Invitrogen, Thermo Fisher Scientific) following the manufacture’s protocol. Briefly, 96-well plates were coated with capture antibody overnight at 4 °C. Subsequently after blocking, 100 µl of cell culture supernatants, standards and blanks were added and incubated overnight at 4 °C. Next, samples were incubated with detection antibody for 1 h at RT, prior addition of streptavidin-coupled horseradish peroxidase and incubation for 30 min at RT. Ultimately, detection was started by adding tetramethylbenzidine substrate solution for 15 min at RT and stopped by 1 M phosphoric acid. Washing of 96-well plate was included between all described steps. Finally, the optical density (OD) at 450 nm was measured and cytokine concentration was calculated from a calibration curve achieved with a recombinant mouse IFN-γ standard.
Differential gene expression analysis was performed using DESeq2 (v1.28.1) (36). Raw read counts were normalized and variance stabilized. FDR was controlled by Benjamini-Hochberg adjustment. DEGs were filtered by FDR < 0.01. Genes that were filtered as DEGs specifically expressed in either Ahr+/+ or Ahr-/- Mϕ under exposure with AhR ligand and hk S.E. were assigned as AhR-dependently affected, whereas genes filtered as DEGs expressed in both Ahr+/+ and Ahr-/- cells were assigned as AhR-independently affected. Genes filtered as DEGs in Ahr+/+ or Ahr-/- Mϕ without AhR ligand exposure where considered as unspecific effects, and therefore excluded. Correlation analyses of gene expression profiles were performed using cor.test function in R base environment.
For statistical analyses of RT-PCR experiments, all targeted genes were normalized to housekeeping genes aminolevulinate synthase 1 (Alas1) and hypoxanthine-guanine phosphoribosyl transferase (Hprt). Relative quantification was performed by means of the LightCycler® 480 software v.1.5 (Roche). Significance of altered relative expression against DMSO-treated sample was determined by Student’s t-test (two-sided, unpaired).
For statistical analyses of ELISA experiments, determined ODs were reduced by mean blank values. Calibration curve was calculated from mean values of log2-titrated IFN-γ standard applying four-parameter Marquardt regression. Concentrations of IFN-γ in cell culture supernatants were calculated from fitting to calibration curve. Significance of altered IFN-γ secretion against vehicle control (DMSO) was determined by Student’s t-test (two-sided, unpaired).
Mϕ are one key player in the innate immune response. To elucidate molecular effects of AhR stimulation on their activation, murine BMMs were exposed to BaP in vitro. Subsequently, conditioned BMMs were activated by hk S.E. for 3 h or 20 h. To determine the therapeutic potential of targeting AhR and to identify ligand-dependency of downstream effects, BMMs were also exposed to the non-toxic AhR-ligand I3C analogously in the same experimental setup.
Overall the expression of 14,549 quality filtered genes was profiled (Supplementary Table 1). To elucidate the modulation of gene expression by AhR ligands, whole expression patterns of pairwise-comparisons of ligand-exposed and hk S.E.-activated to non-exposed and hk S.E.-activated Ahr+/+ BMMs were analysed by gene set enrichment analysis (GSEA). Overall, 467 gene sets were identified as significantly enriched in any ligand/time point combination (Figure 1; Supplementary Table 2). 179 gene sets were constitutively upregulated by means of positive NES. This includes genes of the “AhR pathway” and in “Cellular response to xenobiotics” as proofs of successful AhR activation upon ligand binding. Additionally, genes of the “Regulation of Mϕ activation”, “Toll-like receptor (TLR) cascade”, “Signaling by interleukins”, “Positive regulation of IL-6 production” and “IFN-α production” were increased in expression by AhR activation. In contrast, 19 gene sets were constitutively downregulated by AhR ligand exposure mainly comprising genes in the transcription and translation process, e.g., “RNA splicing”. Further gene sets were identified with a time-specific expression profile. Whilst genes of “Mitotic G2/M-transition checkpoint” were upregulated, genes involved in “Translation elongation” were downregulated at 20 h compared to 3 h post hk S.E. administration. Even more, gene sets with ligand-specific expression profiles were identified. Genes involved in “Protein folding” were upregulated by I3C compared to BaP, but genes of “Antigen processing and presentation” as well as genes involved in both “Signaling by IFN-α/β” and “Signaling by IFN-γ” were upregulated by BaP compared to I3C. Other gene sets, e.g., of “ROS biosynthetic process” and “Positive regulation of phagocytosis” possess ligand and time dependency of expression profiles. In summary, AhR activation modulates the expression of a broad spectrum of genes that are involved in basal cellular processes and innate immunity in a partly ligand-specific manner.
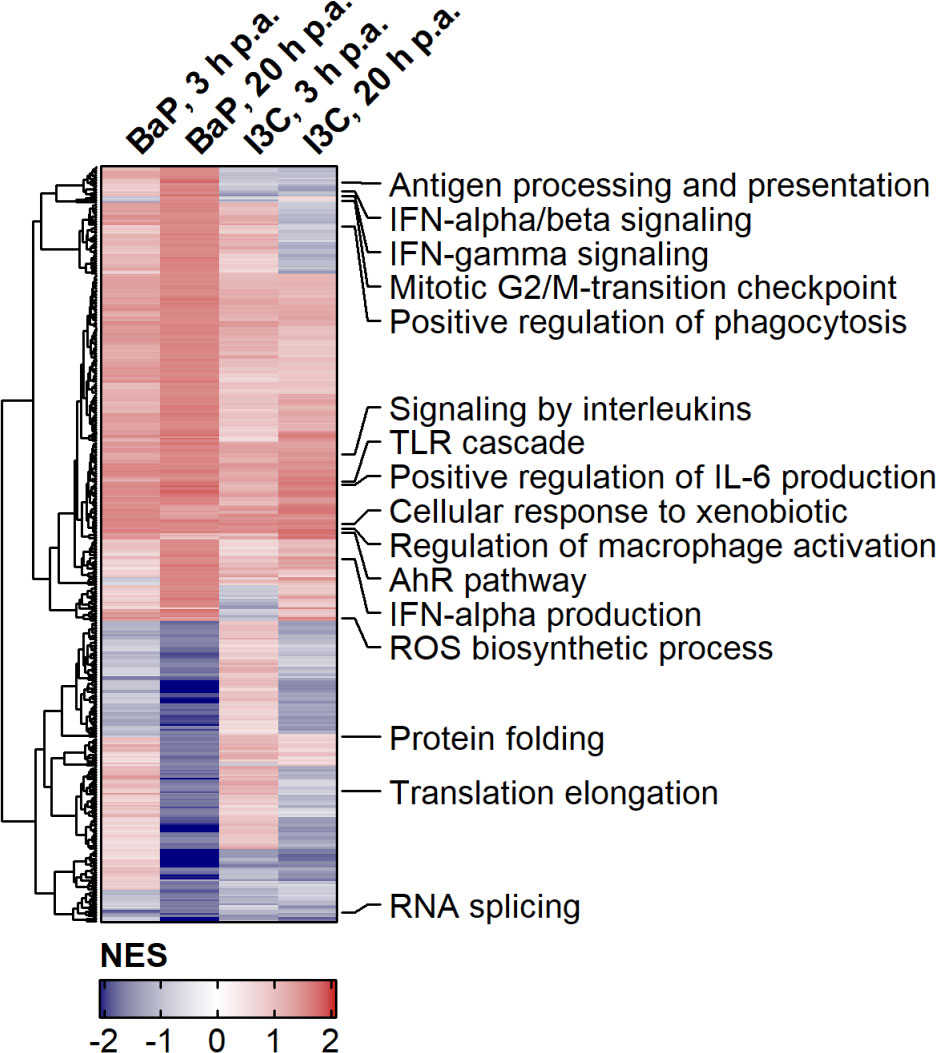
Figure 1 Effects of AhR activation on gene expression profiles during PAMP-induced BMM activation. Murine Ahr+/+ BMMs were exposed to AhR ligands (BaP or I3C) or treated with vehicle control (DMSO) for 6 h (n = 4). Subsequently, cells were activated with hk S.E. for 3 h or 20 h (p.a. – post administration) for PAMP activation. Whole cellular RNA was extracted and analyzed by means of RNA sequencing. Relative quantitative whole gene expression profiles of ligand-exposed vs. DMSO-treated cells retrieved from DESeq2 analysis were inspected via gene set enrichment analysis against biological process category of gene ontology and canonical pathways of databases REACTOME and Wiki Pathways. NES of gene sets were used for Euclidean Clustering. Gene sets were selected to cover innate immunity and representative basal cellular processes and annotated in die figure. Complete results are provided as Supplementary Table 2.
In order to focus on single gene expression and to exclude effects that are triggered by ligand exposure but are independent of AhR, the above-mentioned experimental setup was likewise applied for Ahr+/+ as well as Ahr-/- BMMs and DEGs of ligand exposed to vehicle control treated BMMs were identified. Only genes that were identified as DEGs in Ahr+/+ or Ahr-/- Mϕ but not in both were referred to as AhR-dependent DEGs. Out of 1,197 DEGs in total were found after AhR ligand exposure, 1,108 DEGs could be identified to be AhR-dependently affected. However, the number of AhR-dependent DEGs varied between the both ligands and duration of hk S.E. activation with more genes being affected by BaP and after 20 h of Mϕ activation via PRRs by PAMPs (i.e., hk S.E.), respectively (Figure 2A; Table 2).
