 Filipa D. dos Reis
Filipa D. dos Reis Carmen Jerónimo
Carmen Jerónimo Margareta P. Correia
Margareta P. Correia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 April 2023
Sec. NK and Innate Lymphoid Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1152572
Immunoepigenetics is a growing field, as there is mounting evidence on the key role played by epigenetic mechanisms in the regulation of tumor immune cell recognition and control of immune cell anti-tumor responses. Moreover, it is increasingly acknowledgeable a tie between epigenetic regulation and prostate cancer (PCa) development and progression. PCa is intrinsically a cold tumor, with scarce immune cell infiltration and low inflammatory tumor microenvironment. However, Natural Killer (NK) cells, main anti-tumor effector immune cells, have been frequently linked to improved PCa prognosis. The role that epigenetic-related mechanisms might have in regulating both NK cell recognition of PCa tumor cells and NK cell functions in PCa is still mainly unknown. Epigenetic modulating drugs have been showing boundless therapeutic potential as anti-tumor agents, however their role in immune cell regulation and recognition is scarce. In this review, we focused on studies addressing modulation of epigenetic mechanisms involved in NK cell-mediated responses, including both the epigenetic modulation of tumor cell NK ligand expression and NK cell receptor expression and function in different tumor models, highlighting studies in PCa. The integrated knowledge from diverse epigenetic modulation mechanisms promoting NK cell-mediated immunity in various tumor models might open doors for the development of novel epigenetic-based therapeutic options for PCa management.
Prostate cancer (PCa) is the second most commonly diagnosed cancer in men and the fifth most deadly worldwide (1). There are several well-established risk factors, such as advanced age, African origin, and family history of PCa (2). When diagnosed at early stages, localized PCa is potentially curable by local therapy such as radical prostatectomy (RP) or radiotherapy (RT). However, between 27% and 53% of patients develop prostate-specific antigen (PSA) recurrence, defined by a rising PSA level (2, 3). Regarding advanced hormone-sensitive prostate cancer, the standard treatment is androgen deprivation therapy (ADT), which consists of agents targeting the androgen pathway, such as luteinizing hormone-releasing hormone (LHRH) agonists and antagonists (3). Despite an initial response by most patients, resistance to ADT can occur, leading to castration-resistant PCa (CRPC) and metastatic castration-resistant PCa (mCRPC). CRPC can sustain androgen signaling regardless of the low levels of circulating androgens, through different mechanisms such as increased intra-tumoral hormone synthesis, androgen receptor (AR) amplification, mutations, as well as dysregulated expression of AR coactivators and corepressors. In this setting, treatment options include chemotherapy and second-generation antiandrogens, which are not curative but can increase patient survival, although all patients who are treated for mCRPC will eventually progress (3, 4). ARlow or AR- PCa with neuroendocrine differentiation constitutes a form of aggressive AR-independent CRPC that occurs either de novo or through transdifferentiation, for which therapeutic approaches are limited to chemotherapy (5). This advanced form of disease poses several challenges besides the emerging mechanisms of androgen independence, such as high genomic heterogeneity, epigenetic deregulation, and a pro-immunosuppressive environment. Moreover, PCa therapies targeting angiogenesis are currently being explored. The expression of vascular endothelial growth factor A (VEGF-A), a marker of angiogenic activity, is known to be increased in PCa and associated with poorer prognosis, distant metastasis and advanced tumor grading (6–8). Clinical trials have shown some beneficial effects of targeting VEGF-A in patients with hormone-sensitive recurrent PCa, however, this was not observed in patients with castration-resistant PCa, and these therapies were also associated with adverse effects and high toxicity (8). Ongoing clinical trials intend to meet the need for new treatment strategies concerning these patients, which include immunotherapy in combination with other therapies (4).
The tumor microenvironment (TME) encompasses a network of stromal fibroblasts, myofibroblasts, mesenchymal stem cells (MSCs), endothelial cells, as well as immune cells, and their secreted factors such as chemokines, cytokines, and extracellular matrices (ECMs). TME has an impact on tumor cell survival, proliferation, resistance to conventional therapies, metastatic dissemination, and evasion of immune surveillance (4, 9). Several studies have been conducted to exploit the landscape of immune cell populations of the PCa TME, their possible influence on tumor progression and prognosis, and to understand the changes that occur in prostate cancer cells that could lead to evasion from immune responses (Table 1). PCa is considered a “cold tumor”, with low immune cell infiltration, immunosuppressive microenvironment and infiltrating effector lymphocytes with dysfunctional phenotype (73). Recently, Wu et al. compared in silico RNA sequencing data from normal prostate tissue and PCa tissue, observing increased infiltration of neutrophils, activated mast cells, M1 and M2 macrophages, resting natural killer cells, resting dendritic cells, and naïve B cells in PCa tissues compared to normal tissues. However, monocytes, CD8+ T cells and activated mast cells showed higher infiltration in normal tissues. Notably, T cells accounted for the highest infiltrating cells in both normal and PCa tissues (40).
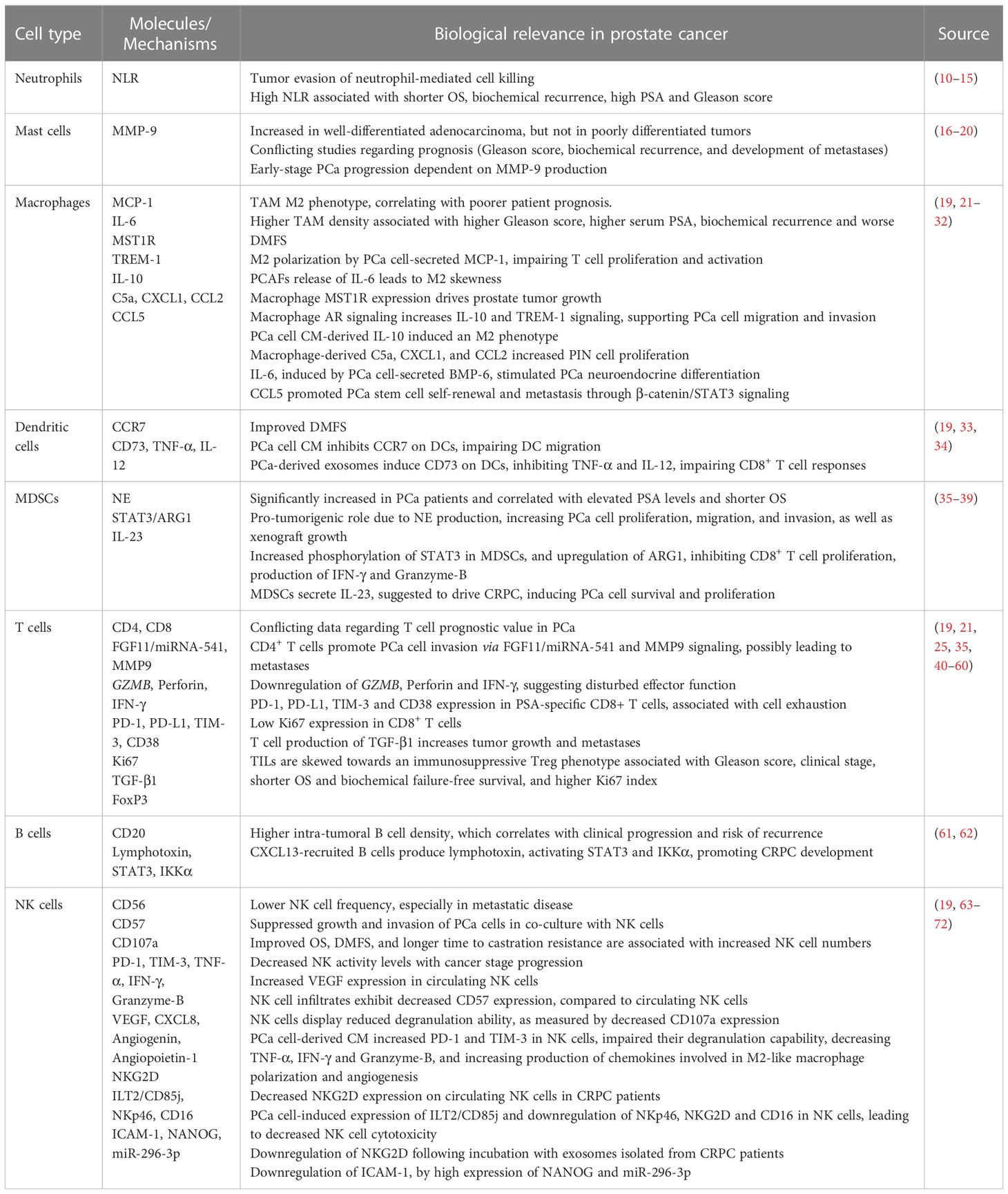
Table 1 Summary of the immune cell landscape in PCa.
Neutrophils are key mediators of innate immunity and have been shown to play a role in inhibiting PCa growth in vitro and in vivo. However, as bone metastatic PCa progressed, tumors were able to evade neutrophil-mediated cell killing (10). In PCa patients, a high neutrophil-to-lymphocyte ratio (NLR) is associated with shorter OS (11), biochemical recurrence after RP, high PSA and Gleason score (12–14), compared to patients with low NLR, being considered as a valid prognostic biomarker for CRPC patients treated with second-line chemotherapy (15).
Mast cells are reportedly increased in well-differentiated (WD) PCa adenocarcinoma, but not in poorly differentiated (PD) tumors (16). The role of mast cells in PCa is still controversial, with some studies indicating that high numbers of mast cells are associated with higher Gleason score, biochemical recurrence and development of metastases after RP (17, 18), while others show improved distant metastasis-free survival (DMFS) (19). These observations might be linked to different tumor subtypes and stages, since in an in vivo study, initial progression of WD PCa tumors was dependent on matrix metalloprotease 9 (MMP-9) production by mast cells, which was no longer essential for post epithelial-to-mesenchymal transition (EMT) stages, where PD tumors were MMP-9 self-competent (16). Besides, intratumoral mast cells appear to negatively regulate both PCa tumor growth and angiogenesis, while peritumoral mast cells seem to contribute to PCa expansion due to production of the angiogenic factor FGF-2 (20). Additional research is still required to further clarify the role of mast cells in PCa.
Macrophages have been identified as one of the most common immune cell populations in PCa (74, 75). These cells can be skewed towards a tumor-suppressive M1 phenotype or to a tumor-promoting M2 phenotype. In PCa, tumor associated macrophages (TAMs) mainly display an M2 phenotype, which correlates with more aggressive disease and poorer patient prognosis (21–23). Moreover, higher TAM density has been associated with higher Gleason score, higher serum PSA, biochemical recurrence, and worse DMFS (19, 24, 25). M2-induced polarization in PCa has been associated with production of monocyte chemotactic protein-1 (MCP-1), leading to a consequent impairment of T lymphocyte proliferation and activation (21), and also with the production of IL-6 by prostate carcinoma-associated fibroblasts (PCAFs) and PCa cells (26). In a recent study, loss of prostate epithelial macrophage stimulating 1 receptor (MST1R) was shown to suppress M2 marker expression, while epithelial MST1R activation upregulated its own expression in macrophages, in a positive feedback mechanism, driving prostate tumor growth (27). Also, AR signaling was demonstrated to upregulate Interleukin-10 (IL-10) and Triggering Receptor Expressed on Myeloid cells-1 (TREM-1) signaling on macrophages, supporting PCa cell migration and invasion (28). In addition, IL-10 induced macrophages toward an M2 phenotype, after incubation with PCa cell conditioned media (CM) (29). Macrophage-secreted molecules have also been associated with PCa development, such as C5a, CXCL1, and CCL2, by increasing prostatic intraepithelial neoplasia (PIN) cell proliferation (30). IL-6, induced by PCa cell-secreted bone morphogenetic protein-6 (BMP-6), stimulated PCa neuroendocrine differentiation (31), and CCL5 promoted PCa stem cell self-renewal and PCa metastasis through activation of β-catenin/STAT3 signaling (32). Thus, further characterization of the TAM population, and development of new treatment strategies targeting its phenotype and function, are key for PCa patient treatment.
Dendritic cells (DCs), as professional antigen presenting cells (APCs), are crucial to activate T cell responses through antigen presentation via major histocompatibility complex (MHC) (33). DCs have an important role in PCa immunity, conferring improved DMFS (19). However, PCa cell-derived CM has been shown to inhibit CC chemokine receptor-7 (CCR7) expression on maturing DCs, impairing chemotactic movement of DCs (33). PCa-derived exosomes are also capable of inducing CD73 expression on DCs, inhibiting Tumor Necrosis Factor (TNF)-α and IL-12 production, ultimately impairing CD8+ T cell responses (34).
Circulating and intra-tumoral myeloid-derived suppressor cells (MDSCs) are significantly increased in PCa patients compared to healthy controls (35, 36). In PCa, MDSCs have been suggested to have a role in establishing an immunosuppressive microenvironment and promoting cancer progression, as increased frequencies of MDSCs correlated with elevated PSA levels and shorter median OS (35), and MDSC depletion significantly decreased tumor xenograft growth/progression (37, 38). Neutrophil elastase (NE), a serine protease, might be involved in the pro-tumorigenic role of MDSCs in PCa since a small-molecule inhibitor of NE was able to significantly decrease PCa cell proliferation, migration, and invasion, as well as tumor xenograft growth (37). Circulating MDSCs from PCa patients showed increasingly activated signal transducer and activator of transcription 3 (STAT3), an immune checkpoint regulator, contributing to arginase-1 (ARG1) upregulation and consequent decrease of CD8+ T cell proliferation, production of Interferon-γ (IFN-γ) and Granzyme-B. Targeting STAT3/ARG1 signaling of PCa patient-derived MDSCs abrogated their immunosuppressive effects (39). Moreover, MDSCs secrete IL-23, shown to be increased in blood and tumor samples of CRPC patients, and suggested to contribute to CRPC development due to AR pathway activation, leading to PCa cell survival and proliferation in androgen deprived conditions. Inhibition of IL-23 was able to restore ADT sensitivity in mice (36).
