
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 19 July 2023
Sec. Systems Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1148684
 Rong Li1†
Rong Li1† Qi Guo2†
Qi Guo2† Jian Zhao1Wenhui Kang1Ruoyu Lu1
Jian Zhao1Wenhui Kang1Ruoyu Lu1 Zichong Long1
Zichong Long1 Lili Huang1
Lili Huang1 Yiting Chen1
Yiting Chen1 Anda Zhao3
Anda Zhao3 Jinhong Wu4
Jinhong Wu4 Yong Yin4*
Yong Yin4* Shenghui Li1*
Shenghui Li1*Background: Accumulating evidence has suggested that gut microbiota dysbiosis is commonly observed in asthmatics. However, it remains unclear whether dysbiosis is a cause or consequence of asthma. We aimed to examine the genetic causal relationships of gut microbiota with asthma and its three phenotypes, including adult-onset asthma, childhood-onset asthma, and moderate-severe asthma.
Methods: To elucidate the causality of gut microbiota with asthma, we applied two sample Mendelian randomization (MR) based on the largest publicly available genome-wide association study (GWAS) summary statistics. Inverse variance weighting meta-analysis (IVW) was used to obtain the main estimates; and Weighted median, MR-Egger, Robust Adjusted Profile Score (MR-RAPS), Maximum likelihood method (ML), and MR pleiotropy residual sum and outlier (MR-PRESSO) methods were applied in sensitivity analyses. Finally, a reverse MR analysis was performed to evaluate the possibility of reverse causation.
Results: In the absence of heterogeneity and horizontal pleiotropy, the IVW method revealed that genetically predicted Barnesiella and RuminococcaceaeUCG014 were positively correlated with the risk of asthma, while the association between genetically predicted CandidatusSoleaferrea and asthma was negative. And for the three phenotypes of asthma, genetically predicted Akkermansia reduced the risk of adult-onset asthma, Collinsella and RuminococcaceaeUCG014 increased the risk of childhood-onset asthma, and FamilyXIIIAD3011group, Eisenbergiella, and Ruminiclostridium6 were correlated with the risk of moderate-severe asthma (all P<0.05). The reverse MR analysis didn’t find evidence supporting the reverse causality from asthma and its three phenotypes to the gut microbiota genus.
Conclusion: This study suggested that microbial genera were causally associated with asthma as well as its three phenotypes. The findings deepened our understanding of the role of gut microbiota in the pathology of asthma, which emphasizes the potential of opening up a new vista for the prevention and diagnosis of asthma.
The human gastrointestinal system is a habitat for trillions of bacteria and archaea that live in mutual symbiosis with an individual’s body, and this symbiotic relationship is crucial to human health (1). Gut microbes are involved in a variety of physiological functions, including the maintenance of metabolic stability, the regulation of immune response, the resistance to infection, and et al (2). The gut microbiota has been confirmed to be implicated in disease susceptibility and progression by an increasing number of studies, and it is now regarded as an endocrine organ (3). Numerous connections between the gut microbiota and other complex features have been discovered with the advancement of high-throughput sequencing technologies and platforms (4).
As one of the most common chronic respiratory diseases, asthma affects around 334 million people worldwide, covering all age groups and usually originating from childhood (5). To date, the pathogenesis of asthma has not been fully elucidated yet, and the contribution of genetic factors was estimated to be ranged from 25% to 80% (6). Recent genome-wide association studies (GWAS) identified 18 asthma-associated genomic loci including five new loci at 5q31.3, 6p22.1, 6q15, 12q13.3, and 17q21.33 (7). And the 22 distinct genome-wide-significant single nucleotide polymorphisms (SNPs) corresponding to 18 loci can explain 3.5% of the variance in asthma liability (7). As regards the phenotypes of asthma, existing evidence revealed that childhood-onset asthma was highly associated with at least two independent loci, 17q12-21 and filaggrin (FLG) locus; and a relative moderate association was also found between adult-onset asthma, moderate-severe asthma and genes, which are largely a subset of those associated with childhood-onset asthma (8).
A growing body of literature has revealed that asthma comprises a range of heterogeneous subtypes with significant variance in clinical characteristics and course, and the heterogeneity should be based, at least partly, on different genetic origins with respect to varied asthma subtypes (9). Therefore, examing subtype-specific genetics, along with identifying the shared genetic factors underlying asthma and its coexistent diseases, should be of importance in clarifying the heterogeneity of asthma. By using the UK Biobank and 23andMe data, two recent studies obtained largely consistent findings that there was not only genetic overlap (eg, IL1RL1, HLA-DQA1) between childhood asthma and adult asthma but also subtype-specific components, providing evidence that the heterogeneity of asthma is related to distinct genetics (10, 11). These research advances bring fresh perspectives on the genetic genesis of asthma.
There is a growing interest in the relationship between gut microbiota and asthma (12, 13). The “hygiene hypothesis” is the initial theory to propose a connection between microbiota and allergy (12), and then followed by the concept of the “gut-lung axis”, which states that a dysbiosis of the gut microbiota might cause airway disease by changing the immune response (13). Asthma is mainly mediated by type I hypersensitivity reactions, accompanied by T helper (Th) subgroup and Th1/Th2 immune imbalance (5). Several studies have confirmed the role of the microbiota in regulating T-cell homeostasis (14–16). Data suggested that Bacteroides fragilis can modulate Th1/Th2 balance, Segmented filamentous bacteria can directly stimulate Th17 cell differentiation, and Clostridium is involved in the induction of Treg production (14–16). Tregs are a subpopulation of T cells that play key roles in modulating immune system activity and in maintaining tolerance to self-antigens (17). A previous study among preschool children diagnosed with asthma provided the evidence of gut microbiota dysbiosis that the reduction of Lachnospira was potentially linked to asthma (18). In particular, the opposite changes in the relative abundance of Lachnospira and Clostridium in the first three months after birth at three months of life play a role in promoting the development of a childhood-onset asthma phenotype (18). Moreover, in clinical studies, the connections between microbial dysbiosis and asthma characteristics have involved adult asthmatics, who have a higher concentration of histamine-secreting bacteria in their gut than healthy subjects, indicating these gut microbes may affect manifestations of allergic asthma (19).
Nevertheless, above findings from observational studies make it difficult to infer true causality, given the presence of reverse causality and the potential confounding factors. With the rapid increase in genetic data on microbiota and complex diseases, Mendelian randomization (MR) has gained widespread use in recent years. MR has a unique advantage in exploring the potential causal relationship between two traits based on mendelian laws of inheritance (20). In this study, we conducted a two sample MR study based on recently published large GWAS summary datasets to investigate the genetic causal link between gut microbiota, asthma and its phenotypes, and to identify particular groupings of pathogenic bacteria (4, 21, 22).
This MR study was undertaken following a framework as delineated in Figure S1. The approach is conducted based on three assumptions: 1) genetic variation used as instrumental variable (IV) is associated with exposure; 2) genetic variation is independent of confounding factors, and 3) genetic variation affects outcome risk only through the exposure of interest and not through other pathways (20).
