
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 21 April 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1154566
This article is part of the Research Topic Expert Opinions and Perspectives in adoptive cell therapy for cancer: 2022 View all 7 articles
 Hanwen Zhang1*
Hanwen Zhang1* Tenzin Passang1
Tenzin Passang1 Sruthi Ravindranathan1Ramireddy Bommireddy2,3Mohammad Raheel Jajja4
Sruthi Ravindranathan1Ramireddy Bommireddy2,3Mohammad Raheel Jajja4 Lily Yang3,5
Lily Yang3,5 Periasamy Selvaraj2,3Chrystal M. Paulos3,5,6
Periasamy Selvaraj2,3Chrystal M. Paulos3,5,6 Edmund K. Waller1,3*
Edmund K. Waller1,3*In the past decades, advances in the use of adoptive cellular therapy to treat cancer have led to unprecedented responses in patients with relapsed/refractory or late-stage malignancies. However, cellular exhaustion and senescence limit the efficacy of FDA-approved T-cell therapies in patients with hematologic malignancies and the widespread application of this approach in treating patients with solid tumors. Investigators are addressing the current obstacles by focusing on the manufacturing process of effector T cells, including engineering approaches and ex vivo expansion strategies to regulate T-cell differentiation. Here we reviewed the current small-molecule strategies to enhance T-cell expansion, persistence, and functionality during ex vivo manufacturing. We further discussed the synergistic benefits of the dual-targeting approaches and proposed novel vasoactive intestinal peptide receptor antagonists (VIPR-ANT) peptides as emerging candidates to enhance cell-based immunotherapy.
Adoptive T-cell therapy (ACT) is a form of cellular immunotherapy in which tumor-reactive T cells recognize and eliminate malignant cells after infusion into patients. Barnes and Loutit initially proposed the ACT concept in 1956, describing the graft-versus-leukemia (GvL) effect of allogeneic hematopoietic stem cell transplantation (HSCT), which represents the earliest clinical example of the adoptive transfer of T cells with anti-cancer activity (1). In the past few decades, cell-based therapies with chimeric antigen receptor (CAR) T cells, engineered T cell receptor (eTCR) T cells, tumor-infiltrating lymphocytes (TILs), and other antigen-specific T cells have rapidly developed and shown enormous clinical potential. CAR T cell therapy, which involves the transfer of allogeneic or autologous T cells modified to express a chimeric antigen receptor (CAR), has gained FDA approval with studies documenting durable remissions in patients with relapsed/refractory (R/R) hematologic malignancies (2–4). However, many patients fail to achieve long-lasting remission due to loss of CAR T cell persistence and functionality (5). Hence, developing methods to counteract T-cell exhaustion and improve functionality is essential to improving ACT efficacy.
T cell exhaustion is a homeostatic mechanism that protects the organism against severe immunopathology from overwhelming CD8 T cell responses (6). Generally, exhausted T (Tex) cells have decreased expression of effector cytokines and increased expression of inhibitory immune checkpoint receptors such as PD-1, TIM-3, LAG-3, TIGHT, and CTLA-4 (7). However, expression of these molecules is also upregulated during early T cell activation, presumably as a homeostatic mechanism that modulates activation downstream of co-stimulatory signaling (8). T cell exhaustion comprises a differentiative process of several stages, accompanied by significant epigenetic reorganization and distinct transcriptional signatures (9, 10). Namely, the expression of TCF1/7, a transcription factor critical to maintaining immunological memory, decreases during the transition from the plastic to the irreversible and fixed dysfunctional chromatin state (11, 12).
Manufacturing of modified T cells is a multi-step process (13). The focus of two main areas of optimizing T-cell therapies are designing optimal genetic modifications of T cells and engineering improved cell activation and culture processes during ex vivo T-cell expansion (14). Adding clinically approved compounds, such as monoclonal antibodies and small molecule inhibitors, during the manufacture of cellular products might be a promising strategy to overcome T-cell exhaustion and enhance T-cell cytotoxicity. Adding drugs ex vivo represents an alternative to in vivo administration as part of a preconditioning regimen or therapy concomitant with T-cell infusion. Preclinical testing of ex vivo manufacturing and expansion approaches can identify strategies that yield a more potent adoptive T-cell therapy product with superior anti-tumor activity and persistence after infusion.
Cultures media supplemented with gamma-chain cytokines during ex vivo manufacturing, including IL-2, IL-7, IL-15, and IL-21, leads to the expansion of CAR T cells with enhanced proliferation, metabolic profiles, and less terminal differentiation (15). More recently, adding small-molecule compounds targeting tumor cell metabolic signaling pathways has also been explored to enhance T-cell function and persistence. (Table 1).
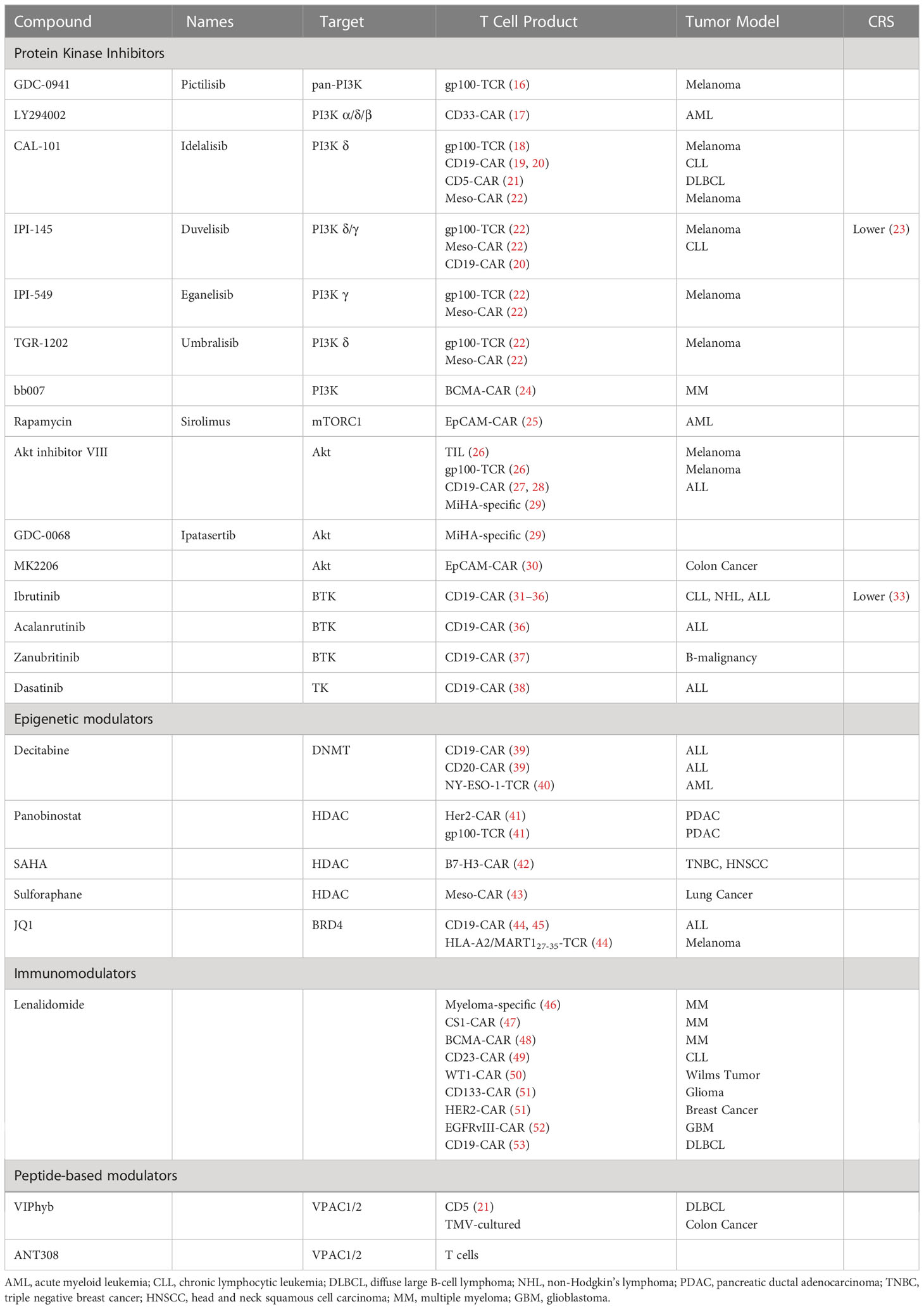
Table 1 Summary of small-molecule drugs enhancing adoptive T cells ex vivo.
