 Fabiola Biasella
Fabiola Biasella Karolina Plössl
Karolina Plössl Paul N. Baird
Paul N. Baird Bernhard H. F. Weber
Bernhard H. F. Weber
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 March 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1147037
This article is part of the Research Topic The Crosstalk Between Oxidative Stress, Altered Metabolic Activity, Immune Activation and Inflammation in Ocular Diseases View all 4 articles
Inherited retinal dystrophies (IRDs) as well as genetically complex retinal phenotypes represent a heterogenous group of ocular diseases, both on account of their phenotypic and genotypic characteristics. Therefore, overlaps in clinical features often complicate or even impede their correct clinical diagnosis. Deciphering the molecular basis of retinal diseases has not only aided in their disease classification but also helped in our understanding of how different molecular pathologies may share common pathomechanisms. In particular, these relate to dysregulation of two key processes that contribute to cellular integrity, namely extracellular matrix (ECM) homeostasis and inflammation. Pathological changes in the ECM of Bruch’s membrane have been described in both monogenic IRDs, such as Sorsby fundus dystrophy (SFD) and Doyne honeycomb retinal dystrophy (DHRD), as well as in the genetically complex age-related macular degeneration (AMD) or diabetic retinopathy (DR). Additionally, complement system dysfunction and distorted immune regulation may also represent a common connection between some IRDs and complex retinal degenerations. Through highlighting such overlaps in molecular pathology, this review aims to illuminate how inflammatory processes and ECM homeostasis are linked in the healthy retina and how their interplay may be disturbed in aging as well as in disease.
Inherited retinal diseases (IRD) confer a diverse group of diseases presenting at varying ages in life. Many phenotypic features appear shared between the different IRDs that can make their clinical classification challenging although application of molecular diagnostic tools and testing can be applied to provide greater clinical certainty (1, 2). What has become clear in this process is that genetic heterogeneity exists between the majority of IRDs, with mutations occurring in different genes giving rise to very similar phenotypes or mutations in the same gene resulting in phenotypically different IRDs (3). Additionally, there appears to be both clinical and genetic overlap between some IRDs, such as Sorsby fundus dystrophy (SFD) and complex genetic disorders such as age-related macular degeneration (AMD) (4–6) These observations infer potential common pathomechanisms, not only between IRDs but also between IRDs and common complex retinal disorders. In particular, two key processes stand out, which contribute to cellular integrity, namely extracellular matrix (ECM) homeostasis and immunity. While much has been discovered in recent years about the immune response in retinal disease (7, 8), it is unclear if this occurs independently or whether it can be exacerbated by an altered ECM – the classic chicken-and-egg problem. A better understanding of the interplay between these two key processes and how they maintain a healthy retina may provide important information as to how aging and disease are impacted by their dysregulation.
The ECM is composed of structural macromolecules such as collagens, fibronectin, laminins, elastin and glycosaminoglycans (GAGs), as well as non-structural extracellular proteins (e.g., matrix metalloproteinases, MMPs; matrikines and matricellular proteins). These have been reported to affect trafficking, local concentration and the function of immune mediators (9, 10). Mechanistically, this is thought to involve an interaction between immune cells and molecules in the extracellular environment (10–12). Accordingly, areas of the eye enriched in ECM components, could represent dynamic multi-link interfaces critical for ocular immune response. In the first part of this article, we review the current understanding of the communication between the ECM and immune factors in the retina and their significance in maintaining homeostasis. We then discuss the regulatory role of matricellular proteins on retinal immune processes, how age and the extracellular environment may impact on retinal immunity and finally the role of the cellular microenvironment and inflammation in ocular disease.
In the retina, structural ECM molecules can be found in basement membranes, the nerve fibre layer, the outer and inner plexiform layers, the interphotoreceptor matrix, and in Bruch’s membrane (BrM) (13). The latter is of particular interest as it represents a pentalaminar ECM structure between the choriocapillaris and the retinal pigment epithelium (RPE). It is formed by a dense core of elastic fibers surrounded by collagen-rich inner and outer layers, flanked by the basement membranes of the RPE and choriocapillaris endothelial cells. Structural elements are mainly glycosaminoglycans (heparan sulfate, chondroitin sulfate, and keratin sulfate), collagen I, III, V, fibronectin, and lipoproteins (14). In addition to its many functions as a structural support for the RPE and outer retinal vascular wall and as a transport pathway for fluids and macromolecules moving between the RPE and the choroid (14), BrM can also act as a modulator of innate immunity and retinal homeostasis. The pentalaminar structure of the BrM and the regular spacing of its ECM components enhances its molecular sieving capacity. It limits the passive diffusion of proteins with a molecular weight above 200 kDa from the choroidal blood supply to the RPE and vice versa (15). Moreover, BrM restricts the distribution of smaller molecules, such as components of the complement system (16, 17). In diffusion experiments with enriched BrM from human donors, Clark and colleagues showed that while some complement proteins (e.g., complement factor D, CFD; factor H-like protein 1, FHL-1; and C5a) diffuse through Bruch’s membrane, other central components such as C3 and C3b, complement factor B (CFB) and the complement inhibitors factor H (CFH) and factor I (CFI) are typically retained (18, 19). This could contribute to maintaining a reduced retinal inflammatory environment, as it physically separates systemic complement factors (along with those produced by the choroid) from those produced locally by the RPE and the neural retina (18).
Nonetheless, the concerted activity of the complement system at the choroid/BrM interface needs to be kept at bay to avoid damage to healthy tissue and subsequent excessive inflammatory responses. Here, CFH acts as the main negative regulator of complement activity, possessing both a cofactor activity for the breakdown of the central complement factor C3b and a cleavage activity against C3-convertase, responsible for the formation of C3 (20). By binding to GAGs, particularly to heparan sulphate (21), CFH and its splice variant FHL-1 protect the retina from inappropriate complement overactivation (19). As a consequence, the variability of heparan sulphate chains in terms of length, degree of sulphation and binding pattern to extracellular proteoglycans (21) are likely to influence the binding affinity of CFH, thus affecting its regulatory function (21). A modified binding of CFH could thus impair its inhibitory effect on complement, eventually leading to dysregulation of complement activity and consequent local chronic inflammation. In a pro-inflammatory milieu, immune cells such as neutrophils, B lymphocytes, monocytes, and platelets, which express receptors for CFH on their cell surface, are also attracted to the chemotactic properties of CFH (22–25). Interestingly, in a study by Schneider and colleagues, it was shown in vitro that CFH directly influences neutrophil activation, migration and spreading only when immobilized on a surface and not in solution (26). It is therefore conceivable that neutrophils may infiltrate a retinal lesion site as a first line of defense in a manner that depends on the composition of the ECM and the contact with CFH adhering to it.
In the ECM, GAGs also appear essential for the function of numerous inflammatory cytokines aimed at tissue growth and repair (27, 28). It is therefore plausible that appropriate abundance of GAGs distributed within the retina, particularly within BrM and the interphotoreceptor matrix (29)contributes to the binding of cytokines to the ECM. The resultant binding limits cytokine diffusion and the promotion of a localized activity of immune cells, including leucocytes (28). The recruitment of immune cells to the ECM is also modulated by the presence of hyaluronic acid, which binds to the CD44 adhesion receptor of leucocytes and macrophages (30). Collagen density may also directly modulate the immunoactivity of leukocytes (31) and macrophages (32). Therefore, its presence in the collagen-rich layers of BrM (but not in the interphotoreceptor matrix) can compartmentalize the migration and activity of immune cells in the retina. Furthermore, the collagen and elastin fibers of BrM may act as docking points for complement factors such as C3 and C4 (Figure 1). In support of this, covalent binding properties of these complement proteins for collagen and elastin fibers have been previously reported in vascular tissue (33).