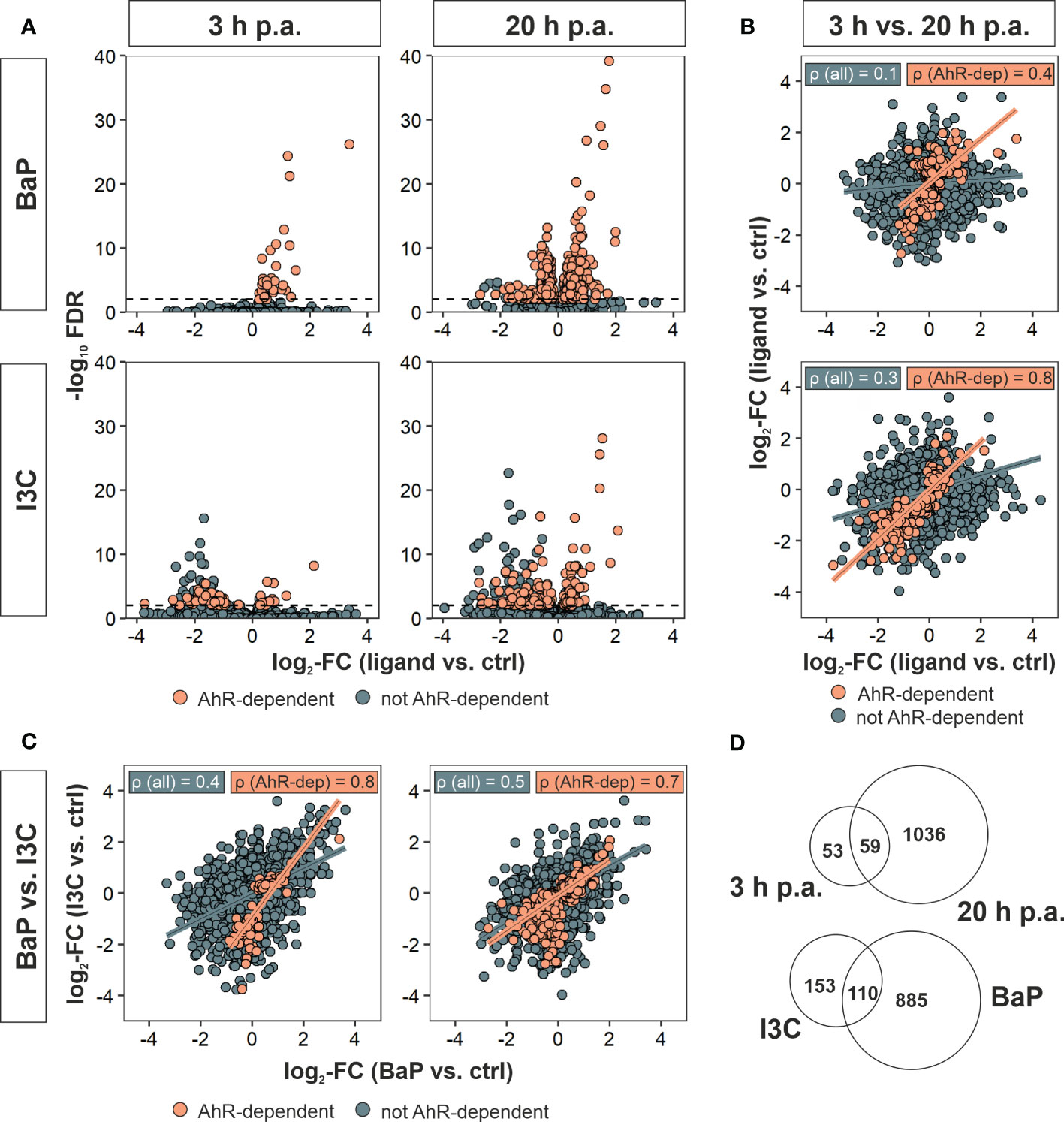
Figure 2 Identification of AhR-dependently differentially expressed genes. Murine Ahr+/+ BMMs were exposed to AhR ligands (BaP or I3C) or treated with vehicle control (DMSO) for 6 h (n = 4). Subsequently, cells were activated with hk S.E. for 3 h or 20 h (p.a. – post administration) for PAMP activation. Whole cellular RNA was extracted and analyzed by means of RNA sequencing. Mapped read counts were analyzed by DESeq2 to identify DEGs (n = 4). (A) Volcano plots representing log2-transformed fold changes (FCs) of gene expression after ligand exposure compared to DMSO-treated BMMs and −log10-transformed adjusted p values (FDR). (B) Correlation of gene expression 3 h to 20 h post administration (p.a.) of hk S.E. for BaP (upper panel) and I3C exposure (lower panel) represented by log2-FCs. (C) Correlation of gene expression after BaP (x-axis) to I3C (y-axis) exposure after 3 h (left panel) and 20 h p.a. of hk S.E. (right panel) represented by log2-FCs. Linear regression is represented by correlation of all genes (blue) and AhR-dependently DEGs (AhR-dep, orange). Corresponding Pearson’s correlation coefficients (ρ) are indicated in the plots. Complete statistics of correlation analyses are provided as Supplementary Table 3. (D) Venn-Euler diagrams of total AhR-dependent DEGs comparing 3 h and 20 h p.a. of hk S.E. (upper panel) or I3C and BaP exposure (lower panel).

Table 2 Number of AhR-dependently differentially expressed genes (DEGs).
Only 54 and 73 genes were AhR-dependently modulated after 3 h of BMM activation through hk S.E. While all of them are upregulated after BaP exposure, both down- and upregulated genes are present after I3C exposure. After 20 h of hk S.E.-induced Mϕ activation, the number of AhR-dependently modulated genes increased up to 972 or 221 upon BaP or I3C exposure, respectively. Pearson’s correlation of gene expression profiles at 3 h to 20 h post hk S.E. administration revealed correlation coefficients (ρ) of 0.1 and 0.3 for BaP and I3C, respectively (Figure 2B). However, when including only AhR-dependent genes ρ increases to 0.4 and 0.8, respectively. Correlations of transcriptome profiles of BaP- to I3C-exposed BMMs resulted in ρ = 0.4 or 0.5 at 3 h or 20 h post hk S.E. administration, respectively (Figure 2C). Similarly, when including only AhR-dependent genes, ρ increased to 0.8 and 0.7, respectively. Thus, similar AhR-dependent effects regardless the ligand used for AhR activation may be anticipated. Interestingly, the increment of the resulting regression function is 0.5 and 0.6, which may indicate an affinity-dependent effect. All tested correlations possess significance with p < 2.2 × 10-16 (Supplementary Table 3). However, in direct comparisons of identified DEGs at specific ligand exposures and time post hk S.E. administrations, only 110 AhR-dependent DEGs are common to BaP and I3C exposure, while 885 and 153 genes are affected specifically by one of the both ligands, respectively. Fifty-nine AhR-dependent DEGs were altered at both time points, whilst 53 and 1,036 genes were affected only at 3 h or 20 h post hk S.E. administration, respectively (Figure 2D).
Hence, whole expression profiles and correlation analyses may point to general AhR-dependent effects that occur at different effect strengths upon the usage of BaP or I3C. However, focussing on single gene expression may allow the identification of ligand-specific target genes.
Among the AhR-dependent DEG, previously described and putative novel AhR-regulated genes were identified, including canonical and non-canonical targets (Table 3). The expression of the canonical AhR-targets Ahrr and Nqo1 was increased by both ligands at both time points in Ahr+/+ but not Ahr-/- BMMs, confirming successful activation of AhR in Ahr+/+ and no AhR signaling in Mϕ without functional AhR. Interestingly, the transcriptional activation of Glutathione S transferase alpha 3 (Gsta3) was slightly induced only by I3C. Expression of Cyp genes was not induced, but possesses overall low expression rates near the detection limit. Matrix metalloproteinases (MMP) gene expression was found to be upregulated for Mmp8 and Mmp27 by both ligands BaP and I3C. Similar evidence was gained for Kunitz type 1 serine protease inhibitor (Spint1), whereas secretory leukocyte protease inhibitor (Slpi) expression was only induced by I3C. Kynurenine-producing indoleamine 2,3-dioxygenase 2 (Ido2) expression was induced only by BaP. However, the expression of the IDO-regulator interferon regulatory factor 1 (Irf1) was induced by both ligands. The transcriptional rates of antibacterial interferon-inducible GTPase 1 (Iigp1) and interferon-induced GTP-binding protein (Mx1) were found to be increased only by BaP. Similar evidence of a BaP-specific expressional induction was gained for cyclic GMP-AMP synthase (Cgas) as well as for the type I interferon receptor alpha and beta chains (Ifnar1, Ifnar2) Moreover, several important functional Mϕ genes were found to be induced following BaP exposure. Among them are the genes encoding for CD64 (Fcgr1), CD16 (Fcgr3), CD84 (Cd84), toll-like receptor 9 (Tlr9), and IL-12Rβ1 subunit (Il12rb1). The transcription of the IL-21R (Il21r) was upregulated by both ligands, BaP and I3C. Interestingly, Spn a gene encoding for sialophorin (leukosialin, CD43) a major sialoglycoprotein expressed on Mϕ but also several other leukocytes, mainly T lymphocytes, was found to be downregulated following BaP exposure. Unexpectedly, no significant changes in Il1b and Il10 transcriptional rates could be detected, although trends were observable for reduction of Il1b but induction of Il10 gene expression through BaP exposure (Supplementary Table 1). However, verification of those results by RT-qPCR revealed a significant and dose-dependent reduction of Il1b but induction of Il10 mRNA expression through exposure with both ligands, BaP and I3C (Figure 3). In terms of chemokines, transcription was reduced in case of CC-chemokine ligand Ccl3 but induced in case of Ccl7 following BaP exposure. Finally, the expression of insulin-like growth factor 1 receptor (Igf1r) was declined by I3C, whilst its ligand Igf1 was induced by BaP.
