
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 July 2023
Sec. Primary Immunodeficiencies
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1155380
This article is part of the Research Topic Mechanisms of Dysregulated Antibody Responses in Inborn Errors of Immunity View all 5 articles
 Qing Min1
Qing Min1 Krisztian Csomos2Yaxuan Li3Lulu Dong3Ziying Hu4Xin Meng3Meiping Yu1
Krisztian Csomos2Yaxuan Li3Lulu Dong3Ziying Hu4Xin Meng3Meiping Yu1 Jolan E. Walter2,5
Jolan E. Walter2,5 Ji-Yang Wang1,3,4,6*
Ji-Yang Wang1,3,4,6*Mutations in the recombination activating gene 1 (RAG1) and RAG2 in humans are associated with a broad spectrum of clinical phenotypes, from severe combined immunodeficiency to immune dysregulation. Partial (hypomorphic) RAG deficiency (pRD) in particular, frequently leads to hyperinflammation and autoimmunity, with several underlying intrinsic and extrinsic mechanisms causing a break in tolerance centrally and peripherally during T and B cell development. However, the relative contributions of these processes to immune dysregulation remain unclear. In this review, we specifically focus on the recently described tolerance break and B cell abnormalities, as well as consequent molecular and cellular mechanisms of autoantibody production in patients with pRD.
A diversified repertoire of antigen receptors is generated by somatic recombination (rearrangement) of variable (V), diversity (D) and joining (J) segments (1–3). V(D)J recombination is initiated by the recombination activating gene 1 (RAG1) and RAG2, which bind to the recombination signal sequences (RSSs) flanking each gene and introduce DNA double strand breaks (DSBs) between the RSS and the adjacent V, D or J gene (1, 4, 5). The resulting DSBs are then processed and joined by the non-homologous end joining (NHEJ) repair pathway (1, 6–8).
B cell development in the bone marrow (BM) proceeds in a stepwise fashion and is accompanied by immunoglobulin (Ig) gene rearrangements (9). Pro-B cells undergo Ig heavy chain D to J and then V to DJ rearrangements and if successful become pre-B cells. Pre-B cells further rearrange their light chain to form IgM+ immature B cells (9). Absence of RAG1 or RAG2 abolishes V(D)J recombination causing a complete block in B cell development at the pro-B stage (10). Hypomorphic RAG mutations with residual recombinase activity lead to reduced V(D)J rearrangements and partial block of BM B cell development, resulting in limited Ig gene diversity and decreased but not absent peripheral B cell count (10, 11).
Due to the random nature of V(D)J recombination and the introduction of non-templated nucleotides during the joining process by NHEJ, it was suggested that up to 80% of the newly generated immature B cells are autoreactive (12, 13). Such autoreactive immature B cells are eliminated by at least three mechanisms, including clonal deletion via apoptosis, receptor editing, and functional inactivation (anergy) (12, 14). Receptor editing changes the specificity of autoreactive immature B cells by inducing a new round of light chain gene rearrangement and has been shown to be the major mechanism of central B cell tolerance (15–17). One study suggested that 25% of peripheral mature B cells in mice underwent receptor editing in the BM (18). Since receptor editing is mediated by RAG1 and RAG2, impaired central B cell tolerance could contribute to the generation of autoreactive B cells in pRD.
In humans, biallelic RAG1 or RAG2 pathogenic mutations lead to a broad spectrum of clinical phenotypes (1, 5, 19). While RAG null mutations cause T-B- severe combined immunodeficiency (SCID) (20), pRD results in CID associated with immune dysregulation, such as Omenn syndrome (OS) (21), atypical or leaky SCID (AS or LS) (22, 23), and delayed onset combined immunodeficiency with granulomas or autoimmunity manifestations (CID-G/AI) (24, 25). pRD can also present as chronic infections with Cytomegalovirus (CMV) or Epstein-Barr virus (EBV) accompanied by γδ T cell expansion (26, 27), idiopathic CD4+ T cell lymphopenia (ICL) (28), common variable immunodeficiency (CVID) (29–31), and selective IgA deficiency (SIgAD) (32). Recent studies have indicated that more than half of the patients with pRD present with hyperinflammatory and/or autoimmune complications (33, 34) and produced a broad range of autoantibodies (33, 35, 36). The clinical phenotype likely depends on the nature of the defect and its quantitative and qualitative effects on V(D)J recombination, genetic modifiers and environmental factors (1, 37, 38).
In this short review, we focus on B cell abnormalities observed in patients with pRD and present an updated version of a recently published model that illustrates the molecular and cellular mechanisms underlying autoantibody production in these patients (36). This review provides additional insights into the diagnosis and treatment of RAG-deficient patients with autoimmune manifestations.
RAG1 and RAG2 initiate V(D)J recombination to generate a diverse repertoire of B cell antigen receptors. In humans, the Ig heavy chain (IgH) genes are located at the telomeric end of chromosome 14 and contain at least 55 functional/ORF VH genes (11 of these are not found in the reference genome), forming seven phylogenetically related subgroups (VH1 to VH7) (39, 40). Downstream of the VH cluster are 23 functional DH genes, followed by 6 JH genes (39).
Recently, there has been an increase in the number of patients diagnosed with pRD and presence of peripheral B cell compartment allowed for the analysis of BCR diversity. Five studies have extensively analyzed BCR repertoire in pRD patients and mouse models (36–38, 41, 42). Lee et al. (41) examined the total B cell compartment and found reduced diversity only in OS and LS but not in CID. However, when naïve B cell compartment was assessed by Csomos et al. (38), the skewing of BCR repertoire was evident even in CID cases. Consistently, the primary repertoire of T and B cells was skewed in mouse models of pRD (42). In general, reduced BCR repertoire diversity and increased clonotypic expansion have been found in most pRD patients.
