 Osman Dadas
Osman Dadas Ayse Ertay2
Ayse Ertay2 Mark S. Cragg
Mark S. Cragg- 1Antibody and Vaccine Group, School of Cancer Sciences, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
- 2School of Cancer Sciences, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
- 3Institute for Life Sciences, University of Southampton, Southampton, United Kingdom
The tumor necrosis factor superfamily (TNFSF) and their receptors (TNFRSF) are important regulators of the immune system, mediating proliferation, survival, differentiation, and function of immune cells. As a result, their targeting for immunotherapy is attractive, although to date, under-exploited. In this review we discuss the importance of co-stimulatory members of the TNFRSF in optimal immune response generation, the rationale behind targeting these receptors for immunotherapy, the success of targeting them in pre-clinical studies and the challenges in translating this success into the clinic. The efficacy and limitations of the currently available agents are discussed alongside the development of next generation immunostimulatory agents designed to overcome current issues, and capitalize on this receptor class to deliver potent, durable and safe drugs for patients.
Introduction
Members of the tumor necrosis factor superfamily (TNFSF) and their receptors (TNFRSF) are important regulators of the immune system. Interaction between these ligands and receptors can mediate proliferation, survival, differentiation, and function of immune cells (1, 2). There are 19 TNFSF ligands and 29 TNFRSF receptors, representing a large and diverse family.
The TNFSF ligands are type II proteins which are characterized by the presence of a C-terminal TNF homology domain (THD) responsible for ligand trimerization and receptor binding (3). In comparison, the TNFRSF receptors have between one to six cysteine rich domains (CRD) in their extracellular region (Figure 1) that are involved in ligand binding and receptor auto-association (5).
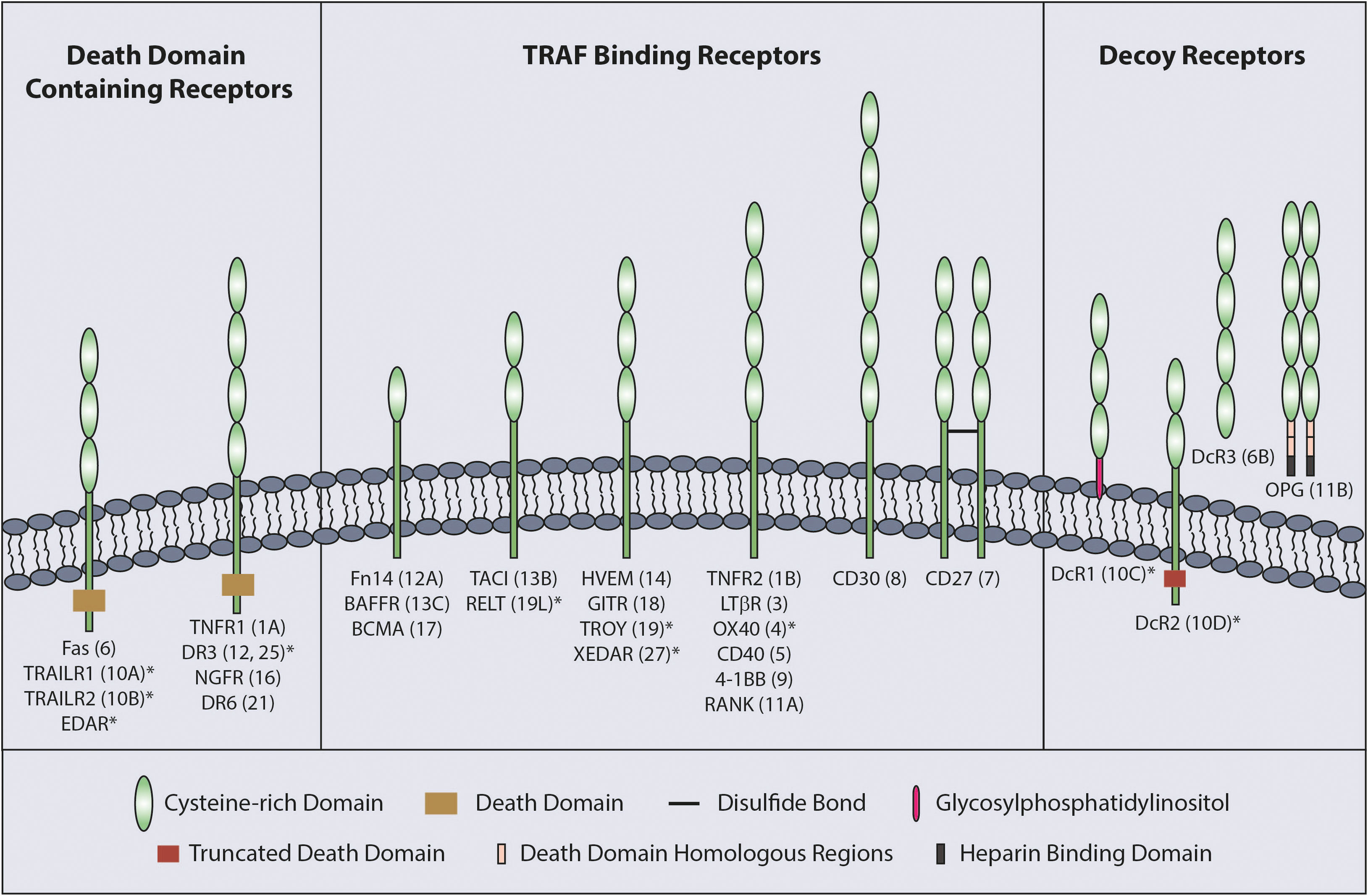
Figure 1 Classification of TNFRSF. The TNFRSF can be classified into three sub-families. All twenty-nine members of the family, grouped into three sub-families, are indicated with the number of CRDs on their extracellular region and TNFRSF number in brackets. CRD domains are defined by Uniprot with the exception of RELT which was published as having two CRDs (4). * indicates the receptors with a truncated CRD domain.
TNFRs can be sub-divided into three groups according to functional and structural differences; death domain (DD) containing receptors, decoy receptors and TNF receptor associated factor (TRAF) binding receptors (Figure 1). The DD is an 80 amino acid domain present in the cytoplasmic tail of the DD containing receptors. Although the DD containing receptors mainly initiate cell death signaling, they can also mediate other outcomes, such as NF-κB signaling (1, 5). The decoy receptors lack signal initiation capacity and consist of glycosylphosphatidylinositol (GPI) tethered receptors, soluble receptors and receptors possessing a non-functioning DD (5). Finally, TRAF binding receptors possess TRAF-interacting motifs (TIF) in their cytoplasmic tail that is responsible for recruiting TRAFs to mediate downstream signaling upon receptor activation.
Following expression on the cell surface, several members of the TNFRSF can self-associate into dimers or multimers prior to ligand binding. Although some members can be found as covalently linked dimers (e.g. CD27 (6)), self-association for others is mainly driven by the pre-ligand assembly domain (PLAD), largely covering the N-terminal CRD1 (7, 8), and glucocorticoid-induced TNFR related protein (GITR) is an exception as dimerization of this TNFR is driven by interactions within CRD3 (9). Formation of receptor dimers or trimers for several members of the TNFRSF before ligand binding has been shown to be crucial for their interaction with ligand. Deletion of the PLAD domain in TNFR1 and TNFR2, significantly reduced TNFα binding to both receptors (7, 10). Although the ligand binding domain is located in CRD2/3, the reduced binding suggested that ligand-independent multimerization, driven by the PLAD domain, is important for ligand binding.
The TNFSF ligands can be found in soluble or membrane bound forms. Although one group of TNFRSF members (category I) can be activated by soluble ligand trimers, others (category II) require interaction with the membrane bound ligand to be fully activated (5). For example, soluble TNFα binds with higher affinity to TNFR1 than TNFR2 and primarily activates TNFR1 signaling, whereas TNFR2 is mainly activated by membrane bound ligand (11, 12). Although CD40 and GITR are both activated by trimeric ligands, activation is further enhanced with higher valency ligands or cross-linking of the trimeric molecules, presumably through induction of higher-order clustering (9, 13, 14). In contrast, CD27 and 4-1BB show minimal activation and require higher-order clustering (13). As described above activation of TNFRSF members can lead to multiple cellular outputs including proliferation, survival and differentiation, several of which may be therapeutically beneficial.
Rationale behind targeting TNFRSF
In addition to T-cell receptor (TCR) interaction with peptide-MHC (major histocompatibility complex), T cells require co-stimulatory signaling to be fully activated and generate an optimal response (15). Co-stimulatory TNFRSF members expressed on T cells include CD27, OX40, 4-1BB, TNFR2 and GITR. Co-stimulatory receptors on antigen presenting cells (APCs) are also important, with molecules such as CD40 playing a critical role in licensing and activation of dendritic cells (DCs) and B lymphocytes during an immune response (16), to elicit appropriate humoral and cellular adaptive immunity. DCs can be excluded from the tumor microenvironment and multiple immunosuppressive mechanisms can suppress their maturation and full activation, preventing effective T-cell responses (17–19). The DCs up-regulate multiple TNFSF ligands after maturation which are required for the optimal co-stimulation of T cells. Thus, targeting the TNFRSF members to provide co-stimulation is an attractive approach to elicit effective T-cell responses.
The majority of the T-cell co-stimulatory receptors are only upregulated and appreciably expressed after TCR activation, e.g. 4-1BB expression on adoptively transferred T cells is detected 12 to 24 hrs after stimulation (20) whereas others, most notably CD27, are constitutively expressed on T cells (2). Once expressed, the various TNFR are available for engagement by their ligands, which themselves also possess specific kinetics of expression (21). Although the downstream signaling pathways of the co-stimulatory TNFRSF members are not identical, signals are mainly initiated after TRAF recruitment to their cytoplasmic tails which leads to NF-κB and JNK pathway activation (22).
Stimulation of these co-stimulatory receptors contributes to enhanced effector function but also survival of the T cells. For instance, CD27 stimulation through engagement of its ligand CD70 leads to expression of cytokines such as IFN-γ, Interleukin-12 (IL-12), IL-5, IL-4 and IL-2 (2, 23), alongside the complementary cytokine receptors including IL-12R and IL-2R. Similar to CD27, stimulation of OX40 leads to upregulation of cytokines and cytokine receptors such as IL-12R and IL-2R on T cells, supporting their activation (24, 25). GITR stimulation also promotes the expression of IFN-γ, IL-2 and IL-2R (26) and is required for optimal CD8+ effector T-cell generation as absence of GITR on CD8+ T cells significantly reduces their expansion following an influenza infection (27). CD27 engagement can alter cellular metabolism to support the rapid expansion of T cells after activation. Here, the expression of the serine threonine kinase Pim-1 is upregulated to facilitate increased aerobic glycolysis and protein translation during proliferation (28–30).
TNFR signaling also supports survival of activated T cells. CD27 increases expression of the anti-apoptotic protein Bcl-XL in T cells, reduces the level of FasL on CD4+ T cells and reduces CD8+ T-cell sensitivity to FasL-stimulated apoptosis (29, 31). Similarly, anti-apoptotic proteins such as Bcl-XL and Bcl-2 are upregulated following OX40 stimulation (32), Bcl-XL and Bfl-1 are upregulated by 4-1BB (33) and Bcl-XL is upregulated after GITR engagement (27).
CD27 signaling induces CD8+ T-cell differentiation into cytotoxic T lymphocytes (CTL) and CD4+ T-cell differentiation into Th1 cells (28). Increased cytotoxic capacity of CTLs is supported by mechanisms such as upregulated of IL-2, important for their survival, and IFN-γ, which is further upregulated by IL-2 signaling. Increased cytotoxic capacity and effector functions of CD8+ T cells has also been shown after 4-1BB stimulation (34). Similar activities are evident on APCs, where CD40 signaling is critical for their ability to induce effective CD8+ T-cell responses. Stimulation of CD40 on DCs is important for their maturation and ability to present antigens to T cells. Activation of CD40 also leads to production of pro-inflammatory cytokines such as IL-12, IL-6 and IL-1β (35). Moreover, CD40 stimulates expression of co-stimulatory ligands such as CD80 and CD86, that interact with the receptors on T cells (e.g. CD28) for further activation.
In addition to the effects during naïve T-cell priming, co-stimulatory receptors of the TNFRSF contribute to the generation of the memory T-cell pool. CD27 signaling during the initial activation phase of CD8+ T cells is required for the development of memory CD8+ T-cell subsets and efficient expansion during the secondary response. Stimulation of CD27 during the initial response leads to IL-7Rα expression on effector CD8+ T cells, which in turn increases the frequency of memory precursor cells (36, 37). Similarly, 4-1BB and OX40 signaling are required for the generation of robust memory T-cell pools (38, 39). Stimulation of antigen specific CD8+ T cells with a 4-1BB agonist during priming leads to the generation of a strong memory CD8+ T-cell pool, resulting in a high secondary response (40). OX40 signaling is also important for T-cell memory. Although the primary expansion of CD8+ T cells was not impaired in OX40L-/- mice following influenza infection, there were defects in the secondary response of the virus specific CD8+ T cells (41). GITR has also been shown to be important for the secondary expansion of memory CD8+ T cells as in vitro generated WT or GITR-/- memory cells showed significantly different expansion capacity in an influenza infection recall response (27).
