 Qiao Li
Qiao Li Yue-Zi Hu
Yue-Zi Hu Shan Gao
Shan Gao Peng-Fei Wang
Peng-Fei Wang Zhao-Lan Hu
Zhao-Lan Hu Ru-Ping Dai
Ru-Ping Dai- 1Department of Anesthesiology, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China
- 2Anesthesia Medical Research Center, Central South University, Changsha, Hunan, China
- 3Clinical Laboratory, The Second Hospital of Hunan University of Chinese Medicine, Changsha, Hunan, China
Immune-mediated inflammatory diseases (IMIDs) consist of a common and clinically diverse group of diseases. Despite remarkable progress in the past two decades, no remission is observed in a large number of patients, and no effective treatments have been developed to prevent organ and tissue damage. Brain-derived neurotrophic factor precursor (proBDNF) and receptors, such as p75 neurotrophin receptor (p75NTR) and sortilin, have been proposed to mediate intracellular metabolism and mitochondrial function to regulate the progression of several IMIDs. Here, the regulatory role of proBDNF and its receptors in seven typical IMIDs, including multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus, allergic asthma, type I diabetes, vasculitis, and inflammatory bowel diseases, was investigated.
1 Introduction
Immune-mediated inflammatory diseases (IMIDs) are a group of highly disabling chronic diseases characterized by immune dysregulation and chronic inflammation as the basic manifestation, which affect different organs and systems (1, 2). The prevalence of IMIDs in well-developed countries is about 5%–8% (1), and its global incidence gradually increases (3, 4). IMIDs consist of more than 100 different types of diseases, such as multiple sclerosis (MS), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), allergic asthma, type I diabetes (T1D), vasculitis, and inflammatory bowel diseases (IBD), involving multiple disciplinary fields (5–8). At present, no effective targeted treatment has been developed for IMIDs; thus, exploring novel therapeutic targets for IMIDs is necessary.
Based on previous reports, targeting brain-derived neurotrophic factor (BDNF), BDNF precursor (proBDNF), and its receptors exert therapeutic effects in IMIDs. The administration of anti-proBDNF monoclonal antibodies (mAb-proB) can effectively improve the neurological score and reduce the number of lymphocytes in the experimental autoimmune encephalomyelitis (EAE) mouse model, a classic model of MS. ProBDNF and its high-affinity receptor, p75 neurotrophin receptor (p75NTR), were upregulated in peripheral blood mononuclear cells (PBMCs) from patients with RA compared with healthy controls. Treatment of the extracellular domain of p75NTR (p75ECD) can significantly relieve inflammatory pain in collagen-induced arthritis (CIA) model mice, which is a standard RA model. MAb-proB could also reduce the production of auto-antibodies and attenuate kidney injury in SLE by altering the mitochondrial respiratory chain complex transcription level and cholesterol metabolism (9, 10).
ProBDNF plays an important role in the mitochondria-mediated release of cytochrome C (cyt C) to regulate cell death by binding the receptor complex of p75NTR and sortilin (11). P75NTR signaling could regulate glucose uptake (12), and sortilin is a crucial regulatory molecule of lipid metabolism (13, 14). In this review, the potential effects of proBDNF and its receptors on glucose, lipid, and mitochondrial metabolism in IMIDs were explored and concluded.
2 ProBDNF and its receptors
2.1 Role of proBDNF signaling in immune cells and IMIDs
BDNF is a widely studied member of the neurotrophin family. The BDNF gene produces preproBDNF protein in the endoplasmic reticulum, which is processed to proBDNF in the Golgi for sorting into either constitutive or regulated secretory vesicles (15). ProBDNF may be cleaved into mature BDNF (mBDNF) intracellularly by furin in the trans-Golgi network or proconvertases in secretory vesicles. ProBDNF and mBDNF can be secreted from neurons in an activity-dependent manner. In addition, proBDNF can be cleaved into mBDNF extracellularly by plasmin or selective matrix metalloproteinases (MMPs), including MMP3, MMP7, and MMP9 (16, 17) (Figure 1). ProBDNF and mBDNF play contrasting biological roles in the synaptic structure, plasticity, transmission, and activity in the central nervous system (CNS) (18). MBDNF binds tyrosine kinase receptor B (TrkB) to promote cell survival, whereas proBDNF induces apoptosis (16, 19–22).
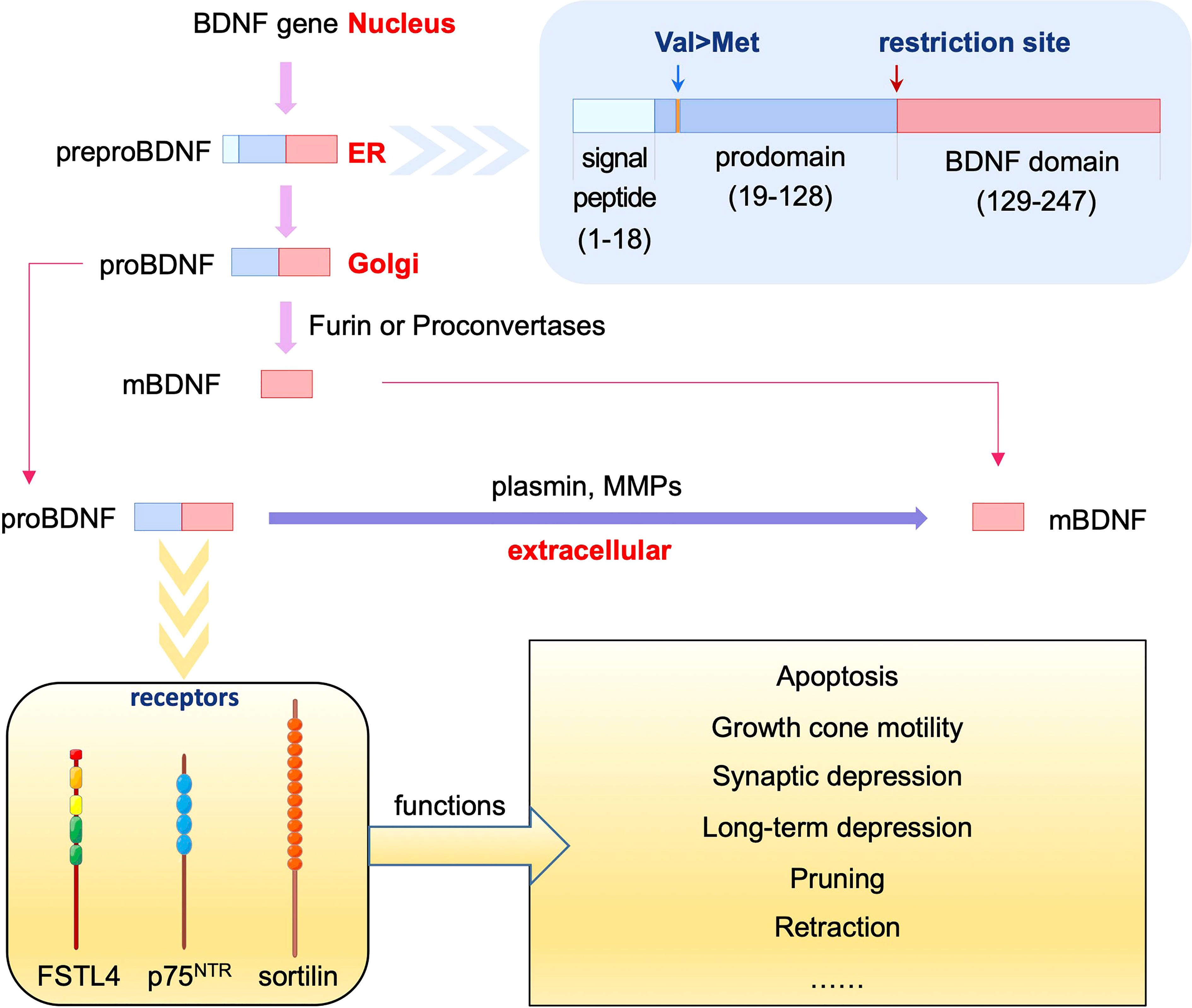
Figure 1 Processing and function of proBDNF and its receptors. The BDNF gene produces preproBDNF protein in the ER, which is processed to proBDNF in Golgi. ProBDNF may be cleaved into mBDNF intracellularly by furin or proconvertases, or extracellularly by plasmin or MMPs. ProBDNF binds to p75NTR, sortilin or FSTL4 to exert different effects including apoptosis, growth cone motility, synaptic depression, long-term depression, pruning, retraction and other functions.
ProBDNF consists of 129 amino acids in the N-terminal prodomain and 118 amino acids in the C-terminal mature domain (23). In the CNS, proBDNF is primarily located in the spinal cord dorsal horn, nuclei tractus solitarius, spinal trigeminal nuclei, spinal trigeminal nuclei, hypothalamus, and amygdala (24). Multiple expressions of proBDNF are also found in peripheral tissues, such as the skin, intestine, adrenal gland, pituitary gland, spinal cord dorsal horn (24), and liver (25). ProBDNF inhibits neural stem cell proliferation, differentiation, and migration, and reduces the number of neurons, oligodendrocytes, and astrocytes, whereas anti-proBDNF antibodies reverse neural stem cell proliferation and differentiation (26). Recombinant proBDNF protein modulates neuronal architecture and alters the long-term plasticity in the hippocampus in vitro; however, the role of endogenous proBDNF remains unclear (27).
ProBDNF can be expressed not only in the CNS and peripheral tissues, but also in immune cells such as monocytes/macrophages (28), T cells (29, 30), and B cells (9). In addition, proBDNF plays an important role in IMIDs. Studies have shown that peripheral macrophages can secrete proBDNF under certain inflammatory conditions (31, 32). Our previous studies also indicated that proBDNF, as a key mediator of neuroinflammation in spinal cord injury and inflammatory pain, is released from infiltrating macrophages, and its activity can be reduced using mAb-proB (31, 32). The increased proBDNF expression on monocytes/macrophages promotes inflammatory response in type A aortic dissection patients with severe systemic inflammation (28). The injection of lipopolysaccharide (LPS) could induce the upregulation of proBDNF in CD4+ T cells (30, 33) and further modulate sepsis-associated encephalopathy (30). The activation of Rac1 and TRPM7 channels in innate immune cells, such as microglia, can mediate the combination of proBDNF and p75NTR, which induced a sustained increase in intracellular Ca2+ concentration and enhanced IFN-γ-induced nitric oxide production (34). Interestingly, proBDNF does not always exert pro-inflammatory effects. Our recent study revealed that endogenous proBDNF in proinflammatory monocytes/macrophages played a protective role by regulating MMP‐9 signaling in acute myocardial infarction (AMI). Administration of mAb-proB skewed monocytes/macrophages into a proinflammatory phenotype after AMI (35).
Studies have shown that the receptors binding to proBDNF include p75NTR (9), sortilin (32, 36), and follistatin Like 4 (FSTL4) (37), which play different roles in nerve-immunity-endocrine network (Figure 1).
P75NTR, a member of the tumor necrosis factor receptor superfamily, is a receptor with high affinity to proBDNF. ProBDNF binds to p75NTR to promote cell death and inhibit long-term potentiation and neuronal axon outgrowth (38, 39), and to be involved in regulating neurotransmitter release in the entorhinal cortex (18). ProBDNF does not always induce cell death but can also be involved in regulating synaptic activity, pruning, and network reorganization (18). In the CNS, proBDNF/p75NTR weakens synaptic transmission under the synergistic effect of sortilin protein, negatively regulating synaptic plasticity; triggering neuronal apoptosis, axon pruning, and axon collapse; and exerting biological effects contrary to mBDNF (27, 39–43).
By the end of the 1980s, studies have found that in addition to neurons, the expression of p75NTR can also be detected on various immune cells such as PBMCs (44). Increased expression of proBDNF and p75NTR could be detected in PBMCs after strenuous exercise in normal adult males (45). This result indicates that when vigorous exercise reaches a certain threshold, immune cells produce proBDNF in an autocrine and/or paracrine manner to regulate apoptotic pathways. ProBDNF binds to p75NTR on monocytes, thereby activating the NF-κB pathway and enhancing the immune function of peripheral lymphocytes (29, 46). In addition, B cells can self-secrete proBDNF, and the combination of proBDNF and p75NTR with the cooperation of sortilin transporter induces apoptosis of B cells (47).