The role played by tumor infiltrating lymphocytes (TILs) in PCa is still ambiguous and further research is necessary to clarify their prognostic value in PCa patients. While a high CD8+ TIL density in PCa has been independently associated with improved OS, PCa‐specific survival (41, 42) and longer time to biochemical recurrence in patients with seminal vesicle invasion (43), other studies have found an association between high CD8+ T cell density, worse clinical progression and PCa malignant degree (40, 44, 45). A recent study of 1567 retrospective radical prostatectomy samples with long-term metastasis and survival outcomes showed that both CD8+ and CD4+ T cells appear to be associated with worse DMFS (19). In addition, infiltrating CD4+ T cells were suggested to promote PCa metastasis via modulation of fibroblast growth factor 11 (FGF11)/miRNA-541 signaling, suppressing androgen receptor signals, which could then increase matrix metallopeptidase 9 (MMP9) expression and promote PCa cell invasion (46). The functional status of TILs is key for their cytotoxic and anti-tumor immune responses. PCa tissues obtained from RP displayed almost no expression of Granzyme-B (GZMB) and Perforin 1 (PRF1), markers of cytolytic activity, indicating that T cells infiltrating prostate tumors are functionally impaired (21). This is in agreement with another study reporting downregulation of Perforin and IFN-γ on tumor infiltrating lymphocytes, showing signs of disturbed effector function, and expression of molecules associated with lymphocyte exhaustion, such as the programmed cell-death-1 (PD-1) receptor and its ligand, PD-L1 (47–49). Moreover, PSA-specific CD8+ T cells of PCa patients showed higher expression of exhaustion and activation markers, T cell immunoglobulin and mucin domain 3 (TIM-3) and CD38, respectively, indicating that PSA-specific T cells are exhausted (50). Additionally, CD8+ T cells accumulated in PCa pelvic node metastases exhibited decreased proliferation, with low Ki67 expression (51). Cytokine production by T cells can also contribute to immunosuppression, as ablation of tumor growth factor β1 (TGF-β1) in T cells (not Treg) enhanced tumor T cell cytotoxic activity in mice, inhibiting tumor growth and metastases, and suggesting that tumor immunosurveillance escape is dependent on TGF-β1 production by conventional T cells (52). Furthermore, PCa tumors have been described to be surrounded by clusters of CD25+ and FoxP3+ regulatory T cells (Tregs) (48). These cells have a suppressive role on innate and adaptive immunity and are significantly increased in the peripheral blood and tumor tissues of PCa patients, especially in metastases, which present a higher abundance of FoxP3+ Tregs when compared to primary lesions (35, 53–55). In PCa, a high proportion of TILs appear to be skewed towards a Treg phenotype (FoxP3+) (56). Treg tumor infiltration has prognostic value in PCa patients, since higher numbers of Tregs are positively correlated with Gleason score and clinical stage (57, 58), shorter OS and biochemical failure-free survival (25, 59, 60), and higher tumor cell Ki67 index (59).
Regarding B cells, a cohort of PCa paraffin-embedded radical prostatectomy specimens displayed higher B cell density in tumor areas compared to extra-tumoral ones, which correlated with clinical progression and higher risk of recurrence (61). A study with PCa murine models reported that B cells recruited by the chemokine CXCL13 produce lymphotoxin, which then activates STAT3 and IκB kinase α (IKKα), promoting CRPC development (62).
Innate Lymphoid Cells (ILCs) are characterized by the lack of rearranged antigen receptors, recognizing tumor cells in an antigen-independent manner. They are composed by the ILC1, ILC2 and ILC3 subgroups (76, 77), and are generally known to be specialized cytokine producers, mirroring the T helper subtypes. Interestingly, ILC2 cells were found to be enriched in the peripheral blood of PCa patients, compared to healthy donors, and associated with advanced tumor stage (77).
Natural Killer (NK) cells are innate lymphocytes, belonging to the ILC1 subgroup. Contrarily to the other ILCs, they rather resemble CD8+ T cells, being cytotoxic and specialized in targeting infected or transformed cells, such as tumor cells. In the next chapter, we will focus on the role of NK cells in prostate cancer (78).
NK cell recognition of target cells relies on the expression of an array of activating and inhibitory receptors that interact with specific ligands on the surface of the target cells, resulting in killing if the stimulatory signals outweigh suppressive signals (78). Kärre and Ljunggren uncovered the missing-self mechanism, by which NK cells can distinguish healthy cells from target cells lacking MHC-I molecules, also known as Human Leukocyte Antigen Class-I (HLA-I) in humans (79). HLA-A, B and C molecules act as inhibitory ligands that bind to Killer-Cell Immunoglobulin-Like Receptors (KIRs), and HLA-E is recognized by the NKG2A transmembrane receptor expressed on the surface of NK cells. Besides the lack of MHC class I, infected or transformed cells (over)express specific ligands that are recognized by specific NK cell activating receptors. NKG2D, DNAM-1 and the natural cytotoxicity receptors (NCRs) NKp46 (NCR1), NKp44 (NCR2) and NKp30 (NCR3), are recognized as major activating NK cell receptors (80). UL16-binding protein (ULBP), MHC class I-related chain A (MICA) and B (MICB) are NKG2D ligands, commonly overexpressed on tumor cells. DNAM-1 recognizes the activating ligands Nectin-2 (CD112) and Polio Virus Receptor (PVR, CD155), capable of increasing NK cell triggering (80). While NCR ligands have not been completely identified in tumor cells, B7-H6 has been described as an activating tumor cell ligand recognized by NKp30 (81). Another key activating receptor is the Fcγ receptor CD16, able to bind to the Fc portion of Immunoglobulin G (IgG) antibodies in target cells, leading to antibody-dependent cell-mediated cytotoxicity (ADCC). When activated, NK cells induce target cell apoptosis via exocytosis of cytolytic granules containing Granzyme-B and Perforin, also releasing effector cytokines such as IFN-γ and TNF-a, and various chemokines that allow for recruitment and communication with other immune cells (78, 82). Importantly, the frequency of circulating NK cells has been found to be significantly lower in PCa patients than in healthy controls, and further reduced in metastatic disease than in localized PCa (63). Moreover, in metastatic PCa patients, increased circulating NK cell numbers confer improved overall survival and longer time to castration resistance (64), and higher intratumoral NK cell numbers have been shown to confer longer distant metastasis-free survival (19). Evidence shows that NK cell function is inhibited at multiple levels in the PCa TME (79). Different studies reported suppressed growth and invasion of PCa cells when co-cultured with NK cells, highlighting the crucial role of NK cells in prostate cancer immunosurveillance (65, 66). Moreover, NK cell activity levels in tumors gradually decrease according to stage progression (67). NK cell infiltrates from PCa tumors display decreased CD57 expression compared to circulating NK cells, associated with an immature phenotype (68). Moreover, reduced degranulation capacity against K562 cells was observed in circulating NK cells from PCa patients, compared to healthy donors (69). A recent study reported that NK cells exposed to CM of different PCa cell lines presented increased expression of the exhaustion markers PD-1 and TIM-3, and displayed impaired degranulation capability, with reduced production of TNF-α, IFN-γ and Granzyme-B. These cells also displayed increased production of chemokines involved in monocyte recruitment and M2-like macrophage polarization (69). Moreover, circulating NK cells isolated from PCa patients have shown increased expression of VEGF, a marker of angiogenic activity, as well as upregulation of CXCL8, associated with inflammation and angiogenesis. Additionally, NK exposure to PCa cell line CM increased production of the pro-angiogenic factors Angiogenin and Angiopoietin-1 (69). Co-culture experiments led to decreased recognition of PCa cells and lower NK cell cytotoxicity due to increased expression of the NK cell inhibitory receptor ILT2/CD85j (68), which recognizes HLA-A, B, C and G molecules (80), and reduction of the activating receptors NKp46, NKG2D, and CD16, by PCa cells (68). Downregulation of NKG2D on NK cells was also found following incubation with exosomes isolated from serum or plasma of CRPC patients (70). Flow cytometry analysis revealed that surface NKG2D expression on circulating NK cells was decreased in CRPC patients, compared to healthy individuals (70). PCa cells can also evade NK cell-mediated death due to high NANOG expression (a pluripotent-related transcription factor), repressing ICAM-1, a cell adhesion molecule that is necessary for the β2 integrin lymphocyte function associated antigen 1 (LFA-1)-dependent early stimulatory signal for NK cell cytotoxicity, whose low expression is correlated with a high recurrence rate in PCa patients (71, 72). ICAM-1 was also shown to be targeted by the micro-RNA 296-3p (miR-296-3p), downregulating its expression. miR-296-3p is frequently upregulated in PCa, and its knockdown in mice decreased resistance to NK cell-mediated killing, as well as PCa-derived circulating tumor cell (CTC) extravasation into the lungs and the number of pulmonary metastases (72). Thus, given the diverse mechanisms that impair NK cell cytotoxicity in PCa and their impact on patient prognosis, there is a rationale for further research on targeting NK cell-mediated immunity to treat PCa patients.
Epigenetics comprises modifications, such as DNA methylation and chromatin remodeling, that do not alter the DNA sequence itself. There is growing evidence implicating epigenetic mechanisms in immune cell function and in the modulation of tumor cell recognition mechanisms (83).
DNA methyltransferases (DNMTs) catalyze the transfer of a methyl group from S-Adenosyl-L-methionine (SAM) to the fifth carbon of a cytosine residue, creating 5-methylcytosine (5mC) and leading to transcriptional repression (83). In addition, histone modifications play a significant role in chromatin remodeling, both in normal and neoplastic processes. Histones may undergo posttranslational modifications at the N-terminal tail, such as acetylation and methylation. Acetylation is catalyzed by histone acetyltransferases (HATs), which enables gene transcription through a decrease in DNA affinity for histones, creating an “open” chromatin conformation. Histone methylation, in which histone methyltransferases (HMTs) catalyze the transfer of the methyl group from SAM to specific lysine residues on histones, is associated with either transcriptional activation or repression, depending on the amino acid residue and the number of methyl groups added (83–85).
Epigenetic modulating agents are known to impact both cancer cells and immune cells, leading to changes in tumor cell recognition and targeting. The expression of genes part of the antigen presentation machinery, such as HLA, TAP1, TAP2, B2M, LMP2 and LMP7 has been upregulated upon treatment with DNMTi and HDACi (86–100). DNMT and HDAC inhibition also leads to increased expression of the co-stimulatory molecules CD40, CD80 and CD86, necessary for antigen presentation (89, 98), as well as cancer-specific antigens (101). Accordingly, dendritic cells treated with DNMTi showed increased CD40 and CD86 expression, while also secreting lower levels of IL-10 (102).
Regarding T cells, increased tumor CD8+ T cell infiltration has been shown in several cancer models, such as breast (96, 103), ovary (87, 104), pancreatic (105) and bladder (95) tumor-bearing mice treated with DNMTi, HDACi, EZH2i, G9a/DNMTi and LSD1i, enhancing anti-tumor immunity. Namely, epigenetic reprogramming of chemokine expression has been reported upon treatment with DNMTi, EZH2i and LSD1, increasing tumor production of CXCL9 and CXCL10 chemokines (103, 104), known to be involved in T cell recruitment to the tumor. Furthermore, increased CD8+ T cell Perforin expression, as well as IFNγ and TNFα production have been observed upon treatment with DNMTi (87) and HDACi (106). Moreover, PCa cell treatment with HDACi has led to increased antigen-specific CD8+ T cell targeting (107).
Additionally, inhibition of DNMTs and EZH2 in mice xenograft models increased CD8+ T to Treg ratio and decreased Treg function, leading to increased T cell activation (108, 109). Interestingly, class I/II HDAC inhibition increased Treg function and proportions in lymphoid tissues (110), while class I HDACi decreased Treg numbers and Foxp3 expression (101, 111). Additionally, EZH2 and BET inhibitors have also been described to decrease Foxp3 levels (109, 112).
Several pre-clinical studies have reported an inhibition of the immunosuppressive MDSC population upon treatment with HDACi, alone or in combination with DNMTi, indicating a reversal of the immunosuppressive TME and resulting in increased survival (100, 113–115). Another study reported that treatment with HDACi increased MDSCs within tumors, however, their immunosuppressive function was impaired, as their ability to inhibit CD8+ T proliferation was decreased (116). Regarding macrophages, DNMTi treatment with 5-aza in an ovarian cancer mouse model revealed an increase in the M1/M2 macrophage ratio (100).
Recently, epigenetic therapy using DNMT, HDAC and EZH2 inhibitors was shown to de-repress endogenous retroviruses (ERVs), inducing double-stranded RNA (dsRNA) that is then sensed by intracellular pattern recognition receptors (PRRs), causing a viral mimicry state that leads to increased IFN secretion and enhanced expression of IFN-stimulated genes, ultimately leading to an inflammatory environment able to recruit immune cells (117–120).
Although an improvement in treatment outcome has been observed in pre-clinical models and early clinical trial results, patients treated with different epigenetic modulating agents have shown increased expression of immune checkpoints such as PD-1/PD-L1 and CTLA-4, which could impair anti-tumor immune responses (101, 121). Thus, several clinical trials are currently focused on the potential of combining checkpoint inhibitor therapies with epigenetic drugs.
Overall, treatment with epigenetic drugs is suggested to turn “cold” tumors into inflamed “hot” tumors through several mechanisms, providing a possibility to modulate immune responses and promote anti-tumor immune targeting.
In the next chapter, we will focus on the epigenetic modulation involved on NK cell-mediated immunity, including regulation of NK cell ligands, NK cell receptors and NK cell function.
Treatment of PCa cells with the class I histone deacetylase inhibitor (HDACi) valproic acid (VPA), alone or in combination with the DNMT inhibitor hydralazine, was able to upregulate MICA/B and ULBPs expression at the transcriptional level (122, 123) due to dimethylation of histone 3 at lysine 4 (H3K4me2) activating mark increase, as confirmed by chromatin immunoprecipitation (ChIP). This led to enhanced NK cell cytotoxicity against PCa cells, minimizing PCa-induced suppression of anti-tumor NK cell responses (122). Cell treatment with the HDAC inhibitor suberoylanilide hydroxamic acid (vorinostat also known as SAHA, a class I and II HDAC inhibitor), trichostatin A (TSA, a class I and class II HDAC inhibitor), or VPA led to decreased B7-H6 transcription and cell surface expression. SAHA-treated target cells in co-culture with NK cells showed impaired NKp30-dependent degranulation. The authors found that HDAC3 and the HAT cyclic adenosine monophosphate response element-binding protein (CREBBP) were required for B7-H6 expression (124).