Additionally, the crucial role of co-stimulatory members of the TNFRSF in generating immune surveillance is evidenced by the development of various pathologies in individuals with TNFR deficiencies/mutations. For example, deficiency of CD27 or CD70 can lead to development of Epstein-Barr virus (EBV)-related immunodeficiency and lymphoproliferative disorders including B-cell malignancies (42, 43). Characterization of the immune response of an individual with CD27 deficiency who had hypogammaglobulinemia and persistent symptomatic EBV viremia revealed impaired IL-2 production in their CD8+ T cells which are the primary immune cells responsible for clearing EBV infections. IL-2 is critical for CD8+ T-cell function and impaired IL-2 production contributes to defective immune responses (42, 44, 45). 4-1BB deficiency can also lead to EBV driven complications and individuals can have persistent EBV viremia and EBV-related lymphoproliferation. CD8+ T cells from 4-1BB deficient individuals showed reduced proliferative and cytotoxic capacity (46). Deficiency in functional OX40 can lead to Kaposi sarcoma development in individuals with human herpes virus 8 infection (47). Similarly, CD40 or CD40 ligand deficiency can lead to immunodeficiency due to impaired APC function, which subsequently leads to impaired T-cell responses (48, 49), alongside an absence of germinal center-mediated somatic hypermutation and class switching in the humoral response known as hyper-IgM syndrome (50, 51). Dysregulation of the TNFRSF co-stimulatory receptor signaling and associated diseases identified to date are illustrated in Table 1. Further, the importance of co-stimulatory TNFRSF members in functional immune response generation is also supported in multiple constitutive and conditional TNFRSF knock out (-/-) models. For example, 4-1BBL deficiency in mice leads to impaired CD8+ T-cell responses against viral infections and predisposes the mice to B-cell lymphoma development (72–74). Similarly, CD27-/- mice have defects in the generation and accumulation of effector T cells at the site of infection following influenza infection, with the memory T-cell pool impaired (75, 76).
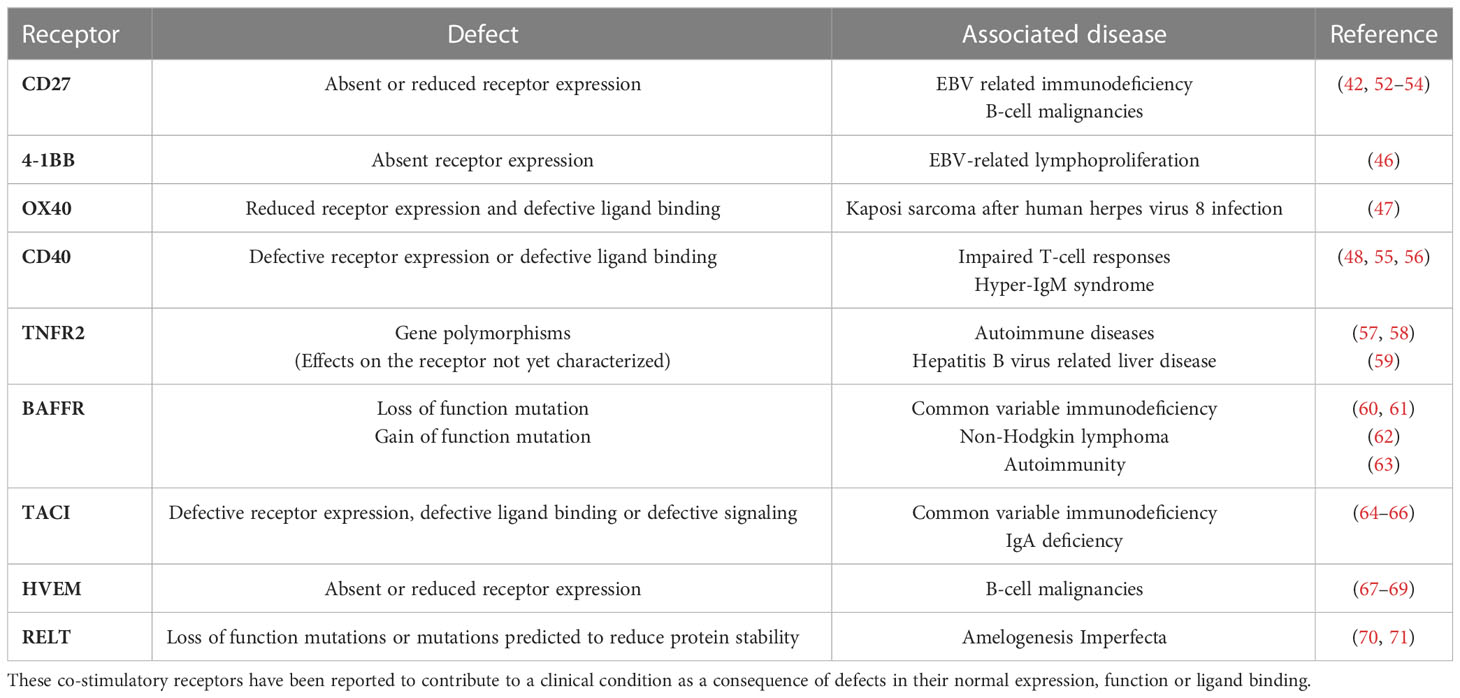
Table 1 TNFRSF co-stimulatory receptor dysregulation and disease development.
As the importance of co-stimulatory TNFRSF members in the development of a functional immune response has become clear, many of these receptors have subsequently been targeted to modulate the immune response in the context of immunotherapy. In this review we have restricted ourselves to discussing findings mainly in the field of cancer immunotherapy. Moreover, as various definitions of agonism exist, here we have defined agonism as activating the target receptor either via Fc gamma receptor (FcγR) dependent or independent mechanisms.
Therapeutic targeting of the TNFRSF
Agonistic targeting of the co-stimulatory members of the TNFRSF has shown to be effective in pre-clinical tumor models. Targeting 4-1BB in tumor models representing liver cancer, floor of mouth squamous cell cancer, colorectal cancer and lymphoma, using monoclonal antibodies (mAb) or recombinant 4-1BBL has generated robust anti-tumor responses (77–80). Buchan et al. demonstrated that two different mechanisms can contribute to a robust anti-tumor response induced by anti-4-1BB antibodies in certain models and contexts; 1) stimulating the effector T cells and 2) depleting T regulatory (Treg) cells. Additionally, depleting Tregs first and then agonizing the effector T cells induced better responses than only depleting the Tregs or agonizing the effector T cells (79). Similar to 4-1BB, targeting OX40 or GITR has been shown to stimulate robust anti-tumor responses in several pre-clinical tumor models, through a similar mechanism of action i.e. agonizing effector T cells or depleting Tregs (81–83). Treatment of solid tumors in a pre-clinical study with an agonistic anti-GITR mAb, increased the infiltration and activity of effector CD4+ and CD8+ T cells (84). In another study with the same agonistic mAb targeting a different solid tumor model however, the effect was mainly through depletion of intra-tumoral Tregs and slight increase in the infiltration of CD8s which resulted in a significantly improved CD8+ to Treg ratio (85). Additionally, the CD8+ T cells exhibited a more activated phenotype. These results indicate that anti-GITR mAbs can also act through different mechanisms and the dominant mechanism of action can vary depending on the tumor model. Moreover, it has been shown for OX40 and GITR targeting that the differential level of expression on effector T cells vs Tregs can lead to preferential depletion of Tregs as a consequence of higher levels of receptor expressed on them, enhancing immunotherapy (86–88).
Targeting CD27 has also been shown to induce significant anti-tumor responses in several pre-clinical models. Agonistic anti-CD27 antibody was efficacious in murine lymphoma models such as BCL1 and A31 (89). In a study where DCs in CD27-/- mice were manipulated to exhibit constitutive expression of CD70, an ovalbumin (OVA) expressing melanoma model (B16-OVA) was rejected following OVA specific (OT-1) CD8+ T cell transfer and OVA challenge whereas adoptive transfer of CD27-/- OT-1 CD8+ T cells did not elicit protective anti-tumor immunity (90) indicating the contribution of CD27/CD70 pathway to anti-tumor response in this model. In theory, targeting CD27 can induce anti-tumor responses by either agonizing the effector cells or depleting the Tregs dependent on the level of expression on individual cell populations (91) similar to targeting other members of the TNFRSF. Additionally, the method of CD27 targeting (modality, engagement of FcγR etc.) is also a key issue determining the mode of action as described in more detail below. Despite providing a strong anti-tumor response, the most agonistic anti-CD27 mAb also induced activation induced cell death in the effector CD8+ T cells (91) indicating that the strength of the stimulation needs to be appropriately tuned to induce a strong primary immune response and not impair other effects such as memory generation.
Another therapeutically exciting TNFR, TNFR2, is expressed on multiple immune cells, including Tregs at high levels and has been shown to be crucial for their survival. Therefore, targeting TNFR2 to deplete Tregs was considered as a potential mechanism to boost effector T-cell responses in anti-tumor immunity. Although several studies demonstrated the possibility of such an approach (92, 93), it has recently been shown that targeting TNFR2 can also work through agonistic mechanisms in pre-clinical models. Tam and colleagues demonstrated that an agonistic anti-TNFR2 mAb could stimulate the expansion of tumor specific CD8+ T cells with improved effector function. The agonistic mAb was efficacious in multiple pre-clinical solid tumor models and agonizing the effector CD8+ T cells was shown to be the main mechanism of action as demonstrated by increased frequency and functionality of antigen specific CD8+ T cells without the depletion of Tregs (94).
Antibody targeting of the TNFRSF
The main method for targeting the TNFRSF to date has been by using mAb. As the TNFRSF members require trimerization and higher-order clustering for optimal activation, one way that canonical bivalent mAbs can achieve this is by concurrently engaging with FcγR (Figure 2A). Depending on their isotype and subclass, mAbs interact with different FcγRs (95, 96). In mouse models, the mIgG1 isotype interacts with the inhibitory FcγRIIB with higher affinity and mediates further TNFR clustering to induce strong agonistic responses. However, depending on the tumor model, anatomical location of the tumor and microenvironmental factors, the availability of FcγRIIB can be limiting, impacting the response. In support of this observation, it has been shown in pre-clinical studies that the agonistic activity of anti-CD40 and anti-4-1BB mIgG1 antibodies relies on the availability of FcγRIIB (79, 97). It was further demonstrated that a two-fold reduction in FcγRIIB expression completely eliminated the agonistic activity of certain agonist anti-TNFR mAbs in vivo (98). In vitro studies support that for CD40 at least, if expressed at sufficient level, all FcγR can mediate increased agonism in line with their relative affinities for the given mAb isotype (97, 99). Cross-linking of the receptors is the most likely explanation for mAb induced agonism with chemical cross-linking of a mIgG2a mAb able to elicit potent agonism in vivo, whereas the native mIgG2a does not (100). Importantly, several studies have shown that downstream signaling from FcγRIIB is not required for its cross-linking activity (97, 98), most recently demonstrated for OX40 mAb in a mouse expressing FcγRIIB with a mutant, non-signaling, ITIM (101). Therefore, why FcγRIIB has this key cross-linking role in mice is not fully clear but perhaps relates to expression in the right place at the right time and the fact that multiple mouse models upregulate FcγRIIB in the tumor microenvironment, potentially due to hypoxia (102). Other variables such as the genetic background of the mouse strain may also contribute. For example, various polymorphisms in FcγRIIB have been shown to lead to reduced expression on macrophages and B cells which can increase the prevalence of autoimmune conditions (103). However, the extent of TNFR mAb agonism has not been compared in these different strains. Additionally, it has been reported in individuals with the autoimmune disorder systemic lupus erythematosus that the level of FcγRIIB expression on B cells is reduced (104) further highlighting that the level of FcγRIIB expression between individuals can vary, which could impact the agonistic activity of mAb in humans.
