
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 June 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1149203
 Tingfen Ji
Tingfen Ji Hequan Li*
Hequan Li*Prosperous advances in understanding the cellular and molecular mechanisms of chronic inflammation and airway remodeling in asthma have been made over the past several decades. Asthma is a chronic inflammatory disease of the airways characterized by reversible airway obstruction that is self-resolving or remits with treatment. Around half of asthma patients are “Type-2-high” asthma with overexpression of type 2 inflammatory pathways and elevated type 2 cytokines. When stimulated by allergens, airway epithelial cells secrete IL-25, IL-33, and TSLP to derive a Th2 immune response. First ILC2 followed by Th2 cells produces a series of cytokines such as IL-4, IL-5, and IL-13. TFH cells control IgE synthesis by secreting IL-4 to allergen-specific B cells. IL-5 promotes eosinophil inflammation, while IL-13 and IL-4 are involved in goblet cell metaplasia and bronchial hyperresponsiveness. Currently, “Type-2 low” asthma is defined as asthma with low levels of T2 biomarkers due to the lack of reliable biomarkers, which is associated with other Th cells. Th1 and Th17 are capable of producing cytokines that recruit neutrophils, such as IFN-γ and IL-17, to participate in the development of “Type-2-low” asthma. Precision medicine targeting Th cells and related cytokines is essential in the management of asthma aiming at the more appropriate patient selection and better treatment response. In this review, we sort out the pathogenesis of Th cells in asthma and summarize the therapeutic approaches involved as well as potential research directions.
Asthma is a heterogeneous disease, characterized by major differences in patients’ clinical manifestations, severity, response to treatment, and allergens stimulation (1). With intensive research on the underlying molecular mechanisms or endotypes of asthma, in recent years, the asthma population has been divided into “Type-2-high” asthma and “Type-2-low” asthma (2). “Type-2-high” asthma is characterized by high secretion of interleukin (IL)-4, IL-5, and IL-13, elevated eosinophil counts, serum periostin, and total IgE, including allergic and eosinophilic asthma (3, 4). “Type-2-low” asthma occurs in the absence of T2 biomarkers or eosinophilic elevation, including neutrophilic, paucigranulocytic, and obesity asthma (4, 5) (Figure 1).
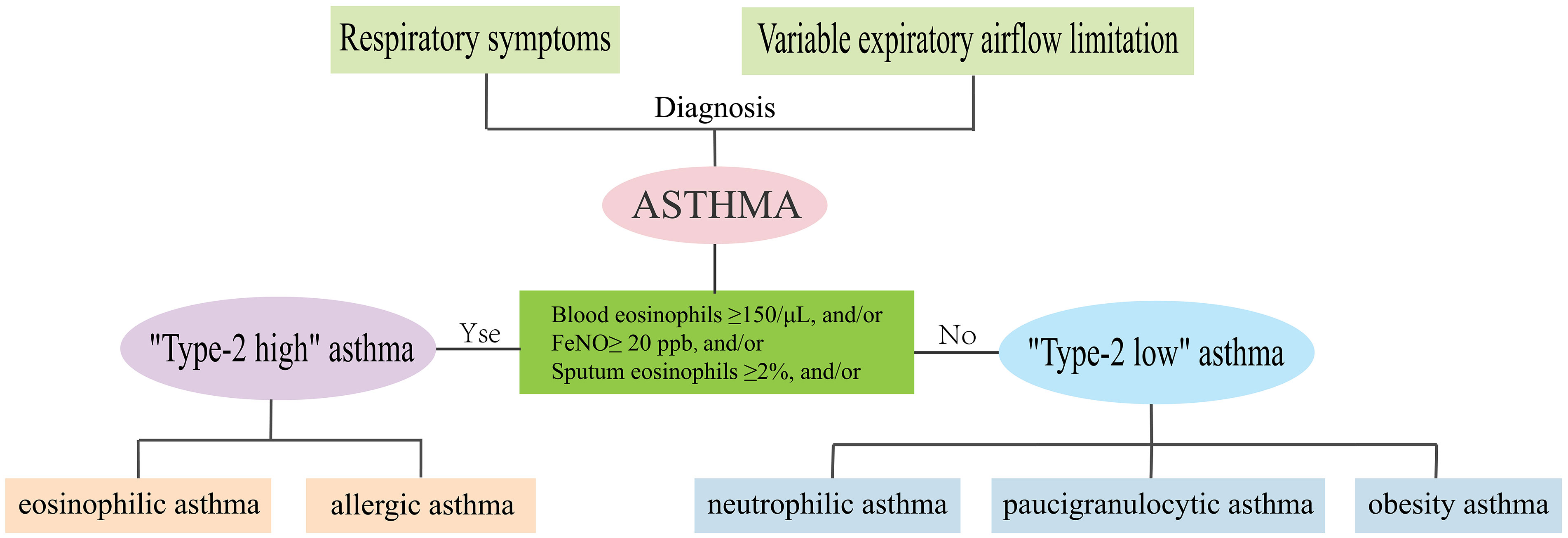
Figure 1 Phenotypes and endotypes of asthma.
The two endotypes of asthma involve different Th cell-driven inflammatory responses and airway remodeling processes, including Th2, T follicular helper (TFH), Th17, and Th1 cells. Activation of Th2-pathways is at the core of type 2 inflammation, producing excessive amounts of the cytokines IL-4, IL-5, and IL-13. IL-5 induce eosinophil activation, maturation, and recruitment, while IL-4 and IL-13 are involved in goblet cell metaplasia, airway smooth muscle(ASM) contractility, and airway hyperresponsiveness(AHR) (6). TFH cells regulate B cell proliferation and conversion of immunoglobulin classes, particularly in germinal center structures (7). These cells produce IL-21 and IL-4, which are essential for B-cell stimulation and play a key role in controlling IgE production in asthma (8, 9). Accumulating evidence suggests that “Type-2-low” asthma is associated with abnormal Th17 or Th1 cell immune responses. Th17 cells producing the IL-17 family are involved in neutrophil inflammation and airway remodeling processes and are responsible for corticosteroid resistance in asthma (10). Th1 cell activation promoting interferon (IFN)-γ and tumor necrosis factor (TNF)-α production also contributes to driving neutrophil inflammation (11). Some other Th cell subsets such as Th9 and Th22 have been described by some laboratories as being involved in the secretion of IL-9 and IL-22, respectively, which are associated with airway inflammation and AHR in asthma (12, 13). However, defining them as distinct subgroups remains controversial. It is unclear if these subsets are stable or are indeed relevant to asthma pathogenesis outside animal models.
Currently, conventional asthma medications include bronchodilators and glucocorticoids. They focus on symptom control, are not curative, and are often ineffective in severe cases. Th cells remain at the forefront of asthma pathogenesis due to chronic airway inflammation and airway remodeling (Figure 2). Precision medicine targeting Th cells and related cytokines is essential in the management of asthma aiming at the more appropriate patient selection and better treatment response. This review focuses on Th cells and their cytokines in the pathogenesis of asthma and the associated targeted emerging therapeutic approaches.
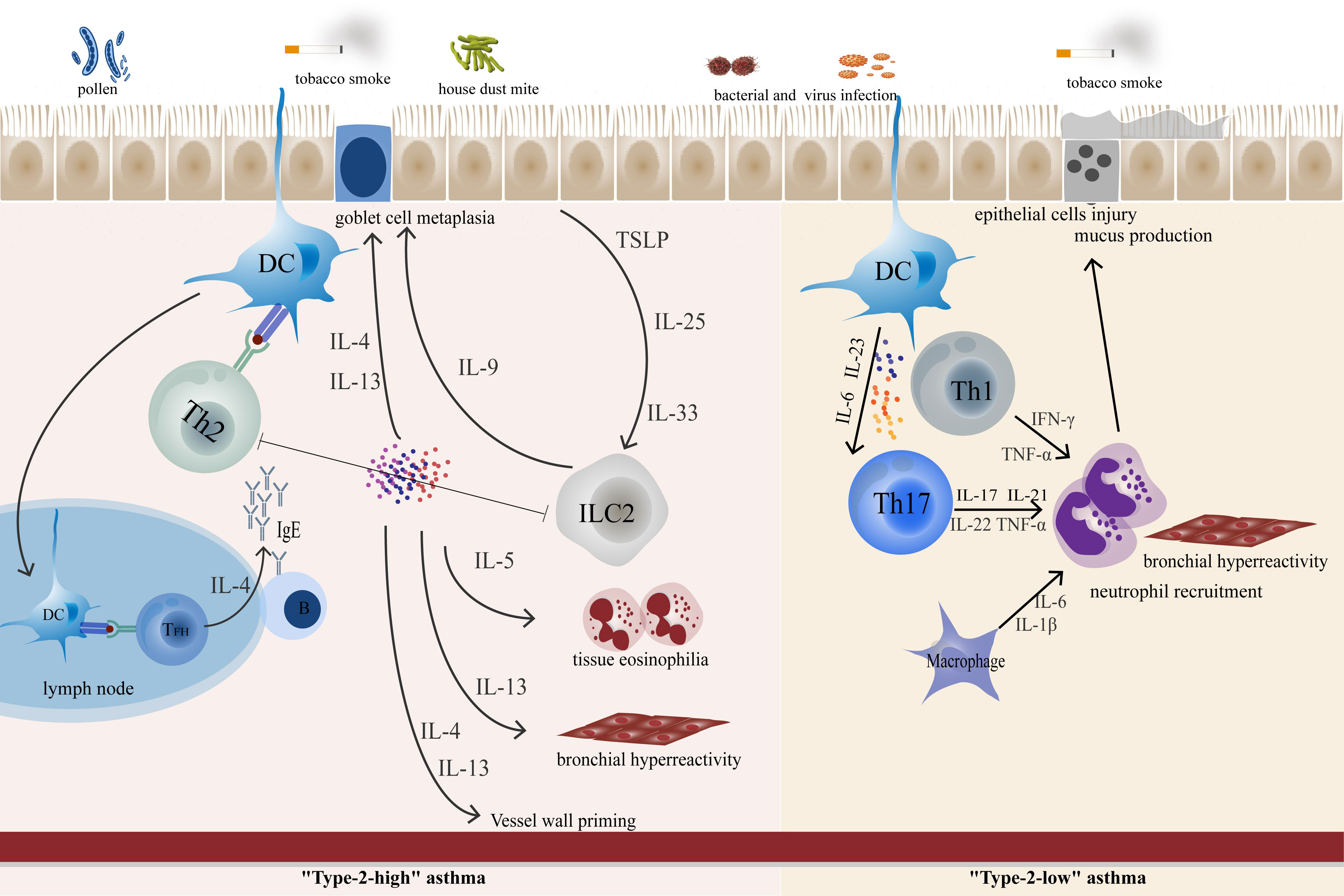
Figure 2 In “Type-2 high” asthma, epithelial-derived TLPS, IL-25, and IL-33 can awaken ILC2. Cytokines produced by epithelial cells promote DC function, polarize CD4+ T cells and promote the polarization of Th2 cells. TFH cells control IgE synthesis by secreting IL-4 to allergen-specific B cells. ILC-derived IL-9, which promotes goblet cell metaplasia. IL-5 promotes eosinophil inflammation, while IL-13 and IL-4 are involved in goblet cell metaplasia and bronchial hyperresponsiveness. In “Type-2-low” asthma, Th17 and Th1 play an important role in airway neutrophil inflammation and airway remodeling.