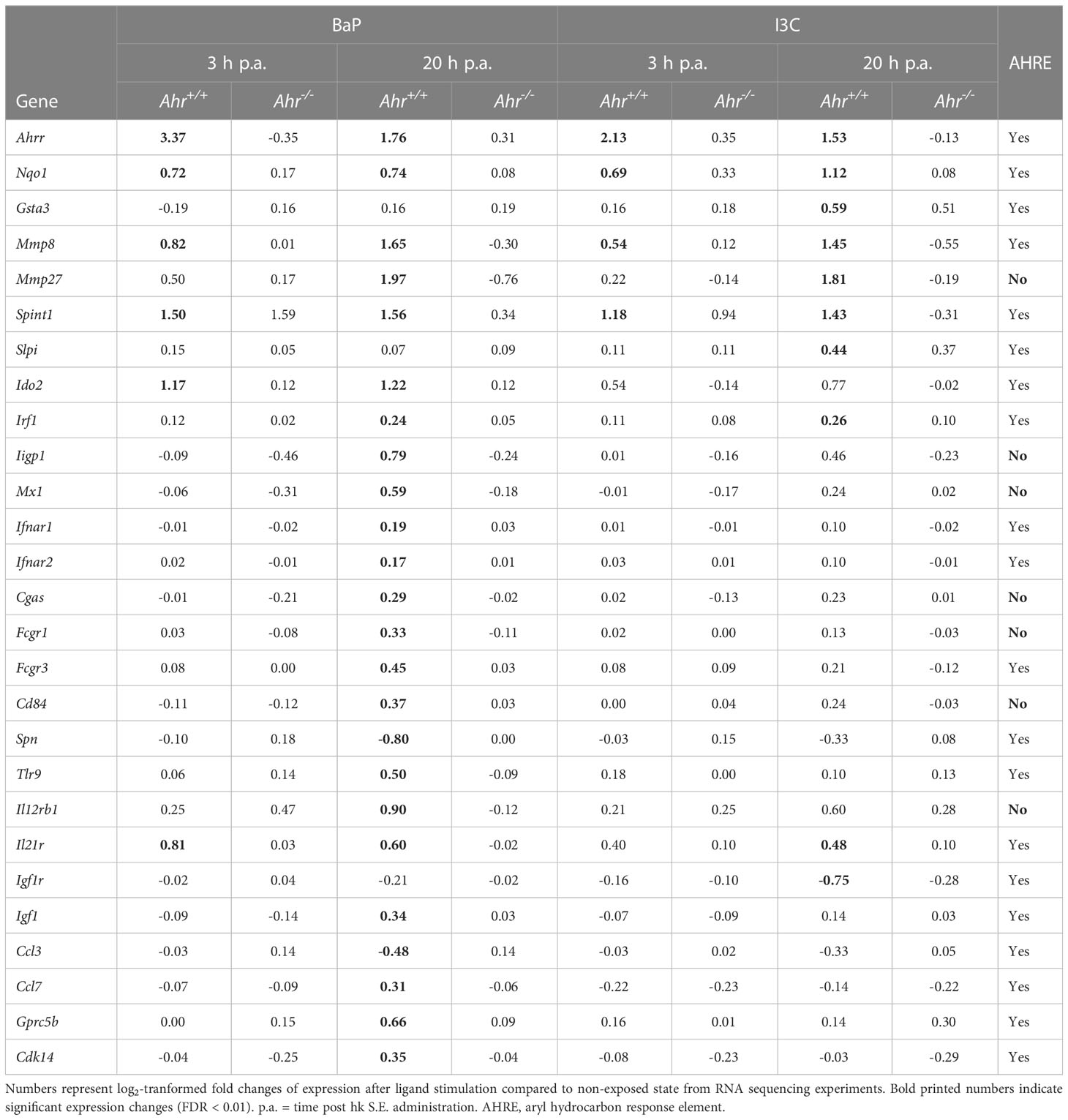
Table 3 Selected AhR-dependently differentially expressed genes.
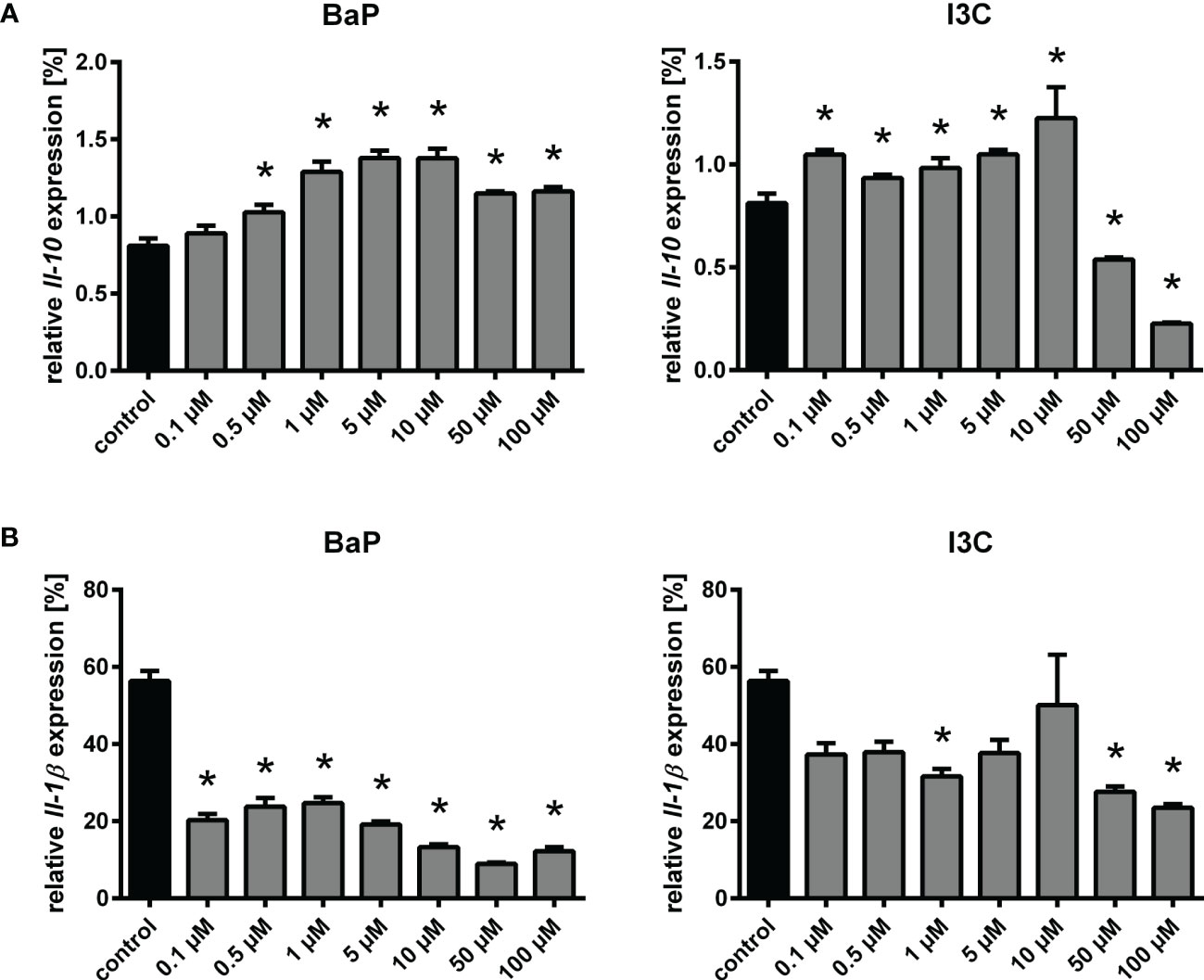
Figure 3 Changes in relative Il10 (A) and Il1b (B) expression following exposure to different concentrations of BaP or I3C. BMMs from Ahr+/+ mice were treated with indicated concentrations of BaP or I3C or vehicle control (DMSO) for 6 h. Subsequently, cells were activated with hk S.E. for 20 h (p.a. – post administration) for PAMP activation. Changes in gene transcription were assessed by real-time RT-PCR. Data represent the mean of the relative cytokine gene expression ± SEM after normalization with housekeeping genes Alas1 and Hprt (n=4). *p ≤ 0.05 indicates significant differences between treated and untreated cells.
Hence, AhR modulates the expression of several genes in part ligand-dependently. Interestingly, however, for most of the genes, that are differentially expressed after BaP but not I3C exposure, the expressional trend could be observed for both ligands, which implies that the observed effects depend on the affinity of the individual AhR ligand. To this end, we selected genes of importance in innate immunity, that were identified as ligand-specifically regulated and performed RT-qPCR experiments to monitor their expressional changes under exposure with different concentrations of both ligands used in this study, i.e., BaP and I3C (Supplementary Figure 1). The ligand-specific upregulation of Gsta3 through I3C observed in the RNA sequencing result was confirmed by the RT-qPCR analysis. Similarly, an expressional repression of Igf1r by I3C was confirmed. Interestingly, an expressional activation of Igf1r through BaP was revealed by RT-qPCR, that was not anticipated from RNA sequencing experiments. RT-qPCR revealed upregulation of Il12rb1, Ido2, and Slpi expression through both ligands, whereas the RNA sequencing data showed only a differential expression for BaP (Il12rb1, Ido2) or I3C (Slpi). However, expressional trends as observed in RT-qPCR were already anticipated from RNA sequencing indicating a relation of the observations to higher sensitivity and accuracy of RT-qPCR over RNA sequencing. However, in contrast to the RNA sequencing data Spn was not downregulated but upregulated in RT-qPCR analysis.
To model the mechanism of AhR regulation the promotor regions of all AhR-dependent DEG were screened for the known AHRE: AHRE I (5’-GCGTG-3’), AHRE II (5’- CATGnnnnnnC(A/T)TG-3’) and RelB AHRE (5’-GGGTGCAT-3’). Of all 1,108 AhR-dependent DEG, 905 genes (82 %) possess at least one of the AHRE in their promotor regions and can be considered as potential canonical AhR targets. However, the proportions and usage of AHRE motifs vary between the used ligands and duration of hk S.E. activation (Table 4). Upon BaP exposure, proportion of AhR-affected genes possessing an AHRE I motif is higher compared to I3C exposure at both time points (85 % and 81 % vs. 49 % and 68 %). While under BaP exposure the proportion of AHRE I-possessing genes was higher after 3 h compared to 20 h of BMM activation by hk S.E., we found a reciprocal result upon I3C exposure. Similarly, also a higher proportion of AHRE II-possessing genes was observed in response to BaP compared to I3C exposure but without changes between duration of Mϕ activation (18 % vs. 14 %). The proportion of RelB AHRE possessing AhR-dependent DEGs did not vary between both ligands but was higher after 20 h compared to 3 h of Mϕ activation (6 % vs. 3 %). However, although the presence of an AHRE does not automatically point to a canonical regulation of the target gene, the absence of an AHRE excludes it definitely. Thus, the highly regulated genes Mmp27, Iigp1, Mx1, Fcgr1, Cd84, and Il12rb1 can be considered as non-canonical targets (Table 3).
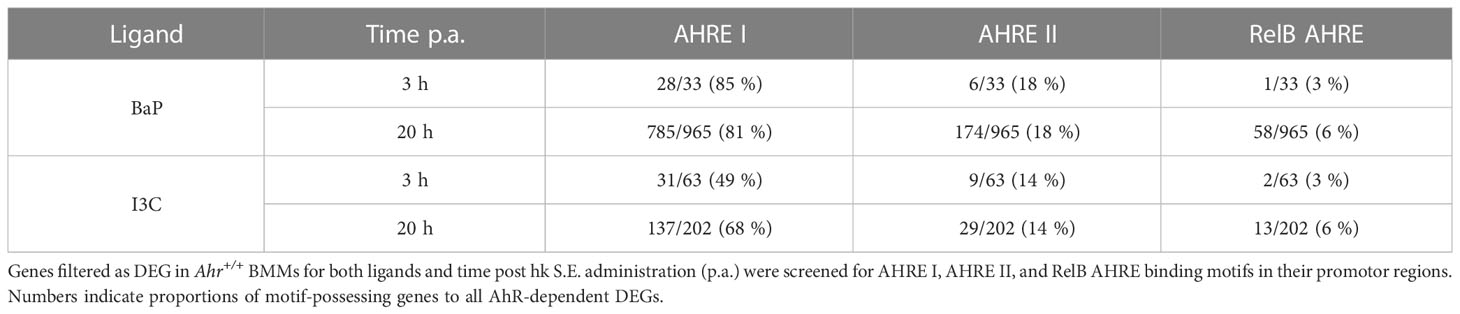
Table 4 Identification of AHRE motifs in AhR-dependently differentially expressed genes.