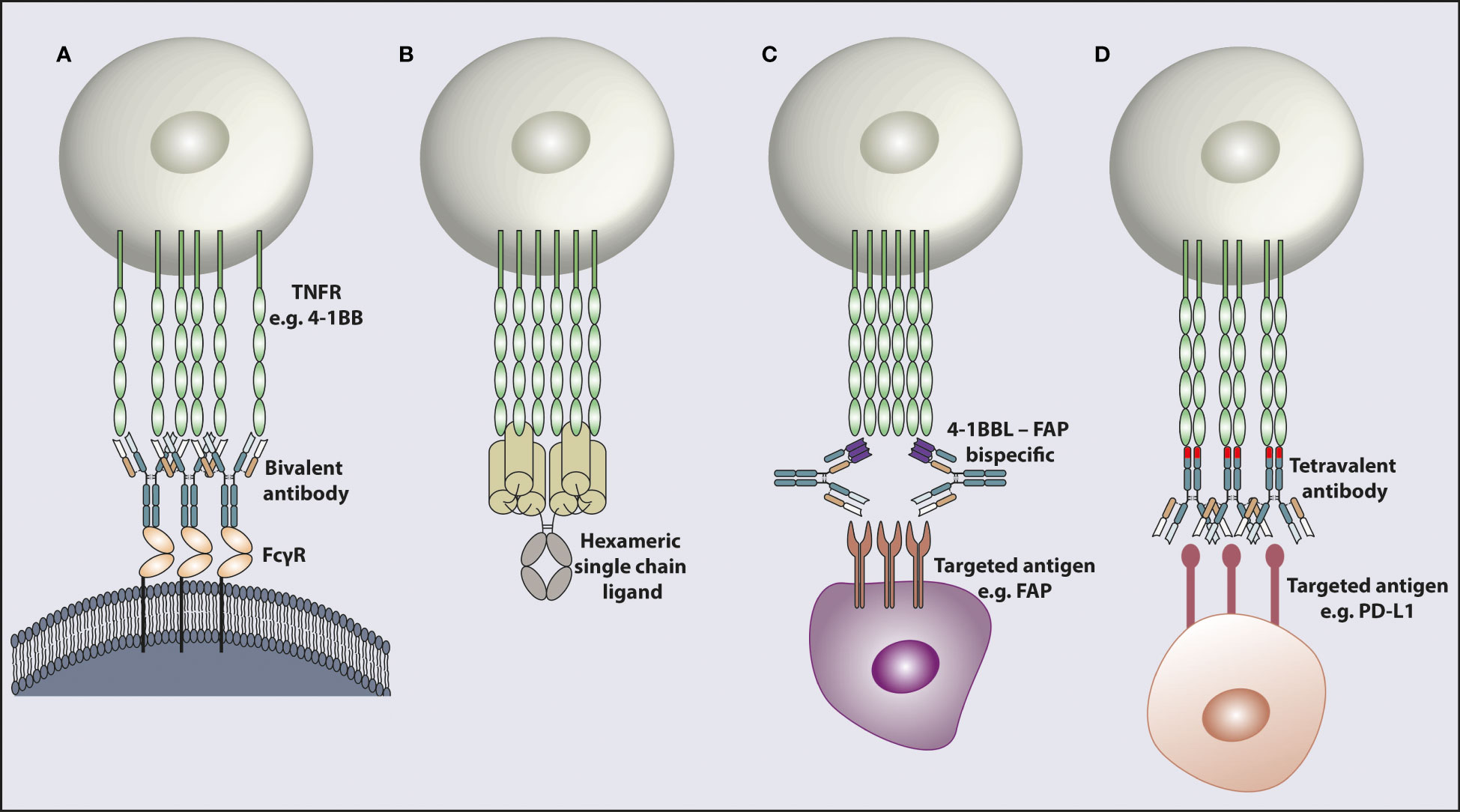
Figure 2 Modalities for targeting and activating TNFR. TNFR cross-linking achieved by different mechanisms. (A) Engagement with FcγR enables bivalent mAb cross-linking leading to target receptor clustering. (B) A recombinant hexameric single chain ligand inducing receptor clustering. The hexameric ligand structure is composed of a full Fc domain and six TNFSF ligand ECDs. (C) An antibody shaped bispecific molecule with one antigen binding arm targeting a TNFR e.g. 4-1BB and the other arm targeting a receptor e.g. FAP in the tumor microenvironment. (D) A bispecific molecule in a tetravalent format with two antigen binding arms targeting one receptor e.g. PD-L1 and the other two antigen binding arms in the opposite end of the molecule binding the TNFR e.g. 4-1BB, to induce receptor clustering. The 4-1BB binding domains inserted into the CH3 domain are indicated as a different color in the CH3 domain. TNFR; tumor necrosis factor receptor, FcγR; Fc gamma receptor, 4-1BBL; 4-1BB ligand, FAP; fibroblast activation protein, PD-L1; programmed death ligand 1.
FcγRIIB engagement however is not the only way to elicit higher-order TNFR cross-linking. In addition to FcγR cross-linking mediated agonistic activity of TNFR mAbs, it has been shown that the human IgG2 isotype can evoke greater clustering of TNFR leading to powerful receptor activation (105). Critically, this agonism is independent of the presence of FcγR and can be achieved in mice lacking all FcγR (106), although other studies indicate that hIgG2 induced agonism may be further augmented by FcγR binding (107, 108). The hIgG2 antibody is known to undergo disulfide switching in its hinge region, producing several different isoforms, including hIgG2A, hIgG2B and hIgG2A/B (109) with the hIgG2B isoform being highly agonistic and the hIgG2A isoform agonistically inert (105). Recent analysis has confirmed that the disulfide bonding pattern in the hinge region of the more agonistic isoforms gives the antibodies a less flexible conformation leading to increased agonism whereas the isoforms with higher flexibility were found to be less agonistic (110). Although initially shown first for anti-CD40 mAb, this capability of the hIgG2(B) isotype has subsequently been confirmed for OX40 and 4-1BB and also CD28 (a member of the immunoglobulin receptor superfamily) (99, 105).
Detailed characterization of several anti-TNFR mAb has also revealed that the level of agonistic activity can depend on which domain of the receptor the antibody binds to. Antibodies binding to CRD1, the membrane distal domain, of the CD40 extracellular region induced higher agonistic activity than antibodies binding to the membrane proximal domains (111). Similar to CD40, mAb binding to membrane distal domain of CD27, CRD1, were more agonistic (112). However, mAb binding to the membrane proximal CRD4 of OX40 were found to be more potent agonists than mAb binding to other CRDs (113). It should also be noted that even within a single domain, activity of antibodies may be markedly different with some far more highly agonistic dependent upon their fine epitope and also in rare cases can be independent of their isotype. For example the anti-CD40 mAb, CP870,893 binds CRD1 (111) and is highly agonistic in any isotype, whereas 341G2, which also binds CRD1, is entirely inert as a hIgG1 and hIgG4 but maximally active and super-agonistic as a hIgG2 (106). Similar observations can also be made with other TNFRs (94). Of interest, most agonistic anti-CD40 mAbs, bind in CRD1 and so do not block ligand binding. In contrast, mAbs binding within CRD2/3 block ligand binding and are less agonistic (111). This observation may support a model whereby optimal agonists bind outside the ligand binding region. However, the above mentioned 341G2 mAb blocks ligand binding but is highly agonistic, indicating this model is incorrect. This observation is supported with other TNFR family members as an agonistic TNFR2 mAb was found to completely block ligand binding but still induce strong agonism (94). These observations suggest that binding to the same epitope as the natural ligand is not a key determinant of mAb-mediated receptor agonism but rather that certain domains and epitopes might be more generally preferable for driving agonism (such as CRD1). However, as detailed above this is likely to differ for individual receptors, according to their structure and biology.
Tolerability and response of agonistic TNFRSF targeting in clinical trials
Several agents targeting the co-stimulatory members of the TNFRSF have been tested in clinical trials. Results have demonstrated that targeting certain receptors is well tolerated whereas targeting others is limited due to toxicity. A list of the agents targeting these receptors can be found in Table 2, with specific examples outlined in further detail below.
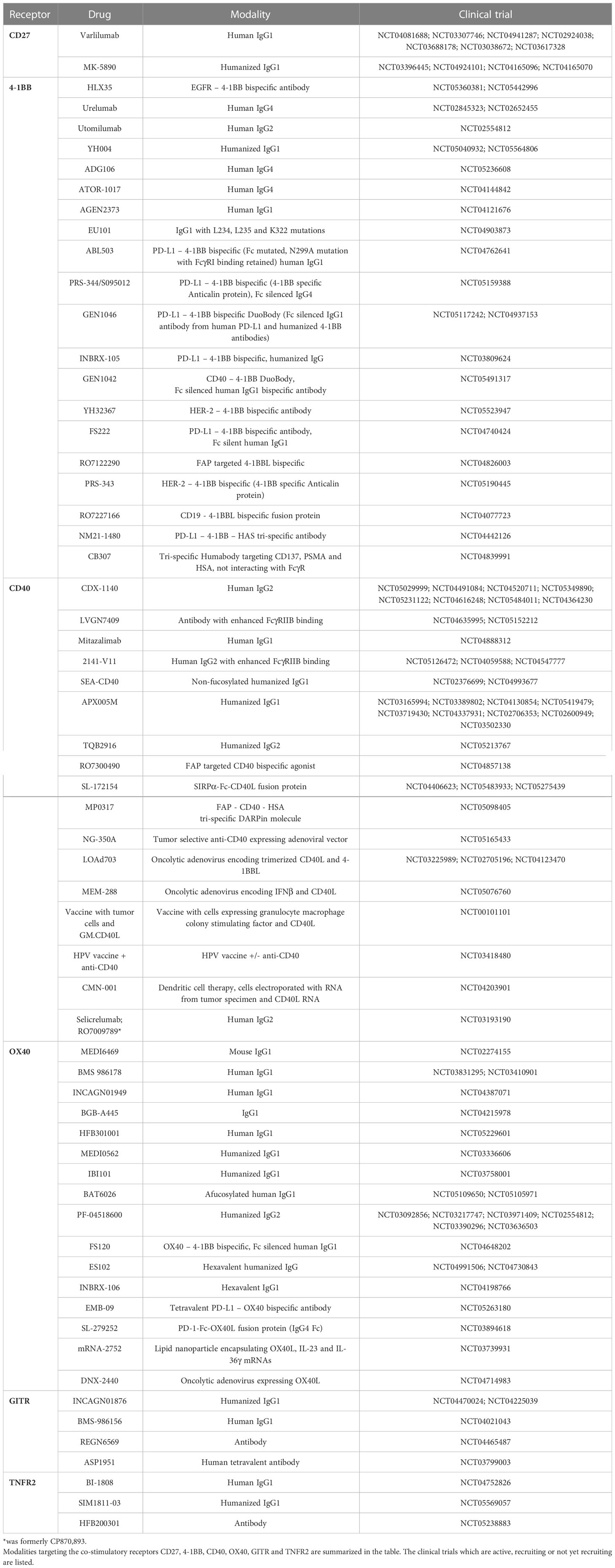
Table 2 Co-stimulatory TNFRSF targeting agents in clinical trials.
Targeting CD27
As discussed above, CD27 is required for generating functional immune responses and targeting this receptor in pre-clinical studies has generated promising results supporting clinical evaluation. Varlilumab is a human IgG1 anti-CD27 antibody. It was well tolerated up to the maximum tested dose of 10 mg/kg with no major adverse events as a monotherapy (114, 115). Most of the toxicity related events were grade 1 or 2 with fatigue, rash, nausea, and diarrhoea the most common. Only 1 out of 56 patients had a transient grade 3 adverse event which was asymptomatic hyponatremia at 1 mg/kg. As a monotherapy, Varlilumab showed biological and clinical efficacy against tumors including hematologic malignancies, melanoma and renal cell carcinoma (114, 115). It stimulated chemokine secretion, increased the number of activated T cells and induced Treg depletion. Overall, 8 out of 56 patients had stable disease (SD) and 1 patient had a partial response (PR) (115). More recently, Varlilumab has been combined with anti-programmed cell death protein-1 (PD-1) checkpoint blockade and no additional toxicities were observed compared to anti-CD27 monotherapy. Although the initial results suggested that the combination treatment was safe and induced SD in 17% of colorectal cancers (CRC), SD in 39% of ovarian cancer (OVAC) patients, PR in 5% of CRC and PR in 10% of OVAC patients (116), more recent results revealed that the objective response rate (ORR) observed in the study was less impressive: 0% for renal cell carcinoma, 5% for CRC, 12.5% for head and neck squamous cell carcinoma and 12.5% for OVAC (117). Following promising results of a pre-clinical study demonstrating that anti-CD27 and anti-CD20 mAb in combination induced robust anti-tumor efficacy in pre-clinical B-cell lymphoma models (118), another clinical study was designed where Varlilumab was combined with the anti-CD20 antibody Rituximab to test efficacy in relapsed or refractory B-cell lymphoma. Combination treatment was in general safe but induced a grade 3 or higher adverse event in 33% of patients. The treatment was efficacious in tumors with T-cell activated status inducing SD in 3 out of 26 patients and PR in 4 out of 26 patients (119). Another CD27 targeting agonistic mAb in development is MK-5890, which is a humanized IgG1 antibody that is being tested in the clinic as a single agent or in combination with PD-1 blocking agents in advanced solid tumors. The pre-clinical characterization of the mAb demonstrated that it could induce anti-tumor responses as a monotherapy or in combination with PD-1 blockade (120). Early results suggest an acceptable safety profile, although 24% of patients in the monotherapy group developed grade 3 or 4 adverse events related to treatment. Combination treatment did not increase the level of adverse events observed with single agent. Early signs of efficacy with MK-5890 monotherapy or combination, stimulating anti-tumor responses in patients, were observed (121). Although the mAb could induce transient upregulation of chemokine levels in patients, it also induced decreases in the level of circulating T cells (120) suggesting that identifying the right dosing regimen will be important for the successful application of this mAb. A recent study in a pre-clinical setting addressed the determinants of agonism for anti-CD27 mAb (112). It demonstrated that agonism is dictated in part by the mAb-binding domain, with the membrane distal, externally facing epitopes delivering the highest level of agonism. Additionally, the agonistic activity of hIgG1 mAb was shown to be improved by Fc engineering through either enhanced binding to FcγRIIB or hIgG2 isotype selection. The anti-CD27 mAb currently in clinic (Table 2) are unmodified hIgG1 antibodies, likely sub-optimal for agonism, and so armed with this encouraging pre-clinical data, the next generation of anti-CD27 mAb may provide greater clinical efficacy.