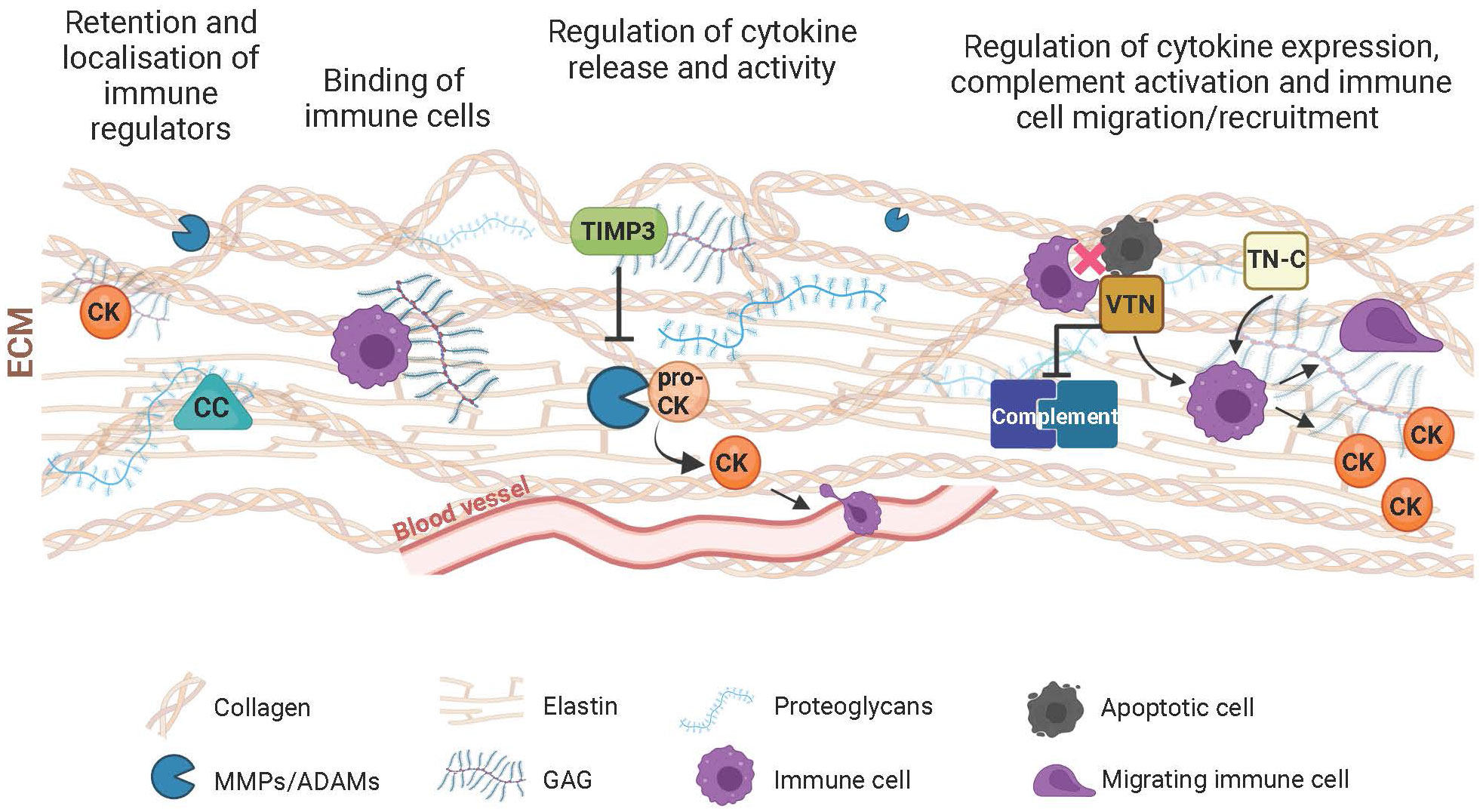
Figure 1 Schematic of components of the extracellular matrix (ECM) and their immunoregulatory role. The ECM participates in the control of the immune response with its structural and functional constituents through various mechanisms. Complement components (CC), cytokines/chemokines (CK) and immune cells are retained by the ECM meshwork or specifically bind to its structural components such as collagen, elastin, proteoglycans or glycosaminoglycans (GAG). This results in the local activity of immune regulators. Through turnover mechanisms involving extracellular proteases (e.g., MMPs or ADAMs), the ECM influences the transformation of proactive (pro-CK) to active CKs, thereby influencing their inflammatory and chemotactic function with consequent influence on the extravasation and migration of immune cells. Inhibitors of extracellular proteases, including the tissue inhibitor of metalloproteases 3 (TIMP3), further modulate immune regulation resulting from the digestion of the ECM and the inflammatory agents residing within. Matricellular proteins such as vitronectin (VTN) or tenascin-C (TN-C), which mediate the interaction between cells and their extracellular environment also influence ECM-associated immunological processes, including complement activation, immune cell migration, phagocytic removal of apoptotic cells, and secretion of inflammatory molecules.
Finally, it has been shown in vitro that changes in structural components of the ECM (e.g., fibronectin, collagen, laminin) deposited by RPE cells can in turn influence the deposition of immune factors, such as complement factors CFH and C3 (34). This supports the idea of a close relationship between structural components forming the retinal extracellular scaffold and immune defense pathways for homeostatic regulation which is highly dynamic in nature. Fragmentation, deposition and reorganization of ECM structural components are crucial for the maintenance of an optimal cellular microenvironment, as well as for native cell morphology and function, angiogenesis, and tissue repair. Metalloproteinases and their inhibitors contained in the ECM are also responsible for managing this dynamic process (35).
Through their proteolytic action on most ECM components, MMPs (also known as matrixins) are crucial for the maintenance and homeostasis of tissues (36), including those of the retina (37). Moreover, MMPs have attracted increasing interest due to their ability to influence migration of immune cells, such as neutrophils and macrophages (38, 39), as well as the activity of inflammatory molecules such as cytokines and chemotactic derivatives (chemokines) by direct proteolysis (40, 41) or modification of their ECM substrates (42, 43) (Figure 1). This suggests an involvement of MMPs in the regulation of immune processes, such as leukocyte extravasation and infiltration and glial activation (44), and thus an influence on inflammation associated with various ocular disorders (45).
In the retina, the presence of various MMPs, mainly MMP-1, MMP-2, MMP-3 and MMP-9 have been identified in the interphotoreceptor matrix, BrM and the RPE (46, 47) and implicated in the regulation of the retinal immune response. For example, mice lacking MMP-3 are subject to acute lipopolysaccharide-induced inflammation, resulting in a reduced number of adherent leucocytes in the retinal vasculature compared to mice expressing MMP-3 (48).The absence of MMP-3 in the murine choroid/RPE complex resulted in reduced protein expression of the intercellular adhesion molecule-1 (ICAM-1), known to mediate leukocyte recruitment and adhesion during inflammation. Lipopolysaccharide treatment has been shown to lead to upregulation of inflammatory molecules (e.g., interleukin-6, tumor necrosis factor alpha, TNF-α; nitric oxide synthase) in the retina and RPE but absence of MMP-3 appears to inhibit this response (48).
Like MMP-3, MMP-9 can impact retinal homeostasis when its function as an immune enhancer involved in the processing, specificity, and receptor utilization of inflammatory molecules is altered (40, 49). This likely includes the potent pro-inflammatory interleukin 8 (IL8), which revealed a 10- to 30-fold increase in neutrophil activation following incubation and proteolytic cleavage by MMP-9 in vitro (40): In particular, neutrophils purified from human blood and incubated with the MMP-digested form of IL8 showed increased migration, activation of intracellular signaling and mobilization of intracellular calcium compared to cells exposed to the unprocessed form of IL8 (35). This suggests a sophisticated mechanism of regulation by extracellular proteases in the interaction between chemokines and their cognate receptors on neutrophils.
Since variations in IL8 production and/or activity are associated with the onset of inflammation and individual susceptibility to retinal diseases such as AMD (50, 51), the effect of MMP-9 on IL8 activity may have a direct influence on the pathogenic role of IL8 in the retina. In line with this, an increased intraocular concentration of both IL8 and MMP-9 has been measured in eyes of AMD patients and those with diabetic macular oedema (52, 53).
In an excellent review by Van Lint and Libert (54), the authors point out that processing of cytokines by MMPs can lead to their potentiation but also to their inactivation. After proteolytic cleavage, truncated cytokines can lose their activity, but they can also counteract the activity of intact cytokines by steric hindrance of the receptors on the surface of cells such as leucocytes. By doing this, they exert an anti-inflammatory effect (54, 55).