Regarding tendency for autoreactive properties in BCR, IJspeert et al. (37) analyzed the IgH repertoire in peripheral blood mononuclear cells and BM mononuclear cells (BMMNC) from 3 patients with similar RAG1 mutations. While they did not find preferential usage of proximal or distal VH or JH genes, in two of the three patients they found increased frequency of VH4-34, which is known to encode intrinsically self-reactive cold agglutinin antibodies that recognize carbohydrate antigens on erythrocytes (43, 44). Lee et al. (41) studied BCR repertoire of circulating B cells in eight patients with pRD (3 CID-G/AI, 2 LS and 3 OS) and found skewed usage of VH, DH and JH genes, especially increased usage of VH3-9, VH4-31, and VH3-23 in most patients. Csomos et al. (38) evaluated BCR repertoire in sorted CD38int, CD38− and CD27+ B cell populations from six patients with pRD and found restricted Ig diversity with VH families being dominated by a few expanded unique clones in the “naïve” CD38int population. They did not find elevated frequency of VH4-34-carrying clones in the patients. Min et al. (36) analyzed IgH repertoire diversity in the BM and peripheral blood of a CID-G/AI patient and a healthy control (HC) and found reduced number of CDR3 clonal peaks in the BM µ and peripheral µ and γ transcripts in the patient compared with the HC, indicating reduced Ig diversity in this patient. Intriguingly, patient BM IgG had much higher diversity than the patient peripheral IgM and IgG. In addition, 7.46% of the peripheral IgG and 1.41% of the BM IgG had identical CDR3 sequences, suggesting that these IgG+ cells shared the same origin (36). Along with the findings that the patient BM contained abundant CD38high plasma cells (PC) and expressed high levels of IgG transcripts, these observations collectively suggest that the patient peripheral IgG+ B cells differentiated into IgG-secreting PC that accumulated in the BM. The analysis did not find increased CDR3 length or preferential usage of VH4-34 gene in the patient B cells. Therefore, reduced BCR repertoire diversity associated with clonotypic expansion, rather than frequent usage of certain VH genes, is a common feature of pRD.
Receptor editing is a RAG-dependent process and serves to change the specificity of autoreactive immature B cells (14, 45). In the BM, autoreactive immature B cells lose BCR surface expression upon self-antigen stimulation and de-differentiate back into small pre-B cells, which re-express RAG to initiate light chain gene rearrangement using available upstream Vκ and downstream Jκ genes (14). Repeated Vκ-Jκ rearrangements eventually lead to exhaustion of the recombination potential at the Vκ gene locus and expression of a λ light chain. Thus, distal Jκ usage and the proportion of Igλ+ B cells serves as indicators of the efficiency of receptor editing (14, 46).
Knock-in mice expressing hypomorphic RAG mutants (Rag2R229Q, Rag1R972Q, Rag1F971L or Rag1R972W) exhibit a markedly reduced proportion of Igλ-expressing splenic B cells, implicating impaired receptor editing in these mice (42, 46). Similarly, a cohort of pRD patients showed a reduced proportion of Igλ-expressing peripheral transitional B (TrB) cells (38). Moreover, reduced usage of distal Jκ4 and Jκ5 segments was observed in patients with OS and CID-G/AI (37, 38). Impaired receptor editing thus appears to be another common feature of pRD.
B cell activating factor of the TNF-family receptor (BAFF; also known as TNFSF13B or B lymphocyte stimulator, Blys) is a membrane-bound protein that can be processed by the membrane-bound protease furin, resulting in a soluble form (47–50). It binds to three receptors, namely, BAFF receptor (BAFFR; also known as TNFRSF13C), B cell maturation antigen (BCMA; also known as TNFRSF17) and transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI; also known as TNFRSF13B) (49). BAFFR, TACI, and BCMA are expressed by B cells at different developmental and differentiation stages (48, 51). BAFFR is first expressed when immature B cells develop into TrB cells, which then receive BAFF-BAFFR dependent pro-survival signals to rescue them from premature cell death (9, 48). TACI is expressed in B cells upon activation (52) and is expressed at higher levels in marginal zone B cells (53), whereas BCMA is constitutively expressed by long-lived PC and primarily functions to support their survival through interaction with the TNF family member APRIL (a proliferation-inducing ligand) (54).
BAFF binds to BAFFR on B cells and under B lymphopenic conditions serum BAFF levels are elevated (14, 55). Assuming that a fixed amount of BAFF is produced by myeloid cells, this observation suggests a possibility that BAFF may be sequestered or degraded after binding to the BAFFR on B cells. It is also possible that chronic infections and inflammations observed in patients with pRD may result in elevated production of BAFF by myeloid cells (38, 56). pRD leads to B cell lymphopenia (33) and consistently BAFF levels have been shown to be increased in both mice and humans with pRD (36, 38, 42, 46, 56). Autoreactive TrB cells normally express low levels of BAFFR and thus have a disadvantage in BAFF-induced survival compared to non-autoreactive TrB cells (48, 57). However, in patients with pRD, serum levels of BAFF are elevated, which allows autoreactive TrB cells to survive and become mature naïve B cells.
Mature naive lymphocytes can undergo antigen independent homeostatic proliferation to sustain lymphocyte numbers in response to lymphopenic environments (58, 59). In the case of mature B cells, this process is independent of T cells (58, 60), and relies on tonic signaling through BCR, BAFF-BAFFR signaling, and recently demonstrated Notch signaling (59, 61). Transfer of resting B cells into B cell-deficient mice resulted in homeostatic expansion of a fraction of the transferred cells, which acquired an activated phenotype and differentiated into IgM-secreting cells (58, 62). In addition, in a mouse model of OS with severe B cell developmental arrest, the few remaining B cells underwent homeostatic expansion and generated a normal or even enlarged compartment of antibody-secreting cells (ASC) (46, 56). These observations in mice suggest that homeostatically expanded B cells acquire an activated phenotype and readily differentiate into ASC. In line with these observations in mice, BLIMP1+CD138+ PCs are detected in lymph node biopsies from two OS patients who are virtually devoid of circulating B cells (46). In addition, Min et al. found abundant PC in the BM of a CID-G/AI patient with few B cells in the peripheral blood (36). More recently, CD38int, CD38- and CD27+ B cell subsets from patients with pRD were found to display increased activation status with higher CD69, CD80 and CD86 expression (38). These observations collectively suggest that, in patients with pRD, homeostatic proliferation allows B cells to acquire an activated phenotype and readily differentiate into PC. Additionally, reduced numbers of regulatory T cells in patients with pRD may further permit the activation and differentiation of the homeostatically expanded B cells (1).