Targeting 4-1BB
4-1BB activation contributes to an optimal immune response and pre-clinical targeting of 4-1BB in mouse tumor models generated robust anti-tumor responses, supporting clinical evaluation. There are two mAbs that have been explored comprehensively in the clinic that target 4-1BB. Utomilumab is a human IgG2 antibody that has been shown to have a favorable safety profile, being well tolerated up to 10 mg/kg. The majority of the adverse events caused by the antibody were grade 1 or 2 including rash, dizziness, decreased appetite and fatigue in less than 10% of the patients in the study. Only 1 patient developed a grade 3/4 fatigue without increased transaminase levels. The overall ORR in solid tumors was 3.8% whereas the ORR in fifteen Merkel cell carcinoma patients was 13.3% with one PR and one complete response (CR) (122). Utomilumab has also been tested in combination with anti-CD20 treatment in patients with relapsed or refractory follicular lymphoma and CD20+ non-Hodgkin lymphoma (NHL). Initial results suggested that the combination did not affect tolerability with the majority of the treatment related adverse events being grade 1 or 2. The combination treatment showed some clinical activity especially in the NHL patients (123). Additionally, safety of Utomilumab in combination with anti-PD-1 blockade was tested in patients with advanced solid tumors and the combination was found to be tolerable with mainly grade 1 or 2 toxicities and PR or CR in 6 out of 23 patients in the study (124). However, despite tolerability, clinical responses have overall been underwhelming.
Urelumab is another 4-1BB targeting agonist antibody which is of human IgG4 isotype. A study testing the safety and tolerability of Urelumab indicated that the maximum tolerated dose (MTD) of antibody given every 3 weeks was 0.1 mg/kg and higher doses induced liver toxicity in a higher percentage of patients and at higher severity above 1 mg/kg dose (125). In another study in which Urelumab was combined with Rituximab, the MTD was again found to be 0.1 mg/kg but the combination did not enhance the effect achieved by Rituximab alone (126). Several pre-clinical studies suggested that the liver toxicity induced by agonist anti-4-1BB antibody could be due to infiltration and activation of macrophages in the liver which leads to infiltration and abnormal activation of T cells, mainly CD8+ T cells, leading to tissue damage (127, 128). Minimizing FcγR interactions through deglycosylation has been shown to reduce these toxicities (129).
Targeting CD40
CD40 signaling is important for APC (DC and B cell) activation and the development of strong T-cell responses. It is one of the most targeted members of the TNFRSF in clinical trials. One of the initial antibodies to be tested in multiple studies was CP870,893 which is a human IgG2 mAb. However, the antibody had to be given at low doses due to the MTD being 0.2 mg/kg. The antibody achieved modest clinical effects as a monotherapy in advanced solid tumor patients potentially due to the low doses not saturating the receptor (16). CP870,893 has also been tested in combination with multiple agents ranging from checkpoint blockade antibodies to chemotherapy. Although a significant improvement in response was not achieved with checkpoint blockade combination, combining anti-CD40 mAb with chemotherapy achieved significant responses in pancreatic ductal adenocarcinoma patients (130). Another human IgG2 anti-CD40 mAb recently developed is CDX1140. Initial studies suggested that the antibody is tolerated up to 1.5 mg/kg as a single agent or in combination with a recombinant dendritic cell growth factor, with the majority of the adverse events being low grade and early suggestion of clinical benefit in advanced solid and hematologic tumor patients (131). The 1.5 mg/kg dose is expected to give better systemic targeting of the receptor and tissue penetration compared to the MTD of CP870,893.
As described above, human IgG2 antibodies can elicit TNFR activation without requiring FcγR mediated cross-linking. However, there is also interest in developing agents with enhanced ability to bind to FcγRIIB to mediate optimal cross-linking of the antibody, leading to greater receptor clustering and activation. APX005M is a humanized IgG1 anti-CD40 antibody possessing the S267E mutation in its Fc domain which enhances the affinity for FcγRIIB binding by 30-fold (132). Combining APX005M with anti-PD-1 blockade to treat anti-PD-1/PD-L1 refractory melanoma patients showed that the combination did not increase toxicity and the majority of adverse events were grade 1 or 2. Early results from the study are promising and indicate that the combination evokes clinical benefit (133).
Targeting OX40
Pre-clinical studies demonstrated the anti-tumor potential of reagents targeting OX40 and agonistic anti-OX40 antibodies have been shown to be well tolerated in patients. However, the response rates as a monotherapy have been low. GSK3174998 was an agonist humanized IgG1 mAb tested against advanced solid tumors but only induced 1 PR and 1 SD in 45 patients as a monotherapy and the combination with the anti-PD-1 mAb Pembroluzimab did not significantly improve the efficacy expected with Pembrolizumab alone (134). A humanized IgG2 mAb PF-04518600 was tested as a monotherapy in advanced solid tumor patients but only 1 out of 25 patients had a PR while 15 out of 25 had SD (135). In a recent study in which PF-04518600 was combined with Utomilumab, early indications were that the combination was found to be well tolerated and 7 out of 10 melanoma patients and 7 out of 20 non-small cell lung cancer (NSCLC) patients experienced SD in addition to only 1 NSCLC patient experiencing a PR (136).
Another agonistic anti-OX40 mAb being tested in clinical trials is MEDI0562 which is a humanized IgG1 antibody. As a monotherapy in advanced solid tumors, MEDI0562 was found to be safe with the majority of adverse events being grade 1 or 2. Despite the favorable safety profile, only 2 out of 55 patients experienced a PR and 24 out of 55 patients experienced SD (137). In another study where MEDI0562 was combined with anti-PD-L1 or anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4) immune checkpoint blockade in advanced solid tumors, early results indicated that the combinations induced grade 3 or 4 adverse events in a high frequency of patients and only 11.5% of patients in the anti-PD-L1 combination group showed PRs. 34.6% of patients in the anti-PD-L1 combination group and 29% in the anti-CTLA-4 combination group experienced SD (138).
Targeting GITR
GITR activation leads to the development of strong T-cell responses and mouse tumor model studies have demonstrated the anti-tumor potential of GITR targeting. Several agonistic antibodies targeting GITR have been tested in clinical trials. MK-1248 is an agonist humanized IgG4 antibody against GITR. In a study investigating the tolerability of MK-1248 as a single agent or in combination with anti-PD-1 blockade in advanced solid tumors, it was found that despite approximately 50% of patients in both arms of the study developing grade 3 or higher adverse events, the clinical benefit was very limited. No objective response was achieved with monotherapy and only 1 CR and 2 PRs were observed in the combination arm. 15% of patients receiving single agent experienced SD whereas 41% of patients receiving combination therapy experienced SD (139). Another agonistic anti-GITR agent is BMS-986156, which is a human IgG1 antibody. BMS-986156 was well tolerated as a single agent in advanced solid tumor patients with no grade 3 or higher adverse events and only 9.3% of patients in combination with anti-PD-1 experiencing grade 3 or 4 adverse events. Despite the favorable safety profile, no response was observed with BMS-986156 as a single agent and the highest ORR in the combination group was only 11.1% (140). MK-4166 is another human IgG1 anti-GITR antibody that has been recently tested in advanced solid tumor patients in combination with anti-PD-1 blockade. Although the treatments were found to be well tolerated, single agent again did not induce any clinical benefit. Comparing the checkpoint blockade treatment naïve versus pre-treated melanoma patients showed that the treatment naïve patients were responsive to MK-4166 and anti-PD-1 combination. 5 out of 13 patients had a CR and 3 out of 13 patients had a PR suggesting that the combination treatment might be efficacious in this particular group of patients (141).
Targeting TNFR2
TNFR2 targeting agonist mAbs can generate strong anti-tumor T-cell immunity but are mainly still in pre-clinical development and only recently starting clinical assessment. MM-401 is an agonist anti-human TNFR2 mAb in development. Using a mouse surrogate version of the antibody, it was found that TNFR2 agonism could generate strong anti-tumor responses by activating CD8+ T cells and NK cells with activity dependent on FcγR interactions, presumably mediated by cross-linking of the receptor. In addition, the antibody synergized with checkpoint blockade (142). BI-1910 is another agonist anti-TNFR2 mAb in development following promising results from a surrogate anti-mouse TNFR2 antibody; this mAb induced strong anti-tumor responses in several pre-clinical tumor models and was effectively combined with checkpoint blockade antibodies. The dominant mechanism of action was expansion of CD8+ T cells and improved CD8+ to Treg ratio in the tumor site (143). BI-1808 is an alternative TNFR2 targeting mAb, classified as a deleting, ligand blocking molecule. However, pre-clinical studies with a mouse surrogate indicated intra-tumoral Treg depletion and effector T-cell expansion leading to an improved CD8:Treg ratio. Similar results were obtained with BI-1808 in pre-clinical characterization. BI-1808 was found to be well tolerated in non-human primates and is in clinical assessment (143, 144). HFB200301 is also an anti-TNFR2 agonist antibody which is already in a phase I clinical trial of advanced solid tumor patients (145). Using human TNFR2 knock-in mouse models, it was suggested that HFB200301 could stimulate anti-tumor responses through expansion of effector T cells and NK cells without depleting the Tregs. The agonistic ability of the antibody was found to be independent of FcγR mediated cross-linking (146). Although much of the data is not yet mature, with peer review lacking for most of the pre-clinical studies, the potential of TNFR2 targeting antibodies in oncology are exciting and the initial results from clinical trials are eagerly awaited by the immuno-oncology community.
Recent approaches in targeting TNFRSF members to overcome current limitations
Fc engineering
As described above, despite success in pre-clinical studies, clinical efficacy of targeting TNFRSF members has been limited. One factor which may help to explain this is the lack of a human antibody isotype equivalent of mIgG1 with preferential binding toward FcγRIIB to facilitate agonistic activity. Therefore, in order to enhance FcγRIIB engagement, Fc engineering approaches have been developed to improve the affinity of antibodies toward hFcγRIIB. Although several mutations such as SE (S267E) and SELF (S267E-L382F) have been identified to improve affinity to hFcγRIIB, those mutations improved affinity to hFcγRIIa as well, due to sequence and structural similarity between the two receptors. Other mutations such as V9 (G237D-P238D-P271G-A330R) and V11 (G237D-P238D-H268D-P271G-A330R) however, were found to specifically improve the affinity of antibodies toward hFcγRIIB by approximately 32 and 96 fold, respectively (107). Comparing WT and Fc engineered anti-human CD40 antibodies in mice expressing hFcγRs, the variant with the V11 mutation was found to be superior to others, indicating the possibility of this approach to be taken forward for further development. Subsequent analysis demonstrated that systemic delivery of the agonistically enhanced variant could pose a risk of inducing toxicity and optimal receptor occupancy might not be reached with the MTD. Delivering the mAb via intra-tumoral injections was shown to ameliorate toxicity, yet retain significant tumor control even at low doses (147) indicating that where this method of delivery is practical (e.g. for localized/accessible lesions) it could provide a solution.
Another approach to overcome the requirement for mAb cross-linking could be via alternative, FcγR-independent, Fc domain engineering which was recently demonstrated for anti-human OX40 mAbs. Building on seminal studies showing that E345R, E345K and E430K single point mutations in the Fc region could promote “on-target” multimerization (once the mAb binds to the receptor) of the mAbs to facilitate optimal engagement of the hexa-headed C1q molecule (148, 149), Zhang et al. showed that E345R single mutation or K248E-T437R double mutations in the Fc region could induce “on-target” multimerization of agonistic OX40 antibodies, leading to activation of the receptor in an FcγR-independent way (150, 151). Although the Fc engineered antibodies were active in the absence of FcγR cross-linking, their activity could be further improved by FcγRIIB mediated cross-linking, suggesting that this approach could provide the possibility of targeting receptors in tissues without FcγRIIB availability but when FcγR are available, the activity will be further boosted.