Furthermore, Starr and colleagues (41) demonstrated that MMPs can influence the binding affinity and release of chemokines such as chemokine (C-C motif) ligand 16 (CCL16) to and from GAGs in the ECM. This, in turn, influences the concentration of chemokines and their effect on migration, recruitment and activation of immune cells in situ (chemotactic potential) (41). As a consequence, cytokines further upregulate the cellular expression and production of MMPs. For example, cytokine-mediated upregulation of MMP expression has been demonstrated in cultured RPE cells (56). Following this line of argument, pharmacological inhibitors of MMPs have been suggested as a potential therapeutic agent to counteract inflammation in the retina (57–60). In particular, for those retinal diseases where upregulation/dysregulation of MMPs is known to occur, e.g., AMD and diabetic retinopathy (DR) (61–64).
In vivo, the activity of MMPs is inhibited by tissue inhibitors of metalloproteinases (TIMPs) (65). In human, the TIMP protein family comprises four members (TIMP-1 to 4). Of these, TIMP3 is the only one to be incorporated into the ECM due to its high affinity for proteoglycans (66). In the retina, TIMP3 is mainly expressed and secreted by the RPE and deposited in BrM (67, 68). There, it participates in regulating the degradation and turnover of structural constituents of the ECM, thus influencing the microenvironment outside the RPE and choroidal cells. In addition to its regulatory functions for MMPs, TIMP3 has also been reported to possess immunoregulatory properties.
In TIMP3-deficient mice the absence of TIMP3 coincided with an infiltration of neutrophils and macrophages in various organs, resulting in an increased inflammation (69–73). This is attributed to the ability of TIMP3 to inhibit the pro-inflammatory transduction signal induced by TNF-α, a pleiotropic cytokine produced predominantly by activated macrophages regulating apoptosis and the inflammatory response upon receptor binding (74). Specifically, TIMP3 inhibits the TNF-α converting enzyme (TACE/ADAM17), which transforms the membrane-bound proTNF-α into a soluble active form (75) (Figure 1). Inappropriate TNF-α production or prolonged activation of TNF-α signaling can predispose to a broad spectrum of inflammatory diseases, including retinal disorders such as glaucoma or DR (76, 77).
Treatment of cultured retinal microvascular endothelial cells with purified TIMP3 was further demonstrated to alter the expression of ICAM-1, which is a known regulator of immune cell transmigration. In addition, a TIMP3-dependent downregulation of cytokine-induced vascular adhesion molecule-1 (VCAM-1), which controls endothelial leukocyte adhesion and recruitment, and a reduction in vascular endothelial growth factor (VEGF)-induced migration and proliferation was also observed in these cells (78). Together, these results suggest a role for TIMP3 in preventing extracellular engagement of circulating anti-inflammatory molecules but also support the hypothesis of its anti-angiogenic effects, which limits the formation of new vessels that would otherwise potentially increase tissue inflammation (79, 80).
During matrix remodeling, proteolysis of ECM structural proteins by extracellular enzymes such as MMPs can lead to the release of small, soluble, bioactive peptide fragments called matrikines. Compared to progenitor molecules, which play a predominantly structural role, matrikines regulate a range of cellular activities, including cell migration, proliferation, protease production, or apoptosis by binding directly to cell membrane receptors (81, 82). For example, the matrikine endostatin has long been known for its anti-angiogenic properties in homeostasis of the ocular vasculature and prevention of retinal neovascularization (83–85) as it inhibits endothelial cell migration and capillary morphogenesis (86, 87). Endostatin represents a cleavage product of collagen XVIII, associated with the basement membrane of almost all epithelia and endothelia, including the choriocapillary basement membrane of BrM (88–90). Liu and colleagues (91), in their work exploring the effect of endostatin on growth, angiogenesis and inflammation in mouse tumorigenesis, suggested a close relationship of this matrikine with the immune system. In particular, they observed a reduction in the expression of immunosuppressive cytokines (IL6, IL10, IL17, TGF-β) and a downregulation of inhibitory immune cells by endostatin. In contrast, an infiltration of immune dendritic cells and cytotoxic T lymphocytes into the tumor was observed following endostatin treatment (91). Although studies on the immune role of endostatin in the retina seem to be scarce, the data available reveal its immunoregulatory effect, which may also have an impact on the retinal microenvironment that merits investigation.
In addition to angiogenesis-regulatory activity, immunomodulatory function appears to be a property shared by many matrikines reported to direct inflammation in various inflammatory diseases, mainly due to their acquired chemoattractive properties (92–95). For example, fragments resulting from the degradation of fibronectin, a structural component of BrM (14) have been shown to have pro-inflammatory properties in vitro. Exposure of murine RPE to high doses of fibronectin fragments induced a significant increase in the RPE expression of the inflammatory cytokine IL6 and of the monocyte chemoattractant protein-1 (MCP-1), a key chemokine regulating the recruitment of monocytes/macrophages (96). Furthermore, fibronectin fragments induced increased expression of MMP-3 and MMP-9 in murine RPE cells. Based on findings that fibronectin expression by the RPE is increased in retinal disease development (97), Austin and colleagues discussed their results in the context of AMD pathogenesis, suggesting that the action of fibronectin fragments may contribute to a chronic RPE-mediated inflammation that may lead to abnormal BrM remodeling, deposit formation and drusenogenesis (96). Similarly, increased elastin-derived matrikine serum levels resulting from fragmentation of elastin in the ECM have been reported in AMD, particularly in the neovascular form, and discussed as potentially contributing to disease complications (98).
Non-structural glycoproteins of the ECM meshwork that mediate cell-ECM communication are referred to as matricellular proteins. Interestingly, many of these proteins are found with low expression in healthy adult tissues, but increase in expression during inflammatory and pathological processes, suggesting their vital role in the immune response (99). This section addresses more closely the immune significance of two representative matricellular proteins namely vitronectin and tenascin-C.
In basement membranes and the ECM, vitronectin is a multifunctional matricellular glycoprotein involved in the regulation of numerous processes, including cell adhesion and migration, matrix deposition and remodeling, fibrinolysis, angiogenesis, and coagulation (100, 101). The association of vitronectin with inflammatory diseases and its upregulation following inflammatory stimuli indicate an involvement of this protein in the immune response (102–109). Indeed, vitronectin has long been reported as a complement inhibitor (Figure 1) that binds complement proteins C5b-7 and C9 to prevent their attraction to the cell membrane. As a result, the membrane attack complex (MAC), which forms cytotoxic pores to drive cell lysis and inflammatory processes, no longer forms on target cells (101, 110).
In more recent years, the role of vitronectin in immune regulation has been extended to areas other than complement regulation. Most notably, vitronectin appears to mediate the recruitment of circulating inflammatory cells by promoting their passage through the endothelium (106, 108, 111, 112). Specifically, it has been shown that in complex with plasminogen activator inhibitor-1 (PAI-1), vitronectin is essential for the binding of neutrophils to microvascular endothelial cells, a crucial step in the immune response towards immune cell extravasation into the perivascular space and thus in inflamed tissues. In vitronectin-deficient mice a drastic reduction in the adhesion of circulating neutrophils to postcapillary endothelial cells of the cremaster muscle subjected to inflammatory stimuli was observed (108). Such an effect of vitronectin was thought to be attributable to its ability to bridge interaction between endothelial cell basement membrane GAGs and the low-density lipoprotein receptor (LRP-1) bound by the vitronectin binding partner PAI-1 on the surface of circulating neutrophils. Upon carrying out their function of killing invading organisms or damaged cells, neutrophils undergo spontaneous apoptosis, preserving the integrity of the neutrophil membrane and preventing the release of toxic cytosolic cell contents from destroying surrounding tissues (113). At this point, macrophages intervene to resolve the inflammation by removing apoptotic neutrophils (a process known as efferocytosis) (113). Both in vitro and in vivo, vitronectin has been found to decrease neutrophil efferocytosis by binding to macrophages and apoptotic neutrophils through interaction with membrane integrins (αvβ3 and αvβ5) and the urokinase plasminogen activator receptor (uPAR), respectively (114). These results presented by Bae and colleagues support the hypothesis that vitronectin binding to different receptors on the two cell surfaces, may inhibit the receptors from interacting with each other directly or via bridging proteins, preventing recognition between macrophages and apoptotic cells and thus the latter from engulfment by macrophages (114) (Figure 1).