Epithelial cytokines play a role as “whistle-blowers” in developing allergic responses at barrier surfaces and are involved in the pathogenesis of type 2 inflammatory diseases, typically asthma. The release of these “alarmins”, including IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), occurs when the allergen stimulates bronchial epithelial cells (14). Although all of them are indispensable cytokines for initiating the Th2 immune response, they do not have the same role. IL-25 expresses at high levels in the lung of sensitized mice, promoting ASM contraction and increasing eosinophil recruitment and antigen-presenting ability (15, 16). In humans, levels of IL-25 receptors were increased in both myeloid cells and plasmacytoid dendritic cells (DCs) in the airways of patients with mild asthma after the allergen challenge (17). Bronchial epithelial IL-25 expression has a major role in triggering type 2 responses, with patients with higher IL-25 levels having greater AHR and more airway and blood eosinophils, higher serum IgE, more subepithelial thickening, and higher expression of Th2 signature genes (18). IL-33 induces signals in immune cells through its receptor IL-1RL1, which leads to the production of type 2 cytokines and chemokines (19). In mouse models, prophylactic or therapeutic blockade of IL-33 release attenuated asthma exacerbations (20). In allergic or eosinophilic asthma patients, IL-33 in bronchoalveolar lavage fluid (BALF) correlates with asthma severity (21). Interestingly, anti-inflammatory IL-37, whose target cells include airway epithelial cells (AECs), Th cells, and DCs, may limit the pro-inflammatory signaling of IL-33 (22). IL-37 reduced Th2 cytokine production in allergen-activated monocytes from wild-type but not IL-1R1-deficient mice and inhibited IL-33-induced Th2 cytokine release (22). In addition, IL-37 down-regulated allergic airway inflammation and attenuated IL-1β and IL-33-induced expression of pro-inflammatory mediators (22).
TSLP is considered to be a primary regulator of type 2 immune response to the respiratory barrier, which activates Th2 cells, innate lymphoid cells (ILCs)-2, mast cells, and other immune cells by inducing costimulatory molecules on DCs (23). Accumulated shreds of evidence are proposed to improve this view. Early studies have shown that compared with healthy controls, the expression of TSLP mRNA in the bronchial epithelium and submucosa of asthmatic patients is significantly increased (24). A large epidemiological study reveals that plasma high TSLP levels were associated with current asthma, poor lung function, and even the persistence of asthma attacks and dyspnoea 10 years later (25). Additionally, a part of patients with high TSLP expression shows hormone resistance indicating that glucocorticoid resistance asthma might be particularly relevant to the interaction of TSLP and ILCs (26). This is possibly due to ILCs expressing higher TSLPR decreasing the apoptosis of ILCs via the activation of STAT5 and the upregulation of (B-cell lymphoma-xl)Bcl-xl (26). In addition, TSLP also has convincing evidence for an effect on “Type-2-low” asthma. Human TSLP promotes the differentiation of Th17 cells with the central memory T cell phenotype through DCs activation (27). TSLP expression in human BALF was closely associated with neutrophil infiltration, and anti-TSLP therapy has been shown to reduce exacerbations in patients with severe, uncontrolled asthma with lower blood eosinophil and FeNO levels (28).
In addition, interactions between cytokines released by Th cells or other cells and AECs contribute to the progression of asthma. While higher levels of activin-A expressed by AECs may promote an increase in Th2 and Th9 cells (29), upregulating type 2 inflammation, IL-4 secreted by Th2 is involved in coordinating the polarization of AECs (30). Furthermore, there are epithelial indicators that are directly dependent on the cytokine profile of asthma patients and may play a part in the management of asthma (31). (The details will be described in turn below)
Th2-driven adaptive immune response and ILC2s-driven innate immune response are currently regarded as two independent but linked pathogenic mechanisms of asthma, both of which produce type 2 cytokines (32). ILC2s primarily respond to epithelial cell-derived “alarmins” with proliferation and activation (33, 34). ILC2s are unique in their high secretion of IL-9 can promote goblet cell metaplasia and promote mast cell growth and survival (35). Th2 cells, which produce IL-4, IL-5, and IL-13 in an antigen-dependent manner, are thought to play a major role in the pathogenesis of asthma (36). An array of those ‘type 2 cytokines’ exerts different and superimposed effects on the airway, which is pivotal to the pathological process of allergic asthma. IL-5 creates lung tissue and systemic eosinophilia, while IL-4 and IL-13 are involved in goblet cell metaplasia and AHR (37, 38). These changes in inflammation and remodeling further increase the susceptibility to exacerbation, thereby stimulating airway damage and subsequent remodeling (39). Besides, the interaction of Th2 cytokines and AECs may also be beneficial in maintaining a type 2 inflammatory environment. On the one hand, IL-4 antagonizes the IFN-γ-inducing genes expressed by AECs (30). On the other hand, IL-4 induces the expression of IL-24 in normal human AECs. IL-24 expression was elevated in nasal scrapings and sputum from patients with allergic rhinitis and seasonal asthma compared to healthy controls (40). IL-24 promotes airway remodeling by modulating epithelial–mesenchymal transition (EMT), and silencing IL-24 treatment attenuates airway inflammation, AHR, and airway remodeling in the asthmatic mouse model (41).
IL-5 (to a certain extent, IL-13 and granulocyte-macrophage colony-stimulating factor (GM-CSF)) controls eosinophils’ maturation, differentiation, and release (42). In the mouse model, IL-5 maintains the survival of lung-resident-like eosinophils, and IL-5 neutralization markedly reduces the percentage and the total number of lung and peripheral blood eosinophils (43). Also, sputum eosinophils and IL-5 were significantly elevated in asthmatic patients with accelerated decline in lung function (44). IL-5 promotes the proliferative properties of inflammatory-like and lung-resident-like eosinophils and the effect may be related to the expression of IL-5R in asthmatic patients (45). Eosinophils highly express IL-5Rα on their surface, and airway inflammation and injury occur once eosinophils are mobilized from the bone marrow and blood pool to the airway (46). Activated eosinophils are associated with endogenous AHR and airway inflammation with the release of reactive oxygen species and granulins, and interact with intraepithelial mast cells to regulate airway inflammation (47). Normally, eosinophils can survive in vitro for about 48 hours, however, IL-5 can prolong the survival time of eosinophils (48).
IL-4 and IL-13 share one type II receptor, directly or indirectly leading to various pathological changes of airway obstruction and AHR. IL-13 and IL-4 induce up-regulation of vascular cell adhesion molecule-1 (VCAM-1) expression on human endothelial cells and are involved in collagen production in fibroblasts (49). In addition, they enhanced the histamine-induced Ca2+ mobilization that was accompanied by increased mRNA expression of histamine H1 and cysteinyl leukotriene CysLT1 receptors, inducing glucocorticoid-insensitive AHR in isolated human small airways (50). Furthermore, more pieces of evidence confirmed that IL-13 mainly mediates physiological functions of airway remolding, including mucous cell metaplasia, smooth muscle cell contraction, goblet cell metaplasia and proliferation, and tissue fibrosis (51, 52).
Accumulating emerging evidence suggests that TFH cells, rather than Th2 cells, drive IgE responses in asthma. Using house dust mites (HDM) and mice, León and her colleagues found that after the first sensitizing exposure, TFH cells reside in the draining mediastinal lymph nodes of the lungs and differentiate into Th2 and migrate to the lungs to exert their effects after a second attack by HDM allergens (53). Meanwhile, studies have also shown pulmonary cDCs effectively induced TFH cells in asthmatic lungs and that the induced TFH cells secreted IL-4 to support the production of IgE antibodies (54, 55). It is worth emphasizing that TFH cells are not only necessary for IgE antibody production but are also sufficient even in the absence of typical Th2 cells or when their effector function is impaired (54). Further evidence suggests that a rare population of IL-13-producing TFH cells is essential for the production of high-affinity IgE antibodies and subsequent allergen-induced allergic responses (9). In humans, the frequency of circulating CXCR5CD4T cells (TFH cells) was elevated in allergic asthma, with increased production of IL-4, and IL-21, and positively correlated with total IgE in the blood (56). After responding to IL-4 and/or IL-13 from TFH cells, memory IgG+ B cells begin to produce IgE (57). The interaction between IgE and FcϵRI on mast cells and basophils is an amplifier of the allergic cascade response, triggering increased vascular permeability and increased cellular recruitment, the release of mediators, lipid mediators, and cytokines (58).
Th9 cells are a unique subpopulation of Th cells that mainly produce IL-9 as a signature cytokine. Increased numbers of Th9 cells were found in lung tissue, draining lymph nodes and airways of asthmatic mouse models, while anti-IL-9 antibody inhibits Th9 cell differentiation and attenuates allergic airway inflammation (59). Transcriptomic analysis revealed increased IL-9 expression in peripheral blood cells of patients with allergic eosinophilic asthma (60). IL-9 is mainly derived from Th9 cells (partly, ILC2 and Th2) and is a key factor in allergic airway inflammation. IL-9 promotes the pathogenesis of asthma by activating and recruiting mast cells and eosinophils, enhancing B-cell IgE production, increasing epithelial cell mucus production, and triggering airway AHR (29, 61). Natural allergen exposure raised local and systemic Th2, Th9, and reduced Tregs cells, but allergen-specific immunotherapy(AIT) inhibited Th2 and Th9 cell frequencies along with transforming growth factor (TGF)-β and IL-9 secretion (62). A crucial pro-inflammatory mediator in allergy reactions, activin A, a TGF-superfamily member connected to TGF-β1, also regulates Th9 development and effector activities (63). Musiol et al. found that targeting the TGF-β pathway may not only inhibit airway remodeling processes but also target pro-inflammatory Th2 and Th9 cells without adversely affecting immune tolerance in the asthmatic model (62). Another study found that activin-A was able to replicate the ability of TGF-β1 to drive Th9 polarization in vitro and in vivo. Crucially, effective inhibition of Th9 function in vivo is entirely dependent on the blockade of TGF-β and activin-A (29).
Targeting these epithelial cytokines and associated signal pathways is considered an effective therapeutic strategy for asthma. Tezepelumab is a human monoclonal antibody that specifically binds to TSLP and blocks its interaction with the receptor complex, and is approved for severe asthma without phenotypic (e.g., eosinophilic or allergic) or biomarker restrictions. In phase 2 and 3 clinical trials, including patients with severe asthma, Tezepelumab (subcutaneous injection) consistently and significantly reduced acute asthma exacerbations regardless of key biomarkers (64, 65). In addition to this, the anti-TSLP treatment improved lung function, reduced airway eosinophil inflammation, and decreased the proportion of patients with AHR in asthmatic patients (66, 67). More in-depth studies on Tezepelumab to improve airway remodeling are possible in the future. Ecleralimab (CSJ117), a novel anti-TSLP monoclonal antibody fragment administered by inhalation, recently showed results from a phase I randomized controlled trial in mild asthma. After 14 weeks of administration (4 mg once daily inhaled), Eculalizumab was shown to be effective in attenuating allergen-induced responses in asthma patients as it significantly attenuated allergen-induced bronchoconstriction and airway inflammation (68).
Clinical trials with anti-IL-25 antibodies have not been conducted, but anti-IL-33 has obtained prospective results in a few trials. Itepekimab, a monoclonal antibody targeting IL-33, causes a reduction in blood eosinophils with a long-lasting effect in patients with moderate asthma (69). Results from another phase 2 trial showed that blocking IL-33 with itepekimab resulted in a lower incidence of uncontrolled asthma events compared to placebo, which was close to dupleuximab (22% vs 19%) and improved lung function (70). Astegolimab, a selective inhibitor of the IL-33 receptor ST2, was shown to attenuate the asthma exacerbation rate at a subcutaneous dose of 70 mg in a phase 2 trial in patients with severe asthma, including those with low eosinophils (71).
