
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 30 May 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1155935
 Altan Rentsendorj1
Altan Rentsendorj1 Koen Raedschelders2
Koen Raedschelders2 Dieu-Trang Fuchs1Julia Sheyn1Vineet Vaibhav2Rebecca A. Porritt3,4
Dieu-Trang Fuchs1Julia Sheyn1Vineet Vaibhav2Rebecca A. Porritt3,4 Haoshen Shi1Jargalsaikhan Dagvadorj5
Haoshen Shi1Jargalsaikhan Dagvadorj5 Juliana de Freitas Germano2Yosef Koronyo1
Juliana de Freitas Germano2Yosef Koronyo1 Moshe Arditi2,4
Moshe Arditi2,4 Keith L. Black1
Keith L. Black1 Bhakta Prasad Gaire1Jennifer E. Van Eyk2Maya Koronyo-Hamaoui1,3,6*
Bhakta Prasad Gaire1Jennifer E. Van Eyk2Maya Koronyo-Hamaoui1,3,6*Introduction: Osteopontin (OPN; also known as SPP1), an immunomodulatory cytokine highly expressed in bone marrow-derived macrophages (BMMΦ), is known to regulate diverse cellular and molecular immune responses. We previously revealed that glatiramer acetate (GA) stimulation of BMMΦ upregulates OPN expression, promoting an anti-inflammatory, pro-healing phenotype, whereas OPN inhibition triggers a pro-inflammatory phenotype. However, the precise role of OPN in macrophage activation state is unknown.
Methods: Here, we applied global proteome profiling via mass spectrometry (MS) analysis to gain a mechanistic understanding of OPN suppression versus induction in primary macrophage cultures. We analyzed protein networks and immune-related functional pathways in BMMΦ either with OPN knockout (OPNKO) or GA-mediated OPN induction compared with wild type (WT) macrophages. The most significant differentially expressed proteins (DEPs) were validated using immunocytochemistry, western blot, and immunoprecipitation assays.
Results and discussion: We identified 631 DEPs in OPNKO or GA-stimulated macrophages as compared to WT macrophages. The two topmost downregulated DEPs in OPNKO macrophages were ubiquitin C-terminal hydrolase L1 (UCHL1), a crucial component of the ubiquitin-proteasome system (UPS), and the anti-inflammatory Heme oxygenase 1 (HMOX-1), whereas GA stimulation upregulated their expression. We found that UCHL1, previously described as a neuron-specific protein, is expressed by BMMΦ and its regulation in macrophages was OPN-dependent. Moreover, UCHL1 interacted with OPN in a protein complex. The effects of GA activation on inducing UCHL1 and anti-inflammatory macrophage profiles were mediated by OPN. Functional pathway analyses revealed two inversely regulated pathways in OPN-deficient macrophages: activated oxidative stress and lysosome-mitochondria-mediated apoptosis (e.g., ROS, Lamp1-2, ATP-synthase subunits, cathepsins, and cytochrome C and B subunits) and inhibited translation and proteolytic pathways (e.g., 60S and 40S ribosomal subunits and UPS proteins). In agreement with the proteome-bioinformatics data, western blot and immunocytochemical analyses revealed that OPN deficiency perturbs protein homeostasis in macrophages—inhibiting translation and protein turnover and inducing apoptosis—whereas OPN induction by GA restores cellular proteostasis. Taken together, OPN is essential for macrophage homeostatic balance via the regulation of protein synthesis, UCHL1-UPS axis, and mitochondria-mediated apoptotic processes, indicating its potential application in immune-based therapies.
Osteopontin (OPN), encoded by the secreted phosphorylated protein 1 (Spp1) gene, is a multifaceted matricellular glycoprotein secreted by various immune cells, such as macrophages and T cells (1–3). OPN is an immunomodulatory cytokine that is highly expressed by bone marrow-derived macrophages (BMMΦ) and regulates diverse cellular immune responses, including migration, communication, and immunological responses (4–6). Notably, OPN has dual roles in peripheral immune cells, influencing both inflammatory and anti-inflammatory responses depending on the acute or chronic inflammatory microenvironment. Although previous studies have reported that OPN is associated with type 1 pro-inflammatory macrophage and T cell polarization (4, 7), other reports demonstrate that OPN is associated with macrophage and T cell polarization towards anti-inflammatory phenotypes (6, 8–10). In addition, macrophage-derived OPN induces an anti-inflammatory immune response during Cryptococcus neoformans infection (11). Furthermore, a recent report revealed that OPN can reduce inflammation, tissue injury, and bacterial loads during concurrent pneumococcal infection in a murine model (10). These studies suggest dual roles for OPN in regulating peripheral immune functions, and strict modulation of OPN function is crucial to accomplishing optimal immune responses.
In the context of the central nervous system (CNS), OPN has been shown to be a critical regulator of neuroprotection in several neurological disorders, such as brain ischemia, stroke, and traumatic brain injury (12–14). Notably, macrophages are the key regulators of immune responses due to their rapid and diverse activities. Macrophages exhibit high plasticity and adaptability to various adverse conditions, which promote repair and restore tissue homeostasis (15–18). Hence, regulating macrophage OPN could have a significant impact on various peripheral and CNS inflammatory conditions.
We recently reported that stimulating BMMΦ with glatiramer acetate (GA; generic name Copaxone®), an FDA-approved drug for the treatment of the neurological autoimmune disease, relapsing-remitting multiple sclerosis, upregulates OPN expression, promoting an anti-inflammatory response (6). Our in vivo and in vitro studies indicated that GA-stimulation of macrophages substantially induces their ability to clear pathogenic forms of amyloid β-protein (Aβ), including Aβ40 and Aβ42 fibrils. Moreover, GA treatment protected both synapses and cognitive function in transgenic murine models of Alzheimer’s disease (AD) (19, 20). We found that GA immunomodulation can lead to increased cerebral infiltration of monocytes, which are directly involved in Aβ plaque clearance (6, 19–22). Furthermore, in BMMΦ cultures, GA exposure improved phagocytosis of fibrillar Aβ via increased expression of surface scavenger receptors (i.e., CD36, Scara-1). These receptors recognize and bind Aβ fibrils to facilitate their uptake (19, 20). Importantly, GA promoted a shift into anti-inflammatory microglia and monocyte/macrophage phenotypes, as evidenced by marked increases in anti-inflammatory interleukin (IL)-10, insulin-like growth factor-1 (IGF-1), matrix metalloproteinase-9 (MMP-9), and OPN production, as well as decreases in pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and IL-12 release (6, 19, 23, 24).
Recent proteomic studies on microglia and macrophages have identified distinct protein expression profiles of pro-inflammatory versus anti-inflammatory-type macrophages related to cell metabolism and signaling (25–29). Additionally, a few whole proteome analyses have been conducted to decipher GA’s effects and mechanisms of action (30, 31). In the current study, we applied quantitative mass spectrometry (MS)-based proteomics profiling to analyze the effects of OPN deficiency vs. GA immune modulation on macrophage protein networks and functional pathways. Top up and down-regulated proteins were defined as differentially expressed proteins (DEPs), whereas protein expression networks were examined using functional clustering analysis. Then, proteins were enriched for canonical pathways to identify specific signaling pathways. Upstream regulator analysis was performed to identify molecules that can influence the changes in downstream signaling. We comprehensively identified and quantified proteins from OPN-deficient and GA-stimulated BMMΦ relative to untreated macrophages through proteomics. In addition, key identified DEPs were validated in vitro by employing immunocytochemistry, immunoprecipitation, Western blot, and mitochondrial tracing analyses. Findings from this study provide new insights into the immune mechanisms of OPN deficiency in macrophages, affecting cellular homeostasis and survival via reduced ubiquitin-proteosome system (UPS) functions.
OPN knock-out (OPNKO) mice from the B6.129S6 (Cg)-Spp1tm1Blh/J strain (Jackson laboratories stock #004936|OPNKO) and aged-matched wild type (WT) controls (Jackson laboratories) were used for in vitro experiments. Animal experiments were performed in accordance with Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC) guidelines under an approved protocol.
Bone marrow-derived macrophages (BMMФ) were collected from 8- to 16-week-old WT or OPNKO mice and cultured as previously described (6). On the sixth day of culture, cells were either plated in a 24-well plate on glass coverslips for immunocytochemistry (1x105 cells per well; 3-6 wells per condition) or a 6-well plate for protein assays (3x105 cells per well; 3 wells per condition). Next, the wells were treated with either 30 µg/mL GA (TEVA; Glatopa), 50 ng/mL recombinant OPN (Peprotech #120-35), 10 µM minocycline (Millipore sigma #M9511), or stealth OPN siRNA (Invitrogen) for 24 hours. The control groups were the untreated WT and the GA-treated OPNKO cells. Each experiment was repeated 2-4 times. Cells were washed three times with 1X PBS before fixation with 4% paraformaldehyde (15 min at room temperature) or ice-cold methanol at -20°C. Then, the cells were rinsed 3 times with 1X PBS and stored at 4°C until further analysis.
Cells plated on glass coverslips were blocked with serum-free protein blocking (Dako Cytomation) for 30 min at room temperature, then hybridized with primary antibodies overnight at 4°C (see Table S1). On the following day, cells were incubated with appropriate secondary polyclonal antibodies for 1 hour at room temperature. Coverslips were then mounted using ProLong Gold with DAPI (Molecular Probes, Life Technologies). Negative controls were processed using the same protocol with the omission of the primary antibody to assess non-specific labeling (Figure S4A).
In cases where cells were stained in parallel with Dihydroethidium (DHE; Sigma-Aldrich #D7008), fluorescent dye was added after the secondary antibody for 30 min at 37°C (10 μM final concentration in PBS) before mounting.
Cells treated with Mitotracker™ (Thermofisher #M7512) were incubated for 15-30 min with 250 μl of the fluorescent dye at 37°C, then washed three times with 1X PBS before fixation with 4% PFA (15 min at room temperature). Cells were rinsed three times with 1X PBS and mounted for microscopy analysis.
Representative images were captured with a Carl Zeiss Axio Imager Z1 fluorescence microscope equipped with ApoTome, AxioCam MRm, and AxioCam HRc cameras (Carl Zeiss MicroImaging, Inc.) using the same exposure time for each channel across all images. Images were analyzed using ImageJ software (NIH), converted to grayscale, and processed with the same post-acquisition threshold for analysis. Five to ten images from each well (n=3-5 wells), covering an average of 100 cells per image, were analyzed.
Cells plated on 6-well plates were lifted with 2 mM EDTA-PBS, collected in tubes, and pelleted (1000 rpm for 5 min). Cell pellets were lysed in RIPA lysis buffer (Thermofisher Scientific, #89900) supplemented with 1% protease inhibitor (Calbiochem #539131) and stored at -80°C until further analysis. Protein concentration was determined using a BCA Protein Assay Kit (Thermofisher Scientific, #23225). Equal amounts of total protein samples were electrophoretically separated onto 4% to 20% Tris-glycine gels (Invitrogen, #XP04205BOX), then transferred to nitrocellulose membranes, blocked for 1 hour at room temperature in Tris-buffered saline with Tween 20 (TBST; 10 mmol/L Tris-HCl buffer, pH 8.0, 150 mmol/L NaCl, and 0.1% Tween 20) containing 5% BSA, and hybridized with appropriate primary antibodies overnight at 4°C. After four washes in TBST, membranes were incubated with HRP-conjugated secondary antibody for 1 hour at room temperature, then washed again four times in TBST prior to development with a chemiluminescence substrate kit (Thermofisher Scientific, #34580). Images were taken on an iBright imaging system (iBright imaging system; Thermofisher Scientific). Protein expressions were analyzed using ImageJ software and normalized to β-Actin or GAPDH. Two to eight experimental repeats were performed for each experiment. Representative blots are shown for each protein.
Cells were lysed in lysis buffer (Pierce™ IP Lysis Buffer, Cat. 87787) on ice for 20 min, and lysates were centrifuged at 8000 rpm for 10 min. The supernatant was collected and incubated with 50 ml of IP beads (TrueBlot Anti-Rabbit) for 30 min on a rocking platform at 4°C. After incubation, the supernatant was collected by centrifugation and added 5 mg of the primary antibody and incubated on a rocking platform overnight at 4°C. After overnight incubation, 50 ml of IP beads was added to the supernatant, and after 1 hour the beads were collected by centrifugation and washed with lysis buffer. Beads were then loaded with 2X loading buffer and run on sodium dodecyl-sulfate polyacrylamide gel electrophoresis.
Cell pellet processing for MS as outlined in Parker et al. (32). Briefly, proteins were reduced with 5 mm TCEP, alkylated with iodoacetamide, digested with sequencing grade modified trypsin (Promega, Madison, WI) at a 1:100 ratio of enzyme/substrate, and the digestion was stopped by the addition of formic acid. Tryptic peptides were desalted using reverse phase cartridges Sep-Pak C18 (Waters, Milford, MA) according to the following procedure. The tryptic peptides were dried using a vacuum centrifuge and then resolubilized in 0.1% formic acid with synthetic iRT calibration peptides (Biognosys, Schlieren, Switzerland) at a 1:20 v/v ratio.