Sortilin belongs to the VPS10 family (48) and exerts a dual function involved in intracellular protein transport and cell signal transduction to regulate neuronal death or survival and the process of immune cells. It is primarily expressed on macrophages and dendritic cells and, to a lesser extent, on B and T cells (49). In neurons, sortilin serves as a membrane-bound coreceptor complex with p75NTR to facilitate the affinity of proBDNF-binding p75NTR to signal cell death (40, 50, 51). Studies have pointed out that sortilin plays a role in the survival and activation of B cells by regulating the transport of BDNF, and silencing sortilin can decrease the secretion of BDNF and increase the apoptosis of B cells (47). The loss of sortilin on cytotoxic T cells can decrease the release of IFN-γ and increase the expression of granzyme A in T cells (52). Rogers et al. found that sortilin binds to p75NTR to mediate NK cell apoptosis, and blocking sortilin with neurotensin could reduce NK cell death (53). In addition, sortilin plays a role in the antigen processing of DCs (49).
FSTL4, also known as SPARC-related protein containing immunoglobulin domains 1 (SPIG1), belongs to the SPARC family (54). It consists of a signal peptide, a follistatin-like domain, an extracellular calcium-binding domain with two EF-hand motifs, and two immunoglobulin-like domains (54, 55). Based on previous reports, FSTL4 negatively regulates BDNF maturation by binding to proBDNF (54). Furthermore, Suzuki et al. confirmed that the extinction of aversive memories was enhanced in Spig1-KO mice, revealing that FSTL4 suppresses synaptic plasticity in the extinction of inhibitory avoidance memory, which might be associated with its negative regulation on BDNF maturation from proBDNF (55). Our study indicated that the proBDNF/FSTL4 pathway contributed to neuronal apoptosis, whereas its downstream signaling remained unknown (37).
2.2 ProBDNF and its receptors mediate the regulation of metabolism and mitochondria
ProBDNF treatment could cause a significant dose-related decrease in mitochondria membrane potential, but it could not alter LDH released from dying or damaged cortical neurons (56). A study reported that proBDNF binds to p75NTR and sortilin to induce mitochondrial apoptosis by inhibiting the PI3K signaling pathway, which contributes to neuronal apoptosis in dorsal root ganglia (57). The age-dependent increase in proBDNF expression was found to be associated with a decrease in mitochondrial metabolism activity and content of epididymal white adipose tissue (eWAT) (58). Therefore, upregulated proBDNF expression in adipose progenitor cells of aged animals triggered the death of adipocytes, leading to the infiltration of immune cells and disruption of metabolic fitness. In addition, val66Met single-nucleotide polymorphism in the prodomain of BDNF induces altered trafficking of BDNF within neurons and decreases the activity-dependent secretion of mBDNF (59). Moreover, this variant could increase food intake in mice, which is consistent with the orexigenic activity of p75NTR (60). Based on these reports, proBDNF may exert stronger control of energy regulation. ProBDNF could be cleaved to BDNF if energy supply is sufficient even excessive, converting the orexigenic activity to anorexigenic.
Three main downstream signaling pathways are identified after p75NTR activation, including RhoA, JNK, and NF-κB pathways (43, 46, 61, 62). Among these, the canonical pathway mediating cell death is JNK signaling activated by the p75NTR/sortilin complex, which causes the activation of proapoptotic Bak and Bax proteins in the Bcl-2 family, cyt C release from the mitochondria, and formation of apoptosome, followed by caspase9/3 activation, which contributes to cell death and synaptic depression (11, 63) (Figure 2A). Although p75NTR signaling has been well-characterized over the years, few studies have well interpreted the signaling pathways involved to exert effects on mitochondrial metabolism. Furthermore, signaling cascades required for the regulation of mitochondrial metabolism remain unclear.
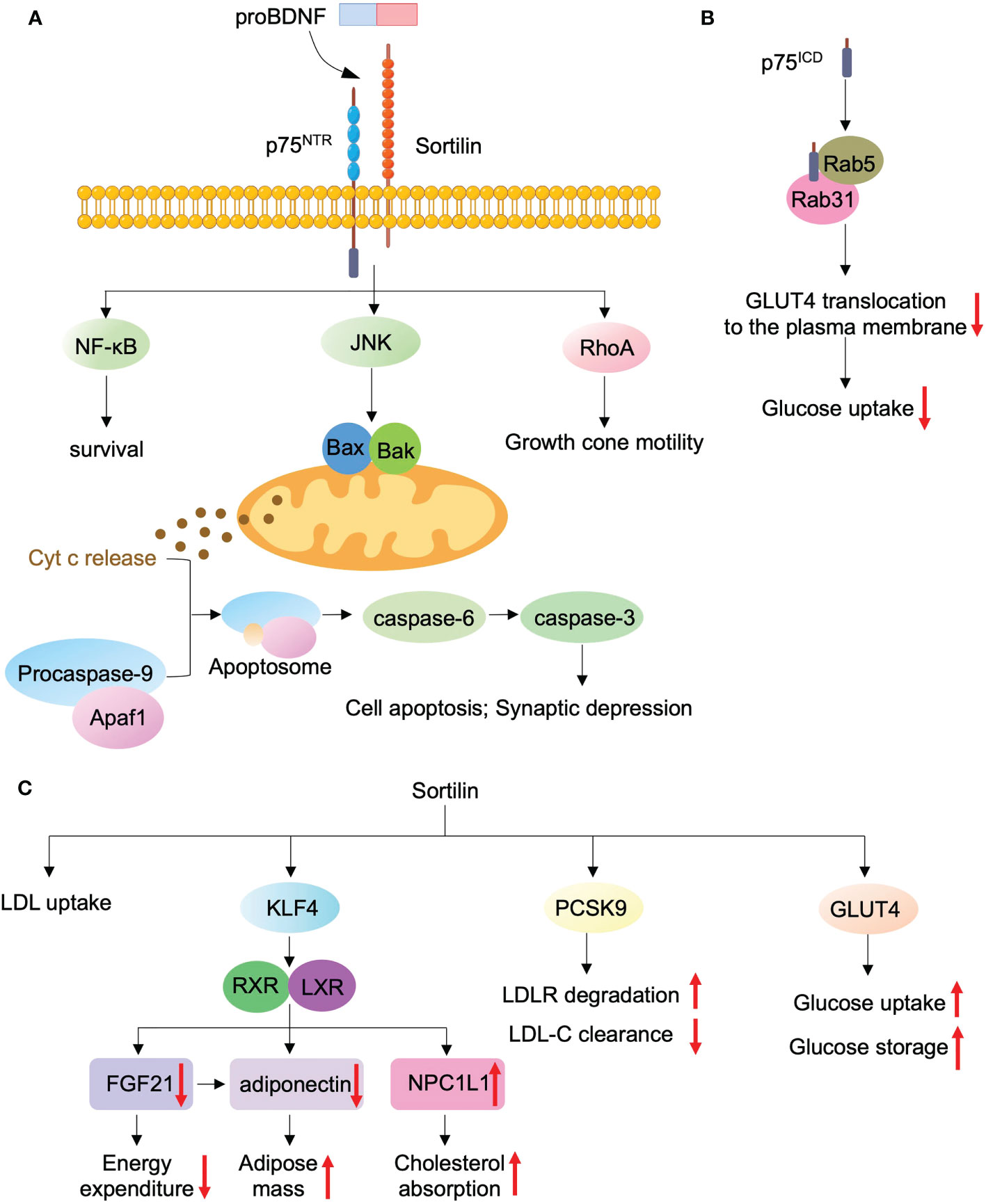
Figure 2 ProBDNF and their receptors mediate the regulation of metabolism and mitochondria. ProBDNF binds specifically to p75NTR through interaction with sortilin, which induces JNK signaling to increase the release of cytochrome C from mitochondria and promote cell apoptosis and synaptic depression. ProBDNF binds p75NTR and sortilin promotes growth cone motility and cell survival via activating RhoA and NF-κB, respectively (A). P75ICD forms a complex with Rab5 and Rab31 GTPases to enhance glucose uptake via promoting GLUT4 translocation to the plasma membrane (B). Sortilin also enhances LDL uptake and LDLR degradation, whereas decreases clearance of LDL-C. Sortilin promotes glucose uptake and storage via interacting with GLUT4 (C).
P75NTR is expressed in WAT, skeletal muscle, and liver, and it serves as a central regulator of glucose metabolism and energy balance (64, 65). P75NTR knockout (p75NTR−/−) mice showed improved glucose tolerance, insulin sensitivity, and inhibited hepatic glucose production (64). In addition, p75NTR−/− mice are protected from high-fat diet (HFD)-induced obesity as a result of enhanced energy expenditure and insulin sensitivity (65). In adipocytes, the intracellular domain of p75NTR (p75ICD) forms a complex with Rab5 and Rab31 GTPases to regulate Glut4 trafficking (Figure 2B) (64). P75NTR also directly interacts with protein kinase A (PKA) and mediates cAMP signaling in adipocytes, thereby inhibiting lipolysis, and thermogenesis. Adipocyte-specific p75NTR knockout or transplantation of WAT from p75NTR−/− into wild-type mice fed a HFD protects mice against weight gain and insulin resistance; thus, the p75NTR/PKA signaling pathway is a potential therapeutic target for metabolic disorders (65). However, p75NTR serves as a neurotrophin receptor; thus, future studies must be conducted to determine whether or not p75NTR regulates PKA signaling in the CNS and the integral mechanism of p75NTR-regulated-metabolism affecting metabolic diseases.
Sort1 (encoding sortilin), a novel lipid gene, could regulate body weight and cholesterol metabolism (66). This gene also has a basic expression in hepatocytes, and it plays a key role in lipid metabolism and glucose metabolism (Figure 2C). Sortilin increases adipose mass and cholesterol absorption and decreases energy expenditure via the KLF4-LXR signaling pathway, leading to downregulated FGF21 and adiponectin and upregulated NPC1L1 (66). Sort1 is also expressed on macrophages. Sort1 deficiency could downregulate macrophage cellular cholesterol levels by reducing LDL uptake. Correspondingly, sortilin overexpression in macrophages increased the uptake of LDL and foam cell formation (14). Thus, a close correlation is observed between macrophage sortilin and lipid metabolism. Sortilin could also interact with PCSK9 to promote the secretion of PCSK9, which induces the degradation of the LDL receptor and reduces the clearance rate of LDL-C (13, 67). In addition, several studies have shown that sortilin plays an essential role in glucose metabolism. Sortilin−/− mice had enhanced glucose uptake and reduced inflammatory cytokine production compared with wild-type mice (68). Sortilin also served as a sorting partner for the glucose transporter GLUT4 to promote glucose storage (69).
3 ProBDNF and its receptors in MS
MS is a chronic inflammatory demyelinating disease of the CNS characterized by axonal degeneration and neurodegeneration (70) as well as an autoimmune inflammatory disease (71). The infiltration of heterogeneous cell populations such as T cells, B cells, macrophages, and microglia triggers chronic inflammatory pathological damage in the CNS (72). Based on the most extensive global study to date, a total of 2.8 million people worldwide has MS (73). MS is classified into three phenotypes: relapsing/remitting MS (RR–MS), secondary progressive MS (SP–MS) and primary progressive MS (PP–MS) (74).
Studies have shown that proBDNF induces apoptosis by activating p75NTR-mediated downstream signaling pathways (75). ProBDNF and mBDNF in the serum of RR–MS patients decreased, whereas truncated BDNF increased compared with healthy controls (76). Thus, low proBDNF in the serum of RR–MS patients may not be sufficient to limit the proliferation of autoreactive T cells; in addition, low mBDNF in the serum could not exert enough neuroprotection (76). Our previous study found that the expression level of proBDNF and p75NTR significantly increased in the peripheral blood, spleen, and spinal cord of patients with MS and EAE model mice and co-localized with T and B cells. The administration of proBDNF-neutralizing antibodies, such as mAb-proB, can effectively improve the neurological score of EAE model mice, inhibit the expression of inflammatory cytokines in the spleen and spinal cord, reduce the percentage of T cells and B cells, and improve demyelinating lesions and inflammatory infiltration of the spinal cord. The mechanism is due to the hyperactivation of downstream NF-κB signaling by proBDNF/p75NTR on peripheral human PBMCs (29). In early 2008, the New England Journal of Medicine reported that about 20.3% of patients with MS would relapse after B cell depletion therapy with anti-CD20 monoclonal antibody (77). Extensive B cell depletion therapy may lead to infusion reactions and increase the risk of serious opportunistic infections, including progressive multifocal leukoencephalopathy, bone marrow suppression, and liver damage (78). MAb-proB would not eliminate all lymphocytes in EAE model mice, indicating that mAb-proB may be a safe and effective clinical candidate drug for MS treatment.