Based on accumulating evidence implicating IL-4, IL-5, and IL-13 are responsible for the occurrence and development of asthma. Several clinical trials targeting type 2 cytokines are currently underway, and some of the relevant biologics have been approved for the treatment of severe asthma. Mepolizumab, Reslizumab (anti-IL-5 mAb), and Benralizumab (anti-IL-5R mAb) were FDA-approved to treat patients with severe eosinophilic asthma (37). Several prospective clinical trials have shown that the use of mepolizumab (administered subcutaneously or intravenously) or reslizumab (administered intravenously) in patients with asthma not adequately controlled by inhaled corticosteroids or severe eosinophilic asthma significantly reduced asthma exacerbations and was associated with improvements in markers of asthma control with a favorable safety profile (72–75). In addition to this, in patients with oral corticosteroid-dependent asthma, the use of reslizumab resulted in fewer new systemic corticosteroid prescriptions and a lower systemic corticosteroid burden (76). Besides, the effectiveness of mepolizumab and reslizumab is consistent with clinical trial results under real-world settings, with significant reductions in exacerbations and daily maintenance of oral corticosteroid dose (77, 78). Another biologic agent on IL-5, benralizumab, a monoclonal antibody directed against the alpha subunit of the IL-5R, has also shown significant clinically relevant benefits in patients with severe asthma (79). In several phase 3 trials, patients with severe asthma who received subcutaneous benralizumab achieved clinical benefits in terms of oral glucocorticoid use, lung function, asthma control, symptoms, asthma-related quality of life, and rate of exacerbation (79, 80). The most frequent adverse events were nasopharyngitis, worsening asthma, and bronchitis (80).
Likewise, the clinical trials of targeted IL-4/IL-13 therapy are also continuously increasing, with mixed results. Concerning IL-13, two biological agents have been studied, Lebrikizumab and Tralokinumab. In 2016, two phase 3 clinical trials of lebrikizumab for asthma (Lavolta I and Lavolta II), Lavolta I enrolled 2,000 patients with severe asthma and showed that lebrikizumab significantly reduced acute asthma exacerbations and also improved lung function in patients, yet similar results were not obtained in Lavolta II (81). A phase 3 trial in uncontrolled adolescent asthma patients found that Lebrikizumab reduced the rate of asthma exacerbation in uncontrolled adolescents, yet this study was terminated prematurely (82). Some trials found that lebrikizumab treatment was associated with improved lung function, especially in patients with high periostin levels (83). Another biologic, tralokinumab, has also not performed satisfactorily in severe asthma. Although tralokinumab reduced the annualized asthma exacerbation rate in participants with severe asthma with baseline FeNO 37 ppb or higher in the phase 3 clinical trial (STRATOS 1), it did not in another (STRATOS 2) (83). Further, William and his colleagues disappointedly found that asthma exacerbation rate and inhaled corticosteroid dosage were not significantly different between tralokinumab and placebo with uncontrolled asthma (84). Anti-IL-4 biologic, Pascolizumab, has not achieved better results in clinical trials of asthma and further studies have been discontinued. Bispecific antibodies that can simultaneously bind IL-4 and IL-13 have also entered clinical trials. Dupilumab is the first fully human anti-IL-4 receptor α monoclonal antibody approved for asthma that blocks both IL-4 and IL-13 signaling (85). In moderate to severe uncontrolled asthma, the incidence of exacerbations was significantly lower in patients treated with dupilumab than in those receiving a placebo, as well as better lung function and asthma control (85). Dupilumab also improved FEV1 and asthma symptom control, with greater efficacy observed in patients with elevated type 2 inflammatory biomarkers at baseline (86). In addition to the control of asthma symptoms and improvement in lung function, a multicenter study demonstrated that dupilumab reduced the use of oral corticosteroids in patients with corticosteroid-dependent severe asthma (87). The long-term efficacy and safety of dupilumab are also noteworthy. Further improvements in exacerbation rate, asthma control and health-related quality of life, and progressive reductions in blood eosinophils and total serum IgE were observed in patients treated with dupilumab at treatment extensions up to 148 weeks (88).
By targeting the Fc fragment of IgE, Omalizumab, an anti-IgE monoclonal antibody, reduces free serum IgE levels and inhibits the binding of IgE to its high-affinity receptors on mast cells and basophils for the treatment of asthma. Compared with the other antibodies, Omalizumab was the first biologic approved by the FDA for the treatment of asthma. A review that included 25 clinical trials showed that omalizumab (mostly, subcutaneous injection) was effective in reducing asthma exacerbations and hospitalizations (89). Evidence from real-world studies suggests that the treatment of omalizumab in patients with asthma improved exacerbation rates and reduced hospitalizations, regardless of biomarker status (90). Dosing of omalizumab is based on body weight and pre-treatment serum total IgE levels, and its immunomodulatory effects persist after discontinuation. Although IgE levels do not accurately predict treatment efficacy, they may help predict cases requiring re-treatment after discontinuation (91). Serum inflammation-protective collagen 4 (COL4A3) levels are elevated in adults and children with asthma and are associated with a more severe, exacerbated allergic asthma phenotype. A cohort study evaluating patients treated with omalizumab using an asthma control test identified COL4A3 as a novel biomarker for predicting response to anti-IgE therapy (31). In biomarker analysis, a subgroup of patients with higher FeNO, peripheral blood eosinophil count, and serum periostin had relatively better treatment responses (92).
IL-9-targeted therapy may offer a new approach to treating patients with asthma, but clinical studies are not well underway. In two randomized phase 2a studies in asthmatic subjects, patients who received MEDI-528, a humanized anti-IL-9 monoclonal antibody, showed a slight improvement in symptoms without a change in lung function (93). Another clinical trial using MEDI-528 in patients with moderate to severe asthma also failed to show efficacy (94). Perhaps this is because the heterogeneity of asthma makes it difficult for MEDI-528-targeted therapy to show significant beneficial effects in non-selected asthma populations (94). Current biological agents in “Type-2 high” asthma are summarized in Table 1.
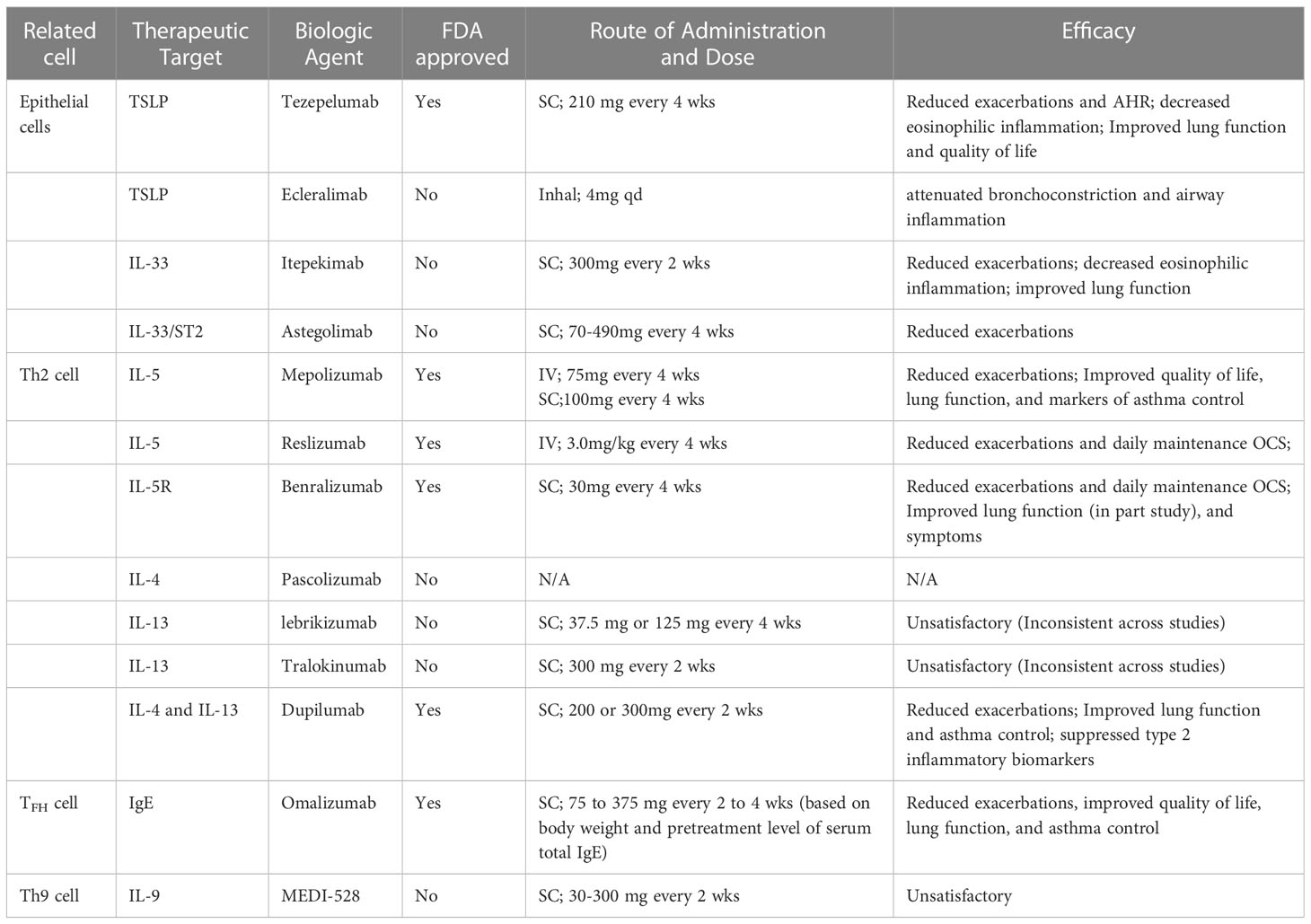
Table 1 Current biological agents in “Type-2 high” asthma.
Th1 inflammation is mainly characterized by an increase in Th1 cells and infiltration of signature cytokines including IFN-γ and TNF-α. Because of the inhibition Th2 inflammatory response, Th1 is considered to have an inhibitory effect on asthmatic airway inflammation in early investigations. Several studies have shown that IFN-γ inhibits Th2 cell function to attenuate the inflammatory response in asthma (95). Further, Mitchell et al. found that IFN-γ acting through the airway epithelium inhibits mucus, chitinases, and eosinophilia, independent of Th2 cell activation (96).
Growing amounts of data are showing a link between Th1 cells and the emergence of “Type-2-low” corticosteroid-resistant asthma. AHR, neutrophilia, and airway remodeling were persistent in mice with elevated Th1 cells during allergic airway inflammation, and these mice were resistant to corticosteroids (97, 98). Wisniewski and colleagues discovered that IFN-γ+CD4+ T cells were concentrated in the BALF of adult patients with severe asthma and that Th1 airway inflammation was evident in children with allergic and non-allergic severe asthma (11, 99). ASM is an important target for corticosteroid therapy and IFN-γ and TNF-α have a significant effect on ASM, which is responsible for the insensitivity to corticosteroids in patients with severe asthma. Rodney et al. used human fetal ASM as an in vitro model and found that the interaction between TNF-α and IFN-γ mediated pathways may promote corticosteroid resistance during ASM development (100). In a mouse model, inhibition of TNF-α resulted in a decrease in airway remodeling-related mediators (101). Raundhal et al. revealed that IFN-γ levels are inversely correlated with secretory leukocyte protease inhibitors (SLPI) expressed by airway epithelial cells, dysregulation of the IFN-γ/SLPI axis involves the AHR and corticosteroid refractoriness (11). The transcriptomic analysis also revealed that combined exposure to TNF-α and IFN-γ affects several gene expressions, which impairs corticosteroid sensitivity in ASM and may lead to persistent symptoms and worsening of severe pediatric asthma (102). Recently, several interesting studies have found that obesity-associated asthma patients are associated with Th1 activation, neutrophil inflammation, and pulmonary function deficits (103). Th cell transcriptome analysis identified upregulation of genes in the cytokinesis cycle 42 pathway (associated with TNF-α and IFN-γ expression) in Th cells of children with obesity-related asthma (5).
Current researches reveal that Th17 cells and their cytokines are linked to some, but not all, “Type-2-low” asthma, particularly in severe asthma with neutrophil predominance.