Naïve B cells can be activated by antigen stimulation, toll-like receptor (TLR) ligands, cytokines and T cell-derived signals such as CD40 ligand. Activated B cells undergo proliferation, followed by differentiation into memory B or PC. CD27 has been presumed to be an exclusive marker for memory B cells, which can be further divided into CD27+IgD+ unswitched and CD27+IgD- switched memory B cells (63). Interestingly, an earlier study identified a CD19+IgD-CD27- double negative (DN) B cell population, which was phenotypically and functionally similar to CD27+ memory B cells, in patients with systemic lupus erythematosus (SLE) (64). Although DN B cells were also present in healthy individuals, their frequency was found to be increased in SLE and other autoimmune/autoinflammatory diseases such as mixed connective tissue disease (65) and multiple sclerosis (66), and in elderly people (67). Memory B cells can quickly differentiate into PC upon stimulation. In fact, a recent study demonstrated that DN B cells in patients with SLE were precursors of PC and upon stimulation with TLR7, IL-21, and IL-10, they efficiently differentiated into PC that secreted autoantibodies (68). It should be noted that T cell differentiation is biased toward memory/effector/helper cells in pRD and this bias may also contribute to the generation, activation and PC differentiation of these memory B-like cells (36, 38).
In some pRD patients, the distribution of B cell subsets, serum Ig levels and autoantibody production have been analyzed (25, 27, 28, 32, 36, 38, 69) (Table 1). These included two CID with γδ T cell expansion (γδ T+ CID), ten CID-G/AI, one ICL, one SIgAD, one AS, and five CID. Nineteen of the twenty patients showed increased proportions of CD27-IgD- DN B and/or CD27+ memory-like B cells. Both of these subsets have the propensity to become plasma cells upon stimulation. Consistently, despite the striking reduction of peripheral B cell numbers, the majority of these patients exhibited normal or even elevated serum Ig levels (Table 1).
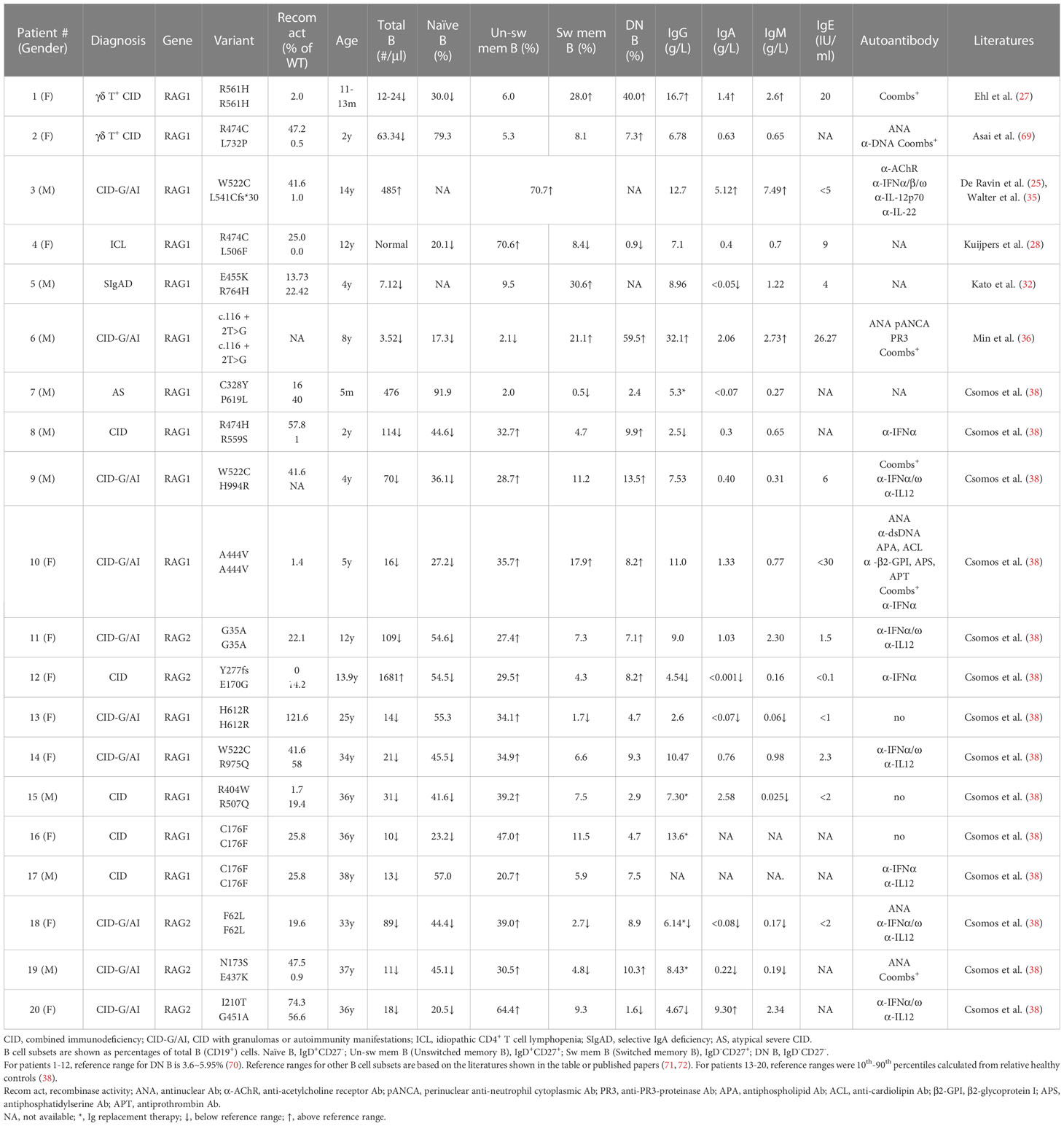
Table 1 B cell abnormalities and dysregulated antibody production in pRD patients.