By the end of the last century, glial cells in MS plaques have been reported to enrich for p75NTR (79). Kust et al. found that the expression level of p75NTR was increased in endothelial cells of the CNS of EAE model mice. The proportion of B cells, monocytes/macrophages, and segmental neutrophils was decreased in the spinal cord of p75NTR−/−-induced EAE. By contrast, the proportion of T cells doubled, and inflammation in the CNS was significantly enhanced. The results indicate that p75NTR in endothelial cells plays a role in protecting the integrity of the blood–brain barrier and regulating immune cells in EAE model mice, particularly the interaction with T cells (80, 81). Studies have found that p75NTR is expressed only on B lymphocytes (B220+ cells) in the brain and spinal cord of EAE model mice, whereas its expression on T lymphocytes is weak (82). P75NTR does not directly act on T cells that infiltrate into the CNS of EAE model mice. In addition, Steven et al. found that p75NTR is expressed on NG2-positive (an integral membrane chondroitin sulfate protein glycan expressed by oligodendrocyte progenitor cells) oligodendrocyte progenitor cells in periventricular plaques, in the subventricular zone adjacent to plaques, and in the corpus callosum of patients with MS (83). The abovementioned studies suggest that p75NTR can participate in the regulation of neuroimmune pathology in EAE model mice through various pathways. In lesioned brain tissues of patients with MS and EAE mice, sortilin was highly expressed on infiltrating macrophages and activated microglia. However, the knockdown of sortilin had no effect on the progression of EAE (49). Therefore, proBDNF may play a role in the neuroimmune inflammation of MS/EAE by binding to p75NTR.
Several independent investigations have demonstrated that the diffused neurodegeneration in patients with MS involves mitochondrial dysfunction, such as mitochondrial respiratory chain deficiency, inadequate ATP production, and accumulated mitochondrial ROS (84–86). The accumulation of ROS and the consequent DNA and protein and lipid damage occur in hypoxia-induced neurotoxicity during MS progression. During hypoxia, p75NTR undergoes oxygen-dependent cleavage by γ-secretase to provide a positive feedforward mechanism as an adaptive response to low oxygen tension (87). The cellular adaptation to hypoxia is mediated by the transcription factor hypoxia-inducible factor-1α (HIF-1α), which controls a group of genes engaging in cell migration, proliferation, metabolism, and inflammation. Hypoxia stimulates the γ-secretase-dependent release of endogenous p75ICD and its interaction with absentia homolog 2 (Siah2), which decreases auto-ubiquitination. Then, Siah2 targets prolyl hydroxylases for proteasomal degradation to stabilize HIF-1α (87). The accumulation of HIF-1α triggers mitochondrial signaling pathways to induce neuronal apoptosis by activating p53 or upregulating the proapoptotic gene NIX (Figure 3). Therefore, targeting the oxygen-dependent cleavage of p75NTR may exert potential therapeutic effects on MS. Compared with the role of proBDNF in promoting cell apoptosis by inducing cyt C release from mitochondria, BDNF contributes to endogenous neurotrophic support in MS plaques by binding to TrkB, thereby inducing the expression of NF-κB (88). Carito et al. found that polyphenolic compounds may exert neuroprotective effects and reduce the risk of MS disease by upregulating BDNF to control oxidative stress, inflammation, apoptosis, and mitochondrial dysfunction (89).
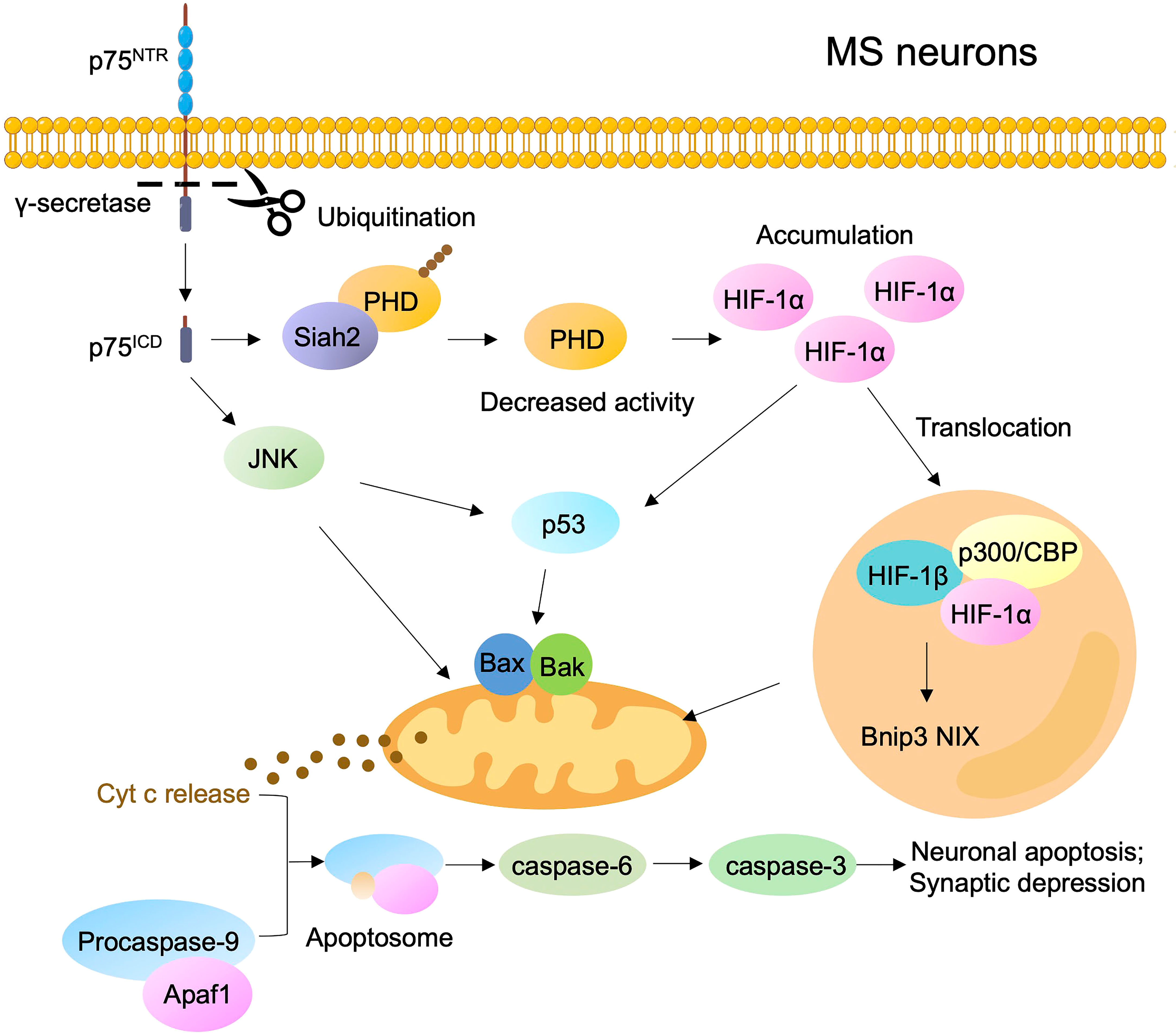
Figure 3 P75NTR activates signaling pathways regulating neuronal death in MS during hypoxia. Hypoxia increases the γ-secretase of p75NTR to release p75ICD and then increases Siah2, which results in the ubiquitination and degradation of prolyl hydroxylases (PHD), thereby promoting stabilization and accumulation of HIF-1α. The pool of HIF-1α triggers mitochondrial signaling pathways to induce neuronal apoptosis by activating p53 or by up-regulating the proapoptotic gene NIX.
4 ProBDNF and its receptors in RA
RA is an autoimmune disease, wherein the immune system attacks the joints of the whole body, thereby causing joint inflammation, which, in severe cases, may lead to permanent joint damage and disability. In addition, RA may induce complications by damaging the heart, lungs, blood vessels, skin, and eyes. The prevalence of RA is approximately 0.5% worldwide, and it is higher in women (90). Our previous studies have demonstrated that mitochondria, as a key disease-related organelle, promote the pathogenesis of RA (91–93).
Recently, a study has shown that increased proBDNF and p75NTR were detected in inflammatory cells of synovial tissue of patients with RA through immunohistochemistry compared with osteoarthritis. Abundant proBDNF was co-localized with p75NTR in RA synovial tissue. In addition, proBDNF was co-localized with CD14+ monocytes and some CD20+ B cells in RA synovial tissue, and p75NTR was primarily expressed in CD4+ T cells of synovial tissue. Moreover, the expression of proBDNF and its receptors, namely, p75NTR, and sortilin, were all higher in PBMCs in patients with RA than in healthy controls. Furthermore, the serum p75NTR and sortilin levels were positively and significantly correlated with the Disease Activity Score in 28 joints (94). Intervention with p75ECD can significantly decrease proinflammatory cytokines and proBDNF/p75NTR/sortilin in the serum and spinal cord of CIA model mice, which indicates that proBDNF/p75NTR/sortilin signaling promotes inflammatory response in RA (94). Our previous study found that in inflammatory pain model mice, the expression level of proBDNF and p75NTR was upregulated in nerve fibers and inflammatory cells of local tissues. An anti-proBDNF antibody can relieve pain in different inflammatory pain mouse models, inhibit inflammatory cell infiltration, and activate proinflammatory cytokines (31). Therefore, we hypothesize that the proBDNF/p75NTR signal derived from immune cells may be closely related to the course of RA, and inhibiting the proBDNF/p75NTR signal may provide a new therapeutic strategy for improving pain in patients with RA.
A recent study using single-cell RNA sequencing has found that fibroblast-like synoviocyte (FLS) from active patients with RA overexpressed p75NTR and sortilin compared with patients with RA in the remission stage (95). P75NTR was significantly enriched in PRG4pos lining FLS and THY1pos COL1A1pos sublining FLS, as well as remarkably expressed in FLS (95). After IL-1β stimulation in vitro, the expression level of p75NTR on FLS of patients with RA was significantly upregulated. Consequently, p75NTR signaling activated the inflammatory response in FLS, and neutralizing, or inhibiting p75NTR could reduce the inflammatory factors IL-6, IL-8, and MCP1 in FLS, which was related to the activation of downstream JNK/p38 MAPK signaling (95). Chronic glucose metabolic changes induced by hypoxia and inflammatory mediators in FLS and synovial T cells will activate many signaling pathways, including MAPK, NF-κB, and PI3K/Akt pathways (96), which are crucial for the expression of adhesion molecules, secretion of cytokines, and inhibition of apoptosis, as well as for migration and invasion (96) (Figure 4). Therefore, proBDNF/p75NTR signaling may regulate glucose metabolism, particularly in FLS and CD4+ T cells during RA inflammatory response. In addition, serum BDNF contributed to proinflammatory responses in patients with RA. Thus, BDNF may also play a similar role in regulating glucose metabolism and mitochondria by releasing proinflammatory cytokines (97).

Figure 4 Mitochondria and glucose metabolism regulation by proBDNF/p75NTR/sortilin signaling in RA FLS. ProBDNF/p75NTR/sortilin signaling promotes the release of inflammatory cytokines, which stimulate Akt phosphorylation and then up-regulate expression and phosphorylation of hexokinase 2 (HK2). Increased binding of HK2 accompanies the phosphorylation of HK2 by Akt to mitochondrial outer membrane voltage-dependent anion channel (VDAC). Binding to VDAC enhances the affinity of hexokinases. Thus, HK2 mitochondria binding promotes glucose metabolism to induce FLS proliferation, migration, invasion and cytokine secretion, contributing to joint destruction in RA. Mitochondrial HK2 might also suppress FLS apoptosis by decreasing cytochrome C release from mitochondria.
In addition, sortilin is highly expressed in nine clusters of synovial macrophages, particularly in SPP1pos macrophage cluster and TREM2pos macrophage cluster. Based on the accumulation of mitochondrially encoded electron transport chain (ETC) subunit genes, TREM2pos macrophages have stronger oxidative phosphorylation (OXPHOS) activity. On the contrary, SPP1pos macrophages are more involved in the glycolytic pathway (98, 99). However, data connecting mitochondrial activity and function and sortilin in different synovial macrophage subsets must be further studied during RA pathogenesis.
5 ProBDNF and its receptors in SLE
SLE is an autoimmune disease of unknown etiology, which is characterized as the deposition of auto-antibodies caused by the hyperactivation of autoreactive B cells and dysregulation of antibody-secreting cells (ASCs) (100–102). In general, SLE is characterized by the inappropriate expansion of ASCs, leading to the production of auto-antibodies such as anti-DNA antibodies and antinuclear antibodies (103). ASCs can develop from multiple types of activated B cells with highly and strongly positive cell surface markers, namely, CD38 and CD27 (104–106). As ASCs develop and mature, they exhibit multiple transient cellular phenotypes to diversify their antibody repertoire and then develop distinct cell-fate endpoints (107–109). For example, ASCs with high expression of CXCR3 migrate to inflamed tissues (110). Furthermore, some subsets of ASCs such as CD27hiHLA-DRhi (111) and TLR4+ CXCR4+ plasma cells (112) contributed to auto-antibody production and glomerulonephritis compared with other plasma cells.