Many well-known cytokines work together for Th17 cell differentiation to occur, with IL-23, IL-6, and TGF-βbeing the main drivers of this process. IL-6 in combination with TGF-β induced differentiation of naïve T cells to Th17 cells, which is the cornerstone in autoimmune diseases (104). Activated Th17 cells release more IL-6 to further promote Th17 cell differentiation (104). IL-23, a member of the IL-12 family of cytokines, is released primarily by DCs, macrophages, B cells, and endothelial cells and is essential for the survival and effector capacity of Th17 cells (105). In the absence of IL-23, Th17 cells stimulated by TGF-β and IL-6 showed impaired pathogenic effects despite increased IL-17 production (106). IL-23 enhances eosinophil inflammation in “Type-2-high” asthma. Inhaled allergen stimulation in asthmatic mice elevated IL-23 expression in the bronchial epithelium and eosinophil infiltration, whereas IL-23 inhibition reduced airway inflammation and hyperresponsiveness (107). Moreover, IL-23 plays an essential part in neutrophil recruitment. According to Wakashin et al., forced expression of IL-23 in sensitized mice’s airways increased the production of IL-17A in response to antigens and the recruitment of neutrophils (108). In comparison to healthy populations, IL-23 activation of peripheral blood neutrophils isolated from asthmatic patients dramatically boosted IL-17A and IL-17F production (109).
Th17 cells secrete IL-17A, IL-17F, IL-21, IL-22, and TNF-α to perform their functions. IL-17A and IL-17F are believed to contribute to asthmatic neutrophil inflammation. Experimental mouse models of asthma suggest that IL-17A enhances the recruitment of bronchial neutrophils through CXCR2 signaling (110). Serum levels of IL-17A were also higher in asthmatic mice than in the wild type, implying that IL-17A may also promote neutrophil transmigration (111). Sorbello et al. found increased expression of IL-17F and IL-17A in the nasal and bronchial lamina propria and higher expression of bronchial neutrophils in patients with severe asthma (112). An immunohistochemical study of bronchial and nasal biopsies found that bronchial and nasal IL-17F correlated with bronchial neutrophil counts, asthma exacerbation rates, and FEV1 (113). Bullone et al. conducted an interesting study in an attempt to characterize the immunopathology of neutrophilic asthma and to identify potential clinical markers for this subtype. They performed bronchial biopsies on 70 asthmatic patients and identified the “high neutrophil” subtype. The final results showed a significant increase in IL-17F-positive cells in patients with high neutrophils. After excluding smokers, an increase in IL-17A was also found in highly neutrophilic patients (114). It has also been found that IL-17A and IL-17F exert different biological effects on the airway inflammation of asthma. Several studies have associated IL-17A with airway remodeling and AHR, the major physiological characteristics of asthma. IL-17A was found to induce mucus cell chemotaxis independent of STAT6 by establishing a mouse model (115). A study quantifying bronchial angiogenesis as well as the number of IL-17A cells and the concentration of angiogenic factors in sputum in subjects with severe asthma, COPD, and healthy subjects found that IL-17A enhances bronchial vascular remodeling by stimulating the synthesis of other angiogenic factors may lead to mucosal congestion resulting in airway narrowing (116). Other studies have found that IL-17F, but not IL-17A, underlies airway inflammation in steroid-insensitive toluene diisocyanate-induced asthma models and that anti-IL-17F ameliorates toluene diisocyanate-induced AHR and airway neutrophilia with a reduced Th17 response (117).
Other cytokines, IL-21 activate Th17 in an autocrine manner (118), while TNF-α is associated with airway inflammation in severe asthma. Patients with neutrophilic asthma were shown to have mucosa-associated invariant T cells that were predisposed to the Th17 subtype, which released higher levels of IL-17A and TNF-α and lower levels of IFN-γ (119). Niessen et al. found that sputum TNF signaling protein was more significantly elevated in neutrophilic asthma and that elevated sputum TNF signaling protein in patients with severe asthma was associated with poorer lung function and worse asthma control (120). In addition, TNF-α in combination with IL-17 altered the bronchial epithelial cell proteome to enhance proteins that promote neutrophil migration (121).
Th17-related cytokines are also relevant to obesity-associated asthma. Late-onset asthma patients with typical features of obesity-associated asthma are mostly female and involve neutrophilic airway inflammation and oxidative stress (122). In mice with obesity-associated asthma, TNF-α increases metalloproteinase (MMP)-9 and in situ gelatinase activity and contributes to airway remodeling (123). According to multiple research, children, and adults with obesity-associated asthma had higher peripheral blood levels of IL-17A, IL-21, and TNF-α than non-obese patients (124, 125).
IL-22 is produced by Th22, but this cytokine can also be secreted by other cells including Th1, Th2, and Th17 (13). Similar to the results in mice, the study found elevated serum and sputum IL-22 levels in asthma patients compared to healthy samples suggesting that IL-22 may be involved in the pathogenesis of asthma (126, 127). Although some studies suggest that IL-22 may exhibit inhibitory effects on airway inflammation in asthma by inhibiting the action of IFN-γ on epithelial cells (13), there are still few studies exploring the mechanisms of IL-22 and human asthma pathophysiology.
TNF-α, which is released by Th1 cells, Th17 cells, mast cells, and macrophages, is thought to be an attractive target in severe asthma. A case report identified that asthma symptoms may be masked by anti-TNF-α therapy, suggesting that anti-TNF-α may have a therapeutic effect on asthma (128). The recombinant protein Etanercept (ETN), which binds TNF with a high affinity, is made up of two molecules of the extracellular region of the p75 TNF-α receptor. In the Holgate et al. randomized controlled study, patients with asthma showed no discernible changes in treatment objectives between ETN and placebo (129). Another study found that administration of ETN for 12 weeks modestly reduced AHR, improved asthma symptom control, quality of life, and lung function, and reduced the expression levels of membrane-bound TNF on peripheral blood mononuclear cells (130). Other anti-TNF-α treatments, such as infliximab, adalimumab, and golimumab, have similarly failed to provide promising outcomes (131). Although anti-TNF-α has been shown to improve asthma control in animal models, patients with severe asthma have not benefited from this therapy because there is limited evidence of improved lung function and reduced asthma exacerbation. More studies are needed to further explore it.
IL-6 can induce Th17 differentiation, and blocking IL-6 may be a potential target for the treatment of asthma. Tocilizumab, a humanized anti-IL-6 receptor (IL-6R) mAb, was used in two patients with recurrent acute exacerbations of asthma, both of whom showed a good clinical response and reduced hospital admissions (132). Clazakizumab, another anti-IL-6 monoclonal antibody, is part of the PrecISE clinical trial, which uses a biomarker-guided approach to identify patient groups most likely to respond to one of six predefined interventions (133). Clazakizumab’s target population is defined as a subgroup characterized by obesity and metabolic dysfunction. Other clinical trials on targeted IL-6 interventions for asthma are not currently well underway, and a phase 2a study evaluating the impact of Sirukumab in subjects with poorly controlled severe asthma was terminated in early may due to safety concerns (NCT02794519). Another study evaluating the efficacy and safety of FB704A in adults with severe asthma is still enrolling patients with uncertain results (NCT05018299).
IL-23 is a pro-inflammatory cytokine regulated in Th17 and targeting IL-23 may be particularly beneficial in neutrophilic asthma. In a randomized, double-blind, placebo-controlled phase 2a study, IL-23 p19 inhibitor, Risankizumab was used in adult patients with severe asthma. However, disappointingly the study did not meet the primary endpoint. time to first asthma worsening was shorter in the risankizumab group than in the placebo group, and the annualized rate of asthma worsening was higher in the risankizumab group than in the placebo group. Lung function, ACQ-5 scores, and the incidence of severe asthma exacerbations were similar in both groups (134).
Targeting IL-17 in neutrophilic asthma is also being studied continuously. Brodalumab is an anti-IL-17 receptor monoclonal antibody for which Busse et al. conducted a placebo-controlled phase 2a clinical trial for the treatment of moderate-to-severe asthma. Subjects requested discontinuation of a long-acting beta-agonist (LABA) for the duration of the study, and inhaled glucocorticoids were available. At week 12, Brodalumab did not produce any statistically significant benefit in terms of ACQ scores, FEV1, or short-acting β2-agonist (SABA) use (135). BITS7201A is a novel humanized bispecific antibody that binds and neutralizes IL-13 and IL-17. A phase I trial investigating the effect of BITS7201A on asthma was terminated early due to high antibodies to therapeutic agents (136). Secukinumab, a fully human anti-IL-17A monoclonal antibody, is now used in rheumatic immune diseases, and a clinical trial has attempted to evaluate its role in patients with asthma not adequately controlled with inhaled corticosteroids but has failed to obtain satisfactory results (NCT01478360). Badi et al. found that the degree of enrichment of similar transcriptomic features in atopic dermatitis, including overexpression of Th17 and Th22-related genes, was strongly associated with severe neutrophilic asthma. Then, patients with severe neutrophilic asthma were identified as potential responders to Fezakinumab (anti-IL-22, currently approved for atopic dermatitis treatment) therapy (137). Although some trials have reported different data against Th17-related cytokines, the safety, and efficacy remain unsatisfactory. Perhaps the selection of a more appropriate “type-2 low” asthma patient for precisely targeted therapy is the key to successful trial progress. Current biological agents in “Type-2 low” asthma are summarized in Table 2.
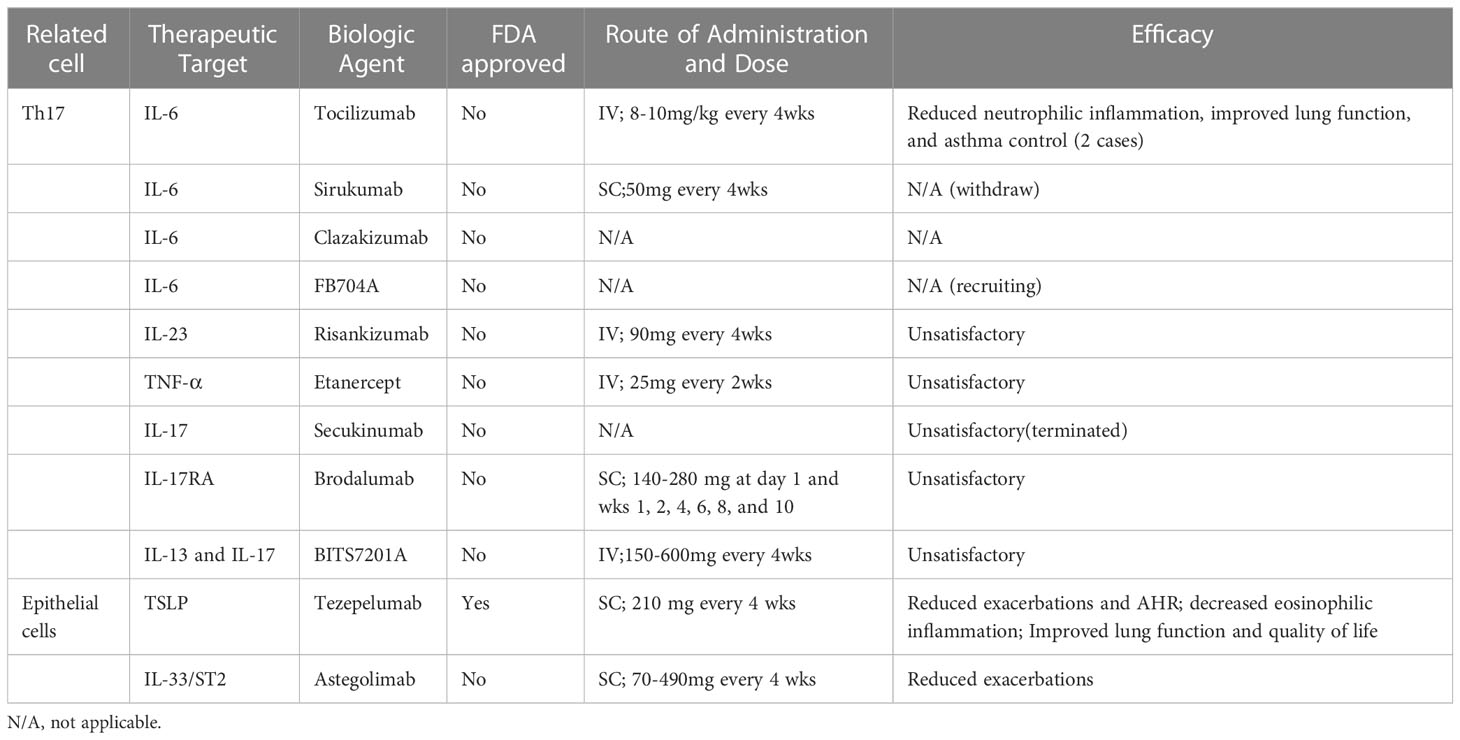
Table 2 Current biological agents in “Type-2 low” asthma.