PVR and Nectin-2 (CD112), DNAM-1 activating ligands, were shown to be upregulated upon PCa cell treatment with both HDAC inhibitors sodium butyrate (NaB) or SAHA. Besides PVR and Nectin-2, SAHA and NaB also upregulated MICA/B and ULBP1-3. This effect was more pronounced for MICA and ULBP2, whose expression increased more than 4-fold, enhancing NK cell-mediated tumor cell killing (125). Similarly, exposure of the PCa cell lines PC-3 and DU-145 to the class I HDAC inhibitor entinostat was able to upregulate MICA, MICB, ULBP1/2/5/6 and PVR expression, as assessed by flow cytometry (126). This is agreement with another study, which revealed that treatment of Ewing sarcoma and rhabdomyosarcoma cell lines with entinostat increased the expression of these ligands, however HLA expression was also increased (127). Treatment of colon carcinoma cells with entinostat led to enhancement of acetylated histone 3 (AcH3) binding to MICA and MICB promoters, increasing their expression in a dose and time-dependent manner, resulting in higher NK cell-mediated tumor cell lysis, as compared to untreated cells. Of note, entinostat treatment did not induce higher NKG2D ligand expression in normal cells (128).
Panobinostat, a pan-HDACi, was able to increase the transcription and surface expression of MICA and ULBP2 in pancreatic cancer cell lines, resulting in a more potent NK-mediated killing of panobinostat treated cells than untreated cells. Xenograft mouse models further demonstrated that enhanced NK cell-mediated tumor surveillance was NKG2D ligand-dependent (129). Panobinostat also upregulated expression of the activating ligands CD80, CD86, and Nectin-2 in tumor cells, promoting NK cell cytolytic effect (130).
Treatment of lung cancer cells with different selective HDACi revealed that only FK228, an HDAC1/2 inhibitor, induced MICA, MICB and ULBP1/3 expression, which led to increased susceptibility to NK cell-mediated lysis. However, it is important to note that FK228 also increased MHC-I inhibitory ligand expression by 1.3-fold (86). Similarly, treatment with Nexturastat A (NextA), an HDAC6i, also increased MHC-I expression, which was further increased in combination with 5-azacytidine (5-aza), a DNMT inhibitor already approved for treatment of high-risk myelodysplastic syndrome (MDS) (100).
HeLa cells treated with different doses of TSA showed increased ULBP1 mRNA and protein levels, as confirmed through real-time PCR and flow cytometry, when compared to untreated cells, leading to enhanced NK cell cytotoxicity. ULBP1 was shown to be repressed by transcription factor Sp3-mediated HDAC3 binding to the ULBP1 promoter, while TSA treatment interfered with this association, decreasing HDAC3 recruitment and increasing ULBP1 expression (131). In agreement with these observations, Rossi et al. also showed TSA-induced upregulation of MICA, MICB and ULBP-1/2 expression, both at the protein and mRNA level (132) (Figure 1 and Table 2).
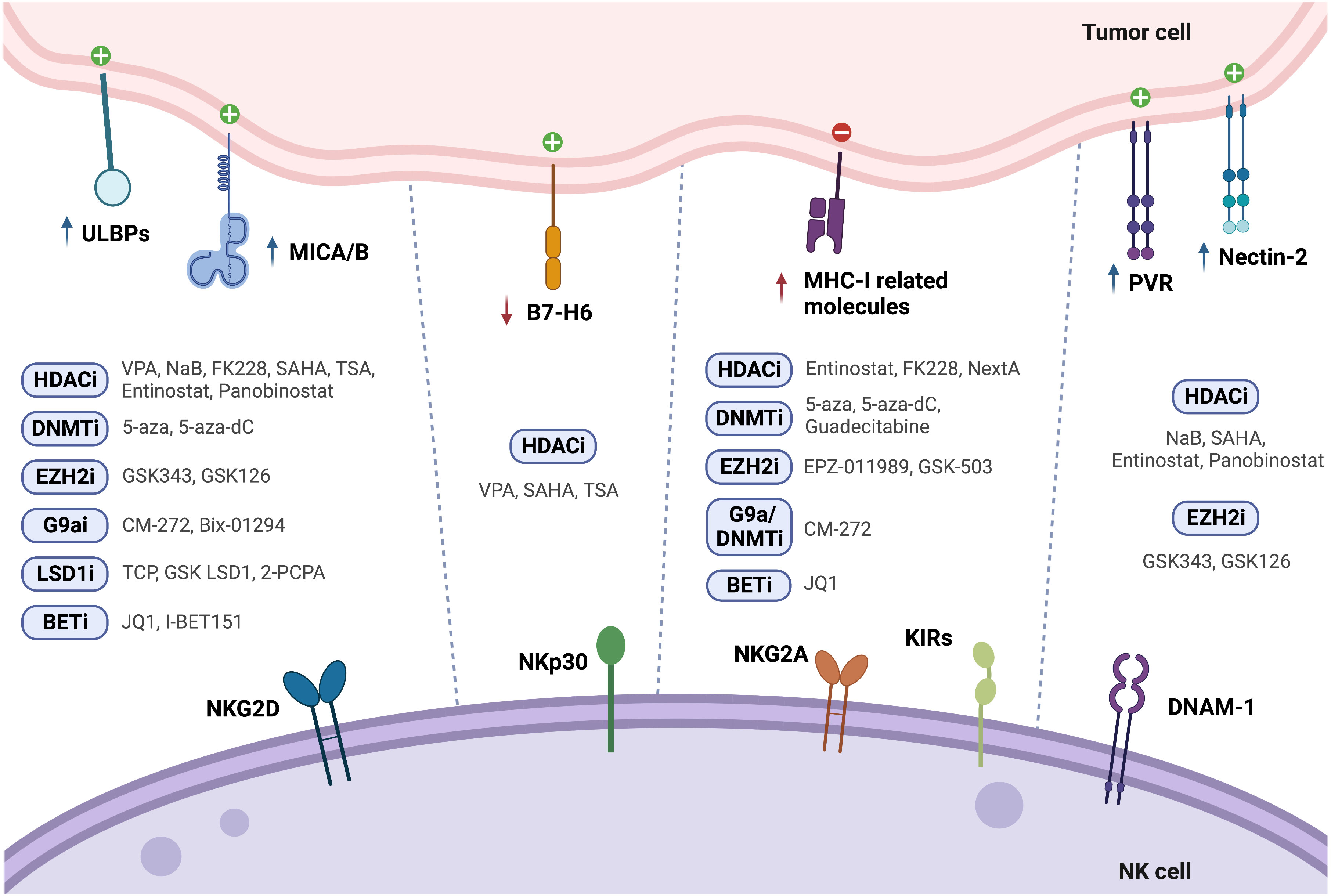
Figure 1 Modulation of NK cell ligands following tumor cell treatment with epigenetic drugs (↑ - upregulation; ↓ - downregulation).
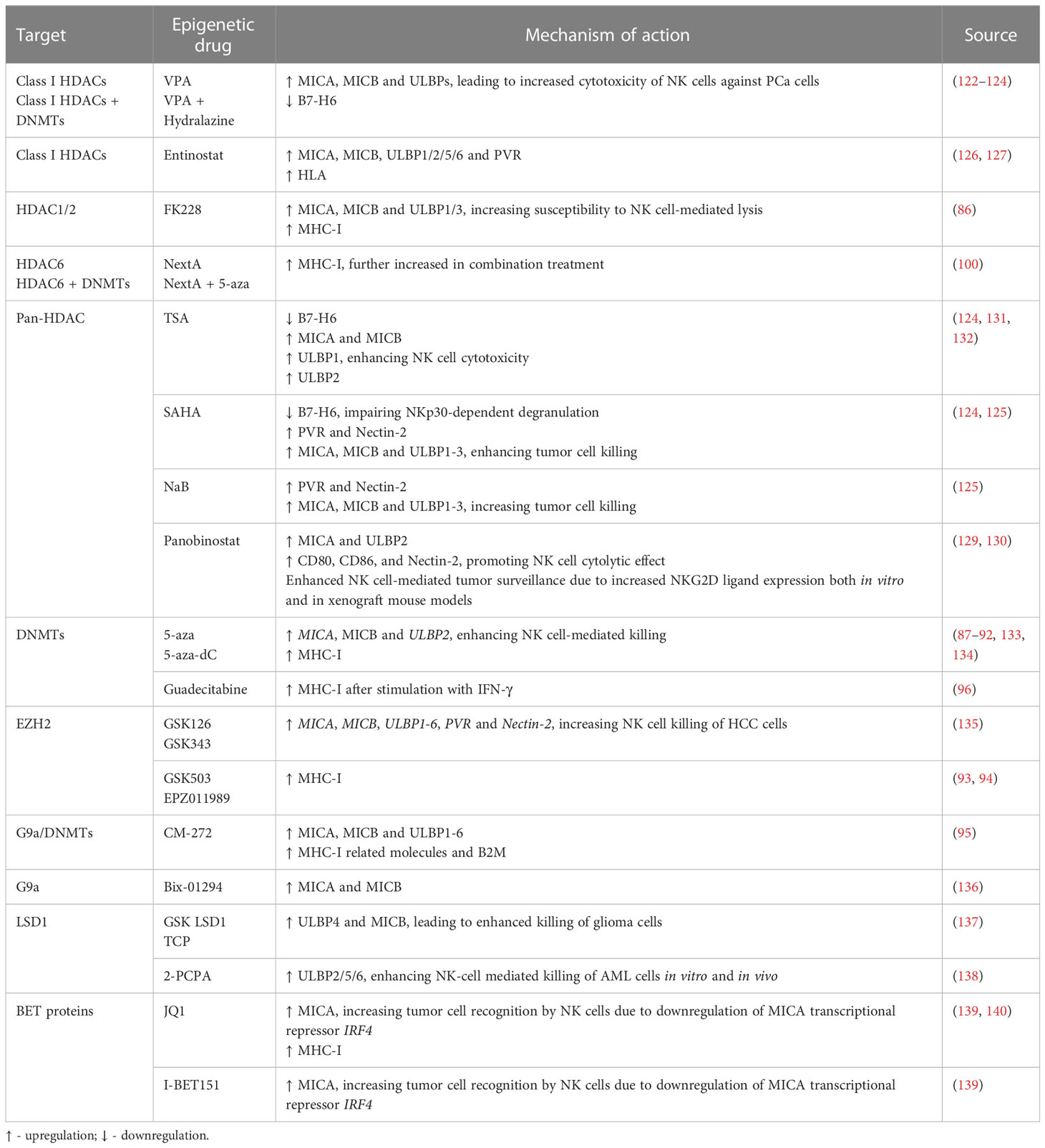
Table 2 Epigenetic modulating agents and their effect on NK cell ligands.
Treatment of HCC cells with 5-Aza-2’-deoxycytidine (5-aza-dC, also known as decitabine), a pyrimidine analogue that incorporates into DNA and inhibits DNMTs, led to increased MICB surface expression, which enhanced susceptibility to NK cell-mediated killing (133). Raneros et al. revealed hypermethylation of the NKG2D ligands MICA, MICB and ULBP2 in acute myeloid leukemia (AML) cell lines and patients, which correlated with absence of transcription. It was further demonstrated that treatment of AML cell lines with 5-aza or 5-aza-dC was able to restore transcription of these ligands, of which ULBP2 was the most susceptible to demethylation, resulting in increased NK cell-mediated lysis of tumor cells (134).
Treatment with 5-aza-dC has shown upregulation of MHC-I surface expression in several tumor models, such as ovarian, melanoma, lymphoma, leukemia, lung, colorectal, liver and breast cancer cells (87–91), which could possibly have a negative impact on NK cell anti-tumor immunity. Moreover, Paulson et al. showed that ex vivo culture of Merkel cell carcinoma (MCC) tumors treated with 5-aza resulted in upregulation of HLA-B, which was previously shown to be downregulated in these tumors (92). In another study, breast cancer cells treated with the next generation DNMT inhibitor guadecitabine also showed upregulation of MHC-I, but only after subsequent stimulation with IFN-γ (96) (Figure 1 and Table 2).
Enhancer of zeste homolog 2 (EZH2) is one of the most studied HMTs. EZH2 is the catalytic subunit of the polycomb repressive complex 2 (PRC2), responsible for the trimethylation of histone 3 at lysine 27 (H3K27me3), leading to heterochromatin formation, epigenetic gene silencing, and transcriptional repression. In this manner, EZH2 regulates cell cycle progression, proliferation and differentiation of both cancer cells and immune cells, affecting immune surveillance and tumor growth (141). EZH2 is overexpressed in PCa, particularly in mCRPC, and has been suggested to promote PCa progression (84, 142). Treatment of hepatocellular carcinoma (HCC) cells with the small-molecule EZH2 inhibitors GSK343 or GSK126 was able to upregulate several activating NK cell ligands, such as ULBP1-6, MICA, MICB, PVR and NECTIN2, which led to a significant increase in NK cell-induced cytotoxicity dependent especially on ULBP1 and MICA. Chromatin immunoprecipitation (ChIP) assays showed EZH2 recruitment to ULBP1 and MICA promoters, increasing H3K27me3 repression mark levels. Moreover, methylated-DNA immunoprecipitation (MeDIP) analysis revealed higher ULBP1 promoter DNA methylation due to EZH2-induced DNMT3A recruitment. GSK343 treatment decreased DNMT3A recruitment to the ULBP1 ligand promoter, reducing promoter DNA methylation and increasing ULBP1 expression, further elucidating the role of EZH2 as an inhibitor of anti-tumor immune responses (135). However, pharmacological EZH2 inhibition in tumor cell lines has also been shown to upregulate cell surface expression of the NK cell-inhibitory ligand MHC-I, following treatment with GSK503 or EPZ011989 (93, 94). This dual role highlights the need for further in vivo research on NK cell tumor cytotoxicity following EZH2 inhibition (Figure 1 and Table 2).