Our recent study has shown that proBDNF and p75NTR were upregulated in ASCs (CD19+ CD27hi CD38hi). Moreover, the expression of proBDNF on ASCs has a significant clinical correlation with the auto-antibody level and disease activity of patients with SLE, indicating that ASCs+ proBDNF+ cells can be used as a clinical marker of SLE. In animal experiments, proBDNF/p75NTR signaling was significantly upregulated in B cells of spontaneous and induced lupus mice. The intraperitoneal administration of mAb-proB can alleviate the condition of spontaneous and induced lupus mice, reduce the proportion of ASCs and production of auto-antibody and proinflammatory cytokines, as well as delay kidney damage (9, 10). Intraperitoneally administering pristane to induce lupus in B cell-specific p75NTR knockout (CD19cre p75f/f) mice showed that the knockout of B cell p75NTR signaling can also alleviate the progression of SLE. RNA-Seq suggested the downregulation of immune-related and antibody secretion-related genes in lymph nodes of CD19cre p75f/f mice. R848 stimulation significantly upregulated the proBDNF/p75NTR signal on B cells in vitro. Moreover, anti-proBDNF antibody intervention or B cell conditional knockout of p75NTR could inhibit R848-induced generation of ASCs. In vitro intervention with mAb-proB inhibited the CpGB-stimulated B cell differentiation and production of IgG and IgM in PBMCs from healthy volunteers and patients with SLE. Therefore, the proBDNF/p75NTR signaling pathway promoted the proliferation of ASCs, which plays a pathogenic role in SLE and may be a potential therapeutic biological target for SLE (9, 113). Our recent research indicated that mAb-proB inhibited the overexpansion of CD3+ B220+ cells and altered transcription levels related to cholesterol metabolism, cell cycle, and cell apoptosis, which may contribute to the attenuation of conditions of MRL/lpr lupus mice (10).
Several studies on SLE have observed mitochondrial damage and dysfunction in SLE B cells and T cells, which are characterized as enhanced mitochondrial membrane depolarization, OXPHOS, and mitochondria mass (114, 115). In addition, mitochondrial dysfunction in B cells is associated with plasmablast differentiation and disease activity in SLE (114). A previous report used spinal cord injury model mice to confirm that p75NTR overexpression induced mitochondrial damage and cell apoptosis in spinal cord neurons by downregulating neurotrophic tyrosine receptor kinase 3 (116). Our study also indicated the significant difference in gene ontology of mitochondrial respiratory chain complex assembly between pristane-immunized CD19cre p75f/f mice and p75f/f control mice by RNA-seq. Therefore, p75NTR signaling may exert potential effects on mitochondrial metabolism in B cells to participate in the regulation of SLE pathogenesis (Figure 5). Similar to the upregulation of proBDNF, BDNF levels in B cells and serum increased in patients with SLE compared with healthy controls (117, 118). However, the level of serum BDNF and the number of BDNF+ B cells were independent of the SLEDAI score (117).
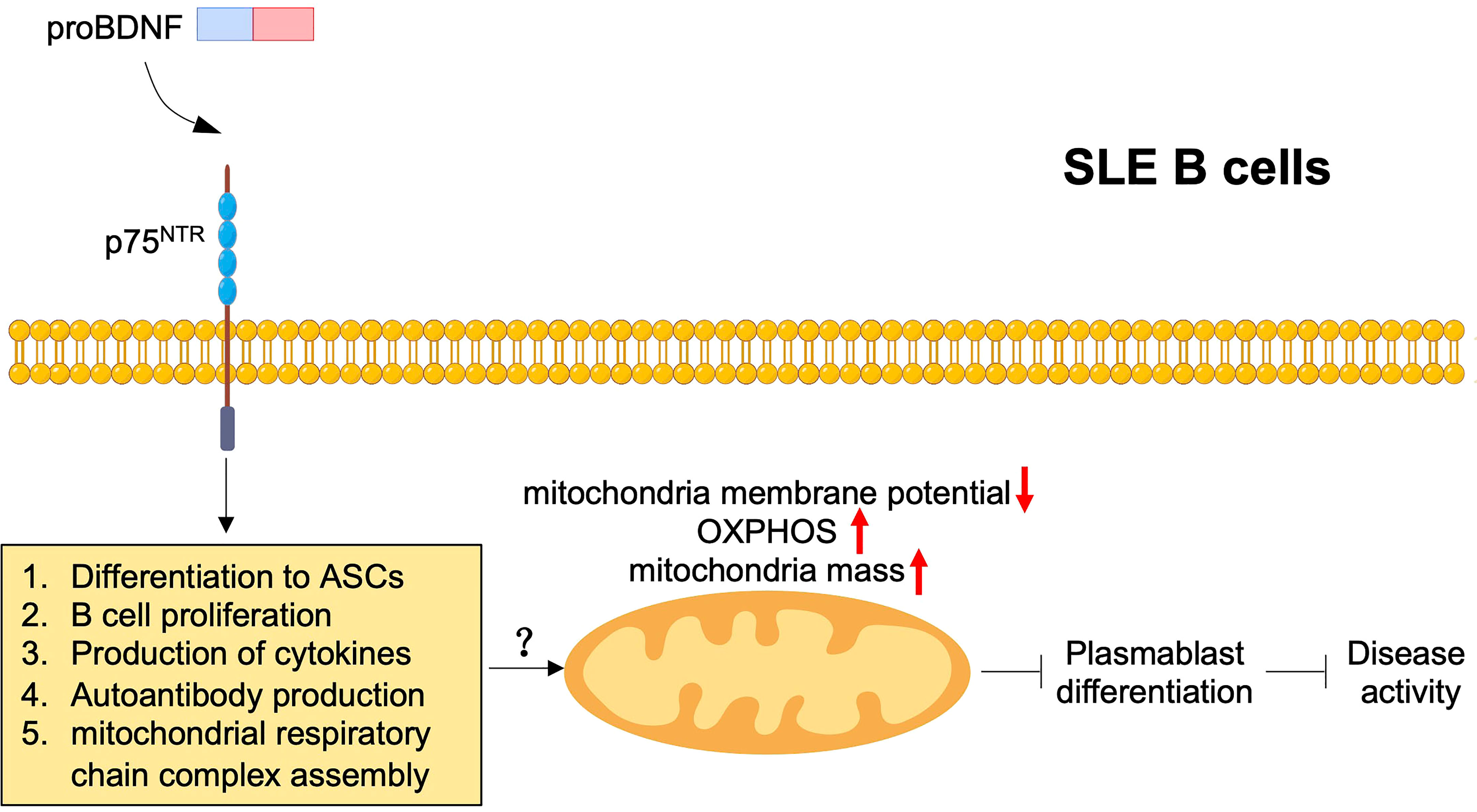
Figure 5 ProBDNF/p75NTR signaling in SLE B cells. ProBDNF/p75NTR signaling promotes B cell differentiation, proliferation, release of proinflammatory cytokines and autoantibody production and mitochondrial respiratory chain complex assembly. Mitochondria damage and dysfunction including decreased mitochondria membrane potential and enhanced OXPHOS and mitochondria mass, which may increase disease activity through inhibiting plasmablast differentiation by proBDNF/p75NTR signaling pathway.
6 ProBDNF and its receptors in allergic asthma
Asthma is an immune-mediated inflammatory condition characterized by increased responsiveness to broncho-constrictive stimuli (6). Inhaled allergens can cause biphasic and reversible airflow obstruction. In the late phase of the response, activated T lymphocytes and eosinophils infiltrate in the airways because of increased bronchial reactivity (119–122). Possible sources of neurotrophins in allergic inflammation include neurons, neuron-associated cells (123–125), and immune cells, such as T cells, B cells, mast cells, macrophages, epithelial cells, and eosinophils (126–133).
Neurotrophins not only affect neurons but also interfere with functions of immune cells associated with allergies, such as mast cell degranulation, Th2 cytokine synthesis, B cell antibody production, and eosinophil survival (130, 134–136). The pathophysiological roles of neurotrophins in allergic asthma are as follows: first, elevated neurotrophin levels in bronchoalveolar fluid (BALF); second, blood neurotrophin levels correlate with airflow limitation; third, the induction of airway hyperactivity and airway obstruction through the production of ROS- and MAPK-mediated allergen-induced airway inflammation, modulation of neurite formation and cellular contractility, and proinflammatory cytokine release; finally, airway smooth muscle proliferation and matrix metalloproteinase induction (137).
Studies in animal models of allergic asthma have shown that p75NTR plays a key role in the accumulation of eosinophils in the lungs. Christina et al. observed that eosinophils in peripheral blood and BALF expressed p75NTR after segmental allergen provocation (138). Therefore, neurotrophins can mediate bronchial eosinophil activation by combining with p75NTR and may play a role in regulating eosinophil inflammation in allergic asthma. In addition, studies showed that p75NTR−/− mice had significantly reduced allergic inflammation and non-increased airway eosinophils (139, 140). Meanwhile, blocking p75NTR by anti-p75NTR antibody treatment can prevent eosinophilic inflammation in the lungs in mouse models (140). The activation of p75NTR signaling plays a dual role in regulating the function of plasmacytoid dendritic cells (pDC), which not only reduced blood glucose levels and delayed the onset of autoimmune diabetes in RIPCD80GP mice but also aggravated graftversushost disease in a xenotransplantation model (141). Its mechanism may be related to the activation of IRF3, IRF7, c-Jun, and IKKα/β, which reveals a novel regulatory circuit in pDC-mediated immune responses. By contrast, during allergic airway inflammation, BDNF plays a central role in modulating airway hyperresponsiveness but not the inflammatory response induced by allergen exposure (142).
7 ProBDNF and its receptors in T1D
Diabetes refers to a group of chronic diseases distinguished by hyperglycemia (143). The two prevalent types of diabetes are T1D and type 2 diabetes (143). The International Diabetes Federation estimates that 1 in 10 adults currently suffers from diabetes, corresponding to 537 million people worldwide. Diabetes is a major health threat, which causes 4 million deaths annually (144). Long-term hyperglycemia may lead to multiple complications, including heart diseases, nerve damage, oral health, vision loss, chronic kidney diseases, hearing loss, and impaired foot health and mental health (145). The prevalence of T1D is highest in children, although T1D can occur at any age (146). T1D is an autoimmune process that begins years before the clinical onset, with autoimmune-mediated selective damage to pancreatic β cells (147). The main clinical manifestation of T1D is hyperglycemia, which initiates polyuria, polydipsia, and weight loss. In severe cases, acute ketoacidosis may occur.
Peripheral neuropathy is a serious but often neglected complication of diabetes. The prevalence of diabetic neuropathy is as high as 50% in patients with diabetes, and it is characterized by damage of neurons, Schwann cells (SCs), and blood vessels in the peripheral nervous system (148). A study on T1D found that pancreatic sympathetic neurons contain abundant p75NTR mRNA, which is directly activated on pancreatic sympathetic axons and is responsible for rapid nerve damage in patients with T1D (149). This degree of nerve damage can significantly suppress glucagon response to sympathetic activation (150, 151). Segmental axonal degeneration is secondary to p75NTR activation on sympathetic axons (152), and p75NTR knockout prevents most islet nerve damage (149).
The effects of oxidative stress and mitochondrial disorders in SCs on neuronal dysfunction during diabetes become more evident. In addition, long-term hyperglycemia is widely considered as a trigger of excessive ROS formation in cells, including SCs (153). Hyperglycemia increases flux through the ETC (154). Schwann cells induced by high glucose cause oxidative stress via intramitochondrial stress, including the overactivation of caspase-9 and Bax, and decrease Bcl-2 (155). Furthermore, a previous study utilized a mouse model of Ngfr-specific deletion in SCs (SC-p75NTR-KO) and RNA sequencing to demonstrate several metabolic pathways activated by p75NTR, including cholesterol metabolism and glycerolipid metabolism (148). Direct evidence has also shown that diabetic peripheral neuropathy is related to the decreased production of neurotrophins and increased p75NTR expression on SCs in humans and mouse models of T1D (156–158). In an in vitro model of hyperglycemia stress, Tan et al. found that high glucose treatment inhibited Cav-1 transcription and protein expression within SCs, which enhanced the mitogenic response of SCs to human recombinant neuregulin-1-β1-(176–246) (NRG1-β1). NGF suppresses the glucose-induced downregulation of Cav-1 transcription and protein expression through p75NTR-mediated JNK activation (159). NGF/p75NTR signaling increases the expression of p53 and promotes its activation by JNK in sympathetic neurons (160, 161). On the contrary, p53 could upregulate the transcription of the human CAV-1 gene (162). Therefore, NGF/p75NTR cassette modulates the response of SCs to neuregulin, which may affect the regenerative/degenerative response of these cells to hyperglycemic stress.