The current focus of asthma treatment remains on pharmacotherapy, and the main drugs can be divided into bronchodilators such as SABA which provide rapid symptomatic relief by relaxing ASM and control drugs that inhibit inflammation represented by inhaled corticosteroids (ICS) (138). In the confrontation of severe asthma, additional biologic therapies can significantly reduce severe acute exacerbations and ICS exposure (138). In addition, other types 2 coexisting conditions (allergic rhinitis, eczema/atopic dermatitis, urticaria, etc.) are associated with a higher risk of exacerbations and lower asthma control, and clinicians may consider assessing the burden of type 2 comorbidities when evaluating patients with asthma (139). Th cells immune response plays an important role in the initiation and persistence of asthma, and biological agents targeting its cytokines and pathways are constantly updated (Figure 3). The vast majority of FDA-approved biologics (including benralizumab, dupilumab, mepolizumab, et al) are for “Type-2 high” asthma, with indications for severe eosinophilic asthma and/or oral corticosteroid-dependent asthma. Typical eligibility criteria for each category, as well as predictors of a good response, can be found in the GINA pocket guide decision tree (140). In the ALLIANCE cohort, patients in all age groups exhibited a T2-high phenotype characterized by eosinophilia and atopy, with the highest prevalence of T2-high asthma in school-aged children and young adults. Using easily accessible biomarkers, patients with T2-high asthma can be identified across all ages delineating a distinct phenotype. These patients may benefit from therapy with biologicals even at a younger age (141). Some other biologics with similar targets have achieved satisfactory clinical results and are progressing to the next stage of the study. In contrast, the progress of biologics for “Type-2 low “ asthma has been bumpy. Tezepelumab, the only indication for severe asthma (not just eosinophilic asthma), was found to be also effective regardless of key biomarkers (67). Astegolimab did a similar effect in clinical trials (71). Other biologics targeting Th1 or Th17 have shown poor results in several clinical trials. It is known that “Th1, Th17” may play essential roles in “Type-2-low “ asthma, and a more in-depth study of the pathological mechanism and the selection of the appropriate patient population based on the corresponding clinical indicators may bring about a turnaround. Larger studies are needed to investigate which asthma phenotypes benefit most from biologics that target Th17 and Th1-related cytokines.
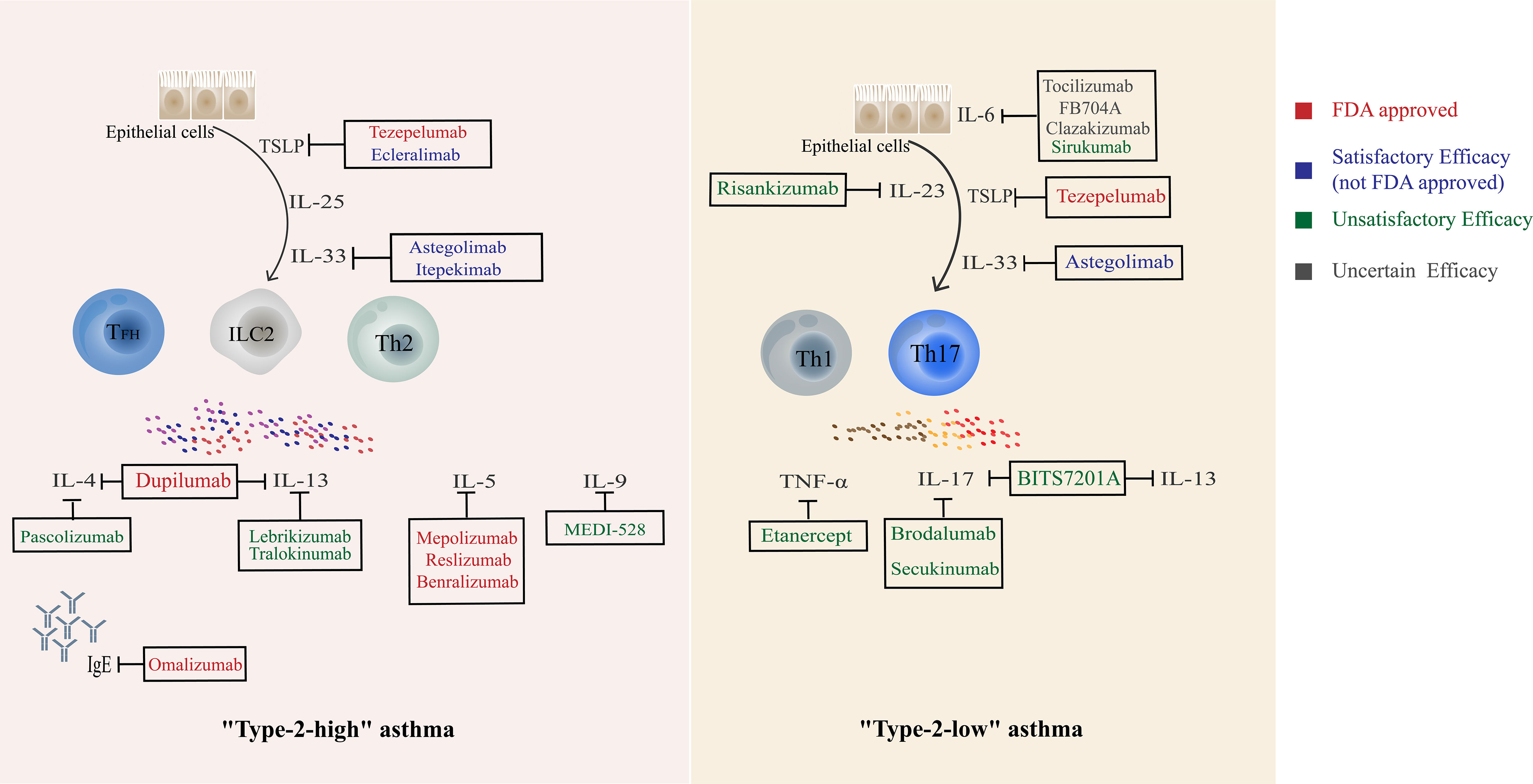
Figure 3 Current biological agents targeting Th cells and related cytokines.
AIT is the only disease-modifying and potentially preventive asthma treatment based on repeated injections or sublingual administration of specific allergens to allergic individuals to induce immune tolerance (142). The mechanism of AIT is primarily based on Treg cells, which downregulate allergic inflammation by producing the cytokines IL-10 and TGF-β (142). AIT slows local airway inflammation and inhibit pro-inflammatory CXCL8, IL24, and CCL26 mRNA expression (143). In addition, chronic allergen exposure resulted in high expression of CTLA-4 and PD-1 in Th2 cells and the prolonged presence of pro-allergic Th2 cells with a depleted phenotype during AIT may explain the long-lasting effect of AIT treatment (144). Several biomarkers were identified that may predict the clinical efficacy of AIT, such as the production of allergen-specific IgG or IgG4 and IgA in serum, suppression of local Th2/ILC2 cell numbers, or secretion of type 2 cytokines, increase in Th1 cell, Treg or Bregs numbers, and induction of a recently introduced anti-inflammatory mediator, secretoglobin 1A1, in the local environment (143, 145). An interesting study hypothesized three consecutive phases of AIT, namely an initiation, a conversion, and a tolerance mounting phase. In this study, the ratio of IL-10 B-cells and Th17 cells during the early initiation phase corresponded to symptom improvement after three years of treatment, representing a potential decision point for treatment adjustment before long-term treatment (146). AIT as an evolving area of asthma treatment still needs to be better understood.
Complex interactions between cellular participants and the innate and adaptive immune systems in response to multiple allergenic stimuli coordinate the immune response in the lung. In Type-2 high asthma, TFH cells control IgE synthesis by secreting IL-4 to allergen-specific B cells. Epithelial-derived TLPS, IL-25, and IL-33 can awaken ILC2 and Th2, while IL-4, IL-5, and IL-13 play a central role in the subsequent series of transformations. In the past few years, significant advances in Th17 and Th1 cell research have revealed new aspects of the “Type-2 low” asthma cellular machinery. Th1 and Th17-related cytokines act as central effector mediators and continuity factors in airway inflammatory storms and airway injury.
At present, the efficacy and safety of various biologics for Th2 responses have been well evaluated. Several biologics have been approved by the FDA for the treatment of “Type 2 high” asthma, such as benralizumab, dupilumab, and mepolizumab. They have demonstrated favorable results in clinical trials and real-world studies on reducing exacerbations, improving quality of life and lung function in patients with severe asthma, and even reducing OCS doses. However, various biologics for Th1 and Th17 are still struggling to move forward. Two biological agents (Tezepelumab and Astegolimab) have shown partial effects on “Type-2 low” asthma by blocking epithelial cell cytokines. More in-depth study of the pathological mechanism and the selection of the appropriate patient population based on the corresponding clinical indicators may bring about a turnaround. There is no silver bullet in asthma treatment. Asthma treatment may be individualized in the future. Perhaps more in-depth research on Th cells and big data analysis to match different clinical manifestations of asthma with specific Th cells for targeted and individualized treatment will be the new direction in the future.
TJ, access to literatures and writing of the manuscript. HL, revising the manuscript. Both authors contributed to the article and approved the submitted version.