The most frequent aberrations in cell signaling associated with tumorigenesis, angiogenesis, cell growth, or metastasis are hyperactive PI3K-AKT-mTOR pathways, exemplified by activating mutations of PIK3CA and the loss of PTEN functionality (54). Hence, the pharmaceutical industry has dedicated significant effort to developing PI3K inhibitors (PI3Ki) as targeted therapies. Mutational activation of PI3K signaling is relatively rare in hematologic malignancies, yet PI3Kδ inhibitors (idelalisib, duvelisib) were approved as a therapy for B-cell malignancies. Aside from suppressing tumor cell growth directly via inhibiting intracellular PI3K signaling (55), the beneficial clinical effects of PI3K inhibition in this setting may also be to indirectly activate immune cells with anti-cancer cytotoxicity in the tumor microenvironment. PI3Kγ and PI3Kδ are selectively expressed in leukocytes and are essential in promoting glycolysis and differentiation (56, 57). Indeed, while PI3K inhibitors may dampen many immune cell functions, blocking regulatory T (Treg) cell-mediated suppression of anti-tumor immune responses shows promise in immunotherapy (58). Initially, pan-PI3K inhibitor Pictilisib (GDC-0941) and PI3Kα/δ/β inhibitor LY294002 were found to delay terminal differentiation and preserve a reservoir of memory T cells (TCM and TEM) (16, 17). Selective inhibition of PI3Kδ with idelalisib (CAL-101), but not PI3Kα or PI3Kβ, promoted the generation of naïve-like (CD45RA+CCR7+) and undifferentiated CD8+ T cells phenotypes (CCR7+CD62L+, CD127, Tcf7) that had enhanced proliferative potential, function, and survival (16, 18). Idelalisib also preferentially inhibits human regulatory T-cell function (59). Subsequent studies showed idelalisib-treated T cells, or CAR T cells persisted longer and engrafted better after adoptive transfer into tumor-bearing mice, resulting in improved anti-tumor immunity (18, 19). These cells expressed fewer exhaustion markers (i.e., lower PD1 expression levels) and had a less senescent phenotype (CD27-CD28-) (21, 60). PI3Kγ, first promoted as a selective immunotherapeutic target in myeloid cells, was later found to be involved in remodeling T-cell differentiation (22, 61, 62). Inhibition of PI3Kγ and PI3Kδ with duvelisib (IPI-145) reprogramed terminal differentiation and the metabolism of CAR T cells to enhance expansion, persistence, and anti-tumor cytotoxicity (20). A recent phase 1 study (NCT03274219) of bb21217, an anti-BCMA CART therapy based on ide-cel that included the PI3K inhibitor bb007 during ex vivo culture showed increased enrichment for CD27+/CCR7+ Tm cells, depletion of CD57+ senescent cells, increased CD127 expression, and higher peak in vivo CAR T expansion, resulting in improved clinical outcomes in MM patients (24, 63). Excitingly, duvelisib also potently inhibits IL-6 production and cytokine release syndrome (CRS) (23). Two clinical trials were initiated to verify enhanced CAR T-cell functionality (NCT04890236, diffuse large B-cell lymphoma (DLBCL)) and CRS prevention (NCT05044039, non-Hodgkin lymphoma (NHL), acute lymphocytic leukemia (ALL)) in Duvelisib-treated DLBCL patients.
Inhibiting the pathway downstream of PI3K showed a similar effect as direct inhibition of PI3K. mTOR acts intrinsically through the mTORC1 (mTOR complex 1) pathway to regulate memory T-cell differentiation (64). The mTORC1 inhibitor rapamycin promoted memory CD8 T-cell survival, maintenance of a less differentiated phenotype, and improved the functional qualities of CD8 T cells (CD127High CD62LHigh Bcl2High KLRG1Low) (64). Furthermore, rapamycin-pretreated EpCAM CAR T cells had upregulated CXCR4, increased infiltration into the bone marrow, and superior elimination of AML cells in leukemia xenograft mouse models (25). Interestingly, CAR-T cell expansion in IL-15 preserved the stem cell memory (Tscm) phenotype and improved metabolic fitness, likely via mTORC1 suppression. However, the inclusion of IL-7 and/or IL-21 in addition to IL15 reduced the beneficial effects of IL-15 on the phenotype and anti-tumor potency of CAR-T (65). Akt functions as a critical signaling node to maintain T cell survival during the effector-to-memory cell transition (66). Like rapamycin, Akt inhibitors, notably Akt-inhibitor VIII and GDC-0068, enhanced the expansion of tumor-specific lymphocytes and promoted the ex vivo generation of stem cell memory-like CD8+ T cells (CD62Lhigh CCR7high CXCR4high) with a unique metabolic profile and cytokine polyfunctionality (26–29). A pre-clinical study utilizing EpCAM CAR T in a T murine AML model showed that Akt inhibition (MK2206) at the initial stage of CAR T manufacture enhanced the expansion of CAR T cells and CART efficacy in vivo (30). Overall, targeting PI3K-AKT-mTOR signaling shows therapeutic potential in improving adoptive T-cell therapy. PI3K inhibition produces a more profound TCF1/7 upregulation than other small molecular TK inhibitors and may elucidate better anti-tumor efficacy in vivo (18).