Receptor cross-linking independent of FcγR
Although improving FcγRIIB affinity of antibodies can augment agonism, as previously mentioned the availability of FcγRIIB at the relevant anatomical site to provide the cross-linking can be a limiting factor. Thus, alternative approaches have been developed to generate agonistic agents without the requirement of FcγR mediated cross-linking. In addition to the hIgG2 isotype, soluble recombinant TNFSF ligands have been explored as a means to replicate the natural multimeric ligand-receptor interaction. The potency of soluble trimeric ligands could be improved by additional cross-linking and this approach was demonstrated for several ligands including OX40L, CD40L and 4-1BBL (13, 152). However, as the soluble trimeric ligands still require additional cross-linking, practicality of this approach in vivo is likely to be challenging due to possible short serum persistence of the trimers and also additional non-native sequences potentially making the products more immunogenic. To overcome this limitation, multimeric forms of soluble trimeric TNFSF ligands such as Fc fusion proteins have been developed. Multimeric ligands do not require the additional cross-linking required by the trimeric forms and the Fc fusion facilitates better in vivo persistence via its interaction with the neonatal Fc receptor (FcRn) (153). A CD27L-Fc fusion protein designed to mimic the natural CD27L activity was found to be active in in vitro and in vivo assays boosting T-cell activation (154). In that study, one CD27L extracellular domain (ECD) was fused to one Fc domain suggesting that the active product consisted of multimeric trimers of the ligand and multimers of Fc domains. More recently, a hexameric human CD27L fusion protein consisting of six CD27L ECDs and a silent human IgG1 Fc domain (not interacting with FcγR) has been reported (155). In this construct, three ECDs of the ligand are linked in a single chain format and fused to the IgG1 Fc domain with the idea of bringing two ligand trimers together upon Fc domain dimerization (Figure 2B). The fusion protein induced activation and proliferation of T cells in in vitro and in vivo experiments independently of FcγR engagement (155). Additionally, the hexameric fusion protein demonstrated anti-tumor efficacy in pre-clinical models. Hexameric Fc fusion ligand proteins in the same format have also been developed for CD40L, GITRL and 4-1BBL (156–158). Despite the promising pre-clinical results, the hexameric ligand proteins have short half-lives in circulation. Although this could be considered as a disadvantage, shorter stimulation of the immune cells can also lead to generation of a strong response and possibly could be better than chronic stimulation, which might have detrimental effects (159, 160). It has been shown in multiple studies that continuous stimulation of CD27 leads to defects in the immune cells. Continuous stimulation of CD27 by constitutive expression of CD70 on B cells resulted in increased apoptosis and depletion in NK cells (161) or T-cell immunodeficiency (159). Similarly, continuous 4-1BB stimulation leads to overactivation of CD8+ T cells and macrophages which eventually results in impaired CD8+ T-cell activity (160). Thus, timing and strength of stimulation are crucial in inducing a strong immune response and avoiding immunopathology. By experimentally determining the correct dose, schedule and treatment routes the hexameric ligands might generate strong immune responses in patients. On the other hand, it is worth noting that agonistic ligand formats, specifically TNFR2 specific recombinant TNF ligand protein, have also been developed with the aim of expanding Tregs in non-cancer contexts. A nonameric version of the recombinant protein was initially found to have suboptimal serum retention in vivo but a newly developed version in which an Fc silent irrelevant IgG molecule is fused to two trimeric ligand units to generate a hexameric ligand showed improved pharmacokinetics and robust Treg expansion in vivo (162).
Recent technological advances in the field have enabled the use of computational methods to design desired structures. Using such approaches, researchers have produced antibody molecules in various oligomeric states, in a format described as “antibody nanocages”. These nanocages were found to activate several receptor targets, including converting an antagonist anti-CD40 mAb into an agonist due to the ability of the designed structure to induce receptor clustering (163). This approach could potentially be applied to a plethora of different receptors to identify the best design for optimal receptor activation in each case.
Reagents targeting tumor microenvironment to induce localized TNFR activation and reduce toxicity
In addition to the variation of the availability of FcγRIIB in target tissue to provide optimal cross-linking of agonistic mAbs, off-target toxicity has also been an issue. Although some agonistic mAbs such as Varlilumab against CD27 was well tolerated, the clinical efficacy was modest. In contrast, the 4-1BB agonist Urelumab was active but found to induce liver toxicity at high doses. The mechanism behind the toxicity of Urelumab is thought to be the activation of the liver resident FcγR-expressing Kupffer cells, with the agonistic cross-linking of the anti-4-1BB mAb enabled by the high level of FcγR expressed on these myeloid cells (98) or other FcγR-expressing cells in the liver, such as FcγRIIB expressing sinusoidal liver endothelial cells (164). Activated Kupffer cells produce IL-27 which is an inflammatory cytokine involved in infiltration and expansion of other immune cells, especially T cells into the tissue (127). Hepatotoxicity following 4-1BB agonism indicated that systemic delivery of the agonistic reagents has the risk of off-target toxicity. Thus, recent efforts have focussed on eliminating the risk associated with systemic delivery in favor of targeted agonism – localizing the mAb to the desired site. One approach has been to generate recombinant proteins with a tumor targeting domain. For example, single chain fragment variable (scFv) domains of an anti-4-1BB mAb have been fused to a trimerization domain (producing a trivalent 4-1BB targeting molecule) with further fusion of a tumor targeting domain on the C-terminus to direct the trimer to the tumor site (165). Although the trimeric protein had short in vivo stability, the anti-tumor response generated in mouse tumor models was similar to an agonistic anti-4-1BB mAb and the trimer did not induce toxicity, which was apparent with the agonistic mAb. Additionally, repeated dosing of the trimeric protein also did not induce off-target toxicity indicating that targeted agonism approach could overcome the non-specific toxicity.
More recently, a tumor antigen targeting 4-1BB bispecific molecule was generated with one arm of the antibody designed to target a tumor antigen and the other designed to form a trimeric h4-1BBL. The bispecific molecule was generated in an Fc silent format to maintain normal antibody-like pharmacokinetics but at the same time eliminating FcγR engagement to prevent off-target toxicity. Binding of the tumor antigen specific arm at the tumor site allows accumulation of 4-1BBL in the tumor tissue to facilitate multimeric interaction between the ligand and receptor (Figure 2C) and activate the T cells in the tumor microenvironment. The bispecific molecule had a favorable pharmacokinetic profile and could accumulate in the tumor site, confirmed in non-human primates (166). Additionally, the bispecific molecule proved to be able to induce activation of T cells from human tumor tissues and also induce anti-tumor immunity in pre-clinical models. However, the main activity was observed when the bispecific molecule was used in combination with another T-cell bispecific agent stimulating the TCR and targeting a tumor antigen (166, 167), indicating that optimal co-stimulation happens in the presence of TCR stimulation. While the bispecific molecule had favorable serum stability, it did not induce toxicity indicating that it could be used in combination with other T-cell inducing treatments. Similar bispecific molecules with a scFv arm targeting a tumor associated antigen and a TNFSF ligand arm targeting a co-stimulatory receptor on the T cells have also been characterized in other studies (168). In these molecules however, a tag was inserted for purification purposes and its immunogenicity will have to be assessed further during in vivo validation of these reagents.
Another approach to induce TNFR clustering involves duokines, where both arms of the bispecific molecule are targeting members of the TNFRSF. Initially, the proteins were developed by either fusing one ECD protomer of a TNFSF ligand to one ECD protomer of another TNFSF ligand to allow trimerization of the ligand molecules by non-covalent interactions or by developing them as a single chain polypeptide in which three ECDs of each ligand were linked onto the same polypeptide chain separated by flexible linkers (169). Depending on the choice of ligands, this approach allows targeting of receptors in cis (on the same cell surface) or trans (on different cells) orientations. The single chain duokines were found to be more stable than non-covalently formed duokines and could induce in vitro and in vivo stimulation of T cells as co-stimulatory molecules. Using 4-1BBL-CD40L as a trans acting duokine or 4-1BBL-CD27L as a cis acting duokine, Fellermeier-Kopf and colleagues showed that both molecules could induce anti-tumor immunity in a pre-clinical melanoma model in combination with a TCR targeting bispecific antibody (169). In a subsequent study, Fc fusion proteins of the duokines were generated to facilitate enhanced stability in circulation with the active protein adopting an antibody structure with each single chain trimeric ligand domain being fused to Fc regions and dimerization of the Fc regions bringing two trimeric ligands together (170). Although the protein was still active in combination with a TCR targeting bispecific antibody, interestingly, the Fc fusion did not improve the pharmacokinetic profile. These data demonstrated the possibility of using these duokines to target two co-stimulatory TNFR molecules to boost the anti-tumor response. By identifying the optimal combination strategies, they could potentially enhance the anti-tumor responses in the clinic.
It has been clearly observed that blocking the immune checkpoint molecules PD-1 or CTLA-4 can generate strong anti-tumor responses but the majority of the patients are either refractory or develop resistance to these therapies. In recent studies, bispecific molecules targeting the checkpoint inhibitory receptors and co-stimulatory members of the TNFRSF have been developed as a means to enhance their activity. There are multiple advantages to this approach: First, the interaction of the inhibitory checkpoint receptor and its ligand is blocked to release the suppression on the immune response. Second, the inhibitory molecules are mainly expressed in the tumor microenvironment and this ensures targeted activation of the co-stimulatory receptor at the tumor microenvironment, avoiding systemic toxicity. Third, the bispecific antibody can be generated in an Fc silent format to avoid potential systemic toxicity with co-stimulatory receptor clustering achieved by the checkpoint receptor targeting arm acting as an anchoring domain. An Fc silent IgG1 bispecific antibody in a tetravalent format with two Fab arms targeting PD-L1 and two 4-1BB targeting domains introduced into the CH3 domains, termed Fc-region with antigen binding, was recently developed (Figure 2D). The mouse surrogate version of the bispecific induced activation of T cells in vitro and induced anti-tumor immunity in vivo without hepatotoxicity. The human version of the protein induced human T-cell activation in vitro and toxicology studies in non-human primates, enabled by cross-reactivity between species, showed that the bispecific was well tolerated (171) and had higher activity than the combination of the single agents. Similar bispecific molecules in tetravalent formats targeting PD-L1 and CD40 or 4-1BBL have also been reported in other studies. In vitro characterization of these products showed PD-1/PD-L1 blockade and target receptor activation in an FcγR independent manner, supporting further validation in in vivo studies (172). In support of these findings with bispecific molecules, it was recently shown that an anti-PD-1/GITRL bispecific molecule induced a different mechanism of action than the combination of single agents and was more efficacious in pre-clinical studies (9). The co-stimulatory antibodies being tested in combination with checkpoint blockade antibodies to date have shown favorable tolerability in clinical trials (see above), and the recent findings support the idea that the bispecific molecules could achieve better results than the combination treatments.
Conclusion
TNFRSF members represent powerful targets for immunomodulation. Promising pre-clinical data of agonistic mAbs targeting TNFRSF has clearly demonstrated their potential to provide anti-tumor efficacy. However, the translation from the pre-clinical studies to the clinic has been difficult and lack of significant response rates or toxicity in the clinic with conventional mAbs has directed researchers to develop new strategies.
Other immunomodulatory agents such as the checkpoint blockade antibodies have shown better success than the agonistic antibodies against TNFRs. However, while the responses thus far are limited, there is an opportunity for combining the two strategies, as has been shown in pre-clinical studies. With new approaches, such as targeted agonism and bispecifics delivering two or more different mechanisms of action with a single agent, success rates may improve. The challenge however remains the same – evoking powerful, curative immune responses while avoiding toxicity. Hopefully, such innovation will finally unlock TNFR targeting for the clinic.
Author contributions
OD researched data and wrote the manuscript with MC, OD, and AE produced the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Cancer Research UK funding received by MC.
Acknowledgments
We would like to thank the members of the Antibody and Vaccine group for helpful discussions related to agonistic antibodies and TNFR targeting over the last decade.