The tryptic peptides were separated using a ChromXP column ((3 um particle size, 150 um x 10 cm) Eksigent technologies) from 3–35% acetonitrile over 60 min gradient using an Eksigent NanoLC Ultra 2D Plus HPLC system coupled to a 5600 TripleTOF mass spectrometer (AB Sciex, Framingham, MA). To build the sample-specific peptide library, the tryptic peptides data was generated using data-dependent acquisition as outlined in Parker et al. (32). Briefly, the top 20 most intense MS1 precursors (collected between 360 and 1460 m/z for 250 msec) with charge states between 2 and 5 were selected for MS2 fragmentation, with a 15 sec exclusion window. Fragment MS2 ions were collected for 100 ms across a 50–2000 m/z range. The raw MS spectra were converted to mzML using AB Sciex converter (v1.3) and subsequently converted to mzXML using msconvert (ProteoWizard, v3.04.238). They were then searched parallelly using OMSSA and X!Tandem algorithms Swiss-Prot–reviewed canonical mouse FASTA database (mouse database, 2013) appended with Biognosys iRT peptides for retention time alignment (Biognosys, Schlieren, Switzerland) and randomized decoy sequences. Search engine results were then converted to pepXML format using omssa2pepXML (v2.1.9) and Tandem2XML (v4.6.0). Peptide spectral match probability scoring was modeled in PeptideProphet (v4.6.0), and the resulting interact.pepXML files of the two search engines were combined in iProphet (v4.6.0). A Peptide assay library was generated with SpectraST (v4.0) from the identified peptides with a Peptide Prophet probability > 0.95. The resulting spectrast.splib file was submitted as input to the custom spectrast2spectrast_irt.py converter script that was used to align RT using iRT peptides. The spectral libraries were then formatted for OpenSWATH first using the custom script spectrast2tsv.py, followed by the OpenSWATH tool ConvertTSVtoTraML. Finally, OpenSwathDecoyGenerator was used to append shuffled decoys to the full assay library.
Each individual sample was analyzed by DIA-MS on the tripleTOF 5600 mass spectrometer in data-independent mode as outlined in Parker et al. (32), and Howlenski et al. (33). In DIA-MS, MS1 was collected with 100 m/z windows across a 400–1200 m/z range, with 0.5 m/z overlap at either end of a given window. Data were analyzed using OpenSWATH workflow (REF). Briefly, raw intensity data for peptide fragments were extracted from the DIA files using the open source openSWATH workflow against the custom-generated library. Target and decoy peptides were then extracted, scored, and analyzed using the mProphet algorithm to determine scoring cut-offs consistent with 1% FDR. The peak group extraction data from each DIA file was combined using the “featurealignment” script, which performed data alignment and modeling analysis across the experimental data set. The normalized transition-level data was then processed using the mapDIA software to perform pairwise comparisons between groups at the peptide level.
Detectable protein hierarchies displayed as heatmaps and principal component analysis (PCA) were created by using ClustVis (https://biit.cs.ut.ee/clustvis/). Volcano plots were created using Prizm 9. Pie chart of Protein annotation through evolutionary relationship (PANTHER) protein classification analysis was created using http://pantherdb.org/geneListAnalysis.do. Data was analyzed using IPA (Ingenuity Pathway Analysis, Qiagen (https://digitalinsights.qiagen.com)). Differentially expressed genes (with corresponding fold-changes and p values) were incorporated in canonical pathways and upstream regulators analyses and were used to generate diagrams.
The 1.2-fold change difference and p ≤ 0.05 were selected as the cutoff since modest numbers of DEPs were observed between the different experimental groups.
Data have been deposited to the MassIVE repository under number MSV000091221 (https://urldefense.com/v3/:http://massive.ucsd.edu/ProteoSAFe/status.jsp?task=05165f2ddf9f4eecbf5465d94e0c3607:;!!KOmnBZxC8_2BBQ!xzewe2MHK3WeGKFzdYro-c2tOjsxpz26J1s-qZmQbIwJkyhY4DBmbuUHbC2NQ0bPQL3yuRBxBpYSM-lz_ervsQaZsDw$). Proteome Exchange ID: PXD039950.
Experimental data were analyzed using GraphPad Prism (GraphPad Software). One-way ANOVA with Tukey’s multiple comparison test was performed in case of three group comparisons. Two-tailed unpaired Student’s t-tests were used for two-group comparisons. The results are shown as means ± standard errors of the mean (SEM). Degree of significance between groups is represented as follows: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. A p-value lower than 0.05 was considered significant.
We previously found that GA stimulation upregulates OPN expression in BMMΦ, promoting an anti-inflammatory macrophage phenotype, whereas OPN deletion in BMMΦs results in pro-inflammatory phenotypes (6). We explored in vitro molecular network effects of OPN deficiency versus GA-mediated OPN induction in BMMΦ using applied MS-based quantitative proteomics profiling on protein extracts from OPNKO versus GA-stimulated BMMΦ compared to untreated wild type (WT) control BMMΦ. The scheme of the experimental procedure, which encompasses biochemical and MS analysis, data curation, and profiling followed by data validation, is outlined in Figure 1A. In brief, bone marrow was isolated from WT or OPNKO mice and cultured for 7 days in MCSF-enriched media to differentiate into macrophages. On day 6, one group of WT cells underwent overnight treatment with GA. On day 7, the cells were collected, pelleted, and processed for quantitative proteomic and bioinformatics analyses, and followed by immunocytochemical (ICC) and Western blot (WB) validation experiments.
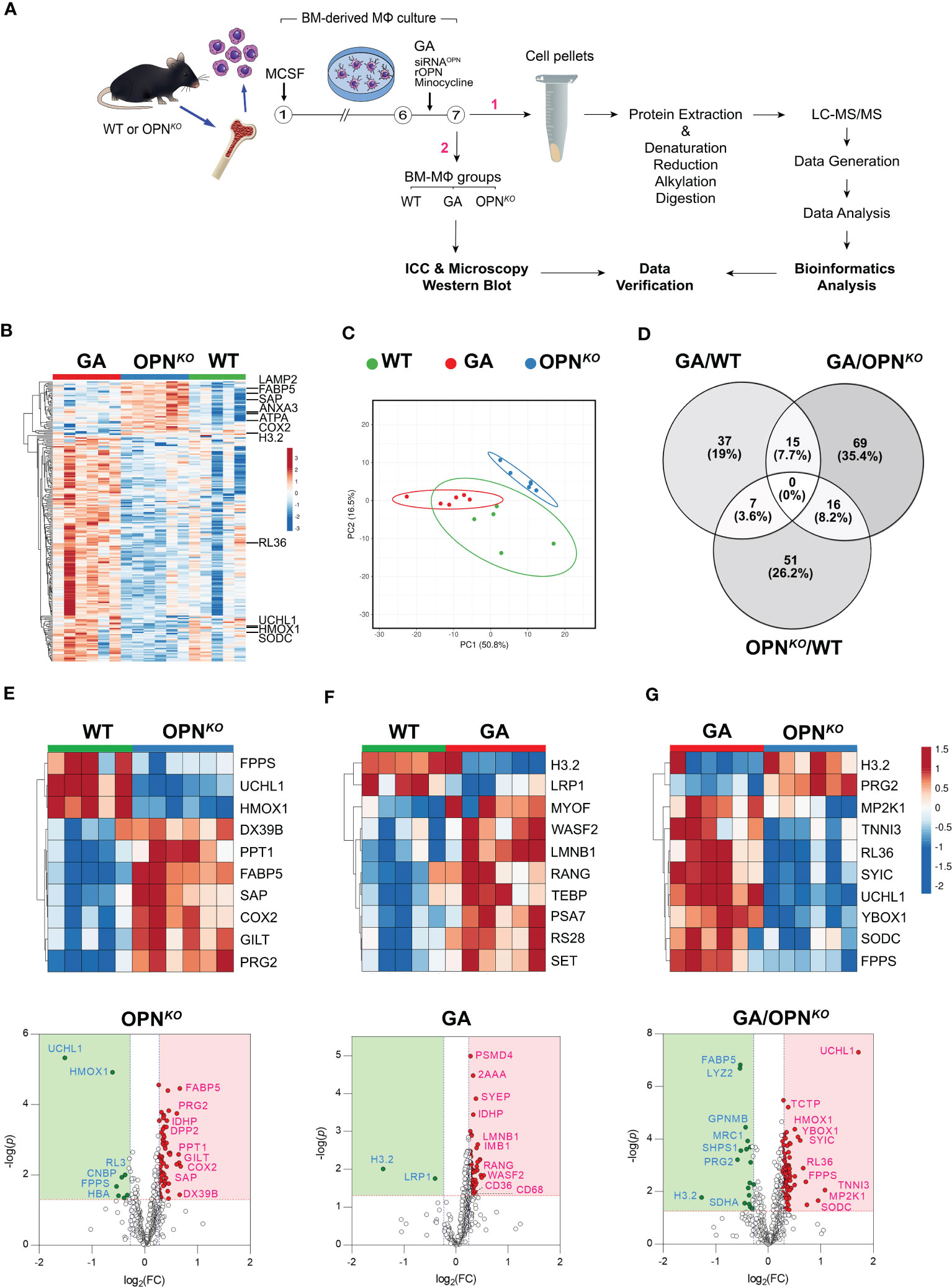
Figure 1 Proteome signatures of OPN-deficient and GA-stimulated macrophages. (A) Schematic illustration of experimental workflow for the proteomics profiling and validation studies. (B) Heatmap with hierarchical clustering analysis of the most abundant up (red)- and down (blue)-regulated DEPs across three experimental groups. (C) Hierarchical clustering of the top up and downregulated 225 DEPs by principal component analysis (PCA performed with ClustVis). Unit variance scaling is applied to rows; SVD with imputation is used to calculate principal components. Prediction ellipses are such that with probability of 0.95, a new observation from the same group will fall inside the ellipse. (D) Venn diagram showing the number and percentage (%) of overlapping DEPs according to statistical significance (p<0.05) and 1.2-fold change threshold criteria in the three analyzed groups. The number of common DEPs between pair groups is also shown. (E–G) Heatmaps and volcano plots of the top 10 most DEPs (FDR < 0.05, ranked by FC) generated by differential analysis of the proteome profiles between (E) OPNKO and (F) GA versus WT macrophages, and (G) GA versus OPNKO macrophages. Significantly upregulated proteins are shown as red dots and down-regulated proteins as green dots. The proteins with fold change >1.2 and p value <0.05 were considered significantly differentially expressed.
Mass spectrometry analysis identified 2505 peptides corresponding to 631 DEPs amongst the three BMMΦ cell groups (Table S2). Of these, 249 proteins were significantly altered exceeding our 1.2-fold change cutoff (Table 1). In OPN-deficient (OPNKO) versus WT BMMΦ, 66 proteins were upregulated, while 16 were downregulated. In GA-treated compared to WT BMMΦ, 56 proteins were upregulated, and only 2 proteins were significantly downregulated. DEPs numbers were further enriched in comparing between GA-stimulated and OPNKO BMMΦ groups, in which 84 proteins were upregulated and 25 were downregulated (Table 1). A heatmap with hierarchical clustering analysis of the most abundant up (red)- and down (blue)-regulated DEPs across three experimental groups exhibited distinct cluster profiles for each group (Figure 1B). Prediction ellipses by principal component and Venn diagram analyses showed that each group has a discrete protein expression profile, and notably the OPNKO group had the least overlapping profile compared to the GA-treated BMMΦ group (Figures 1C, D).
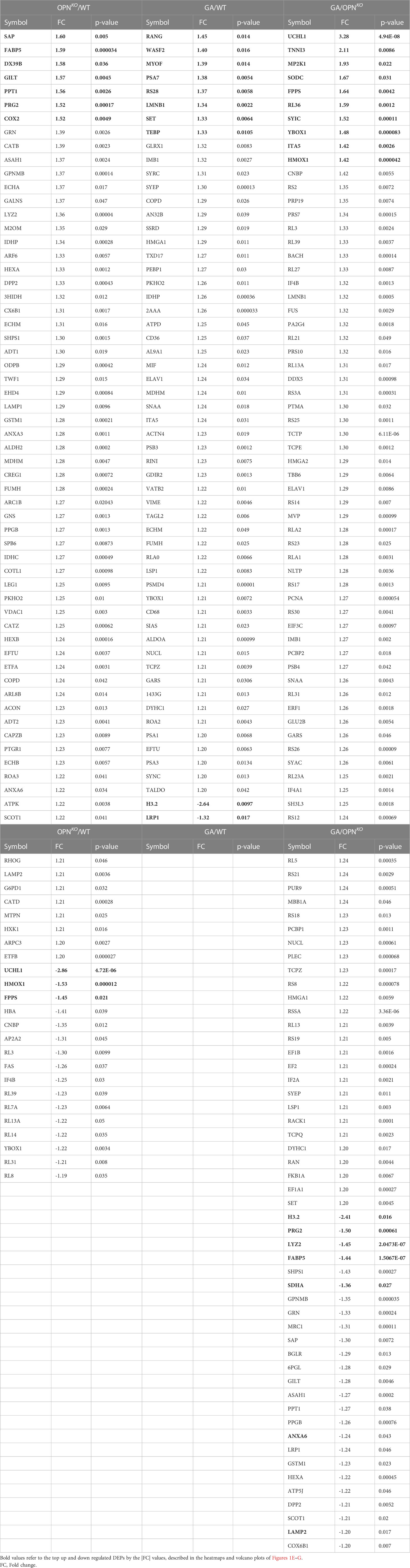
Table 1 Mass spectrometry data showing significantly differentially expressed proteins (P<0.05).
