 Anna E. Brunell
Anna E. Brunell Riitta Lahesmaa
Riitta Lahesmaa Anu Autio2
Anu Autio2 Anil K. Thotakura
Anil K. Thotakura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 April 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1151632
This article is part of the Research TopicMitochondrial Damage and the Road to T-cell Exhaustion in Tumor MicroenvironmentView all 5 articles
T cell exhaustion is an alternative differentiation path of T cells, sometimes described as a dysfunction. During the last decade, insights of T cell exhaustion acting as a bottle neck in the field of cancer immunotherapy have undoubtedly provoked attention. One of the main drivers of T cell exhaustion is prolonged antigen presentation, a prerequisite in the cancer-immunity cycle. The umbrella term “T cell exhaustion” comprises various stages of T cell functionalities, describing the dynamic, one-way exhaustion process. Together these qualities of T cells at the exhaustion continuum can enable tumor clearance, but if the exhaustion acquired timeframe is exceeded, tumor cells have increased possibilities of escaping immune system surveillance. This could be considered a tipping point where exhausted T cells switch from an asset to a liability. In this review, the contrary role of exhausted T cells is discussed.
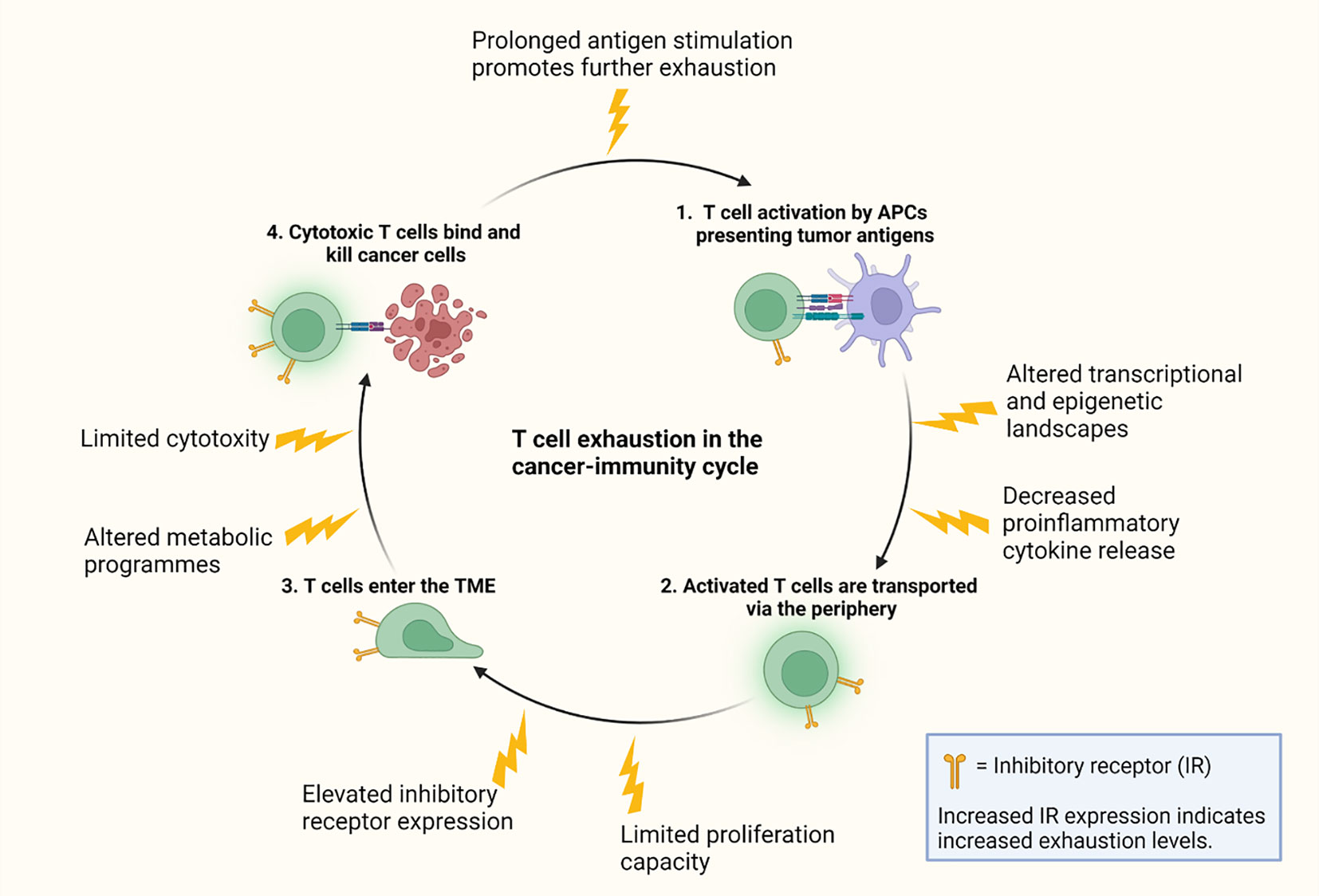
Graphical Abstract
T cell exhaustion can be defined as a physiological state of T cells displaying phenotypical and functional changes, such as eventual loss of proliferation capacity and effector functions, changes in cell metabolism and transcription, decreased production of cytokines and elevated expression of inhibitory receptors (IRs) (1–3). Exhausted T cells are epigenetically altered (4), having approximately 6000 different accessible chromatin regions compared to effector and memory T cells, indicating that exhausted T cells may be considered a distinct cell type (5, 6). The main drivers of T cell exhaustion such as prolonged T cell receptor (TCR) stimulation by antigens, exposure to suppressive cytokines and tumor mediated immunosuppressive metabolic byproducts, were previously thought to be located mainly in the tumor microenvironment (TME) (7). More recent studies provide evidence that the exhaustion journey of T cells is initiated already in the lymph nodes (LNs) through interaction between antigen presenting cell (APC)-presented tumor antigens and T cells (8).
T cell exhaustion is an umbrella term describing various functional states of T cells. Exhausted T cells can be divided into subpopulations based on functionality and phenotype, ranging from stem-like exhausted T cells to terminally exhausted T cells (7, 9, 10). However, there are no universal, clearly defined lines between the exhaustion subpopulations, and hence T cell exhaustion resembles a continuum; T cells shifting from precursor to terminal exhaustion. Some of the main markers used for categorizing populations of exhausted T cells include programmed death receptor 1 (PD-1) and transcription factor 1 (TCF-1) (10, 11). TCF-1 is a transcription factor essential for T cell development (12), and the maintenance of TCF-1 expression in exhausted cells is limited to approximately three rounds of divisions (13), where after its expression is epigenetically silenced (10). PD-1+TCF-1+ precursor exhausted T cells have stem cell-like properties as they have the capacity to proliferate, self-renew and further produce progeny populations of exhausted T cells, including terminally exhausted less proliferative PD-1+TCF-1–T cells (Figure 1) (11, 14). Studies have shown that the epigenetic state of exhausted T cells is irreversible (6, 9), indicating that exhaustion of T cells is a one-way process. However, recent studies suggests that the epigenetic state of exhaustion may be modifiable with the right treatments (15, 16).
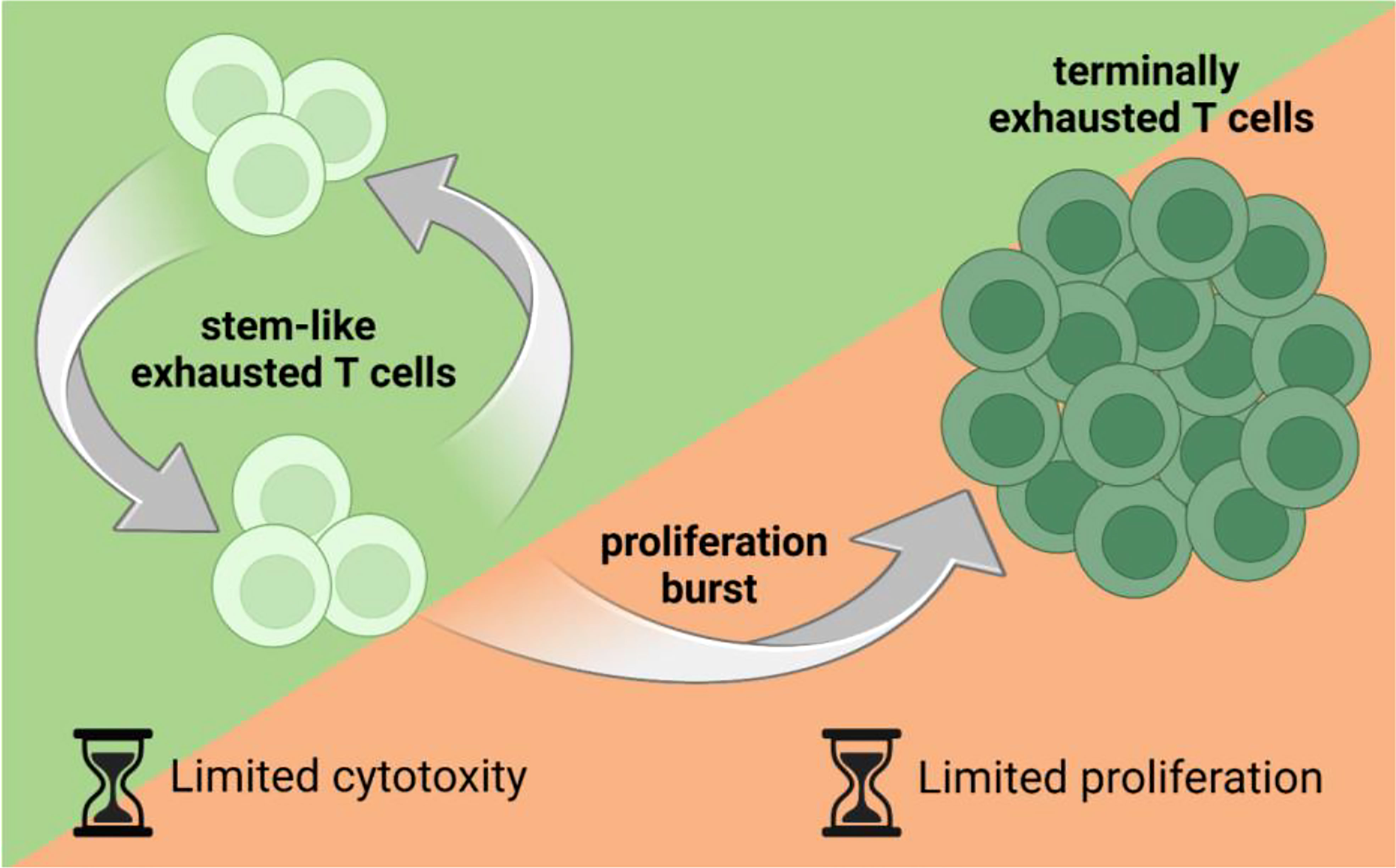
Figure 1 Stem-like, proliferative exhausted T cells produce progeny populations of exhausted T cells, including terminally exhausted T cells. Stem-like exhausted T cells have the capacity to proliferate but have been considered less cytotoxic than terminally exhausted T cells. On the other hand, terminally exhausted T cells have been considered more cytotoxic than stem-like exhausted T cells but have a very limited proliferation capacity. Figure created with BioRender.com.