In the study of diabetic nephropathy (diabetic kidney disease [DKD]), Bryan et al. found that the symptoms of DKD were reduced after treating the streptozotocin-induced diabetic model mice with THX-B, a small-molecule p75NTR antagonist, or a monoclonal antibody neutralizing proNGF. Diabetes increased urea and creatinine levels, decreased albumin levels in plasma, and downregulated p75NTR expression in the kidney, all of which were reversed by THX-B treatment. In addition, microRNAs (miR-21-5p, miR-214-3p, and miR-342-3p) were tightly related to and highly expressed in the diabetic kidney. Moreover, the renal inflammation marker miR-146a and the elevated mRNA level of Fn-1 and Nphs, which are markers of fibrosis and inflammation, were partially, or completely reversed after THX-B or anti-proNGF mAb treatment. Therefore, p75NTR antagonists and antibodies against neurotrophins may be novel tools for treating or alleviating DKD and other diabetes-associated complications (163).
BDNF also played a role in T1D. Mitsugu et al. demonstrated that the intermittent administration of BDNF ameliorated blood glucose levels in diabetic mice. The same team confirmed that BDNF reduced food intake and lowered blood glucose levels in obese diabetic animal models. Furthermore, BDNF had a hypoglycemic action independent of appetite alteration in diabetic mice. Meek et al. further confirmed that BDNF lowered blood glucose levels because of decreased glucose uptake, which is consistent with the role of p75ICD but contrary to the role of sortilin in glucose uptake (164).
8 ProBDNF and its receptors in vasculitis
Vasculitis is a condition that covers a group of rare diseases characterized by inflammation of blood vessels, which causes organ ischemia and damage. Vasculitis usually shows a marked age tropism. For example, giant cell arteritis (GCA) predominantly affects those aged >50 years, whereas Kawasaki disease (KD) primarily affects young children (165).
Masayuki et al. found that sortilin is elevated in the acute phase but decreased in the convalescent phase of KD. The ratio of sortilin to platelet (sortilin/platelet) still increased after initial intravenous high-dose immunoglobulin treatment in unresponsive cases, whereas CRP decreased in unresponsive and responsive cases, indicating that sortilin/platelet may reflect the activity of KD more sensitively than CRP (166). In patients with GCA, the overexpression of BDNF, and their receptors was observed in the temporal artery, which may be related to the presence of proinflammatory cytokines in the inflamed arterial wall (167). Reports have demonstrated that vascular inflammation and innate immunity contribute to cardiovascular diseases such as aortic dissection (28, 168, 169). ProBDNF was upregulated in M2- but not M1-like monocytes in patients with Stanford type A acute aortic dissection (AAD). Furthermore, sera from patients with AAD promoted inflammatory responses in PBMCs from healthy controls, which was attenuated by mAb-proB treatment. Therefore, the upregulation of proBDNF in M2-like monocytes may promote the proinflammatory response in AAD (28). Furthermore, proBDNF, and BDNF, as well as its receptor, may serve as inflammatory biomarkers in vasculitis.
More but smaller mitochondria were observed in cells of the medial layer of the ascending aorta in patients with AAD compared with healthy controls, which may illustrate mitochondrial dysfunction (170). In a fluoroquinolone-induced AAD model, mitochondrial dysfunction produced more ROS and STING to promote cell apoptosis. As previously mentioned, proBDNF/p75NTR signaling regulates mitochondrial function to induce cell apoptosis. Therefore, proBDNF/p75NTR signaling may engage in apoptosis in the aortic wall in AAD (171).
9 ProBDNF and its receptors in IBD
IBD is characterized by chronic immune-mediated intestinal inflammation that is driven by genetic susceptibility as well as environmental and microbial factors, encompassing ulcerative colitis (UC) and Crohn’s disease (CD). Such diseases manifest as a relapsing–remitting course. IBD has become a global disease with increasing incidence rate worldwide in the 21st century (172, 173). Some studies have shown that neurotrophins and receptors play an essential role in intestinal inflammation. Receptors for proBDNF and BDNF are common in neurons of the myenteric and submucosal plexus and mucosal endocrine cells in the gastrointestinal tract (174–176). In CD, the loss of enteric glial cells results in severe inflammation of the intestine. BDNF attenuates apoptosis of glial cells, whereas anti-BDNF antibodies significantly increase apoptosis (176). Johansson et al. (177) reported a strong correlation between massive inflammation with decreased neurotrophin immunoreaction. Furthermore, a high expression level of p75NTR was observed in lamina propria cells of patients with UC (177). However, the expression and role of proBDNF in IBD remain unclear.
10 Conclusion
There has been a great breakthrough in therapies for IMIDs. However, new and urgent problems and difficulties in restoring immune dysregulation to normality, relieving pain more than inflammation, and exploring potential interactions between the immune and neurological system begin to exist. ProBDNF signaling promotes cell apoptosis by inducing cyt C release from mitochondria, whereas BDNF signaling exerts opposite effects by controlling oxidative stress and mitochondrial dysfunction. BDNF decreased glucose uptake in IMIDs, which is consistent with the role of p75ICD but contrary to the role of sortilin in glucose uptake. Traditional studies tend to focus on the role of proBDNF and its receptors in the CNS. With the deepening of research, we have comprehensively understood the role of proBDNF and its receptors in mediating mitochondrial and metabolic pathways and in regulating the peripheral immune system in IMIDs, a large class of diseases. ProBDNF signaling usually exerts proinflammatory effects in IMIDs, whereas it exerts anti-inflammatory effects in AMI. Based on the proBDNF/p75NTR signal, a series of original research was conducted, and an anti-proBDNF monoclonal mouse antibody and humanized mAb-proB with independent intellectual property rights and patents was produced. In addition, we focused on the p75NTR target and prepared p75ECD from prokaryotic and eukaryotic cells. In the future, we will explore the role of proBDNF and its receptors from multiple dimensions and perspectives, particularly in cellular metabolism and mitochondrial homeostasis of different immune cell subsets to further understand its mechanism involved in patients with IMID. Finally, these new insights may promote preclinical research by targeting proBDNF and its receptors in the future.
Author contributions
QL wrote the manuscript and created the figures. ZLH revised the manuscript and figures. YZH participated in literature review and data summary. SG and PFW participated in literature review. RPD was the guarantor and revised the manuscript. All authors reviewed the final manuscript.
Funding
This research was supported by the Natural Science Foundation of Hunan province (2023JJ10088 to ZLH) and the National Natural Science Foundation of China (81901231 and 82271379 to ZLH, 82071347 and 81771354 to RPD).
Acknowledgments
We appreciate Yu-Da Huang for his assistance in editing English language of the revised manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Forbes JD, Van Domselaar G, Bernstein CN. The gut microbiota in immune-mediated inflammatory diseases. Front Microbiol (2016) 7:1081. doi: 10.3389/fmicb.2016.01081
2. McInnes IB, Gravallese EM. Immune-mediated inflammatory disease therapeutics: past, present and future. Nat Rev Immunol (2021) 21(10):680–6. doi: 10.1038/s41577-021-00603-1
3. Forbes JD, Chen CY, Knox NC, Marrie RA, El-Gabalawy H, de Kievit T, et al. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome. (2018) 6(1):221. doi: 10.1186/s40168-018-0603-4
4. Wraith DC. The future of immunotherapy: A 20-year perspective. Front Immunol (2017) 8:1668. doi: 10.3389/fimmu.2017.01668
5. Bellinger DL, Lorton D. Sympathetic nerve hyperactivity in the spleen: Causal for nonpathogenic-driven chronic immune-mediated inflammatory diseases (IMIDs)? Int J Mol Sci (2018) 19(4):1188. doi: 10.3390/ijms19041188
6. Openshaw PJ, Yamaguchi Y, Tregoning JS. Childhood infections, the developing immune system, and the origins of asthma. J Allergy Clin Immunol (2004) 114(6):1275–7. doi: 10.1016/j.jaci.2004.08.024
7. Marrie RA, Walld R, Bolton JM, Sareen J, Walker JR, Patten SB, et al. Increased incidence of psychiatric disorders in immune-mediated inflammatory disease. J Psychosom Res (2017) 101:17–23. doi: 10.1016/j.jpsychores.2017.07.015
8. Carubbi F, Alunno A, Santilli J, Natali L, Mancini B, Di Gregorio N, et al. Immune-mediated inflammatory diseases after anti-SARS-CoV-2 vaccines: new diagnoses and disease flares. RMD Open (2022) 8(2):e002460. doi: 10.1136/rmdopen-2022-002460
9. Shen WY, Luo C, Hurtado PR, Liu XJ, Luo RY, Li H, et al. Up-regulation of proBDNF/p75(NTR) signaling in antibody-secreting cells drives systemic lupus erythematosus. Sci Adv (2022) 8(3):eabj2797. doi: 10.1126/sciadv.abj2797
10. Zha AH, Luo C, Shen WY, Fu D, Dai RP. Systemic blockade of proBDNF inhibited the expansion and altered the transcriptomic expression in CD3(+)B220(+) cells in MRL/lpr lupus mice. Lupus Sci Med (2022) 9(1):e000836. doi: 10.1136/lupus-2022-000836
11. Sankorrakul K, Qian L, Thangnipon W, Coulson EJ. Is there a role for the p75 neurotrophin receptor in mediating degeneration during oxidative stress and after hypoxia? J Neurochem (2021) 158(6):1292–306. doi: 10.1111/jnc.15451
12. Malik SC, Sozmen EG, Baeza-Raja B, Le Moan N, Akassoglou K, Schachtrup C. In vivo functions of p75(NTR): challenges and opportunities for an emerging therapeutic target. Trends Pharmacol Sci (2021) 42(9):772–88. doi: 10.1016/j.tips.2021.06.006
13. Gustafsen C, Kjolby M, Nyegaard M, Mattheisen M, Lundhede J, Buttenschon H, et al. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab (2014) 19(2):310–8. doi: 10.1016/j.cmet.2013.12.006
14. Patel KM, Strong A, Tohyama J, Jin X, Morales CR, Billheimer J, et al. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ Res (2015) 116(5):789–96. doi: 10.1161/CIRCRESAHA.116.305811
15. Ilchibaeva TV, Kondaurova EM, Tsybko AS, Kozhemyakina RV, Popova NK, Naumenko VS. Brain-derived neurotrophic factor (BDNF) and its precursor (proBDNF) in genetically defined fear-induced aggression. Behav Brain Res (2015) 290:45–50. doi: 10.1016/j.bbr.2015.04.041
16. Anastasia A, Deinhardt K, Chao MV, Will NE, Irmady K, Lee FS, et al. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat Commun (2013) 4:2490. doi: 10.1038/ncomms3490
17. Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, et al. Neuronal release of proBDNF. Nat Neurosci (2009) 12(2):113–5. doi: 10.1038/nn.2244
18. Gibon J, Barker PA. Neurotrophins and proneurotrophins: Focus on synaptic activity and plasticity in the brain. Neuroscientist (2017) 23(6):587–604. doi: 10.1177/1073858417697037
19. Kowianski P, Lietzau G, Czuba E, Waskow M, Steliga A, Morys J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol (2018) 38(3):579–93. doi: 10.1007/s10571-017-0510-4
20. Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, et al. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci (2000) 20(18):6888–97. doi: 10.1523/JNEUROSCI.20-18-06888.2000
21. Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci (2005) 6(8):603–14. doi: 10.1038/nrn1726
22. Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol (2014) 220:223–50. doi: 10.1007/978-3-642-45106-5_9
23. Anand SK, Mondal AC. Neuroanatomical distribution and functions of brain-derived neurotrophic factor in zebrafish (Danio rerio) brain. J Neurosci Res (2020) 98(5):754–63. doi: 10.1002/jnr.24536
24. Zhou XF, Song XY, Zhong JH, Barati S, Zhou FH, Johnson SM. Distribution and localization of pro-brain-derived neurotrophic factor-like immunoreactivity in the peripheral and central nervous system of the adult rat. J Neurochem (2004) 91(3):704–15. doi: 10.1111/j.1471-4159.2004.02775.x