MiBioGen is an international consortium dedicating the better understanding to genetic architecture of gut microbiota. It has collected information from 24 population-based cohorts with a total of 18,340 individuals. Each cohort investigated the gut microbiota via 16S rRNA sequencing and participants were genotyped by using full-genome SNP arrays (4). The HRC 1.0 or 1.1 reference panel was employed for genotyping imputation. Then, covariates such as age, sex, technical covariates, and genetic principal components were taken into account, and association analysis was performed by using Spearman correlation (4). The gut microbiota GWAS summary data includes a total of 131 genera; and 119 genera were included in our study as exposures, with the exclusion of 12 unknown genera. Detailed information on the classification of gut microbiota can be seen in Additional file 1: Table S1.
GWAS summary datasets for asthma, adult-onset asthma and childhood-onset asthma were obtained from the largest sample of recent publications, and the source of cases and controls was the UK Biobank (22). Asthma cases in the UK Biobank were determined by subjects’ responses to the question “whether they had been diagnosed with asthma by a doctor” (22). In the asthma GWAS data, a total of 394,283 people of European ancestry were included, comprising 46,802 asthmatics and 347,481 controls, childhood-onset asthma was defined as onset age of asthma ≤12 years (including 9,676 cases and 347,481 controls), whereas adult-onset asthma was defined as onset age ≥26 years (including 22,296 cases and 347,481 controls) (22).
As for moderate to severe asthma, a recent large-scale European ancestry GWAS summary dataset was used, which includes 5,135 moderate-to-severe asthma cases and 25,675 controls (21). The consortia for cases and control sources were the Genetics of Asthma Severity and Phenotype study (GASP), the Unbiased Biomarker Prediction of respiratory diseases outcomes project (U-BIOPRED), and the UK Biobank. Clinical records were used to evaluate patients in GASP and U-BIOPRED in accordance with British Thoracic Society criteria since 2014. Additionally, cases of moderate-severe asthma of moderate-severe asthma in UK Biobank are determined by a doctor’s diagnosis (21).
All datasets were taken from previously published literature, exempt from ethical approval. Details of the GWAS summary datasets for gut microbiota, asthma, and asthma phenotypes can be seen in Additional file 1: Table S2.
The following quality control procedures were used to choose the appropriate genetic IV in order to guarantee the validity and correctness of the conclusions about the causal link between the gut microbiota and asthma risk. First, with reference to most MR studies on gut microbiota (23, 24), we set the significance level at P<1.0×10-5 to identify enough candidate instruments due to the minimal number of loci found for gut microbiota. Second, to identify the independent SNPs assorted randomly during gestation, the clumping process (R2 <0.001, and clumping distance=10,000 kb) was conducted to assess the linkage disequilibrium (LD). Third, palindromic SNPs were removed since the GWAS of gut microbiota did not provide the effective allele frequency so we are unable to determine whether these SNPs were aligned in the same direction for exposure and outcome or not. Four, to reduce the heterogeneity and avoid the pleiotropy, MR Pleiotropy Residual Sum and Outlier (MR-PRESSO) methods were used to identify the horizontal pleiotropic outliers. Finally, the F-statistics (BETA2/SE2) for each IV of the gut microbiota was calculated as a measure of instrument strength (25). Generally, F-statistics > 10 were set as the threshold of strong IVs, otherwise, the IV is considered to have a weak association with exposure and would be therefore excluded. The detailed information on the IVs is displayed in Additional file 2: Table S3.
In the main analysis, we obtained estimates from inverse variance weighting meta-analysis (IVW), which aggregate the Wald values for each SNP and derive the overall estimates of the effect by using meta-analysis (26). The random-effect IVW would be employed if there is heterogeneity among the SNPs included in each analysis (27).
To evaluate the sensitivity of genetic causal effects, Weighted median, MR-Egger, Robust Adjusted Profile Score (MR-RAPS), Maximum likelihood method (ML), and MR-PRESSO methods were applied, which would promise to provide evidence of validity under different conditions (28–33). The PhenoScanner database (http://www.phenoscanner.medschl.cam.ac.uk/) was also searched to investigate whether the selected SNPs are associated with confounding traits (BMI, tobacco or alcohol exposure, and pulmonary function) at a significance level of 1 × 10–5, and we re-run the analysis after dropping these SNPs. Moreover, the Cochran Q statistic and I2 statistic were used to test the heterogeneity. To determine if a specific genetic locus have an impact on random estimates, the leave-one-out sensitivity method was used. Scatterplots, forest plots, and funnel plots were created to further demonstrate the sensitivity of the results.
Power calculations for this MR were performed on the website: mRnd (http://cnsgenomics.com/shiny/mRnd) (34).
The threshold for statistical significance was set at 4×10-4 (P=0.05/119) using a Bonferroni-adjusted P-value. If 4×10-4<P<0.05, this was considered suggestive of evidence for a potential association. All the analyses and relevant figures were made by R 3.6.5, using the “TwoSampleMR (0.5.6)”, “MR-PRESSO (1.0)”, and “mr.raps (0.2)” packages. Reporting follows the STROBE-MR statement (35).
The characteristics of the selected SNPs for each gut microbiota are presented in Additional file 2: Table S3. Since the gut microbiota GWAS summary statistics didn’t report the effective allele frequency and variance explained (R2), the range of possible R2 values was shown, and we calculated the OR detectable with 80% power, detailed information can be seen in Additional file 2: Table S4.
Based on the IVW method, 3 genera of genetically predicted gut microbiota were found to be related to the risk of asthma (Table 1). The MR analysis showed that genetically predicted Barnesiella increased the risk of asthma (OR=1.097; 95% confidence interval [CI]=1.034–1.163; P=2.10E-03), the similar results were also obtained in the ML, MR Egger, RAPS and MR-PRESSO methods. Similarly, genetically predicted RuminococcaceaeUCG014 increased the risk of asthma (OR=1.121; 95%CI=1.039–1.210; P=3.06E-03). And ML, RAPS and MR-PRESSO also yielded the same results. By contrast, genetically predicted CandidatusSoleaferrea was shown to be a protective factor for asthma (OR=0.934; 95%CI=0.884–0.987; P=1.58E-02), and both the ML and MR-PRESSO methods supported the finding.