Most studies investigating T cell exhaustion focus on CD8+ cytotoxic T cells, while exhaustion in CD4+ helper T cells has received less attention. The main role of CD4+ T cells is to support immune responses by activating and recruiting immune cells through cytokine production (17). However, similarly to CD8+ T cells, CD4+ T cells in cancerous conditions show elevated expression of IRs related to exhaustion, such as PD-1 and TIM-3 (18). CD4+ T cells do not typically exert cytotoxic functions, but rather promote inflammation and tumor reactivity by cytokine mediated activation of CD8+ T cells (18). In advanced melanoma murine models, development of a tumor reactive CD4+ T cell population with cytotoxic activity have been identified (19), indicating that CD4+ cells can obtain cytotoxic functions. The potential cytotoxicity of CD4+ T cells has been highlighted by Cachot et al. (20), demonstrating a CD4+ tumor-specific cytotoxic T cell population in cancer patients by mining single-cell RNA-sequencing datasets. The mechanisms behind CD4+ T cell adaptation to cancerous conditions and a possible correlation between cytotoxic CD4+ T cells and T cell exhaustion remains unclear.
Likewise, exhaustion in unconventional γδ T cell lineages have been reported but is less investigated than in CD8+ T cells. The predominant T cell type in the body is αβ T cells (here referred to simply as T cells), and these express αβ TCRs consisting of an α- and a β subunit, while γδ T cells express TCRs consisting of a γ- and a δ subunit (21). Studies provides insights in the acquirement of an exhaustion signature by γδ T cell, which has been extensively reviewed by Chen et al. (22). Furthermore, it has been reported that γδ T cells can have immunosuppressive functions, imposing immune exhaustion of antitumor αβ T cells through PD-1/PD-L1 signaling (23).
To portray the interactions between oncology and immunology, the cancer-immunity cycle has been introduced (24). Cancer cells are defined by genetic and cellular alterations, enabling the immune system to generate T cell responses for recognition and elimination of cancer cells. The cancer-immunity cycle term is used to describe the various necessary steps of T cell-cancer cell interactions, starting from the release of cancer cell antigens. The first step of generating an immune response is tumor antigen release from tumor cells (24). Tumor antigens are originally self-antigens, which makes their recognition by the immune system more challenging compared to foreign antigens such as those originating from pathogens (25). However, tumor antigens can be recognized by the immune system by distinguishing mutations. The higher the mutational burden, the higher the chance of successful recognition by the immune surveillance system (26). Antigens released from the tumor site are then trafficked to the blood stream and captured by APCs, which process and present antigens to T cells (24). Next step involves priming and activation of naïve T cells in LNs. APCs present processed tumor antigens to naïve T cells via major histocompatibility complex (MHC) molecules. Each T cell expresses TCRs, typically consisting of a α and β chain. Stochastic V(D)J gene recombination during T cell maturation results in a broad variety of TCRs, estimated to contain 107-108 unique signatures (27). Different TCRs recognize different epitopes presented by MHCs on the cell surface of surrounding cells. Binding of specific TCRs to MHC-antigen complexes, leads to activation and proliferation of T cells (24, 28). Activated T cells are trafficked to the tumor site via the blood stream guided by chemokine signaling. Chemokines are a subcategory of cytokines providing chemical signals trafficking immune cells to specific destinations (29). Infiltration of T cells to tumors is desired, since high levels of cytotoxic T cells in the TME correlates with a positive antitumor response in cancer patients (30). Via TCR specificity, T cells recognize antigens earlier activated against by APCs in LNs. Once TCRs of T cells bind antigens on cancer cells, T cells can kill cancer cells by releasing perforin and granzyme B (31). Killing of cancer cells triggers release of more antigens, leading to further immune responses, and so the cancer-immunity cycle continues.
The draining lymph node is the first site of APC-T cell interaction (32). Antitumor T cell response-efficacy is dependent on the quality of antigen presentation and possible immunosuppressive conditions of the microenvironment (32–34). Tissue-derived soluble antigens in lymphatic fluid flow into LNs via afferent lymphatic vessels (35). Antigens are captured by LN resident and migratory dendritic cells (DCs) and transported to the LN cortex and paracortex for antigen presentation to T cells (35–37). Cross-presentation of DC internalized tumor antigens to CD8+ T cells is a key step for priming antitumor immunity (38).
Presence of antigens is necessary for adaptive immune responses (39), and the continuous stimulation by tumor-derived antigens can be considered critical giving the ongoing evolution and accumulation of mutations in tumor cells (40–42). However, it has been shown that one of the main factors behind T cell exhaustion is prolonged, consistent antigen stimulation (43), indicating that the factors making robust immune responses against tumor cells possible are also the ones assumed to be dampening immune responses in form of T cell exhaustion. Antigenic peptides with a low mutational burden deliver weaker TCR signals (26), and are thus less potent to induce an immune response but might also be less potent drivers of T cell exhaustion. Weak TCR signaling combined with lack of co-stimulation rather leads to T cell anergy than exhaustion (44, 45), an extended hyporesponsive state considered a tolerance mechanism (46). Dysfunction in form of anergy is induced rapidly after antigen stimulation, while exhaustion is developed progressively over a period of weeks to months (47).
Studies provide evidence of the maintenance of a stem-like TCF-1+ CD8+ exhausted T cell reservoir in tumor-draining lymph nodes (TDLNs), and these stem-like exhausted T cells are necessary for long-term T cell responses and efficacy of immunotherapy (48). Data suggests that T cells differentiate towards a stem-like exhausted state in LNs, becoming prepared for migration towards tumor sites (48). Studies propose that activated CD8+ T cells in human TDLNs are precursors to tumor-resident stem-like CD8+ T cells (48). Prokhnevska et al. (49) used murine tumor models to reveal that tumor-specific CD8+ T cells in TDLNs were activated, but lacked an effector phenotype. When the tumor-specific stem-like CD8+ T cells migrated into the tumor, effector differentiation was driven by additional co-stimulation by APCs in the TME. This tumor-specific CD8+ T cell activation model proposes a two-step activation: initial activation in TDLNs and additional co-stimulation in the tumor, resulting in subsequent effector program acquisition (49).
Stem-like CD8+ T cells are also referred to as progenitor exhausted T cells (TPEX), and characterized to be TCF-1+, CXCR5+, PD-1int and TIM-3- (9). Lugli et al. (50) describes TPEX cells as partially memory, effector, and exhausted T cells. TPEX cells retain memory-like gene expression and preferentially localize in lymphoid tissues, but also display traits ascribed to effector-like T cells. Upon antigen stimulation, TCF-1+ T cells have shown contradictory IFN-γ production capacity. Some studies display increased IFN-γ production by TCF-1+ TPEX cells compared to TCF-1- terminally exhausted T cells (51), while other studies have shown greater production of IFN-γ by terminally exhausted T cells than TPEX cells after in vitro stimulation (9).
T cell exhaustion has been well characterized in chronic infections in lymphocytic choriomeningitis virus (LCMV) models (52). In a LCMV-model study by Im et al. (53), a distinct CXCR5+ LCMV glycoprotein 33–41 epitope (GP33)-specific CD8+ T cell population was found in the spleens of mice chronically infected with the LCMV clone 13 strain. In contrast, in mice infected with the LCMV Armstrong strain having cleared the infection, GP33-specific memory CD8+ T cells did not express CXCR5. The study showed that during early phase chronic infection, both CXCR5+ and CXCR5- CD8+ T were found in the blood, but at later stages on infection (day 30 onwards), only the CXCR5−CD8+ T cells were found in the blood (53). In the same study, the CXCR5+Tim-3− subset displayed to be TCF-1+ whereas the CXCR5−TIM-3+ cells were TCF1−. Lymphoid tissue resident T cells were characterized by TCF-1 expression, while mainly TCF-1-negative cells were found in the periphery (53).
Dammeijer et al. (54) used LNs of ovalbumin (OVA)-expressing AE17 mesothelioma tumor mouse models to analyze the frequencies and phenotype of tumor antigen-specific CD8+ T cells. The study revealed that TDLN-localized tumor-specific PD-1+CD8+ T cells are capable of effectively initiate antitumor immune responses following TDLN-targeted PD-L1-blockade. Following PD-L1-blockade, TDLN-resident T cells induced TPEX cell accumulation at the tumor site, resulting in improved tumor control. These findings support the pivotal role of progenitor exhausted T cells in LNs as an extra-tumoral source of antitumor T cell activity.
These data indicate that the exhaustion journey of T cells is initiated already in the lymph nodes, preparing for later terminal stages of exhaustion (Figure 2). The antigen density in lymph nodes might be enough to initiate exhaustion of T cells, but not enough to promote terminal differentiation of exhausted T cells. Hence, TCF-1+CD8+ TPEX cells migrating from lymph nodes to tumor sites provide a therapeutic window for maintaining a proliferative T cell population and possibly preventing differentiation of a terminal exhausted state of T cells (48, 55).