G9a, a histone methyltransferase that catalyzes H3K9me2, can be targeted with the newly developed dual G9a/DNMT inhibitor, CM-272, described to decrease H3K9me2 and m5C levels in bladder and prostate cancer cells (95, 143). Recently, we found G9a, H3K9me2 and DNMT1 protein expression to be upregulated in a cohort of CRPC patients. Herein, CM-272 was able to significantly impair proliferation and induce apoptosis of PCa cell lines, particularly of the AR-independent DU-145 cell line (143). In vitro assays showed increased expression of activating NK-cell ligands such as ULBP1-6, MICA and MICB after 48h of CM-272 treatment, however, concomitantly increasing inhibitory major histocompatibility genes, such as HLA-A, -B and -C, and beta-2-microglobulin (B2M). These changes in MHC-related genes were also verified in mice treated with CM-272 in combination with cisplatin (CDDP)-based chemotherapy in a bladder cancer model (95). Furthermore, tumor cell lines treated with Bix-01294, a G9a inhibitor, also showed upregulation of MICA and MICB activating ligands (136) (Figure 1 and Table 2).
Histone demethylase (KDM) LSD1 activity can be targeted with catalytic inhibitors which form covalent adducts in the LSD1 demethylation site. Treatment of glioma cells with the catalytic LSD1 inhibitors tranylcypromine (TCP) or GSK LSD1 was able to increase MICB and ULBP4 expression, enhancing human NK-cell mediated lysis of the target cells (137). Furthermore, treatment of AML cells with the LSD1 inhibitor tranylcypromine (2-PCPA) hydrochloride was able to upregulate expression of ULBP2/5/6, leading to increased susceptibility to NK cell-mediated killing, both in vitro and in vivo (138) (Figure 1 and Table 2).
Bromodomain and extra-terminal (BET) proteins recognize acetylated lysine residues in N-terminal tails of histones, modulating gene expression through transcription factor and chromatin-regulating enzymes recruitment. BET small molecule inhibitors, such as the triazolo-benzodiazepine-based JQ1 and the quinolone-based I-BET151, were able to upregulate MICA expression on multiple myeloma cells, due to downregulation of the target gene IRF4, a transcriptional repressor of MICA, increasing their recognition by NK cells (139). However, JQ1 has also been shown to upregulate MHC-I expression in colorectal cancer (CRC) cells (140) (Figure 1 and Table 2).
NK cell mediated immunity can also be modulated through epigenetic regulation of NK receptor expression. NK cell treatment with the HDAC inhibitor entinostat was shown to lead to a significant increase in activating receptors such as NKG2D (126, 128), NKp30 and DNAM-1, although resulting in a decreased expression of NKp46 (126). Furthermore, dual-treatment of NK cells and colon carcinoma cells resulted in higher NK cell mediated cytotoxicity, as compared to either entinostat-treated NK cells or entinostat-treated tumor cells alone (128). In another study, NK cells incubated with entinostat showed increased surface expression of NKG2D, although they also displayed an increase in inhibitory isoforms of KIR2DL1, KIR2DL2 and KIR2DS4, which indicates that entinostat increases not only the expression of activating receptors, but can also enhance surface expression of inhibitory receptors (127). Moreover, treatment with VPA inhibited NK cell cytotoxicity against leukemic cells due to downregulation of NKG2D, since it induced H3K9me2 and DNA methylation in the gene promoter (132, 144). NK cells stimulated with IL-12, IL-15 and IL-18, and treated with TSA or NaB also exhibited decreased NKG2D and NKp46 surface expression (132). In another study, primary NK cells showed decreased CD16, NKp46 and NKG2D expression following panobinostat treatment (145). However, a recent assessment of splenic NK cell populations from panobinostat-treated tumor-bearing mice showed increased surface expression of the activating receptors NKG2D and DNAM-1, while the expression of the inhibitory receptors NKG2A and Ly49I/C was decreased (130), albeit further mechanistic insights are still to be exploited. Panobinostat-treated NK cells showed enhanced expression of CD56, which is a marker of NK cell activation and has an important role in cell-cell contact, inducing activation signals via adhesion molecules (130). Also, NKG2D expression was found to be decreased upon VPA treatment through repression of STAT3 phosphorylation, following HDAC3 inhibition (146) (Figure 2 and Table 3).
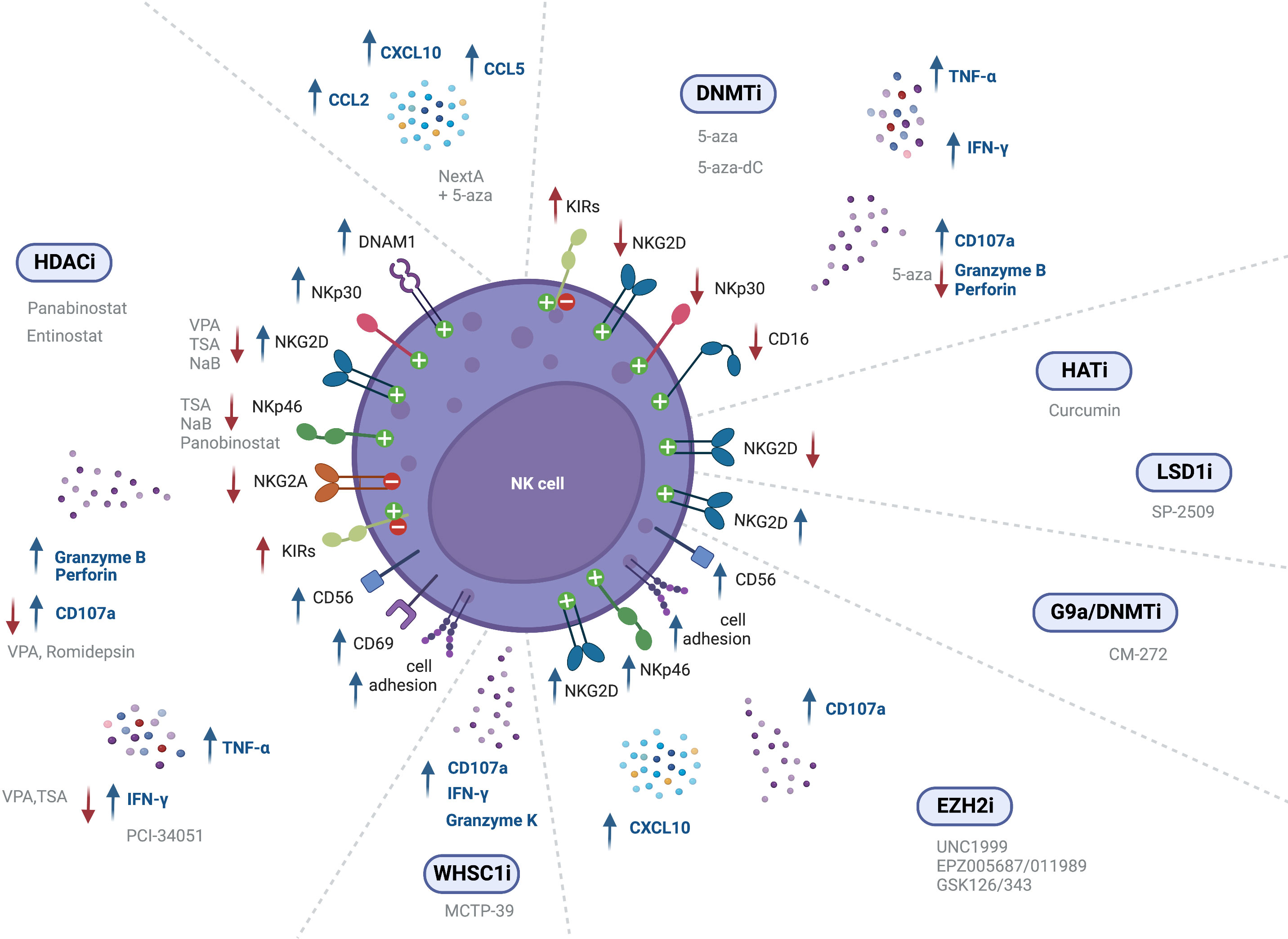
Figure 2 Epigenetic modulating agents and their effect on NK cell receptor expression and cell function (↑ - upregulation; ↓ - downregulation).
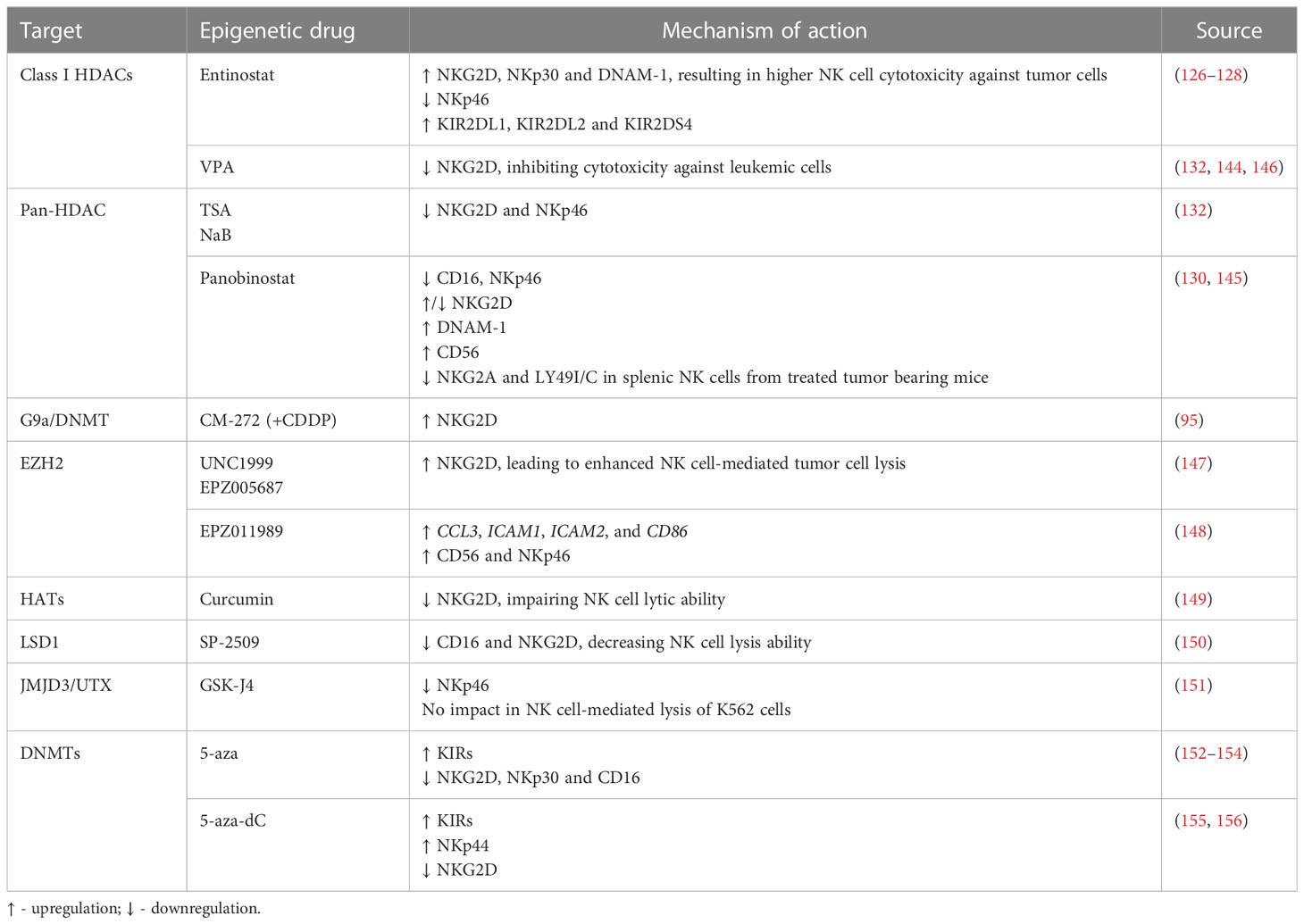
Table 3 Modulation of NK cell receptors through treatment with epigenetic modulating agents.
In vivo treatment with the G9a/DNMT inhibitor CM-272, in combination with cisplatin-based chemotherapy (CDDP), revealed induced expression of the activating cell receptor NKG2D (95). Treatment of NK cells with both EZH2i UNC1999 or EPZ005687 was also able to augment NKG2D expression, leading to enhanced cytolytic activity against a lymphoma cell line. ChIP analysis revealed decreased H3K27me3 at the KLRK1 promoter, that encodes for NKG2D (147). Bladder cancer xenografts treated with EPZ011989, alone and in combination with cisplatin, showed greater expression of NK cell-associated genes CCL3, ICAM1, ICAM2, and CD86, indicating activated NK signaling. Moreover, immunohistochemistry (IHC) analysis revealed greater CD56 and NKp46 expression (148) (Figure 2 and Table 3).
However, treatment with the HAT inhibitor curcumin led to hypoacetylation at the NKG2D gene, as shown by decreased H3K9Ac levels, downregulating NKG2D transcription and impairing the cytolytic ability of NK cells (149) (Figure 2 and Table 3).
Treatment of human expanded NK cells with the LSD1 scaffolding inhibitor SP-2509, decreased CD16 and NKG2D activating receptors expression, as compared to the control. This led to reduced NK cell lysis against K562 and MOLM13 leukemic cells (150). Treatment of NK cells with GSK-J4, an inhibitor of the H3K27 demethylase jumonji domain-containing protein D3 (JMJD3/UTX), led to NKp46 downregulation, demonstrated through flow cytometry. However, co-culture assays did not show any impact in NK cell killing of K562 leukemic cells (151) (Figure 2 and Table 3).