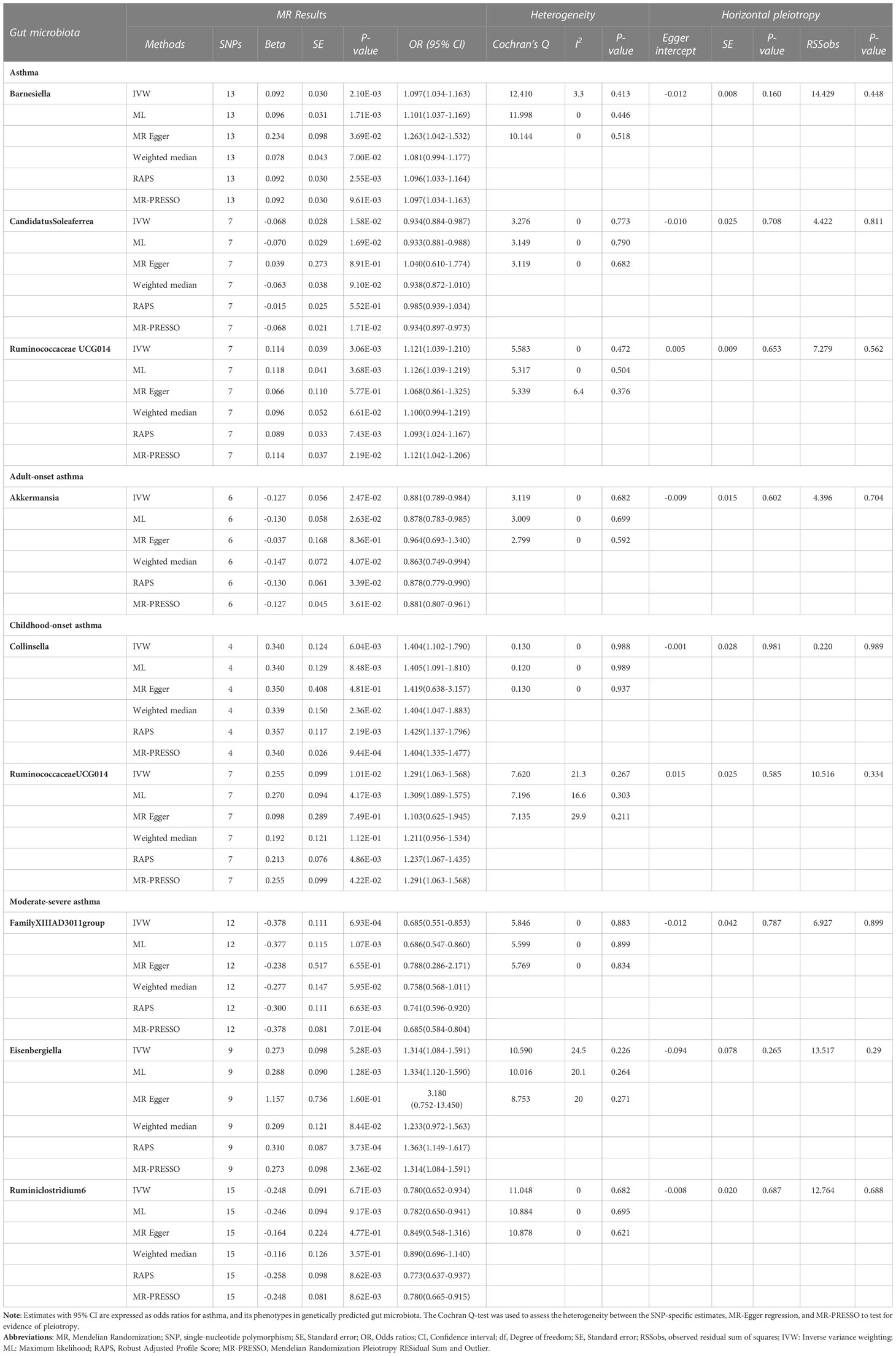
Table 1 MR results of causal relationships between the Gut microbiota and Asthma, and its phenotypes risk.
As for asthma phenotypes, the IVW method supported the causal associations between genetically predicted Akkermansia (OR=0.881; 95%CI=0.789–0.984; P=2.47E-02) and adult-onset asthma, and between genetically predicted Collinsella (OR=1.404; 95%CI=1.102–1.790; P=6.04E-03), RuminococcaceaeUCG014 (OR=1.291; 95%CI=1.063–1.568; P=1.01E-02) and childhood-onset asthma. Similar results were also obtained in the ML, Weighted median, RAPS and MR-PRESSO methods. Furthermore, the decrease in moderate-severe asthma risk could attribute to the increase of genetically predicted FamilyXIIIAD3011group (OR=0.685; 95%CI=0.551–0.853; P=6.93E-04) and Ruminiclostridium6 (OR=0.780; 95%CI=0.652–0.934; P=6.71E-03), and the increase of moderate-severe asthma risk could attribute to the increase of genetically predicted Eisenbergiella (OR=1.314; 95%CI=1.084–1.591; P=5.28E-03); and ML, RAPS, and MR-PRESSO methods also yielded the same results. Detailed information on the MR results can be seen in Additional file 2: Table S5.
As shown in Table 1, the evidence of horizontal pleiotropy for SNPs was not found by using the MR-Egger regression intercept test, Cochran Q statistic and I2 statistic indicated low heterogeneity and more reliability of these SNPs, and MR-PRESSO global test showed that no potential outlier could affect our estimation substantially, detailed information can be seen in Additional file 2: Table S5. Furthermore, the funnel plots displayed general symmetry, indicating little evidence of heterogeneity (Additional file 1: Figure S2). Scatter plots was used to show the estimated effect sizes for SNPs of gut microbiota on asthma and its phenotypes (Figure 1). Beyond which, we used IVW methods to run the leave-one-out sensitivity analysis, and the results were similar after excluding individual SNPs from the study, indicating that no single SNP had an exorbitant influence on the total estimations (Figure 2). In the PhenoScanner search, we found that rs9276029, and rs2548459 were associated with alcohol intake, forced expiratory volume in 1-second, and peak expiratory flow (Additional file 2: Table S6). We re-analyzed after eliminating these SNPs, the results indicated that FamilyXIIIAD3011group (OR=0.659; 95%CI=0.523–0.831; P=4.12E-04) and Ruminiclostridium6 (OR=0.771; 95%CI=0.642–0.927; P=5.70E-03) still reduced the risk of developing moderate to severe asthma, detailed information can be seen in Additional file 2: Table S7. The forest plots were shown in Additional file 1: Figure S3.