The National Natural Science Foundation of China (81970015).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lameire S, Hammad H. Lung epithelial cells: upstream targets in type 2-high asthma. Eur J Immunol (2023) 13:975914. doi: 10.1002/eji.202250106
2. Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity (2019) 50:975–91. doi: 10.1016/j.immuni.2019.03.018
3. Curto E, Mateus-Medina ÉF, Crespo-Lessmann A, Osuna-Gómez R, Ujaldón-Miró C, García-Moral A, et al. Identification of two eosinophil subsets in induced sputum from patients with allergic asthma according to CD15 and CD66b expression. Int J Environ Res Public Health (2022) 19(20):13400. doi: 10.3390/ijerph192013400
4. Flinkman E, Vähätalo I, Tuomisto LE, Lehtimäki L, Nieminen P, Niemelä O, et al. Association between blood eosinophils and neutrophils with clinical features in adult-onset asthma. J Allergy Clin Immunol Pract (2023) 11(3):811–21. doi: 10.1016/j.jaip.2022.11.025
5. Thompson D, Wood LG, Williams EJ, McLoughlin RF, Rastogi D. Endotyping pediatric obesity-related asthma: contribution of anthropometrics, metabolism, nutrients, and CD4+ lymphocytes to pulmonary function. J Allergy Clin Immunol (2022) 150:861–71. doi: 10.1016/j.jaci.2022.04.033
6. Gandhi NA, Bennett BL, Graham NMH, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov (2016) 15:35–50. doi: 10.1038/nrd4624
7. Wei X, Niu X. T Follicular helper cells in autoimmune diseases. J Autoimmun (2023) 134:102976. doi: 10.1016/j.jaut.2022.102976
8. Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, et al. Cytokine-mediated regulation of human b cell differentiation into ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol (2007) 179:8180–90. doi: 10.4049/jimmunol.179.12.8180
9. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science (2019) 365(6456):eaaw6433. doi: 10.1126/science.aaw6433
10. Xie Y, Abel PW, Casale TB, Tu Y. TH17 cells and corticosteroid insensitivity in severe asthma. J Allergy Clin Immunol (2022) 149:467–79. doi: 10.1016/j.jaci.2021.12.769
11. Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High IFN-γ and low SLPI mark severe asthma in mice and humans. J Clin Invest (2015) 125:3037–50. doi: 10.1172/JCI80911
12. Badolati I, Sverremark-Ekström E, van der Heiden M. Th9 cells in allergic diseases: a role for the microbiota? Scand J Immunol (2020) 91(4):e12857. doi: 10.1111/sji.12857
13. Pennino D, Bhavsar PK, Effner R, Avitabile S, Venn P, Quaranta M, et al. IL-22 suppresses IFN-γ-mediated lung inflammation in asthmatic patients. J Allergy Clin Immunol (2013) 131:562–70. doi: 10.1016/j.jaci.2012.09.036
14. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
15. Huang ZX, Qiu ZE, Chen L, Hou XC, Zhu YX, Zhou WL, et al. Cellular mechanism underlying the facilitation of contractile response induced by IL-25 in mouse tracheal smooth muscle. Am J Physiol Lung Cell Mol Physiol (2022) 323:L27–36. doi: 10.1152/ajplung.00468.2021
16. Peng B, Sun L, Zhang M, Yan H, Shi G, Xia Z, et al. Role of IL-25 on eosinophils in the initiation of Th2 responses in allergic asthma. Front Immunol (2022) 13:842500. doi: 10.3389/fimmu.2022.842500
17. Tworek D, Smith SG, Salter BM, Baatjes AJ, Scime T, Watson R, et al. IL-25 receptor expression on airway dendritic cells after allergen challenge in subjects with asthma. Am J Respir Crit Care Med (2016) 193:957–64. doi: 10.1164/rccm.201509-1751OC
18. Cheng D, Xue Z, Yi L, Shi H, Zhang K, Huo X, et al. Epithelial interleukin-25 is a key mediator in Th2-high, corticosteroid-responsive asthma. Am J Respir Crit Care Med (2014) 190:639–48. doi: 10.1164/rccm.201403-0505OC
19. Saikumar Jayalatha AK, Hesse L, Ketelaar ME, Koppelman GH, Nawijn MC. The central role of IL-33/IL-1RL1 pathway in asthma: from pathogenesis to intervention. Pharmacol Ther (2021) 225:107847. doi: 10.1016/j.pharmthera.2021.107847
20. Werder RB, Ullah A, Rahman MM, Simpson J, Lynch JP, Collinson N, et al. Targeting the P2Y13 receptor suppresses IL-33 and HMGB1 release and ameliorates experimental asthma. Am J Respir Crit Care Med (2022) 205:300–12. doi: 10.1164/rccm.202009-3686OC
21. Li Y, Wang W, Lv Z, Li Y, Chen Y, Huang K, et al. Elevated expression of IL-33 and TSLP in the airways of human asthmatics In vivo: a potential biomarker of severe refractory disease. J Immunol (2018) 200:2253–62. doi: 10.4049/jimmunol.1701455
22. Schröder A, Lunding LP, Zissler UM, Vock C, Webering S, Ehlers JC, et al. IL-37 regulates allergic inflammation by counterbalancing pro-inflammatory IL-1 and IL-33. Allergy (2022) 77:856–69. doi: 10.1111/all.15072
23. Lin H, Li H. How does cigarette smoking affect airway remodeling in asthmatics? Tob Induc Dis (2023) 21:1–14. doi: 10.18332/tid/156047
24. Shikotra A, Choy DF, Ohri CM, Doran E, Butler C, Hargadon B, et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J Allergy Clin Immunol (2012) 129:104–11.e1-9. doi: 10.1016/j.jaci.2011.08.031
25. Ibrahim B, Achour D, Zerimech F, De Nadai P, Siroux V, Tsicopoulos A, et al. Plasma thymic stromal lymphopoietin (TSLP) in adults with non-severe asthma: the EGEA study. Thorax (2023) 78:207–10. doi: 10.1136/thorax-2022-219192
26. Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat Commun (2013) 4:1–7. doi: 10.1038/ncomms3675
27. Tanaka J, Watanabe N, Kido M, Saga K, Akamatsu T, Nishio A, et al. Human TSLP and TLR3 ligands promote differentiation of Th17 cells with a central memory phenotype under Th2-polarizing conditions. Clin Exp Allergy (2009) 39:89–100. doi: 10.1111/j.1365-2222.2008.03151.x
28. Mitchell PD, Salter BM, Oliveria JP, El-Gammal A, Tworek D, Smith SG, et al. IL-33 and its receptor ST2 after inhaled allergen challenge in allergic asthmatics. Int Arch Allergy Immunol (2018) 176:133–42. doi: 10.1159/000488015
29. Jones CP, Gregory LG, Causton B, Campbell GA, Lloyd CM. Activin a and TGF-β promote T(H)9 cell-mediated pulmonary allergic pathology. J Allergy Clin Immunol (2012) 129(4):1000–10.e3. doi: 10.1016/j.jaci.2011.12.965
30. Zissler UM, Chaker AM, Effner R, Ulrich M, Guerth F, Piontek G, et al. Interleukin-4 and interferon-γ orchestrate an epithelial polarization in the airways. Mucosal Immunol (2016) 9:917–26. doi: 10.1038/mi.2015.110
31. Weckmann M, Bahmer T, Sand JM, Rønnow SR, Pech M, Vermeulen C, et al. COL4A3 is degraded in allergic asthma and degradation predicts response to anti-IgE therapy. Eur Respir J (2021) 58(6):2003969. doi: 10.1183/13993003.03969-2020
32. Andrea M, Susanna B, Francesca N, Enrico M, Alessandra V. The emerging role of type 2 inflammation in asthma. Expert Rev Clin Immunol (2021) 17:63–71. doi: 10.1080/1744666X.2020.1860755
33. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential “inflammatory” type 2 innate lymphoid cells. Nat Immunol (2015) 16:161–9. doi: 10.1038/ni.3078
34. Mjösberg JM, Trifari S, Crellin NK, Peters CP, Van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12:1055–62. doi: 10.1038/ni.2104
35. Koch S, Sopel N, Finotto S. Th9 and other IL-9-producing cells in allergic asthma. Semin Immunopathol (2017) 39:55–68. doi: 10.1007/s00281-016-0601-1
36. Hynes GM, Hinks TSC. The role of interleukin-17 in asthma: a protective response? ERJ Open Res (2020) 6:00364–2019. doi: 10.1183/23120541.00364-2019
37. Nagase H, Ueki S, Fujieda S. The roles of IL-5 and anti-IL-5 treatment in eosinophilic diseases: asthma, eosinophilic granulomatosis with polyangiitis, and eosinophilic chronic rhinosinusitis. Allergol Int (2020) 69:178–86. doi: 10.1016/j.alit.2020.02.002
38. Gour N, Wills-Karp M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine (2015) 75:68–78. doi: 10.1016/j.cyto.2015.05.014
39. Akdis CA, Arkwright PD, Brüggen MC, et al. Type 2 immunity in the skin and lungs. Allergy Eur J Allergy Clin Immunol (2020) 75:1582–605. doi: 10.1111/all.14318
40. Zissler UM, Ulrich M, Jakwerth CA, Rothkirch S, Guerth F, Weckmann M, et al. Biomatrix for upper and lower airway biomarkers in patients with allergic asthma. J Allergy Clin Immunol (2018) 142:1980–3. doi: 10.1016/j.jaci.2018.07.027
41. Feng K, Meng P, Zou X, Zhang M, Li H, Yang H, et al. IL-37 protects against airway remodeling by reversing bronchial epithelial-mesenchymal transition via IL-24 signaling pathway in chronic asthma. Respir Res (2022) 23(1):244. doi: 10.1186/s12931-022-02167-7
42. Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol (2006) 24:147–74. doi: 10.1146/annurev.immunol.24.021605.090720
43. Dolitzky A, Grisaru-Tal S, Avlas S, Hazut I, Gordon Y, Itan M, et al. Mouse resident lung eosinophils are dependent on IL-5. Allergy (2022) 77:2822–5. doi: 10.1111/all.15362
44. Marc-Malovrh M, Camlek L, Škrgat S, Kern I, Fležar M, Dežman M, et al. Elevated eosinophils, IL5 and IL8 in induced sputum in asthma patients with accelerated FEV1 decline. Respir Med (2020) 162:105875. doi: 10.1016/j.rmed.2020.105875
45. Palacionyte J, Januskevicius A, Vasyle E, Rimkunas A, Bajoriuniene I, Miliauskas S, et al. IL-5 and GM-CSF, but not IL-3, promote the proliferative properties of inflammatory-like and lung resident-like eosinophils in the blood of asthma patients. Cells (2022) 11(23):3804. doi: 10.3390/cells11233804
46. Malm-Erjefält M, Greiff L, Ankerst J, Andersson M, Wallengren J, Cardell LO, et al. Circulating eosinophils in asthma, allergic rhinitis, and atopic dermatitis lack morphological signs of degranulation. Clin Exp Allergy (2005) 35:1334–40. doi: 10.1111/j.1365-2222.2005.02335.x
47. Al-Shaikhly T, Murphy RC, Parker A, Lai Y, Altman MC, Larmore M, et al. Location of eosinophils in the airway wall is critical for specific features of airway hyperresponsiveness and T2 inflammation in asthma. Eur Respir J (2022) 60(2):21018. doi: 10.1183/13993003.01865-2021
48. Ueki S, Mahemuti G, Oyamada H, Kato H, Kihara J, Tanabe M, et al. Retinoic acids are potent inhibitors of spontaneous human eosinophil apoptosis. J Immunol (2008) 181:7689–98. doi: 10.4049/jimmunol.181.11.7689
49. Ying S, Meng Q, Barata LT, Robinson DS, Durham SR, Kay AB. Associations between IL-13 and IL-4 (mRNA and protein), vascular cell adhesion molecule-1 expression, and the infiltration of eosinophils, macrophages, and T cells in allergen-induced late-phase cutaneous reactions in atopic subjects. J Immunol (1997) 158:5050–7. doi: 10.4049/jimmunol.158.10.5050
50. Manson ML, Säfholm J, James A, Johnsson AK, Bergman P, Al-Ameri M, et al. IL-13 and IL-4, but not IL-5 nor IL-17A, induce hyperresponsiveness in isolated human small airways. J Allergy Clin Immunol (2020) 145:808–17.e2. doi: 10.1016/j.jaci.2019.10.037
51. Allahverdian S, Harada N, Singhera GK, Knight DA, Dorscheid DR. Secretion of IL-13 by airway epithelial cells enhances epithelial repair via HB-EGF. Am J Respir Cell Mol Biol (2008) 38:153–60. doi: 10.1165/rcmb.2007-0173OC
52. Grunstein MM, Hakonarson H, Leiter J, Chen M, Whelan R, Grunstein JS, et al. IL-13-dependent autocrine signaling mediates altered responsiveness of IgE-sensitized airway smooth muscle. Am J Physiol Lung Cell Mol Physiol (2002) 38(2):153–60. doi: 10.1152/ajplung.00343.2001
53. Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, León B, et al. T Follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust mite. Immunity (2016) 44:259–73. doi: 10.1016/j.immuni.2015.11.017
54. Kobayashi T, Iijima K, Dent AL, Kita H. Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol (2017) 139:300–13.e7. doi: 10.1016/j.jaci.2016.04.021
55. Sakurai S, Furuhashi K, Horiguchi R, Nihashi F, Yasui H, Karayama M, et al. Conventional type 2 lung dendritic cells are potent inducers of follicular helper T cells in the asthmatic lung. Allergol Int (2021) 70:351–9. doi: 10.1016/j.alit.2021.01.008
56. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Circulating CXCR5+CD4+ T cells participate in the IgE accumulation in allergic asthma. Immunol Lett (2018) 197:9–14. doi: 10.1016/j.imlet.2018.03.001
57. Gong F, Zhu HY, Zhu J, Dong QJ, Huang X, Jiang DJ. Allergen-specific IgG+ memory b cells are temporally linked to IgE memory responses. J Allergy Clin Immunol (2020) 146:180–91. doi: 10.1016/j.jaci.2019.11.046
58. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell (2021) 184:1469–85. doi: 10.1016/j.cell.2021.02.016
59. Zhang X, Ma Y, He Y, Gu W, Yan Y, Ji W, et al. Foxp2 inhibits Th9 cell differentiation and attenuates allergic airway inflammation in a mouse model of ovalbumin-induced asthma. Int Immunopharmacol (2022) 111:109060. doi: 10.1016/j.intimp.2022.109060
60. Seumois G, Ramírez-Suástegui C, Schmiedel BJ, Liang S, Peters B, Sette A, et al. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci Immunol (2020) 5(48):eaba6087. doi: 10.1126/sciimmunol.aba6087
61. Kerzerho J, Maazi H, Speak AO, Szely N, Lombardi V, Khoo B, et al. Programmed cell death ligand 2 regulates TH9 differentiation and induction of chronic airway hyperreactivity. J Allergy Clin Immunol (2013) 131(4):1048–57, 1057.e1-2. doi: 10.1016/j.jaci.2012.09.027
62. Musiol S, Alessandrini F, Jakwerth CA, Chaker AM, Schneider E, Guerth F, et al. TGF-β1 drives inflammatory Th cell but not treg cell compartment upon allergen exposure. Front Immunol (2022) 12:763243. doi: 10.3389/fimmu.2021.763243
63. Chaker AM, Zissler UM, Poulos N, Wagenmann M, Bas M, Gürth F, et al. Activin-a is a pro-inflammatory regulator in type-2-Driven upper airway disease. Int Arch Allergy Immunol (2018) 176:15–25. doi: 10.1159/000487930
64. Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med (2021) 384:1800–9. doi: 10.1056/NEJMoa2034975
65. Diver S, Khalfaoui L, Emson C, Wenzel SE, Menzies-Gow A, Wechsler ME, et al. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med (2021) 9:1299–312. doi: 10.1016/S2213-2600(21)00226-5
66. Sverrild A, Hansen S, Hvidtfeldt M, Clausson CM, Cozzolino O, Cerps S, et al. The effect of tezepelumab on airway hyperresponsiveness to mannitol in asthma (UPSTREAM). Eur Respir J (2021) 59(1):2101296.