Bruton’s tyrosine kinase (BTK) is a nonreceptor tyrosine kinase initially discovered as a critical component of B cell receptor (BCR) signal transduction in both healthy and malignant B lymphocytes (67, 68). The clinical role of BTK extends beyond its effects on normal and malignant B cells. PI3Kγ can activate BTK to promote phospholipase C (PLC) γ-dependent signaling in hematopoietic cells, including myeloid cells (69). A recent study suggested a regulatory role for BTK in T-cell activation. After TCR engagement, BTK was activated and subsequently activated PLCγ1, which amplified downstream TCR signaling and facilitated T-cell activation and expansion (70). Clinically, long-term treatment with ibrutinib, an inhibitor that forms irreversible covalent bonds to BTK, reversed CD8 T cell exhaustion and protected T cells from proliferation-induced senescence in chronic lymphocytic leukemia (B-CLL) patients. In addition, T cells from ibrutinib-treated CLL patients have decreased PD-1, TIM3, and LAG3 expression and increased antigen-specific responses (71–73). Ibrutinib improved CAR T cell expansion in vitro and promoted a less-differentiated less-exhausted naïve-like phenotype by inhibiting interleukin-2-inducible T-cell kinase (ITK) (31, 32). To date, several clinical trials are investigating the regimen of concurrent administration of CAR T cells and ibrutinib in B-cell malignancies (NCT02640209, NCT03960840). Concurrent ibrutinib therapy may improve CD19 CAR T-cell engraftment, enhance anti-tumor efficacy, and decrease CRS severity, leading to high rates of minimal residual disease (MRD)-negative responses, but progression-free survival (PFS) was unchanged (32–35). Second-generation BTKi acalanrutinib and zanubritinib, which are more selective and well-tolerated, have also been examined (74). Acalabrutinib improved CAR T-cell effector function and prolonged survival of tumor-bearing mice when combined with CAR T cells (36), while zanubrutinib lacked these positive effects (37).
Dasatinib, a second-generation tyrosine kinase inhibitor (TKI), was initially approved by FDA to treat Ph+ chronic myeloid leukemia (CML) (75). A recent study showed that dasatinib prevents or reverses CD28/CAR T and 4-1BB/CA T cell differentiation and exhaustion during ex vivo expansion, resulting in profoundly enhanced therapeutic efficacy and in vivo persistence (38). Multiple pathways are involved in this process, including Src phosphorylation, JAK/STAT, MAPK, and PI3K/AKT (38, 76).
Epigenetic modulators represent another promising strategy to enhance T-cell function based on the epigenetic remodeling and chromatin transitions discovered during the process of T-cell exhaustion. DNA methyltransferases and histone deacetylases (HDACs) are activated during T-cell differentiation, resulting in high levels of DNA and histone methylation in exhausted T cells (7, 9). Recent studies revealed that decitabine, a clinical DNA methylation inhibitor, enhances anti-tumor activities, cytokine production, and CAR T cell proliferation in both in vitro and in vivo non-Hodgkin lymphoma (NHL) models (39). Decitabine also promotes the maintenance of effector function and the memory phenotype of NY-ESO-1-specific eTCR T cells leading to greater anti-AML efficacy (40). Likewise, HDAC inhibitors panobinostat, SAHA, and sulforaphane promote the generation of T cells with a central memory phenotype and reduce expression of immunosuppressive markers (PD-1, CTLA-4, TET2) in CAR T-cell, resulting in enhanced anti-tumor response in solid tumor models (41–43).
BRD4 is a member epigenetic modulator of the bromodomain and extra terminal motif (BET) subfamily. BRD4 promotes TEM CD8 T-cell differentiation by regulating BATF expression. Treatment of CAR T cells with BET inhibitor JQ1 promoted the expansion of less differentiated TSCM and TCM, downregulated PD-1 and TET2 exhaustion marker expression, improved persistence and effector function, and augmented T-cell mediated anti-tumor effect in leukemia models (44, 45). Interestingly, the commonly used PI3K inhibitor LY294002 is also an inhibitor of BET bromodomains (77).
Immunomodulatory imide drugs (IMiDs) are thalidomide analogs with pleiotropic anti-myeloma properties. IMiDs act directly on malignant cells and indirectly via enhancing T and NK cell effector functions (78). Early in vitro studies showed that IMiDs induced T-cell proliferation, IL-2 and IFN-γ secretion, and myeloma-specific T-cell responses (46, 79). Myeloma patients treated with lenalidomide had increased numbers of central (TCM) and effector (TEM) memory CD8 T cells with decreased PD-1 expression (80). However, the effect of IMiDs on Treg remains uncertain (81). In general, favorable clinical outcomes with lenalidomide were observed from either induction or post-autologous stem cell transplant (ASCT) consolidation and maintenance (81). Aligned with previous findings, lenalidomide-treated CAR T cells acquired a memory phenotype, enhanced polyfunctional cytokine secretion, and increased immune synapse formation (47). CS1 CAR T cells expanded in vitro with lenalidomide had improved anti-tumor efficacy and in vivo persistence in murine myeloma models (47). Preclinical studies showed treatment with lenalidomide during the early phase of in vivo CAR T cell expansion recapitulated the effects of ex vivo lenalidomide exposure in multiple hematologic and solid tumor mouse models (48–52). As expected, several clinical trials investigating the combination of CAR T cells with lenalidomide have been initiated (NCT03070327, NCT05032820, NCT04923893, NCT04002401). Preliminary data has shown that early lenalidomide infusion enhances CAR T-cell response in patients with R/R DLBCL (53, 82).
Vasoactive intestinal peptide (VIP) is a 28-amino acid neuropeptide isolated in 1970 from porcine duodenum that induces vasodilation and hypotension (83, 84). The immunosuppressive properties of VIP were described in the early 2000s by Delgado, who noted that VIP promoted the survival of Th2 effectors, the generation of memory Th2 cells, and enhanced Treg function (85–87). More recent studies showed that VIP enhances M2 macrophage polarization and promotes macrophages with a less-inflammatory physiologic profile that promotes tissue repair (88–90). We recently noted that VIP produced by activated T cells limits their proliferation in vitro (91), and VIP produced by donor plasmacytoid dendritic cells (pDCs) limits Graft-versus-Host Disease (GVHD) in vivo (92). The emerging immunoregulatory role of VIP on innate and adaptive immune functions makes it a candidate immunotherapy target.
VIP-hybrid (VIPhyb) is a VIP-receptor antagonist synthesized by replacing the six N-terminal residues of VIP with highly charged residues from the N-terminal peptide sequence of neurotensin (93). VIPhyb acts as a competitive antagonist, binding to VIP receptors VPAC1 and VPAC2 without activating the downstream VIP-receptor signaling pathway (94). In the past decade, our group showed that inhibiting VIP signaling could enhance CD8 T-cell proliferation and function, leading to favorable T-cell-dependent anti-viral and anti-cancer responses in murine models of CMV infection and acute leukemia, respectively (91, 95–98). To further improve the efficacy of VIP-receptor antagonists as immuno-modulatory drugs, we have developed a series of peptides, including ANT008, ANT308, and ANT195, that are predicted to have increased binding affinity to human VIP receptors VPAC1 and VPAC2 and have enhanced ability to elicit T cell-dependent antileukemia responses in mice (Li unpublished). Recently, we published that VIP-receptor antagonists (ANT008, ANT308) were synergistic when added to anti-PD1 antibodies in enhancing T-cell mediated anti-tumor response to multiple murine models of pancreatic ductal carcinoma (PDAC) (99). These exciting findings validated using VIP-receptor antagonists as anti-cancer immunotherapy agents. In addition, we further investigated the feasibility of using VIP-receptor antagonists in adoptive T-cell therapy.