Conflict of interest
MC acts as a consultant for a number of biotech companies, being retained as a consultant for BioInvent International and has received research funding from BioInvent International, GSK, UCB, iTeos, and Roche and receives institutional payments and royalties from patents and licenses relating to antibody immunotherapy. OD has received research funding from BioInvent International, outside of the current work.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF family of ligands and receptors: Communication modules in the immune system and beyond. Physiol Rev (2019) 99(1):115–60. doi: 10.1152/physrev.00045.2017
2. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9(4):271–85. doi: 10.1038/nri2526
3. Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci (2002) 27(1):19–26. doi: 10.1016/S0968-0004(01)01995-8
4. Sica GL, Zhu G, Tamada K, Liu D, Ni J, Chen L. RELT, a new member of the tumor necrosis factor receptor superfamily, is selectively expressed in hematopoietic tissues and activates transcription factor NF-kappaB. Blood (2001) 97(9):2702–7. doi: 10.1182/blood.V97.9.2702
5. Kucka K, Wajant H. Receptor oligomerization and its relevance for signaling by receptors of the tumor necrosis factor receptor superfamily. Front Cell Dev Biol (2020) 8:615141. doi: 10.3389/fcell.2020.615141
6. Camerini D, Walz G, Loenen WA, Borst J, Seed B. The T cell activation antigen CD27 is a member of the nerve growth factor/tumor necrosis factor receptor gene family. J Immunol (1991) 147(9):3165–9. doi: 10.4049/jimmunol.147.9.3165
7. Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science (2000) 288(5475):2351–4. doi: 10.1126/science.288.5475.2351
8. Chan FK. Three is better than one: pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine (2007) 37(2):101–7. doi: 10.1016/j.cyto.2007.03.005
9. Chan S, Belmar N, Ho S, Rogers B, Stickler M, Graham M, et al. An anti-PD-1-GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy. Nat Cancer. (2022) 3(3):337–54. doi: 10.1038/s43018-022-00334-9
10. Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, et al. Solution of the structure of the TNF-TNFR2 complex. Sci Signal (2010) 3(148):ra83. doi: 10.1126/scisignal.2000954
11. Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell (1995) 83(5):793–802. doi: 10.1016/0092-8674(95)90192-2
12. Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A. (1998) 95(2):570–5. doi: 10.1073/pnas.95.2.570
13. Wyzgol A, Müller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, et al. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol (2009) 183(3):1851–61. doi: 10.4049/jimmunol.0802597
14. Haswell LE, Glennie MJ, Al-Shamkhani A. Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur J Immunol (2001) 31(10):3094–100. doi: 10.1002/1521-4141(2001010)31:10<3094::AID-IMMU3094>3.0.CO;2-F
15. June CH, Bluestone JA, Nadler LM, Thompson CB. The B7 and CD28 receptor families. Immunol Today (1994) 15(7):321–31. doi: 10.1016/0167-5699(94)90080-9
16. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res (2013) 19(5):1035–43. doi: 10.1158/1078-0432.CCR-12-2064
17. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
18. Marciscano AE, Anandasabapathy N. The role of dendritic cells in cancer and anti-tumor immunity. Semin Immunol (2021) 52:101481. doi: 10.1016/j.smim.2021.101481
19. Belderbos RA, Aerts J, Vroman H. Enhancing dendritic cell therapy in solid tumors with immunomodulating conventional treatment. Mol Ther Oncol (2019) 13:67–81. doi: 10.1016/j.omto.2019.03.007
20. Dawicki W, Watts TH. Expression and function of 4-1BB during CD4 versus CD8 T cell responses in vivo. Eur J Immunol (2004) 34(3):743–51. doi: 10.1002/eji.200324278
21. Nguyen J, Pettmann J, Kruger P, Dushek O. Quantitative contributions of TNF receptor superfamily members to CD8(+) T-cell responses. Mol Syst Biol (2021) 17(11):e10560. doi: 10.15252/msb.202110560
22. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol (2003) 3(9):745–56. doi: 10.1038/nri1184
23. Grant EJ, Nüssing S, Sant S, Clemens EB, Kedzierska K. The role of CD27 in anti-viral T-cell immunity. Curr Opin Virol (2017) 22:77–88. doi: 10.1016/j.coviro.2016.12.001
24. Williams CA, Murray SE, Weinberg AD, Parker DC. OX40-mediated differentiation to effector function requires IL-2 receptor signaling but not CD28, CD40, IL-12Rbeta2, or T-bet. J Immunol (2007) 178(12):7694–702. doi: 10.4049/jimmunol.178.12.7694
25. Ruby CE, Montler R, Zheng R, Shu S, Weinberg AD. IL-12 is required for anti-OX40-mediated CD4 T cell survival. J Immunol (2008) 180(4):2140–8. doi: 10.4049/jimmunol.180.4.2140
26. Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, et al. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol (2004) 34(3):613–22. doi: 10.1002/eji.200324804
27. Snell LM, McPherson AJ, Lin GH, Sakaguchi S, Pandolfi PP, Riccardi C, et al. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol (2010) 185(12):7223–34. doi: 10.4049/jimmunol.1001912
28. van de Ven K, Borst J. Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: rationale and potential. Immunotherapy (2015) 7(6):655–67. doi: 10.2217/imt.15.32
29. Peperzak V, Veraar EA, Keller AM, Xiao Y, Borst J. The pim kinase pathway contributes to survival signaling in primed CD8+ T cells upon CD27 costimulation. J Immunol (2010) 185(11):6670–8. doi: 10.4049/jimmunol.1000159
30. Amaravadi R, Thompson CB. The survival kinases akt and pim as potential pharmacological targets. J Clin Invest. (2005) 115(10):2618–24. doi: 10.1172/JCI26273
31. Dolfi DV, Boesteanu AC, Petrovas C, Xia D, Butz EA, Katsikis PD. Late signals from CD27 prevent fas-dependent apoptosis of primary CD8+ T cells. J Immunol (2008) 180(5):2912–21. doi: 10.4049/jimmunol.180.5.2912
32. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes bcl-xL and bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity (2001) 15(3):445–55. doi: 10.1016/S1074-7613(01)00191-1
33. Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of bcl-xL and bfl-1. J Immunol (2002) 169(9):4882–8. doi: 10.4049/jimmunol.169.9.4882
34. Laderach D, Movassagh M, Johnson A, Mittler RS, Galy A. 4-1BB co-stimulation enhances human CD8(+) T cell priming by augmenting the proliferation and survival of effector CD8(+) T cells. Int Immunol (2002) 14(10):1155–67. doi: 10.1093/intimm/dxf080
35. Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol (2009) 21(5):265–72. doi: 10.1016/j.smim.2009.05.010
36. Dong H, Franklin NA, Roberts DJ, Yagita H, Glennie MJ, Bullock TN. CD27 stimulation promotes the frequency of IL-7 receptor-expressing memory precursors and prevents IL-12-mediated loss of CD8(+) T cell memory in the absence of CD4(+) T cell help. J Immunol (2012) 188(8):3829–38. doi: 10.4049/jimmunol.1103329
37. Dong H, Buckner A, Prince J, Bullock T. Frontline science: Late CD27 stimulation promotes IL-7Rα transcriptional re-expression and memory T cell qualities in effector CD8(+) T cells. J Leukoc Biol (2019) 106(5):1007–19. doi: 10.1002/JLB.1HI0219-064R
38. Bertram EM, Dawicki W, Sedgmen B, Bramson JL, Lynch DH, Watts TH. A switch in costimulation from CD28 to 4-1BB during primary versus secondary CD8 T cell response to influenza in vivo. J Immunol (2004) 172(2):981–8. doi: 10.4049/jimmunol.172.2.981
39. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x
40. Willoughby JE, Kerr JP, Rogel A, Taraban VY, Buchan SL, Johnson PW, et al. Differential impact of CD27 and 4-1BB costimulation on effector and memory CD8 T cell generation following peptide immunization. J Immunol (2014) 193(1):244–51. doi: 10.4049/jimmunol.1301217
41. Hendriks J, Xiao Y, Rossen JW, van der Sluijs KF, Sugamura K, Ishii N, et al. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J Immunol (2005) 175(3):1665–76. doi: 10.4049/jimmunol.175.3.1665
42. van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol (2012) 129(3):787–93.e6. doi: 10.1016/j.jaci.2011.11.013
43. Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med (2017) 214(1):73–89. doi: 10.1084/jem.20160784
44. Makedonas G, Hutnick N, Haney D, Amick AC, Gardner J, Cosma G, et al. Perforin and IL-2 upregulation define qualitative differences among highly functional virus-specific human CD8 T cells. PloS Pathog (2010) 6(3):e1000798. doi: 10.1371/journal.ppat.1000798
45. Savoldo B, Huls MH, Liu Z, Okamura T, Volk HD, Reinke P, et al. Autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for the treatment of persistent active EBV infection. Blood (2002) 100(12):4059–66. doi: 10.1182/blood-2002-01-0039
46. Alosaimi MF, Hoenig M, Jaber F, Platt CD, Jones J, Wallace J, et al. Immunodeficiency and EBV-induced lymphoproliferation caused by 4-1BB deficiency. J Allergy Clin Immunol (2019) 144(2):574–83.e5. doi: 10.1016/j.jaci.2019.03.002
47. Byun M, Ma CS, Akçay A, Pedergnana V, Palendira U, Myoung J, et al. Inherited human OX40 deficiency underlying classic kaposi sarcoma of childhood. J Exp Med (2013) 210(9):1743–59. doi: 10.1084/jem.20130592
48. Murguia-Favela L, Sharfe N, Karanxha A, Bates A, Dadi H, Cimpean L, et al. CD40 deficiency: a unique adult patient with hyper immunoglobulin m syndrome and normal expression of CD40. LymphoSign J (2017) 4(2):70–6. doi: 10.14785/lymphosign-2017-0004
49. Leite LFB, Máximo TA, Mosca T, Forte WCN. CD40 ligand deficiency. Allergol Immunopathol (2020) 48(4):409–13. doi: 10.1016/j.aller.2019.08.005
50. Durandy A, Revy P, Fischer A. Human models of inherited immunoglobulin class switch recombination and somatic hypermutation defects (hyper-IgM syndromes). Adv Immunol (2004) 82:295–330. doi: 10.1016/S0065-2776(04)82007-8
51. Qamar N, Fuleihan RL. The hyper IgM syndromes. Clin Rev Allergy Immunol (2014) 46(2):120–30. doi: 10.1007/s12016-013-8378-7
52. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: Clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol (2015) 136(3):703–12.e10. doi: 10.1016/j.jaci.2015.02.022
53. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica (2013) 98(3):473–8. doi: 10.3324/haematol.2012.068791
54. Ghosh S, Köstel Bal S, Edwards ESJ, Pillay B, Jiménez Heredia R, Erol Cipe F, et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood (2020) 136(23):2638–55. doi: 10.1182/blood.2020006738
55. Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci U S A. (2001) 98(22):12614–9. doi: 10.1073/pnas.221456898
56. Mishra A, Italia K, Gupta M, Desai M, Madkaikar M. Hyperimmunoglobulin syndrome due to CD40 deficiency: possibly the first case from India. J Postgrad Med (2015) 61(1):46–8. doi: 10.4103/0022-3859.147053
57. Barton A, John S, Ollier WE, Silman A, Worthington J. Association between rheumatoid arthritis and polymorphism of tumor necrosis factor receptor II, but not tumor necrosis factor receptor I, in caucasians. Arthritis Rheumatol (2001) 44(1):61–5. doi: 10.1002/1529-0131(200101)44:1<61::AID-ANR9>3.0.CO;2-Q
58. Sashio H, Tamura K, Ito R, Yamamoto Y, Bamba H, Kosaka T, et al. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and crohn’s disease, respectively. Immunogenetics (2002) 53(12):1020–7. doi: 10.1007/s00251-001-0423-7
59. Ma L, Chen S, Mao X, Lu Y, Zhang X, Lao X, et al. The association between TNFR gene polymorphisms and the risk of hepatitis b virus-related liver diseases in Chinese population. Sci Rep (2018) 8(1):9240. doi: 10.1038/s41598-018-27623-7
60. Sevdali E, Block Saldana V, Speletas M, Eibel H. BAFF receptor polymorphisms and deficiency in humans. Curr Opin Immunol (2021) 71:103–10. doi: 10.1016/j.coi.2021.06.008
61. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Böhm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. (2009) 106(33):13945–50. doi: 10.1073/pnas.0903543106
62. Hildebrand JM, Luo Z, Manske MK, Price-Troska T, Ziesmer SC, Lin W, et al. A BAFF-r mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-r signaling. J Exp Med (2010) 207(12):2569–79. doi: 10.1084/jem.20100857
63. Ntellas P, Dardiotis E, Sevdali E, Siokas V, Aloizou AM, Tsinti G, et al. TNFRSF13C/BAFFR P21R and H159Y polymorphisms in multiple sclerosis. Mult Scler Relat Disord (2020) 37:101422. doi: 10.1016/j.msard.2019.101422
64. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet (2005) 37(8):829–34. doi: 10.1038/ng1601
65. Lee JJ, Rauter I, Garibyan L, Ozcan E, Sannikova T, Dillon SR, et al. The murine equivalent of the A181E TACI mutation associated with common variable immunodeficiency severely impairs b-cell function. Blood (2009) 114(11):2254–62. doi: 10.1182/blood-2008-11-189720
66. Kakkas I, Tsinti G, Kalala F, Farmaki E, Kourakli A, Kapousouzi A, et al. TACI mutations in primary antibody deficiencies: A nationwide study in Greece. Medicina (Kaunas) (2021) 57(8):827. doi: 10.3390/medicina57080827
67. Manso BA, Wenzl K, Asmann YW, Maurer MJ, Manske M, Yang ZZ, et al. Whole-exome analysis reveals novel somatic genomic alterations associated with cell of origin in diffuse large b-cell lymphoma. Blood Cancer J (2017) 7(4):e553. doi: 10.1038/bcj.2017.33
68. Cheung KJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res (2010) 70(22):9166–74. doi: 10.1158/0008-5472.CAN-10-2460
69. Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, et al. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell (2016) 167(2):405–18.e13. doi: 10.1016/j.cell.2016.08.032
70. Nikolopoulos G, Smith CEL, Brookes SJ, El-Asrag ME, Brown CJ, Patel A, et al. New missense variants in RELT causing hypomineralised amelogenesis imperfecta. Clin Genet (2020) 97(5):688–95. doi: 10.1111/cge.13721
71. Kim JW, Zhang H, Seymen F, Koruyucu M, Hu Y, Kang J, et al. Mutations in RELT cause autosomal recessive amelogenesis imperfecta. Clin Genet (2019) 95(3):375–83. doi: 10.1111/cge.13487
72. Middendorp S, Xiao Y, Song JY, Peperzak V, Krijger PH, Jacobs H, et al. Mice deficient for CD137 ligand are predisposed to develop germinal center-derived b-cell lymphoma. Blood (2009) 114(11):2280–9. doi: 10.1182/blood-2009-03-208215
73. Tan JT, Whitmire JK, Ahmed R, Pearson TC, Larsen CP. 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J Immunol (1999) 163(9):4859–68. doi: 10.4049/jimmunol.163.9.4859
74. Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol (2002) 168(8):3777–85. doi: 10.4049/jimmunol.168.8.3777
75. Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1(5):433–40. doi: 10.1038/80877
76. Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med (2003) 198(9):1369–80. doi: 10.1084/jem.20030916
77. Gauttier V, Judor JP, Le Guen V, Cany J, Ferry N, Conchon S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int J Cancer. (2014) 135(12):2857–67. doi: 10.1002/ijc.28943
78. Adappa ND, Sung CK, Choi B, Huang TG, Genden EM, Shin EJ. The administration of IL-12/GM-CSF and ig-4-1BB ligand markedly decreases murine floor of mouth squamous cell cancer. Otolaryngol Head Neck Surg (2008) 139(3):442–8. doi: 10.1016/j.otohns.2008.05.001
79. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity (2018) 49(5):958–70.e7. doi: 10.1016/j.immuni.2018.09.014
80. Houot R, Goldstein MJ, Kohrt HE, Myklebust JH, Alizadeh AA, Lin JT, et al. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by treg depletion. Blood (2009) 114(16):3431–8. doi: 10.1182/blood-2009-05-223958
81. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: Putting the pedal to the metal. Front Oncol (2015) 5:34. doi: 10.3389/fonc.2015.00034
82. Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: Structure and function - what questions remain? Mol Immunol (2017) 83:13–22. doi: 10.1016/j.molimm.2017.01.006
83. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer. (2016) 67:1–10. doi: 10.1016/j.ejca.2016.06.028
84. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med (2005) 202(7):885–91. doi: 10.1084/jem.20050940
85. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PloS One (2010) 5(5):e10436. doi: 10.1371/journal.pone.0010436
86. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med (2013) 210(9):1685–93. doi: 10.1084/jem.20130573
87. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol (2014) 92(6):475–80. doi: 10.1038/icb.2014.26
88. Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and fc receptors on the activity of immunomodulatory antibodies. Trends Immunol (2014) 35(7):290–8. doi: 10.1016/j.it.2014.05.002
89. French RR, Taraban VY, Crowther GR, Rowley TF, Gray JC, Johnson PW, et al. Eradication of lymphoma by CD8 T cells following anti-CD40 monoclonal antibody therapy is critically dependent on CD27 costimulation. Blood (2007) 109(11):4810–5. doi: 10.1182/blood-2006-11-057216
90. Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity (2008) 29(6):934–46. doi: 10.1016/j.immuni.2008.10.009
91. Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J Immunol (2017) 199(12):4110–23. doi: 10.4049/jimmunol.1700606
92. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated tregs. Sci Signal (2017) 10(462):eaaf8608.