Notably, the binding of vitronectin to αv integrins on macrophages also induces nuclear transcription factor kappa B (NF-κB)-mediated expression of the inflammatory cytokines TNF-α, IL1β and IL6 (Figure 1), as demonstrated by experiments with bone marrow-derived murine macrophages (109). Vitronectin-dependent expression of IL6 and leukemia inhibitory factor (LIF) was also observed in cultured endothelial and glial cells as well as in mouse brains after treatment with recombinant vitronectin (115). An effect of vitronectin on microglial activation was also detected in a mouse model of experimental autoimmune encephalomyelitis. Immunohistochemical analysis revealed a link between perivascular deposits of vitronectin (as well as fibronectin) in the murine brain and the number of activated microglial cells (105). The investigations of Chakravarty and colleagues on the role of vitronectin in the atherosclerotic inflammatory process suggested an alternative mechanism. This attributes an increase in inflammation with the recruitment of the pro-inflammatory molecules NF-κB, ICAM-1 and VCAM-1 as a consequence of systemic vitronectin deficiency (116). These latter findings raise the possibility of vitronectin as a quencher or promoter of inflammation-related processes.
In the retina, the immunomodulatory properties of vitronectin have been scarcely studied. Vitronectin is produced by RPE cells, photoreceptors, and ganglion cells, but is found mainly in BrM and in the retinal vasculature (14, 117, 118). Studies on cultured immortalized RPE cells (ARPE-19) have shown that treating cells with commercial human complement serum (119) or inducing MAC formation (120) results in a significant increase in vitronectin production. Given the inhibitory properties of vitronectin on complement, these results suggest that increased vitronectin production may defend RPE cells from uncontrolled complement activation (120). Consequently, vitronectin may mediate chronic low-grade inflammation in the retina, such as that arising with ageing (121). This could be related to the accumulation of vitronectin, along with other complement components, in drusen (sub-RPE and subretinal extracellular deposits) that have also been associated with the age-related manifestations in AMD pathogenesis (122, 123).
Finally, vitronectin may indirectly be involved in the immune regulation of the retina. We have previously shown that deposition of vitronectin in the ECM produced by ARPE-19 cells visibly affects the accumulation of structural matrix components such as collagen, fibronectin, and elastin (124). The altered production of vitronectin in the retina could therefore influence the immunoregulatory functions of the structural components of the ECM. Consequently, it can affect the exposure of neo-epitopes by the ECM in the binding of complement components or influence the accumulation of chemotactic proteolytic ECM fragments, as discussed above. Furthermore, we observed an increase in complement components C9 and CFH in the ECM of ARPE-19 cells containing vitronectin compared to the ECM lacking detectable amounts of this protein, which could further support the immunoregulatory role of vitronectin in the retina. Interestingly, the rs704 polymorphism in the vitronectin gene (VTN) has been associated with an increased risk of AMD (6). Studies have shown that this polymorphism leads to increased vitronectin protein expression (124, 125), which in turn could result in an exacerbation/dysregulation of its functional activities. Hypothetically, this could affect the immunoregulatory activities of vitronectin and contribute to retinal immune dysregulation, such as that associated with AMD (17, 126).
The matricellular protein tenascin-C is a ligand of toll-like receptor 4 (TLR4) and integrins (αvβ3 and α9β1) and is located on the membrane of several cell types, e.g., macrophages, dendritic cells and microglia. Its manifold immunoregulatory functions have been excellently summarized by Marzeda and Midwood (127). By binding to cell surface receptors, tenascin-C exerts mainly pro-inflammatory effects stimulating intracellular signaling towards the production of inflammatory cytokines (IL6, IL8, TNF-α), as well as the migration and polarization of immune cells, particularly in the context of neuroinflammation (Figure 1) (128). In contrast, an anti-inflammatory property was shown for tenascin-C conferred to either by itself or through its ability to bind and increase the local concentration of the immunosuppressive cytokine TGF-β. In mouse models of tenascin-C deficiency, the absence of this matricellular protein correlated mainly with altered immune cell infiltration and inflammatory response (127). For example, Manrique-Castano and colleagues demonstrated that, upon cerebral ischemia, tenascin-C-deficient mice exhibited reduced microglial coverage and increased leucocyte infiltration of the ischemic region compared to wildtype mice (129). Interestingly, the absence of tenascin-C seems to protect mice from the formation of amyloid deposits associated with Alzheimer disease and from experimental autoimmune encephalomyelitis (127).
In the retina, tenascin-C is predominantly expressed by amacrine and horizontal cells, thereby concentrating the matricellular protein in the outer and inner plexiform layers. Moreover, a considerable amount of tenascin-C is produced by astrocytes of the optic nerve (13, 130–132). The role of tenascin-C in retinal homeostasis is emerging through studies aimed at understanding pathogenic mechanisms such as the relationship between ECM remodeling and immunological processes in ocular diseases such as glaucoma, which is characterized by progressive retinal ganglion cell death and optic nerve damage (133), AMD, and DR (as reviewed by Reinhard and colleagues (134). In glaucoma, immune dysregulation is also discussed among potential causes that predispose to this disease (135, 136).
Wiemann and colleagues recently demonstrated that mouse models of experimental autoimmune glaucoma show differences in glial response and cytokine release depending on whether tenascin-C is expressed or not (137). Specifically, ganglion cell degeneration was induced in these mice by immunization with optic nerve homogenate antigens. Immunostaining and western blot analysis of retina and optic nerve sections showed an increase in astrocytes labelled with glial fibrillary acidic protein (GFAP) in the ganglion cell layer of wildtype mice, while tenascin C-deficient mice subjected to the same treatment showed a reduced microglial response. Likewise, tenascin C-deficient mice exhibited a reduction in Iba-1-positive microglial cells (137). Moreover, in the analysis of retinal and optic nerve mRNA expression, the absence of tenascin-C coincided with a reduction in the pro-inflammatory cytokine TNF-α and an increase in the anti-inflammatory cytokine TGF-β (137). In line with this and other studies (138–140), a dramatic upregulation of tenascin-C in murine retinal tissues of intraocular pressure-dependent glaucoma models has been described as part of an extensive remodeling of ECM molecules associated with glaucomatous neurodegeneration (141).
Tenascin-C upregulation has also been associated with ischemia-associated retinal degeneration (142) and vascular disorder associated-retinal diseases such DR (143–147) with the latter known to affect the retinal microvascular circulation (148). Tenascin-C has been described as increased in the basement membrane of diabetic retinal endothelial cells, where it is thought to influence ECM formation/remodeling and vascularization associated with DR, potentially interacting with other extracellular molecules such as fibronectin and periostin (145–147). Similarly, tenascin-C has been detected in choroidal neovascular membranes (CNVs) and BrM of patients with neovascular AMD (149–152). Here, it is thought to exert an anti-adhesive effect on RPE cells (152) and likely promotes neovascularization via integrin αv-mediated promotion of endothelial adhesion, migration, and neo-angiogenesis, as demonstrated in vitro with cultured endothelial cells (human dermal microvascular endothelial cells, HMVECs) (151). In line with this, tenascin C-deficient mice showed a significant reduction in the extent of choroidal vascularization (151).
It remains to be determined whether tenascin-C and its angiogenic function play a significant role in the immune dysregulation and inflammation that affects the aforementioned retinal diseases such as DR and AMD. The stimulation of tenascin-C production by retinal endothelial cells through inflammatory cytokines TNF-α and IL1β does appear to suggest a potential involvement of this matricellular protein in the immune response in both homeostasis and diseases of the retinal endothelium (146).