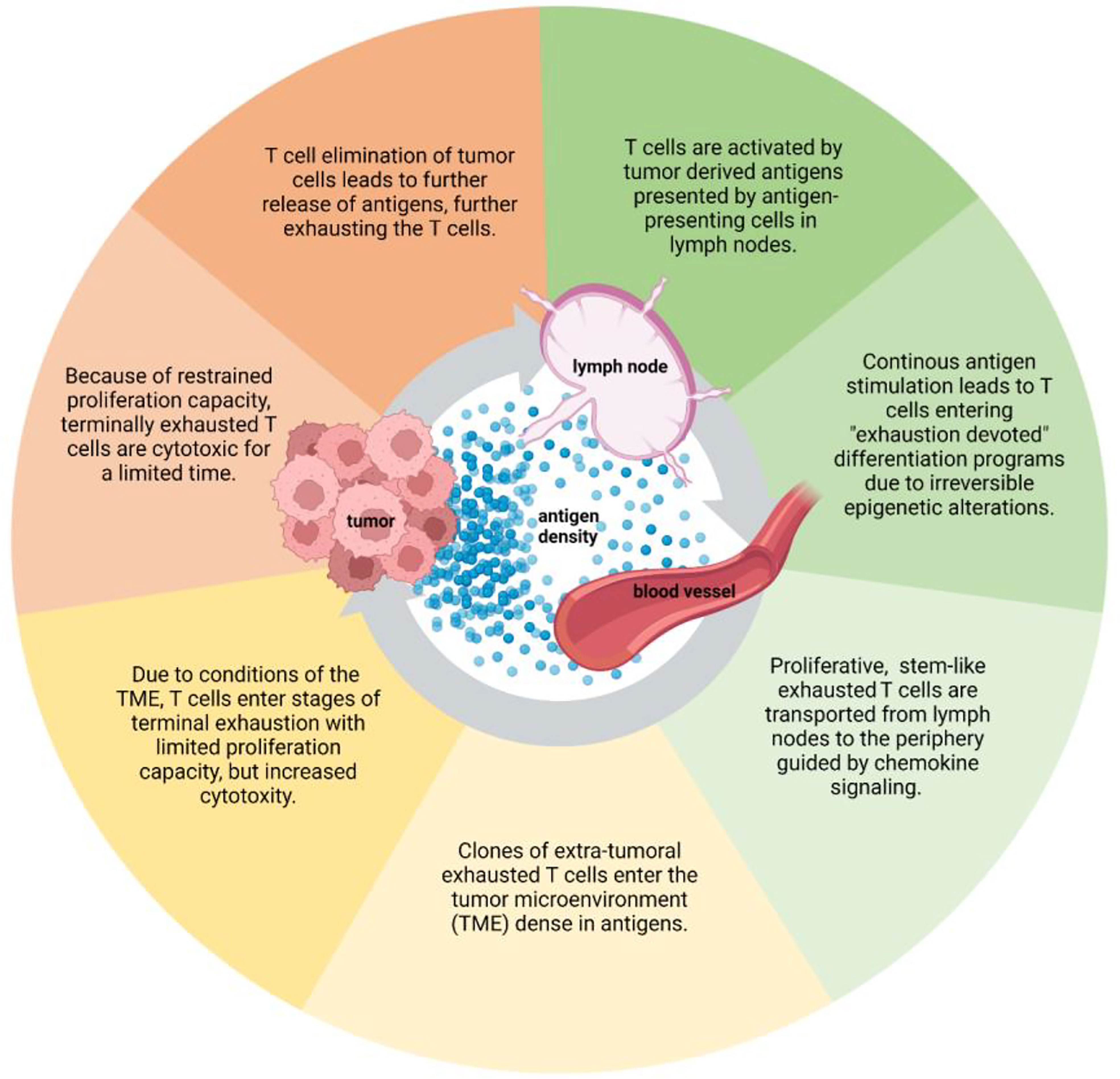
Figure 2 The development of T cell exhaustion throughout the cancer-immunity cycle. The exhaustion journey of T cells starts from tumor antigen-T cell interaction in lymph nodes. The exhaustion continues as the cancer-immunity cycles goes on, impacting the tumor cell elimination capacity of T cells. Figure created with BioRender.com.
Activated T cells are trafficked to tumor sites via the blood stream guided by chemokine signaling (Figure 2) (29). Chemokines are secreted by various cell types in the TME, including tumor cells and immune cells. Loss of CCR7, a lymph node homing chemokine receptor, allows effector T cells to migrate to non-lymphoid tissues (33), and chemokines associated with tumor homing of T cells attract T cells to the TME. CXCL9, CXCL10 and CCL5 are positively associated with immune cell infiltration to tumors, and likewise CX3CL1, CCL3, CCL4, CCL11, and CXCL11 have been associated with T cell infiltration (55). Most chemokine profiles described above are inducible by IFN-γ, which is one of the key proinflammatory molecules secreted by effector T cells. Presence of activated T cells secreting IFN-γ in the TME cause a positive feedback loop, since IFN-γ leads to production of various chemokines attracting T cells to tumors (55). T cells reaching terminal exhaustion and thus produce less IFN-γ (2, 51), could possibly interfere with the positive feedback loop and reduce T cell attraction to the TME.
T cell trafficking is facilitated by dynamic associations between T cells, endothelial cells, and adhesion molecules (33). Lymphocyte function-associated antigen-1 (LFA-1) is an adhesion molecule expressed by activated T cells (56). Binding of LFA-1 to its ligand ICAM-1 expressed by endothelial cells, APCs and tumor cells, facilitates endothelium adhesion, prolonged contact with APCs, and tumor cell binding (57). Hence, LFA-1-ICAM-1 interactions can impact the cancer-immunity cycle from beginning to end. In lymph nodes, ICAM-1 expression on APCs is essential to guide T cell migration throughout the lymph node (58), and LFA-1-ICAM-1 engagement is necessary for complete T cell activation and differentiation (59). Studies have investigated the effect of PD-1/PD-L1 immune checkpoint blockade (ICB) on adhesion molecule expression and T cell motility. In chronic infection LCMV models, exhausted T cells had reduced motility capacity, and PD-1 blockade was observed to restore T cell motility leading to increased clearance of viruses (60). Whether the same applies for PD-1/PD-L1 blockade in tumor models remains to be investigated.
Several studies provide evidence that anti-PD-1 ICB treatment drives peripheral T cell expansion and recruitment of de novo responses. A study by Nagasaki et al. (61) underlines that clonotypes in exhausted tumor infiltrating lymphocyte (TIL) populations rarely overlap with non-exhausted peripheral blood leukocytes (PBLs), indicating that TDLNs might be the primary source of newly infiltrating exhausted T cells in clusters promoted by PD-1 blockade. Similar conclusions were made by Yost et al. (62), providing evidence that both pre-existing and new CD8+ T cell clones infiltrate tumors following PD-1 blockade. Beltra et al. (10) have identified two distinct circulating states of exhausted T cells: TCF1+CD69- Texprog2 and TCF1-CD69-T-bethi Texint. The Texint population expanded upon PD-1 pathway blockade, but ultimately differentiated into a TCF1-CD69+Eomeshi terminally exhausted subset. Upon PD-L1 blockade, TCF1+CD69+ Texprog1 and TCF1-CD69+ Texterm cells increased 2.1- and 2.2-fold, while Texprog2 and Texint accumulated heavily and increased 17- and 10-fold. Results of these studies call attention to circulating exhausted T cells as the main source of expanding T cells upon ICB treatment.
Studies have described a shift from precursor to terminally exhausted T cell as T cells migrate from the lymphoid tissues to the tumor site (10). During this shift a change in transcriptional programs inducing expression alterations of transcription factors BATF, IRF4, NR4A, EOMES, NFATC1 and TOX have been identified (63–65). Especially TOX have been reported as a critical TF for exhaustion development and differentiation. TOX expression correlates with an exhausted transcriptional program of T cells, including co-expression of PD-1, TIM-3, and CD244 (66, 67). Interestingly, deletion of TOX lead to chromatin inaccessible gene regions coding for the IRs Pdcd1, Entpd1, Havcr2, Cd244 and Tigit and thus reduced IR expression. Even though TOX-deleted T cells showed reduced IR expression, they have been observed to be dysfunctional in terms of effector functions such as cytokine release (66).
Studies provide evidence of pre-existing TILs having limited reinvigoration potential following ICB treatment, and the majority of ICB responsive cells originating from outside tumors (62). Profiling of T cells in human basal and squamous cell carcinomas prior and post anti-PD-1 treatment displayed a more robust clonal expansion of CD8+ T cell with an exhausted phenotype compared to other TIL populations (62). Additionally, clonal expansion of T cells in response to immunotherapy was derived from extra-tumoral clones of T cells, and the effect was specific to T cells characterized as exhausted (62). This study demonstrates a tumor infiltration capacity of particularly exhausted T cells. However, where on the exhaustion continuum such capacity is at its most optimal remains to be investigated.
TPEX CD8+ TILs are able to control tumor growth and can respond to anti-PD-1 therapy, while terminally exhausted TILs cannot (9). As pre-existing TILs characterized as terminally exhausted have been considered resistant to ICB immunotherapy (9, 62, 68), recent studies have investigated whether these terminally exhausted T cells are involved in generation of an immunosuppressive TME (68). Vignali et al. (68) have identified intratumoral CD8+ terminally exhausted T cells with transcriptional features of CD4+Foxp3+ regulatory T (Treg) cells and show that these cells are capable of directly suppressing T cell proliferation ex vivo.
Eradication of tumor cells can be considered the final goal of the cancer-immunity cycle. Cytotoxic mechanisms of T cells rely on two distinct pathways: the perforin-granzyme-induced apoptosis pathway (granule exocytosis) and Fas/Fas ligand (FasL) pathway (death ligands) (69, 70). Out of these two pathways, granule exocytosis is considered the main pathway to eliminate cancer cells (69, 70). Perforin is a pore-forming protein facilitating the delivery of granzymes into target cells (71), and granzymes belong to a family of serine proteases known for mediating cytotoxic T cell elimination of infected cells and tumor cells (72). A study by Wu et al. (73) demonstrated defects in CD8+ TIL perforin expression in colorectal cancer (CRC), even though these cells stored the highest levels of granzyme B. Furthermore, PD-1 expression correlated with impaired perforin production, and the intact perforin expression was restricted to tumor resident T cells (73). Yan et al. (74) have identified an effector memory phenotype PD-1+CXCR1+ CD8+ T cells population able to withstand chemotherapy and expand after chemo immunotherapy with cytolytic activity, defined by granzyme B and perforin release. Hurkmans et al. (75) studied granzyme B serum levels in metastatic non-small cell lung cancer (NSCLC) patients post PD-1 blockade by Nivolumab. Lower serum levels of granzyme B correlated with worse clinical outcome, and interestingly, serum levels of granzyme B positively correlated with peripheral abundance of T cell populations expressing PD-1 and TIM-3. Together these findings show sustained and elevated granzyme B release capacity of exhausted T cells, while perforin production remains questionable. It is widely assumed that both perforin and granzyme B are necessary for efficient cytotoxicity of T cells. Interestingly, perforin knockout OT-I T cells have been shown to efficiently kill MC38Ova and B16Ova cells in prolonged presence (>18 hours) of antigens (76), indicating cytolytic activity of T cells in absence of perforin.