67. Corren J, Pham TH, Garcia Gil E, Sałapa K, Ren P, Parnes JR, et al. Baseline type 2 biomarker levels and response to tezepelumab in severe asthma. Allergy (2022) 77:1786–96. doi: 10.1111/all.15197
68. Gauvreau GM, Hohlfeld JM, FitzGerald MJ, Boulet L-P, Cockcroft DW, Davis BE, et al. Inhaled anti-TSLP antibody fragment, ecleralimab, blocks responses to allergen in mild asthma. Eur Respir J (2023) 61(3):2201193. doi: 10.1183/13993003.01193-2022
69. Kosloski MP, Kalliolias GD, Xu CR, Harel S, Lai CH, Zheng W, et al. Pharmacokinetics and pharmacodynamics of itepekimab in healthy adults and patients with asthma: phase I first-in-human and first-in-patient trials. Clin Transl Sci (2022) 15:384–95. doi: 10.1111/cts.13157
70. Wechsler ME, Ruddy MK, Pavord ID, Israel E, Rabe KF, Ford LB, et al. Efficacy and safety of itepekimab in patients with moderate-to-Severe asthma. N Engl J Med (2021) 385:1656–68. doi: 10.1056/NEJMoa2024257
71. Kelsen SG, Agache IO, Soong W, Israel E, Chupp GL, Cheung DS, et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J Allergy Clin Immunol (2021) 148:790–8. doi: 10.1016/j.jaci.2021.03.044
72. Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med (2014) 371:1198–207. doi: 10.1056/NEJMoa1403290
73. Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, Bardin P, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med (2015) 3:355–66. doi: 10.1016/S2213-2600(15)00042-9
74. Chupp GL, Bradford ES, Albers FC, Bratton DJ, Wang-Jairaj J, Nelsen LM, et al. Efficacy of mepolizumab add-on therapy on health-related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): a randomised, double-blind, placebo-controlled, parallel-group, multicentre, phase 3b trial. Lancet Respir Med (2017) 5:390–400. doi: 10.1016/S2213-2600(17)30125-X
75. Jackson DJ, Bacharier LB, Gergen PJ, Gagalis L, Calatroni A, Wellford S, et al. Mepolizumab for urban children with exacerbation-prone eosinophilic asthma in the USA (MUPPITS-2): a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet (London England) (2022) 400:502–11. doi: 10.1016/S0140-6736(22)01198-9
76. Nair P, Bardin P, Humbert M, Murphy KR, Hickey L, Garin M, et al. Efficacy of intravenous reslizumab in oral corticosteroid-dependent asthma. J Allergy Clin Immunol Pract (2020) 8:555–64. doi: 10.1016/j.jaip.2019.09.036
77. Harrison T, Canonica GW, Chupp G, Lee J, Schleich F, Welte T, et al. Real-world mepolizumab in the prospective severe asthma REALITI-a study: initial analysis. Eur Respir J (2020) 56(4):2000151. doi: 10.1183/13993003.00151-2020
78. Pilette C, Canonica GW, Chaudhuri R, Chupp G, Lee FE-H, Lee JK, et al. REALITI-a study: real-world oral corticosteroid-sparing effect of mepolizumab in severe asthma. J Allergy Clin Immunol Pract (2022) 10(10):2646–56. doi: 10.1016/j.jaip.2022.05.042
79. FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (London England) (2016) 388:2128–41. doi: 10.1016/S0140-6736(16)31322-8
80. Nair P, Wenzel S, Rabe KF, Bourdin A, Lugogo NL, Kuna P, et al. Oral glucocorticoid-sparing effect of benralizumab in severe asthma. N Engl J Med (2017) 376:2448–58. doi: 10.1056/NEJMoa1703501
81. Hanania NA, Korenblat P, Chapman KR, Bateman ED, Kopecky P, Paggiaro P, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med (2016) 4:781–96. doi: 10.1016/S2213-2600(16)30265-X
82. Szefler SJ, Roberts G, Rubin AS, Zielen S, Kuna P, Alpan O, et al. Efficacy, safety, and tolerability of lebrikizumab in adolescent patients with uncontrolled asthma (ACOUSTICS). Clin Transl Allergy (2022) 12(7):e12176. doi: 10.1002/clt2.12176
83. Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med (2011) 365:1088–98. doi: 10.1056/NEJMoa1106469
84. Busse WW, Brusselle GG, Korn S, Kuna P, Magnan A, Cohen D, et al. Tralokinumab did not demonstrate oral corticosteroid-sparing effects in severe asthma. Eur Respir J (2019) 53(2):1800948. doi: 10.1183/13993003.00948-2018
85. Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab efficacy and safety in moderate-to-Severe uncontrolled asthma. N Engl J Med (2018) 378:2486–96. doi: 10.1056/NEJMoa1804092
86. Corren J, Castro M, O’Riordan T, Hanania NA, Pavord ID, Quirce S, et al. Dupilumab efficacy in patients with uncontrolled, moderate-to-Severe allergic asthma. J Allergy Clin Immunol Pract (2020) 8:516–26. doi: 10.1016/j.jaip.2019.08.050
87. Sher LD, Wechsler ME, Rabe KF, Maspero JF, Daizadeh N, Mao X, et al. Dupilumab reduces oral corticosteroid use in patients with corticosteroid-dependent severe asthma: an analysis of the phase 3, open-label extension TRAVERSE trial. Chest (2022) 162:46–55. doi: 10.1016/j.chest.2022.01.071
88. Wechsler ME, Ford LB, Maspero JF, Pavord ID, Papi A, Bourdin A, et al. Long-term safety and efficacy of dupilumab in patients with moderate-to-severe asthma (TRAVERSE): an open-label extension study. Lancet Respir Med (2022) 10:11–25. doi: 10.1016/S2213-2600(21)00322-2
89. Normansell R, Walker S, Milan SJ, Walters EH, Nair P. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev (2014) 1:CD003559. doi: 10.1002/14651858.CD003559.pub4
90. Bousquet J, Humbert M, Gibson PG, Kostikas K, Jaumont X, Pfister P, et al. Real-world effectiveness of omalizumab in severe allergic asthma: a meta-analysis of observational studies. J Allergy Clin Immunol Pract (2021) 9:2702–14. doi: 10.1016/j.jaip.2021.01.011
91. Arslan B, Çetin GP, Türk M, Gülmez I, Yllmaz I. Discontinuing omalizumab treatment in super-responder patients with allergic severe asthma: can the baseline total IgE level be used as a biological marker to decide discontinuing omalizumab treatment? Int Arch Allergy Immunol (2022) 183:1071–7. doi: 10.1159/000525723
92. Hanania NA, Wenzel S, Roseń K, Hsieh HJ, Mosesova S, Choy DF, et al. Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med (2013) 187:804–11. doi: 10.1164/rccm.201208-1414OC
93. Parker JM, Oh CK, LaForce C, Miller SD, Pearlman DS, Le C, et al. Safety profile and clinical activity of multiple subcutaneous doses of MEDI-528, a humanized anti-interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm Med (2011) 11:14. doi: 10.1186/1471-2466-11-14
94. Oh CK, Leigh R, McLaurin KK, Kim K, Hultquist M, Molfino NA. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir Res (2013) 14(1):93. doi: 10.1186/1465-9921-14-93
95. Watanabe A, Mishima H, Renzi PM, Xu LJ, Hamid Q, Martin JG. Transfer of allergic airway responses with antigen-primed CD4+ but not CD8+ T cells in brown Norway rats. J Clin Invest (1995) 96:1303–10. doi: 10.1172/JCI118165
96. Mitchell C, Provost K, Niu N, Homer R, Cohn L. IFN-γ acts on the airway epithelium to inhibit local and systemic pathology in allergic airway disease. J Immunol (2011) 187:3815–20. doi: 10.4049/jimmunol.1100436
97. Kobayashi M, Ashino S, Shiohama Y, Wakita D, Kitamura H, Nishimura T. IFN-γ elevates airway hyper-responsiveness via up-regulation of neurokinin a/neurokinin-2 receptor signaling in a severe asthma model. Eur J Immunol (2012) 42:393–402. doi: 10.1002/eji.201141845
98. Oriss TB, Raundhal M, Morse C, Huff RE, Das S, Hannum R, et al. IRF5 distinguishes severe asthma in humans and drives Th1 phenotype and airway hyperreactivity in mice. JCI Insight (2017) 2(10):e91019. doi: 10.1172/jci.insight.91019
99. Wisniewski JA, Muehling LM, Eccles JD, Capaldo BJ, Agrawal R, Shirley DA, et al. TH1 signatures are present in the lower airways of children with severe asthma, regardless of allergic status. J Allergy Clin Immunol (2018) 141:2048–60.e13. doi: 10.1016/j.jaci.2017.08.020
100. Britt RD, Thompson MA, Sasse S, Pabelick CM, Gerber AN, Prakash YS. Th1 cytokines TNF-α and IFN-γ promote corticosteroid resistance in developing human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol (2019) 316:L71–81. doi: 10.1152/ajplung.00547.2017
101. Namakanova OA, Gorshkova EA, Zvartsev RV, Nedospasov SA, Drutskaya MS, Gubernatorova EO. Therapeutic potential of combining IL-6 and TNF blockade in a mouse model of allergic asthma. Int J Mol Sci (2022) 23(7):3521. doi: 10.3390/ijms23073521
102. Jackson D, Walum J, Banerjee P, Lewis BW, Prakash YS, Sathish V, et al. Th1 cytokines synergize to change gene expression and promote corticosteroid insensitivity in pediatric airway smooth muscle. Respir Res (2022) 23(1):126. doi: 10.1186/s12931-022-02046-1
103. Rastogi D. Pediatric obesity-related asthma: a prototype of pediatric severe non-T2 asthma. Pediatr Pulmonol (2020) 55:809–17. doi: 10.1002/ppul.24600
104. Aliyu M, Zohora FT, Anka AU, Ali K, Maleknia S, Saffarioun M, et al. Interleukin-6 cytokine: an overview of the immune regulation, immune dysregulation, and therapeutic approach. Int Immunopharmacol (2022) 111:109130. doi: 10.1016/j.intimp.2022.109130
105. Taherian M, Razavi AR, Izad M, Boghozian R, Namdari H, Ghayedi M, et al. The role of interleukin-23 in stability of in vitro T helper-17 cells. Iran J Allergy Asthma Immunol (2014) 13:131–7.