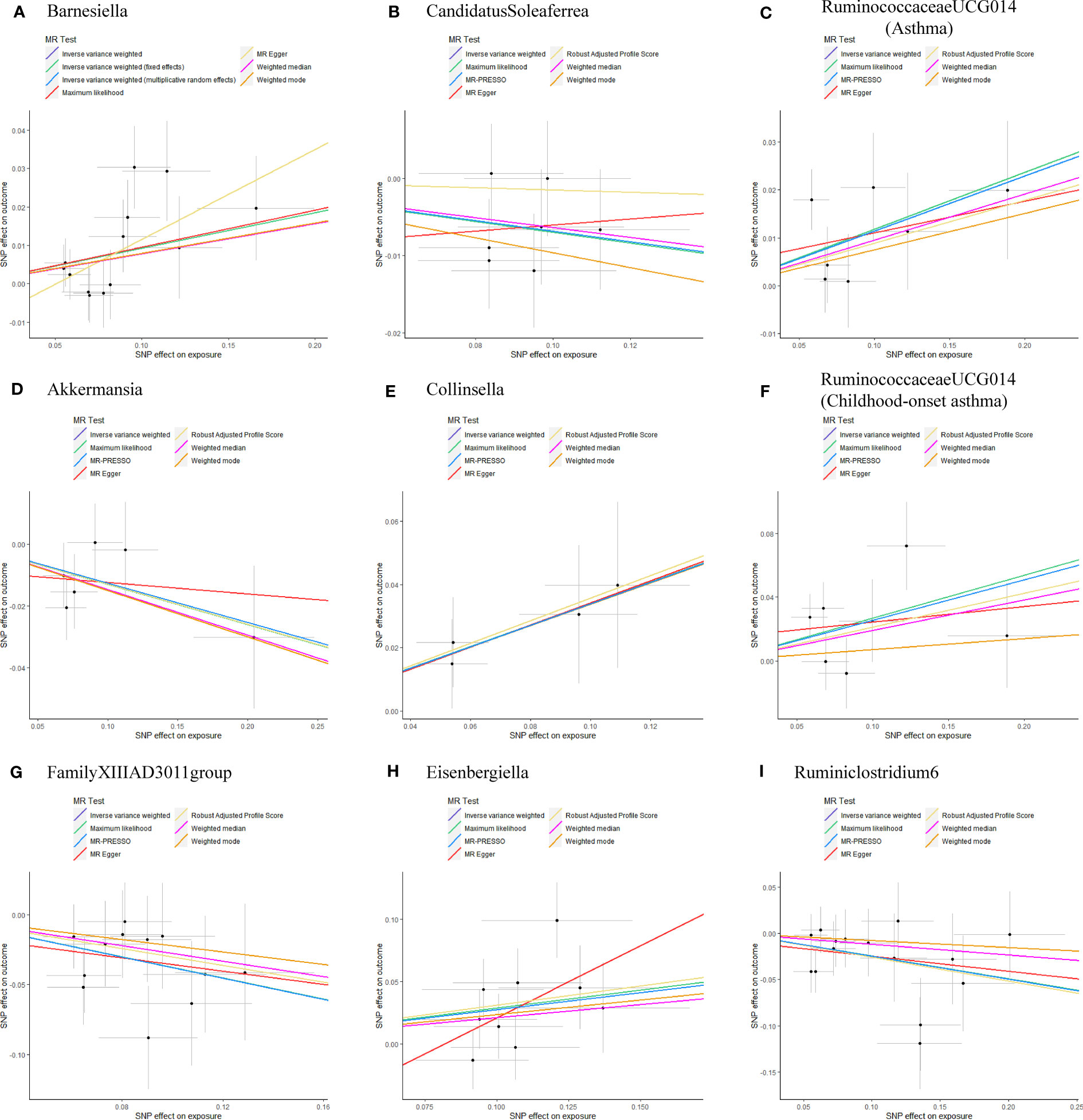
Figure 1 Scatterplots of potential effects of SNPs on Gut microbiota versus Asthma and its phenotypes (A) Barnesiella; (B) CandidatusSoleaferrea; (C) RuminococcaceaeUCG014(Asthma); (D) Akkermansia; (E) Collinsella; (F) RuminococcaceaeUCG014(Childhood-onset asthma); (G) FamilyXIIIAD3011group; (H) Eisenbergiella; (I) Ruminiclostridium6. Scatter plots presented the per-allele association with outcome risk plotted against the per-allele association with one standard deviation of exposure (with vertical and horizontal purple lines showing the 95% CI for each SNP). Analyses were conducted using the Inverse Variance Weighting (IVW), Weighted median, Wald ratio, Robust Adjusted Profile Score (RAPS), MR Egger, MR-PRESSO, and Maximum likelihood (ML) methods. The slope of each line corresponding to the estimated MR effect per method. CI, confidence interval; SNP, single-nucleotide polymorphism; MR-PRESSO, Mendelian Randomization Pleiotropy RESidual Sum and Outlier.
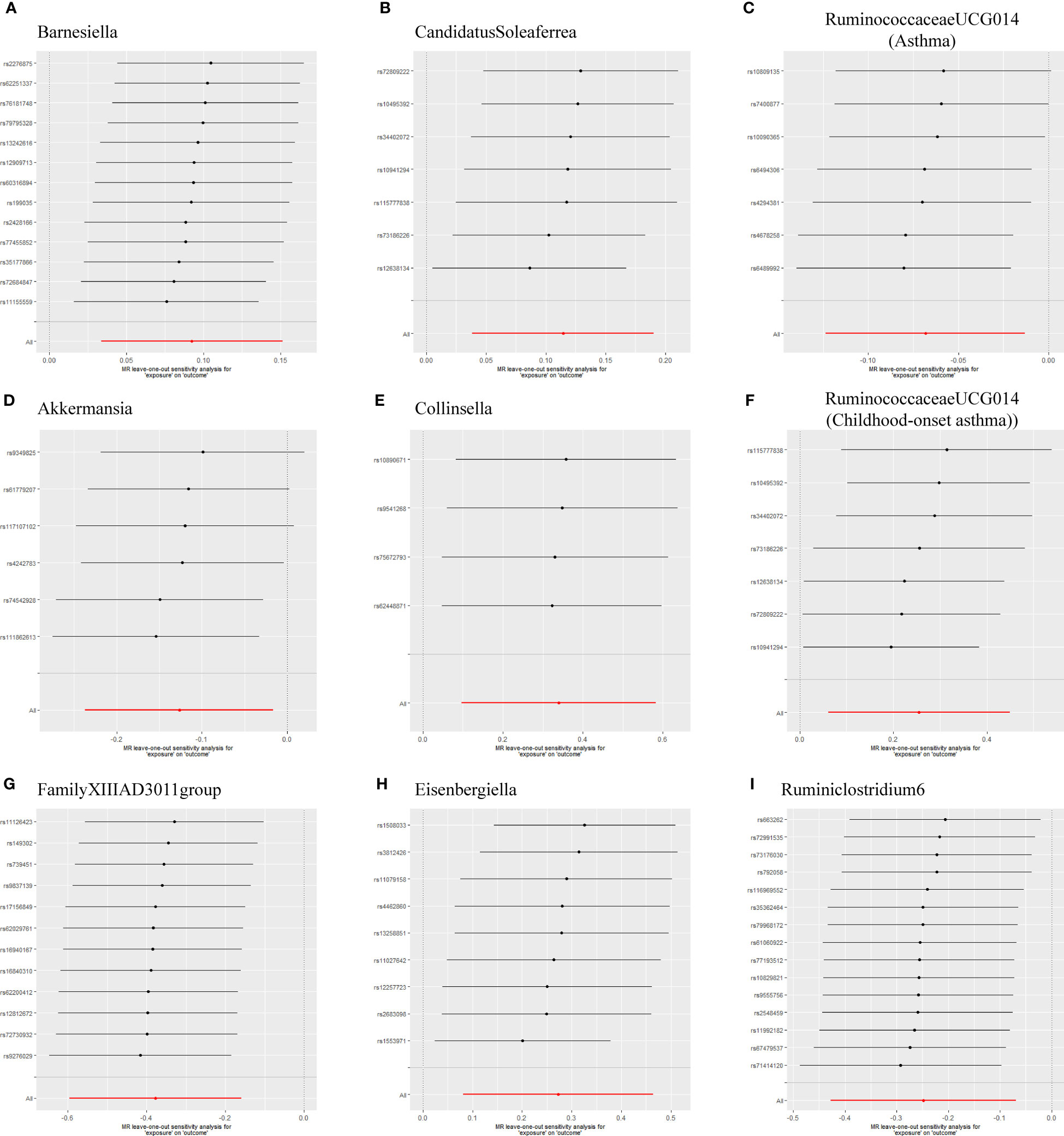
Figure 2 Leave-one-out sensitivity based on IVW model for Gut microbiota on Asthma and its phenotypes (A) Barnesiella; (B) CandidatusSoleaferrea; (C) RuminococcaceaeUCG014(Asthma); (D) Akkermansia; (E) Collinsella; (F) RuminococcaceaeUCG014(Childhood-onset asthma); (G) FamilyXIIIAD3011group; (H) Eisenbergiella; (I) Ruminiclostridium6. The IVW causal estimate and how the overall estimate (red horizontal line) was disproportionately driven, which is influenced by the removal of a single variant (black horizontal line), were visualized. There was no evidence of obvious heterogeneity, indicating that no specific SNP alone accounted for the association of gut microbiota on Periodontitis. The results suggested that there was no individual SNP with a strong influence on the overall effect. SNP, single-nucleotide polymorphism; IVW, Inverse variance weighted.