93. Torrey H, Khodadoust M, Tran L, Baum D, Defusco A, Kim YH, et al. Targeted killing of TNFR2-expressing tumor cells and t(regs) by TNFR2 antagonistic antibodies in advanced sézary syndrome. Leukemia (2019) 33(5):1206–18. doi: 10.1038/s41375-018-0292-9
94. Tam EM, Fulton RB, Sampson JF, Muda M, Camblin A, Richards J, et al. Antibody-mediated targeting of TNFR2 activates CD8(+) T cells in mice and promotes antitumor immunity. Sci Transl Med (2019) 11(512):eaax0720. doi: 10.1126/scitranslmed.aax0720
95. Bournazos S, Wang TT, Ravetch JV. The role and function of fcγ receptors on myeloid cells. Microbiol Spectr. (2016) 4(6). doi: 10.1128/microbiolspec.MCHD-0045-2016
96. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood (2012) 119(24):5640–9. doi: 10.1182/blood-2012-01-380121
97. White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, et al. Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol (2011) 187(4):1754–63. doi: 10.4049/jimmunol.1101135
98. Li F, Ravetch JV. Antitumor activities of agonistic anti-TNFR antibodies require differential FcγRIIB coengagement in vivo. Proc Natl Acad Sci U.S.A. (2013) 110(48):19501–6. doi: 10.1073/pnas.1319502110
99. Yu X, James S, Felce JH, Kellermayer B, Johnston DA, Chan HTC, et al. TNF receptor agonists induce distinct receptor clusters to mediate differential agonistic activity. Commun Biol (2021) 4(1):772. doi: 10.1038/s42003-021-02309-5
100. White AL, Dou L, Chan HT, Field VL, Mockridge CI, Moss K, et al. Fcγ receptor dependency of agonistic CD40 antibody in lymphoma therapy can be overcome through antibody multimerization. J Immunol (2014) 193(4):1828–35. doi: 10.4049/jimmunol.1303204
101. Simpson AP, Roghanian A, Oldham RJ, Chan HTC, Penfold CA, Kim HJ, et al. FcγRIIB controls antibody-mediated target cell depletion by ITIM-independent mechanisms. Cell Rep (2022) 40(3):111099. doi: 10.1016/j.celrep.2022.111099
102. Hussain K, Liu R, Smith RCG, Müller KTJ, Ghorbani M, Macari S, et al. HIF activation enhances FcγRIIb expression on mononuclear phagocytes impeding tumor targeting antibody immunotherapy. J Exp Clin Cancer Res (2022) 41(1):131. doi: 10.1186/s13046-022-02294-5
103. Pritchard NR, Cutler AJ, Uribe S, Chadban SJ, Morley BJ, Smith KG. Autoimmune-prone mice share a promoter haplotype associated with reduced expression and function of the fc receptor FcgammaRII. Curr Biol (2000) 10(4):227–30. doi: 10.1016/S0960-9822(00)00344-4
104. Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, et al. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol (2007) 178(5):3272–80. doi: 10.4049/jimmunol.178.5.3272
105. White AL, Chan HT, French RR, Willoughby J, Mockridge CI, Roghanian A, et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell (2015) 27(1):138–48. doi: 10.1016/j.ccell.2014.11.001
106. Yu X, Chan HTC, Fisher H, Penfold CA, Kim J, Inzhelevskaya T, et al. Isotype switching converts anti-CD40 antagonism to agonism to elicit potent antitumor activity. Cancer Cell (2020) 37(6):850–66.e7. doi: 10.1016/j.ccell.2020.04.013
107. Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell (2016) 29(6):820–31. doi: 10.1016/j.ccell.2016.05.001
108. Richman LP, Vonderheide RH. Role of crosslinking for agonistic CD40 monoclonal antibodies as immune therapy of cancer. Cancer Immunol Res (2014) 2(1):19–26. doi: 10.1158/2326-6066.CIR-13-0152
109. Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem (2008) 283(23):16206–15. doi: 10.1074/jbc.M709988200
110. Orr CM, Fisher H, Yu X, Chan CH, Gao Y, Duriez PJ, et al. Hinge disulfides in human IgG2 CD40 antibodies modulate receptor signaling by regulation of conformation and flexibility. Sci Immunol (2022) 7(73):eabm3723. doi: 10.1126/sciimmunol.abm3723
111. Yu X, Chan HTC, Orr CM, Dadas O, Booth SG, Dahal LN, et al. Complex interplay between epitope specificity and isotype dictates the biological activity of anti-human CD40 antibodies. Cancer Cell (2018) 33(4):664–75.e4. doi: 10.1016/j.ccell.2018.02.009
112. Heckel F, Turaj AH, Fisher H, Chan HTC, Marshall MJE, Dadas O, et al. Agonistic CD27 antibody potency is determined by epitope-dependent receptor clustering augmented through fc-engineering. Commun Biol (2022) 5(1):229. doi: 10.1038/s42003-022-03182-6
113. Griffiths J, Hussain K, Smith HL, Sanders T, Cox KL, Semmrich M, et al. Domain binding and isotype dictate the activity of anti-human OX40 antibodies. J Immunother Cancer (2020) 8(2):e001557. doi: 10.1136/jitc-2020-001557
114. Burris H, Ansell S, Neumanitis J, Weiss G, Sikic B, Northfelt D, et al. A phase I study of an agonist anti-CD27 human antibody (CDX-1127) in patients with advanced hematologic malignancies or solid tumors. J ImmunoTher Cancer (2013) 1(1):P127. doi: 10.1186/2051-1426-1-S1-P127
115. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-Class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol (2017) 35(18):2028–36. doi: 10.1200/jco.2016.70.1508
116. Sanborn RE, Pishvaian MJ, Callahan MK, Weise AM, Sikic BI, Rahma OE, et al. Anti-CD27 agonist antibody varlilumab (varli) with nivolumab (nivo) for colorectal (CRC) and ovarian (OVA) cancer: Phase (Ph) 1/2 clinical trial results. J Clin Oncol (2018) 36(15_suppl):3001. doi: 10.1200/JCO.2018.36.15_suppl.3001
117. Sanborn RE, Pishvaian MJ, Callahan MK, Weise A, Sikic BI, Rahma O, et al. Safety, tolerability and efficacy of agonist anti-CD27 antibody (varlilumab) administered in combination with anti-PD-1 (nivolumab) in advanced solid tumors. J Immunother Cancer (2022) 10(8):e005147. doi: 10.1136/jitc-2022-005147
118. Turaj AH, Hussain K, Cox KL, Rose-Zerilli MJJ, Testa J, Dahal LN, et al. Antibody tumor targeting is enhanced by CD27 agonists through myeloid recruitment. Cancer Cell (2017) 32(6):777–91.e6. doi: 10.1016/j.ccell.2017.11.001
119. Lim SH, Sow HS, Wignall C, Mercer K, Caddy J, Boxall C, et al. Clinical and biological effects of combined CD27 and CD20 antibody therapy in Relapsed/Refractory b-cell lymphoma: The Riva trial. Blood (2021) 138:715. doi: 10.1182/blood-2021-148332
120. Guelen L, Fischmann TO, Wong J, Mauze S, Guadagnoli M, Bąbała N, et al. Preclinical characterization and clinical translation of pharmacodynamic markers for MK-5890: a human CD27 activating antibody for cancer immunotherapy. J ImmunoTher Cancer. (2022) 10(9):e005049. doi: 10.1136/jitc-2022-005049
121. Shapira-Frommer R, Dongen M, Dobrenkov K, Chartash E, Liu F, Li C, et al. O83 phase 1 study of an anti-CD27 agonist as monotherapy and in combination with pembrolizumab in patients with advanced solid tumors. J ImmunoTher Cancer. (2020) 8(Suppl 1):A2–A. doi: 10.1136/LBA2019.3
122. Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res (2018) 24(8):1816–23. doi: 10.1158/1078-0432.CCR-17-1922
123. Gopal AK, Levy R, Houot R, Patel SP, Popplewell L, Jacobson C, et al. First-in-Human study of utomilumab, a 4-1BB/CD137 agonist, in combination with rituximab in patients with follicular and other CD20(+) non-Hodgkin lymphomas. Clin Cancer Res (2020) 26(11):2524–34. doi: 10.1158/1078-0432.CCR-19-2973
124. Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, et al. Phase ib study of utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res (2017) 23(18):5349–57. doi: 10.1158/1078-0432.CCR-17-1243
125. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res (2017) 23(8):1929–36. doi: 10.1158/1078-0432.CCR-16-1272
126. Timmerman J, Herbaux C, Ribrag V, Zelenetz AD, Houot R, Neelapu SS, et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory b-cell lymphoma. Am J Hematol (2020) 95(5):510–20. doi: 10.1002/ajh.25757
127. Bartkowiak T, Jaiswal AR, Ager CR, Chin R, Chen CH, Budhani P, et al. Activation of 4-1BB on liver myeloid cells triggers hepatitis via an interleukin-27-Dependent pathway. Clin Cancer Res (2018) 24(5):1138–51. doi: 10.1158/1078-0432.CCR-17-1847
128. Zhang J, Song K, Wang J, Li Y, Liu S, Dai C, et al. S100A4 blockage alleviates agonistic anti-CD137 antibody-induced liver pathology without disruption of antitumor immunity. Oncoimmunology (2018) 7(4):e1296996. doi: 10.1080/2162402X.2017.1296996
129. Reitinger C, Ipsen-Escobedo A, Hornung C, Heger L, Dudziak D, Lux A, et al. Modulation of urelumab glycosylation separates immune stimulatory activity from organ toxicity. Front Immunol (2022) 13:970290. doi: 10.3389/fimmu.2022.970290
130. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res (2013) 19(22):6286–95. doi: 10.1158/1078-0432.CCR-13-1320
131. Sanborn R, Hauke R, Gabrail N, O’Hara M, Bhardwaj N, Bordoni R, et al. 405 CDX1140–01, a phase 1 dose-escalation/expansion study of CDX-1140 alone (Part 1) and in combination with CDX-301 (Part 2) or pembrolizumab (Part 3). J ImmunoTher Cancer. (2020) 8(Suppl 3):A246–A. doi: 10.1136/jitc-2020-SITC2020.0405
132. Filbert EL, Björck PK, Srivastava MK, Bahjat FR, Yang X. APX005M, a CD40 agonist antibody with unique epitope specificity and fc receptor binding profile for optimal therapeutic application. Cancer Immunol Immunother (2021) 70(7):1853–65. doi: 10.1007/s00262-020-02814-2
133. Weiss S, Sznol M, Shaheen M, Berciano-Guerrero M-Á, Felip E, Rodríguez-Abreu D, et al. 389 phase II of CD40 agonistic antibody sotigalimab (APX005M) in combination with nivolumab in subjects with metastatic melanoma with confirmed disease progression on anti-PD-1 therapy. J ImmunoTher Cancer. (2021) 9(Suppl 2):A422–A. doi: 10.1136/jitc-2021-SITC2021.389
134. Postel-Vinay S, Lam VK, Ros W, Bauer TM, Hansen AR, Cho DC, et al. Abstract CT150: A first-in-human phase I study of the OX40 agonist GSK3174998 (GSK998) +/- pembrolizumab in patients (Pts) with selected advanced solid tumors (ENGAGE-1). Cancer Res (2020) 80(16_Supplement):CT150–CT. doi: 10.1158/1538-7445.AM2020-CT150
135. Diab A, El-Khoueiry A, Eskens FA, Ros W, Thompson JA, Konto C, et al. A first-in-human (FIH) study of PF-04518600 (PF-8600) OX40 agonist in adult patients (pts) with select advanced malignancies. Ann Oncol (2016) 27:vi361. doi: 10.1093/annonc/mdw378.08