Min et al. have recently reported a CID-G/AI patient caused by a c.116 + 2T>G homozygous splice site mutation in the first intron of RAG1 (Table 1, Patient 6) (36). The patient had few B cells in the peripheral blood, with a remarkable 59.5% being DN B cells. Despite B cell lymphopenia, the patient BM contained a higher proportion of CD38high PC and expressed elevated levels of transcripts for PRDM1 (encoding BLIMP1) and SDC1 (encoding CD138) compared to two HCs. An enzyme-linked immunospot (ELISPOT) assay confirmed the presence of increased number of PC in the patient BM. Interestingly, PC from the patient BM secreted higher amounts of IgM and IgG than those from the HCs. To investigate whether PCs can be generated from DN B cells, DN and naïve B cells were sorted from the peripheral blood of two patients with inborn errors of immunity (IEI) and cultured in the presence of TLR7 agonist and cytokines. Remarkably, it was found that DN B cells secreted 70-fold more IgG than did naïve B cells (36).
To explore whether the increased proportion of DN B and/or memory B cells observed in B cell lymphopenic pRD is also a general feature in patients with other IEI, Min et al. analyzed 25 immunodeficient patients with known causative genes (including RAG1/2, PIK3CD, PIK3R1, TACI, BTK, WAS, ADA2, IKBKG, SH2D1A, CD40LG, MHCII, AICDA) and 27 IEI patients with unknown causative genes. They found a strong association between the increased proportion of DN B and memory B (CD27+IgD-) cells and a decreased number and proportion of naive B cells in this diverse cohort (36). Notably, the proportions of DN B and memory B cells exhibited a striking increase when the count of naive B cells fell below 100 counts/µl. Therefore, the increased proportion of DN B and/or memory B cells appears to be a common characteristic in patients with IEI and B cell lymphopenia.
The pRD patients analyzed by Csomos et al. (Table 1) exhibited a substantial expansion of CD21lowCD19high B cell population, which is a clinically established hallmark of immune dysregulation (73–76). Moreover, CD11chiCXCR5lo B cell population, which is part of the CD21lowCD19high cells and resembles murine age-associated B cells (ABCs) (77), was present at a higher frequency at several B cell developmental stages in these patients (38). Consistently, the expression of T-bet, a transcription factor found in ABCs and in B cells of patients with immune dysregulation (77–79), was increased in the CD21lowCD11chigh compartment (38). In addition, such CD21low or CD11chigh B cells were found to be expanded in CVID and IEI caused by loss-of-function mutations in CTLA4, LRBA, AICDA, ADA2, NFKB1 or gain-of-function mutations in PIK3CD, STAT1, STAT3 and TLR7 (80–83).
Recent studies suggest that a subset of DN B (DN2, CD27-IgD-CXCR5-), unswitched and switched memory-like B, CD21lowCD19high B, CD11chighCXCR5low B and T-bethighCD21low B cells represent a group of related B cell subsets that share the T-bet+CD21lowCD11c+ phenotype (82). In vitro and in vivo studies indicate that T-bet is the critical transcription factor required for the generation of the CD11chighCD21low B cells (84) and that the expansion of the T-bethighCD21low B cells depend on BCR- and helper T cell-derived signals (81). Although the precise origin of and the relationship among these atypical B cell subsets are not fully understood, a common feature of these B cells is that they are prone to differentiate into PC upon stimulation, such as infections, and may produce autoantibodies (36, 68, 77, 82, 85, 86). These B cells may also serve as antigen-presenting cells by their high levels of CD80, CD86 and HLA-DR expression and activate autoreactive T cells (77, 82, 86), and may contribute to the pathogenesis of autoimmune/autoinflammatory diseases.
Based on the available data, Min et al. have recently proposed a model for autoantibody production in patients with pRD (36). An updated version of the model is shown in Figure 1. RAG mutations impair receptor editing (37, 38, 42, 46), allowing some autoreactive immature B cells to exit BM and become TrB cells. Since serum BAFF levels are elevated in the patients with B cell lymphopenia and/or inflammation (36, 38, 56), autoreactive TrB cells can survive and become mature naive B cells. These naive B cells undergo homeostatic proliferation due to the lymphopenic environment and the presence of high levels of BAFF, resulting in the generation of DN, CD11chighTbet+, or memory B cells (25, 27, 28, 32, 36, 38, 69). Additionally, recurrent infection and chronic inflammation in pRD may also contribute to the generation and/or accumulation of these abnormal B cell subsets. DN and CD11chighTbet+ B cells may represent two populations with similar characteristics rather than two distinct subsets. Upon stimulation with TLR ligands and cytokines or with self- or foreign antigens, these B cells can efficiently differentiate into ASC and migrate to the BM where they secrete large amounts of antibodies, including autoantibodies.