25. Yang B, Ren Q, Zhang JC, Chen QX, Hashimoto K. Altered expression of BDNF, BDNF pro-peptide and their precursor proBDNF in brain and liver tissues from psychiatric disorders: rethinking the brain-liver axis. Transl Psychiatry (2017) 7(5):e1128. doi: 10.1038/tp.2017.95
26. Li JY, Liu J, Manaph NPA, Bobrovskaya L, Zhou XF. ProBDNF inhibits proliferation, migration and differentiation of mouse neural stem cells. Brain Res (2017) 1668:46–55. doi: 10.1016/j.brainres.2017.05.013
27. Yang J, Harte-Hargrove LC, Siao CJ, Marinic T, Clarke R, Ma Q, et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep (2014) 7(3):796–806. doi: 10.1016/j.celrep.2014.03.040
28. Shen WY, Luo C, Reinaldo Hurtado P, Hurtado-Perez E, Luo RY, Hu ZL, et al. The regulatory role of ProBDNF in monocyte function: Implications in Stanford type-a aortic dissection disease. FASEB J (2020) 34(2):2541–53. doi: 10.1096/fj.201901905RR
29. Hu ZL, Luo C, Hurtado PR, Li H, Wang S, Hu B, et al. Brain-derived neurotrophic factor precursor in the immune system is a novel target for treating multiple sclerosis. Theranostics. (2021) 11(2):715–30. doi: 10.7150/thno.51390
30. Luo RY, Luo C, Zhong F, Shen WY, Li H, Hu ZL, et al. ProBDNF promotes sepsis-associated encephalopathy in mice by dampening the immune activity of meningeal CD4(+) T cells. J Neuroinflammation (2020) 17(1):169. doi: 10.1186/s12974-020-01850-0
31. Luo C, Zhong XL, Zhou FH, Li JY, Zhou P, Xu JM, et al. Peripheral brain derived neurotrophic factor precursor regulates pain as an inflammatory mediator. Sci Rep (2016) 6:27171. doi: 10.1038/srep27171
32. Wong I, Liao H, Bai X, Zaknic A, Zhong J, Guan Y, et al. ProBDNF inhibits infiltration of ED1+ macrophages after spinal cord injury. Brain Behav Immun (2010) 24(4):585–97. doi: 10.1016/j.bbi.2010.01.001
33. Wang Z, Wu JL, Zhong F, Liu Y, Yu YQ, Sun JJ, et al. Upregulation of proBDNF in the mesenteric lymph nodes in septic mice. Neurotox Res (2019) 36(3):540–50. doi: 10.1007/s12640-019-00081-3
34. Mizoguchi Y, Ohgidani M, Haraguchi Y, Murakawa-Hirachi T, Kato TA, Monji A. ProBDNF induces sustained elevation of intracellular Ca(2+) possibly mediated by TRPM7 channels in rodent microglial cells. Glia. (2021) 69(7):1694–708. doi: 10.1002/glia.23996
35. Li JN, Luo RY, Luo C, Hu ZL, Zha AH, Shen WY, et al. Brain-derived neurotrophic factor precursor contributes to a proinflammatory program in Monocytes/Macrophages after acute myocardial infarction. J Am Heart Assoc (2023) 12(6):e028198. doi: 10.1161/JAHA.122.028198
36. Xiong J, Zhou L, Yang M, Lim Y, Zhu YH, Fu DL, et al. ProBDNF and its receptors are upregulated in glioma and inhibit the growth of glioma cells in vitro. Neuro Oncol (2013) 15(8):990–1007. doi: 10.1093/neuonc/not039
37. Liu Y, Zou GJ, Tu BX, Hu ZL, Luo C, Cui YH, et al. Corticosterone induced the increase of proBDNF in primary hippocampal neurons Via endoplasmic reticulum stress. Neurotox Res (2020) 38(2):370–84. doi: 10.1007/s12640-020-00201-4
38. Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science (2001) 294(5548):1945–8. doi: 10.1126/science.1065057
39. Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, et al. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci (2005) 8(8):1069–77. doi: 10.1038/nn1510
40. Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci (2005) 25(22):5455–63. doi: 10.1523/JNEUROSCI.5123-04.2005
41. Deinhardt K, Chao MV. Shaping neurons: Long and short range effects of mature and proBDNF signalling upon neuronal structure. Neuropharmacology (2014) 76 Pt C(0 0):603–9. doi: 10.1016/j.neuropharm.2013.04.054
43. Sun Y, Lim Y, Li F, Liu S, Lu JJ, Haberberger R, et al. ProBDNF collapses neurite outgrowth of primary neurons by activating RhoA. PloS One (2012) 7(4):e35883. doi: 10.1371/journal.pone.0035883
44. Morgan B, Thorpe LW, Marchetti D, Perez-Polo JR. Expression of nerve growth factor receptors by human peripheral blood mononuclear cells. J Neurosci Res (1989) 23(1):41–5. doi: 10.1002/jnr.490230106
45. Brunelli A, Dimauro I, Sgro P, Emerenziani GP, Magi F, Baldari C, et al. Acute exercise modulates BDNF and pro-BDNF protein content in immune cells. Med Sci Sports Exerc (2012) 44(10):1871–80. doi: 10.1249/MSS.0b013e31825ab69b
46. Charalampopoulos I, Vicario A, Pediaditakis I, Gravanis A, Simi A, Ibanez CF. Genetic dissection of neurotrophin signaling through the p75 neurotrophin receptor. Cell Rep (2012) 2(6):1563–70. doi: 10.1016/j.celrep.2012.11.009
47. Fauchais AL, Lalloue F, Lise MC, Boumediene A, Preud'homme JL, Vidal E, et al. Role of endogenous brain-derived neurotrophic factor and sortilin in b cell survival. J Immunol (2008) 181(5):3027–38. doi: 10.4049/jimmunol.181.5.3027
48. Lane RF, St George-Hyslop P, Hempstead BL, Small SA, Strittmatter SM, Gandy S. Vps10 family proteins and the retromer complex in aging-related neurodegeneration and diabetes. J Neurosci (2012) 32(41):14080–6. doi: 10.1523/JNEUROSCI.3359-12.2012
49. Reuter E, Weber J, Paterka M, Ploen R, Breiderhoff T, van Horssen J, et al. Role of sortilin in models of autoimmune neuroinflammation. J Immunol (2015) 195(12):5762–9. doi: 10.4049/jimmunol.1403156
50. Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature. (2004) 427(6977):843–8. doi: 10.1038/nature02319
51. Skeldal S, Sykes AM, Glerup S, Matusica D, Palstra N, Autio H, et al. Mapping of the interaction site between sortilin and the p75 neurotrophin receptor reveals a regulatory role for the sortilin intracellular domain in p75 neurotrophin receptor shedding and apoptosis. J Biol Chem (2012) 287(52):43798–809. doi: 10.1074/jbc.M112.374710
52. Herda S, Raczkowski F, Mittrucker HW, Willimsky G, Gerlach K, Kuhl AA, et al. The sorting receptor sortilin exhibits a dual function in exocytic trafficking of interferon-gamma and granzyme a in T cells. Immunity. (2012) 37(5):854–66. doi: 10.1016/j.immuni.2012.07.012
53. Rogers ML, Bailey S, Matusica D, Nicholson I, Muyderman H, Pagadala PC, et al. ProNGF mediates death of natural killer cells through activation of the p75NTR-sortilin complex. J Neuroimmunol (2010) 226(1-2):93–103. doi: 10.1016/j.jneuroim.2010.05.040
54. Suzuki R, Matsumoto M, Fujikawa A, Kato A, Kuboyama K, Yonehara K, et al. SPIG1 negatively regulates BDNF maturation. J Neurosci (2014) 34(9):3429–42. doi: 10.1523/JNEUROSCI.1597-13.2014
55. Suzuki R, Fujikawa A, Komatsu Y, Kuboyama K, Tanga N, Noda M. Enhanced extinction of aversive memories in mice lacking SPARC-related protein containing immunoglobulin domains 1 (SPIG1/FSTL4). Neurobiol Learn Mem (2018) 152:61–70. doi: 10.1016/j.nlm.2018.05.010
56. Kailainathan S, Piers TM, Yi JH, Choi S, Fahey MS, Borger E, et al. Activation of a synapse weakening pathway by human Val66 but not Met66 pro-brain-derived neurotrophic factor (proBDNF). Pharmacol Res (2016) 104:97–107. doi: 10.1016/j.phrs.2015.12.008
57. Wang X, Ma W, Wang T, Yang J, Wu Z, Liu K, et al. BDNF-TrkB and proBDNF-p75NTR/Sortilin signaling pathways are involved in mitochondria-mediated neuronal apoptosis in dorsal root ganglia after sciatic nerve transection. CNS Neurol Disord Drug Targets (2020) 19(1):66–82. doi: 10.2174/1871527319666200117110056
58. Song HD, Kim SN, Saha A, Ahn SY, Akindehin S, Son Y, et al. Aging-induced brain-derived neurotrophic factor in adipocyte progenitors contributes to adipose tissue dysfunction. Aging Dis (2020) 11(3):575–87. doi: 10.14336/AD.2019.0810
59. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell (2003) 112(2):257–69. doi: 10.1016/s0092-8674(03)00035-7
60. Ieraci A, Barbieri SS, Macchi C, Amadio P, Sandrini L, Magni P, et al. BDNF Val66Met polymorphism alters food intake and hypothalamic BDNF expression in mice. J Cell Physiol (2020) 235(12):9667–75. doi: 10.1002/jcp.29778
61. Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates rho activity and axonal outgrowth. Neuron (1999) 24(3):585–93. doi: 10.1016/s0896-6273(00)81114-9
62. Harrington AW, Kim JY, Yoon SO. Activation of rac GTPase by p75 is necessary for c-jun n-terminal kinase-mediated apoptosis. J Neurosci (2002) 22(1):156–66. doi: 10.1523/JNEUROSCI.22-01-00156.2002
63. Numakawa T, Odaka H. Brain-derived neurotrophic factor signaling in the pathophysiology of alzheimer's disease: Beneficial effects of flavonoids for neuroprotection. Int J Mol Sci (2021) 22(11):5719. doi: 10.3390/ijms22115719
64. Baeza-Raja B, Li P, Le Moan N, Sachs BD, Schachtrup C, Davalos D, et al. p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proc Natl Acad Sci U S A (2012) 109(15):5838–43. doi: 10.1073/pnas.1103638109
65. Baeza-Raja B, Sachs BD, Li P, Christian F, Vagena E, Davalos D, et al. p75 neurotrophin receptor regulates energy balance in obesity. Cell Rep (2016) 14(2):255–68. doi: 10.1016/j.celrep.2015.12.028
66. Hagita S, Rogers MA, Pham T, Wen JR, Mlynarchik AK, Aikawa M, et al. Transcriptional control of intestinal cholesterol absorption, adipose energy expenditure and lipid handling by sortilin. Sci Rep (2018) 8(1):9006. doi: 10.1038/s41598-018-27416-y
67. Kjolby M, Nielsen MS, Petersen CM. Sortilin, encoded by the cardiovascular risk gene SORT1, and its suggested functions in cardiovascular disease. Curr Atheroscler Rep (2015) 17(4):496. doi: 10.1007/s11883-015-0496-7
68. Rabinowich L, Fishman S, Hubel E, Thurm T, Park WJ, Pewzner-Jung Y, et al. Sortilin deficiency improves the metabolic phenotype and reduces hepatic steatosis of mice subjected to diet-induced obesity. J Hepatol (2015) 62(1):175–81. doi: 10.1016/j.jhep.2014.08.030
69. Blondeau N, Beraud-Dufour S, Lebrun P, Hivelin C, Coppola T. Sortilin in glucose homeostasis: From accessory protein to key player? Front Pharmacol (2018) 9:1561. doi: 10.3389/fphar.2018.01561
70. McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol (2007) 8(9):913–9. doi: 10.1038/ni1507
71. Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol (2005) 252(Suppl 5):v3–9. doi: 10.1007/s00415-005-5002-7
72. Prat A, Antel J. Pathogenesis of multiple sclerosis. Curr Opin Neurol (2005) 18(3):225–30. doi: 10.1097/01.wco.0000169737.99040.31
73. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the atlas of MS, third edition. Mult Scler (2020) 26(14):1816–21. doi: 10.1177/1352458520970841
74. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Primers (2018) 4(1):43. doi: 10.1038/s41572-018-0041-4
75. Koshimizu H, Hazama S, Hara T, Ogura A, Kojima M. Distinct signaling pathways of precursor BDNF and mature BDNF in cultured cerebellar granule neurons. Neurosci Lett (2010) 473(3):229–32. doi: 10.1016/j.neulet.2010.02.055
76. Tongiorgi E, Sartori A, Baj G, Bratina A, Di Cola F, Zorzon M, et al. Altered serum content of brain-derived neurotrophic factor isoforms in multiple sclerosis. J Neurol Sci (2012) 320(1-2):161–5. doi: 10.1016/j.jns.2012.07.016
77. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med (2008) 358(7):676–88. doi: 10.1056/NEJMoa0706383
78. Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: Entering an era. Ann Neurol (2018) 83(1):13–26. doi: 10.1002/ana.25119
79. Dowling P, Ming X, Raval S, Husar W, Casaccia-Bonnefil P, Chao M, et al. Up-regulated p75NTR neurotrophin receptor on glial cells in MS plaques. Neurology (1999) 53(8):1676–82. doi: 10.1212/wnl.53.8.1676
80. Copray S, Kust B, Emmer B, Lin MY, Liem R, Amor S, et al. Deficient p75 low-affinity neurotrophin receptor expression exacerbates experimental allergic encephalomyelitis in C57/BL6 mice. J Neuroimmunol (2004) 148(1-2):41–53. doi: 10.1016/j.jneuroim.2003.11.008
81. Kust B, Mantingh-Otter I, Boddeke E, Copray S. Deficient p75 low-affinity neurotrophin receptor expression does alter the composition of cellular infiltrate in experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol (2006) 174(1-2):92–100. doi: 10.1016/j.jneuroim.2006.01.020
82. Delivanoglou N, Boziki M, Theotokis P, Kesidou E, Touloumi O, Dafi N, et al. Spatio-temporal expression profile of NGF and the two-receptor system, TrkA and p75NTR, in experimental autoimmune encephalomyelitis. J Neuroinflammation (2020) 17(1):41. doi: 10.1186/s12974-020-1708-9
83. Petratos S, Gonzales MF, Azari MF, Marriott M, Minichiello RA, Shipham KA, et al. Expression of the low-affinity neurotrophin receptor, p75(NTR), is upregulated by oligodendroglial progenitors adjacent to the subventricular zone in response to demyelination. Glia. (2004) 48(1):64–75. doi: 10.1002/glia.20056