Treatment of NK cells derived from healthy donors and MDS patients with 5-aza was shown to significantly increase KIR expression (152). Likewise, Gao et al. reported increased KIR expression in NK-92MI cells following treatment with 5-aza (153). Moreover, Santourlidis et al. observed induction of KIR expression on several NK cell lines following treatment with 5-aza-dC, indicating that repression of KIRs is dependent on DNA methylation (155). These results are consistent with another study showing increased surface expression of KIRs in NK cells treated with 5-aza-dC, while also observing increased NKp44 expression, and decreased NKG2D surface expression, as confirmed through flow cytometry (156). Furthermore, 5-aza treatment of NK cells resulted in a slight reduction in NKG2D, NKp30 and CD16 expression (154) (Figure 2 and Table 3).
Besides NK cell ligands and receptors, epigenetic agents may also modulate NK cell function through several mechanisms. CD107a is localized within the NK cell vesicles which contain cytotoxic granules and is brought to the cell surface upon granzyme exocytosis, thus representing a mark of degranulation. Entinostat-treated NK cells subsequently co-cultured with untreated tumor cells showed a significant increase in NK cell CD107a-positivity (127, 128). Importantly, RNA sequencing data of entinostat-treated human NK cells before co-culture showed transcriptional changes, revealing upregulation of molecules related to NK cell cytotoxic functions, such as GZMB, PRF1, IFNG, and TNFA. Thus, HDAC inhibition in NK cells led to significant transcriptional profile alterations, independently of NK cell ligand modulation or co-culture with tumor cells (127). Additionally, treatment with entinostat enhanced NK cell cytotoxicity against tumor cells (127, 128). Likewise, assessment of splenic NK cells from panobinostat-treated tumor-bearing mice also showed a significant increase in the percentage of CD107a+ NK cells, as well as positive for CD69, a surface marker related NK cell activation, when compared to the control. Furthermore, increased expression of cell adhesion molecules and tight junction genes was shown following panobinostat treatment, resulting in increased conjugation between NK and target cells, and subsequent enhancement of NK cell mediated-tumor cell lysis (130). In an ovarian cancer mouse model, treatment with NextA, an HDAC6 inhibitor, in combination with 5-aza led to increased expression of interferon-stimulated genes (ISGs), and of CCL2, CCL5 and CXCL10, resulting in increased NK cell activation and infiltration into the TME (100). Moreover, selective HDAC8 inhibition with PCI-34051 increased the percentage of IFN-γ producing NK cells, as assessed by intracellular IFN-γ flow cytometry prior to co-culture with tumor cells. However, no significant changes were observed regarding NK cell killing of K562 cells (157). On the other hand, NK cell treatment with VPA impaired CD107a degranulation as well as IFN-γ secretion (132, 144). NK cells isolated from healthy donors, stimulated with IL-12, IL-15 and IL-18, and treated with TSA also exhibited decreased IFN-γ secretion, degranulation and cytotoxicity against K562 leukemic cells (132). Inhibition of NK cell degranulation was also found upon treatment of primary NK cells with panobinostat or romidepsin, resulting in decreased CD107a expression, and reduced killing of K562 leukemic cells (145). Moreover, entinostat treatment was shown to increase NK cell proliferation, as measured by Ki67 (126) (Figure 2 and Table 4).
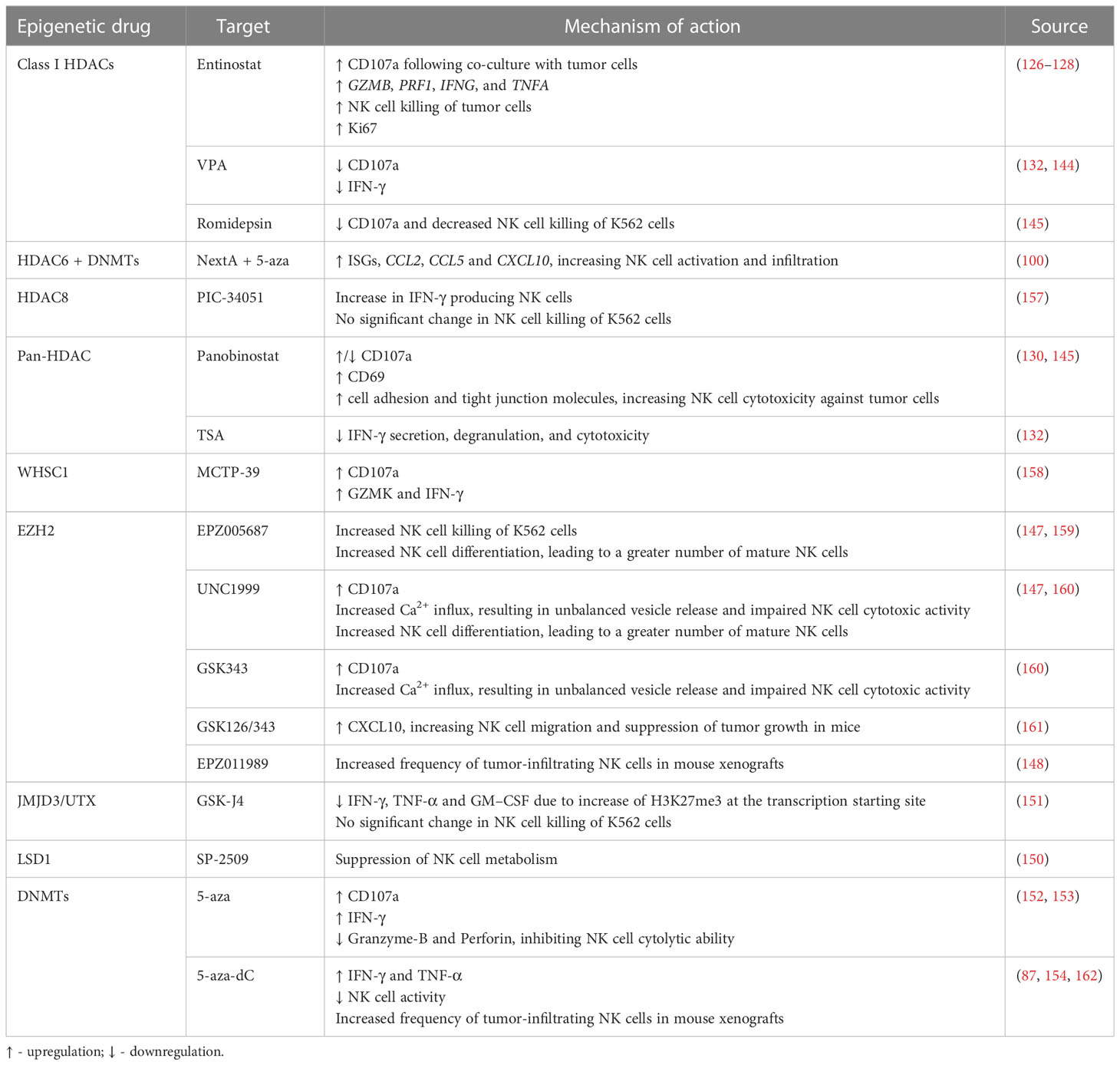
Table 4 Effect of epigenetic modulating agents on NK cell function, infiltration, and development.
Mice grafted with prostate adenocarcinoma cells treated with MCTP-39, an inhibitor of the HMT WHSC1, showed increased NK cell effector function, CD107a, Granzyme-K (GZMK) and IFN-γ expression (158). Treatment with EZH2 inhibitors has also led to increased NK function in several studies. The small-molecule EZH2 inhibitor EPZ005687 resulted in enhanced NK cell cytotoxicity against K562 cells (159). A recent study reported that NK cell treatment with the EZH2 inhibitors UNC1999 or GSK343 enhanced CD107a levels, while NK cell cytotoxicity against K562 erythroleukemia cells was unexpectedly lower than in the control group. Results indicated that EZH2i treatment upregulated the expression of the calcium channel PKD2 and increased Ca2+ influx, resulting in an unbalanced vesicle release, where a large number of cytotoxic particles were released to fewer target cells within a short period of time, ultimately impairing NK cell cytotoxic activity (160). A recent report revealed that treatment of hepatocellular carcinoma cells with EZH2i GSK126/343 was able to increase CXCL10 expression by tumor cells, which induced NK cell migration to the tumor site and suppressed tumor growth in a mouse model. This study also disclosed that EZH2 recruitment to the CXCL10 promoter was dependent on HDAC10 (161). Moreover, treatment with EZH2i EPZ011989 alone and in combination with cisplatin resulted in increased frequency of tumor-infiltrating NK cells (148). Moreover, NK cell development can also be targeted with epigenetic modulating agents, since treatment of Hematopoietic Stem and Progenitor Cells (HSPCs) with the small-molecule inhibitors UNC1999 and EPZ005687 decreased H3K27me3 levels and accelerated differentiation into CD122+NKp46+ phenotype in treated cells, leading to a greater number of mature NK cells as compared to control. Thus, EZH2 inhibition is suggested to promote NK cell development in vitro (147, 159) (Figure 2 and Table 4).
GSK-J4 has been shown to reduce cytokine levels in IL-15-stimulated NK cells, such as IFN-γ, TNF-α and granulocyte–macrophage colony-stimulating factor (GM–CSF), since it increased the repressive H3K27me3 mark around the transcription starting site of those genes. Despite this, GSK-J4 did not impact NK cell killing of K562 leukemic cells (151). Targeting the histone demethylase LSD1 with the scaffolding inhibitor SP-2509 was able to suppress NK cell metabolism, since treatment of NK cells derived from several donors abolished oxidative phosphorylation in NK cells, through glutathione depletion (150) (Figure 2 and Table 4).
NK cells from 5-aza-dC-treated mice exhibited upregulation of IFN-γ and TNF-α, which inhibited tumorigenicity in vivo, indicating that 5-aza-dC promotes NK cell antitumor activity (87). However, Triozzi et al. showed that, in an in vivo melanoma model, treatment with low-dose 5-aza-dC, either alone or in combination with IL-2, decreased NK cell activity (162). Co-culture of tumor cells with NK cells pre-treated with the DNMTi 5-aza induced higher degranulation and IFN-γ production in response to target cells, indicating increased NK cell function following 5-aza treatment (152). On the contrary, 5-aza treatment of human polyclonal NK cells has been reported to decrease Granzyme-B and Perforin secretion by NK cells, inhibiting NK cell cytolytic ability against K562 leukemic cells. The authors pose the hypothesis that this loss of NK activity may be due to excessive demethylation following 5-aza treatment (153). In a different study, although 5-aza-dC was not able to directly stimulate NK cells, it increased NK cell responsiveness to IL-2 stimulation, and induced IFN-γ production upon co-culture with tumor cells (154). An increase in the frequency of tumor-infiltrating NK cells after treatment with 5-aza-dC has also been reported (87) (Figure 2 and Table 4).
The understanding of the immune landscape in prostate cancer and its impact on patient prognosis is increasing, however insights on the mechanisms involved in its regulation are still lacking. NK cells in PCa have shown increased expression of exhaustion markers PD-1 and TIM-3, impairing their degranulation ability and reducing production of TNF-α, IFN-γ and Granzyme-B. In addition, in PCa, NK cells seem to decrease in frequency, also displaying fewer activating receptors NKG2D, CD16 and NKp46 and impaired anti-tumor activity (Figure 3). These findings highlight the potential and need for additional studies on targeting NK cell-mediated immunity in PCa patients.
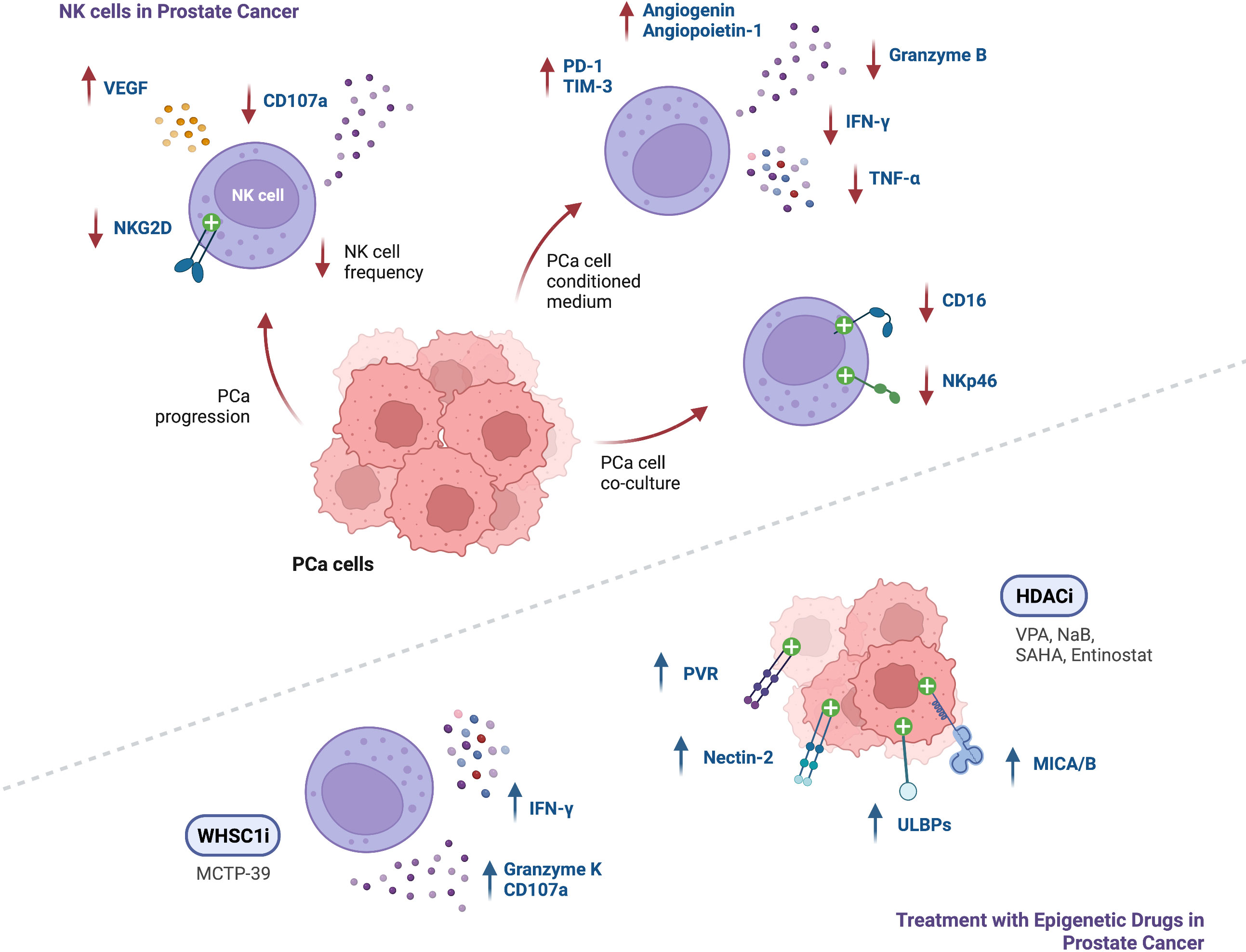
Figure 3 Summary of findings regarding NK cell function in the context of PCa, and modulation of NK cell anti-tumor responses following treatment with epigenetic modulating agents (↑ - upregulation; ↓ - downregulation).