To evaluate mechanisms beyond canonical AhR signaling the expression profiles were filtered for AhR-dependent DEGs and analyzed via IPA to predict the activity of potential upstream regulators. Only profiles of BMMs exposed to BaP and activated for 20 h with hk S.E. resulted in sufficient analysis depth and will be focused in the further section. The activity of 36 potential upstream regulators could be modeled (Supplementary Table 4). Of those, 24 were found to regulate 14 downstream targets not possessing any of the screened AHRE motifs, and thus cannot be targeted directly by canonical AhR signaling (Figure 4A). Among the putative upstream regulators are type I (IFN-β) and type II (IFN-γ) interferons and their associated transcriptional regulators (IRF3, IRF7) as well as transmembrane receptors (IL-10Rα, TLR3, TLR9), kinases (e.g., CRKL, MARK2, MAPK9) and others (e.g., DOCK8, SOCS1, TICAM1). Several proinflammatory regulators (e.g., IFN-γ, IFN-β, TLR3, TLR9, TICAM1) were predicted to be activated. Four of the modeled upstream regulators are predicted to be inhibited. Among those are the anti-inflammatory regulators IL-10Rα, SOCS1 and CITED2. The most non-AHRE motif-possessing AhR-dependent DEGs are known to be directly or indirectly regulated by IFN-γ including Iigp1, Mx1, Casp1, Il12rb1, Lcp2, Ly6a, and Runx3. All but Lcp2 were found in upregulated expression states. Ten out of 14 genes are targeted by two or more upstream regulators. For example, Iigp1 is regulated by 17 and Mx1 by 10 upstream elements, representing highly complex regulation mechanisms.
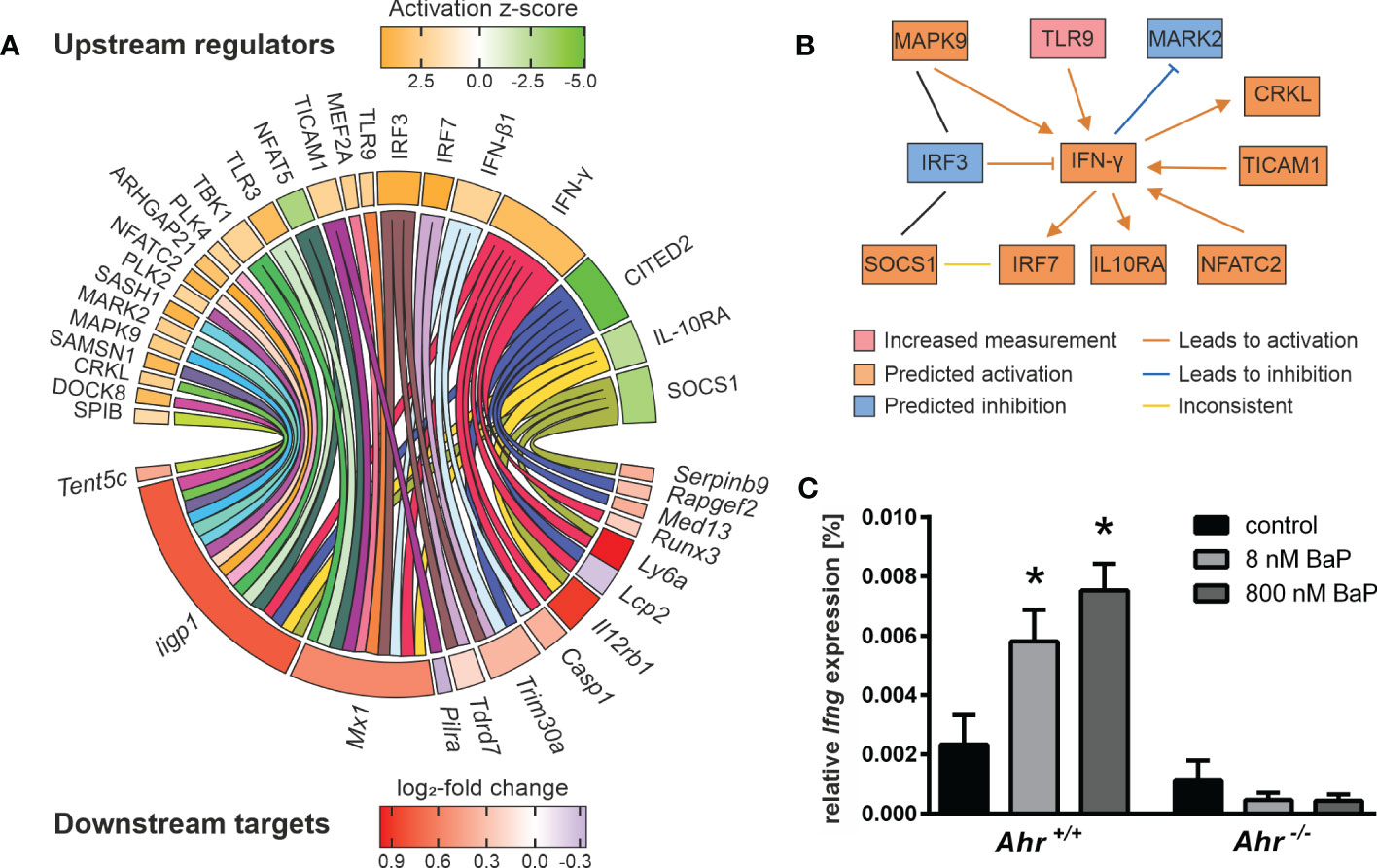
Figure 4 Putative mechanisms in AhR-dependent gene expression. Murine Ahr+/+ BMMs were exposed to BaP or treated with vehicle control (DMSO) for 6 h. Subsequently, cells were activated with hk S.E. for 20 h for PAMP activation. Whole cellular RNA was extracted and analyzed by means of RNA sequencing. Mapped read counts were analyzed by DESeq2 to identify DEGs (n = 4). (A) Potential upstream regulators were predicted from AhR-dependent gene expression to identify 24 regulator elements (upper panel) that target 14 non-AHRE motif-possessing genes (lower panel). Circle plot illustrating functional interactions of upstream regulators to downstream targets. Outer grid color represents predicted activation z-score (for upstream regulators) and measured log2-fold change (for downstream targets). (B) Upstream regulator interaction network modified from IPA output (Supplementary Figure 2). Nodes represent measured or predicted upstream regulator activities and interactions based on experimental results from BaP-exposed BMMs activated for 20 h with hk S.E. (C) BMMs from Ahr+/+ and Ahr-/- mice were exposed to BaP (8 nM or 800 nM) or DMSO (vehicle control) for 6 h. Changes in gene transcription of Ifng were assessed by quantitative real-time RT-PCR after activation of BMMs with hk S.E. for 20 h. Data represent the mean of the relative expression ± SEM after normalization with housekeeping genes Alas1 and Hprt (n = 3). *p ≤ 0.05 indicates significant differences between treated and untreated cells.
The computed upstream regulators itself are interconnected. Remarkably, a functional network that connects IFN-γ to upstream and downstream effects, is modeled to be activated. The IFN-γ expression and secretion is regulated by TICAM1, TLR9 and NFATC2. IFN-γ itself regulates the activity of MARK2, CRKL, SOCS1, and MAPK9. The latter two interact with IRF3. Further, IFN-γ regulates the expression of IRF3, IRF7, and IL-10Rα. These findings suggest a crucial impact of AhR activation, i.e., through BaP, on IFN-γ production and signaling in Mϕ (Figure 4B). Ifng itself does not possess any of the screened AHREs and its expression was not detected by RNA sequencing. Due to the ongoing controversy of IFN-γ secretion by Mϕ, IFN-γ transcription in response to AhR ligation was additionally proved by RT-qPCR, revealing AhR- and BaP-dependent transcription of Ifng (Figure 4C). This result implies IFN-γ synthesis in BMMs, that involves a BaP-triggered, non-canonical AhR-dependent mechanism. In turn, autocrine and paracrine activation of BMMs through IFN-γ may partly explain the gene expression profiles found in this study.
This study complements and extends previous approaches elucidating the molecular mechanisms of AhR-mediated effects by means of (i) comparing Ahr+/+ and Ahr-/- cells, (ii) comparing prototypic high-affinity and low-affinity ligands, and (iii) transcriptomics in combination with bioinformatic genomic analyses (37–39). The aim of the present study was, to identify potential differences in the global gene expression pattern of activated murine BMMs subsequently to exposure with high-affinity vs. low-affinity AhR ligands represented by BaP and I3C, respectively, by means of mRNA sequencing. As an in-vitro model well-established in our laboratory we utilized BMMs from Ahr-+/+ and Ahr-/- C57BL/6 mice (11, 23). The latter do not possess any remaining AhR activity, and are therefore suitable to study AhR-dependent effects in vivo and in vitro (26). For our in-vitro model, i.e., BMMs from Ahr-/- mice, functional AhR deficiency was confirmed by absent Ahrr expression upon incubation with AhR ligands (data not shown). BMMs were alternatively exposed to both AhR ligands, BaP or I3C, for 6 h prior to either 3- or 20-h activation through a Salmonella antigen preparation (i.e., hk S.E.), that comprises several PAMPs (i.e., LPS, flagellin, CpG, NOD1/2) and is therefore optimally suitable to stimulate different pattern recognition receptors. Using this approach, we intended to get new insights into ligand-specific molecular effects of latent AhR activation prior to an innate inflammatory response. This in turn should contribute to unravel the mechanism(s) of action how AhR activation may control acute and chronic inflammatory conditions in infection, cancer, or immune-mediated diseases as shown by many authors (38, 40–46).