The Fas/FasL pathway plays an essential role in regulating apoptosis and T cell activation (77). The Fas/FasL signaling in exhausted antigen-specific CD8+ T cells during tumor immune response have been studied in C57BL/6 mice inoculated with EG.7. The study showed that the number of activated antigen-specific CD8+ T cells decreased via apoptosis during prolonged tumor immune responses, but the number of T cells in FasL-dysfunctional gld mice were higher than in control mice (78). Thus, the Fas/FasL signaling pathway is critical for survival of exhausted CD8+ T cells during tumor immune response. In chronic infection LCMV mice models, the FasL mRNA expression is reported to be substantially elevated in exhausted CD8+ T cells compared to naive, effector, and memory CD8+ T cells, and the Fas/FasL pathway is discussed to provide alternate cytolytic mechanisms during chronic infection (1). In CD8+ T cells specific for chronic persistent virus (HIV), PD-1 expression has been observed to be associated with spontaneous and Fas-induced apoptosis (79). Studies provide insights of an altered Fas/FasL pathway in exhausted T cells, but the precise outcomes of these alterations in cancerous conditions remain to be investigated.
Studies describe various ICAM-expression levels by exhausted T cells, and its impact on tumor eradication remains unclear. LFA-1-ICAM-1 binding enables T cell-tumor cell binding prior to contact killing by cytotoxic elimination. The importance of proper immune synapse formation prior to target cell elimination has been highlighted in cancer models of hematological malignancies. Ramsay et al. (80) show that F-actin polymerization was suppressed upon T cell chronic lymphocytic leukemia (CLL)-B cell encounter, leading to impaired immune synapse formation, and hindered antitumor activity. Studies show that loss of ICAM-1 expression as a possible consequence of reduced proinflammatory cytokine production (81, 82), could lead to elevated granzyme B release (83). Loss of ICAM-1 expression has been observed to result in elevated levels of IFN-γ and granzyme B, as well as enhanced cytotoxicity (83). T cell activation without classical immune synapse formation but via TCR microclusters has been reported (84, 85), providing evidence of maintained T cell activation and effector functions regardless expression levels of LFA-1 and ICAM-1.
To adapt to hypoxic conditions in the TME, T cells undergo a metabolic switch upon activation. Metabolic reprogramming is essential to meet the increasing energy demand necessary for effector T cell functions (86). Naïve T cells exhibit lower energy demands than effector T cells and rely on oxidative phosphorylation (OXPHOS) derived ATP as the main source of energy (87). Effector T cells utilize aerobic glycolysis as their primary metabolic program, increasing the glucose uptake and oxidative consumption (88). T cells can also use fatty acids as an energy source, and the use of fatty acid β-oxidation (FAO) pathway is linked to memory T cell differentiation (89). Furthermore, FAO is an important pathway of survival in metabolically stressed cells (89), and Tregs generally rely upon FAO for their metabolic needs (90). The metabolic plasticity and regulation of T cell exhaustion has been extensively reviewed by Li et al. (91), focusing on T cell exhaustion in chronic infections. FAO and OXPHOS are discussed to be the primary energy sources for TPEX, while terminally exhausted T cells rely on glycolytic metabolism, but having reduced glucose uptake and an inability to effectively utilize OXPHOS to provide energy (91).
The high consumption of glucose by cancer cells creates glucose derivatized, acidic extracellular conditions in the TME (92), caused by lactate accumulation (93) known to suppress the proliferation and effector functions of cytotoxic T cells (94). Effector T cells exhibit decreased mitochondrial respiratory activity, and studies show exhausted T cells exhibiting suppressed glycolysis and mitochondrial respiration, causing poor metabolic fitness and exhaustion (95, 96). A study by Chang et al. (88) displays the role of aerobic glycolysis as a metabolically regulated signaling mechanism, and activated T cells blocked from aerobic glycolysis have a compromised ability to produce IFN-γ (88), possible explaining decreased IFN-γ production of terminally exhausted T cells in the TME. A study by Scharping et al. (97) point out that progressive loss of mitochondrial function and mass correlates with decreased cytokine production in solid tumor infiltrating T cells. Expression of the glucose transporter-1 (Glut1) has been observed to increase in PD-1hi exhausted T cells in hepatitis B virus (HBV) chronic infection, and these cells were dependent on glucose supplies. In contrast, non-chronic infection cytomegalovirus (CMV)-specific T cells that could utilize OXPHOS in the absence of glucose to optimize their energy supply (98). Similar metabolic deficiencies in exhausted T cells have not yet been investigated in tumor models.
The metabolism of T cells has been studied in murine melanoma models, and a positive correlation between immune checkpoints expression levels on CD8+ T cells and total cholesterol content in the cells has been observed. Lung B16 tumor-infiltrating PD-1high2B4high CD8+ T cells had significantly higher cholesterol content than PD-1med/low2B4 med/lowCD8+ T cells. The same pattern was observed in LN and spleen resident T cells, although these cells had significantly lower cholesterol content than the tumor-infiltrating T cells (99). Cholesterol is a tumor metabolic byproduct inducing metabolic stress in T cells, and the study demonstrated that cholesterol in the TME induces CD8+ T cell exhaustion via activation of the endoplasmic reticulum (ER)-stress sensor XBP1. XBP1 can directly increase PD-1, TIM-3, and LAG-3 expression, thus immunosuppression of T cells (99). Patsoukis et al. (100) discovered the incapability of activated T cells to engage in glycolysis upon PD-1 ligation, and their studies show that PD-1 ligation increase the FAO rate. FAO supports T cell persistence, but not necessarily T cell function. Altogether these findings support the theory of an altered metabolic program in exhausted T cells, causing altered effector functions, but precise locations and timelines of metabolic switches in exhausted T cells during the cancer-immunity cycle remain to be explored.
T cell exhaustion has been discussed to be an evolutionarily conserved adaptation to chronic antigen stimulation, important for restricting autoreactivity and immunopathology. RNA-sequencing and ATAC-sequencing data generated from circulating T cell subsets from healthy donors show that circulating PD1+ CD39+ T cells had the highest enrichment of tumor exhausted T cell genes, providing transcriptional evidence of recent proliferation and characteristics of intermediate exhausted T cells (101). This study provides evidence of circulating exhausted T cell populations present also in healthy donors, suggesting that PD-1+CD39+CD8+ exhausted T cells are not restricted to cancer or chronic infections (101). Their existence might point towards an alternative fate to deletion for autoreactive T cells, hence enabling a broader diversity of T cell populations, but their precise role in healthy donors remains to be explored. Galletti et al. (102) have identified two different subsets of human stem-like CD8+ memory T cell progenitors in healthy donors: CCR7+PD-1−TIGIT− stem-like T (TSTEM) cells and CCR7+ PD-1+TIGIT+ T progenitor exhausted T (TPEX) cells. The TPEX population gave rise to a cell linage with reduced functionality compared to TSTEM cells. Interestingly, the TPEX cells had memory-like features, suggesting that exhaustion-like and memory-like characteristics could coexist in healthy individuals. This further indicates that T cell exhaustion could be a functional adaption to persistent antigen stimulation aimed at minimizing the risk of immunopathology, and simultaneously maintain a memory-like population. The role of exhaustion in limiting immunopathology is underlined in a study by McKinney et al. (103), displaying reduced clinical autoimmunity as T cell exhaustion gets more severe.
Expression of members of the transcription factor (TF) family NR4A is regulated downstream of TCR signaling (104). Beltra et al. (10) have defined a four-cell-stage developmental framework for exhausted T cells. In studies combining motif enrichment analysis with RNA expression, NR4A2 was predicted to be the most enriched TF in terminally exhausted T cells. TCR stimulation induces expression of the NR4A family (105), indicating that terminally differentiated exhausted T cells are subjects of heavy TCR signaling. Nr4a triple knockout (TKO) in CAR T cells have been shown to prolong the survival of tumor-bearing mice, and NR4A inhibition has been pointed out as a promising cancer immunotherapy strategy (14). Expression of mRNAs encoding effector proteins such as granzymes, TNF and IL-2Rα were increased in Nr4a TKO TILs (14), indicating that dense antigen environments and heavy TCR signaling eventually leads to decreased cytotoxicity of T cells. Together these findings point out that prolonged antigen-TCR stimulation indeed leads to a terminally exhausted state of T cells and eventual loss of cytotoxicity, limiting the risk of immunopathology but also limiting tumor cell clearance. These studies draw attention to the importance of eliminating tumor cells within an exhaustion restricted time frame.
Studies suggest that varying tumor types might generate distinct exhaustion patterns in T cells. Woroniecka et al. (106) performed a study with mouse-implanted SMA-560 malignant glioma, CT2A malignant glioma, E0771 breast medullary adenocarcinoma, B16F10 melanoma, and Lewis Lung Carcinoma (LLC), comparing exhaustion patterns according to cell line. They found distinct functional programs of exhausted T cells depending on tumor type, displaying variations in proinflammatory cytokine production and expression of IRs. Tumors causing greater functional impairment of T cells showed higher levels of alternative IRs such as TIGIT, CD39 and 2B4, indicating that greater functional impairment indeed provides opportunities for alternative immunotherapy strategies. Distinct exhaustion programs according to tumor type explains varying response to PD-1 pathway inhibitors, which have shown greatest results in melanoma, NSCLC, RCC, and metastatic bladder cancer (107). These concluding remarks point out the need to understand not only how T cell exhaustion impacts the cancer-immunity cycle, but the varying T cell exhaustion cycles according to specific tumor types. Distinct exhaustion programs according to tumor type calls attention to the dynamic features of T cell exhaustion, adapting to specific tumor conditions.