The function of the ECM and its structural and non-structural constituents in modulating immune responses can be drastically impaired by age-dependent changes in the extracellular environment. As extensively reviewed by Moreau and colleagues (153),age-related changes in the ECM, including increased cross-linking and stiffness of collagen, would be a major contributor to so-called immunosenescence, an age-related loss of immune efficacy. These changes have been reported to primarily affect the mobility of immune cells, particularly tissue-resident lymphocytes, and neutrophils, which is essential for their effective mediated immunity. It has been discussed that the movement of these cells within a much stiffer matrix due to ageing causes damage to the cell membrane and nucleus, ultimately leading to immune cell death and immunodeficiency (153). In contrast, neutrophils seeded in vitro on very dense and rigid matrices showed increased activation in terms of formation of neutrophil extracellular traps, a network of DNA/histone complexes and proteins with pro-inflammatory and microbiocidal effects. Such effects are enhanced by the presence of ECM proteins, including fibronectin and vitronectin, which mediate the activation of focal adhesion kinase upon integrin-mediated neutrophil interaction (154).
In vitro, macrophage immune function was also found to be sensitive to microenvironmental changes. In particular, culturing macrophages on stiffer matrices alone was sufficient to increase cell spreading as a result of the increased presence of integrin adhesion receptors, protruding filopodia and denser F-actin organization, as well as differentiation towards an anti-inflammatory phenotype with pro-tissue repair functions (155–159). These include the secretion of cytokines (IL10, IL1β), chemokines and growth factors (VEGF, TGF-β), as well as ECM remodeling proteases, which further modify the content and turnover of the ECM (160).
Similarly, Wong et al. showed that mesenchymal stromal cells respond to changes in matrix stiffness with cytoskeletal changes and surface receptor reorganization (161). In contrast to rigid matrices, in soft matrices these cells reacted with actin polymerization and clustering of TNF-α receptors resulting in increased TNF-α binding and, in turn, activation of TNF-α-induced signaling pathways that are likely implicated in the production, differentiation, and recruitment of leukocytes (161). The adhesion of the latter to endothelial cells, necessary for their extravasation into perivascular tissues, also appears to be affected by matrix stiffness (162, 163). With aging, hardening of the subendothelial matrix leads to an increase in the permeability of the endothelial monolayer due to increased cellular contractility and loss of cellular junctions. This in turn could promote increased leukocyte transmigration (162).
The retinal ECM undergoes reorganization with aging. For example, BrM develops a significant stiffening and thickening due to the accumulation of lipids, GAGs, collagen, and elastin (14). This may have implications for immune cell activation and function in the immune response. In addition, due to its thickening, BrM shows an age-dependent decrease in permeability (14), possibly leading to abnormalities in the compartmentalization of complement factors. In vitro, aging of BrM is simulated by nitrite modification of the ECM produced by RPE cells in culture. The growth of RPE cells on this type of matrix appears to influence the secretome of RPE cells. Indeed, these cells were found to secrete significantly higher levels of complement factors such as C3a than cells grown on unaged matrices (164). The effect of nitrite-modified matrices on complement gene expression was also evident in RPE derived from human induced-pluripotent stem cells (iPSCs) from AMD patients. When these cells were grown on nitrite-modified matrices a significantly increased expression of complement immune genes such as C3, CFB and CFH was observed when compared with age-matched controls (165). This suggests that changes in the ECM may strongly contribute to or underlie an altered/increased immune response in retinal diseases such as AMD. Based on such findings, the reduced collagen solubility and loss of elastin in BrM associated with aging (14) can potentially affect immune cell recruitment and activity, binding and activation of complement factors, and propagation of inflammatory cytokines. In line with this, Kenan and colleagues found a reduced amount of heparan sulfate in BrM and neurosensory retina in macular tissue sections from older probands compared to younger donors (166). Again, the results point to a role of alterations in ECM components impacting on the immune response associated with retinal aging and thus susceptibility to disease.
It should be noted that in ageing BrM, increased binding of extracellular MMPs to the ECM appears to underlie their sequestration and thus reduction of proteolytic potential (47, 167). In addition, this impacts the remodeling and composition of the ECM, resulting in reduction in the degradative capacity of MMPs in aged BrM and subsequent consequences for the release, enhancement, or inactivation of cytokines. Finally, age-dependent alterations in the expression and secretion of matricellular proteins may also contribute to changes in the extracellular environment with aging. By analyzing the expression of the VTN gene in a large-scale collection of 311 healthy retinal tissue samples, we found a statistically significant increase in the expression of this matricellular protein in retinal samples aged 60 years or older (168). This implies an increased accumulation of vitronectin in retinal extracellular spaces, such as BrM, with consequences for its immunoregulatory functions.
Dystrophies of the retina and the RPE represent broad clinical entities with many sharing a number of clinical features often rendering differential diagnosis demanding. Mutations in more than 300 different genes have been reported to be associated with degenerative changes in the retina and the RPE (169), although so far, causal genetic variants can be identified in only about 40-70% of all retinal patients. Many retinal dystrophies are monogenic in nature, i.e., they are caused by defined mutations with strong effect sizes in a single gene. In contrast, age-related macular degeneration (AMD), the third most common cause of legal blindness globally is a complex disorder, where disease risk is influenced by both genetic and environmental factors (170, 171). While causes and phenotypic presentations may greatly vary within the group of retinal conditions, a growing number are recognized to involve the ECM and a dysregulation of the immune response.
The following section provides an overview on the involvement of ECM homeostasis and immune responses in the pathological processes of both monogenic and complex retinal disorders (Figure 2).
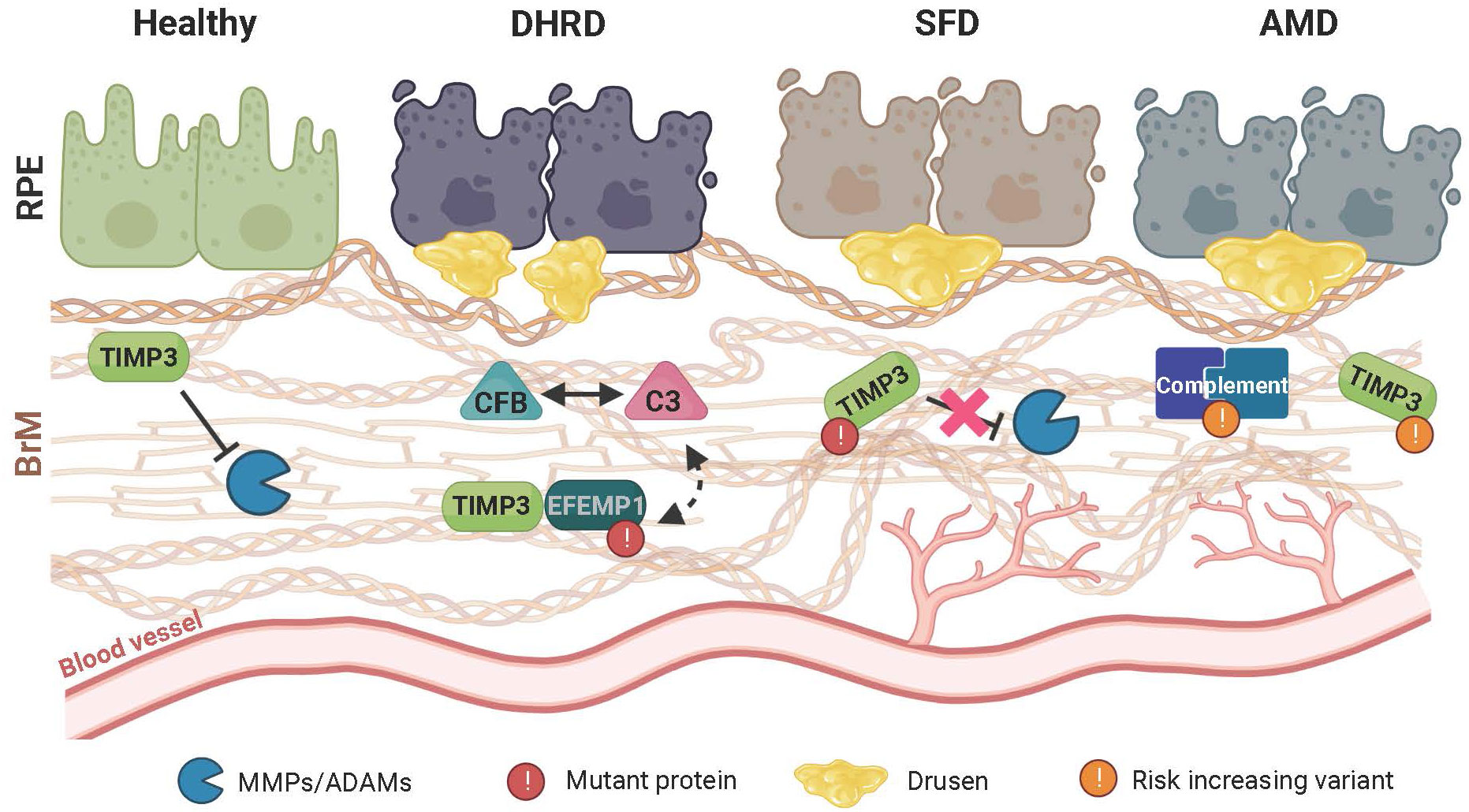