In order to address the impact of the AhR ligand’s nature, that means high-affinity (i.e., BaP) vs. low-affinity (i.e., I3C) compound, on the AhR-dependent gene expression in Mϕ, we have identified and evaluated DEGs that are specifically expressed in response to either BaP or I3C and, in addition, DEGs that were significantly up- or downregulated by both types of AhR ligands. While the majority of AhR-dependently affected genes (885 DEGs) were found to be affected only by the high-affinity AhR ligand BaP, only 153 DEGs were influenced by the low-affinity AhR ligand I3C. Only a minority of 110 DEGs where affected by both AhR ligands, BaP and I3C. According to our expectations, this result confirmed a stronger influence of the high-affinity AhR ligand onto the functional activity of Mϕ. Interestingly, this result also reveals that high affinity of an AhR ligand is not sufficient to affect the entire potential repertoire of DEGs in Mϕ, suggesting that the individual chemical structure, and thus the individual binding mode in the AhR binding pocket seems to be a critical condition for modifying the expression of certain Mϕ genes.
We identified several DEGs, previously related to innate immunity, that possess known AHRE motifs, and thus are putatively under canonical AhR regulation. Among those genes are Il21r, Spint1, Slpi, Irf1, and Ido2, that were found to be expressionally activated by both ligands, BaP and I3C, in RNA-seq or RT-qPCR experiments. IL-21 inhibited LPS-induced expression of IL-1β, TNF-α, and IL-6 in peritoneal Mϕ, but not of IFN-γ, IL-10, CCL5, or CXCL2 via inhibiting the phosphorylation of ERK and translocation of NF-κB (47). Further, IL-21R-mediated signaling is involved in M2‐like Mϕ polarization by decreasing the expression of CD86, iNOS, and TLR4 and by increasing STAT3 phosphorylation that promotes the secretion of IL-10 by Mϕ (47–49). Irf1 expression is regulated by IFN-γ and was previously shown to induce the expression of Ido2 (50). AhR itself is required to induce Ido2 expression, that encodes for an immunosuppressive enzyme that catabolizes tryptophan into the endogenous AhR ligand kynurenine. In turn, AhR-mediated increased IDO levels, either directly or indirectly via IRF-1, contribute to decreased innate immunogenicity as previously shown for DC and Mϕ (40, 50). To the best of our knowledge, no AhR-mediated function was previously described for the here observed Slpi expressional induction. However, SLPI was shown to inhibit IL-1β maturation in human monocytes further contributing to an AhR-mediated decreased proinflammatory phenotype (51). Interestingly, Gsta3 and Igf1r both possess AHRE-motifs in their promotor regions and were previously shown to be either transcriptionally induced or functionally activated by AhR stimulation (37, 41). Here, we present an I3C-specific induction of Gsta3 and contrary expressional regulation of Igf1r, implying either ligand-specific or further AhR-independent regulatory mechanisms to be involved. Additionally, DEGs not possessing any known AHRE were identified, including Fcgr1 (encoding for the high-affinity IgG receptor FcγR1/CD64), Cd84, and Il12rb1. CD64-enriched CD11+/F4/80+ Mϕ have previously been detected in the peritoneal cavity of BaP-exposed mice in a systemic Salmonella enterica infection model (45). Cd84 was linked to inhibition of IL-1β production contributing to declined proinflammatory effects (52). However, Wang et al. reported an I3C-mediated expressional repression for Cd84 that was only observable in undifferentiated but not phorbol 12-myristate 13-acetate-induced differentiated THP-1 cells. Here, we point to a BaP-specific expressional induction of Cd84 in BMMs, implying a species-, cell type- or differentiation state-specific regulation. Il12rb1 was found to be expressionally activated by both ligands, BaP and I3C, in our study. The mature IL-12Rβ1 acts as subunit in both the IL-12 and IL-23 receptor complexes that bind the corresponding proinflammatory cytokines. IL12Rβ1 deficiency has been associated with childhood-onset and recurrence of salmonellosis, tuberculosis, and candidiasis indicating its role in the prevention of bacterial and fungal infection (53–56). However, to the best of our knowledge, no previous AhR-mediated regulation of Il12rb1 has been described. It can be speculated, that IL12Rβ1 expression might represent a mechanism for autocrine or paracrine activation of Mϕ resulting in the activation of STAT4 via TYK2 and JAK2 and subsequently in the induction of IFN-γ production; this process might be supported by IL-18 (57).
Overall, we gained evidence, that AhR activation by both ligands, BaP and I3C, reduces proinflammatory effects in PAMP-activated Mϕ. This is in line with previous observations in vitro and in vivo. BaP seems to dampen acute proinflammatory responses by suppressing proinflammatory cytokines (e.g., IL-1β) but induce anti-inflammatory cytokines (e.g., IL-10), and thus prevent septic shock (11, 45). However, other studies reported increased levels of IL-1β production upon AhR ligand exposure, for example in the human synovial fibroblast cell line MH7A by 3-methylcholanthrene (58) or in the human monocyte cell line THP-1 in response to AhR activation by polychlorinated biphenyls (59). In fact, most studies focus on direct effects of AhR ligands on resting cells. Here, we investigated the modulation of gene expression in PAMP-activated Mϕ. Thus, different observations of IL-1β production rates might be related to activation state of the cell but also to the cell type or species differences. Nonetheless, BaP may also directly or indirectly exacerbate Th2-driven inflammatory responses by suppressing proinflammatory cytokines that induce Th1 responses, i.e., IL-12 (42–44). Another important immunoregulatory effect of BaP concerns to reciprocal regulation of IL-22 and IL-17, suggesting an impact of BaP on the homeostasis at epithelial barriers (60). Notably, these immunomodulatory effects of BaP occur preferentially at lower BaP doses. Previous studies found, that BaP represents a high-affinity ligand with a relative AhR ligand binding affinity of 617 nM (indicated as IC50 – competition of [3H]TCDD; for comparison: IC50 of TCDD and FICZ = 1 nM) while I3C possesses a very low relative binding affinity to AhR (IC50 = 26 mM) (61, 62). Moreover, Tagliabue et al. previously proofed, that ligand binding affinity and potency to stimulate AhR nuclear translocation/DNA binding were relatively well correlated (EC50) (61). Thus, the stronger effect of BaP in terms of the high number of affected genes found in our study might be due to its higher binding affinity and EC50 values compared to I3C, and thus to its higher relative concentration applied in this study. Although, I3C was used at a 10-fold higher concentration than BaP, its affinity to the AhR is estimated to be about 42,000-fold lower compared to BaP resulting in a dose advantage of factor 4,200 as applied for BaP. Given, that the BaP concentration of 1 µM applied in the present study is considered as “low-dose” from previous studies (11, 45), BaP may be seen as a highly potent immunoregulatory AhR ligand. However, genes that are affected either by BaP or I3C might reflect differential responses caused by the individual steric localization of an individual AhR ligand inside the binding cavity of the receptor, as can be presumed from docking studies using different AhR ligands (61) rather than determined by the affinity of the AhR ligand. Hence, further putative AhR ligands, e.g., quercetin (46), that combine non-toxic properties with higher affinities or different binding modes should be tested in future studies for their utilization in immunomodulatory therapies.
In addition to (i) the concentration and (ii) the individual chemical structure of an AhR ligand, the number of AhR-affected genes also critically depends (iii) on the duration of PRR-mediated Mϕ activation. Using a transcriptomics approach, the present study identified 18 times vs. 3 times more DEGs at 20 h vs. 3 h of activation for BaP or I3C, respectively. This implies an even stronger dependency of AhR-driven immunomodulation on the activation state of the Mϕ as already previously shown by our group by investigating the mRNA or protein expression of individual genes of interest using real-time RT-PCR or flow cytometry, respectively (11, 23). This underlines the value of applying untargeted analyses such as transcriptomics in screening approaches. However, the individual nature of the genes differentially regulated at 3 h vs. 20 h of activation revealed different clusters of genes, reflecting specific early or late occurring cellular processes. For example, genes of “Mitotic G2/M-transition checkpoint” were upregulated, whereas genes involved in “Translation elongation” were downregulated at 20 h compared to 3 h of PAMP-induced Mϕ activation. Furthermore, the chemical stability and metabolic turnover of the used ligands are crucial to affect gene expression, especially at different time scales. BaP is chemically inert and requires metabolic activity to be degraded (63). This is mainly achieved by CYP1A1 and NQO1 that were shown to be active in a variety of organoid cultures (64). In the present study, we did not observe an upregulation of Cyp expression and overall low expression rates of these main BaP-degrading enzymes. However, Nqo1 was upregulated in expression by both ligands indicating only a limited degradation of BaP in the here used experimental setup. In contrast, I3C is chemically condensed to e.g., 3,3’-diindolylmethane (DIM) and 5,11-dihydroindolo-[3,2-b]carbazole (ICZ) at acidic conditions, that is mainly achieved in the stomach at in-vivo conditions (65). Those metabolites were shown to possess even higher affinity to AhR as I3C itself (62). However, we and others have clearly shown, that I3C induces AhR activation in vitro without acidification, likely through the convertion to diindolylmethane (DIM) (66). Although, I3C possesses very low affinity to the AhR it induces several genes via both the canonical and the non-canonical pathway as shown in our work but also by other authors. Thus, it might be anticipated, that the effects observed upon I3C exposure are based on AhR activation by I3C itself as well as its metabolites.
Independent of their nature as AhR ligands, both BaP and I3C affect other pathways and biological processes that are not mediated via the AhR. For instance, BaP was shown to increase TNF-α production in human primary macrophages via ERK1/2 and independent of AhR (67), whereas I3C directly binds to a variety of intracellular receptors, as reviewed previously (68). However, by including Ahr-/- controls into this study we only reported AhR-dependent effects.
Although multiple AhR-dependent effects were described in the last decades, the diverse modes of action are not yet completely understood. Early attempts applied experimental and in silico approaches to screen for canonical targets on whole genome scales. Several hundred putative targets were predicted for human, mouse and rat based on position weight matrices and similarity scores (69). Further, extensive longitudinal expression screening of BaP-exposed cells revealed 81 primary responding genes and more than 1,000 side or secondary effects (70). Those studies found that several transcriptional regulators possess AHRE motifs and primarily respond to AhR activation. Thus, a transcriptional cascade starting at AhR and accounting for multiple phenotypical effects depending on the cell type and activation state was previously proposed. Indeed, AHRE were identified by genome screening in the promotors of the transcription factors CEBP-β, NF-κB1, NF-κB2, NF-κB3, RelB, c-Rel, Jun, IRF-1, IRF-4, and STAT3 (71). Those transcription factors regulate the expression of cytokines in activated Mϕ and may account for the expressional activation of genes involved in the IFN and IL-6 production observed in this study. However, genome-wide screening for known AHRE motifs may not be sufficient as the presence of AhR binding motifs in promotor regions does not necessarily indicates its accessibility for an activated AhR complex, and thus transcriptional activation. Hence, such in-silico screening approaches should be accompanied with advanced bioinformatic approaches for sequence homologies and conservations across different species (72) or by experimental validation applying chromatin immunoprecipitation DNA sequencing (73) in future studies. In contrast to the above-mentioned studies, we did not focus to identify canonical AhR targets based on AHRE motif presence but excluded them to gain insights into non-canonical or secondary effects of AhR in the PRR-mediated Mϕ activation. Therefore, only DEGs not possessing any known AHRE were included for upstream regulator prediction and functional network analysis. Such bioinformatic modeling has previously successfully been applied to identify IRF-7 as a key regulator in LPS-mediated activation in human alveolar Mϕ via TLR4 (74).