Among the top 10 DEPs in OPNKO vs. WT BMMΦ, we focused on the two most significantly downregulated proteins (Figure 1E). These include the ubiquitin carboxyl-terminal hydrolase isozyme 1 (UCHL1; 2.86-fold downregulation; Table 1), a deubiquitinating enzyme of the UPS system with an important role in maintaining ubiquitin homeostasis and protein synthesis turnover (34); Heme oxygenase 1 (HMOX1; 1.53-fold downregulation), a heme degrading enzyme that produces antioxidant and anti-inflammatory compounds (35). The Volcano plot (Figure 1E) highlights additional downregulated proteins that include the 60S ribosomal proteins RL3, cellular nucleic acid-binding protein (CNBP), and Farnesyl pyrophosphate synthase (FPPS). These data indicate that macrophages lacking OPN exhibit deficiencies in proteins involved in ubiquitin-proteosome system and translation.
Conversely, among the top 10 DEPs in OPNKO vs. WT BMMΦ, we detected seven upregulated proteins: including signaling lymphocytic activation molecule-associated protein (SAP), fatty acid-binding protein 5 (FABP5), DExD-Box Helicase 39B (DX39B), gamma-interferon-inducible lysosomal thiol reductase (GILT), palmitoyl-protein thioesterase 1 (PPT1), proteoglycan 2 (PRG2), and cyclooxygenase (COX2) (Figure 1E). These findings implicate the involvement of OPN in lysosomal organization, mitochondrial functions, oxidative stress, as well as metabolic and proteolytic responses in BMMΦ.
In GA-treated macrophages, the top 10 DEPs highlight the histone H3.2 variant as markedly downregulated by 2.64- and 2.41-folds in GA-stimulated macrophages compared to WT and OPNKO macrophages, respectively (Figures 1F, G heatmaps, Table 1). The H3.2 histone protein is a DNA replication-dependent nucleosome assembly protein and associated with replication and/or cell division (36, 37). Volcano plot analysis further shows that the low-density lipoprotein receptor-related protein 1 (LRP1), which is involved in the positive regulation of lysosomal protein catabolic process and cell death (38, 39), was downregulated by GA treatment vs. WT controls (1.32-folds; Table 1, Figure 1F). These results suggest that GA can reverse detrimental lysosomal catabolism and cell death-mediated signaling. Additionally, two other proteins, Lyz2 (involved in cytolysis) and SDHA (a mitochondrial electron transport protein) were downregulated following GA stimulation compared to OPNKO BMMΦ (Figure 1G).
Notably, as described in Table 1 and Figure 1F, G volcano plots, other important proteins were upregulated in macrophages by GA stimulation, including the scavenger receptors CD36 and CD68, which are involved in facilitating uptake and clearance of Aβ and were also previously shown to increase expression following GA stimulation (6, 19, 20).
Furthermore, several anti-inflammatory and antioxidant proteins were upregulated by GA treatments. These include UCHL1, superoxide dismutase C (SODC), HMOX1, troponin I3 (TNNI3), Y-box binding protein 1 (YBox1), which negatively regulates of apoptotic processes and promotes cell proliferation, and ribosomal proteins like RL36 (Figures 1F, G, Table 1). Importantly, the GA-influenced topmost upregulated proteins were anti-inflammatory and antioxidant-related proteins (Table 1). Taken together, GA-immunomodulation downregulated lysosomal and apoptotic cell death-related proteins while upregulating anti-inflammatory and antioxidant proteins, at least partially through increased OPN production in macrophages.
What are the leading biological pathways that OPN deficiency triggers in macrophages? To address this question, we studied the relative distribution of functionally classified DEPs in OPN-deficient and GA-immunomodulated BMMΦ. Pie charts based on the PANTHER functional classification, display the proportions between the 19 PANTHER clusters for each OPNKO vs. WT as well as the GA vs. OPNKO BMMΦ (Figure 2A). When we compared these 2 pie charts, the most substantial difference occurred in the ‘translational protein’ class with 6.6-fold differences, upregulated from 2.7% in OPNKO to 18% in GA-treated BMMΦ. While the most notable protein cluster changes in OPNKO BMMΦ were related to metabolic and protein modifying enzymes, in GA-treated BMMΦ the prominent clusters were related to metabolites, translational and cytoskeletal proteins. Further comparison between the pie clusters of GA vs. OPNKO BMMΦ revealed more clusters related to defense immunity, chromatin binding, and chaperone and cell adhesion class proteins; while fewer clusters were related to intercellular signaling, extracellular matrix, and transmembrane signal receptor following GA treatment in macrophages (Figure 2A).
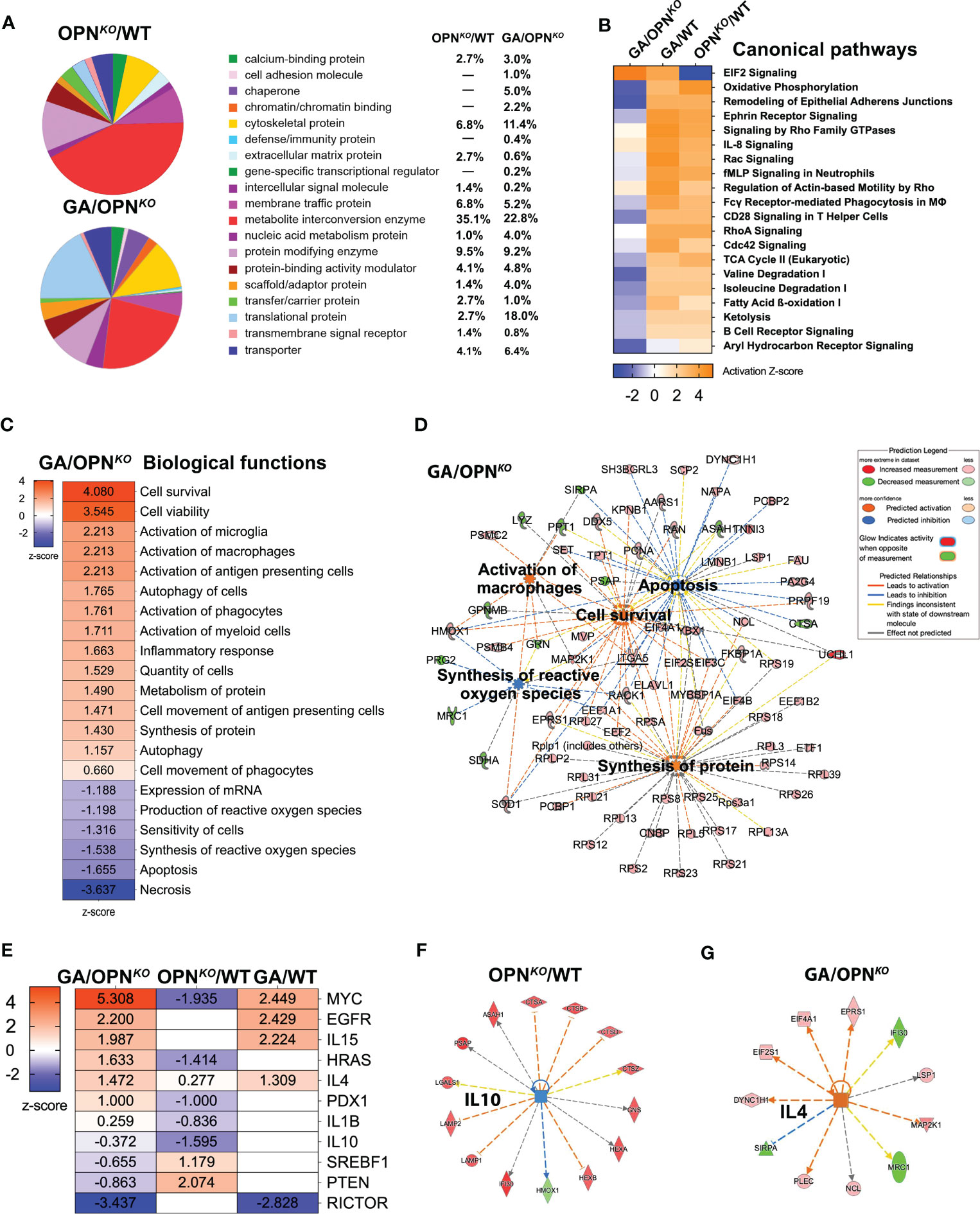
Figure 2 Functional and canonical pathways in OPN-deficient and GA-stimulated macrophages. (A) PANTHER analysis Pie chart by Protein Class distribution in percentages showing fraction and percentage of significant DEPs (up- or downregulated proteins) grouped by protein class category in the OPNKO/WT and GA/OPNKO. (B) IPA was performed to identify canonical pathways, most differentially regulated pathways in all three groups. (C) IPA of top up and down-regulated biological functions in GA/OPNKO group and (D) network displaying five important biological functions and their interactions with each other. (E) IPA of upstream regulator analysis in all three groups and network displays of upstream regulators for (F) IL-10 in OPNKO/WT and (G) IL4 in GA/OPNKO.
Next, ingenuity pathway analysis (IPA) identified the principal canonical pathways and downstream signaling in OPN-depleted and GA-stimulated macrophages (Figure 2B). A top altered pathway was inhibited in OPNKO BMMΦ while activated in GA-stimulated, the EIF2 signaling (Figures 2B, S1); which is essential for most forms of translation initiation and protein synthesis (40–42). Another top altered pathway that was activated in OPNKO and conversely inhibited in GA-stimulated BMMΦ was related to oxidative phosphorylation (Figures 2B, S2). Oxidative phosphorylation is a metabolic pathway in mitochondria that is associated with cellular proliferation (43). Protein Interaction Network Extractor (PINE) analysis (Figure S3) displays the top five canonical ontology networks that were identified across the experimental macrophage groups. In alignment with our aforementioned results, the most significantly altered signaling pathways were related to the innate immune system, translation, and translation initiation.
To gain a deeper understanding of the biological pathways that are altered following GA stimulation in macrophages, we conducted IPA for GA vs. OPNKO BMMΦ (Figures 2C, D). The top activated biological pathways were related to cell survival, cell viability (Z scores = 3.54–4.08), and microglia/macrophage activation, cell movement, and protein synthesis (Z scores = 2.21–1.40). In contrast, the top inhibited functions were necrosis (Z-score = -3.637), apoptosis (Z-score = - 1.655), and synthesis of reactive oxygen species (ROS; Z score = -1.538) (Figure 2C). The top interconnecting molecular networks are displayed in Figure 2D. Opposite IPA functional directions of inhibited cell survival, viability, and macrophage activation and activation of necrotic, apoptotic, and ROS markers were detected for OPNKO vs. GA-treated BMMΦ (not shown). These results were further validated by IPA and gene ontology (GO) analyses (Tables 2, 3). Next, we identified the specific upstream regulators across all three comparison groups (Figures 2E–G). Previous literature supports IL-4 as one of the top upstream activated regulators in GA-treated BMMΦ (Z-score = 1.472), promoting an anti-inflammatory phenotype in macrophages (24). IL-10 is another anti-inflammatory cytokine that was a top inhibited upstream regulator of OPNKO BMMΦ (Z-score = -1.595), which supports a pro-inflammatory phenotype for OPN-deficient macrophages.