Epigenetic modulation of anti-tumor responses can be achieved through exposure to several pharmacological agents. Numerous studies conducted in several tumor models confirmed upregulation of NK cell activating ligands after cancer cell treatment with DNMT, HDAC, HMT, KDM and BET inhibitors, leading to enhanced NK cell cytotoxicity following co-culture with tumor cells. To date, in prostate cancer, it has been shown upregulation of NK-cell activating ligands such as MICA, MICB, ULBPs, PVR and Nectin-2 upon treatment with HDAC inhibitors such as VPA (alone or in combination with the DNMT inhibitor hydralazine), NaB, SAHA and entinostat (Figure 3). Hence, studies with other epigenetic modulating drugs in PCa are still lacking. Of note, it has been reported in other cancers, upregulation of the inhibitory ligand MHC-I after exposure to some epigenetic agents, including DNMTi, HDACi, EZH2i and G9a inhibitors. Moreover, downregulation of the activating NKp30 ligand, B7-H6, was observed upon treatment with HDACi and HATi. Thus, it should be considered the effect that an epigenetic drug might have in the complex balance between expression of activating and inhibitory ligands.
Several studies in tumor models other than PCa demonstrated that direct modulation of NK cells with different epigenetic agents can drive increased expression of activating receptors such as NKG2D, NKp30, and DNAM-1. Conversely, decreased expression of NKG2D, NKp46 and CD16, has also been shown after treatment with some HDACi, DNMTi, and LSD1i. Moreover, expression of inhibitory receptors, such as KIRs, was upregulated following NK cell treatment with entinostat and 5-aza-dC. The intricacy of these findings suggests that different agents can have different effects on NK cells, supporting the need of further research. In PCa, NK cell receptor modulation effect on tumor cell recognition with epigenetic agents is still unknown.
NK cell activity was found to be enhanced after treatment with agents such as entinostat, panobinostat, NextA, 5-aza, 5-aza-dC, MCTP-39, and EZH2 inhibitors, as demonstrated by increased expression of Perforin and Granzyme-B, and enhanced secretion of TNF-α and IFN-γ, which resulted in increased NK cell-killing of tumor cells. In fact, PCa mice xenografts treated with MCTP-39 showed increased NK cell effector function, as well as upregulation of IFN-γ and Granzyme-K (Figure 3).
Several NK cell-based immunotherapies have been studied and some are currently undergoing clinical trials for cancer treatment. These approaches include autologous and allogenic NK cell transfer following ex vivo expansion and activation with cytokines, NK cells derived from induced pluripotent stem cells (iPSCs), and genetically modified chimeric antigen receptor (CAR) NK cells targeting specific antigens on the surface of tumor cells (163). However, limited infiltration, immunosuppressive TME, and downregulation of tumor antigens and NK ligands, are factors restraining the clinical efficacy of those approaches (163, 164). Thus, we propose that combination with epigenetic modulating agents may represent a promising strategy to improve the effectiveness of NK cell-based immunotherapy.
Overall, despite some challenges, emerging data suggests exciting potential in modulating NK cell-mediated immunity. The therapeutic usage of epigenetic modulating agents for prostate cancer in order to render tumor cells more immunogenic and increase NK cell anti-tumor potential, is an exciting possibility to be pursued, opening new venues for further investigation.
FDdR and MPC designed and wrote the manuscript. MPC and CJ revised the paper. All authors contributed to the article and approved the submitted version.
CJ and MPC research are supported by the Research Center of Portuguese Oncology Institute of Porto (FB-CBEG_CI-IPOP-27-2016 and EpImmunoPCa_PI 143-CI-IPOP-131-2020. MPC is funded by FCT—Fundação para a Ciência e Tecnologia (CEECINST/00091/2018).
The authors would like to thank all the scientists who contributed with relevant work mentioned in this review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Mottet N, van den Bergh RCN, Briers E, van den Broeck T, Cumberbatch MG, de Santis M, et al. EAU-EANM-ESTRO-ESUR-SIOG guidelines on prostate cancer–2020 update. part 1: Screening, diagnosis, and local treatment with curative intent. Eur Urol (2021) 79:243–62. doi: 10.1016/j.eururo.2020.09.042
3. Cornford P, van den Bergh RCN, Briers E, Van den Broeck T, Cumberbatch MG, De Santis M, et al. EAU-EANM-ESTRO-ESUR-SIOG guidelines on prostate cancer. part II–2020 update: Treatment of relapsing and metastatic prostate Cancer[Formula presented]. Eur Urol (2021) 79:263–82. doi: 10.1016/j.eururo.2020.09.046
4. Wang G, Zhao D, Spring DJ, Depinho RA. Genetics and biology of prostate cancer. Genes Dev (2018) 32:1105–40. doi: 10.1101/gad.315739.118
5. Wang Y, Wang Y, Ci X, Choi SYC, Crea F, Lin D, et al. Molecular events in neuroendocrine prostate cancer development. Nat Rev Urol (2021) 18:581–96. doi: 10.1038/s41585-021-00490-0
6. Solimando AG, Kalogirou C, Krebs M. Angiogenesis as therapeutic target in metastatic prostate cancer – narrowing the gap between bench and bedside. Front Immunol (2022) 13:842038. doi: 10.3389/fimmu.2022.842038
7. van Moorselaar RJA, Voest EE. Angiogenesis in prostate cancer: its role in disease progression and possible therapeutic approaches . Available at: www.elsevier.com/locate/mce.
8. Melegh Z, Oltean S. Targeting angiogenesis in prostate cancer. Int J Mol Sci (2019) 20(11):11. doi: 10.3390/ijms20112676
9. Bahmad HF, Jalloul M, Azar J, Moubarak MM, Samad TA, Mukherji D, et al. Tumor microenvironment in prostate cancer: Toward identification of novel molecular biomarkers for diagnosis, prognosis, and therapy development. Front Genet (2021) 12:652747. doi: 10.3389/fgene.2021.652747
10. Costanzo-Garvey DL, Keeley T, Case AJ, Watson GF, Alsamraae M, Yu Y, et al. Neutrophils are mediators of metastatic prostate cancer progression in bone. Cancer Immunol Immunother (2020) 69:1113–30. doi: 10.1007/s00262-020-02527-6
11. Chong W, Zhang Z, Luo R, Gu J, Lin J, Wei Q, et al. Integration of circulating tumor cell and neutrophil-lymphocyte ratio to identify high-risk metastatic castration-resistant prostate cancer patients. BMC Cancer (2021) 21:1–11. doi: 10.1186/s12885-021-08405-3
12. Lee H, Jeong SJ, Hong SK, Byun SS, Lee SE, Oh JJ. High preoperative neutrophil–lymphocyte ratio predicts biochemical recurrence in patients with localized prostate cancer after radical prostatectomy. World J Urol (2016) 34:821–7. doi: 10.1007/s00345-015-1701-6
13. Wang Y, Dong X, Qu Z, Peng K, Sun X, Chen R. Correlation between peripheral blood neutrophil-lymphocyte ratio and CD34 expression in prostate cancer. BMC Cancer (2020) 20:1–12. doi: 10.1186/s12885-020-07382-3
14. Wang H, Gu L, Wu Y, Feng D, Duan J, Wang X, et al. The values of neutrophil-lymphocyte ratio and/or prostate-specific antigen in discriminating real Gleason score≥7 prostate cancer from group of biopsy-based Gleason score ≤ 6. BMC Cancer (2017) 17:1–8. doi: 10.1186/s12885-017-3614-9
15. Lorente D, Mateo J, Templeton AJ, Zafeiriou Z, Bianchini D, Ferraldeschi R, et al. Baseline neutrophil-lymphocyte ratio (NLR) is associated with survival and response to treatment with second-line chemotherapy for advanced prostate cancer independent of baseline steroid use. Ann Oncol (2015) 26:750–5. doi: 10.1093/annonc/mdu587
16. Pittoni P, Tripodo C, Piconese S, Mauri G, Parenza M, Rigoni A, et al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res (2011) 71:5987–97. doi: 10.1158/0008-5472.CAN-11-1637
17. Nonomura N, Takayama H, Nishimura K, Oka D, Nakai Y, Shiba M, et al. Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. Br J Cancer (2007) 97:952–6. doi: 10.1038/sj.bjc.6603962
18. Sullivan HH, Heaphy CM, Kulac I, Cuka N, Lu J, Barber JR, et al. High extratumoral mast cell counts are associated with a higher risk of adverse prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev (2020) 29:668–75. doi: 10.1158/1055-9965.EPI-19-0962
19. Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y, et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J Natl Cancer Inst (2019) 111:301–10. doi: 10.1093/jnci/djy141
20. Johansson A, Rudolfsson S, Hammarsten P, Halin S, Pietras K, Jones J, et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am J Pathol (2010) 177:1031–41. doi: 10.2353/ajpath.2010.100070
21. Lundholm M, Hägglöf C, Wikberg ML, Stattin P, Egevad L, Bergh A, et al. Secreted factors from colorectal and prostate cancer cells skew the immune response in opposite directions. Sci Rep (2015) 5:1–12. doi: 10.1038/srep15651
22. Erlandsson A, Carlsson J, Lundholm M, Fält A, Andersson SO, Andrén O, et al. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate (2019) 79:363–9. doi: 10.1002/pros.23742
23. Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med (2018) 24:165–75. doi: 10.1038/nm.4463
24. Gollapudi K, Galet C, Grogan T, Zhang H, Said JW, Huang J, et al. Association between tumor-associated macrophage infiltration, high grade prostate cancer, and biochemical recurrence after radical prostatectomy. Am J Cancer Res (2013) 3:523–9.