136. Chiappori A, Thompson J, Eskens F, Spano J-P, Doi T, Hamid O, et al. P860 results from a combination of OX40 (PF-04518600) and 4–1BB (utomilumab) agonistic antibodies in melanoma and non-small cell lung cancer in a phase 1 dose expansion cohort. J ImmunoTher Cancer. (2020) 8(Suppl 1):A9–A10. doi: 10.1136/LBA2019.14
137. Glisson BS, Leidner RS, Ferris RL, Powderly J, Rizvi NA, Keam B, et al. Safety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Clin Cancer Res (2020) 26(20):5358–67. doi: 10.1158/1078-0432.CCR-19-3070
138. Goldman JW, Piha-Paul SA, Curti BD, Pedersen K, Bauer TM, Groenland SL, et al. Safety and tolerability of MEDI0562 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. J Clin Oncol (2020) 38(15_suppl):3003. doi: 10.1200/JCO.2020.38.15_suppl.3003
139. Geva R, Voskoboynik M, Dobrenkov K, Mayawala K, Gwo J, Wnek R, et al. First-in-human phase 1 study of MK-1248, an anti-glucocorticoid-induced tumor necrosis factor receptor agonist monoclonal antibody, as monotherapy or with pembrolizumab in patients with advanced solid tumors. Cancer (2020) 126(22):4926–35. doi: 10.1002/cncr.33133
140. Heinhuis KM, Carlino M, Joerger M, Di Nicola M, Meniawy T, Rottey S, et al. Safety, tolerability, and potential clinical activity of a glucocorticoid-induced TNF receptor–related protein agonist alone or in combination with nivolumab for patients with advanced solid tumors: A phase 1/2a dose-escalation and cohort-expansion clinical trial. JAMA Oncol (2020) 6(1):100–7. doi: 10.1001/jamaoncol.2019.3848
141. Papadopoulos KP, Autio K, Golan T, Dobrenkov K, Chartash E, Chen Q, et al. Phase I study of MK-4166, an anti-human glucocorticoid-induced TNF receptor antibody, alone or with pembrolizumab in advanced solid tumors. Clin Cancer Res (2021) 27(7):1904–11. doi: 10.1158/1078-0432.CCR-20-2886
142. Richards J, Wong C, Koshkaryev A, Fulton R, Camblin A, Sampson J, et al. Abstract 4846: MM-401, a novel anti-TNFR2 antibody that induces T cell co-stimulation, robust anti-tumor activity and immune memory. Cancer Res (2019) 79(13_Supplement):4846. doi: 10.1158/1538-7445.AM2019-4846
143. Mårtensson L, Cleary K, Semmrich M, Kovacek M, Holmkvist P, Svensson C, et al. Abstract 936: Targeting TNFR2 for cancer immunotherapy: Ligand blocking depletors versus receptor agonists. Cancer Res (2020) 80(16_Supplement):936. doi: 10.1158/1538-7445.AM2020-936
144. Mårtensson L, Cleary K, Holmkvist P, Kovacek M, Svensson C, Semmrich M, et al. Abstract 4156: BI-1808 - a first in class ligand-blocking αTNFR2 antibody for cancer immunotherapy. Cancer Res (2022) 82(12_Supplement):4156. doi: 10.1158/1538-7445.AM2022-4156
145. Spira AI, Naing A, Babiker HM, Borad MJ, Garralda E, Leventakos K, et al. Phase I study of HFB200301, a first-in-class TNFR2 agonist monoclonal antibody in patients with solid tumors selected via drug intelligent science (DIS). J Clin Oncol (2022) 40(16_suppl):TPS2670–TPS. doi: 10.1200/JCO.2022.40.16_suppl.TPS2670
146. Wei S, Fulton R, Lu Y-Y, Zhang Q, Zhou H, Raue A, et al. Abstract 1883: Mechanism of action and biomarker strategy for HFB200301, an anti-TNFR2 agonist antibody for the treatment of cancer. Cancer Res (2021) 81(13_Supplement):1883. doi: 10.1158/1538-7445.AM2021-1883
147. Knorr DA, Dahan R, Ravetch JV. Toxicity of an fc-engineered anti-CD40 antibody is abrogated by intratumoral injection and results in durable antitumor immunity. Proc Natl Acad Sci U S A. (2018) 115(43):11048–53. doi: 10.1073/pnas.1810566115
148. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science (2014) 343(6176):1260–3. doi: 10.1126/science.1248943
149. de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, et al. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PloS Biol (2016) 14(1):e1002344. doi: 10.1371/journal.pbio.1002344
150. Zhang D, Goldberg MV, Chiu ML. Fc engineering approaches to enhance the agonism and effector functions of an anti-OX40 antibody. J Biol Chem (2016) 291(53):27134–46. doi: 10.1074/jbc.M116.757773
151. Zhang D, Armstrong AA, Tam SH, McCarthy SG, Luo J, Gilliland GL, et al. Functional optimization of agonistic antibodies to OX40 receptor with novel fc mutations to promote antibody multimerization. MAbs (2017) 9(7):1129–42. doi: 10.1080/19420862.2017.1358838
152. Müller N, Wyzgol A, Münkel S, Pfizenmaier K, Wajant H. Activity of soluble OX40 ligand is enhanced by oligomerization and cell surface immobilization. FEBS J (2008) 275(9):2296–304. doi: 10.1111/j.1742-4658.2008.06382.x
153. Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: The architect behind the immune and nonimmune functions of IgG and albumin. J Immunol (2015) 194(10):4595–603. doi: 10.4049/jimmunol.1403014
154. Rowley TF, Al-Shamkhani A. Stimulation by soluble CD70 promotes strong primary and secondary CD8+ cytotoxic T cell responses in vivo. J Immunol (2004) 172(10):6039–46. doi: 10.4049/jimmunol.172.10.6039
155. Thiemann M, Richards DM, Heinonen K, Kluge M, Marschall V, Merz C, et al. A single-Chain-Based hexavalent CD27 agonist enhances T cell activation and induces anti-tumor immunity. Front Oncol (2018) 8:387. doi: 10.3389/fonc.2018.00387
156. Merz C, Sykora J, Marschall V, Richards DM, Heinonen K, Redondo Müller M, et al. The hexavalent CD40 agonist HERA-CD40L induces T-cell-mediated antitumor immune response through activation of antigen-presenting cells. J Immunother (2018) 41(9):385–98. doi: 10.1097/CJI.0000000000000246
157. Richards DM, Marschall V, Billian-Frey K, Heinonen K, Merz C, Redondo Müller M, et al. HERA-GITRL activates T cells and promotes anti-tumor efficacy independent of FcγR-binding functionality. J Immunother Cancer. (2019) 7(1):191. doi: 10.1186/s40425-019-0671-4
158. Thiemann M, Sykora J, Richards DM, Merz C, Marschall V, Mueller MR, et al. Abstract 3754: The novel hexavalent human CD137/4-1BB agonist HERA-CD137L promotes anti-cancer immunity by activating CD8 T cells while regulatory T cells are not affected. Cancer Res (2018) 78(13_Supplement):3754. doi: 10.1158/1538-7445.AM2018-3754
159. Tesselaar K, Arens R, van Schijndel GM, Baars PA, van der Valk MA, Borst J, et al. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol (2003) 4(1):49–54. doi: 10.1038/ni869
160. Kim S-H, Singh R, Han C, Cho E, Kim YI, Lee DG, et al. Chronic activation of 4-1BB signaling induces granuloma development in tumor-draining lymph nodes that is detrimental to subsequent CD8+ T cell responses. Cell Mol Immunol (2021) 18(8):1956–68. doi: 10.1038/s41423-020-00533-3
161. De Colvenaer V, Taveirne S, Hamann J, de Bruin AM, De Smedt M, Taghon T, et al. Continuous CD27 triggering in vivo strongly reduces NK cell numbers. Eur J Immunol (2010) 40(4):1107–17. doi: 10.1002/eji.200939251
162. Vargas JG, Wagner J, Shaikh H, Lang I, Medler J, Anany M, et al. A TNFR2-specific TNF fusion protein with improved In vivo activity. Front Immunol (2022) 13:888274. doi: 10.3389/fimmu.2022.888274
163. Divine R, Dang HV, Ueda G, Fallas JA, Vulovic I, Sheffler W, et al. Designed proteins assemble antibodies into modular nanocages. Science (2021) 372(6537):eabd9994. doi: 10.1126/science.abd9994
164. Ganesan LP, Kim J, Wu Y, Mohanty S, Phillips GS, Birmingham DJ, et al. FcγRIIb on liver sinusoidal endothelium clears small immune complexes. J Immunol (2012) 189(10):4981–8. doi: 10.4049/jimmunol.1202017
165. Compte M, Harwood SL, Muñoz IG, Navarro R, Zonca M, Perez-Chacon G, et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun (2018) 9(1):4809. doi: 10.1038/s41467-018-07195-w
166. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med (2019) 11(496). doi: 10.1126/scitranslmed.aav5989
167. Trüb M, Uhlenbrock F, Claus C, Herzig P, Thelen M, Karanikas V, et al. Fibroblast activation protein-targeted-4-1BB ligand agonist amplifies effector functions of intratumoral T cells in human cancer. J Immunother Cancer (2020) 8(2):e000238. doi: 10.1136/jitc-2019-000238
168. Warwas KM, Meyer M, Gonçalves M, Moldenhauer G, Bulbuc N, Knabe S, et al. Co-Stimulatory bispecific antibodies induce enhanced T cell activation and tumor cell killing in breast cancer models. Front Immunol (2021) 12:719116. doi: 10.3389/fimmu.2021.719116
169. Fellermeier-Kopf S, Gieseke F, Sahin U, Müller D, Pfizenmaier K, Kontermann RE. Duokines: a novel class of dual-acting co-stimulatory molecules acting in cis or trans. Oncoimmunology (2018) 7(9):e1471442. doi: 10.1080/2162402X.2018.1471442
170. Aschmoneit N, Kocher K, Siegemund M, Lutz MS, Kühl L, Seifert O, et al. Fc-based duokines: dual-acting costimulatory molecules comprising TNFSF ligands in the single-chain format fused to a heterodimerizing fc (scDk-fc). Oncoimmunology (2022) 11(1):2028961. doi: 10.1080/2162402X.2022.2028961
171. Lakins MA, Koers A, Giambalvo R, Munoz-Olaya J, Hughes R, Goodman E, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res (2020) 26(15):4154–67. doi: 10.1158/1078-0432.CCR-19-2958
Keywords: TNFR, agonism, co-stimulation, cancer, immunotherapy
Citation: Dadas O, Ertay A and Cragg MS (2023) Delivering co-stimulatory tumor necrosis factor receptor agonism for cancer immunotherapy: past, current and future perspectives. Front. Immunol. 14:1147467. doi: 10.3389/fimmu.2023.1147467
Received: 18 January 2023; Accepted: 27 March 2023;
Published: 25 April 2023.
Edited by:
Nataša Obermajer, Janssen Pharmaceutica NV, BelgiumReviewed by:
Michael Croft, La Jolla Institute for Immunology (LJI), United StatesBruce Hostager, The University of Iowa, United States
Isabell Lang, University Hospital Würzburg, Germany
Copyright © 2023 Dadas, Ertay and Cragg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark S. Cragg, bXNjQHNvdG9uLmFjLnVr; Osman Dadas, Ty5EYWRhc0Bzb3Rvbi5hYy51aw==
†Present address: Osman Dadas, Faculty of Health Sciences, European University of Lefke, Lefke, Cyprus