In the reverse MR analysis, 86 genome-wide significance level (P < 5 × 10–8) and independent SNPs were proposed as IVs for asthma, and 12 SNPs for moderate-severe asthma (R2 < 0.001 and clump window > 10,000 kb) Bidirectional analysis for the adult-onset asthma and childhood asthma could not be conducted due to insufficient independent SNPs as IVs (0 and 1 SNP, respectively),detailed information can be seen in Additional file 2: Table S8.In the absence of heterogeneity and pleiotropy, the results suggest that genetically predicted asthma does not increase or decrease the risk of developing Barnesiella, RuminococcaceaeUCG014, and CandidatusSoleaferrea. Likewise, genetically predicted moderate-severe asthma was not associated with FamilyXIIIAD3011group, Ruminiclostridium6, and Eisenbergiella (Table 2).
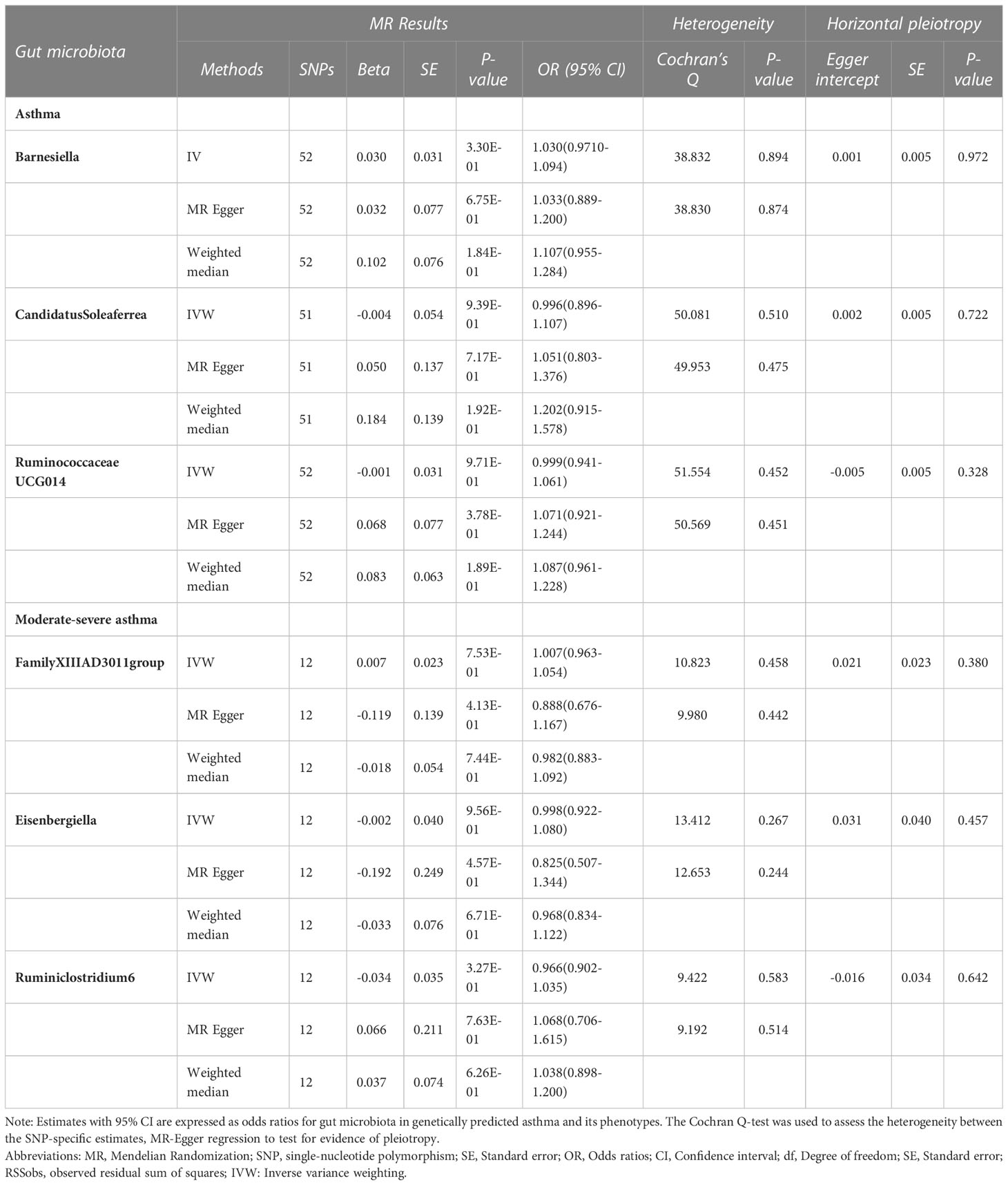
Table 2 MR results of causal relationships between Asthma and the Gut microbiota.
To our knowledge, this two-sample MR study, for the first time, examined the causal associations between gut microbiota and asthma by using publicly available genetic databases. Our findings revealed that several microbial genera were causally associated with asthma and its phenotypes, which enhanced the understanding regarding the role of gut microbiota in the pathology of asthma, providing new insights into the prevention and diagnosis of asthma.