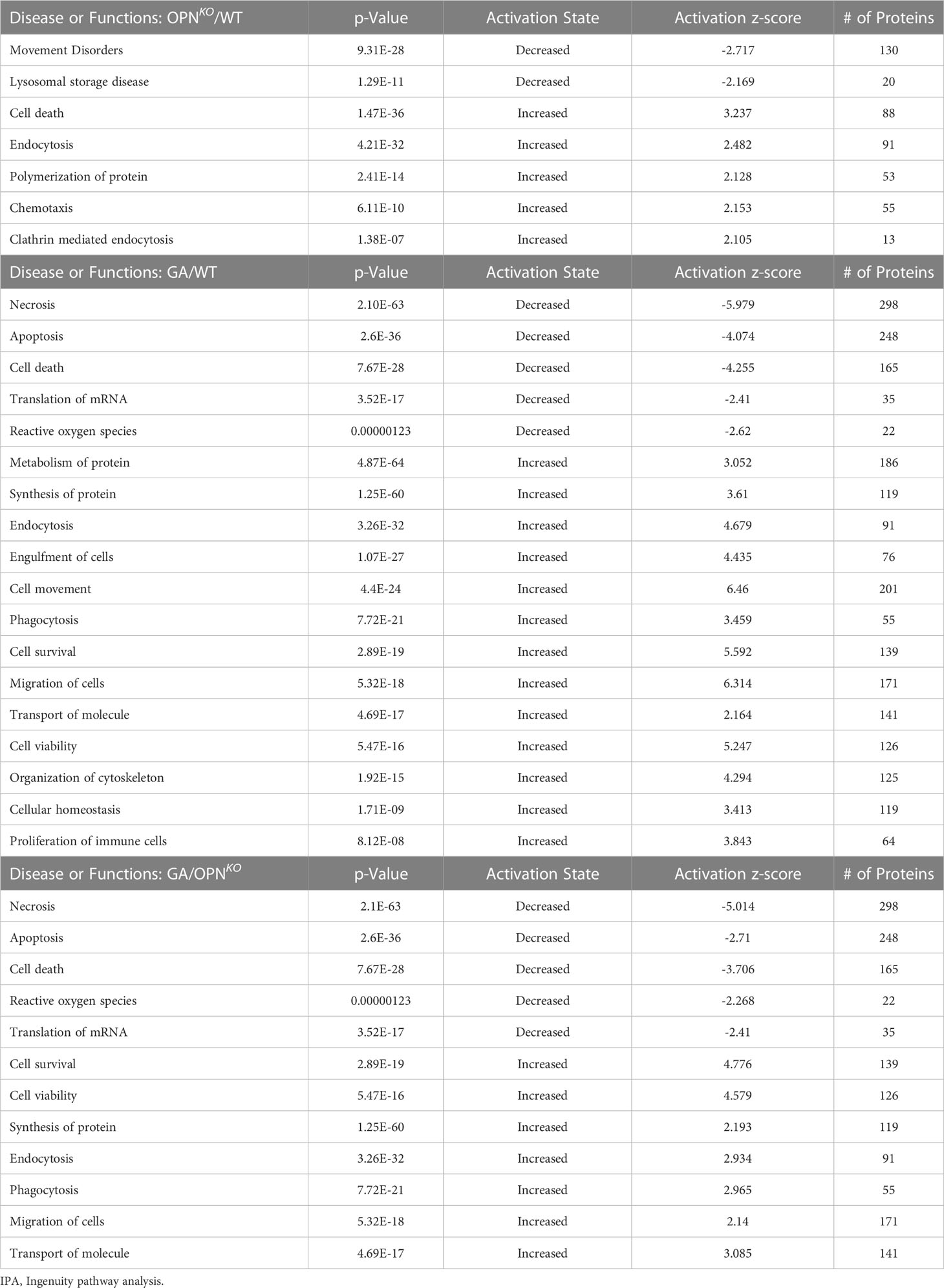
Table 2 Disease or Functions by IPA analysis.
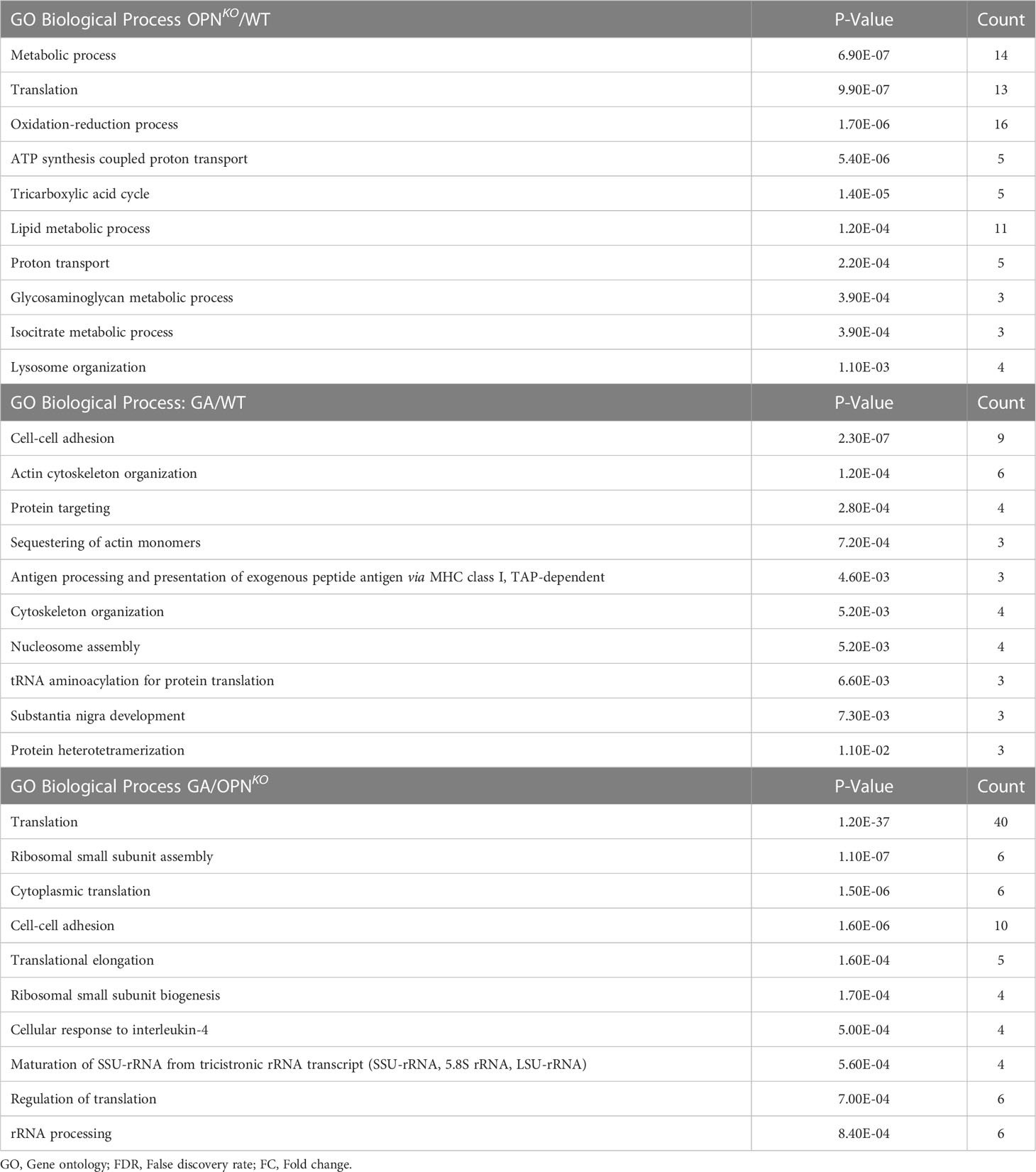
Table 3 Top 10 significant GO Biological processes identified for proteins differentially expressed (FDR<0.05, FC>1.2).
Overall, the combined canonical pathway and network analyses suggests that proteins associated with translation, protein synthesis, metabolism, and apoptosis are oppositely regulated in OPN-deficient and GA-stimulated macrophages, emphasizing their role in cellular homeostasis.
Our proteomics data identified UCHL1 as the most downregulated protein in OPN-deficient BMMΦ (Figure 1E, Table 1) and the most upregulated protein upon GA stimulation compared to OPNKO macrophages (Figure 1G, Table 1). We confirmed these findings by immunocytochemistry and Western blot analyses (Figures 3A–G, S4A–D). These studies revealed that UCHL1 was expressed by CD36-positive BMMΦ cells, and concomitant with enhanced OPN expression, UCHL1 was 2.4-fold upregulated following GA stimulation for 24 hours (Figures 3A–G). On the other hand, along with OPN deficiency in macrophages, UCHL1 expression was nearly absent in OPNKO BMMΦ cultures. Notably, GA-stimulation of OPNKO macrophages did not modify neither OPN nor UCHL1 expression, demonstrating that UCHL1 upregulation by GA is OPN dependent. Additionally, we validated these findings by inhibiting OPN expression using siRNAOPN or minocycline (Figures S4B–D), further suggesting that OPN regulates UCHL1 expression in macrophages even after 2 hours of GA stimulation.
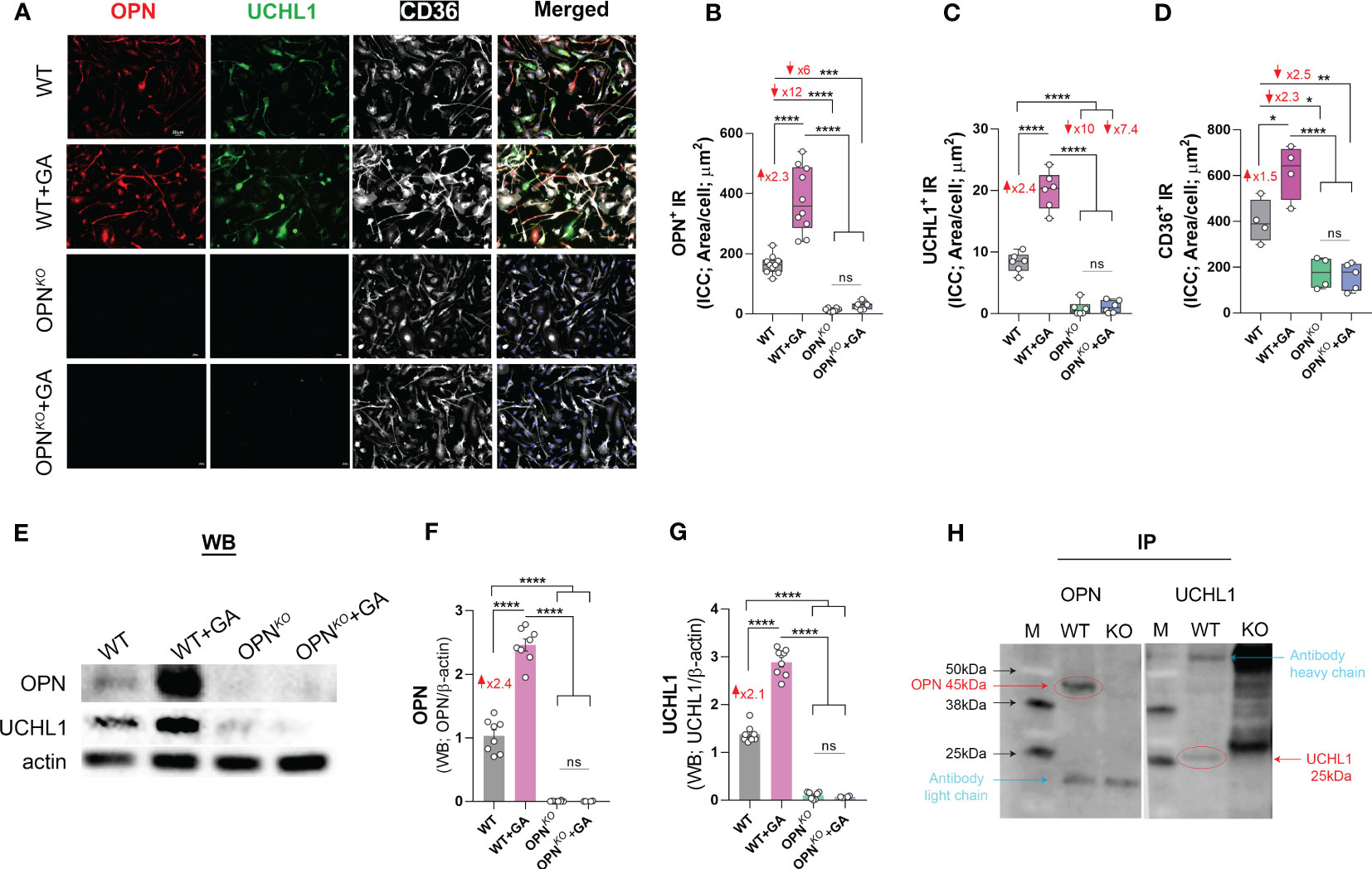
Figure 3 OPN interacts with UCHL1 and regulates its expression in BM-derived macrophages. (A) Representative fluorescent micrographs of BMMΦ cells, either GA-treated or untreated OPNKO or WT, immunostained for OPN (red), UCHL1 (green), CD36 (white) and DAPI (blue for nuclei of cells); scale bar: 20µm. Quantitative analysis of immunoreactive (IR) area of (B) OPN, (C) UCHL1 and (D) CD36 expression in BMMΦs. (E) Western blot (WB) analysis of cell lysates 24 hours after treatment, and quantitative analysis of (F) OPN and (G) UCHL1 bands. (H) Immunoprecipitation (IP) of OPN and UCHL1. IP demonstrates that OPN and UCHL1 interact with each other in protein complex. Individual data points, group means, and SEMs are shown. ns means Not Significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc multiple comparison test.
To explore a possible interaction between OPN and UCHL1 proteins, we performed an immunoprecipitation (IP) and found that both OPN and UCHL1 were pulled down in the same protein complex (Figure 3H). These results suggest that OPN and UCHL1 may directly interact with each other in a protein complex. In conclusion, we identified UCHL1 expression in BMMΦ and found that UCHL1 expression is OPN-dependent.