Although non-AHRE possessing DEGs account for only a minority of all DEGs (< 20 %), one of the most notable results of the here presented approach concerns to the role of the production and signaling of interferons in the activation of Mϕ in innate immunity. We gained multiple evidence in AhR interference in the release of type I (i.e., IFN-α, IFN-β) and type 2 interferons (i.e., IFN-γ). As such, the genes of the IFN-α/β-inducing Irf1 and Cgas were expressionally increased, whereas IRF3 and IRF7 were modeled to be activated from gene expression profiles upon BaP exposure. In sum, transcriptomics profiles pointed to an induced type I interferon production upon AhR activation as revealed from gene set enrichment analysis. The induction of IFN-α/β expression and secretion by Mϕ after PRR activation by bacterial PAMPs is a well-known concept (75, 76). Here, we propose further evidence of modulation of the type I interferon production through AhR activation. The production of IFN-γ by Mϕ has been a matter of debate until Munder and colleagues delivered convincing evidence that Mϕ are capable of producing IFN-γ in response to combined stimulation with IL-12 and IL-18 (77). Later, Schindler et al. demonstrated that IL-12/IL-18 induced IFN-γ production requires STAT4 signaling (78). It was previously shown that AhR activation modulated the IFN-γ production in Mϕ upon viral infection and autoimmune disease both ex vivo and in vivo (25, 79). Here, we confirmed an AhR- and ligand-dependent induction of Ifng expression by RT-qPCR. The finding that the Ifng gene does not possess any of the screened AHREs let assume that AhR ligands may trigger IFN-γ synthesis and secretion in Mϕ via a non-canonical AhR-dependent mechanism. Additionally, we provide evidence for induced autocrine/paracrine effects of the increased production of interferons. For instance, Ifnar1 and Ifnar2 were expressionally activated upon AhR activation. Subsequently, IFN-α/β but also IFN-γ signaling was modeled as upregulated based on gene set enrichment and upstream regulator analysis. This was, among others, based on expressional activation of Iigp, Mx1, Trim30a, Casp1, Il12rb1, Lcp2, Ly6a, and Runx3. None of these genes does possess any of the screened AHRE motifs implying a non-canonical or secondary, autocrine/paracrine interferon-driven effect. An AhR-dependent modulation of IFN-α signaling in antiviral immunity in Mϕ has previously been demonstrated and linked to IRF7 targeting by AhR interacting protein (AIP) (24, 80).
In conclusion, the here presented study extends the knowledge of molecular effects and mechanisms of AhR activation in the modulation of PAMP-induced Mϕ activation (Figure 5). More than 1,000 genes were identified to be modulated in an AhR- and ligand-dependent manner. The majority of those genes, such as Il21r, Irf1, Spint1, Slpi, and Ido2 possess an AHRE motif, and thus represent putative canonical AhR targets. However, further key immune genes, e.g., Il12rb1, Fcgr1, and Cd84 were induced upon AhR activation and do not possess an AHRE motif, indicating yet unknown non-canonical and secondary mechanisms to be involved. This study gained multiple evidence that an AhR-dependent expressional induction and autocrine/paracrine signaling of type I and type II interferons may contribute to the here described modulation of Mϕ activation. However, the majority of DEGs is only significantly affected by BaP, but high correlation of whole-genome expression patterns might point to affinity and concentration effects of the used ligands. Nonetheless, it cannot be excluded that the effects are due to different binding modes of BaP (rigid aromatic 5-ring structure) and I3C (smaller, more flexible indole structure) in the AhR’s ligand binding pocket. Evidence for different binding models has been delivered by Tagliabue et al. using computational modeling, among others (61). In particular, some DEGs, i.e., Gsta3 and Igf1r, possess specific or even contrary modes of regulation. Both, yet unknown non-canonical and secondary mechanisms as well as ligand-specific induction of target genes indicate, that more in-depth study is needed to completely understand the molecular mechanisms of AhR signaling in order to be prospectively utilized in immunomodulatory therapies.
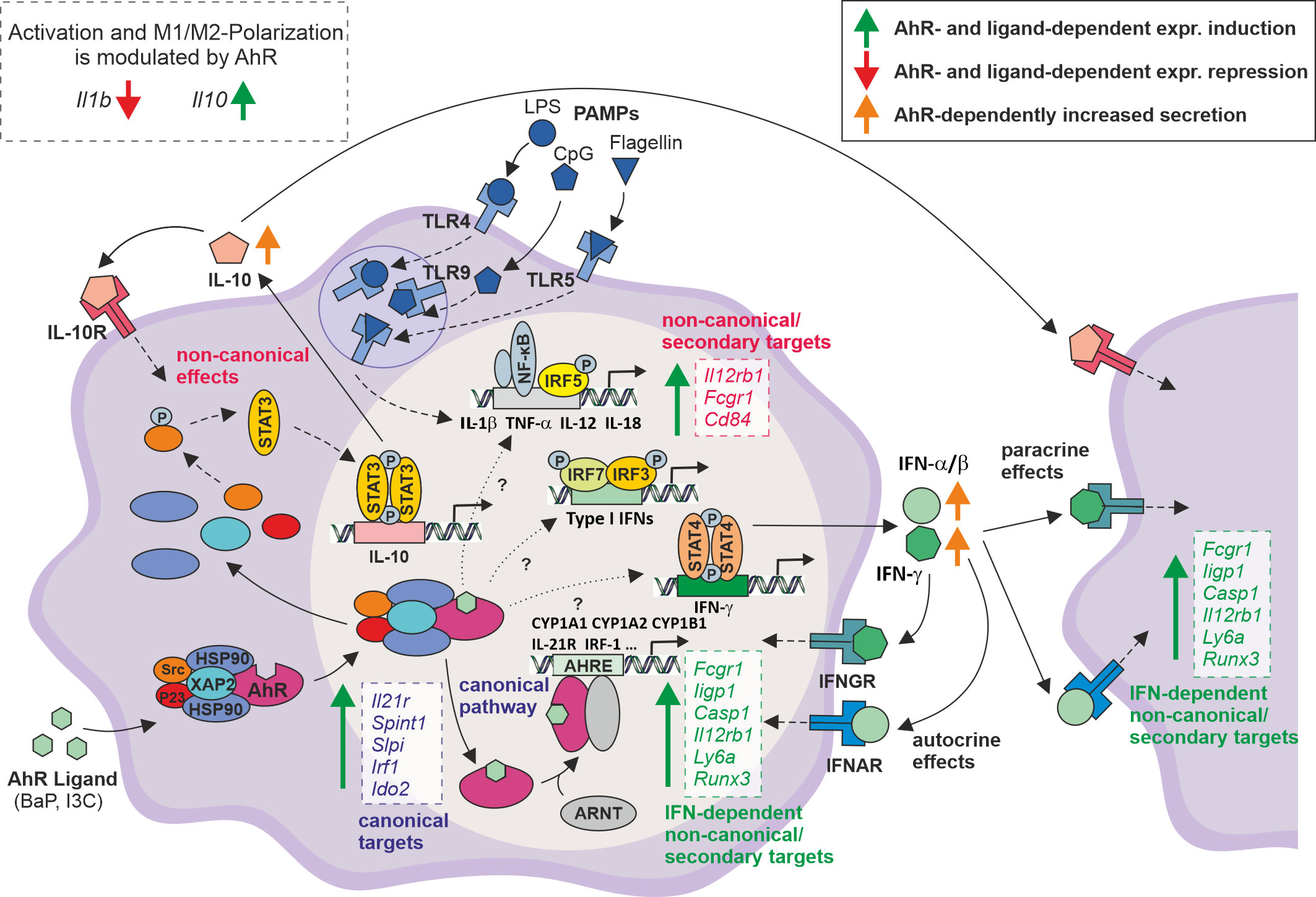
Figure 5 Proposed model for the modulation of PAMP-induced Mϕ activation through AhR ligand exposure. After entering the cytoplasm, AhR ligands such as BaP or I3C bind to a complex of several proteins, containing AhR, HSP90, XAP2, P23, and Src. AhR ligand binding induces a conformational change in this protein complex resulting in translocation of the whole complex into the nucleus where it is degraded to release the AhR molecule. Inside the nucleus, AhR associates with the Aryl hydrocarbon nuclear translocator (ARNT) and binds to the Aryl hydrocarbon response element (AHRE) in the promotor region of target genes, i.e., Cyp1a1, Cyp1a2, Cyp1b1, inducing their transcriptional activity (➪ canonical, genomic targets). This study confirms or suggest the expressional activation of several key immune genes, i.e., Il21r, Irf1, but also Spint1, Slpi, and Ido2 as putative AHRE motif-possessing canonical targets. In the cytoplasm, the AhR complex included src tyrosine kinase phosphorylates STAT3 leading to its dimerization and nuclear translocation. In the nucleus STAT3 binds to a response element in the Il10 promotor, and thus inducing IL-10 production (81). This is the most prominent non-canonical AhR-dependent pathway. If IL-10 is secreted, it exerts autocrine and paracrine anti-inflammatory effects, leading to the suppression of gene expression of proinflammatory cytokines, i.e., Il1b, Tnfa, Il12a/b. Further, this study suggests the AhR-dependent expressional induction of key immune genes for Mϕ activation, such as Il12rb1, Fcgr1, Cd84, that do possess an AHRE motif and thus, are regulated by non-canonical or secondary mechanisms. Of note, from several expressional profiles it is anticipated that the transcriptional activation of type I (IFN-α, IFN-β) and type II (IFN-γ) interferons is regulated by AhR via so far unknown molecular mechanisms possibly including both canonical and non-canonical pathways. Gene expressional profiles and bioinformatic approaches suggest an autocrine/paracrine effect of secreted interferons partly explaining the here presented results. Solid lines ⟹ known mechanisms covered in the figure; dashed lines ⟹ known mechanisms not covered in the figure; dotted line ⟹ unkown mechanisms. Boxes indicate genes expressionally induced in the study.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE223122 [GEO; (82)].