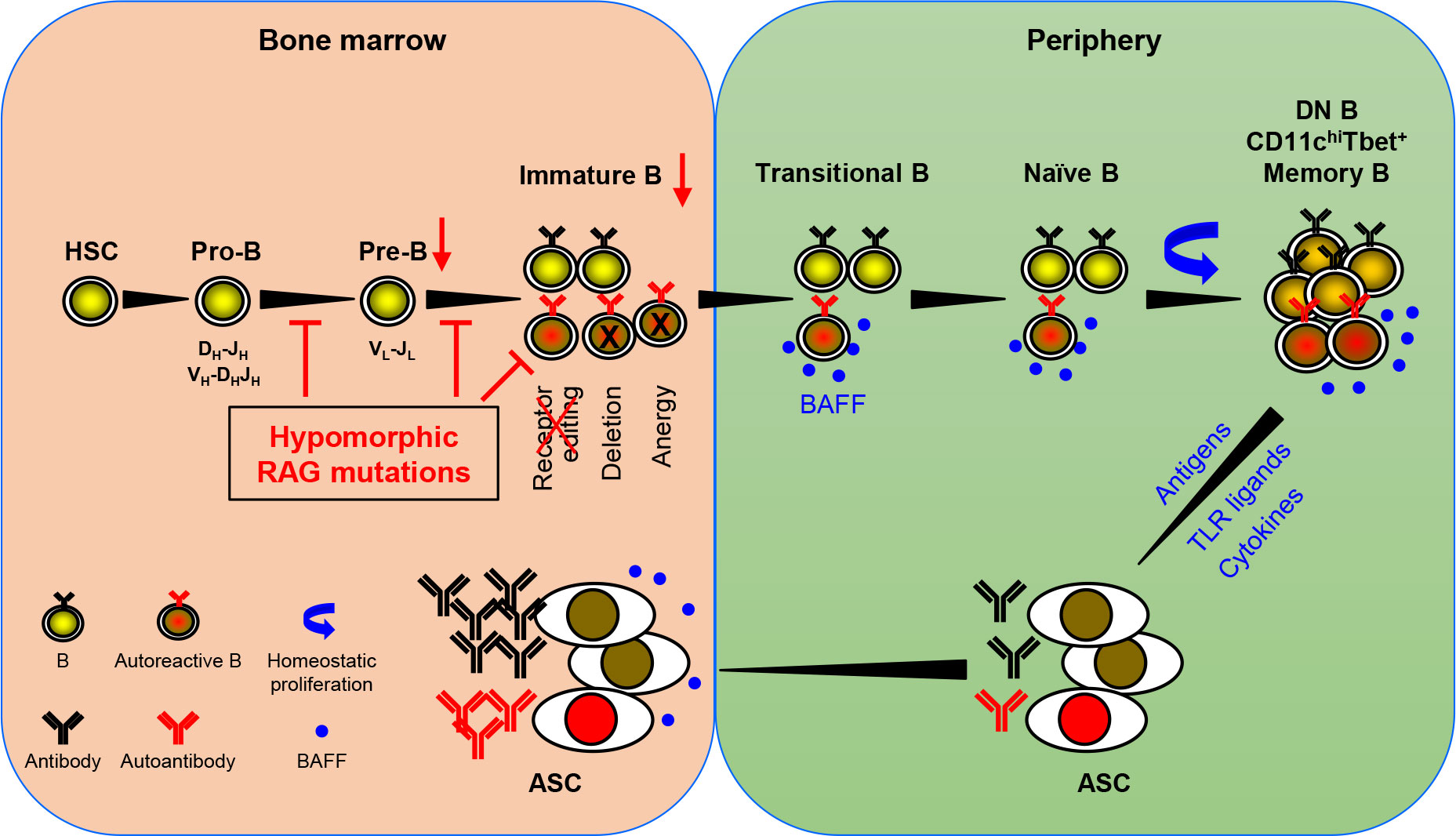
Figure 1 A model for autoantibody production in patients with pRD. pRD leads to limited V(D)J recombination and incomplete block in B cell development at both the pro-B to pre-B and the pre-B to immature B stages, resulting in reduced number of pre-B and immature B cells. In addition, pRD impairs receptor editing, allowing some autoreactive immature B cells to exit BM and become transitional B (TrB) cells in the periphery. Autoreactive TrB can survive in the presence of high levels of BAFF and become mature naïve B cells. These naïve B cells undergo homeostatic proliferation and differentiate into CD27-IgD- DN, CD11chiTbet+ or CD27+ memory B cells. Note that DN B and CD11chiTbet+ B cells may represent two populations with similar characteristics rather than two distinct subsets. Such more differentiated B cells can efficiently become ASC upon stimulation with TLR ligands and cytokines or with foreign or self-antigens, and home to the BM. The elevated levels of serum BAFF may also promote the generation and/or survival of ASC.
Patients with pRD present later in life and often pose a diagnostic dilemma with clinical features combining immune deficiency and dysregulation. Recent studies have found that the immune dysregulation represents a negative prognostic factor for survival in patients with pRD, even for those who are eventually treated with hematopoietic stem cell transplantation (HSCT) (87, 88). Therefore, early diagnosis and treatment are critical to improve patient outcomes (88). Intravenous immunoglobulin (IVIG), steroids and B cell depletion using rituximab are frequently used to treat pRD patients with autoimmunity and hyperinflammation manifestations. However, these therapies often fail to control the disease (33). The model shown in Figure 1 suggests that BAFF plays a key role in mediating the survival of autoreactive TrB cells, promoting mature B cell homeostatic expansion, and enhancing the generation of PC. Indeed, it has been reported that BAFFR blockade reduces serum levels of nucleic acid-specific autoantibodies and significantly ameliorates inflammatory tissue damage in hypomorphic RAG2-deficient mice (46). Therefore, the BAFF-neutralizing monoclonal antibody, belimumab, could be an effective treatment for pRD patients who respond poorly to the current therapeutics. This model also suggests that eliminating autoreactive ASC could be another potential approach. These therapies could be of great importance in controlling immune dysregulation and bridging the patients to HSCT, which is considered a definitive therapy.
QM provided a draft of the manuscript. KC, YL, LD, ZH, XM, MY, and JW reviewed and corrected the manuscript. J-YW designed the outline and made the final corrections to the manuscript. All the authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (grant #32270932 to J-YW), Projects of International Cooperation and Exchanges NSFC (grant #82011540008 to J-YW), National Natural Science Foundation for Young Scholar (grant #82202013 to QM), China Postdoctoral Science Foundation Grant (grant #2022M720782 to QM), the National Institutes of Health (NIH National Institute of Allergy and Infectious Diseases (NIAID) (R01AI153830-05, JW), Robert A. Good Endowment, University of South Florida (JW), Jeffrey Modell Foundation (JW) and Johns Hopkins All Children’s Hospital Foundation (JW).