Given that the proteomic analyses indicated marked changes in translation, lysosomal, and mitochondrial proteins in OPNKO and in GA-stimulated BMMΦ (Figures 4A–F), we aimed to further verify this data by immunolabeling macrophages with antibodies against late lysosomal marker (Lamp1) and mitochondrial markers cytochrome C (CytC) and ATP-β (Figure 4G). ICC quantification in OPNKO BMMΦ showed significant increases in the expression of Lamp1 (2-fold), ATP-β (2.2-fold), and CytC (1.5-fold; Figures 4H–J). Conversely, their expressions were downregulated in GA-treated BMMΦ. These results were further confirmed via Western blot assays. Quantitation of WB bands, including those from the oxidative phosphorylation (OXPHOS) mitochondrial-targeted cocktail containing five antibodies, revealed 1.3-1.5-fold increases in CytC, mitochondrial encoded cytochrome c oxidase I (MTCO1), and ATP synthase complex V (ATP5A) expressions in the OPNKO macrophages, while they were 1.4-2.4-fold reduced in the GA-stimulated macrophages (Figures 4K–M; the OXPHOS gel is displayed in Figure S4E).
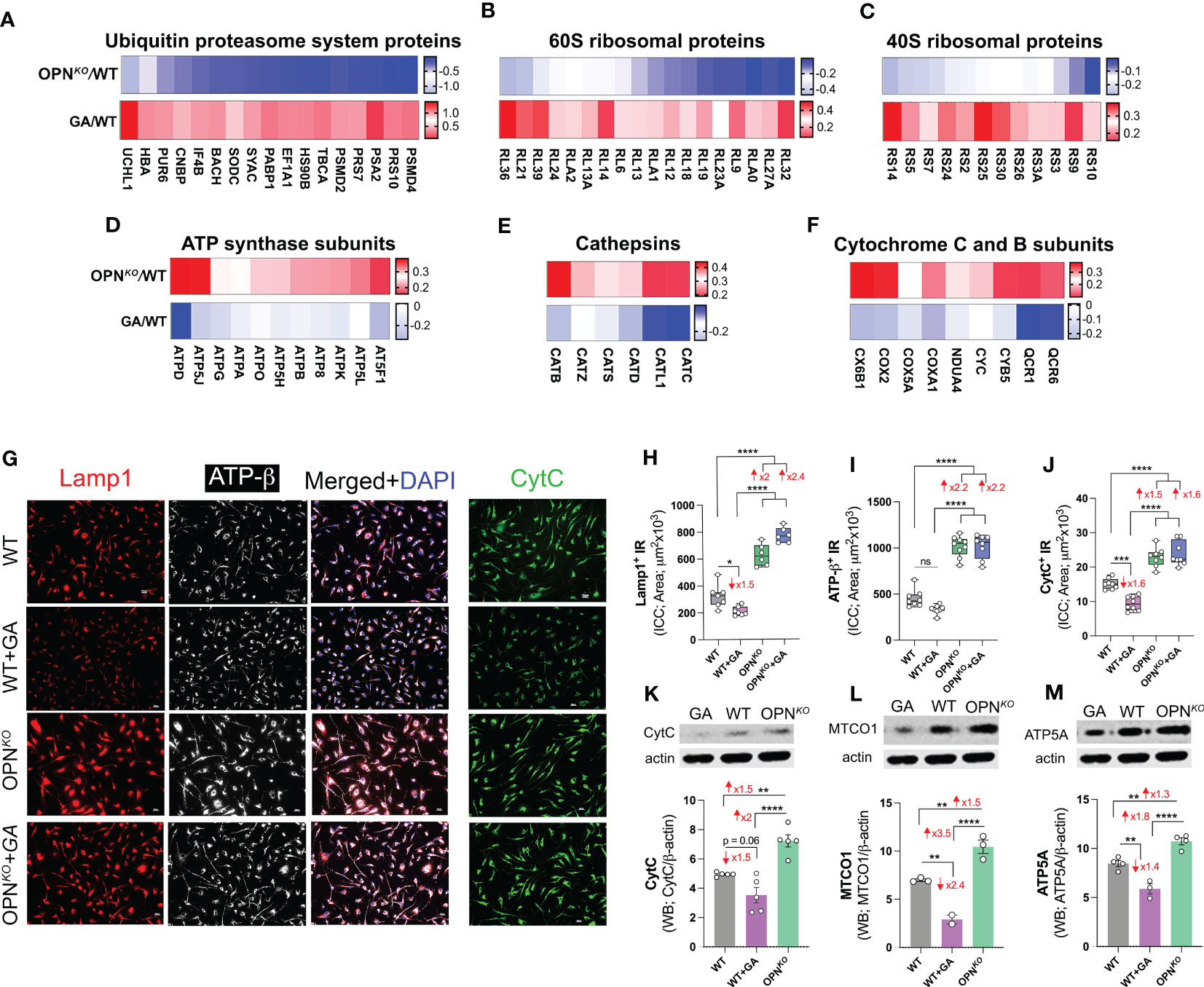
Figure 4 OPN deficiency upregulates lysosomal and mitochondrial markers in macrophages. (A–F) Heatmaps of (A) UPS proteins, (B) 60S ribosomal proteins, (C) 40S ribosomal proteins in GA/WT vs. OPNKO/WT, (D) ATP-synthase subunits, (E) cathepsins and (F) cytochrome C and B subunits in OPNKO/WT vs. GA/WT. (G) Representative fluorescent images of BMMΦ cells, either GA-treated or untreated OPNKO or WT, immunostained for Lamp1+ (red), ATP-β+ (white) and Cytochrome C+ (green); scale bar: 20µm. (H–J) Quantitative analysis of IR of (H) Lamp1, (I) ATP-β and (J) CytC expression in BMMΦs. (K–M) Western blot assay and densitometric analysis of cell lysates using CytC and OXPHOS antibody cocktail containing five mouse antibodies against: CI subunit NDUFB8, CII SDHB, CIII-Core protein 2 UQCRC2, CIV subunit I MTCO1 and CV alpha subunit ATP5A. Full gel is displayed in Figure S4E. Densitometric analysis of (K) CytC, (L) MTCO1 and (M) ATP5A bands. Individual data points, group means, and SEMs are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc multiple comparison test.
Collectively, OPN deficiency was associated with lysosomal and mitochondrial imbalance in BMMΦ, suggesting a role for OPN-primed UCHL1 in regulating mitochondrial functions and cellular homeostasis. Combined with our bioinformatics analysis (Figure S2), we revealed that this dual OPN-UCHL1 deficiency is associated with oxidative phosphorylation and mitochondrial dysfunctions in macrophages.
Given that OPN-primed UCHL1 expression was linked to cell homeostasis and protection, we assessed whether such protective effects in macrophages were, at least in part, associated with the expression of heme oxygenase 1 (HMOX1) and the superoxide dismutase (SODC, encoded by the SOD1 gene). In proteomics, we found that HMOX1 and SOD1 are two top upregulated DEPs in GA-stimulated vs. OPNKO BMMΦ, which were downregulated in OPNKO BMMΦ (Figures 1G, E, Table 1). HMOX1 is a heme degrading enzyme that catalyzes the production of antioxidant and anti-inflammatory compounds and provides beneficial effects for cellular homeostasis and switching macrophages to an anti-inflammatory phenotype (35, 44–46). HMOX1 deficiency renders macrophages sensitive to cell death. Our quantitative ICC analysis showed a substantial 1.9-fold reduction of HMOX1 in OPNKO vs. WT BMMΦ (Figures 5A, B). SOD1 is another potent antioxidant that has pleiotropic biological functions to counter oxidative stress in mitochondria (47, 48). We further reaffirmed the proteomics findings of SOD1 in OPNKO BMMΦ by employing ICC analysis (Figures 5A, C). Likewise, our WB analysis showed that the expression of these two antioxidant enzymes (SOD1 and HMOX1) were upregulated upon GA stimulation (1.3-1.4-fold) and decreased in OPNKO compared to WT BMMΦ (1.3- and 2-fold, respectively; Figures 5D, E).
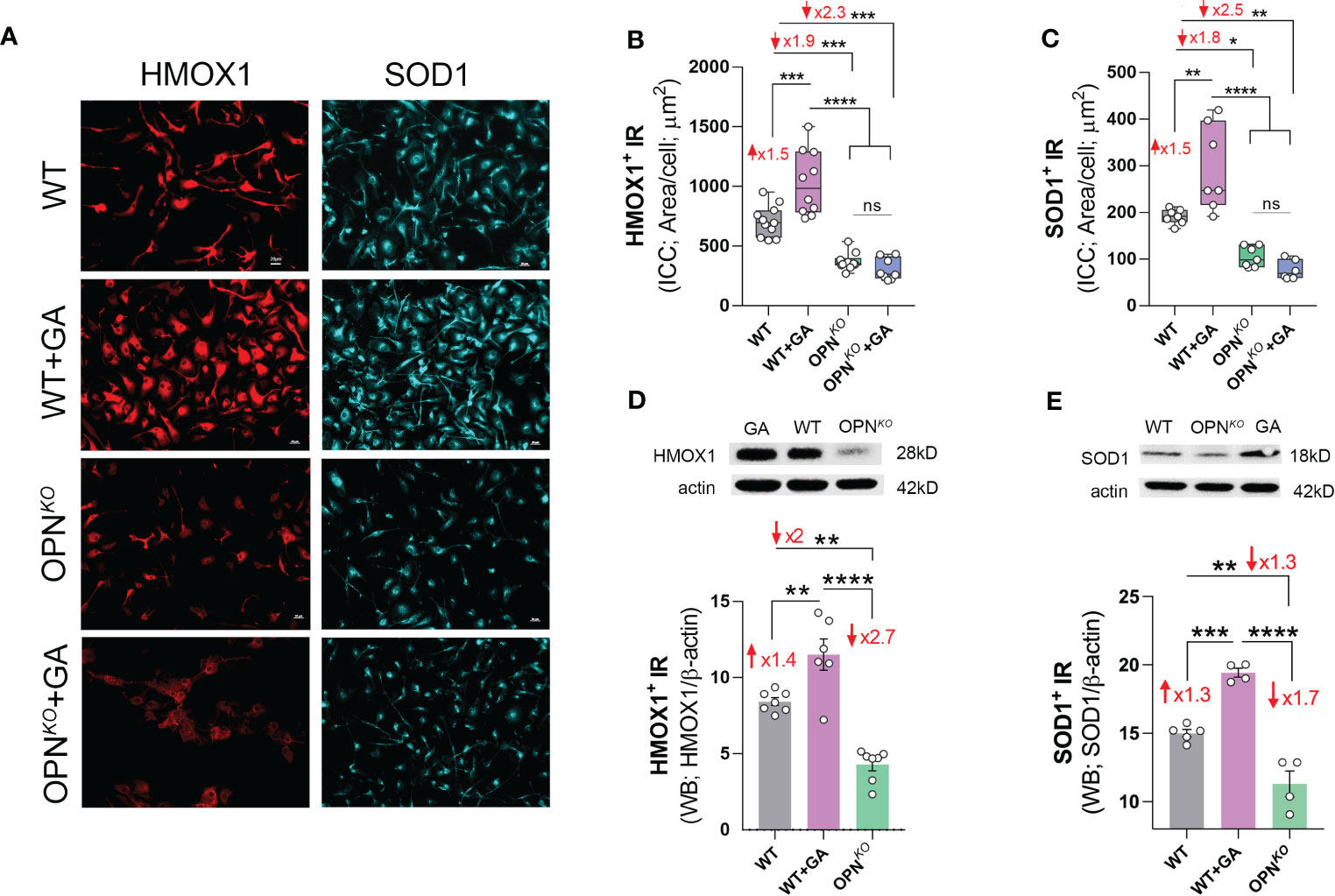
Figure 5 Anti-inflammatory and antioxidant enzymes were downregulated in OPN- deficient BMMФ. (A) Representative fluorescent micrographs of BMMΦ cells, either WT or OPNKO untreated or pretreated with GA, and immunostained for HMOX1+ (red), and SOD1+ (cyan); scale bar: 20µm. Quantitative analysis of ICC reveals increased level of (B) HMOX1 and (C) SOD1 following GA treatment, but reduced expression in OPNKO cells. Western blot and quantitative analysis of (D) HMOX1 and (E) SOD1 bands from cell pellets. Individual data points, group means, and SEMs are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc multiple comparison test.
We sought to determine whether GA stimulation vs. OPN deficiency in macrophages leads to decreased oxidative stress. Indeed, IPA revealed that GA-stimulated vs. OPN-deficient BMMΦ had inhibited the synthesis and production of ROS as well as reduced necrosis and apoptosis pathways (Figures 2C, D). To directly evaluate ROS production, we further employed fluorescence dye-based detection method using dihydroethidium (DHE) staining. As shown in Figure 6A, ROS accumulation was much stronger in OPNKO than in WT BMMΦ. This effect was reversed upon GA treatment. Quantitative analysis of ROS (DHE)-immunoreactive area (Figure 6B) showed a 2-fold increase in ROS levels in OPNKO BMMΦ, whereas GA treatment reduced it by 1.3-fold. Additionally, ICC labeling with annexin-V antibody (an apoptotic marker), demonstrated 1.7-fold increase in OPN-deficient BMMΦ that was 1.4-fold decreased following GA stimulation (Figures 6C, D).