84. Barcelos IP, Troxell RM, Graves JS. Mitochondrial dysfunction and multiple sclerosis. Biol (Basel). (2019) 8(2):37. doi: 10.3390/biology8020037
85. Su K, Bourdette D, Forte M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front Physiol (2013) 4:169. doi: 10.3389/fphys.2013.00169
86. Ohl K, Tenbrock K, Kipp M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp Neurol (2016) 277:58–67. doi: 10.1016/j.expneurol.2015.11.010
87. Le Moan N, Houslay DM, Christian F, Houslay MD, Akassoglou K. Oxygen-dependent cleavage of the p75 neurotrophin receptor triggers stabilization of HIF-1alpha. Mol Cell (2011) 44(3):476–90. doi: 10.1016/j.molcel.2011.08.033
88. Nociti V, Ma O. What is the role of brain derived neurotrophic factor in multiple sclerosis neuroinflammation? Neuroimmunology Neuroinflammation (2020) 7:291–300. doi: 10.20517/2347-8659.2020.25
89. Carito V, Ceccanti M, Tarani L, Ferraguti G, Chaldakov GN, Fiore M. Neurotrophins' modulation by olive polyphenols. Curr Med Chem (2016) 23(28):3189–97. doi: 10.2174/0929867323666160627104022
90. Smith MH, Berman JR. What is rheumatoid arthritis? JAMA. (2022) 327(12):1194. doi: 10.1001/jama.2022.0786
91. Weyand CM, Wu B, Huang T, Hu Z, Goronzy JJ. Mitochondria as disease-relevant organelles in rheumatoid arthritis. Clin Exp Immunol (2023) 211(3):208–23. doi: 10.1093/cei/uxac107
92. Hu Z, Zhao TV, Huang T, Ohtsuki S, Jin K, Goronzy IN, et al. The transcription factor RFX5 coordinates antigen-presenting function and resistance to nutrient stress in synovial macrophages. Nat Metab (2022) 4(6):759–74. doi: 10.1038/s42255-022-00585-x
93. Wu B, Zhao TV, Jin K, Hu Z, Abdel MP, Warrington KJ, et al. Mitochondrial aspartate regulates TNF biogenesis and autoimmune tissue inflammation. Nat Immunol (2021) 22(12):1551–62. doi: 10.1038/s41590-021-01065-2
94. Yang CR, Ding HJ, Yu M, Zhou FH, Han CY, Liang R, et al. proBDNF/p75NTR promotes rheumatoid arthritis and inflammatory response by activating proinflammatory cytokines. FASEB J (2022) 36(3):e22180. doi: 10.1096/fj.202101558R
95. Farina L, Minnone G, Alivernini S, Caiello I, MacDonald L, Soligo M, et al. Pro nerve growth factor and its receptor p75NTR activate inflammatory responses in synovial fibroblasts: A novel targetable mechanism in arthritis. Front Immunol (2022) 13:818630. doi: 10.3389/fimmu.2022.818630
96. de Oliveira PG, Farinon M, Sanchez-Lopez E, Miyamoto S, Guma M. Fibroblast-like synoviocytes glucose metabolism as a therapeutic target in rheumatoid arthritis. Front Immunol (2019) 10:1743. doi: 10.3389/fimmu.2019.01743
97. Lai NS, Yu HC, Huang Tseng HY, Hsu CW, Huang HB, Lu MC. Increased serum levels of brain-derived neurotrophic factor contribute to inflammatory responses in patients with rheumatoid arthritis. Int J Mol Sci (2021) 22(4):1841. doi: 10.3390/ijms22041841
98. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med (2020) 26(8):1295–306. doi: 10.1038/s41591-020-0939-8
99. Clayton SA, MacDonald L, Kurowska-Stolarska M, Clark AR. Mitochondria as key players in the pathogenesis and treatment of rheumatoid arthritis. Front Immunol (2021) 12:673916. doi: 10.3389/fimmu.2021.673916
100. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
101. Tipton CM, Hom JR, Fucile CF, Rosenberg AF, Sanz I. Understanding b-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: A b-cell immunomics approach. Immunol Rev (2018) 284(1):120–31. doi: 10.1111/imr.12660
102. Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus-an update. Curr Opin Immunol (2012) 24(6):651–7. doi: 10.1016/j.coi.2012.10.004
103. Pisetsky DS, Lipsky PE. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat Rev Rheumatol (2020) 16(10):565–79. doi: 10.1038/s41584-020-0480-7
104. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15(3):160–71. doi: 10.1038/nri3795
105. Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma cell differentiation pathways in systemic lupus erythematosus. Front Immunol (2018) 9:427. doi: 10.3389/fimmu.2018.00427
106. Quach TD, Rodriguez-Zhurbenko N, Hopkins TJ, Guo X, Hernandez AM, Li W, et al. Distinctions among circulating antibody-secreting cell populations, including b-1 cells, in human adult peripheral blood. J Immunol (2016) 196(3):1060–9. doi: 10.4049/jimmunol.1501843
107. Bortnick A, Murre C. Cellular and chromatin dynamics of antibody-secreting plasma cells. Wiley Interdiscip Rev Dev Biol (2016) 5(2):136–49. doi: 10.1002/wdev.213
108. Garimalla S, Nguyen DC, Halliley JL, Tipton C, Rosenberg AF, Fucile CF, et al. Differential transcriptome and development of human peripheral plasma cell subsets. JCI Insight (2019) 4(9):e126732. doi: 10.1172/jci.insight.126732
109. Savage HP, Baumgarth N. Characteristics of natural antibody-secreting cells. Ann N Y Acad Sci (2015) 1362(1):132–42. doi: 10.1111/nyas.12799
110. Pattanapanyasat K, Khowawisetsut L, Chuansumrit A, Chokephaibulkit K, Tangnararatchakit K, Apiwattanakul N, et al. B cell subset alteration and the expression of tissue homing molecules in dengue infected patients. J BioMed Sci (2018) 25(1):64. doi: 10.1186/s12929-018-0467-8
111. Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, et al. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis (2010) 69(1):305–8. doi: 10.1136/ard.2008.096495
112. Ma K, Li J, Wang X, Lin X, Du W, Yang X, et al. TLR4(+)CXCR4(+) plasma cells drive nephritis development in systemic lupus erythematosus. Ann Rheum Dis (2018) 77(10):1498–506. doi: 10.1136/annrheumdis-2018-213615
113. Phillips R. proBDNF blockade modulates SLE. Nat Rev Rheumatol (2022) 18(4):185. doi: 10.1038/s41584-022-00766-8
114. Sumikawa MH, Iwata S, Zhang M, Miyata H, Ueno M, Todoroki Y, et al. An enhanced mitochondrial function through glutamine metabolism in plasmablast differentiation in systemic lupus erythematosus. Rheumatol (Oxford) (2022) 61(7):3049–59. doi: 10.1093/rheumatology/keab824
115. Takeshima Y, Iwasaki Y, Fujio K, Yamamoto K. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheumatol (2019) 48(6):1142–5. doi: 10.1016/j.semarthrit.2019.04.006
116. Tan W, Dong L, Shi X, Tang Q, Jiang D. P75NTR exacerbates SCI-induced mitochondrial damage and neuronal apoptosis depending on NTRK3. Curr Neurovasc Res (2021) 18(5):552–64. doi: 10.2174/1567202619666211231091834
117. Fauchais AL, Lise MC, Marget P, Lapeybie FX, Bezanahary H, Martel C, et al. Serum and lymphocytic neurotrophins profiles in systemic lupus erythematosus: a case-control study. PloS One (2013) 8(11):e79414. doi: 10.1371/journal.pone.0079414
118. Shobeiri P, Maleki S, Amanollahi M, Habibzadeh A, Teixeira AL, Rezaei N. Blood levels of brain-derived neurotrophic factor (BDNF) in systemic lupus erythematous (SLE): a systematic review and meta-analysis. Adv Rheumatol (2023) 63(1):8. doi: 10.1186/s42358-023-00291-6
119. Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol (1999) 17:255–81. doi: 10.1146/annurev.immunol.17.1.255
120. Howarth PH. The airway inflammatory response in allergic asthma and its relationship to clinical disease. Allergy (1995) 50(22 Suppl):13–21. doi: 10.1111/j.1398-9995.1995.tb02730.x
121. Rothenberg ME. Eosinophilia. N Engl J Med (1998) 338(22):1592–600. doi: 10.1056/NEJM199805283382206
122. De Monchy JG, Kauffman HF, Venge P, Koeter GH, Jansen HM, Sluiter HJ, et al. Bronchoalveolar eosinophilia during allergen-induced late asthmatic reactions. Am Rev Respir Dis (1985) 131(3):373–6. doi: 10.1164/arrd.1985.131.3.373
123. Thoenen H. Neurotrophins and neuronal plasticity. Science (1995) 270(5236):593–8. doi: 10.1126/science.270.5236.593
124. Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, et al. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. (1989) 341(6238):149–52. doi: 10.1038/341149a0
125. Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci (1996) 19:289–317. doi: 10.1146/annurev.ne.19.030196.001445
126. Barouch R, Appel E, Kazimirsky G, Braun A, Renz H, Brodie C. Differential regulation of neurotrophin expression by mitogens and neurotransmitters in mouse lymphocytes. J Neuroimmunol (2000) 103(2):112–21. doi: 10.1016/s0165-5728(99)00233-7
127. Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, et al. Activated human T cells, b cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med (1999) 189(5):865–70. doi: 10.1084/jem.189.5.865
128. Leon A, Buriani A, Dal Toso R, Fabris M, Romanello S, Aloe L, et al. Mast cells synthesize, store, and release nerve growth factor. Proc Natl Acad Sci U S A (1994) 91(9):3739–43. doi: 10.1073/pnas.91.9.3739
129. Tam SY, Tsai M, Yamaguchi M, Yano K, Butterfield JH, Galli SJ. Expression of functional TrkA receptor tyrosine kinase in the HMC-1 human mast cell line and in human mast cells. Blood. (1997) 90(5):1807–20. doi: 10.1016/S0950-3536(97)80018-2
130. Braun A, Appel E, Baruch R, Herz U, Botchkarev V, Paus R, et al. Role of nerve growth factor in a mouse model of allergic airway inflammation and asthma. Eur J Immunol (1998) 28(10):3240–51. doi: 10.1002/(SICI)1521-4141(199810)28:10<3240::AID-IMMU3240>3.0.CO;2-U
131. Braun A, Lommatzsch M, Mannsfeldt A, Neuhaus-Steinmetz U, Fischer A, Schnoy N, et al. Cellular sources of enhanced brain-derived neurotrophic factor production in a mouse model of allergic inflammation. Am J Respir Cell Mol Biol (1999) 21(4):537–46. doi: 10.1165/ajrcmb.21.4.3670
132. Lommatzsch M, Braun A, Mannsfeldt A, Botchkarev VA, Botchkareva NV, Paus R, et al. Abundant production of brain-derived neurotrophic factor by adult visceral epithelia. implications for paracrine and target-derived neurotrophic functions. Am J Pathol (1999) 155(4):1183–93. doi: 10.1016/S0002-9440(10)65221-2
133. Kobayashi H, Gleich GJ, Butterfield JH, Kita H. Human eosinophils produce neurotrophins and secrete nerve growth factor on immunologic stimuli. Blood. (2002) 99(6):2214–20. doi: 10.1182/blood.v99.6.2214
134. Horigome K, Pryor JC, Bullock ED, Johnson EM Jr. Mediator release from mast cells by nerve growth factor. neurotrophin specificity and receptor mediation. J Biol Chem (1993) 268(20):14881–7. doi: 10.1016/S0021-9258(18)82415-2
135. Kimata H, Yoshida A, Ishioka C, Kusunoki T, Hosoi S, Mikawa H. Nerve growth factor specifically induces human IgG4 production. Eur J Immunol (1991) 21(1):137–41. doi: 10.1002/eji.1830210121
136. Hamada A, Watanabe N, Ohtomo H, Matsuda H. Nerve growth factor enhances survival and cytotoxic activity of human eosinophils. Br J Haematol (1996) 93(2):299–302. doi: 10.1046/j.1365-2141.1996.5151055.x
137. Manti S, Brown P, Perez MK, Piedimonte G. The role of neurotrophins in inflammation and allergy. Vitam Horm (2017) 104:313–41. doi: 10.1016/bs.vh.2016.10.010
138. Nassenstein C, Braun A, Erpenbeck VJ, Lommatzsch M, Schmidt S, Krug N, et al. The neurotrophins nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 are survival and activation factors for eosinophils in patients with allergic bronchial asthma. J Exp Med (2003) 198(3):455–67. doi: 10.1084/jem.20010897
139. Tokuoka S, Takahashi Y, Masuda T, Tanaka H, Furukawa S, Nagai H. Disruption of antigen-induced airway inflammation and airway hyper-responsiveness in low affinity neurotrophin receptor p75 gene deficient mice. Br J Pharmacol (2001) 134(7):1580–6. doi: 10.1038/sj.bjp.0704411
140. Kerzel S, Path G, Nockher WA, Quarcoo D, Raap U, Groneberg DA, et al. Pan-neurotrophin receptor p75 contributes to neuronal hyperreactivity and airway inflammation in a murine model of experimental asthma. Am J Respir Cell Mol Biol (2003) 28(2):170–8. doi: 10.1165/rcmb.4811
141. Bandola J, Richter C, Ryser M, Jamal A, Ashton MP, von Bonin M, et al. Neurotrophin receptor p75NTR regulates immune function of plasmacytoid dendritic cells. Front Immunol (2017) 8:981. doi: 10.3389/fimmu.2017.00981
142. Britt RD Jr., Thompson MA, Wicher SA, Manlove LJ, Roesler A, Fang YH, et al. Smooth muscle brain-derived neurotrophic factor contributes to airway hyperreactivity in a mouse model of allergic asthma. FASEB J (2019) 33(2):3024–34. doi: 10.1096/fj.201801002R
143. Dyer BP, Rathod-Mistry T, Burton C, van der Windt D, Bucknall M. Diabetes as a risk factor for the onset of frozen shoulder: a systematic review and meta-analysis. BMJ Open (2023) 13(1):e062377. doi: 10.1136/bmjopen-2022-062377
144. International Diabetes Federation. IDF diabetes atlas. 10th ed. (2021). Available at: https://diabetesatlas.org/. [Accessed Dec 10, 2022].