As the cancer-immunity cycle points out, the balance between stimulating factors and inhibitors is critical (24). Tumor cells can exploit immune escape mechanisms such as loss of antigenicity, loss of immunogenicity or generation of immunosuppressive microenvironments, to avoid elimination by the immune system (108). In a similar manner, T cell exhaustion could be regarded as an alternative mechanism of the immune system, a tool to control cancer. Exhausted T cells aren’t suitable tools for all types of cancer types and conditions, hence having a contrary role, in some circumstances acting as an asset and in some as a liability.
Immune checkpoint blockade (ICB) therapies such as antibodies targeting PD-1, PD-L1, CTLA-4 and LAG-3 aim to activate T cells by interrupting inhibitory signals (Table 1). Today, the focus area of controlling T cell exhaustion lies within preventing exhaustion and reactivating already exhausted T cells (131). Combination with traditional ICBs PD-1, PD-L1 and CTLA-4 and second-generation checkpoint targets such as LAG-3, TIM-3 and TIGIT are being investigated (131). Since the first ICB therapy was approved in 2011, the US Food and Drug Administration (FDA) have issued over 65 approvals for 20 types of neoplasms (132). Studies from the past decade provide evidence of checkpoint therapies being most efficient when administering in an early state of exhaustion (107). Despite increased knowledge in targeting T cell exhaustion and the large number of FDA approvals, only 20.2% of patients receiving ICBs achieve objective response, and out of these only 13% of patients achieve durable responses for multiple years (132). To improve patient responses, combination therapies with ICBs can be utilized. FDA approved therapies to be combined with ICBs include chemotherapy, radiation, anti-angiogenic agents, cancer vaccines and adoptive cell therapies (ACT) (133).
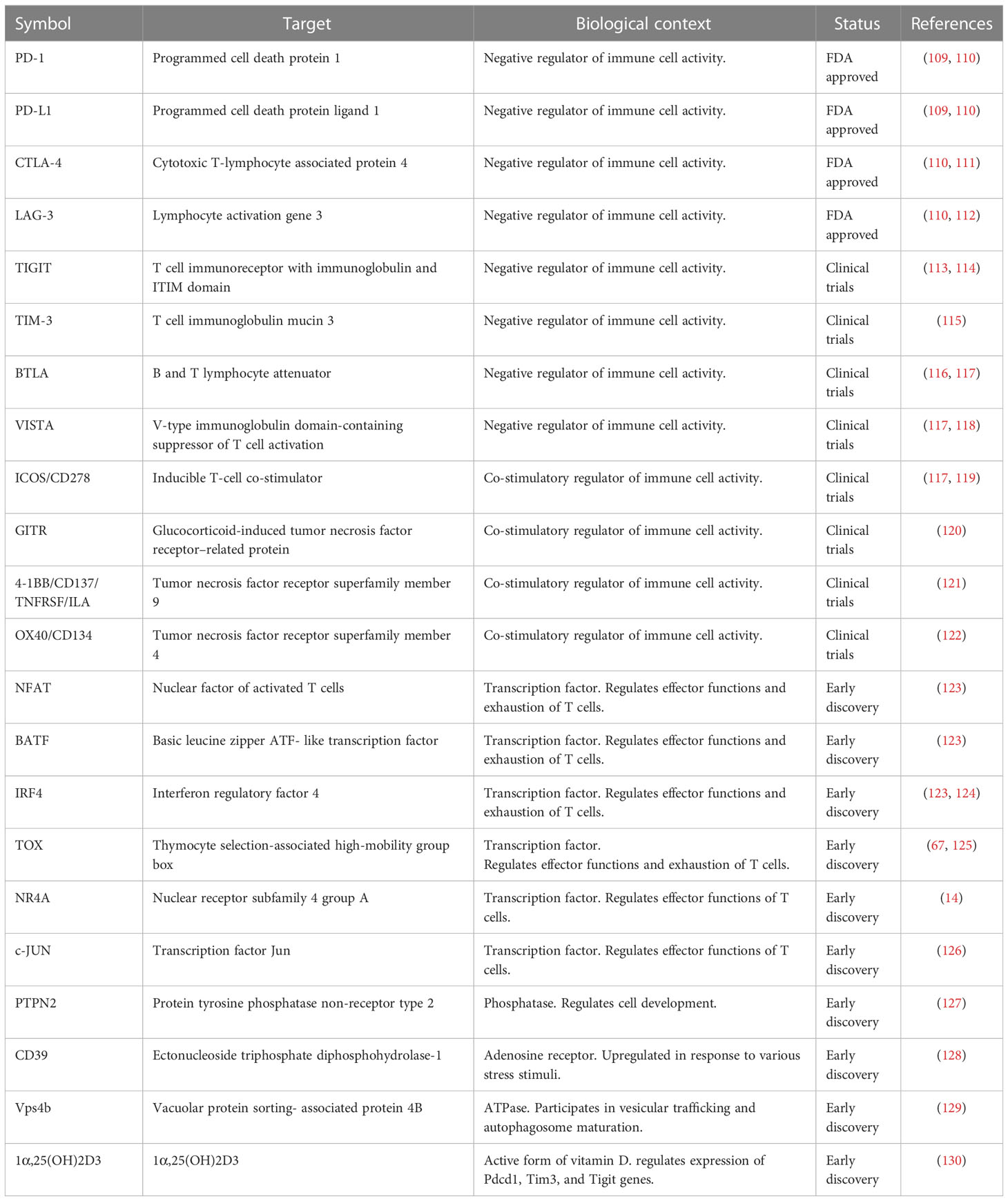
Table 1 Current and potential future immunotherapeutic T cell exhaustion targets.
Some tumors have been shown particularly challenging to target due to T cell exhaustion. Chimeric antigen receptor (CAR) T cells have been proven to be a potent tool for targeting blood cancer, but the dense antigen environment in solid tumor often generates severe exhaustion of these cells, causing restricted efficacy. Studies have shown that combination therapies with ICBs and CAR T cells can improve T cell expansion and tumor reactivity (134).
To further improve CAR T cell efficacy, studies have targeted the exhaustion inducing transcription factor protein TOX (135). TOX is upregulated and co-expressed with PD-1, TIM-3, and CD244 in non-Hodgkin lymphoma (136), multiple myeloma (137), and in acute myeloid leukemia patients (138). Depletion of the Tox gene in CAR T cells proved to improve persistence and tumor reactivity in tumor inoculated mice (139). Recent studies point out the transcription factor MYB as an orchestrator of T cell exhaustion. MYB mediates differentiation of a CD62L+ TPEX cell stem-like population. Upon PD-1 ICB treatment this population is responsible for the proliferative burst, and thus, MYB could play a central role in therapeutic checkpoint blockade success (140). Other transcriptions factors found to promote exhaustion are NFAT and NR4A, while BATF attenuates exhaustion (141). The transcription factor IKZF3 is a repressor of IL-2 (142), and Hay et al. (2022) used an in vitro model to show that exhausted T cells partially can be rescued by treatment with Lenalidomide, an IKZF3 small molecule inhibitor, either as a single treatment or in combination with ICB treatment.
As exhausted T cells display dysregulation in mitochondrial function, strategies for improving mitochondrial biogenesis have raised an interest. In B16OVA tumor microenvironments, PGC1α overexpressing OT-I T cells, leading to increased mitochondrial mass and OXPHOS, are enriched (97). These cells displayed enhanced tumor efficacy, demonstrating a preservation of T cell function in the TME by reprogramming these cells to favor mitochondrial biogenesis. Metabolic stress, such as hypoxia and glucose deprivation in the TME of solid tumors, have shown to lower tumor immunogenicity (143). To improve responses to ICB, signaling pathways such as the PI3K/AKT pathway, could be targeted to restore tumor cell recognition by T cells (143). Thus, addressing metabolic and signaling pathways hold promising potential to improve immunotherapy responses.
The posttranscriptional mechanisms regulating exhaustion of T cells remains poorly understood. Using Affymetrix miR arrays, the microRNA (miR) expression in isolated LCMV DbGP33–41–specific CD8+ T cells have been examined (144). MiR-29a was identified as an exhaustion attenuating molecule in chronic infection and could also play a role in decreasing exhaustion in cancer. Data suggests that miR-29a attenuates TCR signaling pathways involved in driving exhaustion and thus, enhanced expression of miR-29a favor a durable T memory cell-like rather than exhausted T cell-like differentiation in conditions of persistent antigen stimulation (144). Hence, targeting microRNAs in combination therapies could represent novel solutions for improved immunotherapy efficacy.
The development and maintenance of exhausted T cells can be controlled by manipulation of molecular pathways, by utilizing methods such as CRISPR gene editing. Using CRISPR, synthetic DNA sequences can be inserted to generate next-generation autologous T cell therapies (145), and there is an interest in modifying T cell exhaustion promotors such as Tox for augmented tumor control (131). Other proposed CRISPR targets attracting attention by biotech and pharmaceutical companies are PTPN2, NR4A and c-JUN (126, 127, 131). Unlike most molecules and genes of interested currently being targeted to prevent T cell exhaustion, deletion of Ptpn2 has shown to scale up the population of terminally exhausted T cells whilst preserving the population of self-renewable progenitor exhausted T cells (127).
To increase and prolong patient response and survival, bispecific antibodies (BsAbs) have been introduced to the field of immunotherapy. BsAbs can bind two distinct targets on the same cell at the same time, enabling additive effects and novel therapeutic approaches (146). Dual BsAbs targets include immune checkpoints, signaling pathways, tumor associated antigens and cytokines (147). OSE Immunotherapeutics have developed a bispecific antibody checkpoint inhibitor BiCKI® platform, with the BiCKI®-IL-7 program targeting PD-1 and simultaneously releasing IL-7, promoting TCF-1+ stem-like T cell expansion (148). In September 2022, Roche announced taking over Good Therapeutics’ PD1-regulated IL-2 receptor agonist program (149). Similarly to IL-7, IL-2 have potential immunostimulating and antineoplastic functions, but unregulated IL-2 release can cause toxicity. Linkage of anti-PD-1 to an alternative form of IL-2 (IL-2v) enables precise IL-2 stimulation, thus minimizing side effects and toxicity (150).
Throughout the cancer-immunity cycle, T cells are subjects of a variety of factors such as prolonged antigen stimulation and altered conditions of the TME, unavoidably shifting the T cells towards an exhausted state (7). Studies discuss the possibility of separate differentiation programs of T cells, one initiating the “classical” effector/memory T cells pathway and the other one initiating an exhaustion program of T cells (102). Insights display the close integration of the cancer-immunity cycle and T cell exhaustion, pointing out the need of a deeper understanding of the T cell exhaustion cycle to enable discovery of novel immunotherapies.