Figure 2 Schematic representation of retinal disease pathology on extracellular matrix (ECM) homeostasis and immune regulation. In the healthy retina, a finely tuned balance maintains the structural integrity of the extracellular Bruch’s membrane (BrM) to prevent inflammation and sustain a beneficial cellular environment for the retinal pigment epithelium (RPE) and the underlying choroidal blood vessels. Remodeling of BrM by MMPs and their regulation through TIMP3 plays an important role in maintaining this homeostasis. In Doyne honeycomb retinal dystrophy (DHRD), which is caused by a single mutation in the epidermal growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1) gene, an interplay between ECM remodeling and an aberrant activation of the complement system e.g., due to a concomitant increased protein expression of complement component 3 (C3) and complement factor B (CFB), leads to structural changes in BrM and formation of drusen-like deposits, ultimately resulting in RPE atrophy. In Sorsby fundus dystrophy (SFD), which is caused by mutations in the TIMP metallopeptidase inhibitor3 (TIMP3) gene, ECM remodeling and BrM homeostasis is altered leading to RPE atrophy. Subsequently, drusen deposition ensues, which is often accompanied by formation and invasion of new blood vessels towards the RPE. While pathological changes in DHRD and SDF are caused by mutations in single genes, age-related macular degeneration (AMD) is a common disease involving many genetic risk variants with a complex interplay of risk increasing alleles in several components of the complement cascade as well as ECM remodeling enzymes such as TIMP3. Of note, pathological changes seen in AMD closely resemble those seen in SFD.
DHRD, also known as Malattia Leventinese (ML), is an example of a rare autosomal dominant monogenic disorder caused by a single mutation, namely the R345W missense mutation in the gene encoding the epidermal growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1), an extracellular glycoprotein also known as fibulin-3 (172). DHRD/ML patients do not usually become symptomatic until after age 30 with clinical characteristics of the disease including macular sub-RPE deposits/drusen and RPE degeneration which in the advanced stages can also be associated with choroidal neovascular membranes and geographic atrophy.
The molecular pathomechanism in DHRD/ML remains elusive, although the EFEMP1 R345W mutation is speculated to result in increased protein misfolding which may lead to disturbances in the proteostasis network resulting in protein aggregation and drusen formation (173, 174). Molecular constituents of drusen in DHRD/ML include vitronectin, amyloid P, TIMP3 and a number of complement proteins, all components also found in drusen of AMD patients (175, 176). Animal models have greatly fostered our knowledge about the molecular pathomechanism of DHRD/ML disease. Efemp1R345W/R345W knock-in mice mimic the phenotype seen in human patients and show formation of sub-RPE deposits which do not form in wildtype littermates or Efemp1-/- knock-out mice of the same age (177). In fact, Efemp1-deficient- mice intriguingly appear to be protected from drusen formation, suggesting a role of murine Efemp1 in generating the drusen-like deposits (178). Early work applying proteomic analyses of BrM in Efemp1R345W/R345W mice indicated that basal deposits contain normal ECM components, but these are present in abnormal quantities. These studies further revealed significant changes in proteins with immune related function, including those of the complement cascade as part of the immune system (175). Additionally, cultured primary RPE cells from Efemp1R345W/R345W mice have been shown to secrete increased amounts of complement factor 3 (C3) (179). Together, these findings further strengthen the notion that the immune system and inflammation are associated with drusen formation.
To clarify the mutual interplay between changes in ECM/BrM and inflammation, three double-mutant mouse lines were generated including an Efemp1R345W/R345W/C3-/- double mutant mouse (175). The authors demonstrated the requirement of C3 for drusenogenesis, as the double-mutant animals failed to produce sub-RPE deposits. Such findings support the idea that the complement system plays a key role in basal deposit formation, likely through recognition of abnormal ECM. C3 is a key player in the complement system, as it is the first node where the three different upstream activation mechanisms (the alterative, classical and lectin pathways) converge and from thereon follow an identical downstream pathway ultimately culminating in MAC formation.
By analyzing Efemp1R345W/R345W/C5-/- double mutant mice, Garland and colleagues aimed to elucidate whether C3 rather than C5 function is required for drusen formation in DHRD/ML. They demonstrated that genetic ablation of C5 did not prevent the formation of sub-RPE deposits, but rather increased drusen formation in the mutant animals (175). In ex vivo cell cultures strong immunostaining for C3 was demonstrated in RPE cells from Efemp1R345W/R345W compared to RPE cells derived from wildtype animals. In addition, Efemp1 protein colocalized with murine Cfh in the sub-RPE deposits. Based on their results, Garland and colleagues proposed that increased activation of C3 occurs in Efemp1R345W/R345W mediated by the alternative complement pathway activation mechanism of tick-over via the deposition of C3b on abnormal ECM (180). It remains to be shown whether mutant Efemp1 causes presentation of ligands that ultimately activate the classical pathway.
Extending these initial studies, additional data were presented recently demonstrating that complement factor B is critical for sub-RPE deposit formation in the Efemp1R345W/R345W mouse model (177). Quantitative RNA sequencing and subsequent proteomics demonstrated that expression of inflammatory pathway genes was increased in neural retina and posterior eyecups in 17-month-old Efemp1R345W/R345W mice compared to wildtype mice. Efemp1R345W/R345W/Cfb-/- double mutant mice revealed partial restoration of the changes that were seen in Efemp1R345W/R345W single mutant animals, suggesting a key role for murine Cfb in sub-RPE deposit formation. This was further supported by applying a small molecule inhibitor of Cfb reducing sub-RPE deposition by 65%. This reduction, however, was only observed in female mice, but not in male animals (177). Interestingly, loss-of-function alleles in the CFB gene in humans are associated with a decreased risk of developing AMD (181, 182).
In summary, various animal studies suggest that mutant Efemp1 protein leads to activation of the alternative complement pathway, which then drives the formation of sub-RPE deposits. In addition to mouse models, patient-derived cell lines, such as iPSCs-derived RPE cells have become a valuable model to study DRHD/ML. iPSC-RPE cells harboring the EFEMP1 R345W mutation show an increased number of drusen-like deposits compared to controls after only 90 days of cultivation on transwell filters. Additionally, mRNA expression of complement genes C5 and MCP is significantly upregulated in DHRD/ML cells and CFB was also shown to be elevated but failed to reach statistical significance (183).