106. Wu AY, Peebles RS. The emerging role of IL-23 in asthma and its clinical implications. Expert Rev Clin Immunol (2023) 19:1–5. doi: 10.1080/1744666X.2023.2125380
107. Lee HS, Park DE, Lee JW, Chang Y, Kim HY, Song WJ, et al. IL-23 secreted by bronchial epithelial cells contributes to allergic sensitization in asthma model: role of IL-23 secreted by bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol (2017) 312:L13–21. doi: 10.1152/ajplung.00114.2016
108. Wakashin H, Hirose K, Maezawa Y, Kagami SI, Suto A, Watanabe N, et al. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med (2008) 178:1023–32. doi: 10.1164/rccm.200801-086OC
109. Halwani R, Sultana A, Vazquez-Tello A, Jamhawi A, Al-Masri AA, Al-Muhsen S, et al. Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J Asthma (2017) 54:893–904. doi: 10.1080/02770903.2017.1283696
110. Mizutani N, Nabe T, Yoshino S. IL-17A promotes the exacerbation of IL-33-induced airway hyperresponsiveness by enhancing neutrophilic inflammation via CXCR2 signaling in mice. J Immunol (2014) 192:1372–84. doi: 10.4049/jimmunol.1301538
111. Hong Y, Chu Z, Kong J, Li Q, Li N, Liu L, et al. IL-17A aggravates asthma-induced intestinal immune injury by promoting neutrophil trafficking. J Leukoc Biol (2022) 112:425–35. doi: 10.1002/JLB.3MA0622-426RR
112. Sorbello V, Ciprandi G, Di Stefano A, Massaglia GM, Favatà G, Conticello S, et al. Nasal IL-17F is related to bronchial IL-17F/neutrophilia and exacerbations in stable atopic severe asthma. Allergy (2015) 70:236–40. doi: 10.1111/all.12547
113. Ricciardolo FLM, Sorbello V, Folino A, Gallo F, Massaglia GM, Favatà G, et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol (2017) 140:395–406. doi: 10.1016/j.jaci.2016.10.034
114. Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Stefano Di A, et al. Elevated serum IgE, oral corticosteroid dependence and IL-17/22 expression in highly neutrophilic asthma. Eur Respir J (2019) 54(5):1900068. doi: 10.1183/13993003.00068-2019
115. Newcomb DC, Boswell MG, Sherrill TP, Polosukhin VV, Boyd KL, Goleniewska K, et al. IL-17A induces signal transducers and activators of transcription-6-independent airway mucous cell metaplasia. Am J Respir Cell Mol Biol (2013) 48:711–6. doi: 10.1165/rcmb.2013-0017OC
116. Panariti A, Baglole CJ, Sanchez V, Eidelman DH, Hussain S, Olivenstein R, et al. Interleukin-17A and vascular remodelling in severe asthma; lack of evidence for a direct role. Clin Exp Allergy (2018) 48:365–78. doi: 10.1111/cea.13093
117. Chen R, Zhang Q, Chen S, Tang H, Huang P, Wei S, et al. IL-17F, rather than IL-17A, underlies airway inflammation in a steroid-insensitive toluene diisocyanate-induced asthma model. Eur Respir J (2019) 53(4):1801510. doi: 10.1183/13993003.01510-2018
118. Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature (2007) 448:484–7. doi: 10.1038/nature05970
119. Wen X, Nian S, Wei G, Kang P, Yang Y, Li L, et al. Changes in the phenotype and function of mucosal-associated invariant T cells in neutrophilic asthma. Int Immunopharmacol (2022) 106:108606. doi: 10.1016/j.intimp.2022.108606
120. Niessen NM, Gibson PG, Baines KJ, Barker D, Yang IA, Upham JW, et al. Sputum TNF markers are increased in neutrophilic and severe asthma and are reduced by azithromycin treatment. Allergy (2021) 76:2090–101. doi: 10.1111/all.14768
121. Altieri A, Piyadasa H, Hemshekhar M, Osawa N, Recksiedler B, Spicer V, et al. Combination of IL-17A/F and TNF-α uniquely alters the bronchial epithelial cell proteome to enhance proteins that augment neutrophil migration. J Inflamm (Lond) (2022) 19(1):26. doi: 10.1186/s12950-022-00323-w
122. Suzuki Y, Aono Y, Akiyama N, Horiike Y, Naoi H, Horiguchi R, et al. Involvement of autophagy in exacerbation of eosinophilic airway inflammation in a murine model of obese asthma. Autophagy (2022) 18:2216–28. doi: 10.1080/15548627.2022.2025571
123. Vieira CP, de Oliveira LP, Da Silva MB, Majolli Andre D, Tavares EBG, Pimentel ER, et al. Role of metalloproteinases and TNF-α in obesity-associated asthma in mice. Life Sci (2020) 259:118191. doi: 10.1016/j.lfs.2020.118191
124. Al-ayed MS, Al-shaibari KS, Alshehri D, Alzahrani MJ, Nasser I, Alaamri HS, et al. Serum ghrelin levels in Saudi obese asthmatic school-Children-Correlation with interleukin-4, interleukin-5, and interleukin-21. Int J Environ Res Public Health (2020) 17(5):1656. doi: 10.3390/ijerph17051656
125. Leija-Martínez JJ, Del-Río-Navarro BE, Sanchéz-Muñoz F, Muñoz-Hernández O, Hong E, Giacoman-Martínez A, et al. Associations of TNFA, IL17A, and RORC mRNA expression levels in peripheral blood leukocytes with obesity-related asthma in adolescents. Clin Immunol (2021) 229:108715. doi: 10.1016/j.clim.2021.108715
126. Besnard AG, Sabat R, Dumoutier L, Renauld JC, Willart M, Lambrecht B, et al. Dual role of IL-22 in allergic airway inflammation and its cross-talk with IL-17A. Am J Respir Crit Care Med (2011) 183:1153–63. doi: 10.1164/rccm.201008-1383OC
127. Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, et al. The β-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J Immunol (2012) 189:3653–60. doi: 10.4049/jimmunol.1201797
128. Guilleminault L, Carré P, Beau-Salinas F, Taillé C, Dieudé P, Crestani B, et al. Asthma unmasked with tumor necrosis factor-α-blocking drugs. Chest (2011) 140:1068–71. doi: 10.1378/chest.10-2350
129. Holgate ST, Noonan M, Chanez P, Busse W, Dupont L, Pavord I, et al. Efficacy and safety of etanercept in moderate-to-severe asthma: a randomised, controlled trial. Eur Respir J (2011) 37:1352–9. doi: 10.1183/09031936.00063510
130. Morjaria JB, Chauhan AJ, Babu KS, Polosa R, Davies DE, Holgate ST. The role of a soluble TNFalpha receptor fusion protein (etanercept) in corticosteroid refractory asthma: a double blind, randomised, placebo controlled trial. Thorax (2008) 63:584–91. doi: 10.1136/thx.2007.086314
131. Ahmad S, Noor NM, Engku Nur Syafirah EAR, Irekeola AA, Shueb RH, Chan YY, et al. Anti-tumor necrosis factor for supplementary management in severe asthma: a systematic review and meta-analysis. J Interferon Cytokine Res (2023) 43:77–85. doi: 10.1089/jir.2022.0211
132. Esty B, Harb H, Bartnikas LM, Charbonnier LM, Massoud AH, Leon-Astudillo C, et al. Treatment of severe persistent asthma with IL-6 receptor blockade. J Allergy Clin Immunol Pract (2019) 7:1639–42.e4. doi: 10.1016/j.jaip.2019.02.043
133. Georas SN, Wright RJ, Ivanova A, Israel E, LaVange LM, Akuthota P, et al. The precision interventions for severe and/or exacerbation-prone (PrecISE) asthma network: an overview of network organization, procedures, and interventions. J Allergy Clin Immunol (2022) 149:488–516.e9. doi: 10.1016/j.jaci.2021.10.035
134. Brightling CE, Nair P, Cousins DJ, Louis R, Singh D. Risankizumab in severe asthma - a phase 2a, placebo-controlled trial. N Engl J Med (2021) 385:1669–79. doi: 10.1056/NEJMoa2030880
135. Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med (2013) 188:1294–302. doi: 10.1164/rccm.201212-2318OC
136. Staton TL, Peng K, Owen R, Choy DF, Cabanski CR, Fong A, et al. A phase I. Randomized, observer-blinded, single and multiple ascending-dose study to investigate the safety, pharmacokinetics, and immunogenicity of BITS7201A, a bispecific antibody targeting IL-13 and IL-17, in healthy volunteers. BMC Pulm Med (2019) 19(1):5. doi: 10.1186/s12890-018-0763-9
137. Badi YE, Pavel AB, Pavlidis S, Riley JH, Bates S, Kermani NZ, et al. Mapping atopic dermatitis and anti-IL-22 response signatures to type 2-low severe neutrophilic asthma. J Allergy Clin Immunol (2022) 149:89–101. doi: 10.1016/j.jaci.2021.04.010
138. Thomas D, McDonald VM, Pavord ID, Gibson PG. Asthma remission: what is it and how can it be achieved? Eur Respir J (2022) 60(5):2102583. doi: 10.1183/13993003.02583-2021
139. Price D, Menzies-Gow A, Bachert C, Canonica GW, Kocks J, Khan AH, et al. Association between a type 2 inflammatory disease burden score and outcomes among patients with asthma. J Asthma Allergy (2021) 14:1173–83. doi: 10.2147/JAA.S321212
140. Reddel HK, Bacharier LB, Bateman ED, Brightling CE, Brusselle GG, Buhl R, et al. Global initiative for asthma strategy 2021 executive summary and rationale for key changes. Am J Respir Crit Care Med (2022) 205:17–35. doi: 10.1164/rccm.202109-2205PP
141. Maison N, Omony J, Illi S, Thiele D, Skevaki C, Dittrich AM, et al. T2-high asthma phenotypes across lifespan. Eur Respir J (2022) 60(3):2102288. doi: 10.1183/13993003.02288-2021
142. Diamant Z, van Maaren M, Muraro A, Jesenak M, Striz I. Allergen immunotherapy for allergic asthma: the future seems bright. Respir Med (2023) 210:107125. doi: 10.1016/j.rmed.2023.107125
143. Zissler UM, Jakwerth CA, Guerth F, Lewitan L, Rothkirch S, Davidovic M, et al. Allergen-specific immunotherapy induces the suppressive secretoglobin 1A1 in cells of the lower airways. Allergy (2021) 76:2461–74. doi: 10.1111/all.14756
144. Wang SH, Zissler UM, Buettner M, Heine S, Heldner A, Kotz S, et al. An exhausted phenotype of TH 2 cells is primed by allergen exposure, but not reinforced by allergen-specific immunotherapy. Allergy (2021) 76:2827–39. doi: 10.1111/all.14896
145. Zissler UM, Schmidt-Weber CB. Predicting success of allergen-specific immunotherapy. Front Immunol (2020) 11:1826. doi: 10.3389/fimmu.2020.01826
Keywords: bronchial asthma, cell, Th2, cell, Th17, helper T cells, molecular targeted therapy
Citation: Ji T and Li H (2023) T-helper cells and their cytokines in pathogenesis and treatment of asthma. Front. Immunol. 14:1149203. doi: 10.3389/fimmu.2023.1149203
Received: 21 January 2023; Accepted: 30 May 2023;
Published: 12 June 2023.
Edited by:
Susetta Finotto, University Hospital Erlangen, GermanyReviewed by:
Xinyue Hu, Central South University, ChinaCopyright © 2023 Ji and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hequan Li, bGloZXF1YW5Aemp1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.