The gut is the most colonized human organ with up to 100 trillion microbes, approximately 10 times the number of human cells (36), gut microbes are typically dominated by five phyla, including Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, and Tenericutes (37). They confer diverse functions to the host, including vitamin production, absorption of ions, protection against pathogens, histological development, and enhanced immune functions (38). Although the pathways through which the gut microbiota affects the lung microbiota are not fully understood, it appears that intestinal and respiratory disorders show similar clinical alterations, and an inflammatory transfer from the gut to the lung may take place (39). As a result, the disturbances of this bidirectional exchange should be connected to the increased emergence of airway illnesses such as asthma (40). Accumulating evidence highlighted the importance of the cross-talk between gut and lung, the so-called gut-lung axis, in the maintenance of immune homeostasis (41). It seemed that gut microbiota metabolites could be an important entry point in the exploration of asthma pathogenesis, short chain fatty acids, polyunsaturated fatty acids and bile acids have been shown to influence immune function by promoting the growth or maturity of certain immune cell populations (42).
The mechanisms underlying the link between gut microbiota and asthma involve complex metabolic and immune interactions of microbes, metabolites, and host immune responses (40). It was suggested that the commensal bacteria in the gut help to shape the maturation of both innate and adaptive immunity in early life, which would produce profound effects on individual’s asthma susceptibility and pathobiology. An early in vivo study of mouse model showed polysaccharide A from Bacteroides fragilis could induce and ligate TLR2 on plasmacytoid dendritic cells, which is priming IL-10 producing T cells with potential of anti-inflammatory properties (43). Additionally, another in vivo study using an HDM-challenged mouse model found that high-fiber diet increases the abundance of Bacteroides and Bifidobacterium, which can digest the fiber and produce SCFAs, such as butyrate (44). And butyrate can decrease excessive inflammation through downregulating the secretion of pro-inflammatory mediators and activating IL-10 producing T cells and macrophages (45). A very recent study also demonstrated that infants who are breastfed and given Bifidobacterium infantis EVC001 has reduced intestinal TH2 and TH17 cytokines while increased IFN-β level (44).
Previous observational and genetic studies have shown an association between adult BMI and adult-onset asthma (46). In China, recent study identified nine genome-wide significant novel loci for FEV1, six for FVC and three for FEV1/FVC in the CKB. FEV1 and FVC showed significant negative genetic correlation with obesity traits in both the CKB and UKB (47). However, one study has shown that the shared genetic component between childhood BMI and childhood-onset asthma is not driven by the genetic component of adult BMI. They identified one shared causal genomic region between BMI and asthma in childhood that mapped to gene AMN (46). To sum up, obesity is a potential confounder between gut microbiome and asthma/lung function. Our study performed statistical analysis by excluding SNPS associated with obesity (PhenoScanner database), which makes our results even more credible.
Alterations in gut microbial composition may have a notable effect on respiratory diseases, such as asthma, by shaping microbial communities and modulating the metabolic and immune response, growing evidence suggests that gut microbiota in early life is associated with childhood asthma development (18, 48, 49). Data from Canadian Healthy Infant Longitudinal Development Study (CHILD) demonstrated the relative abundance of the genera Lachnospira, Faecalibacterium, Veillonella (Phylum Firmicutes) and Rothia (Phylum Actinobacteria) was considerably lowered in the gut microbiome of infants at risk for asthma in the first 100 days of life (48). Another cohort study found that the opposing shift in the relative abundance of Lachnospira and Clostridium (Phylum Firmicutes) in the early life stage was associated with asthma development at preschool age (18). At 3 months the abundance of the genera, Lachnospira was decreased, whereas Clostridium was increased in asthmatics, and the Lachnospira/Clostridium ratio may serve as a potential early life biomarker to predict asthma development (18). A recent metabolomics-based study made a comparative analysis of stool samples from asthma children and healthy children both aged 4 to 7 years, indicating that children with allergic airway illnesses tended to have a much lower abundance of the Firmicutes (50). The results suggested that childhood rhinitis and asthma may be caused by a decrease in certain gut microbes in Firmicutes, which are involved in the up-regulation of fecal amino acids (50). All above studies suggest that taxa belonging to the phyla Firmicutes or Actinobacteria should play an important role in the pathogenesis of asthma. Particularly, our present study is the first to provide genetic causal evidence to confirm and enrich the current knowledge. We found that Candidatus, Soleaferrea and RuminococcaceaeUCG014 (Phylum Firmicutes, Class Clostridia, order Clostridiales) were correlated with the risk of asthma, among which, Collinsella (Phylum Actinobacteria) and RuminococcaceaeUCG014 increased the risk of childhood-onset asthma, and Eisenbergiella and Ruminiclostridium6 (Phylum Firmicutes, class Clostridia) were associated with the risk of moderate-severe asthma. In South of China, a cross-sectional study investigating the gut microbiome profile in adults found that Ruminococcus gnavus were enriched in newly diagnosed asthmatic patients compared to healthy controls (51). In a twin cohort study, researchers discovered a correlation between the abundance of fecal Ruminococcus gnavus and the development of allergies, particularly respiratory allergies (52). Likewise, RuminococcaceaeUCG014 has a positive effect against asthma as well as childhood-onset asthma in the present study. There are fewer studies on Collinsella and allergic disease, a recent case-control study observed significant increases in the numbers of Collinsella, Ruminococcus, and Akkermansia in the food allergy group compared to the control group, at the genus level (53), and another epidemiological study found that indoor Collinsella was positively associated with asthma, rhinitis and eczema among preschool children (54). All these findings are quite similar to ours. A study from the US birth cohort found that the neonates with the lowest relative abundance of Akkermansia have the highest risk of developing asthma at age of four years, and we got the consistent findings among adults in the present study (55). Eisenbergiella was found to be associated with nasal symptoms in patients with allergy rhinitis (56), our result demonstrated that Eisenbergiella was correlated with the risk of moderate-severe asthma. Furthermore, we found genetic causality between Barnesiella, CandidatusSoleaferrea and asthma, and between FamilyXIIIAD3011group, Ruminiclostridium6 and moderate-severe asthma. Research on these genera is still relatively scarce, and more research is needed on their role in allergic diseases in the future.
This MR study has the following advantages. The use of the MR technique reduced the interference of confounding factors and false causality in the results, which is the first strength. To the best of our knowledge, this is the first MR analysis investigating the causal association between gut microbiota and asthma. Our results offer a theoretical foundation for the subsequent investigation into the regulation mechanism of a single strain on asthma. Second, the current analysis makes full use of the comprehensive GWAS data that are publicly available, which promised a high sample sizes; making our study have necessary power to estimate reliable and lifelong causality. Third, because we conducted MR analysis on gut microbiota at the genus level, it should be possible to pinpoint specific bacterial strains that are indeed causally related to asthma. Our study does have some limitations, though. First, our findings were not robust to Bonferroni-adjusted significance, but MR analysis serves as a hypothesis-driven study testing epidemiologically established associations based on enough physiological evidence. Second, the number of genetic loci found in gut microbiota GWAS is still small, our IVs are suspected of being weak tool variables, which may reduce the statistical power of our MR study. Third, a tiny percentage of the microbiota data were of different races, despite the fact that the majority of the data included in our analysis were European, which may to some extent throw off our estimates.