145. Centers for disease control and prevention (2022). In: Prevent diabetes complications: Centers for disease control and prevention. Available at: https://www.cdc.gov/diabetes/managing/problems.html. [Accessed Dec 12, 2022].
146. Harding JL, Wander PL, Zhang X, Li X, Karuranga S, Chen H, et al. The incidence of adult-onset type 1 diabetes: A systematic review from 32 countries and regions. Diabetes Care (2022) 45(4):994–1006. doi: 10.2337/dc21-1752
147. Petrelli A, Giovenzana A, Insalaco V, Phillips BE, Pietropaolo M, Giannoukakis N. Autoimmune inflammation and insulin resistance: Hallmarks so far and yet so close to explain diabetes endotypes. Curr Diabetes Rep (2021) 21(12):54. doi: 10.1007/s11892-021-01430-3
148. Goncalves NP, Jager SE, Richner M, Murray SS, Mohseni S, Jensen TS, et al. Schwann cell p75 neurotrophin receptor modulates small fiber degeneration in diabetic neuropathy. Glia. (2020) 68(12):2725–43. doi: 10.1002/glia.23881
149. Taborsky GJ Jr., Mei Q, Bornfeldt KE, Hackney DJ, Mundinger TO. The p75 neurotrophin receptor is required for the major loss of sympathetic nerves from islets under autoimmune attack. Diabetes (2014) 63(7):2369–79. doi: 10.2337/db13-0778
150. Taborsky GJ Jr., Mei Q, Hackney DJ, Figlewicz DP, LeBoeuf R, Mundinger TO. Loss of islet sympathetic nerves and impairment of glucagon secretion in the NOD mouse: relationship to invasive insulitis. Diabetologia (2009) 52(12):2602–11. doi: 10.1007/s00125-009-1494-5
151. Mundinger TO, Mei Q, Figlewicz DP, Lernmark A, Taborsky GJ Jr. Impaired glucagon response to sympathetic nerve stimulation in the BB diabetic rat: effect of early sympathetic islet neuropathy. Am J Physiol Endocrinol Metab (2003) 285(5):E1047–54. doi: 10.1152/ajpendo.00136.2003
152. Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME, Kaplan DR, et al. Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci (2008) 11(6):649–58. doi: 10.1038/nn.2114
153. Goncalves NP, Vaegter CB, Andersen H, Ostergaard L, Calcutt NA, Jensen TS. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat Rev Neurol (2017) 13(3):135–47. doi: 10.1038/nrneurol.2016.201
154. Giacco F, Du X, Carratu A, Gerfen GJ, D'Apolito M, Giardino I, et al. GLP-1 cleavage product reverses persistent ROS generation after transient hyperglycemia by disrupting an ROS-generating feedback loop. Diabetes. (2015) 64(9):3273–84. doi: 10.2337/db15-0084
155. Han J, Tan P, Li Z, Wu Y, Li C, Wang Y, et al. Fuzi attenuates diabetic neuropathy in rats and protects schwann cells from apoptosis induced by high glucose. PloS One (2014) 9(1):e86539. doi: 10.1371/journal.pone.0086539
156. Apfel SC, Arezzo JC, Brownlee M, Federoff H, Kessler JA. Nerve growth factor administration protects against experimental diabetic sensory neuropathy. Brain Res (1994) 634(1):7–12. doi: 10.1016/0006-8993(94)90252-6
157. Scarpini E, Conti G, Chianese L, Baron P, Pizzul S, Basellini A, et al. Induction of p75NGFR in human diabetic neuropathy. J Neurol Sci (1996) 135(1):55–62. doi: 10.1016/0022-510x(95)00260-9
158. Conti G, Stoll G, Scarpini E, Baron PL, Bianchi R, Livraghi S, et al. p75 neurotrophin receptor induction and macrophage infiltration in peripheral nerve during experimental diabetic neuropathy: possible relevance on regeneration. Exp Neurol (1997) 146(1):206–11. doi: 10.1006/exnr.1997.6521
159. Tan W, Rouen S, Barkus KM, Dremina YS, Hui D, Christianson JA, et al. Nerve growth factor blocks the glucose-induced down-regulation of caveolin-1 expression in schwann cells via p75 neurotrophin receptor signaling. J Biol Chem (2003) 278(25):23151–62. doi: 10.1074/jbc.M212986200
160. Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, et al. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol (1998) 143(6):1691–703. doi: 10.1083/jcb.143.6.1691
161. Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, et al. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol (1998) 140(4):911–23. doi: 10.1083/jcb.140.4.911
162. Bist A, Fielding CJ, Fielding PE. p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry (2000) 39(8):1966–72. doi: 10.1021/bi991721h
163. Luu BE, Mossa AH, Cammisotto PG, Uri Saragovi H, Campeau L. Modulation of diabetic kidney disease markers by an antagonist of p75(NTR) in streptozotocin-treated mice. Gene (2022) 838:146729. doi: 10.1016/j.gene.2022.146729
164. Meek TH, Wisse BE, Thaler JP, Guyenet SJ, Matsen ME, Fischer JD, et al. BDNF action in the brain attenuates diabetic hyperglycemia via insulin-independent inhibition of hepatic glucose production. Diabetes (2013) 62(5):1512–8. doi: 10.2337/db12-0837
165. Jiang L, Tang K, Levin M, Irfan O, Morris SK, Wilson K, et al. COVID-19 and multisystem inflammatory syndrome in children and adolescents. Lancet Infect Dis (2020) 20(11):e276–e88. doi: 10.1016/S1473-3099(20)30651-4
166. Nagasawa M, Ashiarai M KM, Okada M, Imai M. Soluble sortilin in elevated in the acute phase of Kawasaki disease. GSR J Pediatr (2019) 1(1):1–8.
167. Ly KH, Regent A, Molina E, Saada S, Sindou P, Le-Jeunne C, et al. Neurotrophins are expressed in giant cell arteritis lesions and may contribute to vascular remodeling. Arthritis Res Ther (2014) 16(6):487. doi: 10.1186/s13075-014-0487-z
168. Tieu BC, Lee C, Sun H, Lejeune W, Recinos A 3rd, Ju X, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest (2009) 119(12):3637–51. doi: 10.1172/JCI38308
169. del Porto F, Proietta M, Tritapepe L, Miraldi F, Koverech A, Cardelli P, et al. Inflammation and immune response in acute aortic dissection. Ann Med (2010) 42(8):622–9. doi: 10.3109/07853890.2010.518156
170. Gutierrez PS, Marques Piubelli ML, Naal KG, Dias RR, Borges LF. Mitochondria in aneurysms and dissections of the human ascending aorta. Cardiovasc Pathol (2020) 47:107207. doi: 10.1016/j.carpath.2020.107207
171. Jun C, Fang B. Current progress of fluoroquinolones-increased risk of aortic aneurysm and dissection. BMC Cardiovasc Disord (2021) 21(1):470. doi: 10.1186/s12872-021-02258-1
172. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet (2017) 390(10114):2769–78. doi: 10.1016/S0140-6736(17)32448-0
173. Liu S, Zhao W, Lan P, Mou X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell (2021) 12(5):331–45. doi: 10.1007/s13238-020-00745-3
174. Grider JR, Piland BE, Gulick MA, Qiao LY. Brain-derived neurotrophic factor augments peristalsis by augmenting 5-HT and calcitonin gene-related peptide release. Gastroenterology (2006) 130(3):771–80. doi: 10.1053/j.gastro.2005.12.026
175. Esteban I, Hannestad J, Levanti B, Del Valle ME, Naves FJ, Vega JA. Neurotrophin receptor proteins immunoreactivity in human gastrointestinal endocrine cells. Brain Res Bull (1995) 38(6):539–43. doi: 10.1016/0361-9230(95)02025-9
176. Sochal M, Ditmer M, Gabryelska A, Bialasiewicz P. The role of brain-derived neurotrophic factor in immune-related diseases: A narrative review. J Clin Med (2022) 11(20):6023. doi: 10.3390/jcm11206023
Keywords: ProBDNF, p75NTR, sortilin, immune-mediated inflammatory diseases, mitochondria, metabolism
Citation: Li Q, Hu Y-Z, Gao S, Wang P-F, Hu Z-L and Dai R-P (2023) ProBDNF and its receptors in immune-mediated inflammatory diseases: novel insights into the regulation of metabolism and mitochondria. Front. Immunol. 14:1155333. doi: 10.3389/fimmu.2023.1155333
Received: 31 January 2023; Accepted: 28 March 2023;
Published: 18 April 2023.
Edited by:
Michael R. Elliott, University of Virginia, United StatesReviewed by:
Alexander G. Gabibov, Institute of Bioorganic Chemistry (RAS), RussiaChangwei Wei, Beijing Chaoyang Hospital, Capital Medical University, China
Copyright © 2023 Li, Hu, Gao, Wang, Hu and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ru-Ping Dai, eHlleXlydXBpbmdkYWlAY3N1LmVkdS5jbg==; Zhao-Lan Hu, aHV6aGFvbGFuQGNzdS5lZHUuY24=
†These authors have contributed equally to this work