We thank the clinicians in the Department of Clinical Immunology, Children’s Hospital of Fudan University and the members in Wang lab for their helpful suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol (2016) 16(4):234–46. doi: 10.1038/nri.2016.28
2. Ru H, Zhang P, Wu H. Structural gymnastics of RAG-mediated DNA cleavage in V(D)J recombination. Curr Opin Struct Biol (2018) 53:178–86. doi: 10.1016/j.sbi.2018.11.001
3. Liu C, Zhang Y, Liu CC, Schatz DG. Structural insights into the evolution of the RAG recombinase. Nat Rev Immunol (2022) 22(6):353–70. doi: 10.1038/s41577-021-00628-6
4. Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet (2011) 45:167–202. doi: 10.1146/annurev-genet-110410-132552
5. Villa A, Notarangelo LD. RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev (2019) 287(1):73–90. doi: 10.1111/imr.12713
6. Lescale C, Deriano L. The RAG recombinase: beyond breaking. Mech Ageing Dev (2017) 165(Pt A):3–9. doi: 10.1016/j.mad.2016.11.003
7. Lu H, Schwarz K, Lieber MR. Extent to which hairpin opening by the Artemis : DNA-PKcs complex can contribute to junctional diversity in V(D)J recombination. Nucleic Acids Res (2007) 35(20):6917–23. doi: 10.1093/nar/gkm823
8. Desiderio SV, Yancopoulos GD, Paskind M, Thomas E, Boss MA, Landau N, et al. Insertion of n regions into heavy-chain genes is correlated with expression of terminal deoxytransferase in b cells. Nature (1984) 311(5988):752–5. doi: 10.1038/311752a0
9. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131(4):959–71. doi: 10.1016/j.jaci.2013.01.046
10. Delmonte OM, Schuetz C, Notarangelo LD. RAG deficiency: two genes, many diseases. J Clin Immunol (2018) 38(6):646–55. doi: 10.1007/s10875-018-0537-4
11. Bulkhi AA, Dasso JF, Schuetz C, Walter JE. Approaches to patients with variants in RAG genes: from diagnosis to timely treatment. Expert Rev Clin Immunol (2019) 15(10):1033–46. doi: 10.1080/1744666X.2020.1670060
12. Melchers F. Checkpoints that control b cell development. J Clin Invest (2015) 125(6):2203–10. doi: 10.1172/JCI78083
13. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human b cell precursors. Science (2003) 301(5638):1374–7. doi: 10.1126/science.1086907
14. Nemazee D. Mechanisms of central tolerance for b cells. Nat Rev Immunol (2017) 17(5):281–94. doi: 10.1038/nri.2017.19
15. Ait-Azzouzene D, Verkoczy L, Peters J, Gavin A, Skog P, Vela JL, et al. An immunoglobulin c kappa-reactive single chain antibody fusion protein induces tolerance through receptor editing in a normal polyclonal immune system. J Exp Med (2005) 201(5):817–28. doi: 10.1084/jem.20041854
16. Duong BH, Ota T, Aoki-Ota M, Cooper AB, Ait-Azzouzene D, Vela JL, et al. Negative selection by IgM superantigen defines a b cell central tolerance compartment and reveals mutations allowing escape. J Immunol (2011) 187(11):5596–605. doi: 10.4049/jimmunol.1102479
17. Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of b cell tolerance toward membrane antigens. Nat Immunol (2004) 5(6):645–50. doi: 10.1038/ni1076
18. Casellas R, Shih TA, Kleinewietfeld M, Rakonjac J, Nemazee D, Rajewsky K, et al. Contribution of receptor editing to the antibody repertoire. Science (2001) 291(5508):1541–4. doi: 10.1126/science.1056600
19. Gennery A. Recent advances in understanding RAG deficiencies. F1000Research (2019) 8:148. doi: 10.12688/f1000research.17056.1
20. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human b cell-negative SCID. Science (1996) 274(5284):97–9. doi: 10.1126/science.274.5284.97
21. Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to omenn syndrome. Cell (1998) 93(5):885–96. doi: 10.1016/S0092-8674(00)81448-8
22. Giblin W, Chatterji M, Westfield G, Masud T, Theisen B, Cheng HL, et al. Leaky severe combined immunodeficiency and aberrant DNA rearrangements due to a hypomorphic RAG1 mutation. Blood (2009) 113(13):2965–75. doi: 10.1182/blood-2008-07-165167
23. Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol (2011) 141(1):73–82. doi: 10.1016/j.clim.2011.05.007
24. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. New Engl J Med (2008) 358(19):2030–8. doi: 10.1056/NEJMoa073966
25. De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic rag mutations can cause destructive midline granulomatous disease. Blood (2010) 116(8):1263–71. doi: 10.1182/blood-2010-02-267583
26. de Villartay JP, Lim A, Al-Mousa H, Dupont S, Déchanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest (2005) 115(11):3291–9. doi: 10.1172/JCI25178
27. Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kühr J, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest (2005) 115(11):3140–8. doi: 10.1172/JCI25221
28. Kuijpers TW, Ijspeert H, van Leeuwen EM, Jansen MH, Hazenberg MD, Weijer KC, et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood (2011) 117(22):5892–6. doi: 10.1182/blood-2011-01-329052
29. Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol (2014) 134(6):1375–80. doi: 10.1016/j.jaci.2014.04.042
30. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol (2015) 35(2):119–24. doi: 10.1007/s10875-014-0121-5
31. Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol (2018) 141(6):2303–6. doi: 10.1016/j.jaci.2018.02.007
32. Kato T, Crestani E, Kamae C, Honma K, Yokosuka T, Ikegawa T, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol (2015) 35(3):280–8. doi: 10.1007/s10875-015-0146-4
33. Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol In practice (2019) 7(6):1970–85.e4. doi: 10.1016/j.jaip.2019.02.038
34. Geier CB, Farmer JR, Foldvari Z, Ujhazi B, Steininger J, Sleasman JW, et al. Vasculitis as a major morbidity factor in patients with partial RAG deficiency. Front Immunol (2020) 11:574738. doi: 10.3389/fimmu.2020.574738
35. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2015) 125(11):4135–48. doi: 10.1172/JCI80477
36. Min Q, Meng X, Zhou Q, Wang Y, Li Y, Lai N, et al. RAG1 splicing mutation causes enhanced b cell differentiation and autoantibody production. JCI Insight (2021) 6(19):e148887. doi: 10.1172/jci.insight.148887
37. IJ H, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol (2014) 133(4):1124–33. doi: 10.1016/j.jaci.2013.11.028
38. Csomos K, Ujhazi B, Blazso P, Herrera JL, Tipton CM, Kawai T, et al. Partial RAG deficiency in humans induces dysregulated peripheral lymphocyte development and humoral tolerance defect with accumulation of T-bet(+) b cells. Nat Immunol (2022) 23(8):1256–72. doi: 10.1038/s41590-022-01271-6
39. Watson CT, Breden F. The immunoglobulin heavy chain locus: genetic variation, missing data, and implications for human disease. Genes immunity (2012) 13(5):363–73. doi: 10.1038/gene.2012.12
40. Calis JJ, Rosenberg BR. Characterizing immune repertoires by high throughput sequencing: strategies and applications. Trends Immunol (2014) 35(12):581–90. doi: 10.1016/j.it.2014.09.004
41. Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA, et al. Characterization of T and b cell repertoire diversity in patients with RAG deficiency. Sci Immunol (2016) 1(6):eaah6109. doi: 10.1126/sciimmunol.aah6109
42. Ott de Bruin LM, Bosticardo M, Barbieri A, Lin SG, Rowe JH, Poliani PL, et al. Hypomorphic Rag1 mutations alter the preimmune repertoire at early stages of lymphoid development. Blood (2018) 132(3):281–92. doi: 10.1182/blood-2017-12-820985
43. Pascual V, Victor K, Lelsz D, Spellerberg MB, Hamblin TJ, Thompson KM, et al. Nucleotide sequence analysis of the V regions of two IgM cold agglutinins. evidence that the VH4-21 gene segment is responsible for the major cross-reactive idiotype. J Immunol (1991) 146(12):4385–91.