The animal study was reviewed and approved by Landesdirektion Sachsen Referat 25 Braustraße 2 04107 Leipzig.
JL was the principle investigator, conceptualized and designed the study, interpreted the data, drafted and revised the manuscript. JS and JH contributed equally, designed the study, performed cell culture and validation experiments, analyzed and interpreted the data, created the figures, drafted the initial manuscript, and share first authorship. SR performed cell culture and RT-qPCR experiments, CB and DL performed RNA-seq experiments, whereas KR and CK analyzed RNA-seq data. JS and KR designed the computational screen for known AHRE motifs in promoter regions. JS conducted the AHRE motif discovery, analyzed and interpreted data. SR, CB, DL, KR and CK critically reviewed and revised the manuscript. UK and SK interpreted the data and critically reviewed and revised the manuscript. All authors approved the final manuscript and agree to be accountable for all aspects of the work. All authors contributed to the article and approved the submitted version.
This work was kindly supported by the Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V., especially by the Fraunhofer Cluster of Excellence Immune-Mediated Diseases (Project CIMD_CED-AhR).
The authors gratefully acknowledge the excellent technical assistance by Ulrike Scholz and Sarah Leitenroth. Furthermore, we thank Dr. Christopher Bradfield (University of Wisconsin, Madison, WI, USA) for kindly providing the original breeding pairs of Ahr knockout mice.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1156493/full#supplementary-material
1. Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol (2000) 40:519–61. doi: 10.1146/annurev.pharmtox.40.1.519
2. Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci (2011) 124(1):1–22. doi: 10.1093/toxsci/kfr218
3. Ge NL, Elferink CJ. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin Signaling to Cell Cycle J Biol Chem (1998) 273(35):22708–13. doi: 10.1074/jbc.273.35.22708
4. Gutiérrez-Vázquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity (2018) 48(1):19–33. doi: 10.1016/j.immuni.2017.12.012
5. Naseri-Nosar P, Nogalski MT, Shenk T. The aryl hydrocarbon receptor facilitates the human cytomegalovirus-mediated G1/S block to cell cycle progression. Proc Natl Acad Sci USA (2021) 118(12):e2026336118. doi: 10.1073/pnas.2026336118
6. Omiecinski CJ, Vanden Heuvel JP, Perdew GH, Peters JM. Xenobiotic metabolism, disposition, and regulation by receptors: from biochemical phenomenon to predictors of major toxicities. Toxicol Sci (2011) 120 Suppl 1(Suppl 1):S49–75. doi: 10.1093/toxsci/kfq338
7. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol (2014) 32:403–32. doi: 10.1146/annurev-immunol-032713-120245
8. Whitlock JP. Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol (1999) 39:103–25. doi: 10.1146/annurev.pharmtox.39.1.103
9. Frericks M, Temchura VV, Majora M, Stutte S, Esser C. Transcriptional signatures of immune cells in aryl hydrocarbon receptor (AHR)-proficient and AHR-deficient mice. Biol Chem (2006) 387(9):1219–26. doi: 10.1515/BC.2006.151
10. Vogel CFA, Khan EM, Leung PSC, Gershwin ME, Chang WLW, Wu D, et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-κB. J Biol Chem (2014) 289(3):1866–75. doi: 10.1074/jbc.M113.505578
11. Fueldner C, Kohlschmidt J, Riemschneider S, Schulze F, Zoldan K, Esser C, et al. Benzo(a)pyrene attenuates the pattern-recognition-receptor induced proinflammatory phenotype of murine macrophages by inducing IL-10 expression in an aryl hydrocarbon receptor-dependent manner. Toxicology (2018) 409:80–90. doi: 10.1016/j.tox.2018.07.011
12. Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol (2009) 30(9):447–54. doi: 10.1016/j.it.2009.06.005
13. Wright EJ, de Castro KP, Joshi AD, Elferink CJ. Canonical and non-canonical aryl hydrocarbon receptor signaling pathways. Curr Opin Toxicol (2017) 2:87–92. doi: 10.1016/j.cotox.2017.01.001
14. Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med (2009) 206(9):2027–35. doi: 10.1084/jem.20090560
15. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci USA (2008) 105(28):9721–6. doi: 10.1073/pnas.0804231105
16. Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem (1999) 274(1):510–5. doi: 10.1074/jbc.274.1.510
17. Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene (2000) 19(48):5498–506. doi: 10.1038/sj.onc.1203945
18. Vogel CFA, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol (2007) 21(12):2941–55. doi: 10.1210/me.2007-0211
19. Ishihara Y, Kado SY, Bein KJ, He Y, Pouraryan AA, Urban A, et al. Aryl hydrocarbon receptor signaling synergizes with TLR/NF-κB-Signaling for induction of IL-22 through canonical and non-canonical AhR pathways. Front Toxicol (2021) 3:787360. doi: 10.3389/ftox.2021.787360
20. Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature (2007) 446(7135):562–6. doi: 10.1038/nature05683
21. Enan E, Matsumura F. Identification of c-src as the integral component of the cytosolic ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem Pharmacol (1996) 52(10):1599–612. doi: 10.1016/S0006-2952(96)00566-7
22. Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol (2018) 11(4):1024–38. doi: 10.1038/s41385-018-0019-2
23. Riemschneider S, Kohlschmidt J, Fueldner C, Esser C, Hauschildt S, Lehmann J. Aryl hydrocarbon receptor activation by benzo(a)pyrene inhibits proliferation of myeloid precursor cells and alters the differentiation state as well as the functional phenotype of murine bone marrow-derived macrophages. Toxicol Lett (2018) 296:106–13. doi: 10.1016/j.toxlet.2018.07.050
24. Yamada T, Horimoto H, Kameyama T, Hayakawa S, Yamato H, Dazai M, et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat Immunol (2016) 17(6):687–94. doi: 10.1038/ni.3422
25. Neff-LaFord H, Teske S, Bushnell TP, Lawrence BP. Aryl hydrocarbon receptor activation during influenza virus infection unveils a novel pathway of IFN-gamma production by phagocytic cells. J Immunol (2007) 179(1):247–55. doi: 10.4049/jimmunol.179.1.247
26. Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine ahr null allele: involvement of the ah receptor in hepatic growth and development. Proc Natl Acad Sci USA (1996) 93(13):6731–6. doi: 10.1073/pnas.93.13.6731
27. Lehmann J, Bellmann S, Werner C, Schröder R, Schütze N, Alber G. IL-12p40-dependent agonistic effects on the development of protective innate and adaptive immunity against salmonella enteritidis. J Immunol (2001) 167(9):5304–15. doi: 10.4049/jimmunol.167.9.5304
28. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol (2019) 37(8):907–15. doi: 10.1038/s41587-019-0201-4
29. Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, et al. GENCODE 2021. Nucleic Acids Res (2021) 49(D1):D916–23. doi: 10.1093/nar/gkaa1087
30. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics (2015) 31(2):166–9. doi: 10.1093/bioinformatics/btu638
31. Kämpf C, Specht M, Scholz A, Puppel S-H, Doose G, Reiche K, et al. Uap: reproducible and robust HTS data analysis. BMC Bioinf (2019) 20(1):664. doi: 10.1186/s12859-019-3219-1
32. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA (2005) 102(43):15545–50. doi: 10.1073/pnas.0506580102
33. Schmoldt A, Benthe HF, Haberland G. Digitoxin metabolism by rat liver microsomes. Biochem Pharmacol (1975) 24(17):1639–41. doi: 10.1016/0006-2952(75)90094-5
34. Nguyen NTT, Contreras-Moreira B, Castro-Mondragon JA, Santana-Garcia W, Ossio R, Robles-Espinoza CD, et al. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res (2018) 46(W1):W209–14. doi: 10.1093/nar/gky317
35. Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics (2014) 30(4):523–30. doi: 10.1093/bioinformatics/btt703
36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
37. Großkopf H, Walter K, Karkossa I, von Bergen M, Schubert K. Non-genomic AhR-signaling modulates the immune response in endotoxin-activated macrophages after activation by the environmental stressor BaP. Front Immunol (2021) 12:620270. doi: 10.3389/fimmu.2021.620270
38. Franchini AM, Myers JR, Jin G-B, Shepherd DM, Lawrence BP. Genome-wide transcriptional analysis reveals novel AhR targets that regulate dendritic cell function during influenza a virus infection. Immunohorizons (2019) 3(6):219–35. doi: 10.4049/immunohorizons.1900004
39. Walter K, Grosskopf H, Karkossa I, von Bergen M, Schubert K. Proteomic characterization of the cellular effects of AhR activation by microbial tryptophan catabolites in endotoxin-activated human macrophages. Int J Environ Res Public Health (2021) 18(19):10336. doi: 10.3390/ijerph181910336
40. Kenison JE, Wang Z, Yang K, Snyder M, Quintana FJ, Sherr DH. The aryl hydrocarbon receptor suppresses immunity to oral squamous cell carcinoma through immune checkpoint regulation. Proc Natl Acad Sci USA (2021) 118(19):e2012692118. doi: 10.1073/pnas.2012692118
41. Sekine H, Mimura J, Oshima M, Okawa H, Kanno J, Igarashi K, et al. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol Cell Biol (2009) 29(24):6391–400. doi: 10.1128/MCB.00337-09
42. Wang E, Liu X, Tu W, Do DC, Yu H, Yang L, et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy (2019) 74(9):1675–90. doi: 10.1111/all.13784
43. Tajima H, Tajiki-Nishino R, Watanabe Y, Fukuyama T. Direct activation of aryl hydrocarbon receptor by benzoapyrene elicits T-helper 2-driven proinflammatory responses in a mouse model of allergic dermatitis. J Appl Toxicol (2019) 39(7):936–44. doi: 10.1002/jat.3782