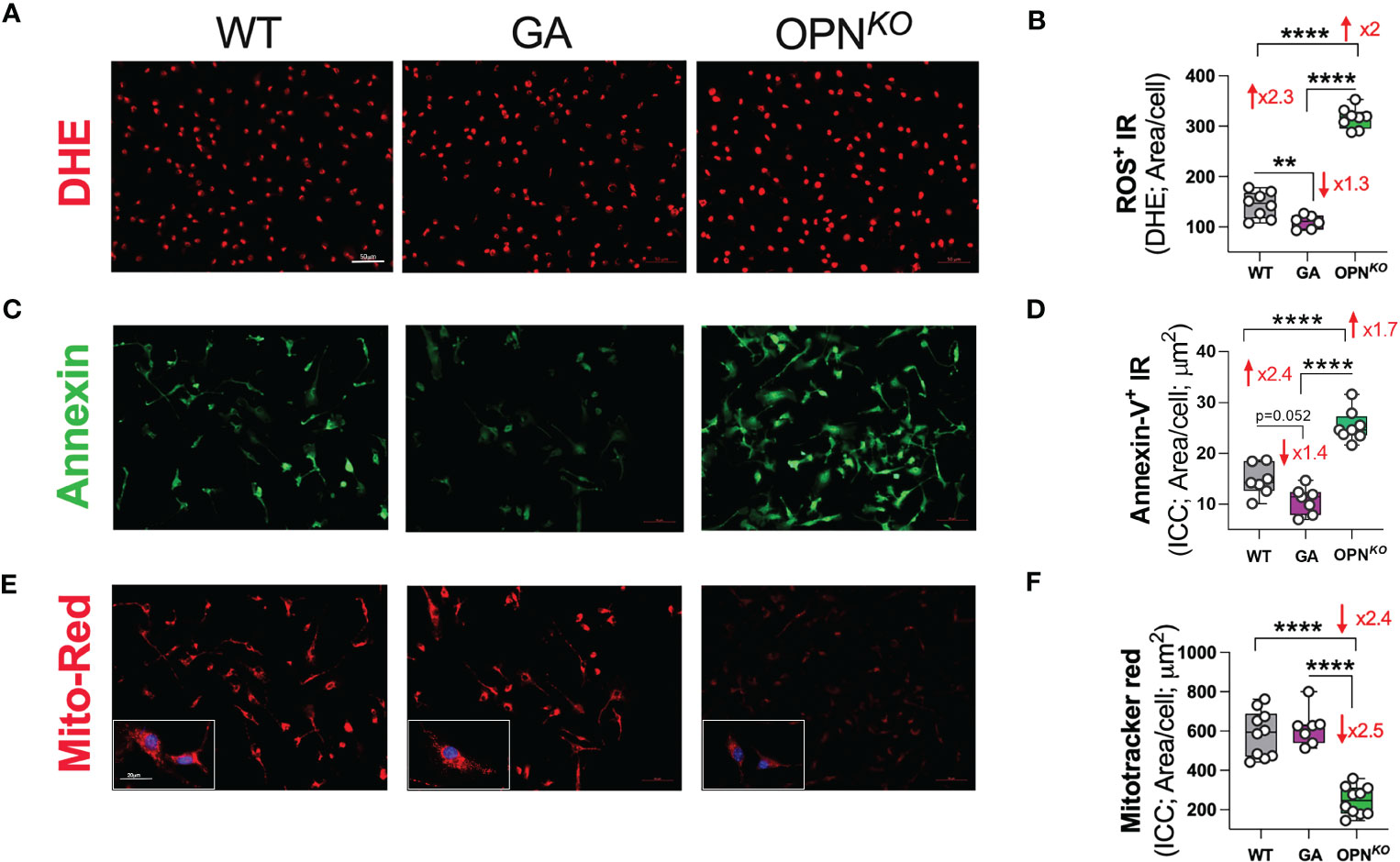
Figure 6 OPN deficiency induces ROS production and subsequent apoptosis in BMMФ. (A, B) Representative fluorescent micrographs of BMMΦ cells stained for DHE and quantitative analysis of ROS/DHE IR area in BMMΦ. (C, D) Representative fluorescent micrographs of BMMΦ cells immune-stained for Annexin-V and quantitative analysis of Annexin-V IR area in BMMΦ. (E, F) Representative fluorescent micrographs of BMMΦ cells immune-stained for Mitotracker-red and quantitative analysis of Mitotracker-red IR area in BMMΦ. Scale bar: 50µm. High power (x40) image inserts are shown. Scale bar: 20µm. Individual data points, group means, and SEMs are shown. **p < 0.01, and ****p < 0.0001 by one-way ANOVA with Tukey’s post-hoc multiple comparison test.
Finally, to determine levels of mitochondrial membrane potential (MMP) in live BMMΦ, we employed Mitotracker-red assay. The MMP of healthy cells is relatively high and shows a red fluorescence, while MMP of apoptotic cells is decreased and shows a diminished fluorescence (Figures 6E, F). The red fluorescence of GA-treated cells did not significantly change, implying preserved mitochondrial membrane potential after GA treatment. By contrast, in OPNKO versus WT macrophages, the ratio of red fluorescence was diminished by 2.4-fold, indicating impaired mitochondrial membranes in OPNKO cells that undergo apoptosis. In conclusion, OPN deficiency in macrophages induces ROS production, mitochondrial membrane potential damage, and subsequent apoptosis. In contrast, GA treatment decreases ROS and preserves MMP integrity.
Figures 7A, B summarizes the effects of OPN-deficiency compared to GA-stimulation on the phenotypes of BMMΦ. We found that OPN deficiency perturbs cellular homeostasis in macrophages through increased ROS production, and lysosome-mitochondria-mediated apoptosis, whereas OPN-primed GA immunomodulation restores cellular homeostasis by markedly increasing UCHL1 expression, inducing anti-inflammatory phenotype and cell survival, reducing ROS production, and mitochondrial dysfunction.
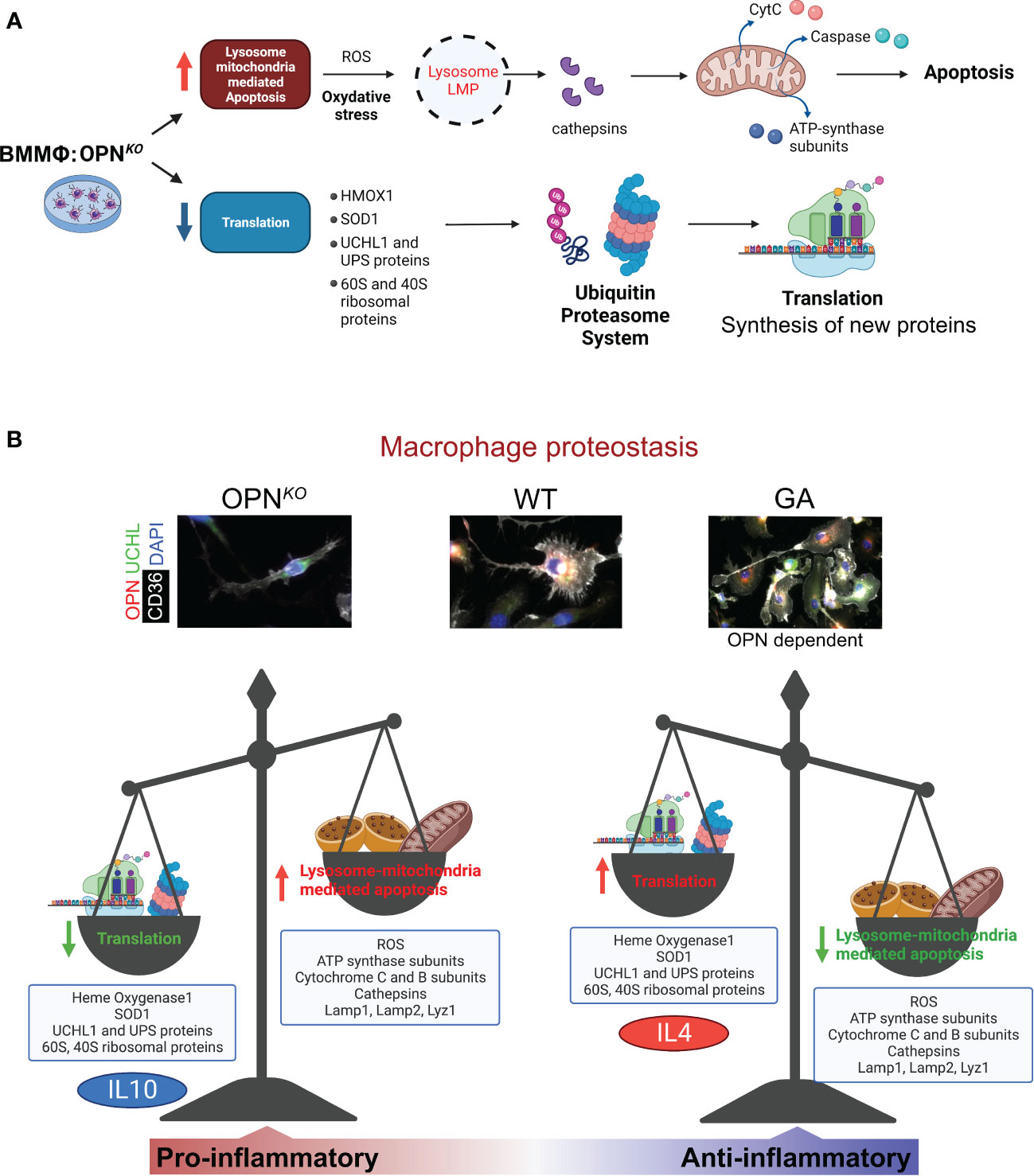
Figure 7 OPN/Spp1 deficiency in BMMФ triggers lysosome-mitochondria-mediated apoptosis accompanied by reduced translation and recycling. (A) Illustration summarizing lysosome-mitochondria-mediated apoptosis accompanied by reduced translation and UPS recycling in OPN-deficient macrophages. (B) Illustration of macrophage proteostasis emphasizing up- and downregulated proteins/groups and functions in OPNKO vs GA-stimulated macrophages in vitro. Illustration made with BioRender.com.
In this study, we identified a novel role for OPN in macrophages related to UPS and proteostasis. We employed both OPN-deletion loss-of-function and GA-stimulated OPN gain-of-function in vitro paradigms. Proteomics profiling by mass spectrometry analysis detected 630 DEPs when comparing between OPNKO, GA drug-stimulated, and WT macrophages. OPN deletion in macrophages induced activation of pro-inflammatory, mitochondrial dysfunction, apoptotic, and lysosomal signaling-related pathways, while inhibiting UPS, translation, and antioxidant-related processes, resulting in disrupted cellular homeostasis. Notably, GA stimulation reversed most of these adverse pathways through induction of OPN expression in macrophages, thus restoring an anti-inflammatory phenotype and macrophage proteostasis. These findings were further confirmed via Western blot and immunocytochemistry analyses, as well as immunoprecipitation and mitochondrial tracing assays. Overall, this study provides substantial evidence for the essential role of OPN in maintaining macrophage homeostasis.
One of the most notable findings of this study is the direct connection between OPN and UCHL1 in macrophages. UCHL1 was the most significantly downregulated protein in OPN-deficient macrophages. Surprisingly, while UCHL1 is recognized as a neuronal-specific protein (49–51), we found that UCHL1 was expressed by macrophages. Importantly, our finding that UCHL1 was downregulated upon OPN deficiency and upregulated by OPN enhancement through GA stimulation indicates that UCHL1 expression is OPN-dependent. Moreover, we show that OPN interacts with UCHL1 in protein complex by co-immunoprecipitation, further affirming that UCHL1 expression is tightly regulated by OPN. Two recent studies indicating that OPN and UCHL1 were intracellularly localized to mitochondria (52, 53), align with our results, suggesting a potential interaction in the mitochondria to regulate oxidative reactions and maintain mitochondrial function.
UCHL1 is a deubiquitinating enzyme (DUB), a key cellular proteolytic enzyme responsible for the removal of oxidized and/or damaged proteins, and a prominent component of the UPS (54, 55). The UPS plays a key role in preserving cellular homeostasis through the regulation of key cellular processes such as transcription, protein quality control and degradation, cell stress responses, cell cycle progression, and apoptosis (55, 56); such pleotropic cellular functions make UPS a central regulator of diverse cellular processes. A recent study showed that DUBs inhibition in macrophages resulted in acute perturbation of cellular ubiquitin homeostasis and induction of oxidative stress through ROS (57). This indicates that DUBs are responsible for mitigating oxidative damage and maintaining cellular homeostasis in macrophages. Furthermore, crosstalk between the UPS and mitochondrial proteins are reported to play a significant role in cellular homeostasis (58–60).
Indeed, a lower level of UCHL1 has been detected in apoptotic cells with severe loss of the mitochondrial membrane potential (61). Another report found that UCHL1 inhibition increased neuronal and apoptotic cell death (62) and UCHL1 deficiency in skeletal muscle cells resulted in altered mitochondrial oxidative phosphorylation (59). Accordingly, in the current study, we found that UCHL1 deficiency in OPNKO macrophages was associated with harmful cellular processes. These processes include lower expression of potent antioxidant enzymes, such as SOD1 and HMOX1 (47, 63), along with increased ROS production as well as, elevated lysosomal, mitochondrial, and apoptotic proteins as lysozyme C-2, ATP-synthase subunits, cathepsins, and cytochrome C and B subunits, and annexin-V. Moreover, there was also a severe loss of mitochondrial membrane potential measured in UCHL1-deficient OPNKO macrophages. Therefore, our data supports a role for the UCHL1-OPN axis in regulation of mitochondrial oxidative stress in macrophages.