Previously, our team has shown that idelalisib and VIPhyb synergically increased the transduction and expansion of anti-CD5 CAR T cells manufactured from DLBCL patients (21). The addition of idelalisib and VIPhyb to cultured T cells reduced terminal differentiation, enhanced cytokine expression, and preserved expression of costimulatory molecules CD27 and CD28 (21). These agents target distinct signaling pathways, and their combinatorial synergy might be applicable for manufacturing CAR T therapy in patients with hematological and in adoptive T cell for patients with solid tumor malignancies. However, applying adoptive T cell therapy to solid tumor patients is constrained by the low frequency of tumor-infiltrating lymphocytes and the high molecular heterogeneity of solid tumors lacking expression of public (shared) tumor antigens. Therefore, we tested the feasibility of expanding patient tumor-specific T cells ex vivo by adding idelalisib and VIPhyb, using matched tumor and PBMCs from consented metastatic colon cancer patients. The source of tumor antigens were tumor membrane vesicles (TMVs) manufactured from that patient’s tumor and decorated with IL-12 and B7-1 (100, 101). Activating TMV-stimulated autologous T cells in the presence of idelalisib, VIPhyb, and anti-CD3 Dyna beads expanded cancer-specific T cells (Figure S1). After 14 days of ex vivo culture, T cells expanded with decorated TMV, VIPhyb, and idelalisib had 25% of IFN gamma-expressing CD8 T cells compared to 13% IFN gamma-expressing CD8 T cells cultured with IL12/B7-1 decorated TMV without VIPhyb and idelalisib (Figure 1A). Notably, this effect was not observed in CD4 T cells, which may attribute to the lack of VPAC1 expression in CD4 subset (Passang unpublished). The tumor-antigen-stimulated T cells expanded with VIPhyb and idelalisib were more effective in controlling the growth of patient-derived colon cancer xenografts (PDX) following infusion into tumor-bearing immunodeficient NSG mice than T cells expanded with TMV but without the addition of VIPhyb + idelalisib, or T cells expanded with neither TMV nor VIPhyb plus idelalisib (Figure 1A).
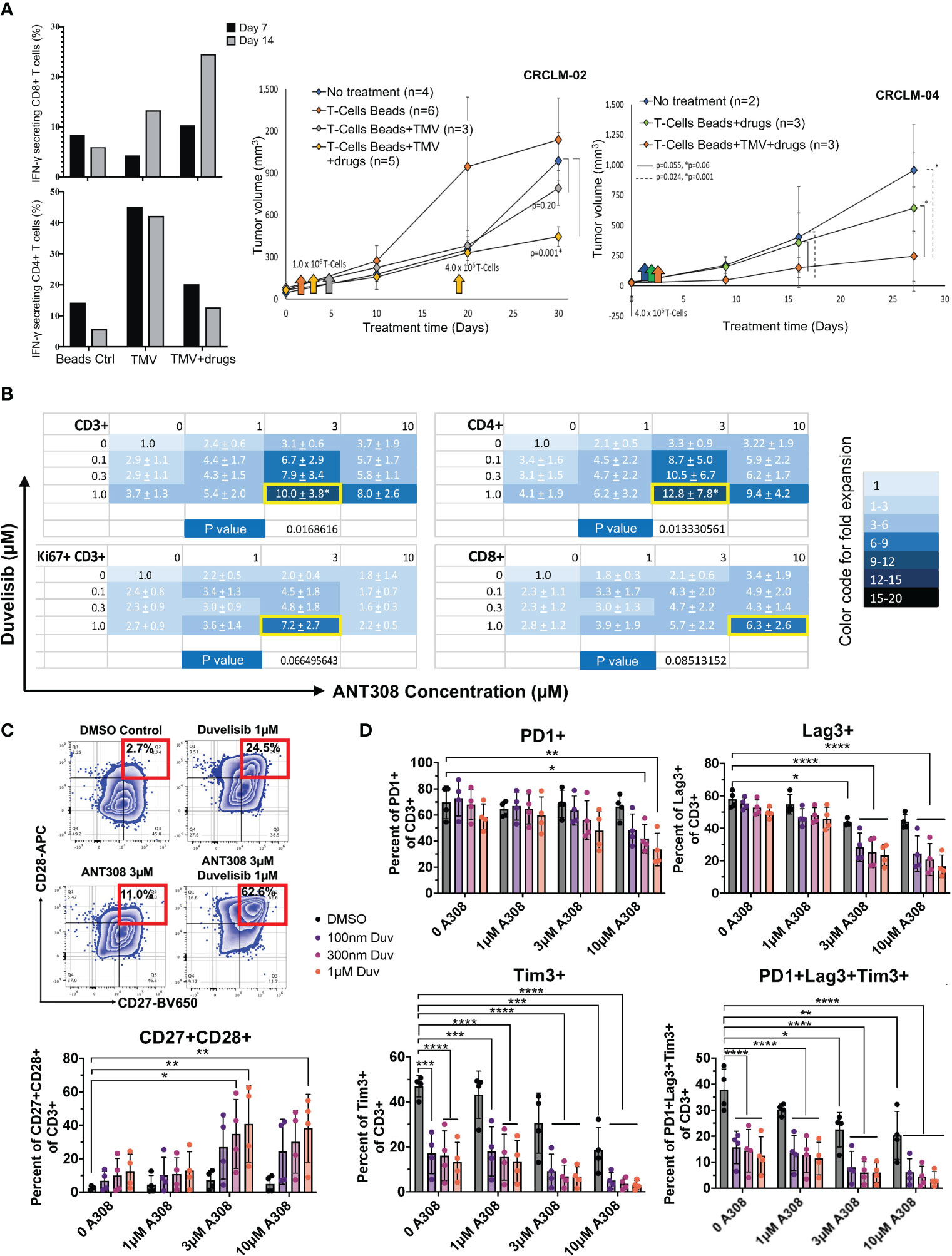
Figure 1 Pharmacological blockade of PI3K and VIPR signaling improves T-cell expansion and function in vivo ((A) adoptive antigen-specific T cells in metastatic colon cancer PDX model) and in vitro ((B–D), human T cells). (A) Left: Increased IFN-γ secreting CD8+ T cells using decorated TMV with VIPhyb and idelalisib (CRCLM-02, n=1); Right: Decreased tumor growth in PDX (CRCLM-02 and CRCLM-04) mice receiving T cells expanded with beads+TMV+drugs. ANOVA was used to determine significance. The standard error (SE) was shown. (B) Total CD3+ T cells, the actively proliferating Ki67+CD3+ subset, CD4+CD3+ T cells, and CD8+CD3+ T cells are synergistically expanded in vitro by the combination of ANT308 and duvelisib. The mean +/- SD fold increase in cell expansion over control cultures containing neither added ANT308 nor duvelisib is shown (n=4), with color shading according to the relative increase. The pair of concentrations yielded the maximal increase in mean fold expansion is shown with a yellow border around the cell. (C) Frequencies of CD27+CD28+ T cells in cultures with duvelisib and ANT308 led to the highest average expansion for that subset of T cells (n=4). An example of gating is on the left. (D) ANT308 and duvelisib demonstrated synergy in decreasing PD1+, Lag3+, Tim3+, and PD1+Lag3+Tim3+ cells (n=4). Figures were plotted with Microsoft Excel and Prism 9. Paired two-sided student t-test was used to determine significance. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Later we investigated the synergy effect of PI3Kγ/δ inhibitor duvelisib and leading VIPR-ANT peptide ANT308. The actively proliferating T cells (Ki67+CD3+), CD4+, and CD8+ T-cell subsets synergistically expanded in vitro with the combinatorial use of duvelisib and ANT308 (Figure 1B). We also found that ANT308 as a single agent, could promote a less senescent T-cell phenotype (CD27+CD28+), decrease exhausted T cells (PD1+Lag3+Tim3+) and reduce the expression of PD-1, LAG3, and TIM3. Adding duvelisib to T cells cultured with ANT308 or ANT195 synergistically enhanced the expansion of central memory T cells. The percentage of T cells co-expressing CD27 and CD28 increased from 2.59% to 7.16% with single-agent ANT308 (3 μM) and 12.5% with single-agent duvelisib (1 µM). The percentage of CD27+CD28+ T cells further increased to 40.83% with the combination of ANT308 and duvelisib (Figure 1C). Similarly, the percentage of T cells with an exhausted phenotype (PD1+Lag3+Tim3+) decreased from 37.8% in control cultures with neither ANT308 nor duvelisib, to 22.53% with only ANT308, to 12.56% with single-agent Duvelisib. Adding both drugs together further decreased the frequency of exhausted T cells to 5.93% (Figure 1D). Comparable synergistic effects of adding the VIP-receptor antagonist ANT195 to duvelisib effect were observed (Figures S2-3).