In summary, by performing two sample MR analyses, our study is the first to provide comprehensive screening data regarding the causal associations of gut microbes with asthma as well as its three phenotypes; by contrast, the reverse MR analysis didn’t establish reverse causal link from asthma and its three phenotypes to the gut microbiota genus. The findings underscore the critical role of gut microbiota in asthma’s pathogenesis, which would be of significance in clinical application; the gut microbes identified may be the potential therapeutic targets for asthma preventing and clinical treating.
All data used in the present study were obtained from genome-wide association study summary statistics which were publicly released by genetic consortia. Gut microbiota: https://www.ebi.ac.uk/gwas/publications/33462485. Asthma, adult-onset asthma and childhood-onset asthma: https://www.ebi.ac.uk/gwas/publications/31619474. Moderate-severe asthma: https://www.ebi.ac.uk/gwas/publications/30552067. All datasets generated for this study are included in the article/Additional files.
RL, QG, and SL designed the study, contributed to the data analysis, and wrote the manuscript. RL, QG, JZ, WK, RYL, LH, ZL, YC, and AZ contributed to the data analysis and data interpretation. JW, YY, and SL contributed to manuscript writing and revision of the manuscript. All authors contributed to the article and approved the submitted version.
This was not an industry-supported study. The study was funded by grants from the National Natural Science Foundation of China (82273651, 81874266, 81673183), special grant for Preschool Children’s Health Management from the Shanghai Municipal Education Commission, Key Project from Shanghai Municipal Science and Technology Commission (18411951600), Major Science and Technology Projects of Hainan Province (ZDKJ2019010), and Key Science and Technology Cooperation Projects of Hainan Province (ZDYF2020210).
We are grateful to the participants in the MiBioGen, UK Biobank, and other consortiums or studies and to all the researchers who worked on the data collection.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1148684/full#supplementary-material
1. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. (2010) 464(7285):59–65. doi: 10.1038/nature08821
2. Adak A, Khan MR. An insight into gut microbiota and its functionalities. Cell Mol Life Sci (2019) 76(3):473–93. doi: 10.1007/s00018-018-2943-4
3. Busnelli M, Manzini S, Chiesa G. The gut microbiota affects host pathophysiology as an endocrine organ: a focus on cardiovascular disease. Nutrients. (2019) 12(1):79. doi: 10.3390/nu12010079
4. Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-Scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet (2021) 53(2):156–65. doi: 10.1038/s41588-020-00763-1
5. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Lancet. (2018) 391(10122):783–800. doi: 10.1016/s0140-6736(17)33311-1
6. Duffy DL, Martin NG, Battistutta D, Hopper JL, Mathews JD. Genetics of asthma and hay fever in Australian twins. Am Rev Respir Dis (1990) 142(6 Pt 1):1351–8. doi: 10.1164/ajrccm/142.6_Pt_1.1351
7. Demenais F, Margaritte-Jeannin P, Barnes KC, Cookson WOC, Altmüller J, Ang W, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet (2018) 50(1):42–53. doi: 10.1038/s41588-017-0014-7
8. Schoettler N, Rodríguez E, Weidinger S, Ober C. Advances in asthma and allergic disease genetics: is bigger always better? J Allergy Clin Immunol (2019) 144(6):1495–506. doi: 10.1016/j.jaci.2019.10.023
9. Zhu Z, Hasegawa K, Camargo CA Jr., Liang L. Investigating asthma heterogeneity through shared and distinct genetics: insights from genome-wide cross-trait analysis. J Allergy Clin Immunol (2021) 147(3):796–807. doi: 10.1016/j.jaci.2020.07.004
10. Ferreira MAR, Mathur R, Vonk JM, Szwajda A, Brumpton B, Granell R, et al. Genetic architectures of childhood- and adult-onset asthma are partly distinct. Am J Hum Genet (2019) 104(4):665–84. doi: 10.1016/j.ajhg.2019.02.022
11. Pividori M, Schoettler N, Nicolae DL, Ober C, Im HK. Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: genome-wide and transcriptome-wide studies. Lancet Respir Med (2019) 7(6):509–22. doi: 10.1016/s2213-2600(19)30055-4
12. Frati F, Salvatori C, Incorvaia C, Bellucci A, Di Cara G, Marcucci F, et al. The role of the microbiome in asthma: the Gut-Lung axis. Int J Mol Sci (2018) 20(1):123. doi: 10.3390/ijms20010123
13. Penders J, Stobberingh EE, van den Brandt PA, Thijs C. The role of the intestinal microbiota in the development of atopic disorders. Allergy. (2007) 62(11):1223–36. doi: 10.1111/j.1398-9995.2007.01462.x
14. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139(3):485–98. doi: 10.1016/j.cell.2009.09.033
15. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science. (2011) 331(6015):337–41. doi: 10.1126/science.1198469
16. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. (2005) 122(1):107–18. doi: 10.1016/j.cell.2005.05.007
17. Lee HS, Park HW, Song WJ, Jeon EY, Bang B, Shim EJ, et al. TNF-α enhance Th2 and Th17 immune responses regulating by IL23 during sensitization in asthma model. Cytokine. (2016) 79:23–30. doi: 10.1016/j.cyto.2015.12.001
18. Stiemsma LT, Arrieta MC, Dimitriu PA, Cheng J, Thorson L, Lefebvre DL, et al. Shifts in lachnospira and clostridium sp. in the 3-month stool microbiome are associated with preschool age asthma. Clin Sci (Lond) (2016) 130(23):2199–207. doi: 10.1042/cs20160349
19. Barcik W, Pugin B, Westermann P, Perez NR, Ferstl R, Wawrzyniak M, et al. Histamine-secreting microbes are increased in the gut of adult asthma patients. J Allergy Clin Immunol (2016) 138(5):1491–1494.e7. doi: 10.1016/j.jaci.2016.05.049
20. VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in mendelian randomization. Epidemiology. (2014) 25(3):427–35. doi: 10.1097/ede.0000000000000081
21. Shrine N, Portelli MA, John C, Soler Artigas M, Bennett N, Hall R, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med (2019) 7(1):20–34. doi: 10.1016/s2213-2600(18)30389-8
22. Zhu Z, Zhu X, Liu CL, Shi H, Shen S, Yang Y, et al. Shared genetics of asthma and mental health disorders: a large-scale genome-wide cross-trait analysis. Eur Respir J (2019) 54(6):1901507. doi: 10.1183/13993003.01507-2019
23. Yu XH, Yang YQ, Cao RR, Bo L, Lei SF. The causal role of gut microbiota in development of osteoarthritis. Osteoarthritis Cartilage (2021) 29(12):1741–50. doi: 10.1016/j.joca.2021.08.003
24. Xiang K, Wang P, Xu Z, Hu YQ, He YS, Chen Y, et al. Causal effects of gut microbiome on systemic lupus erythematosus: a two-sample mendelian randomization study. Front Immunol (2021) 12:667097. doi: 10.3389/fimmu.2021.667097
25. Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int J Epidemiol (2011) 40(3):740–52. doi: 10.1093/ije/dyq151
26. Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for mendelian randomization. Stat Methods Med Res (2017) 26(5):2333–55. doi: 10.1177/0962280215597579
27. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample mendelian randomization analyses using MR-egger regression: the role of the I2 statistic. Int J Epidemiol (2016) 45(6):1961–74. doi: 10.1093/ije/dyw220
28. Zhao Q, Chen Y, Wang J, Small DS. Powerful three-sample genome-wide design and robust statistical inference in summary-data mendelian randomization. Int J Epidemiol (2019) 48(5):1478–92. doi: 10.1093/ije/dyz142
29. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet (2018) 50(5):693–8. doi: 10.1038/s41588-018-0099-7
30. Milligan BG. Maximum-likelihood estimation of relatedness. Genetics. (2003) 163(3):1153–67. doi: 10.1093/genetics/163.3.1153
31. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol (2017) 46(6):1985–98. doi: 10.1093/ije/dyx102
32. Burgess S, Thompson SG. Interpreting findings from mendelian randomization using the MR-egger method. Eur J Epidemiol (2017) 32(5):377–89. doi: 10.1007/s10654-017-0255-x
33. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol (2016) 40(4):304–14. doi: 10.1002/gepi.21965
34. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in mendelian randomization studies. Int J Epidemiol (2013) 42(5):1497–501. doi: 10.1093/ije/dyt179
35. Skrivankova VW, Richmond RC, Woolf BAR, Davies NM, Swanson SA, VanderWeele TJ, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): explanation and elaboration. Bmj. (2021) 375:n2233. doi: 10.1136/bmj.n2233
36. Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. (2006) 124(4):837–48. doi: 10.1016/j.cell.2006.02.017
37. Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The role of lung and gut microbiota in the pathology of asthma. Immunity. (2020) 52(2):241–55. doi: 10.1016/j.immuni.2020.01.007
38. Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell. (2016) 164(3):337–40. doi: 10.1016/j.cell.2016.01.013
39. Tulic MK, Piche T, Verhasselt V. Lung-gut cross-talk: evidence, mechanisms and implications for the mucosal inflammatory diseases. Clin Exp Allergy (2016) 46(4):519–28. doi: 10.1111/cea.12723
40. Hufnagl K, Pali-Schöll I, Roth-Walter F, Jensen-Jarolim E. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin Immunopathol (2020) 42(1):75–93. doi: 10.1007/s00281-019-00775-y
41. Dang AT, Marsland BJ. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol (2019) 12(4):843–50. doi: 10.1038/s41385-019-0160-6
42. Lee-Sarwar KA, Lasky-Su J, Kelly RS, Litonjua AA, Weiss ST. Gut microbial-derived metabolomics of asthma. Metabolites. (2020) 10(3):97. doi: 10.3390/metabo10030097
43. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe (2014) 15(4):413–23. doi: 10.1016/j.chom.2014.03.006
44. Liu C, Makrinioti H, Saglani S, Bowman M, Lin LL, Camargo CA Jr, et al. Microbial dysbiosis and childhood asthma development: integrated role of the airway and gut microbiome, environmental exposures, and host metabolic and immune response. Front Immunol (2022) 13:1028209. doi: 10.3389/fimmu.2022.1028209
45. Chen J, Vitetta L. The role of butyrate in attenuating pathobiont-induced hyperinflammation. Immune Netw (2020) 20(2):e15. doi: 10.4110/in.2020.20.e15
46. Han X, Zhu Z, Xiao Q, Li J, Hong X, Wang X, et al. Obesity-related biomarkers underlie a shared genetic architecture between childhood body mass index and childhood asthma. Commun Biol (2022) 5(1):1098. doi: 10.1038/s42003-022-04070-9
47. Zhu Z, Li J, Si J, Ma B, Shi H, Lv J, et al. A large-scale genome-wide association analysis of lung function in the Chinese population identifies novel loci and highlights shared genetic aetiology with obesity. Eur Respir J (2021) 58(4):2100199. doi: 10.1183/13993003.00199-2021
48. Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med (2015) 7(307):307ra152. doi: 10.1126/scitranslmed.aab2271
49. Hu C, van Meel ER, Medina-Gomez C, Kraaij R, Barroso M, Kiefte-de Jong J, et al. A population-based study on associations of stool microbiota with atopic diseases in school-age children. J Allergy Clin Immunol (2021) 148(2):612–20. doi: 10.1016/j.jaci.2021.04.001
50. Chiu CY, Cheng ML, Chiang MH, Kuo YL, Tsai MH, Chiu CC, et al. Gut microbial-derived butyrate is inversely associated with IgE responses to allergens in childhood asthma. Pediatr Allergy Immunol (2019) 30(7):689–97. doi: 10.1111/pai.13096
51. Zou XL, Wu JJ, Ye HX, Feng DY, Meng P, Yang HL, et al. Associations between gut microbiota and asthma endotypes: a cross-sectional study in south China based on patients with newly diagnosed asthma. J Asthma Allergy (2021) 14:981–92. doi: 10.2147/jaa.S320088
52. Chua HH, Chou HC, Tung YL, Chiang BL, Liao CC, Liu HH, et al. Intestinal dysbiosis featuring abundance of ruminococcus gnavus associates with allergic diseases in infants. Gastroenterology. (2018) 154(1):154–67. doi: 10.1053/j.gastro.2017.09.006
53. Chen CC, Chen KJ, Kong MS, Chang HJ, Huang JL. Alterations in the gut microbiotas of children with food sensitization in early life. Pediatr Allergy Immunol (2016) 27(3):254–62. doi: 10.1111/pai.12522
54. Sun Y, Meng Y, Ou Z, Li Y, Zhang M, Chen Y, et al. Indoor microbiome, air pollutants and asthma, rhinitis and eczema in preschool children - a repeated cross-sectional study. Environ Int (2022) 161:107137. doi: 10.1016/j.envint.2022.107137
55. Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med (2016) 22(10):1187–91. doi: 10.1038/nm.4176
Keywords: Mendelian randomization, gut microbiota, asthma, childhood-onset asthma, causality
Citation: Li R, Guo Q, Zhao J, Kang W, Lu R, Long Z, Huang L, Chen Y, Zhao A, Wu J, Yin Y and Li S (2023) Assessing causal relationships between gut microbiota and asthma: evidence from two sample Mendelian randomization analysis. Front. Immunol. 14:1148684. doi: 10.3389/fimmu.2023.1148684
Received: 20 January 2023; Accepted: 28 June 2023;
Published: 19 July 2023.
Edited by:
Can Yang, Hong Kong University of Science and Technology, Hong Kong SAR, ChinaReviewed by:
Zhaozhong Zhu, Massachusetts General Hospital and Harvard Medical School, United StatesCopyright © 2023 Li, Guo, Zhao, Kang, Lu, Long, Huang, Chen, Zhao, Wu, Yin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenghui Li, bHNoOTkwN0AxNjMuY29t; c3VibWlzc2lvbjk5MDdAMTYzLmNvbQ==; Yong Yin, eWlueW9uZzk5OTlAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.