A summary of our hypothesis on OPN-deficiency and OPN-induction effects in macrophages is illustrated in Figure 7. Following lysosomal membrane permeabilization (LMP), lysosomal proteases, such as cathepsins, are released into the cytosol and mediate cytochrome-C release and caspase activation. This ultimately triggers mitochondria-mediated apoptotic cascade (64). During apoptosis, caspase-dependent cleavage of translation-initiation factors leads to the inhibition of protein synthesis and translational shutdown (65). OPN also influences lysosomal protein degradation and protease activation as it triggers Lamp1 production. The current study reveals that loss of OPN resulted in reduced expression of UCHL1 that ultimately triggered apoptotic markers. As mentioned above, lower UCHL1 levels were detected in apoptotic cells with severe loss of mitochondrial membrane potential (61). The collective data clearly indicates that OPN-UCHL1 signaling can maintain cellular homeostasis by controlling apoptotic processes. We also found that OPN was involved in the regulation of protein synthesis and translation. Since UCHL1 expression in macrophages is found to be regulated by OPN, among other UPS proteins, OPN has a crucial role in protein translation and turnover, and cellular homeostasis. This result is supported by a previous report that found OPN to regulate the homeostasis and function of natural killer cells (66).
Our data also revealed that OPN deficient macrophages were exhibiting a pro-inflammatory phenotype. In fact, our data indicates that OPN may inhibit inducible nitric oxide synthase, thus, inhibiting nitric oxide production in macrophages leading to suppressed oxidative stress and inflammation (67). Indeed, we recently reported the potential anti-inflammatory effects of OPN as it can resolve inflammatory cascades through promoting macrophages polarization towards their anti-inflammatory and highly phagocytic phenotypes (6). Similarly, anti-apoptotic functions of OPN have been previously reported as inhibiting OPN by monoclonal antibody and dramatically promoted the apoptosis of activated T cells (68). These independent studies suggest the antioxidant, anti-inflammatory, and anti-apoptotic effects of OPN in different immune cells.
This study highlights the role of OPN in immune modulation of macrophage phenotype. Previous studies showed that OPN interacts with immune cells and extracellular matrix (ECM) proteins through adhesive integrin binding motifs: the adhesive RGD domain (binding to αv-containing integrins and α5β1 integrins) and the SLAYGLR sequence (binding to α4β1, α4β7 and α9β1 integrins) (69). Notably, our macrophage proteome data indicated that out of 249 identified DEPs only ITA5, an integrin α5 coded by the ITGA5 gene (shown in Table 1), was significantly upregulated by GA stimulation (1.24-fold change, p = 0.031) and further upregulated as compared with OPNKO macrophages (1.42-fold change, p = 0.0026). GO analysis showed activation of cell to cell adhesion by GA (Table 3). Also, in IPA functional analysis, the top interconnecting molecular networks predicted ITGA5 activation in GA-stimulated versus OPNKO macrophages with direct involvement in macrophage activation (Figure 2D, underlined). OPN has been known to interact with the cell surface adhesion receptor CD44 in RGD in an independent manner (4, 5), however, our proteome profiling showed only marginal downregulation of CD44 in OPNKO macrophages (Table s2). Interestingly, we observed that OPNKO macrophages have some tendency to detach from the surface by day 7-8 after culturing, possibly due to lack of OPN’s adhesive integrin binding domains. Following human recombinant OPN supplement (data not shown), the OPNKO macrophages behaved almost like the WT macrophages, restoring their surface adhesive propensity. These findings suggest that OPN impacts macrophage inflammatory phenotype and adhesive properties through its integrin binding sites. Future studies should investigate the molecular mechanisms that governs OPN-dependent effects on macrophage inflammatory and surface adhesion phenotype.
Importantly, our current study also revealed the key OPN-associated functions of GA, as GA treatment not only increased the OPN production in macrophages, but it also reversed the expression of almost all proteins influenced by the OPN deficiency. Furthermore, GA-stimulation of OPNKO macrophages did not change both OPN and UCHL1 expression, demonstrating that GA effects on macrophage profile is OPN dependent. As an immunomodulator, GA has been reported to influence pleiotropic cellular functions in multiple immune cells such as macrophages, microglia, and T cells both in vivo and in vitro disease models of neurodegenerative disorders including AD. We previously reported that administration of GA to transgenic murine models of AD enhanced brain recruitment of blood-borne monocytes, which were involved in the clearance and degradation of Aβ plaques, reduced neuroinflammation, and induced production of neurotrophic factors. This resulted in significant neuroprotection and cognitive improvements (19–22, 70–76). In addition, GA-primed neuroprotection against the pathology associated with AD in transgenic mice model was linked with the induction of a phenotypic shift in microglia, which typically expressed IGF-1. Such microglia were involved in the phagocytosis of cerebral Aβ plaques and regulating detrimental chronic inflammation (24). These effects of GA on immune cells leading to neuroprotection against AD pathogenesis is, at least in part, associated with increased production of OPN in immune cells influencing their anti-inflammatory and neuroprotective functions. Furthermore, a recent report demonstrated that OPN promoted neuroprotection by inhibiting NLRP3 inflammasome and inflammatory microglial activation following focal ischemic brain injury in mice as well as LPS-stimulated rat primary microglia (77), further affirming the strong anti-inflammatory potential of OPN.
Although this study has many strengths and provides new insights, we acknowledge a few limitations. The immunoprecipitation assay did not include the GA-stimulated macrophages. Hence, the effects of OPN deficiency and GA stimulation on UCHL1-OPN interaction in a protein complex need to be investigated under diverse macrophage activation states in future studies. Further research is warranted to determine the physiological role of OPN and UCHL1 in macrophages under both healthy and disease conditions
This study reveals that OPN deficiency in macrophages is associated with homeostatic imbalance by affecting multiple downstream signaling pathways such as UCHL1-UPS downregulation, ROS production, oxidative stress, mitochondrial-related dysfunctions, and subsequent apoptosis. Reciprocally, the induction of apoptosis leads to a substantial inhibition of protein synthesis and translation, which was a feature of OPNKO macrophages. On the other hand, GA appears to maintain cellular homeostasis through the induction of OPN production, which in turn regulates UPS, protein recycling, and protein synthesis. Our proteomics data in conjunction with bioinformatics analysis offer the molecular insights into phenotypic changes that occur in OPN-deficient and GA-stimulated macrophages. We revealed the importance of OPN modulation in cellular proteostasis and ROS production. On aggregate, this study indicates that targeting of UPS and lysosome-mitochondrial pathways may hold therapeutic potential for enhanced macrophage function and immunomodulation therapies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: MSV000091221(MassIVE) and Proteome Exchange ID: PXD039950.
The animal study was reviewed and approved by Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC).
AR, MKH: Study conception, design, and supervision. AR, DTF, JS: Major experimental contribution, planning, data acquisition and analysis. KR, VV, RP, AR, JS, JVE: Mass spectrometry and analysis. HS, JD, YK: additional experiments. AR, KR, DTF, JS, VV, RP, HS, JD, JDFG, YK, MA, KB, BPG, MKH: Data interpretation and edits. AR, DTF, BPG, MKH: Figure preparation, manuscript writing and editing, and revision. All authors approved the final version.
This work was funded by the National Institutes of Health (NIH)/National Institute on Aging (NIA) grant numbers: R01AG056478, R01AG055865, AG056478-04S1, and R01AG075998 (MK-H). In addition, we received support from the Tom Gordon, Haim Saban, and Wilstein Foundations.
We thank Samuel Fuchs for illustrating Figure 1A and Elijiah Maxfield for assisting with editing. We also thank the Cedars-Sinai Medical Center Proteomics and Metabolomics Core and the Advanced Clinical BioSystems Research Institute for their assistance and support in the proteomics analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1155935/full#supplementary-material
1. Franzen A, Heinegard D. Isolation and characterization of two sialoproteins present only in bone calcified matrix. Biochem J (1985) 232(3):715–24. doi: 10.1042/bj2320715
2. Patarca R, Freeman GJ, Singh RP, Wei FY, Durfee T, Blattner F, et al. Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. definition of a novel T cell-dependent response associated with genetic resistance to bacterial infection. J Exp Med (1989) 170(1):145–61. doi: 10.1084/jem.170.1.145
3. Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol (2007) 27(11):2302–9. doi: 10.1161/ATVBAHA.107.144824
4. Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal (2009) 3(3-4):311–22. doi: 10.1007/s12079-009-0068-0
5. Rittling SR. Osteopontin in macrophage function. Expert Rev Mol Med (2011) 13:e15. doi: 10.1017/S1462399411001839
6. Rentsendorj A, Sheyn J, Fuchs DT, Daley D, Salumbides BC, Schubloom HE, et al. A novel role for osteopontin in macrophage-mediated amyloid-beta clearance in alzheimer's models. Brain Behav Immun (2018) 67:163–80. doi: 10.1016/j.bbi.2017.08.019
7. Shinohara ML, Jansson M, Hwang ES, Werneck MB, Glimcher LH, Cantor H. T-Bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci U S A. (2005) 102(47):17101–6. doi: 10.1073/pnas.0508666102
8. Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, et al. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science (2000) 287(5454):860–4. doi: 10.1126/science.287.5454.860
9. Hansel NN, Cheadle C, Diette GB, Wright J, Thompson KM, Barnes KC, et al. Analysis of CD4+ T-cell gene expression in allergic subjects using two different microarray platforms. Allergy (2008) 63(3):366–9. doi: 10.1111/j.1398-9995.2007.01540.x
10. Kasetty G, Bhongir RKV, Papareddy P, Tufvesson E, Stenberg H, Bjermer L, et al. Osteopontin protects against pneumococcal infection in a murine model of allergic airway inflammation. Allergy (2019) 74(4):663–74. doi: 10.1111/all.13646
11. Hansakon A, Png CW, Zhang Y, Angkasekwinai P. Macrophage-derived osteopontin influences the amplification of cryptococcus neoformans-promoting type 2 immune response. J Immunol (2021) 207(8):2107–17. doi: 10.4049/jimmunol.2100202
12. Chan JL, Reeves TM, Phillips LL. Osteopontin expression in acute immune response mediates hippocampal synaptogenesis and adaptive outcome following cortical brain injury. Exp Neurol (2014) 261:757–71. doi: 10.1016/j.expneurol.2014.08.015
13. Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia (2015) 63(12):2198–207. doi: 10.1002/glia.22885
14. Meller R, Stevens SL, Minami M, Cameron JA, King S, Rosenzweig H, et al. Neuroprotection by osteopontin in stroke. J Cereb Blood Flow Metab (2005) 25(2):217–25. doi: 10.1038/sj.jcbfm.9600022
15. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi: 10.1038/nri1733
16. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci (2014) 10(5):520–9. doi: 10.7150/ijbs.8879
17. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122(3):787–95. doi: 10.1172/JCI59643
18. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
19. Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V, et al. Therapeutic effects of glatiramer acetate and grafted CD115(+) monocytes in a mouse model of alzheimer's disease. Brain (2015) 138(Pt 8):2399–422. doi: 10.1093/brain/awv150
20. Li S, Hayden EY, Garcia VJ, Fuchs DT, Sheyn J, Daley DA, et al. Activated bone marrow-derived macrophages eradicate alzheimer's-related Abeta42 oligomers and protect synapses. Front Immunol (2020) 11:49. doi: 10.3389/fimmu.2020.00049
21. Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse alzheimer's disease model. Eur J Neurosci (2007) 26(2):413–6. doi: 10.1111/j.1460-9568.2007.05652.x
22. Doustar J, Rentsendorj A, Torbati T, Regis GC, Fuchs DT, Sheyn J, et al. Parallels between retinal and brain pathology and response to immunotherapy in old, late-stage alzheimer's disease mouse models. Aging Cell (2020) 19(11):e13246. doi: 10.1111/acel.13246
23. Weber MS, Prod'homme T, Youssef S, Dunn SE, Rundle CD, Lee L, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med (2007) 13(8):935–43. doi: 10.1038/nm1620
24. Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, et al. Glatiramer acetate fights against alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A. (2006) 103(31):11784–9. doi: 10.1073/pnas.0604681103
25. Li P, Hao Z, Wu J, Ma C, Xu Y, Li J, et al. Comparative proteomic analysis of polarized human THP-1 and mouse RAW264. 7 Macrophages. Front Immunol (2021) 12:700009. doi: 10.3389/fimmu.2021.700009
26. Kamal AHM, Fessler MB, Chowdhury SM. Comparative and network-based proteomic analysis of low dose ethanol- and lipopolysaccharide-induced macrophages. PloS One (2018) 13(2):e0193104. doi: 10.1371/journal.pone.0193104
27. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep (2021) 37(5):109955. doi: 10.1016/j.celrep.2021.109955
28. Flowers A, Bell-Temin H, Jalloh A, Stevens SM Jr., Bickford PC. Proteomic anaysis of aged microglia: shifts in transcription, bioenergetics, and nutrient response. J Neuroinflammation. (2017) 14(1):96. doi: 10.1186/s12974-017-0840-7
29. Murthy S, Karkossa I, Schmidt C, Hoffmann A, Hagemann T, Rothe K, et al. Danger signal extracellular calcium initiates differentiation of monocytes into SPP1/osteopontin-producing macrophages. Cell Death Dis (2022) 13(1):53. doi: 10.1038/s41419-022-04507-3
30. Criscuolo C, Cianflone A, Lanzillo R, Carrella D, Carissimo A, Napolitano F, et al. Glatiramer acetate modulates ion channels expression and calcium homeostasis in b cell of patients with relapsing-remitting multiple sclerosis. Sci Rep (2019) 9(1):4208. doi: 10.1038/s41598-018-38152-8
31. Hasson T, Kolitz S, Towfic F, Laifenfeld D, Bakshi S, Beriozkin O, et al. Functional effects of the antigen glatiramer acetate are complex and tightly associated with its composition. J Neuroimmunol. (2016) 290:84–95. doi: 10.1016/j.jneuroim.2015.11.020
32. Parker SJ, Rost H, Rosenberger G, Collins BC, Malmstrom L, Amodei D, et al. Identification of a set of conserved eukaryotic internal retention time standards for data-independent acquisition mass spectrometry. Mol Cell Proteomics. (2015) 14(10):2800–13. doi: 10.1074/mcp.O114.042267
33. Holewinski RJ, Parker SJ, Matlock AD, Venkatraman V, Van Eyk JE. Methods for SWATH: data independent acquisition on TripleTOF mass spectrometers. Methods Mol Biol (2016) 1410:265–79. doi: 10.1007/978-1-4939-3524-6_16
34. Reinicke AT, Laban K, Sachs M, Kraus V, Walden M, Damme M, et al. Ubiquitin c-terminal hydrolase L1 (UCH-L1) loss causes neurodegeneration by altering protein turnover in the first postnatal weeks. Proc Natl Acad Sci U S A. (2019) 116(16):7963–72. doi: 10.1073/pnas.1812413116
35. Vijayan V, Wagener F, Immenschuh S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem Pharmacol (2018) 153:159–67. doi: 10.1016/j.bcp.2018.02.010
36. Jansen LE, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol (2007) 176(6):795–805. doi: 10.1083/jcb.200701066
37. Alabert C, Barth TK, Reveron-Gomez N, Sidoli S, Schmidt A, Jensen ON, et al. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev (2015) 29(6):585–90. doi: 10.1101/gad.256354.114
38. Laatsch A, Panteli M, Sornsakrin M, Hoffzimmer B, Grewal T, Heeren J. Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein e. PloS One (2012) 7(1):e29385. doi: 10.1371/journal.pone.0029385
39. Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and akt activation. Arterioscler Thromb Vasc Biol (2010) 30(4):787–95. doi: 10.1161/ATVBAHA.109.202051
40. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol (2010) 11(2):113–27. doi: 10.1038/nrm2838
41. Oliveira MM, Lourenco MV, Longo F, Kasica NP, Yang W, Ureta G, et al. Correction of eIF2-dependent defects in brain protein synthesis, synaptic plasticity, and memory in mouse models of alzheimer's disease. Sci Signal (2021) 14(668):eabc5429. doi: 10.1126/scisignal.abc5429
42. Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci (2013) 70(19):3493–511. doi: 10.1007/s00018-012-1252-6
43. Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife (2019) 8:e41351. doi: 10.7554/eLife.41351
44. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood (2010) 116(26):6054–62. doi: 10.1182/blood-2010-03-272138
45. Weis N, Weigert A, von Knethen A, Brune B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell (2009) 20(5):1280–8. doi: 10.1091/mbc.e08-10-1005
46. Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys (2014) 564:83–8. doi: 10.1016/j.abb.2014.09.005
47. Eleutherio ECA, Silva Magalhaes RS, de Araujo Brasil A, Monteiro Neto JR, de Holanda Paranhos L. SOD1, more than just an antioxidant. Arch Biochem Biophys (2021) 697:108701. doi: 10.1016/j.abb.2020.108701
48. Sun J, Ren X, Simpkins JW. Sequential upregulation of superoxide dismutase 2 and heme oxygenase 1 by tert-butylhydroquinone protects mitochondria during oxidative stress. Mol Pharmacol (2015) 88(3):437–49. doi: 10.1124/mol.115.098269
49. Day IN, Thompson RJ. UCHL1 (PGP 9.5): neuronal biomarker and ubiquitin system protein. Prog Neurobiol (2010) 90(3):327–62. doi: 10.1016/j.pneurobio.2009.10.020
50. Doran JF, Jackson P, Kynoch PA, Thompson RJ. Isolation of PGP 9.5, a new human neurone-specific protein detected by high-resolution two-dimensional electrophoresis. J Neurochem (1983) 40(6):1542–7. doi: 10.1111/j.1471-4159.1983.tb08124.x
51. Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science (1989) 246(4930):670–3. doi: 10.1126/science.2530630
52. Gao H, Antony R, Srinivasan R, Wu P, Wang X, Li Y. UCHL1 regulates oxidative activity in skeletal muscle. PloS One (2020) 15(11):e0241716. doi: 10.1371/journal.pone.0241716
53. Sugiyama Y, Oishi T, Yamashita A, Murata Y, Yamamoto T, Takashima I, et al. Neuronal and microglial localization of secreted phosphoprotein 1 (osteopontin) in intact and damaged motor cortex of macaques. Brain Res (2019) 1714:52–64. doi: 10.1016/j.brainres.2019.02.021
54. Tramutola A, Di Domenico F, Barone E, Perluigi M, Butterfield DA. It is all about (U)biquitin: role of altered ubiquitin-proteasome system and UCHL1 in Alzheimer disease. Oxid Med Cell Longev (2016) 2016:2756068. doi: 10.1155/2016/2756068
55. Bishop P, Rocca D, Henley JM. Ubiquitin c-terminal hydrolase L1 (UCH-L1): structure, distribution and roles in brain function and dysfunction. Biochem J (2016) 473(16):2453–62. doi: 10.1042/BCJ20160082
56. Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front Cell Dev Biol (2018) 6:128. doi: 10.3389/fcell.2018.00128
57. Charbonneau ME, Passalacqua KD, Hagen SE, Showalter HD, Wobus CE, O'Riordan MXD. Perturbation of ubiquitin homeostasis promotes macrophage oxidative defenses. Sci Rep (2019) 9(1):10245. doi: 10.1038/s41598-019-46526-9
58. Bragoszewski P, Turek M, Chacinska A. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol (2017) 7(4):170007. doi: 10.1098/rsob.170007
59. Cerqueira FM, von Stockum S, Giacomello M, Goliand I, Kakimoto P, Marchesan E, et al. A new target for an old DUB: UCH-L1 regulates mitofusin-2 levels, altering mitochondrial morphology, function and calcium uptake. Redox Biol (2020) 37:101676. doi: 10.1016/j.redox.2020.101676
60. Ross JM, Olson L, Coppotelli G. Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin? Int J Mol Sci (2015) 16(8):19458–76. doi: 10.3390/ijms160819458
61. Shen H, Sikorska M, Leblanc J, Walker PR, Liu QY. Oxidative stress regulated expression of ubiquitin carboxyl-terminal hydrolase-L1: role in cell survival. Apoptosis (2006) 11(6):1049–59. doi: 10.1007/s10495-006-6303-8
62. Guglielmotto M, Monteleone D, Vasciaveo V, Repetto IE, Manassero G, Tabaton M, et al. The decrease of uch-L1 activity is a common mechanism responsible for abeta 42 accumulation in alzheimer's and vascular disease. Front Aging Neurosci (2017) 9:320. doi: 10.3389/fnagi.2017.00320
63. Syapin PJ. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br J Pharmacol (2008) 155(5):623–40. doi: 10.1038/bjp.2008.342
64. Joy B, Sivadasan R, Abraham TE, John M, Sobhan PK, Seervi M, et al. Lysosomal destabilization and cathepsin b contributes for cytochrome c release and caspase activation in embelin-induced apoptosis. Mol Carcinog. (2010) 49(4):324–36. doi: 10.1002/mc.20599
65. Bushell M, Stoneley M, Sarnow P, Willis AE. Translation inhibition during the induction of apoptosis: RNA or protein degradation? Biochem Soc Trans (2004) 32(Pt 4):606–10. doi: 10.1042/BST0320606
66. Leavenworth JW, Verbinnen B, Wang Q, Shen E, Cantor H. Intracellular osteopontin regulates homeostasis and function of natural killer cells. Proc Natl Acad Sci U S A. (2015) 112(2):494–9. doi: 10.1073/pnas.1423011112
67. Rollo EE, Laskin DL, Denhardt DT. Osteopontin inhibits nitric oxide production and cytotoxicity by activated RAW264.7 macrophages. J Leukoc Biol (1996) 60(3):397–404. doi: 10.1002/jlb.60.3.397
68. Fan K, Dai J, Wang H, Wei H, Cao Z, Hou S, et al. Treatment of collagen-induced arthritis with an anti-osteopontin monoclonal antibody through promotion of apoptosis of both murine and human activated T cells. Arthritis Rheumatol (2008) 58(7):2041–52. doi: 10.1002/art.23490
69. Lund SA, Wilson CL, Raines EW, Tang J, Giachelli CM, Scatena M. Osteopontin mediates macrophage chemotaxis via alpha4 and alpha9 integrins and survival via the alpha4 integrin. J Cell Biochem (2013) 114(5):1194–202. doi: 10.1002/jcb.24462
70. Kasindi A, Fuchs DT, Koronyo Y, Rentsendorj A, Black KL, Koronyo-Hamaoui M. Glatiramer acetate immunomodulation: evidence of neuroprotection and cognitive preservation. Cells (2022) 11(9):1578. doi: 10.3390/cells11091578
71. Koronyo-Hamaoui M, Gaire BP, Frautschy SA, Alvarez JI. Editorial: role of inflammation in neurodegenerative diseases. Front Immunol (2022) 13:958487. doi: 10.3389/fimmu.2022.958487
72. Koronyo-Hamaoui M, Sheyn J, Hayden EY, Li S, Fuchs DT, Regis GC, et al. Peripherally derived angiotensin converting enzyme-enhanced macrophages alleviate Alzheimer-related disease. Brain (2020) 143(1):336–58. doi: 10.1093/brain/awz364
73. Zuroff LR, Torbati T, Hart NJ, Fuchs DT, Sheyn J, Rentsendorj A, et al. Effects of IL-34 on macrophage immunological profile in response to alzheimer's-related Abeta(42) assemblies. Front Immunol (2020) 11:1449. doi: 10.3389/fimmu.2020.01449
74. Koronyo-Hamaoui M, Ko MK, Koronyo Y, Azoulay D, Seksenyan A, Kunis G, et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: local elevation of IL-10 and MMP-9. J Neurochem (2009) 111(6):1409–24. doi: 10.1111/j.1471-4159.2009.06402.x
75. Bakalash S, Pham M, Koronyo Y, Salumbides BC, Kramerov A, Seidenberg H, et al. Egr1 expression is induced following glatiramer acetate immunotherapy in rodent models of glaucoma and alzheimer's disease. Invest Ophthalmol Vis Sci (2011) 52(12):9033–46. doi: 10.1167/iovs.11-7498
76. Bernstein KE, Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Lopes DH, et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents alzheimer's-like cognitive decline. J Clin Invest. (2014) 124(3):1000–12. doi: 10.1172/JCI66541
Keywords: secreted phosphoprotein 1, early T-lymphocyte activation (ETA-1), bone/sialoprotein I (BSP-1 or BNSP), monocytes, innate immunity, mitochondrial dysfunction, reactive oxygen species
Citation: Rentsendorj A, Raedschelders K, Fuchs D-T, Sheyn J, Vaibhav V, Porritt RA, Shi H, Dagvadorj J, de Freitas Germano J, Koronyo Y, Arditi M, Black KL, Gaire BP, Van Eyk JE and Koronyo-Hamaoui M (2023) Osteopontin depletion in macrophages perturbs proteostasis via regulating UCHL1-UPS axis and mitochondria-mediated apoptosis. Front. Immunol. 14:1155935. doi: 10.3389/fimmu.2023.1155935
Received: 01 February 2023; Accepted: 19 May 2023;
Published: 30 May 2023.
Edited by:
Toshio Hattori, Kibi International University, JapanReviewed by:
Shigeyuki Kon, Fukuyama University, JapanCopyright © 2023 Rentsendorj, Raedschelders, Fuchs, Sheyn, Vaibhav, Porritt, Shi, Dagvadorj, de Freitas Germano, Koronyo, Arditi, Black, Gaire, Van Eyk and Koronyo-Hamaoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya Koronyo-Hamaoui, bWF5YS5rb3JvbnlvQGNzbWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.