DHRD/ML not only shares striking phenotypic similarities with AMD but also with the seemingly unrelated SFD. The latter monogenic maculopathy was first described by Arnold Sorsby and colleagues in 1949 (4) and was reported to ultimately result in bilateral loss of central vision. SFD is inherited in an autosomal dominant mode and is classified as a rare disease with a prevalence of 1:220,000. In contrast to AMD, the onset of SFD is much earlier between the second and fourth decade of life (4). Early symptoms of SFD include nyctalopia (night blindness) or loss of visual acuity, with later complications such as oedema, hemorrhages and macular scaring. Further progression of the disease culminates in retinal and choroidal atrophy (chorioretinal atrophy) and vessel sclerosis, which leads to severe central and peripheral vision loss about 35 years after disease onset (4, 184). In contrast to DHRD/ML with only a single mutation in EFEMP1, a growing number of independent genetic variants in the TIMP3 gene are causative for SFD. Most of these variants result in an uneven number of cysteine residues in the mature TIMP3 protein. In healthy eyes, TIMP3 is expressed by the RPE and accumulates in BrM (5). A major role for TIMP3 relates to its involvement in the regulation of BrM thickness (185). Mutant TIMP3 protein is suggested to be more resistant to turnover and clearance in BrM, which ultimately results in accumulation of TIMP3 in drusen-like deposits (186–189). An increasing thickening of BrM results in impaired exchange of nutrients and molecules between RPE cells and the choroid and finally leads to RPE atrophy (185, 187).
Enhanced RPE autofluorescence due to oxidation products of unsaturated fatty acids is caused by the intracellular accumulation of lipofuscin, which is often referred to as a “wear-and-tear” pigment (190). Increased lipofuscin content of RPE cells is a normal feature of aging cells but has been linked to decreased lysosomal activity and promotion of local retinal inflammation (191–193). Activation of the complement system and secretion of pro-inflammatory cytokines appear to be a consequence of lipofuscin accumulation in the RPE. These findings indirectly link TIMP3 malfunction to immune responses as a consequence of BrM thickening. TIMP3 has also been implicated in the regulation of inflammatory processes in the diseased retina in a more direct manner. As mentioned above, TIMP3 not only inhibits MMPs, but also proteins of the ADAM and ADAMTS families. Specifically, ADAM17, which is also known as TACE (TNF-a-converting enzyme) is known to be directly inhibited by TIMP3 binding (194). ADAM17 is responsible for the cleavage of inactive pro-TNFα to its active form and hence plays a crucial role in TNFα-mediated signaling. This is further supported by the fact that a pronounced inflammation of the liver accompanied by increased levels of both TACE and TNFa was observed in Timp3 knockout mice (69). TIMP3 functions in the retina and other tissues have been recently reviewed (195). Further evidence for an involvement of TIMP3 in immunity and inflammation was obtained from studies in iPSC-RPE cells. iPSC-RPE cells derived from SFD patients showed increased levels of complement genes C1R, C1S, C3, MCP and SERPING1 (183).
In contrast to DHRD/ML and SFD, AMD is a complex disorder caused by both environmental and genetic factors. While monogenic disorders are typically caused by mutations in a single gene, the genetic susceptibility to complex disorders is generally influenced by genetic variants at multiple loci. In the largest genome-wide association study of AMD to date 52 variants in 34 loci were identified as being significantly associated with the disease at the genome wide significance level (6). Of note, 19 of these variants are located within or in proximity to genes coding for components of the complement cascade whereby the most striking association is seen with components of the alternative pathway. Both the highly significant enrichment of complement genes and the fact that several complement components have been found in drusen from AMD patient donor eyes (196–198) suggest an important role of complement dysregulation in AMD pathobiology. As an in-depth discussion of the complement system and its role in AMD pathogenesis is beyond the scope of this article, the reader is referred to two excellent recent reviews on this topic (199, 200). Another cellular feature which shows a marked enrichment in the 34 AMD associated loci is ECM remodeling and homeostasis, again linking the two topics of ECM homeostasis and immune regulation. Although the detailed interplay between inflammatory processes, drusen formation and disturbances in BrM homeostasis remains elusive at present, recent research, as discussed in the next paragraph, has proposed some hypotheses as to how these two key features of cellular maintenance may influence each other.
Not only in disease but also in physiological aging, BrM undergoes structural changes including increased thickness, altered permeability and deposit formation (drusen, hard and soft, basal linear (BlinD) and laminar (BlamD) deposits). To mimic an aged BrM, the use of non-enzymatically crosslinked nitrite-modified ECM has been explored in vitro (165, 201, 202). This is a particularly suitable model to study molecular pathomechanisms in AMD, as cigarette smoking leads to an increase in nitric oxide and represents a key environmental risk factor for the disease (203). Using ARPE-19 cells Fields and colleagues demonstrated that an altered ECM structure modifies the expression of CD46, which is a negative regulator of the complement pathway (202). A progressive decrease in CD46 has also been observed in RPE from GA patients, while Cd46 knock-out mice were shown to be more susceptible to CNV, likely due to impaired complement inhibition (204, 205). The same group confirmed their initial findings in iPSC-RPE grown on nitrite-modified ECM by demonstrating increased levels of bioactive C3a in the conditioned media of the cells (164). Another in vitro model for diseased BrM/ECM interestingly is based on the DHRD/ML-causing R345W variant of EFEMP1. Expressing the EFEMP-R345W variant in ARPE-19 cells caused the production of ECM with altered properties compared to cells expressing normal EFEMP1 (34). Due to the overlapping pathophysiological features of DHRD/ML and AMD, using this altered ECM has become an established system to mimic “diseased” ECM. These alterations can be attributed to increased MMP2 activation in cells expressing the R345W variant of EFEMP1. Analysis of complement components deposited in the altered ECM revealed increased C3b and CFH. When primary human RPE cells were cultivated on the altered ECM, they showed a significantly lower transepithelial electrical resistance and prominent basal deposits after 2 weeks in culture (34). These findings were subsequently corroborated by extending the model with iPSC-RPE cells which harbor different EFEMP1 genotypes. Like primary RPE, iPSC-RPE showed an unwanted activation of the complement cascade when cultured on EFEMP1-R345W ECM derived from ARPE-19 cells. By using a sophisticated assay design and combinations of EFPEP1 and C3 single and double mutant iPSC-RPE lines it was shown that complement activation via the mechanism of tick-over is sufficient to induce basal deposit formation. Additionally, a siRNA knockdown of C3 suppresses the formation of abnormal ECM, which corroborates the data obtained for deposit formation in DHRD/ML (206). Interestingly, an effect of C3a on ECM structure was also reported. Stimulation of human fetal RPE with C3a for two weeks resulted in an increased basal accumulation of collagens IV and VI mirroring ECM alteration, decreased proteasome activity and increased MMP-2 activity (34). While these models used EFEMP1 indirectly to mirror AMD-like pathological changes in vitro, there actually is also a more direct link between the EFEMP1 protein and AMD pathobiology. One of the genetic loci with the strongest association to an increased AMD risk at 10q26 comprises the gene HTRA1 (High Temperature Requirement A Serine Peptidase 1). HTRA1 is a secreted protease which is implicated in a number of biological processes including cell signaling, organization of the ECM, and skeletal development and osteogenesis. One noteworthy target to be cleaved by HTRA1 is EFEMP1 (207). Even though there is no direct link between EFEMP1 and AMD pathogenesis, this interaction between HTRA1 and EFEMP1 yet again points towards a common disease mechanism possibly shared between different retinal dystrophies.
Taken together the findings gathered through research on different, yet similar pathologies, one could propose a model for the order of events in drusenogenesis as follows: in aging or disease, alterations in the structure of the ECM, or in particular in BrM occur, which then leads to the activation of the complement cascade which in turn initiates drusen formation. This order of events would also be in line with a model on drusen formation proposed by Anderson and colleagues who suggest that cellular debris can trigger an inflammatory signal, and which then gives rise to a potential “nucleation” site for drusen formation (208).
DR is the leading cause of vision loss in the adult population and arises as a comorbidity of diabetes (209). Even though GWAS have identified a few hundred risk loci for diabetes, the contribution of the overall genetic risk to disease development is less clear than for example in AMD (210). A potential dialogue between ECM changes and immune responses has also been highlighted in the progression of DR, which results in visual impairment due to altered retinal vascular structure and macular oedema in hyperglycemic conditions. While the aforementioned diseases of DHRD, SFD and AMD primarily result in disturbances of BrM and the RPE, DR most prominently affects the retinal vasculature and to our knowledge, there is no evidence of altered BrM in DR. While the RPE may contribute to DR pathogenesis, it is not clear if it is the primary site of pathology in DR and more evidence is required (211). Recent advances in our understanding of ECM remodeling and inflammation in DR have excellently been summarized in two recent review articles (212, 213). We will therefore only touch upon some of the most current literature on implications of immune dysregulation and ECM alterations in DR.