T cell exhaustion has potential benefits, but also consequences. A stem-like TCF-1+ exhausted T cell state maintain a memory-like population with proliferation capacity (10, 11). Stem-like TCF-1+ T cells giving rise to terminally exhausted cytotoxic T cells could serve as a type of safety mechanism. Excessive antigen density in the TME leading to short-term cytotoxic states restrain cytotoxicity to tumors sites and thus limits immunopathology in healthy tissues. Since terminally exhausted T cells have reduced proliferation capacity (10, 11), the damage caused by this population is limited. On the other hand, high cytotoxicity is desired for tumor elimination, and thus short-term cytotoxicity and lacking proliferation capacity simultaneously serves as a liability. Restricted rounds of proliferation by stem-like exhausted T cells and restricted periods of cytotoxicity creates a time frame of tumor cell clearance. If events in the exhaustion cycle exceed these time frames, the consequences of T cell exhaustion could result in tumor immune escape, rather than tumor elimination. To fully utilize the potential assets of exhausted T cells in terms of immunotherapies, the master mechanisms and regulators behind T cell exhaustion needs to be further elucidated. Nevertheless, it might be useful considering T cell exhaustion as a functional adaptation rather than simply a dysfunction.
AB performed the literature review and wrote the manuscript. RL, AA, and AT provided critical discussion, revised, and approved the manuscript. AT conceived the review article.
RL was supported by the Academy of Finland (AoF) grants 329277, 331793 and 31444 and by grants from the Sigrid Jusélius Foundation; the Jane and Aatos Erkko Foundation, the Finnish Cancer Foundation and InFLAMES Flagship Programme of the Academy of Finland (decision number: 337530).
Authors AA and AT were employed by company Orion Corporation. Author AB was affiliated with Orion Corporation at the time of study.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of Cd8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27(4):670–84. doi: 10.1016/j.immuni.2007.09.006
3. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T Cell dysfunction and exhaustion in cancer. Front Cell Dev Biol (2020) 8:17. doi: 10.3389/fcell.2020.00017
4. Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, et al. Epigenetic signature of pd-1+ Tcf1+ Cd8 T cells that act as resource cells during chronic viral infection and respond to pd-1 blockade. Proc Natl Acad Sci U.S.A. (2019) 116(28):14113–8. doi: 10.1073/pnas.1903520116
5. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354(6316):1165–9. doi: 10.1126/science.aae0491
6. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by pd-1 blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
7. McLane LM, Abdel-Hakeem MS, Wherry EJ. Cd8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol (2019) 37(1):457–95. doi: 10.1146/annurev-immunol-041015-055318
8. Fitzgerald B, Connolly KA, Cui C, Fagerberg E, Mariuzza DL, Hornick NI, et al. A mouse model for the study of anti-tumor T cell responses in kras-driven lung adenocarcinoma. Cell Rep Methods (2021) 1(5). doi: 10.1016/j.crmeth.2021.100080
9. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted Cd8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
10. Beltra J-C, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted Cd8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity (2020) 52(5):825–41.e8. doi: 10.1016/j.immuni.2020.04.014
11. Wang D, Fang J, Wen S, Li Q, Wang J, Yang L, et al. A comprehensive profile of Tcf1(+) progenitor and Tcf1(-) terminally exhausted pd-1(+)Cd8(+) T cells in head and neck squamous cell carcinoma: Implications for prognosis and immunotherapy. Int J Oral Sci (2022) 14(1):8. doi: 10.1038/s41368-022-00160-w
12. Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, et al. An hmg-Box-Containing T-cell factor required for thymocyte differentiation. Nature (1995) 374(6517):70–4. doi: 10.1038/374070a0
13. Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH, et al. Cd4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J Exp Med (2017) 214(1):39–47. doi: 10.1084/jem.20161046
14. Chen J, López-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. Nr4a transcription factors limit car T cell function in solid tumours. Nature (2019) 567(7749):530–4. doi: 10.1038/s41586-019-0985-x
15. Codarri Deak L, Nicolini V, Hashimoto M, Karagianni M, Schwalie PC, Lauener L, et al. Pd-1-Cis il-2r agonism yields better effectors from stem-like Cd8+ T cells. Nature (2022) 610(7930):161–72. doi: 10.1038/s41586-022-05192-0
16. Kong W, Dimitri A, Wang W, Jung I-Y, Ott CJ, Fasolino M, et al. Bet bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J Clin Invest (2021) 131(16):1–16. doi: 10.1172/JCI145459
17. Luckheeram RV, Zhou R, Verma AD, Xia B. Cd4+ T cells: Differentiation and functions. Clin Dev Immunol (2012) 2012:1–12. doi: 10.1155/2012/925135
18. Yuan L, Xu B, Yuan P, Zhou J, Qin P, Han L, et al. Tumor-infiltrating Cd4+ T cells in patients with gastric cancer. Cancer Cell Int (2017) 17(1). doi: 10.1186/s12935-017-0489-4
19. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive Cd4+ T cells develop cytotoxic activity and eradicate Large established melanoma after transfer into lymphopenic hosts. J Exp Med (2010) 207(3):637–50. doi: 10.1084/jem.20091918
20. Cachot A, Bilous M, Liu Y-C, Li X, Saillard M, Cenerenti M, et al. Tumor-specific cytolytic Cd4 T cells mediate immunity against human cancer. Sci Adv (2021) 7(9):eabe3348. doi: 10.1126/sciadv.abe3348
21. Morath A, Schamel WW. Αβ and Γδ T cell receptors: Similar but different. J Leukoc Biol (2020) 107(6):1045–55. doi: 10.1002/jlb.2mr1219-233r
22. Chen D, Guo Y, Jiang J, Wu P, Zhang T, Wei Q, et al. Γδ T cell exhaustion: Opportunities for intervention. J Leukoc Biol (2022) 112(6):1669–76. doi: 10.1002/jlb.5mr0722-777r
23. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. Γδ T cells support pancreatic oncogenesis by restraining Αβ T cell activation. Cell (2016) 166(6):1485–99.e15. doi: 10.1016/j.cell.2016.07.046
24. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
25. Houghton AN, Guevara-Patiño JA. Immune recognition of self in immunity against cancer. J Clin Invest (2004) 114(4):468–71. doi: 10.1172/jci22685
26. Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell (2021) 184(3):596–614.e14. doi: 10.1016/j.cell.2021.01.002
27. Turner SJ, La Gruta NL, Kedzierska K, Thomas PG, Doherty PC. Functional implications of T cell receptor diversity. Curr Opin Immunol (2009) 21(3):286–90. doi: 10.1016/j.coi.2009.05.004
28. Chandrasekaran S, King M. Microenvironment of tumor-draining lymph nodes: Opportunities for liposome-based targeted therapy. Int J Mol Sci (2014) 15(11):20209–39. doi: 10.3390/ijms151120209
29. Zhang Y, Guan XY, Jiang P. Cytokine and chemokine signals of T-cell exclusion in tumors. Front Immunol (2020) 11:594609. doi: 10.3389/fimmu.2020.594609
30. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic Cd8+ T cells in cancer and cancer immunotherapy. Br J Cancer (2021) 124(2):359–67. doi: 10.1038/s41416-020-01048-4
31. Yost KE, Chang HY, Satpathy AT. Recruiting T cells in cancer immunotherapy. Science (2021) 372(6538):130–1. doi: 10.1126/science.abd1329
32. Loo CP, Nelson NA, Lane RS, Booth JL, Loprinzi Hardin SC, Thomas A, et al. Lymphatic vessels balance viral dissemination and immune activation following cutaneous viral infection. Cell Rep (2017) 20(13):3176–87. doi: 10.1016/j.celrep.2017.09.006
33. du Bois H, Heim TA, Lund AW. Tumor-draining lymph nodes: At the crossroads of metastasis and immunity. Sci Immunol (2021) 6(63):eabg3551. doi: 10.1126/sciimmunol.abg3551
34. Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Invest (2016) 126(9):3389–402. doi: 10.1172/jci79434
35. Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity (2015) 42(1):172–85. doi: 10.1016/j.immuni.2014.12.024
36. Woodruff MC, Turley SJ. Chemokine 'Grooming' by clecs directs dc migration. Nat Immunol (2014) 15(7):595–6. doi: 10.1038/ni.2926
37. Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, et al. Cd169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity (2011) 34(1):85–95. doi: 10.1016/j.immuni.2010.12.011
38. Kanda Y, Okazaki T, Katakai T. Motility dynamics of T cells in tumor-draining lymph nodes: A rational indicator of antitumor response and immune checkpoint blockade. Cancers (Basel) (2021) 13(18). doi: 10.3390/cancers13184616
39. Lee SC, Srivastava RM, López-Albaitero A, Ferrone S, Ferris RL. Natural killer (Nk): Dendritic cell (Dc) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res (2011) 50(2-3):248–54. doi: 10.1007/s12026-011-8231-0
40. Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res (1974) 34(9):2311–21.
41. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature (2009) 458(7239):719–24. doi: 10.1038/nature07943
42. Fonseca C, Dranoff G. Capitalizing on the immunogenicity of dying tumor cells. Clin Cancer Res (2008) 14(6):1603–8. doi: 10.1158/1078-0432.Ccr-07-2245
43. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
44. Shah K, Al-Haidari A, Sun J, Kazi JU. T Cell receptor (Tcr) signaling in health and disease. Signal Transduct Target Ther (2021) 6(1):412. doi: 10.1038/s41392-021-00823-w
45. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T Cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25(2):214–21. doi: 10.1016/j.coi.2012.12.003
46. Schwartz RH. T Cell anergy. Annu Rev Immunol (2003) 21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110
47. Schietinger A, Greenberg PD. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol (2014) 35(2):51–60. doi: 10.1016/j.it.2013.10.001
48. Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, et al. A reservoir of stem-like Cd8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol (2021) 6(64):eabg7836. doi: 10.1126/sciimmunol.abg7836
49. Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, et al. Cd8(+) T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity (2023) 56(1):107–24.e5. doi: 10.1016/j.immuni.2022.12.002
50. Lugli E, Galletti G, Boi SK, Youngblood BA. Stem, effector, and hybrid states of memory Cd8(+) T cells. Trends Immunol (2020) 41(1):17–28. doi: 10.1016/j.it.2019.11.004
51. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell factor 1-expressing memory-like Cd8(+) T cells sustain the immune response to chronic viral infections. Immunity (2016) 45(2):415–27. doi: 10.1016/j.immuni.2016.07.021
52. Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature (1993) 362(6422):758–61. doi: 10.1038/362758a0
53. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining Cd8+ T cells that provide the proliferative burst after pd-1 therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
54. Dammeijer F, Van Gulijk M, Mulder EE, Lukkes M, Klaase L, Van Den Bosch T, et al. The pd-1/Pd-L1-Checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell (2020) 38(5):685–700.e8. doi: 10.1016/j.ccell.2020.09.001
55. van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: A roadmap for T cells. Trends Cancer (2017) 3(11):797–808. doi: 10.1016/j.trecan.2017.09.006
56. Varga G, Nippe N, Balkow S, Peters T, Wild MK, Seeliger S, et al. Lfa-1 contributes to signal I of T-cell activation and to the production of T(H)1 cytokines. J Invest Dermatol (2010) 130(4):1005–12. doi: 10.1038/jid.2009.398
57. Walling BL, Kim M. Lfa-1 in T cell migration and differentiation. Front Immunol (2018) 9:952. doi: 10.3389/fimmu.2018.00952
58. Petruzzelli L, Maduzia L, Springer TA. Differential requirements for lfa-1 binding to icam-1 and lfa-1-Mediated cell aggregation. J Immunol (1998) 160(9):4208–16.
59. Verma NK, Fazil MH, Ong ST, Chalasani ML, Low JH, Kottaiswamy A, et al. Lfa-1/Icam-1 ligation in human T cells promotes Th1 polarization through a Gsk3β signaling-dependent notch pathway. J Immunol (2016) 197(1):108–18. doi: 10.4049/jimmunol.1501264
60. Zinselmeyer BH, Heydari S, Sacristán C, Nayak D, Cammer M, Herz J, et al. Pd-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med (2013) 210(4):757–74. doi: 10.1084/jem.20121416
61. Nagasaki J, Inozume T, Sax N, Ariyasu R, Ishikawa M, Yamashita K, et al. Pd-1 blockade therapy promotes infiltration of tumor-attacking exhausted T cell clonotypes. Cell Rep (2022) 38(5):110331. doi: 10.1016/j.celrep.2022.110331
62. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following pd-1 blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
63. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor Irf4 promotes Cd8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity (2017) 47(6):1129–41.e5. doi: 10.1016/j.immuni.2017.11.021
64. Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor nfat promotes exhaustion of activated Cd8⁺ T cells. Immunity (2015) 42(2):265–78. doi: 10.1016/j.immuni.2015.01.006
65. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. Tox reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature (2019) 571(7764):265–9. doi: 10.1038/s41586-019-1326-9
66. Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. Tox is a critical regulator of tumour-specific T cell differentiation. Nature (2019) 571(7764):270–4. doi: 10.1038/s41586-019-1324-y
67. Liang C, Huang S, Zhao Y, Chen S, Li Y. Tox as a potential target for immunotherapy in lymphocytic malignancies. biomark Res (2021) 9(1):20. doi: 10.1186/s40364-021-00275-y
68. Vignali PDA, DePeaux K, Watson MJ, Ye C, Ford BR, Lontos K, et al. Hypoxia drives Cd39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat Immunol (2023) 24(2):267–79. doi: 10.1038/s41590-022-01379-9
69. Trapani JA, Smyth MJ. Functional significance of the Perforin/Granzyme cell death pathway. Nat Rev Immunol (2002) 2(10):735–47. doi: 10.1038/nri911
70. Martínez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res (2015) 21(22):5047–56. doi: 10.1158/1078-0432.Ccr-15-0685
71. Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: Structure, function, and role in human immunopathology. Immunol Rev (2010) 235(1):35–54. doi: 10.1111/j.0105-2896.2010.00896.x
72. Grossman WJ, Revell PA, Lu ZH, Johnson H, Bredemeyer AJ, Ley TJ. The orphan granzymes of humans and mice. Curr Opin Immunol (2003) 15(5):544–52. doi: 10.1016/s0952-7915(03)00099-2
73. Wu X, Zhang H, Xing Q, Cui J, Li J, Li Y, et al. Pd-1(+) Cd8(+) T cells are exhausted in tumours and functional in draining lymph nodes of colorectal cancer patients. Br J Cancer (2014) 111(7):1391–9. doi: 10.1038/bjc.2014.416
74. Yan Y, Cao S, Liu X, Harrington SM, Bindeman WE, Adjei AA, et al. Cx3cr1 identifies pd-1 therapy-responsive Cd8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight (2018) 3(8). doi: 10.1172/jci.insight.97828
75. Hurkmans DP, Basak EA, Schepers N, Oomen-De Hoop E, van der Leest CH, El Bouazzaoui S, et al. Granzyme b is correlated with clinical outcome after pd-1 blockade in patients with stage iv non-Small-Cell lung cancer. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2020-000586
76. Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of tnf sensitivity. Sci Immunol (2018) 3(23). doi: 10.1126/sciimmunol.aar3451
77. Volpe E, Sambucci M, Battistini L, Borsellino G. Fas-fas ligand: Checkpoint of T cell functions in multiple sclerosis. Front Immunol (2016) 7:382. doi: 10.3389/fimmu.2016.00382
78. Yajima T, Hoshino K, Muranushi R, Mogi A, Onozato R, Yamaki E, et al. Fas/Fasl signaling is critical for the survival of exhausted antigen-specific Cd8(+) T cells during tumor immune response. Mol Immunol (2019) 107:97–105. doi: 10.1016/j.molimm.2019.01.014
79. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. Pd-1 is a regulator of virus-specific Cd8+ T cell survival in hiv infection. J Exp Med (2006) 203(10):2281–92. doi: 10.1084/jem.20061496
80. Ramsay AG, Johnson AJ, Lee AM, Gorgün G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest (2008) 118(7):2427–37. doi: 10.1172/JCI35017
81. Lim W-S, Gan M-S-Y, Ong M-H-L, Chew C-H. Lauric acid abolishes interferon-gamma (Ifn-Γ)-Induction of intercellular adhesion molecule-1 (Icam-1) and vascular cell adhesion molecule-1 (Vcam-1) expression in human macrophages. Asian Pac J Reprod (2015) 4(3):217–21. doi: 10.1016/j.apjr.2015.06.005
82. Rothlein R, Czajkowski M, O'Neill MM, Marlin SD, Mainolfi E, Merluzzi VJ. Induction of intercellular adhesion molecule 1 on primary and continuous cell lines by pro-inflammatory cytokines. regulation by pharmacologic agents and neutralizing antibodies. J Immunol (1988) 141(5):1665–9.
83. Zumwalde NA, Domae E, Mescher MF, Shimizu Y. Icam-1-Dependent homotypic aggregates regulate Cd8 T cell effector function and differentiation during T cell activation. J Immunol (2013) 191(7):3681–93. doi: 10.4049/jimmunol.1201954
84. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci U.S.A. (2018) 115(9):E2068–e76. doi: 10.1073/pnas.1716266115
85. Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and slp-76. Nat Immunol (2005) 6(12):1253–62. doi: 10.1038/ni1272
86. Bental M, Deutsch C. Metabolic changes in activated T cells: An nmr study of human peripheral blood lymphocytes. Magn Reson Med (1993) 29(3):317–26. doi: 10.1002/mrm.1910290307
87. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol (2004) 172(8):4661–5. doi: 10.4049/jimmunol.172.8.4661
88. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
89. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing Cd8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
90. Kurniawan H, Soriano-Baguet L, Brenner D. Regulatory T cell metabolism at the intersection between autoimmune diseases and cancer. Eur J Immunol (2020) 50(11):1626–42. doi: 10.1002/eji.201948470
91. Li F, Liu H, Zhang D, Ma Y, Zhu B. Metabolic plasticity and regulation of T cell exhaustion. Immunology (2022) 167(4):482–94. doi: 10.1111/imm.13575
92. Gullino PM, Grantham FH. The vascular space of growing tumors. Cancer Res (1964) 24(10):1727–32.
93. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res (2000) 60(4):916–21.
94. Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res (2001) 61(16):6020–4.
95. Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted Cd8(+) T cells by il-10 enhances anti-tumor immunity. Nat Immunol (2021) 22(6):746–56. doi: 10.1038/s41590-021-00940-2
96. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab (2020) 2(10):1001–12. doi: 10.1038/s42255-020-00280-9
97. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45(2):374–88. doi: 10.1016/j.immuni.2016.07.009
98. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific Cd8 t cells in the same host. Cell Rep (2016) 16(5):1243–52. doi: 10.1016/j.celrep.2016.06.078
99. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces Cd8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–56.e5. doi: 10.1016/j.cmet.2019.04.002
100. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. Pd-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
101. Giles JR, Manne S, Freilich E, Oldridge DA, Baxter AE, George S, et al. Human epigenetic and transcriptional T cell differentiation atlas for identifying functional T cell-specific enhancers. Immunity (2022) 55(3):557–74.e7. doi: 10.1016/j.immuni.2022.02.004
102. Galletti G, De Simone G, Mazza EMC, Puccio S, Mezzanotte C, Bi TM, et al. Two subsets of stem-like Cd8(+) memory T cell progenitors with distinct fate commitments in humans. Nat Immunol (2020) 21(12):1552–62. doi: 10.1038/s41590-020-0791-5
103. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-Cell exhaustion, Co-stimulation and clinical outcome in autoimmunity and infection. Nature (2015) 523(7562):612–6. doi: 10.1038/nature14468
104. Jennings E, Elliot TAE, Thawait N, Kanabar S, Yam-Puc JC, Ono M, et al. Nr4a1 and Nr4a3 reporter mice are differentially sensitive to T cell receptor signal strength and duration. Cell Rep (2020) 33(5):108328. doi: 10.1016/j.celrep.2020.108328
105. Odagiu L, May J, Boulet S, Baldwin TA, Labrecque N. Role of the orphan nuclear receptor Nr4a family in T-cell biology. Front Endocrinol (Lausanne) (2020) 11:624122. doi: 10.3389/fendo.2020.624122
106. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res (2018) 24(17):4175–86. doi: 10.1158/1078-0432.Ccr-17-1846
107. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36(4):265–76. doi: 10.1016/j.it.2015.02.008
108. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21(4):687–92. doi: 10.1158/1078-0432.Ccr-14-1860
109. Han Y, Liu D, Li L. Pd-1/Pd-L1 pathway: Current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
110. AmericanCancerSociety. Immune checkpoint inhibitors and their side effects. Available at: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy/immune-checkpoint-inhibitors.html.