The field of cell-based immunotherapy is growing exponentially. However, efforts are still needed to improve the clinical response rate. Recent studies have focused on developing strategies to optimize efficiency in manufacturing T-cells for ACT therapy and the efficacy of T cells in vivo. We have reviewed current strategies to enhance T-cell expansion, persistence, and functionality during ex vivo manufacturing. Moreover, we further discussed the synergistic benefits of approaches that target multiple signaling pathways. Besides the conventional small-molecule drugs, novel VIPR-ANT peptides are promising immunotherapeutic candidates. Future studies will define the immunoregulatory role of VIP in ACT and its feasibility in clinical application.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Emory University Institutional Animal Care and Use Committee.
HZ conceived, drafted, and wrote the manuscript. TP performed the in vitro experiments and provided the data of T-cells treated with Duvelisib and ANT308. SR led the adoptive T cell therapy project in metastatic colon cancer and conducted in vivo PDX-model experiments. RB and PS produced TMV and incorporated the molecules by protein transfer. MJ and LY established the PDX model. CP critically reviewed and revised the manuscript. EW edited and revised the manuscript and provided funding support. All authors contributed to the article and approved the submitted version.
The cancer immunotherapy research in EW’s laboratory is funded by The Katz Foundation (PI: Waller), Robert W. Woodruff Health Sciences Fund (PI: Waller), The Coulter Foundation (project 60934), NIGMS MARC program (#T34GM105550), the National Institutes of Health (NIH) fund (1R01AI145231-01A1, PI: Waller). The in vivo studies in NSG mice were also funded by the NIH grant (R01 CA202846, PI: Yang).
We thank healthy volunteers and patients for blood and tumor sample donation. We thank current and former colleagues for contributing to the adoptive T-cell therapy research. We thank Metaclipse Therapeutics Corporation for providing purified GPI-IL-12 and GPI-B7-1 molecules.
Intellectual property related to the use of peptide antagonists to vasoactive intestinal polypeptides to treat cancer is the subject of US patent applications, which are licensed to Cambium Oncology, LLC. EW is a co-founder and has equity in Cambium Oncology. Duvelisb was a commercial product of Verastem Oncology now a product of Secura Bio. EW was on the Verastem Scientific Advisory Board. PS holds shares in Metaclipse Therapeutics Corporation, a company that is planning to use GPI-anchored molecules to develop a membrane-based TMV cancer vaccine in the future. A conflict-of-interest management plan has been reviewed and approved by Emory University.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1154566/full#supplementary-material
ACT, adoptive T-cell therapy; GvL, graft-versus-leukemia; GVHD, graft-versus-host disease; HSCT, hematopoietic stem cell transplantation; CAR, chimeric antigen receptor; TCR, T cell receptor; BCR, B cell receptor; TIL, tumor-infiltrating lymphocyte; CRS, cytokine release syndrome; VIP, vasoactive intestinal peptide; VIPR-ANT, vasoactive intestinal peptide receptor antagonist; R/R, relapsed/refractory; ALL, acute lymphocytic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; NHL, non-Hodgkin’s lymphoma; PDAC, pancreatic ductal adenocarcinoma; TNBC, triple negative breast cancer; HNSCC, head and neck squamous cell carcinoma; MM, multiple myeloma; GBM, glioblastoma; MRD, minimal residual disease; PFS, progression-free survival; PDX, patient-derived xenograft; TMV, tumor membrane vesicle.
1. Barnes DW, Corp MJ, Loutit JF, Neal FE. Treatment of murine leukaemia with X rays and homologous bone marrow; preliminary communication. Br Med J (1956) 2:626–7. doi: 10.1136/bmj.2.4993.626
2. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory Large b-cell lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
3. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse Large b-cell lymphoma. N Engl J Med (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
4. Gill S, Brudno JN. CAR T-cell therapy in hematologic malignancies: clinical role, toxicity, and unanswered questions. Am Soc Clin Oncol Educ Book (2021), e246–65. doi: 10.1200/EDBK_320085
5. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
6. Cornberg M, Kenney LL, Chen AT, Waggoner SN, Kim SK, Dienes HP, et al. Clonal exhaustion as a mechanism to protect against severe immunopathology and death from an overwhelming CD8 T cell response. Front Immunol (2013) 4:475. doi: 10.3389/fimmu.2013.00475
7. Gumber D, Wang LD. Improving CAR-T immunotherapy: overcoming the challenges of T cell exhaustion. EBioMedicine (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
8. Jiang W, He Y, He W, Wu G, Zhou X, Sheng Q, et al. Exhausted CD8+T cells in the tumor immune microenvironment: new pathways to therapy. Front Immunol (2020) 11:622509. doi: 10.3389/fimmu.2020.622509
9. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity (2020) 52:825–841.e828. doi: 10.1016/j.immuni.2020.04.014
10. Chu T, Zehn D. Charting the roadmap of T cell exhaustion. Immunity (2020) 52:724–6. doi: 10.1016/j.immuni.2020.04.019
11. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) 545:452–6. doi: 10.1038/nature22367
12. Zhang J, Lyu T, Cao Y, Feng H. Role of TCF-1 in differentiation, exhaustion, and memory of CD8(+) T cells: a review. FASEB J (2021) 35:e21549. doi: 10.1096/fj.202002566R
13. Blache U, Popp G, Dünkel A, Koehl U, Fricke S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat Commun (2022) 13:5225. doi: 10.1038/s41467-022-32866-0
14. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-T persistence. biomark Res (2022) 10:86. doi: 10.1186/s40364-022-00434-9
15. Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rangel Rivera GO, Arhontoulis DC, et al. Fueling cancer immunotherapy with common gamma chain cytokines. Front Immunol (2019) 10:263. doi: 10.3389/fimmu.2019.00263
16. Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced therapeutic efficacy and memory of tumor-specific CD8 T cells by ex vivo PI3K-δ inhibition. Cancer Res (2017) 77:4135–45. doi: 10.1158/0008-5472.CAN-16-1925
17. Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia (2018) 32:1157–67. doi: 10.1038/s41375-017-0008-6
18. Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. PI3Kδ inhibition enhances the antitumor fitness of adoptively transferred CD8(+) T cells. Front Immunol (2017) 8:1221. doi: 10.3389/fimmu.2017.01221
19. Stock S, Übelhart R, Schubert ML, Fan F, He B, Hoffmann JM, et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int J Cancer (2019) 145:1312–24. doi: 10.1002/ijc.32201
20. Funk CR, Wang S, Chen KZ, Waller A, Sharma A, Edgar CL, et al. PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood (2022) 139:523–37. doi: 10.1182/blood.2021011597
21. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
22. Dwyer CJ, Arhontoulis DC, Rangel Rivera GO, Knochelmann HM, Smith AS, Wyatt MM, et al. Ex vivo blockade of PI3K gamma or delta signaling enhances the antitumor potency of adoptively transferred CD8(+) T cells. Eur J Immunol (2020) 50:1386–99. doi: 10.1002/eji.201948455
23. Amatya PN, Carter AJ, Ritchey JK, Niswonger J, Cooper ML, Pachter JA, et al. The dual PI3Kδγ inhibitor duvelisib potently inhibits IL-6 production and cytokine release syndrome (CRS) while maintaining CAR-T function in vitro and. In Vivo. Blood (2020) 136:1–2. doi: 10.1182/blood-2020-139904
24. Alsina M, Shah N, Raje NS, Jagannath S, Madduri D, Kaufman JL, et al. Updated results from the phase I CRB-402 study of anti-bcma CAR-T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma: correlation of expansion and duration of response with T cell phenotypes. Blood (2020) 136:25–6. doi: 10.1182/blood-2020-140410
25. Nian Z, Zheng X, Dou Y, Du X, Zhou L, Fu B, et al. Rapamycin pretreatment rescues the bone marrow AML cell elimination capacity of CAR-T cells. Clin Cancer Res (2021) 27:6026–38. doi: 10.1158/1078-0432.CCR-21-0452
26. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res (2015) 75:296–305. doi: 10.1158/0008-5472.CAN-14-2277
27. Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight (2017) 2(23):e95103. doi: 10.1172/jci.insight.95103
28. Urak R, Walter M, Lim L, Wong CW, Budde LE, Thomas S, et al. Ex vivo akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J ImmunoTherapy Cancer (2017) 5:26. doi: 10.1186/s40425-017-0227-4
29. Mousset CM, Hobo W, Ji Y, Fredrix H, De Giorgi V, Allison RD, et al. Ex vivo AKT-inhibition facilitates generation of polyfunctional stem cell memory-like CD8+ T cells for adoptive immunotherapy. OncoImmunology (2018) 7:e1488565. doi: 10.1080/2162402X.2018.1488565
30. Zhang Q, Ding J, Sun S, Liu H, Lu M, Wei X, et al. Akt inhibition at the initial stage of CAR-T preparation enhances the CAR-positive expression rate, memory phenotype and. Vivo efficacy. Am J Cancer Res (2019) 9:2379–96.
31. Fan F, Yoo HJ, Stock S, Wang L, Liu Y, Schubert M-L, et al. Ibrutinib for improved chimeric antigen receptor T-cell production for chronic lymphocytic leukemia patients. Int J Cancer (2021) 148:419–28. doi: 10.1002/ijc.33212
32. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
33. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood (2020) 135:1650–60. doi: 10.1182/blood.2019002936
34. Gill S, Vides V, Frey NV, Hexner EO, Metzger S, O'Brien M, et al. Anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia. Blood Adv (2022) 6:5774–85. doi: 10.1182/bloodadvances.2022007317
35. Liu M, Deng H, Mu J, Li Q, Pu Y, Jiang Y, et al. Ibrutinib improves the efficacy of anti-CD19-CAR T-cell therapy in patients with refractory non-Hodgkin lymphoma. Cancer Sci (2021) 112:2642–51. doi: 10.1111/cas.14915
36. Qin JS, Johnstone TG, Baturevych A, Hause RJ, Ragan SP, Clouser CR, et al. Antitumor potency of an anti-CD19 chimeric antigen receptor T-cell therapy, lisocabtagene maraleucel in combination with ibrutinib or acalabrutinib. J Immunother (2020) 43:107–20. doi: 10.1097/CJI.0000000000000307
37. Ye X, Liu M, Lv C, Li Y, Chen L, Zhang J, et al. Synergistic effects of zanubrutinib combined with CD19 CAR-T cells in raji cells in vitro and in vivo. Technol Cancer Res Treat (2022) 21:15330338221133224. doi: 10.1177/15330338221133224
38. Zhang H, Hu Y, Shao M, Teng X, Jiang P, Wang X, et al. Dasatinib enhances anti-leukemia efficacy of chimeric antigen receptor T cells by inhibiting cell differentiation and exhaustion. J Hematol Oncol (2021) 14:113. doi: 10.1186/s13045-021-01117-y
39. Wang Y, Tong C, Dai H, Wu Z, Han X, Guo Y, et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun (2021) 12:409. doi: 10.1038/s41467-020-20696-x
40. Kang S, Wang L, Xu L, Wang R, Kang Q, Gao X, et al. Decitabine enhances targeting of AML cells by NY-ESO-1-specific TCR-T cells and promotes the maintenance of effector function and the memory phenotype. Oncogene (2022) 41:4696–708. doi: 10.1038/s41388-022-02455-y
41. Ali AI, Wang M, von Scheidt B, Dominguez PM, Harrison AJ, Tantalo DGM, et al. A histone deacetylase inhibitor, panobinostat, enhances chimeric antigen receptor T-cell antitumor effect against pancreatic cancer. Clin Cancer Res (2021) 27:6222–34. doi: 10.1158/1078-0432.CCR-21-1141
42. Lei X, Ou Z, Yang Z, Zhong J, Zhu Y, Tian J, et al. A pan-histone deacetylase inhibitor enhances the antitumor activity of B7-H3–specific CAR T cells in solid tumors. Clin Cancer Res (2021) 27:3757–71. doi: 10.1158/1078-0432.CCR-20-2487
43. Shen C, Zhang Z, Tian Y, Li F, Zhou L, Jiang W, et al. Sulforaphane enhances the antitumor response of chimeric antigen receptor T cells by regulating PD-1/PD-L1 pathway. BMC Med (2021) 19:283. doi: 10.1186/s12916-021-02161-8
44. Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest (2016) 126:3479–94. doi: 10.1172/JCI86437
45. Kong W, Dimitri A, Wang W, Jung IY, Ott CJ, Fasolino M, et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J Clin Invest (2021) 131(16):e145459. doi: 10.1172/JCI145459
46. Krämer I, Engelhardt M, Fichtner S, Neuber B, Medenhoff S, Bertsch U, et al. Lenalidomide enhances myeloma-specific T-cell responses. Vivo vitro. Oncoimmunology (2016) 5:e1139662. doi: 10.1080/2162402X.2016.1139662
47. Wang X, Walter M, Urak R, Weng L, Huynh C, Lim L, et al. Lenalidomide enhances the function of CS1 chimeric antigen receptor–redirected T cells against multiple myeloma. Clin Cancer Res (2018) 24:106–19. doi: 10.1158/1078-0432.CCR-17-0344