44. Yanagisawa R, Koike E, Win-Shwe T-T, Ichinose T, Takano H. Effects of lactational exposure to low-dose BaP on allergic and non-allergic immune responses in mice offspring. J Immunotoxicol (2018) 15(1):31–40. doi: 10.1080/1547691X.2018.1442379
45. Fueldner C, Riemschneider S, Haupt J, Jungnickel H, Schulze F, Zoldan K, et al. Aryl hydrocarbon receptor activation by benzoapyrene prevents development of septic shock and fatal outcome in a mouse model of systemic salmonella enterica infection. Cells (2022) 11(4):737. doi: 10.3390/cells11040737
46. Riemschneider S, Hoffmann M, Slanina U, Weber K, Hauschildt S, Lehmann J. Indol-3-Carbinol and quercetin ameliorate chronic DSS-induced colitis in C57BL/6 mice by AhR-mediated anti-inflammatory mechanisms. Int J Environ Res Public Health (2021) 18(5):2262. doi: 10.3390/ijerph18052262
47. Li S-n, Wang W, Fu S-p, Wang J-f, Liu H-m, Xie S-s, et al. IL-21 modulates release of proinflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Mediators Inflamm (2013) 2013:548073. doi: 10.1155/2013/548073
48. Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF, Cheever AW, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest (2006) 116(7):2044–55. doi: 10.1172/JCI27727
49. Jian L, Li C, Sun L, Ma Z, Wang X, Yu R, et al. IL-21 regulates macrophage activation in human monocytic THP-1-derived macrophages. Rheumatol Autoimmun (2021) 1(1):18–29. doi: 10.1002/rai2.12000
50. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA (2010) 107(46):19961–6. doi: 10.1073/pnas.1014465107
51. Zakrzewicz A, Richter K, Zakrzewicz D, Siebers K, Damm J, Agné A, et al. SLPI inhibits ATP-mediated maturation of IL-1β in human monocytic leukocytes: a novel function of an old player. Front Immunol (2019) 10:664. doi: 10.3389/fimmu.2019.00664
52. Wang TTY, Pham Q, Kim YS. Elucidating the role of CD84 and AHR in modulation of LPS-induced cytokines production by cruciferous vegetable-derived compounds indole-3-Carbinol and 3,3’-diindolylmethane. Int J Mol Sci (2018) 19(2):339. doi: 10.3390/ijms19020339
53. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Med (Baltimore) (2010) 89(6):381–402. doi: 10.1097/MD.0b013e3181fdd832
54. Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science (1998) 280(5368):1432–5. doi: 10.1126/science.280.5368.1432
55. de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, et al. Severe mycobacterial and salmonella infections in interleukin-12 receptor-deficient patients. Science (1998) 280(5368):1435–8. doi: 10.1126/science.280.5368.1435
56. Ouederni M, Sanal O, Ikinciogullari A, Tezcan I, Dogu F, Sologuren I, et al. Clinical features of candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis (2014) 58(2):204–13. doi: 10.1093/cid/cit722
57. Lawless VA, Zhang S, Ozes ON, Bruns HA, Oldham I, Hoey T, et al. Stat4 regulates multiple components of IFN-gamma-inducing signaling pathways. J Immunol (2000) 165(12):6803–8. doi: 10.4049/jimmunol.165.12.6803
58. Tamaki A, Hayashi H, Nakajima H, Takii T, Katagiri D, Miyazawa K, et al. Polycyclic aromatic hydrocarbon increases mRNA level for interleukin 1 beta in human fibroblast-like synoviocyte line via aryl hydrocarbon receptor. Biol Pharm Bull (2004) 27(3):407–10. doi: 10.1248/bpb.27.407
59. Wang C, Petriello MC, Zhu B, Hennig B. PCB 126 induces monocyte/macrophage polarization and inflammation through AhR and NF-κB pathways. Toxicol Appl Pharmacol (2019) 367:71–81. doi: 10.1016/j.taap.2019.02.006
60. Plé C, Fan Y, Ait Yahia S, Vorng H, Everaere L, Chenivesse C, et al. Polycyclic aromatic hydrocarbons reciprocally regulate IL-22 and IL-17 cytokines in peripheral blood mononuclear cells from both healthy and asthmatic subjects. PLoS One (2015) 10(4):e0122372. doi: 10.1371/journal.pone.0122372
61. Tagliabue SG, Faber SC, Motta S, Denison MS, Bonati L. Modeling the binding of diverse ligands within the ah receptor ligand binding domain. Sci Rep (2019) 9(1):10693. doi: 10.1038/s41598-019-47138-z
62. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci USA (1991) 88(21):9543–7. doi: 10.1073/pnas.88.21.9543
63. Lu D, Harvey RG, Blair IA, Penning TM. Quantitation of benzoapyrene metabolic profiles in human bronchoalveolar (H358) cells by stable isotope dilution liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. Chem Res Toxicol (2011) 24(11):1905–14. doi: 10.1021/tx2002614
64. Caipa Garcia AL, Kucab JE, Al-Serori H, Beck RSS, Fischer F, Hufnagel M, et al. Metabolic activation of benzoapyrene by human tissue organoid cultures. Int J Mol Sci (2023) 24(1):606. doi: 10.3390/ijms24010606
65. Amare DE, Bovee TFH, Mulder PPJ, Hamers A, Hoogenboom RLAP. Acid condensation products of indole-3-carbinol and their in-vitro (anti)estrogenic, (anti)androgenic and aryl hydrocarbon receptor activities. Arabian J Chem (2020) 13(9):7199–211. doi: 10.1016/j.arabjc.2020.08.002
66. Bradlow HL, Zeligs MA. Diindolylmethane (DIM) spontaneously forms from indole-3-carbinol (I3C) during cell culture experiments. In Vivo (2010) 24(4):387–91. Available at: https://iv.iiarjournals.org/content/invivo/24/4/387.full.pdf.
67. Lecureur V, Le Ferrec E, N’diaye M, Le Vee M, Gardyn C, Gilot D, et al. ERK-dependent induction of TNFalpha expression by the environmental contaminant benzo(a)pyrene in primary human macrophages. FEBS Lett (2005) 579(9):1904–10. doi: 10.1016/j.febslet.2005.01.081
68. Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle (2005) 4(9):1201–15. doi: 10.4161/cc.4.9.1993
69. Sun YV, Boverhof DR, Burgoon LD, Fielden MR, Zacharewski TR. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res (2004) 32(15):4512–23. doi: 10.1093/nar/gkh782
70. Michaelson JJ, Trump S, Rudzok S, Gräbsch C, Madureira DJ, Dautel F, et al. Transcriptional signatures of regulatory and toxic responses to benzo-a-pyrene exposure. BMC Genomics (2011) 12:502. doi: 10.1186/1471-2164-12-502
71. Furman DP, Oshchepkova EA, Oshchepkov DY, Shamanina MY, Mordvinov VA. Promoters of the genes encoding the transcription factors regulating the cytokine gene expression in macrophages contain putative binding sites for aryl hydrocarbon receptor. Comput Biol Chem (2009) 33(6):465–8. doi: 10.1016/j.compbiolchem.2009.10.004
72. Krieger G, Lupo O, Wittkopp P, Barkai N. Evolution of transcription factor binding through sequence variations and turnover of binding sites. Genome Res (2022) 32(6):1099–111. doi: 10.1101/gr.276715.122
73. Yang SY, Ahmed S, Satheesh SV, Matthews J. Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)- and aryl hydrocarbon receptor repressor (AHRR)-binding sites in human breast cancer cells. Arch Toxicol (2018) 92(1):225–40. doi: 10.1007/s00204-017-2022-x
74. Pinilla-Vera M, Xiong Z, Zhao Y, Zhao J, Donahoe MP, Barge S, et al. Full spectrum of LPS activation in alveolar macrophages of healthy volunteers by whole transcriptomic profiling. PLoS One (2016) 11(7):e0159329. doi: 10.1371/journal.pone.0159329
75. Falk LA, Vogel SN. Differential production of IFN-alpha/beta by CSF-1- and GM-CSF-derived macrophages. J Leukoc Biol (1990) 48(1):43–9. doi: 10.1002/jlb.48.1.43
76. Kumaran Satyanarayanan S, El Kebir D, Soboh S, Butenko S, Sekheri M, Saadi J, et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat Commun (2019) 10(1):3471. doi: 10.1038/s41467-019-10903-9
77. Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med (1998) 187(12):2103–8. doi: 10.1084/jem.187.12.2103
78. Schindler H, Lutz MB, Röllinghoff M, Bogdan C. The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J Immunol (2001) 166(5):3075–82. doi: 10.4049/jimmunol.166.5.3075
79. Mohammadi S, Memarian A, Sedighi S, Behnampour N, Yazdani Y. Immunoregulatory effects of indole-3-carbinol on monocyte-derived macrophages in systemic lupus erythematosus: a crucial role for aryl hydrocarbon receptor. Autoimmunity (2018) 51(5):199–209. doi: 10.1080/08916934.2018.1494161
80. Zhou Q, Lavorgna A, Bowman M, Hiscott J, Harhaj EW. Aryl hydrocarbon receptor interacting protein targets IRF7 to suppress antiviral signaling and the induction of type I interferon. J Biol Chem (2015) 290(23):14729–39. doi: 10.1074/jbc.M114.633065
81. Zhu J, Luo L, Tian L, Yin S, Ma X, Cheng S, et al. Aryl hydrocarbon receptor promotes IL-10 expression in inflammatory macrophages through src-STAT3 signaling pathway. Front Immunol (2018) 9:2033. doi: 10.3389/fimmu.2018.02033
Keywords: aryl hydrocarbon receptor, macrophage activation, innate immunity, immunomodulation, transcriptomics, benzo[a]pyrene, indol-3-carbinol, type I/II interferons
Citation: Schmidt JR, Haupt J, Riemschneider S, Kämpf C, Löffler D, Blumert C, Reiche K, Koehl U, Kalkhof S and Lehmann J (2023) Transcriptomic signatures reveal a shift towards an anti-inflammatory gene expression profile but also the induction of type I and type II interferon signaling networks through aryl hydrocarbon receptor activation in murine macrophages. Front. Immunol. 14:1156493. doi: 10.3389/fimmu.2023.1156493
Received: 01 February 2023; Accepted: 09 May 2023;
Published: 23 May 2023.
Edited by:
Marianna Kulka, University of Alberta, CanadaReviewed by:
Christoph F. A. Vogel, University of California, Davis, United StatesCopyright © 2023 Schmidt, Haupt, Riemschneider, Kämpf, Löffler, Blumert, Reiche, Koehl, Kalkhof and Lehmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jörg Lehmann, am9lcmcubGVobWFubkBpemkuZnJhdW5ob2Zlci5kZQ==
†These authors have contributed equally to this work and share first authorship
‡ORCID: Stefan Kalkhof, orcid.org/0000-0001-6121-7105
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.