111. NIH. Ctla4 cytotoxic T-lymphocyte associated protein 4 . Available at: https://www.ncbi.nlm.nih.gov/gene/1493.
112. Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. Lag-3: From molecular functions to clinical applications. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-001014
113. Liu L, Wang A, Liu X, Han S, Sun Y, Zhang J, et al. Blocking Tigit/Cd155 signalling reverses Cd8+ T cell exhaustion and enhances the antitumor activity in cervical cancer. J Transl Med (2022) 20(1). doi: 10.1186/s12967-022-03480-x
114. Mullard A. Roche’s anti-tigit drug suffers a phase iii cancer setback. Available at: https://www.nature.com/articles/d41573-022-00068-4.
115. He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch F. Tim-3, a promising target for cancer immunotherapy. Onco Targets Ther (2018) 11:7005–9. doi: 10.2147/ott.s170385
116. Ning Z, Liu K, Xiong H. Roles of btla in immunity and immune disorders. Front Immunol (2021) 12:654960. doi: 10.3389/fimmu.2021.654960
117. Lee JB, Ha SJ, Kim HR. Clinical insights into novel immune checkpoint inhibitors. Front Pharmacol (2021) 12:681320. doi: 10.3389/fphar.2021.681320
118. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. Vista is an immune checkpoint molecule for human T cells. Cancer Res (2014) 74(7):1924–32. doi: 10.1158/0008-5472.Can-13-1504
119. Quaratino S, Sainson R, Thotakura A, Henderson SJ, Pryke K, Newton A, et al. A first-in-Human study of Ky1044, a fully human anti-icos Igg1 antibody as monotherapy and in combination with atezolizumab in patients with selected advanced malignancies. J Clin Oncol (2019) 37(15_suppl):TPS2644–TPS. doi: 10.1200/JCO.2019.37.15_suppl.TPS2644
120. Chan S, Belmar N, Ho S, Rogers B, Stickler M, Graham M, et al. An Anti-Pd-1–Gitr-L bispecific agonist induces gitr clustering-mediated T cell activation for cancer immunotherapy. Nat Cancer (2022) 3(3):337–54. doi: 10.1038/s43018-022-00334-9
121. Cheng LS, Cheng YF, Liu WT, Shen A, Zhang D, Xu T, et al. A humanized 4-1bb-Targeting agonistic antibody exerts potent antitumor activity in colorectal cancer without systemic toxicity. J Transl Med (2022) 20(1):415. doi: 10.1186/s12967-022-03619-w
122. Glisson BS, Leidner RS, Ferris RL, Powderly J, Rizvi NA, Keam B, et al. Safety and clinical activity of Medi0562, a humanized Ox40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Clin Cancer Res (2020) 26(20):5358–67. doi: 10.1158/1078-0432.Ccr-19-3070
123. Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio C-WJ, et al. Batf and Irf4 cooperate to counter exhaustion in tumor-infiltrating car T cells. Nat Immunol (2021) 22(8):983–95. doi: 10.1038/s41590-021-00964-8
124. Harberts A, Schmidt C, Schmid J, Reimers D, Koch-Nolte F, Mittrücker H-W, et al. Interferon regulatory factor 4 controls effector functions of Cd8+ memory T cells. Proc Natl Acad Sci U.S.A. (2021) 118(16):e2014553118. doi: 10.1073/pnas.2014553118
125. Yu X, Li Z. Tox gene: A novel target for human cancer gene therapy. Am J Cancer Res (2015) 5(12):3516–24.
126. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-jun overexpression in car T cells induces exhaustion resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
127. LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, et al. Ptpn2 regulates the generation of exhausted Cd8(+) T cell subpopulations and restrains tumor immunity. Nat Immunol (2019) 20(10):1335–47. doi: 10.1038/s41590-019-0480-4
128. Allard D, Allard B, Stagg J. On the mechanism of anti-Cd39 immune checkpoint therapy. J Immunother Cancer (2020) 8(1):e000186. doi: 10.1136/jitc-2019-000186
129. Frey N, Tortola L, Egli D, Janjuha S, Rothgangl T, Marquart KF, et al. Loss of Rnf31 and Vps4b sensitizes pancreatic cancer to T cell-mediated killing. Nat Commun (2022) 13(1). doi: 10.1038/s41467-022-29412-3
130. Li P, Zhu X, Cao G, Wu R, Li K, Yuan W, et al. 1α,25(Oh)2d3 reverses exhaustion and enhances antitumor immunity of human cytotoxic T cells. J Immunother Cancer (2022) 10(3):e003477. doi: 10.1136/jitc-2021-003477
131. Dolgin E. Immunotherapy takes aim at exhausted T cells. Nat Biotechnol (2020) 38(1):3–5. doi: 10.1038/s41587-019-0381-y
132. Revisiting checkpoint blockade. Nat Biotechnol (2022) 40(7):981. doi: 10.1038/s41587-022-01407-x
133. Vafaei S, Zekiy AO, Khanamir RA, Zaman BA, Ghayourvahdat A, Azimizonuzi H, et al. Combination therapy with immune checkpoint inhibitors (Icis); a new frontier. Cancer Cell Int (2022) 22(1):2. doi: 10.1186/s12935-021-02407-8
134. Kverneland AH, Pedersen M, Westergaard MCW, Nielsen M, Borch TH, Olsen LR, et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget (2020) 11(22):2092–105. doi: 10.18632/oncotarget.27604
135. Gumber D, Wang LD. Improving car-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine (2022) 77. doi: 10.1016/j.ebiom.2022.103941
136. Huang S, Liang C, Zhao Y, Deng T, Tan J, Lu Y, et al. Increased tox expression concurrent with pd-1, Tim-3, and Cd244 in T cells from patients with non-Hodgkin lymphoma. Asia Pac J Clin Oncol (2022) 18(1):143–9. doi: 10.1111/ajco.13545
137. Zhao Y, Liao P, Huang S, Deng T, Tan J, Huang Y, et al. Increased tox expression associates with exhausted T cells in patients with multiple myeloma. Exp Hematol Oncol (2022) 11(1):12. doi: 10.1186/s40164-022-00267-0
138. Huang S, Liang C, Zhao Y, Deng T, Tan J, Zha X, et al. Increased tox expression concurrent with pd-1, Tim-3, and Cd244 expression in T cells from patients with acute myeloid leukemia. Cytometry B Clin Cytom (2022) 102(2):143–52. doi: 10.1002/cyto.b.22049
139. Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. Tox and Tox2 transcription factors cooperate with Nr4a transcription factors to impose Cd8(+) T cell exhaustion. Proc Natl Acad Sci U.S.A. (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
140. Tsui C, Kretschmer L, Rapelius S, Gabriel SS, Chisanga D, Knöpper K, et al. Myb orchestrates T cell exhaustion and response to checkpoint inhibition. Nature (2022) 609(7926):354–60. doi: 10.1038/s41586-022-05105-1
141. Chen C, Liu J, Chen Y, Lin A, Mou W, Zhu L, et al. Application of atac-seq in tumor-specific T cell exhaustion. Cancer Gene Ther (2022) 30:1–10. doi: 10.1038/s41417-022-00495-w
142. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell–like and Foxp3+ regulatory T cells. Nat Immunol (2010) 11(9):846–53. doi: 10.1038/ni.1915
143. Marijt KA, Sluijter M, Blijleven L, Tolmeijer SH, Scheeren FA, van der Burg SH, et al. Metabolic stress in cancer cells induces immune escape through a Pi3k-dependent blockade of ifnγ receptor signaling. J Immunother Cancer (2019) 7(1):152. doi: 10.1186/s40425-019-0627-8
144. Stelekati E, Cai Z, Manne S, Chen Z, Beltra J-C, Buchness LA, et al. Microrna-29a attenuates Cd8 T cell exhaustion and induces memory-like Cd8 T cells during chronic infection. Proc Natl Acad Sci U.S.A. (2022) 119(17):e2106083119. doi: 10.1073/pnas.2106083119
145. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of car T-cells with crispr-Cas9 gene editing. Mol Cancer (2022) 21(1). doi: 10.1186/s12943-022-01559-z
146. Nie S, Wang Z, Moscoso-Castro M, D'Souza P, Lei C, Xu J, et al. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib Ther (2020) 3(1):18–62. doi: 10.1093/abt/tbaa003
147. Zhou Y, Penny HL, Kroenke MA, Bautista B, Hainline K, Chea LS, et al. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J Immunother Cancer (2022) 10(4). doi: 10.1136/jitc-2021-004225
148. OSEImmunotherapeutics. Ose immunotherapeutics presents update on bicki®-Il-7v and clec-1 immuno-oncology programs at international conferences. Available at: https://www.ose-immuno.com/wp-content/uploads/2022/10/EN_221012_Pre-SITC.pdf.
149. Masson G. Roche Snaps up pd-1-Focused good therapeutics for $250m; 'Sky's the limit' for spinout. Available at: https://www.fiercebiotech.com/biotech/roche-snaps-good-therapeutics-shape-shifting-program-250m-skys-limit-spinout.
150. NIH. Pd-1-Targeted il-2 variant antibody fusion protein Ro7284755. Available at: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/pd-1-targeted-il-2-variant-antibody-fusion-protein-ro7284755.
Keywords: T cell exhaustion, cancer-immunity cycle, immunotherapy, functional adaption, T cell dysfunction
Citation: Brunell AE, Lahesmaa R, Autio A and Thotakura AK (2023) Exhausted T cells hijacking the cancer-immunity cycle: Assets and liabilities. Front. Immunol. 14:1151632. doi: 10.3389/fimmu.2023.1151632
Received: 26 January 2023; Accepted: 27 March 2023;
Published: 14 April 2023.
Edited by:
Lewis Zhichang Shi, University of Alabama at Birmingham, United StatesReviewed by:
Ujjaldeep Jaggi, Cedars Sinai Medical Center, United StatesCopyright © 2023 Brunell, Lahesmaa, Autio and Thotakura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil K. Thotakura, YW5pbC50aG90YWt1cmFAb3Jpb25waGFybWEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.