48. Works M, Soni N, Hauskins C, Sierra C, Baturevych A, Jones JC, et al. Anti–b-cell maturation antigen chimeric antigen receptor T cell function against multiple myeloma is enhanced in the presence of lenalidomide. Mol Cancer Ther (2019) 18:2246–57. doi: 10.1158/1535-7163.MCT-18-1146
49. Tettamanti S, Rotiroti MC, Giordano Attianese GMP, Arcangeli S, Zhang R, Banerjee P, et al. Lenalidomide enhances CD23.CAR T cell therapy in chronic lymphocytic leukemia. Leukemia Lymphoma (2022) 63:1566–79. doi: 10.1080/10428194.2022.2043299
50. Zhang L, Jin G, Chen Z, Yu C, Li Y, Li Y, et al. Lenalidomide improves the antitumor activity of CAR-T cells directed toward the intracellular wilms tumor 1 antigen. Hematology (2021) 26:818–26. doi: 10.1080/16078454.2021.1981534
51. Wang Z, Zhou G, Risu N, Fu J, Zou Y, Tang J, et al. Lenalidomide enhances CAR-T cell activity against solid tumor cells. Cell Transplant (2020) 29:0963689720920825. doi: 10.1177/0963689720920825
52. Kuramitsu S, Ohno M, Ohka F, Shiina S, Yamamichi A, Kato A, et al. Lenalidomide enhances the function of chimeric antigen receptor T cells against the epidermal growth factor receptor variant III by enhancing immune synapses. Cancer Gene Ther (2015) 22:487–95. doi: 10.1038/cgt.2015.47
53. Thieblemont C, Chevret S, Allain V, Di Blasi R, Morin F, Vercellino L, et al. Lenalidomide enhance CAR T-cells response in patients with Refractory/Relapsed Large b cell lymphoma experiencing progression after infusion. Blood (2020) 136:16–7. doi: 10.1182/blood-2020-136279
54. Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discovery (2016) 6:1090–105. doi: 10.1158/2159-8290.CD-16-0716
55. Okkenhaug K, Burger JA. PI3K signaling in normal b cells and chronic lymphocytic leukemia (CLL). Curr Top Microbiol Immunol (2016) 393:123–42. doi: 10.1007/82_2015_484
56. Handi J, Patterson S, Levings M. The role of the PI3K signaling pathway in CD4+ T cell differentiation and function. Front Immunol (2012) 3. doi: 10.3389/fimmu.2012.00245
57. Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol (2013) 4. doi: 10.3389/fimmu.2013.00020
58. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature (2014) 510:407–11. doi: 10.1038/nature13444
59. Chellappa S, Kushekhar K, Munthe LA, Tjønnfjord GE, Aandahl EM, Okkenhaug K, et al. The PI3K p110δ isoform inhibitor idelalisib preferentially inhibits human regulatory T cell function. J Immunol (2019) 202:1397–405. doi: 10.4049/jimmunol.1701703
60. Worel N, Grabmeier-Pfistershammer K, Kratzer B, Schlager M, Tanzmann A, Rottal A, et al. The frequency of differentiated CD3(+)CD27(-)CD28(-) T cells predicts response to CART cell therapy in diffuse large b-cell lymphoma. Front Immunol (2022) 13:1004703. doi: 10.3389/fimmu.2022.1004703
61. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
62. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature (2016) 539:443–7. doi: 10.1038/nature20554
63. Chandrasekaran S, Funk CR, Kleber T, Paulos CM, Shanmugam M, Waller EK. Strategies to overcome failures in T-cell immunotherapies by targeting PI3K-δ and -γ. Front Immunol (2021) 12:718621. doi: 10.3389/fimmu.2021.718621
64. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. doi: 10.1038/nature08155
65. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo C-F, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res (2019) 7:759–72. doi: 10.1158/2326-6066.CIR-18-0466
66. Rogel A, Willoughby JE, Buchan SL, Leonard HJ, Thirdborough SM, Al-Shamkhani A. Akt signaling is critical for memory CD8(+) T-cell development and tumor immune surveillance. Proc Natl Acad Sci U.S.A. (2017) 114:E1178–e1187. doi: 10.1073/pnas.1611299114
67. Pal Singh S, Dammeijer F, Hendriks RW. Role of bruton’s tyrosine kinase in b cells and malignancies. Mol Cancer (2018) 17:57. doi: 10.1186/s12943-018-0779-z
68. Hendriks RW, Yuvaraj S, Kil LP. Targeting bruton's tyrosine kinase in b cell malignancies. Nat Rev Cancer (2014) 14:219–32. doi: 10.1038/nrc3702
69. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discovery (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
70. Xia S, Liu X, Cao X, Xu S. T-Cell expression of bruton’s tyrosine kinase promotes autoreactive T-cell activation and exacerbates aplastic anemia. Cell Mol Immunol (2020) 17:1042–52. doi: 10.1038/s41423-019-0270-9
71. Parry HM, Mirajkar N, Cutmore N, Zuo J, Long H, Kwok M, et al. Long-term ibrutinib therapy reverses CD8+ T cell exhaustion in b cell chronic lymphocytic leukaemia. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02832
72. Davis JE, Sharpe C, Mason K, Tam CS, Koldej RM, Ritchie DS, et al. Ibrutinib protects T cells in patients with CLL from proliferation-induced senescence. J Trans Med (2021) 19:473. doi: 10.1186/s12967-021-03136-2
73. Baptista MJ, Baskar S, Gaglione EM, Keyvanfar K, Ahn IE, Wiestner A, et al. Select antitumor cytotoxic CD8(+) T clonotypes expand in patients with chronic lymphocytic leukemia treated with ibrutinib. Clin Cancer Res (2021) 27:4624–33. doi: 10.1158/1078-0432.CCR-20-4894
74. Wen T, Wang J, Shi Y, Qian H, Liu P. Inhibitors targeting bruton’s tyrosine kinase in cancers: drug development advances. Leukemia (2021) 35:312–32. doi: 10.1038/s41375-020-01072-6
75. Vener C, Banzi R, Ambrogi F, Ferrero A, Saglio G, Pravettoni G, et al. First-line imatinib vs second- and third-generation TKIs for chronic-phase CML: a systematic review and meta-analysis. Blood Adv (2020) 4:2723–35. doi: 10.1182/bloodadvances.2019001329
76. Schade AE, Schieven GL, Townsend R, Jankowska AM, Susulic V, Zhang R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood (2008) 111:1366–77. doi: 10.1182/blood-2007-04-084814
77. Dittmann A, Werner T, Chung CW, Savitski MM, Fälth Savitski M, Grandi P, et al. The commonly used PI3-kinase probe LY294002 is an inhibitor of BET bromodomains. ACS Chem Biol (2014) 9:495–502. doi: 10.1021/cb400789e
78. Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia (2010) 24:22–32. doi: 10.1038/leu.2009.236
79. Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai Y-T, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood (2001) 98:210–6. doi: 10.1182/blood.V98.1.210