25. Andersen LB, Nørgaard M, Rasmussen M, Fredsøe J, Borre M, Ulhøi BP, et al. Immune cell analyses of the tumor microenvironment in prostate cancer highlight infiltrating regulatory T cells and macrophages as adverse prognostic factors. J Pathol (2021) 255:155–65. doi: 10.1002/path.5757
26. Comito G, Giannoni E, Segura CP, Barcellos-De-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene (2014) 33:2423–31. doi: 10.1038/onc.2013.191
27. Sullivan C, Brown NE, Vasiliauskas J, Pathrose P, Starnes SL, Waltz SE. Prostate epithelial RON signaling promotes M2 macrophage activation to drive prostate tumor growth and progression Camille. Mol Cancer Res (2020) 18(8), 11244–1254. doi: 10.1158/1541-7786.MCR-20-0060
28. Cioni B, Zaalberg A, van Beijnum JR, Melis MHM, van Burgsteden J, Muraro MJ, et al. Androgen receptor signalling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat Commun (2020) 11:1–17. doi: 10.1038/s41467-020-18313-y
29. Solís-Martínez R, Cancino-Marentes M, Hernández-Flores G, Ortiz-Lazareno P, Mandujano-Álvarez G, Cruz-Gálvez C, et al. Regulation of immunophenotype modulation of monocytes-macrophages from M1 into M2 by prostate cancer cell-culture supernatant via transcription factor STAT3. Immunol Lett (2018) 196:140–8. doi: 10.1016/j.imlet.2018.02.009
30. Thomas MU, Messex JK, Dang T, Abdulkadir SA, Jorcyk CL, Liou GY. Macrophages expedite cell proliferation of prostate intraepithelial neoplasia through their downstream target ERK. FEBS J (2021) 288:1871–86. doi: 10.1111/febs.15541
31. Lee GT, Kwon SJ, Lee JH, Jeon SS, Jang KT, Choi HY, et al. Macrophages induce neuroendocrine differentiation of prostate cancer cells via BMP6-IL6 loop. Prostate (2011) 71:1525–37. doi: 10.1002/pros.21369
32. Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating β-catenin/STAT3 signaling. Cell Death Dis (2020) 11(234):234. doi: 10.1038/s41419-020-2435-y
33. Youlin K, Weiyang H, Simin L, Xin G. Prostaglandin E2 inhibits prostate cancer progression by countervailing tumor microenvironment-induced impairment of dendritic cell migration through LXRα/CCR7 pathway. J Immunol Res (2018) 2018:5808962. doi: 10.1155/2018/5808962
34. Salimu J, Webber J, Gurney M, Al-Taei S, Clayton A, Tabi Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J Extracell Vesicles (2017) 6:1. doi: 10.1080/20013078.2017.1368823
35. Idorn M, Køllgaard T, Kongsted P, Sengeløv L, thor Straten P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol Immunother (2014) 63:1177–87. doi: 10.1007/s00262-014-1591-2
36. Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, et al. IL23 secreted by myeloid cells drives castration resistant prostate cancer. Nature (2019) 559:363–9. doi: 10.1038/s41586-018-0266-0.IL23
37. Lerman I, de la Luz Garcia-Hernandez M, Rangel-Moreno J, Chiriboga L, Pan C, Nastiuk KL, et al. Infiltrating myeloid cells exert pro-tumorigenic actions via neutrophil elastase. Mol Cancer Res (2017) 15(9), 1138–52. doi: 10.1158/1541-7786.MCR-17-0003
38. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discovery (2016) 6:80–95. doi: 10.1158/2159-8290.CD-15-0224
39. Hossain DMS, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin Cancer Res (2015) 21(16), 3771–82. doi: 10.1158/1078-0432.CCR-14-3145
40. Wu Z, Chen H, Luo W, Zhang H, Li G, Zeng F, et al. The landscape of immune cells infiltrating in prostate cancer. Front Oncol (2020) 10:517637. doi: 10.3389/fonc.2020.517637
41. Yang Y, Attwood K, Bshara W, Mohler JL, Guru K, Xu B, et al. High intratumoral CD8+ T-cell infiltration is associated with improved survival in prostate cancer patients undergoing radical prostatectomy. Prostate (2021) 81:20–8. doi: 10.1002/pros.24068
42. Sorrentino C, Musiani P, Pompa P, Cipollone G, Di Carlo E. Androgen deprivation boosts prostatic infiltration of cytotoxic and regulatory T lymphocytes and has no effect on disease-free survival in prostate cancer patients. Clin Cancer Res (2011) 17:1571–81. doi: 10.1158/1078-0432.CCR-10-2804
43. Yanai Y, Kosaka T, Mikami S, Hongo H, Yasumizu Y, Takeda T, et al. CD8-positive T cells and CD204-positive M2-like macrophages predict postoperative prognosis of very high-risk prostate cancer. Sci Rep (2021) 11:1–7. doi: 10.1038/s41598-021-01900-4
44. Ness N, Andersen S, Valkov A, Nordby Y, Donnem T, Al-Saad S, et al. Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate (2014) 74:1452–61. doi: 10.1002/pros.22862
45. Petitprez F, Fossati N, Vano Y, Freschi M, Becht E, Lucianò R, et al. PD-L1 expression and CD8 + T-cell infiltrate are associated with clinical progression in patients with node-positive prostate cancer. Eur Urol Focus (2019) 5:192–6. doi: 10.1016/j.euf.2017.05.013
46. Hu S, Li L, Yeh S, Cui Y, Li X, Chang HC, et al. Infiltrating T cells promote prostate cancer metastasis via modulation of FGF11→miRNA-541→androgen receptor (AR)→MMP9 signaling. Mol Oncol (2015) 9:44–57. doi: 10.1016/j.molonc.2014.07.013
47. Ebelt K, Babaryka G, Figel AM, Pohla H, Buchner A, Stief CG, et al. Dominance of CD4+ lymphocytic infiltrates with disturbed effector cell characteristics in the tumor microenvironment of prostate carcinoma. Prostate (2008) 68:1–10. doi: 10.1002/pros.20661
48. Ebelt K, Babaryka G, Frankenberger B, Stief CG, Eisenmenger W, Kirchner T, et al. Prostate cancer lesions are surrounded by FOXP3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur J Cancer (2009) 45:1664–72. doi: 10.1016/j.ejca.2009.02.015
49. Ozbek B, Ertunc O, Erickson A, Vidal ID, Gomes-Alexandre C, Guner G, et al. Multiplex immunohistochemical phenotyping of T cells in primary prostate cancer. Prostate (2022) 82:706–22. doi: 10.1002/pros.24315
50. Japp AS, Kursunel MA, Meier S, Mälzer JN, Li X, Rahman NA, et al. Dysfunction of PSA-specific CD8+ T cells in prostate cancer patients correlates with CD38 and Tim-3 expression. Cancer Immunol Immunother (2015) 64:1487–94. doi: 10.1007/s00262-015-1752-y
51. Sharma V, Dong H, Kwon E, Karnes RJ. Positive pelvic lymph nodes in prostate cancer harbor immune suppressor cells to impair tumor-reactive T cells. Eur Urol Focus (2018) 4:75–9. doi: 10.1016/j.euf.2016.09.003
52. Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, et al. T Cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-β1 cytokine. Immunity (2011) 35:123–34. doi: 10.1016/j.immuni.2011.04.019
53. Gao Z, Tao Y, Lai Y, Wang Q, Li Z, Peng S, et al. Immune cytolytic activity as an indicator of immune checkpoint inhibitors treatment for prostate cancer. Front Bioeng Biotechnol (2020) 8:930. doi: 10.3389/fbioe.2020.00930
54. Huen N-Y, Pang A, Tucker JA, Lee T-L, Vergati M, Jochems C, et al. Up-regulation of proliferative and migratory genes in regulatory T cells from patients with metastatic castration-resistant prostate cancer. Int J Cancer (2013) 133:373–82. doi: 10.1002/ijc.28026
55. Vidotto T, Saggioro FP, Jamaspishvili T, Chesca DL, Picanço de Albuquerque CG, Reis RB, et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate (2019) 79:969–79. doi: 10.1002/pros.23808
56. Sfanos K, Bruno T, Maris C, Xu L, Thoburn C, DeMarzo AM, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH 17 and treg skewing. Clin Cancer Res (2008) 14(11), 3254–61. doi: 10.1158/1078-0432.CCR-07-5164
57. Watanabe M, Kanao K, Suzuki S, Muramatsu H, Morinaga S, Kajikawa K, et al. Increased infiltration of CCR4-positive regulatory T cells in prostate cancer tissue is associated with a poor prognosis. Prostate (2019) 79:1658–65. doi: 10.1002/pros.23890
58. Laheurte C, Thiery-Vuillemin A, Calcagno F, Legros A, Simonin H, Boullerot L, et al. Metronomic cyclophosphamide induces regulatory T cells depletion and PSA-specific T cells reactivation in patients with biochemical recurrent prostate cancer. Int J Cancer (2020) 147:1199–205. doi: 10.1002/ijc.32803
59. Flammiger A, Weisbach L, Huland H, Tennstedt P, Simon R, Minner S, et al. High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur J Cancer (2013) 49:1273–9. doi: 10.1016/j.ejca.2012.11.035
60. Nardone V, Botta C, Caraglia M, Martino EC, Ambrosio MR, Carfagno T, et al. Tumor infiltrating T lymphocytes expressing FoxP3, CCR7 or PD-1 predict the outcome of prostate cancer patients subjected to salvage radiotherapy after biochemical relapse. Cancer Biol Ther (2016) 17:1213–20. doi: 10.1080/15384047.2016.1235666
61. Woo JR, Liss MA, Muldong MT, Palazzi K, Strasner A, Ammirante M, et al. Tumor infiltrating b-cells are increased in prostate cancer tissue. J Transl Med (2014) 12:1–9. doi: 10.1186/1479-5876-12-30
62. Ammirante M, Luo J-L, Grivennikov S, Nedospasov S, Karin M. B cell–derived lymphotoxin promotes castration-resistant prostate cancer. Nature (2010) 464(7286), 302–5. doi: 10.1038/nature08782
63. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest (2013) 123:4410–22. doi: 10.1172/JCI69369
64. Pasero C, Gravis G, Granjeaud S, Guerin M, Thomassin-Piana J, Rocchi P, et al. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget (2015) 6:14360–73. doi: 10.18632/oncotarget.3965
65. Park MH, Song MJ, Cho MC, Moon DC, Yoon DY, Han SB, et al. Interleukin-32 enhances cytotoxic effect of natural killer cells to cancer cells via activation of death receptor 3. Immunology (2012) 135:63–72. doi: 10.1111/j.1365-2567.2011.03513.x
66. Lin SJ, Chou FJ, Li L, Lin CY, Yeh S, Chang C. Natural killer cells suppress enzalutamide resistance and cell invasion in the castration resistant prostate cancer via targeting the androgen receptor splicing variant 7 (ARv7). Cancer Lett (2017) 398:62–9. doi: 10.1016/j.canlet.2017.03.035
67. Koo KC, Shim DH, Yang CM, Lee SB, Kim SM, Shin TY, et al. Reduction of the CD16-CD56bright NK cell subset precedes NK cell dysfunction in prostate cancer. PloS One (2013) 8:1–8. doi: 10.1371/journal.pone.0078049
68. Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P, et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res (2016) 76:2153–65. doi: 10.1158/0008-5472.CAN-15-1965
69. Gallazzi M, Baci D, Mortara L, Bosi A, Buono G, Naselli A, et al. Prostate cancer peripheral blood NK cells show enhanced CD9, CD49a, CXCR4, CXCL8, MMP-9 production and secrete monocyte-recruiting and polarizing factors. Front Immunol (2021) 11:586126. doi: 10.3389/fimmu.2020.586126
70. Lundholm M, Schröder M, Nagaeva O, Baranov V, Widmark A, Mincheva-Nilsson L, et al. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PloS One (2014) 9:9. doi: 10.1371/journal.pone.0108925
71. Saga K, Park J, Nimura K, Kawamura N, Ishibashi A, Nonomura N, et al. NANOG helps cancer cells escape NK cell attack by downregulating ICAM1 during tumorigenesis. J Exp Clin Cancer Res (2019) 38:1–13. doi: 10.1186/s13046-019-1429-z
72. Liu X, Chen Q, Yan J, Wang Y, Zhu C, Chen C, et al. MiRNA-296-3p-ICAM-1 axis promotes metastasis of prostate cancer by possible enhancing survival of natural killer cell-resistant circulating tumour cells. Cell Death Dis (2013) 4:1–15. doi: 10.1038/cddis.2013.458
73. Boettcher AN, Usman A, Morgans A, VanderWeele DJ, Sosman J, Wu JD. Past, current, and future of immunotherapies for prostate cancer. Front Oncol (2019) 9:884. doi: 10.3389/fonc.2019.00884
74. Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun (2021) 12:1–16. doi: 10.1038/s41467-021-21615-4
75. Shao N, Tang H, Mi Y, Zhu Y, Wan F, Ye D. A novel gene signature to predict immune infiltration and outcome in patients with prostate cancer. Oncoimmunology (2020) 9:1–10. doi: 10.1080/2162402X.2020.1762473
76. Wang S, Wu P, Chen Y, Chai Y. Ambiguous roles and potential therapeutic strategies of innate lymphoid cells in different types of tumor (Review). Oncol Lett (2020) 20:1513–25. doi: 10.3892/ol.2020.11736
77. Trabanelli S, Chevalier MF, Martinez-Usatorre A, Gomez-Cadena A, Salomé B, Lecciso M, et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat Commun (2017) 8:593. doi: 10.1038/s41467-017-00678-2
78. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol (2021) 18:85–100. doi: 10.1038/s41571-020-0426-7
79. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of h-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319, 3–6. doi: 10.1038/319675a0
80. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
81. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med (2009) 206:1495–503. doi: 10.1084/jem.20090681
82. Xia M, Wang B, Wang Z, Zhang X, Wang X. Epigenetic regulation of NK cell-mediated antitumor immunity. Front Immunol (2021) 12:672328. doi: 10.3389/fimmu.2021.672328
83. Jerónimo C, Bastian PJ, Bjartell A, Carbone GM, Catto JWF, Clark SJ, et al. Epigenetics in prostate cancer: Biologic and clinical relevance. Eur Urol (2011) 60:753–66. doi: 10.1016/j.eururo.2011.06.035
84. Graça I, Pereira-Silva E, Henrique R, Packham G, Crabb SJ, Jerónimo C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin Epigenet (2016) 8:98. doi: 10.1186/s13148-016-0264-8
85. Neganova ME, Klochkov SG, Aleksandrova YR, Aliev G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin Cancer Biol (2020) 452–71. doi: 10.1016/j.semcancer.2020.07.015
86. Cho HY, Son WC, Lee YS, Youn EJ, Kang CD, Park YS, et al. Differential effects of histone deacetylases on the expression of nkg2d ligands and nk cell-mediated anticancer immunity in lung cancer cells. Molecules (2021) 26:3952. doi: 10.3390/molecules26133952
87. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res (2015) 3:1030–41. doi: 10.1158/2326-6066.CIR-15-0073
88. Serrano A, Tanzarella S, Lionello I, Mendez R, Traversari C, Ruiz-Cabello F, et al. Expression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2′-deoxycytidine treatment. Int J Cancer (2001) 94:243–51. doi: 10.1002/ijc.1452
89. Dubovsky JA, McNeel DG, Powers JJ, Gordon J, Sotomayor EM, Pinilla-Ibarz JA. Treatment of chronic lymphocytic leukemia with a hypomethylating agent induces expression of NXF2, an immunogenic cancer testis antigen. Clin Cancer Res (2009) 15:3406–15. doi: 10.1158/1078-0432.CCR-08-2099
90. Bourne CM, Mun SS, Dao T, Aretz ZEH, Molvi Z, Gejman RS, et al. Unmasking the suppressed immunopeptidome of EZH2-mutated diffuse large b-cell lymphomas through combination drug treatment. Blood Adv (2022) 6:4107–21. doi: 10.1182/bloodadvances.2021006069
91. Son CH, Lee HR, Koh EK, Shin DY, Bae JH, Yang K, et al. Combination treatment with decitabine and ionizing radiation enhances tumor cells susceptibility of T cells. Sci Rep (2016) 6:32470. doi: 10.1038/srep32470
92. Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun (2018) 9:3868. doi: 10.1038/s41467-018-06300-3
93. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell (2019) 36:385–401.e8. doi: 10.1016/j.ccell.2019.08.008
94. Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep (2017) 20:854–67. doi: 10.1016/j.celrep.2017.07.007
95. Segovia C, San José-Enériz E, Munera-Maravilla E, Martínez-Fernández M, Garate L, Miranda E, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat Med (2019) 25:1073–81. doi: 10.1038/s41591-019-0499-y
96. Luo N, Nixon MJ, Gonzalez-ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA Methyltransferase inhibition upregulates responses in breast cancer. Nat Commun (2018) 9:1–11. doi: 10.1038/s41467-017-02630-w
97. Manning J, Indrova M, Lubyova B, Pribylova H, Bieblova J, Hejnar J, et al. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology (2008) 123:218–27. doi: 10.1111/j.1365-2567.2007.02689.x
98. Khan ANH, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother (2008) 57:647–54. doi: 10.1007/s00262-007-0402-4
99. Kitamura H, Torigoe T, Asanuma H, Honma I, Sato N, Tsukamoto T. Down-regulation of HLA class I antigens in prostate cancer tissues and up-regulation by histone deacetylase inhibition. J Urol (2007) 178:692–6. doi: 10.1016/j.juro.2007.03.109
100. Moufarrij S, Srivastava A, Gomez S, Hadley M, Palmer E, Austin PT, et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci Rep (2020) 10:1–12. doi: 10.1038/s41598-020-60409-4
101. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery (2020) 19:776–800. doi: 10.1038/s41573-020-0077-5
102. Frikeche J, Clavert A, Delaunay J, Brissot E, Grégoire M, Gaugler B, et al. Impact of the hypomethylating agent 5-azacytidine on dendritic cells function. Exp Hematol (2011) 39:1056–63. doi: 10.1016/j.exphem.2011.08.004
103. Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene (2019) 38:390–405. doi: 10.1038/s41388-018-0451-5
104. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527:249–53. doi: 10.1038/nature15520
105. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res (2018) 6:1561–77. doi: 10.1158/2326-6066.CIR-18-0070
106. Murakami T, Sato A, Chun NAL, Hara M, Naito Y, Kobayashi Y, et al. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol (2008) 128:1506–16. doi: 10.1038/sj.jid.5701216
107. Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget (2016) 7:7390–7402. doi: 10.18632/oncotarget.7180
108. Hong YK, Li Y, Pandit H, Li SP, Pulliam Z, Zheng Q, et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol (2019) 336:66–74. doi: 10.1016/j.cellimm.2018.12.010
109. Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep (2018) 23:3262–74. doi: 10.1016/j.celrep.2018.05.050
110. Tao R, de Zoeten EF, Özkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13:1299–307. doi: 10.1038/nm1652
111. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PloS One (2012) 7:1. doi: 10.1371/journal.pone.0030815
112. Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non–small cell lung cancer. Cancer Discovery (2017) 7:852–67. doi: 10.1158/2159-8290.CD-16-1020
113. Li X, Su X, Liu R, Pan Y, Fang J, Cao L, et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene (2021) 40:1836–50. doi: 10.1038/s41388-020-01636-x
114. Wang HF, Ning F, Liu ZC, Wu L, Li ZQ, Qi YF, et al. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol Immunother (2017) 66:355–66. doi: 10.1007/s00262-016-1935-1
115. Kim YD, Park SM, Ha HC, Lee AR, Won H, Cha H, et al. HDAC inhibitor, CG-745, enhances the anti-cancer effect of anti-PD-1 immune checkpoint inhibitor by modulation of the immune microenvironment. J Cancer (2020) 11:4059–72. doi: 10.7150/jca.44622
116. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res (2017) 23:5187–201. doi: 10.1158/1078-0432.CCR-17-0741
117. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell (2015) 162:974–86. doi: 10.1016/j.cell.2015.07.011
118. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell (2015) 162:961–73. doi: 10.1016/j.cell.2015.07.056
119. Morel KL, Sheahan AV, Burkhart DL, SC B, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA–STING–interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer (2021) 2:444–56. doi: 10.1038/s43018-021-00185-w
120. Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U.S.A. (2017) 114:E10981–90. doi: 10.1073/pnas.1712514114
121. Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer (2021) 20(171):171. doi: 10.1186/s12943-021-01464-x
122. Chávez-Blanco A, de la Cruz-Hernández E, Domínguez GI, Rodríguez-Cortez O, Alatorre B, Pérez-Cárdenas E, et al. Upregulation of NKG2D ligands and enhanced natural killer cell cytotoxicity by hydralazine and valproate. Int J Oncol (2011) 39:1491–9. doi: 10.3892/ijo.2011.1144
123. Bhat J, Dubin S, Dananberg A, Quabius ES, Fritsch J, Marie Dowds C, et al. Histone deacetylase inhibitor modulates NKG2D receptor expression and memory phenotype of human gamma/delta T cells upon interaction with tumor cells. Front Immunol (2019) 10:569. doi: 10.3389/fimmu.2019.00569
124. Fiegler N, Textor S, Arnold A, Rölle A, Oehme I, Breuhahn K, et al. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood (2013) 122:684–93. doi: 10.1182/blood-2013-02-482513
125. Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, Beck JF, et al. Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett (2008) 272:110–21. doi: 10.1016/j.canlet.2008.06.027
126. Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. Oncoimmunology (2018) 7:1–17. doi: 10.1080/2162402X.2018.1466018
127. Idso JM, Lao S, Schloemer NJ, Knipstein J, Burns R, Thakar MS, et al. Entinostat augments NK cell functions via epigenetic upregulation of IFIT1-STING-STAT4 pathway. Oncotarget (2020) 11:1799–815. doi: 10.18632/oncotarget.27546
128. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res (2015) 32:779–92. doi: 10.1007/s11095-013-1231-0
129. Ponath V, Frech M, Bittermann M, Al Khayer R, Neubauer A, Brendel C, et al. The oncoprotein ski acts as a suppressor of nk cell-mediated immunosurveillance in PDAC. Cancers (Basel) (2020) 12:1–10. doi: 10.3390/cancers12102857
130. Afolabi LO, Bi J, Li X, Adeshakin AO, Adeshakin FO, Wu H, et al. Synergistic tumor cytolysis by NK cells in combination with a pan-HDAC inhibitor, panobinostat. Front Immunol (2021) 12:701671. doi: 10.3389/fimmu.2021.701671
131. López-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: Potential implications for the immunosurveillance of cancer. Oncogene (2009) 28:2370–82. doi: 10.1038/onc.2009.117
132. Rossi LE, Avila DE, Spallanzani RG, Ziblat A, Fuertes MB, Lapyckyj L, et al. Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J Leukoc Biol (2012) 91:321–31. doi: 10.1189/jlb.0711339
133. Tang KF, He CX, Zeng GL, Wu J, Bin SG, YS S, et al. Induction of MHC class I-related chain b (MICB) by 5-aza-2′-deoxycytidine. Biochem Biophys Res Commun (2008) 370:578–83. doi: 10.1016/j.bbrc.2008.03.131
134. Baraganõ Raneros A, Martín-Palanco V, Fernandez AF, Rodriguez RM, Fraga MF, Lopez-Larrea C, et al. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun (2015) 16:71–82. doi: 10.1038/gene.2014.58
135. Bugide S, Green MR, Wajapeyee N. Inhibition of enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U.S.A. (2018) 115:E3509–18. doi: 10.1073/pnas.1802691115
136. Nakajima NI, Niimi A, Isono M, Oike T, Sato H, Nakano T, et al. Inhibition of the HDAC/Suv39/G9a pathway restores the expression of DNA damage-dependent major histocompatibility complex class I-related chain a and b in cancer cells. Oncol Rep (2017) 38:693–702. doi: 10.3892/or.2017.5773
137. Bailey CP, Figueroa M, Gangadharan A, Yang Y, Romero MM, Kennis BA, et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro Oncol (2020) 22:1302–14. doi: 10.1093/neuonc/noaa058
138. Liu M, Du M, Yu J, Qian Z, Gao Y, Pan W, et al. CEBPA mutants down-regulate AML cell susceptibility to NK-mediated lysis by disruption of the expression of NKG2D ligands, which can be restored by LSD1 inhibition. Oncoimmunology (2022) 11:1–13. doi: 10.1080/2162402X.2021.2016158
139. Abruzzese MP, Bilotta MT, Fionda C, Zingoni A, Soriani A, Vulpis E, et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J Hematol Oncol (2016) 9:1–19. doi: 10.1186/s13045-016-0362-2
140. Wang H, Liu G, Jin X, Song S, Chen S, Zhou P, et al. BET inhibitor JQ1 enhances anti-tumor immunity and synergizes with PD-1 blockade in CRC. J Cancer (2022) 13:2126–37. doi: 10.7150/jca.69375
141. Shao FF, Chen BJ, Wu GQ. The functions of EZH2 in immune cells: Principles for novel immunotherapies. J Leukoc Biol (2021) 110:77–87. doi: 10.1002/JLB.1RU0520-311R
142. Lobo J, Rodrigues Â, Antunes L, Graça I, Ramalho-Carvalho J, Vieira FQ, et al. High immunoexpression of Ki67, EZH2, and SMYD3 in diagnostic prostate biopsies independently predicts outcome in patients with prostate cancer. Urol Oncol: Semin Original Investigations (2018) 36:161.e7–161.e17. doi: 10.1016/j.urolonc.2017.10.028
143. Moreira-Silva F, Outeiro-Pinho G, Lobo J, Guimarães R, Gaspar VM, Mano JF, et al. G9a inhibition by CM-272: Developing a novel anti-tumoral strategy for castration-resistant prostate cancer using 2D and 3D in vitro models. Biomed Pharmacother (2022) 150:0–3. doi: 10.1016/j.biopha.2022.113031
144. Shi X, Li M, Cui M, Niu C, Xu J, Zhou L, et al. Epigenetic suppression of the antitumor cytotoxicity of NK cells by histone deacetylase inhibitor valproic acid. Am J Cancer Res (2016) 6:600–14.
145. Pace M, Williams J, Kurioka A, Gerry AB, Jakobsen B, Klenerman P, et al. Histone deacetylase inhibitors enhance CD4 T cell susceptibility to NK cell killing but reduce NK cell function. PloS Pathog (2016) 12:1–22. doi: 10.1371/journal.ppat.1005782
146. Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C, et al. The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3. Sci Rep (2017) 7:1–9. doi: 10.1038/srep45266
147. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci U.S.A. (2015) 112:15988–93. doi: 10.1073/pnas.1521740112
148. Ramakrishnan S, Granger V, Rak M, Hu Q, Attwood K, Aquila L, et al. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ (2019) 26:2100–14. doi: 10.1038/s41418-019-0278-9
149. Fernández-Sánchez A, Raneros AB, Palao RC, Sanz AB, Ortiz A, Ortega F, et al. DNA Demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics (2013) 8:66–78. doi: 10.4161/epi.23115
150. Bailey CP, Figueroa M, Gangadharan A, Lee DA, Chandra J. Scaffolding LSD1 inhibitors impair NK cell metabolism and cytotoxic function through depletion of glutathione. Front Immunol (2020) 11:2196. doi: 10.3389/fimmu.2020.02196
151. Cribbs A, Hookway ES, Wells G, Lindow M, Obad S, Oerum H, et al. Inhibition of histone H3K27 demethylases selectively modulates inflammatory phenotypes of natural killer cells. J Biol Chem (2018) 293:2422–37. doi: 10.1074/jbc.RA117.000698
152. Sohlberg E, Pfefferle A, Andersson S, Baumann BC, Hellström-Lindberg E, Malmberg KJ. Imprint of 5-azacytidine on the natural killer cell repertoire during systemic treatment for high-risk myelodysplastic syndrome. Oncotarget (2015) 6:34178–90. doi: 10.18632/oncotarget.6213
153. Gao X-n, Lin J, Wang L-l, Yu L. Demethylating treatment suppresses natural killer cell cytolytic activity. Mol Immunol (2009) 46:2064–70. doi: 10.1016/j.molimm.2009.02.033
154. Schmiedel BJ, Arélin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer (2011) 128:2911–22. doi: 10.1002/ijc.25635
155. Santourlidis S, Trompeter H-I, Weinhold S, Eisermann B, Meyer KL, Wernet P, et al. Crucial role of DNA methylation in determination of clonally distributed killer cell ig-like receptor expression patterns in NK cells. J Immunol (2002) 169:4253–61. doi: 10.4049/jimmunol.169.8.4253
156. Kopp LM, Ray A, Denman CJ, Senyukov VS, Somanchi SS, Zhu S, et al. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol Immunol (2013) 54:296–301. doi: 10.1016/j.molimm.2012.12.012
157. Watters JM, Wright G, Smith MA, Shah B, L. Wright K. Histone deacetylase 8 inhibition suppresses mantle cell lymphoma viability while preserving natural killer cell function. Biochem Biophys Res Commun (2021) 534, 773–9. doi: 10.1016/j.bbrc.2020.11.001
158. Want MY, Karasik E, Gillard B, McGray AJR, Battaglia S. Inhibition of whsc1 allows for reprogramming of the immune compartment in prostate cancer. Int J Mol Sci (2021) 22:1–15. doi: 10.3390/ijms22168742
159. Yu M, Su Z, Huang X, Zhou Y, Zhang X, Wang B, et al. Histone methyltransferase Ezh2 negatively regulates NK cell terminal maturation and function. J Leukoc Biol (2021) 110:1033–45. doi: 10.1002/JLB.1MA0321-155RR
160. Li Y, Yu M, Yin J, Yan H, Wang X, Choice A, et al. Enhanced calcium signal induces NK cell degranulation but inhibits its cytotoxic activity. J Immunol (2022) 208:347–57. doi: 10.4049/jimmunol.2001141
161. Bugide S, Gupta R, Green MR, Wajapeyee N. EZH2 inhibits NK cell–mediated antitumor immunity by suppressing CXCL10 expression in an HDAC10-dependent manner. Proc Natl Acad Sci U.S.A. (2021) 118(30):32. doi: 10.1073/pnas.2102718118
162. Triozzi PL, Aldrich W, Achberger S, Ponnazhagan S, Alcazar O, Saunthararajah Y. Differential effects of low-dose decitabine on immune effector and suppressor responses in melanoma-bearing mice. Cancer Immunol Immunother (2012) 61:1441–50. doi: 10.1007/s00262-012-1204-x
163. Shin MH, Kim J, Lim SA, Kim J, Kim SJ, Lee KM. NK cell-based immunotherapies in cancer. Immune Netw (2020) 20(2):2. doi: 10.4110/in.2020.20.e14
Keywords: NK cells, prostate cancer, epigenetics, epigenetic modulating drugs, tumor microenvironment (TME), immune landscape
Citation: dos Reis FD, Jerónimo C and Correia MP (2023) Epigenetic modulation and prostate cancer: Paving the way for NK cell anti-tumor immunity. Front. Immunol. 14:1152572. doi: 10.3389/fimmu.2023.1152572
Received: 27 January 2023; Accepted: 06 March 2023;
Published: 05 April 2023.
Edited by:
Cinzia Fionda, Sapienza University of Rome, ItalyReviewed by:
Duane Hamilton, National Cancer Institute, United StatesCopyright © 2023 dos Reis, Jerónimo and Correia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margareta P. Correia, bWFyZ2FyZXRhLmNvcnJlaWFAaXBvcG9ydG8ubWluLXNhdWRlLnB0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.