44. Silberstein LE, Jefferies LC, Goldman J, Friedman D, Moore JS, Nowell PC, et al. Variable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood (1991) 78(9):2372–86. doi: 10.1182/blood.V78.9.2372.2372
45. Ramsey LB, Vegoe AL, Miller AT, Cooke MP, Farrar MA. Tonic BCR signaling represses receptor editing via raf- and calcium-dependent signaling pathways. Immunol Lett (2011) 135(1-2):74–7. doi: 10.1016/j.imlet.2010.09.018
46. Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of omenn syndrome. J Exp Med (2010) 207(7):1525–40. doi: 10.1084/jem.20091928
47. Kowalczyk-Quintas C, Chevalley D, Willen L, Jandus C, Vigolo M, Schneider P. Inhibition of membrane-bound BAFF by the anti-BAFF antibody belimumab. Front Immunol (2018) 9:2698. doi: 10.3389/fimmu.2018.02698
48. Smulski CR, Eibel H. BAFF and BAFF-receptor in b cell selection and survival. Front Immunol (2018) 9:2285. doi: 10.3389/fimmu.2018.02285
49. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9(7):491–502. doi: 10.1038/nri2572
50. Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and b lymphocyte stimulator. Science (1999) 285(5425):260–3. doi: 10.1126/science.285.5425.260
51. Vigolo M, Chambers MG, Willen L, Chevalley D, Maskos K, Lammens A, et al. A loop region of BAFF controls b cell survival and regulates recognition by different inhibitors. Nat Commun (2018) 9(1):1199. doi: 10.1038/s41467-018-03323-8
52. Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med (2007) 204(8):1959–71. doi: 10.1084/jem.20062567
53. Figgett WA, Fairfax K, Vincent FB, Le Page MA, Katik I, Deliyanti D, et al. The TACI receptor regulates T-cell-independent marginal zone b cell responses through innate activation-induced cell death. Immunity (2013) 39(3):573–83. doi: 10.1016/j.immuni.2013.05.019
54. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi: 10.1084/jem.20031330
55. Kreuzaler M, Rauch M, Salzer U, Birmelin J, Rizzi M, Grimbacher B, et al. Soluble BAFF levels inversely correlate with peripheral b cell numbers and the expression of BAFF receptors. J Immunol (2012) 188(1):497–503. doi: 10.4049/jimmunol.1102321
56. Walter JE, Rucci F, Patrizi L, Recher M, Regenass S, Paganini T, et al. Expansion of immunoglobulin-secreting cells and defects in b cell tolerance in rag-dependent immunodeficiency. J Exp Med (2010) 207(7):1541–54. doi: 10.1084/jem.20091927
57. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged b cells due to increased dependence on BAFF. Immunity (2004) 20(4):441–53. doi: 10.1016/S1074-7613(04)00079-2
58. Woodland RT, Schmidt MR. Homeostatic proliferation of b cells. Semin Immunol (2005) 17(3):209–17. doi: 10.1016/j.smim.2005.02.006
59. Gómez Atria D, Gaudette BT, Londregan J, Kelly S, Perkey E, Allman A, et al. Stromal notch ligands foster lymphopenia-driven functional plasticity and homeostatic proliferation of naive b cells. J Clin Invest (2022) 132(13):e158885. doi: 10.1172/JCI158885
60. Cabatingan MS, Schmidt MR, Sen R, Woodland RT. Naive b lymphocytes undergo homeostatic proliferation in response to b cell deficit. J Immunol (2002) 169(12):6795–805. doi: 10.4049/jimmunol.169.12.6795
61. Tchao NK, Turka LA. Lymphodepletion and homeostatic proliferation: implications for transplantation. Am J Transplant (2012) 12(5):1079–90. doi: 10.1111/j.1600-6143.2012.04008.x
62. Agenès F, Freitas AA. Transfer of small resting b cells into immunodeficient hosts results in the selection of a self-renewing activated b cell population. J Exp Med (1999) 189(2):319–30. doi: 10.1084/jem.189.2.319
63. Anolik JH, Looney RJ, Lund FE, Randall TD, Sanz I. Insights into the heterogeneity of human b cells: diverse functions, roles in autoimmunity, and use as therapeutic targets. Immunologic Res (2009) 45(2-3):144–58. doi: 10.1007/s12026-009-8096-7
64. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the b cell memory compartment in systemic lupus erythematosus. J Immunol (2007) 178(10):6624–33. doi: 10.4049/jimmunol.178.10.6624
65. Hajas A, Barath S, Szodoray P, Nakken B, Gogolak P, Szekanecz Z, et al. Derailed b cell homeostasis in patients with mixed connective tissue disease. Hum Immunol (2013) 74(7):833–41. doi: 10.1016/j.humimm.2013.04.007
66. Fraussen J, Marquez S, Takata K, Beckers L, Montes Diaz G, Zografou C, et al. Phenotypic and ig repertoire analyses indicate a common origin of IgD(-)CD27(-) double negative b cells in healthy individuals and multiple sclerosis patients. J Immunol (2019) 203(6):1650–64. doi: 10.4049/jimmunol.1801236
67. Bulati M, Buffa S, Martorana A, Candore G, Lio D, Caruso C, et al. Trafficking phenotype and production of granzyme b by double negative b cells (IgG(+)IgD(-)CD27(-)) in the elderly. Exp gerontol (2014) 54:123–9. doi: 10.1016/j.exger.2013.12.011
68. Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct effector b cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity (2018) 49(4):725–39.e6. doi: 10.1016/j.immuni.2018.08.015
69. Asai E, Wada T, Sakakibara Y, Toga A, Toma T, Shimizu T, et al. Analysis of mutations and recombination activity in RAG-deficient patients. Clin Immunol (2011) 138(2):172–7. doi: 10.1016/j.clim.2010.11.005
70. Li Y, Li Z, Hu F. Double-negative (DN) b cells: an under-recognized effector memory b cell subset in autoimmunity. Clin Exp Immunol (2021) 205(2):119–27. doi: 10.1111/cei.13615
71. Ding Y, Zhou L, Xia Y, Wang W, Wang Y, Li L, et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China. J Allergy Clin Immunol (2018) 142(3):970–3.e8. doi: 10.1016/j.jaci.2018.04.022
72. Garcia-Prat M, Álvarez-Sierra D, Aguiló-Cucurull A, Salgado-Perandrés S, Briongos-Sebastian S, Franco-Jarava C, et al. Extended immunophenotyping reference values in a healthy pediatric population. Cytometry Part B Clin cytometry (2019) 96(3):223–33. doi: 10.1002/cyto.b.21728
73. Warnatz K, Wehr C, Dräger R, Schmidt S, Eibel H, Schlesier M, et al. Expansion of CD19(hi)CD21(lo/neg) b cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology (2002) 206(5):502–13. doi: 10.1078/0171-2985-00198
74. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low b cells in common variable immunodeficiency resemble tissue homing, innate-like b cells. Proc Natl Acad Sci United States America (2009) 106(32):13451–6. doi: 10.1073/pnas.0901984106
75. Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive b cells contain mostly autoreactive unresponsive clones. Blood (2010) 115(24):5026–36. doi: 10.1182/blood-2009-09-243071
76. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The T(H)1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol (2018) 141(2):730–40. doi: 10.1016/j.jaci.2017.04.041
77. Cancro MP. Age-associated b cells. Annu Rev Immunol (2020) 38:315–40. doi: 10.1146/annurev-immunol-092419-031130
78. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T-Box transcription factor T-bet, a key player in a unique type of b-cell activation essential for effective viral clearance. Proc Natl Acad Sci United States America (2013) 110(34):E3216–24. doi: 10.1073/pnas.1312348110
79. Kačar M, Markelj G, Avčin T. Autoimmune and autoinflammatory manifestations in inborn errors of immunity. Curr Opin Allergy Clin Immunol (2022) 22(6):343–51. doi: 10.1097/ACI.0000000000000860
80. Fernando SL, Jang HS, Li J. The immune dysregulation of common variable immunodeficiency disorders. Immunol letters (2021) 230:21–6. doi: 10.1016/j.imlet.2020.12.002
81. Keller B, Strohmeier V, Harder I, Unger S, Payne KJ, Andrieux G, et al. The expansion of human T-bet(high)CD21(low) b cells is T cell dependent. Sci Immunol (2021) 6(64):eabh0891. doi: 10.1126/sciimmunol.abh0891
82. Keller B, Warnatz K. T-bet(high)CD21(low) b cells: the need to unify our understanding of a distinct b cell population in health and disease. Curr Opin Immunol (2023) 82:102300. doi: 10.1016/j.coi.2023.102300
83. Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature (2022) 605(7909):349–56. doi: 10.1038/s41586-022-04642-z
84. Yang R, Avery DT, Jackson KJL, Ogishi M, Benhsaien I, Du L, et al. Human T-bet governs the generation of a distinct subset of CD11c(high)CD21(low) b cells. Sci Immunol (2022) 7(73):eabq3277. doi: 10.1126/sciimmunol.abq3277
85. Mouat IC, Goldberg E, Horwitz MS. Age-associated b cells in autoimmune diseases. Cell Mol Life sciences: CMLS (2022) 79(8):402. doi: 10.1007/s00018-022-04433-9
86. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest (2017) 127(4):1392–404. doi: 10.1172/JCI91250
87. Delmonte OM, Villa A, Notarangelo LD. Immune dysregulation in patients with RAG deficiency and other forms of combined immune deficiency. Blood (2020) 135(9):610–9. doi: 10.1182/blood.2019000923
Keywords: RAG deficiency, B cell tolerance, BAFF, homeostatic proliferation, double negative B, CD11chighTbet+ B, autoantibody production
Citation: Min Q, Csomos K, Li Y, Dong L, Hu Z, Meng X, Yu M, Walter JE and Wang J-Y (2023) B cell abnormalities and autoantibody production in patients with partial RAG deficiency. Front. Immunol. 14:1155380. doi: 10.3389/fimmu.2023.1155380
Received: 31 January 2023; Accepted: 15 June 2023;
Published: 05 July 2023.
Edited by:
Dinakantha Suramya Kumararatne, Addenbrooke’s Hospital, United KingdomReviewed by:
David Andrew Fulcher, Australian National University, AustraliaCopyright © 2023 Min, Csomos, Li, Dong, Hu, Meng, Yu, Walter and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Yang Wang, d2FuZ0BmdWRhbi5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.