80. Busch A, Zeh D, Janzen V, Mügge LO, Wolf D, Fingerhut L, et al. Treatment with lenalidomide induces immunoactivating and counter-regulatory immunosuppressive changes in myeloma patients. Clin Exp Immunol (2014) 177:439–53. doi: 10.1111/cei.12343
81. D'Souza C, Prince HM, Neeson PJ. Understanding the role of T-cells in the antimyeloma effect of immunomodulatory drugs. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.632399
82. Lemoine J, Morin F, Di Blasi R, Vercellino L, Cuffel A, Benlachgar N, et al. Lenalidomide exposure at time of CAR T-cells expansion enhances response of Refractory/Relapsed aggressive Large b-cell lymphomas. Blood (2021) 138:1433–3. doi: 10.1182/blood-2021-151109
83. Said SI, Mutt V. Potent peripheral and splanchnic vasodilator peptide from normal gut. Nature (1970) 225:863–4. doi: 10.1038/225863a0
84. Said SI, Mutt V. Polypeptide with broad biological activity: isolation from small intestine. Science (1970) 169:1217–8. doi: 10.1126/science.169.3951.1217
85. Delgado M. VIP: A very important peptide in T helper differentiation. Trends Immunol (2003) 24:221–4. doi: 10.1016/S1471-4906(03)00069-3
86. Delgado M, Ganea D. Vasoactive intestinal peptide: a neuropeptide with pleiotropic immune functions. Amino Acids (2013) 45:25–39. doi: 10.1007/s00726-011-1184-8
87. Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev (2004) 56:249–90. doi: 10.1124/pr.56.2.7
88. Carrión M, Pérez-García S, Martínez C, Juarranz Y, Estrada-Capetillo L, Puig-Kröger A, et al. VIP Impairs acquisition of the macrophage proinflammatory polarization profile. J Leukoc Biol (2016) 100:1385–93. doi: 10.1189/jlb.3A0116-032RR
89. Kittikulsuth W, Nakano D, Kitada K, Uyama T, Ueda N, Asano E, et al. Vasoactive intestinal peptide blockade suppresses tumor growth by regulating macrophage polarization and function in CT26 tumor-bearing mice. Sci Rep (2023) 13:927. doi: 10.1038/s41598-023-28073-6
90. Azevedo MCS, Fonseca AC, Colavite PM, Melchiades JL, Tabanez AP, Codo AC, et al. Macrophage polarization and alveolar bone healing outcome: despite a significant M2 polarizing effect, VIP and PACAP treatments present a minor impact in alveolar bone healing in homeostatic conditions. Front Immunol (2021) 12:782566. doi: 10.3389/fimmu.2021.782566
91. Li JM, Petersen CT, Li JX, Panjwani R, Chandra DJ, Giver CR, et al. Modulation of immune checkpoints and graft-versus-Leukemia in allogeneic transplants by antagonizing vasoactive intestinal peptide signaling. Cancer Res (2016) 76:6802–15. doi: 10.1158/0008-5472.CAN-16-0427
92. Zhu J, Wang Y, Li J, Das PK, Zhang H, Passang T, et al. Donor plasmacytoid dendritic cells limit graft-versus-host disease through vasoactive intestinal polypeptide expression. Blood (2022) 140:1431–47. doi: 10.1182/blood.2021012561
93. Gozes I, McCune SK, Jacobson L, Warren D, Moody TW, Fridkin M, et al. An antagonist to vasoactive intestinal peptide affects cellular functions in the central nervous system. J Pharmacol Exp Ther (1991) 257:959–66.
94. Zia H, Hida T, Jakowlew S, Birrer M, Gozes Y, Reubi JC, et al. Breast cancer growth is inhibited by vasoactive intestinal peptide (VIP) hybrid, a synthetic VIP receptor antagonist. Cancer Res (1996) 56:3486–9.
95. Li JM, Southerland L, Hossain MS, Giver CR, Wang Y, Darlak K, et al. Absence of vasoactive intestinal peptide expression in hematopoietic cells enhances Th1 polarization and antiviral immunity in mice. J Immunol (2011) 187:1057–65. doi: 10.4049/jimmunol.1100686
96. Li JM, Hossain MS, Southerland L, Waller EK. Pharmacological inhibition of VIP signaling enhances antiviral immunity and improves survival in murine cytomegalovirus-infected allogeneic bone marrow transplant recipients. Blood (2013) 121:2347–51. doi: 10.1182/blood-2012-06-437640
97. Li JM, Darlak KA, Southerland L, Hossain MS, Jaye DL, Josephson CD, et al. VIPhyb, an antagonist of vasoactive intestinal peptide receptor, enhances cellular antiviral immunity in murine cytomegalovirus infected mice. PloS One (2013) 8:e63381. doi: 10.1371/journal.pone.0063381
98. Petersen CT, Li JM, Waller EK. Administration of a vasoactive intestinal peptide antagonist enhances the autologous anti-leukemia T cell response in murine models of acute leukemia. Oncoimmunology (2017) 6:e1304336. doi: 10.1080/2162402X.2017.1304336
99. Ravindranathan S, Passang T, Li J-M, Wang S, Dhamsania R, Ware MB, et al. Targeting vasoactive intestinal peptide-mediated signaling enhances response to immune checkpoint therapy in pancreatic ductal adenocarcinoma. Nat Commun (2022) 13:6418. doi: 10.1038/s41467-022-34242-4
100. Bommireddy R, Munoz LE, Kumari A, Huang L, Fan Y, Monterroza L, et al. Tumor membrane vesicle vaccine augments the efficacy of anti-PD1 antibody in immune checkpoint inhibitor-resistant squamous cell carcinoma models of head and neck cancer. Vaccines (Basel) (2020) 8(2):182. doi: 10.3390/vaccines8020182
101. Pack CD, Bommireddy R, Munoz LE, Patel JM, Bozeman EN, Dey P, et al. Tumor membrane-based vaccine immunotherapy in combination with anti-CTLA-4 antibody confers protection against immune checkpoint resistant murine triple-negative breast cancer. Hum Vaccin Immunother (2020) 16:3184–93. doi: 10.1080/21645515.2020.1754691
Keywords: adoptive cell therapy (ACT), chimeric antigen receptors (CAR), ex vivo manufacturing, protein kinase inhibitor, PI3K, vasoactive intestinal peptide (VIP), small-molecule drugs, peptide-based drugs
Citation: Zhang H, Passang T, Ravindranathan S, Bommireddy R, Jajja MR, Yang L, Selvaraj P, Paulos CM and Waller EK (2023) The magic of small-molecule drugs during ex vivo expansion in adoptive cell therapy. Front. Immunol. 14:1154566. doi: 10.3389/fimmu.2023.1154566
Received: 30 January 2023; Accepted: 10 April 2023;
Published: 21 April 2023.
Edited by:
Beatriz Martín-Antonio, University Hospital Fundación Jiménez Díaz, SpainReviewed by:
Eduardo Anguita, San Carlos University Clinical Hospital, SpainCopyright © 2023 Zhang, Passang, Ravindranathan, Bommireddy, Jajja, Yang, Selvaraj, Paulos and Waller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanwen Zhang, aGFud2VuLnpoYW5nMkBlbW9yeS5lZHU=; Edmund K. Waller, ZXdhbGxlckBlbW9yeS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.