In the last years convincing evidence has been gathered suggesting that DR represents a neurodegenerative disorder in its early stages. Retinal Müller glia cells are of outmost importance for retinal health and it has become apparent that they also play a role in DR (214). Using a diabetic pig model Sagmeister and colleagues conducted an in-depth proteomic analysis of changes induced in Müller cells under high glucose conditions (215). Interestingly, the majority of proteins found to be differentially expressed were proteins of the ECM. These findings are in line with data from many others, suggesting that alterations of the ECM are a very early events in DR pathogenesis. In DR, capillary ECM becomes fibrotic and is characterized by altered collagen IV structure as well as fibronectin and tenascin-C deposition. Functional consequences of altered ECM structure and composition observed in cell cultures under high glucose conditions include increased stiffness and higher permeability (213). While it is broadly accepted that ECM alterations occur in DR, it remains to be elucidated which molecular pathways trigger the ECM remodeling process. To deepen our understanding of this topic, Giblin and colleagues conducted a study in which they subjected human retinal microvascular endothelial cells (hRMEC) and human pericytes (hPR) to hyperglycemia, dyslipidemia and inflammation. These conditions are considered to represent three major insults in diabetic conditions. Of note, the most prominent and consistent changes in ECM expression were seen upon treatment with cytokines TNF-α and IL1β. The authors further showed, that treating hRMEC with conditioned media from IL1β treated Müller cells caused a marked increase in collagen IV and agrin expression (216)
As already mentioned, the interplay between ECM homeostasis and inflammation is by no means a one-way road, and while inflammation can result in ECM alterations, ECM-related processes can also regulate inflammation. In the context of DR, this was nicely demonstrated by a recent study by Abu El-Asrar and colleagues who suggest that intravitreal administration of TIMP3 in a DR rat model alleviates retinal inflammation by stimulating anti-inflammatory events (78). These events include reduced retinal vascular permeability and an attenuation of TNF-α-dependent retinal expression of the pro-inflammatory NF-κB and the adhesion molecule ICAM-1. Of note, this study also included Müller cells, in which treatment with TIMP3 counteracted induced upregulation of VEGF under various experimental conditions mimicking a diabetic environment. These findings once more highlight the manifold functions of TIMP3 in retinal homeostasis.
Monogenic IRDs such as DHRD/ML or SFD overlap and share several (histo)pathological and genetic features with more complex retinal diseases like AMD. In monogenic IRDs, great efforts have gone into gaining knowledge of the role of disease-causing genetic variation using both in vivo animal as well as in vitro cell culture models. The findings from such models may thus offer avenues for transfer to more complex retinal disorders. Here, we have summarized current knowledge on the commonalities between the divergent clinical conditions and their underlying pathogenicities demonstrating that alteration in the ECM/BrM turnover likely initiates or sustains similar immune responses in both monogenic and complex diseases (Figure 3). This insight may help to broaden our approaches in addressing the modulation of disease mechanisms and may potentially lead to novel designs in therapy of these devastating ocular diseases.
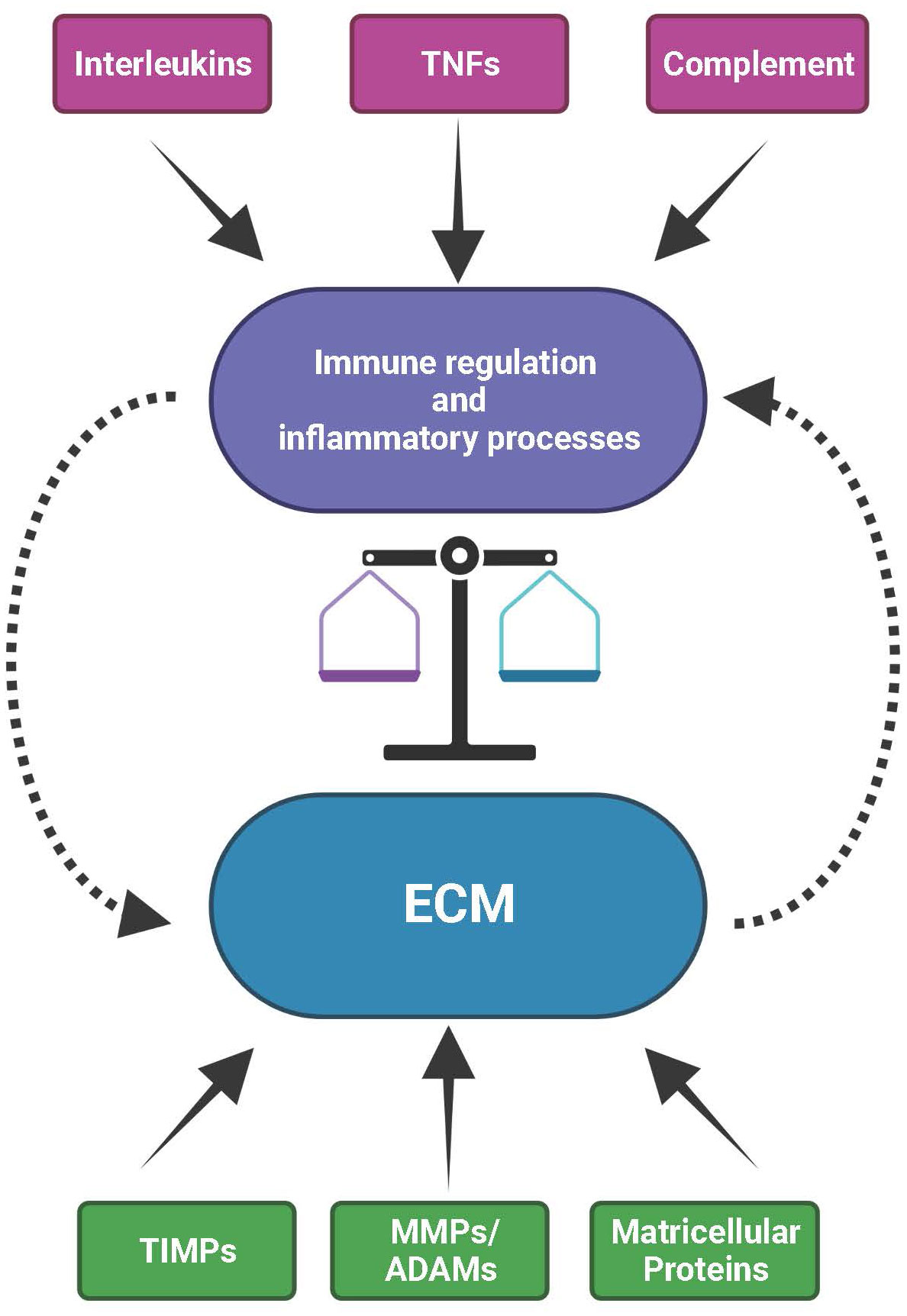
Figure 3 The mutual interplay between the extracellular matrix (ECM) and immunological processes. The ECM with its structural constituents but also its functional components, such as ECM remodeling enzymes (MMPs, ADAMs and TIMPs) and matricellular proteins, provides the microenvironment for the activity of immune and inflammatory processes. Vice versa, immunological/inflammatory activities contributed by e.g., interleukins, tumor necrosis factors (TNFs) or complement can influence the structural and functional organization of the ECM. As a consequence, disturbances in the expression or activity of factors that are part of the ECM or of the immune system affect the balance between inflammatory events and ECM homeostasis. These can eventually impact natural ageing, environmental insult or high genetic risk, together eventually initiating and sustaining onset and progression of progressive disease processes in the retina.
Of note, TIMP3 could highlight a nexus between monogenic and complex diseases with genetic variants in this gene not only being causative for the autosomal dominant SFD but also with non-coding variants in proximity of the gene being significantly associated with risk of AMD (6). Mutant TIMP3 protein is thought to assemble into pathologic aggregates in the SFD condition, very similar to AMD. In the latter situation, the aggregation is not caused by mutated protein, but is likely due to risk-dependent increased expression and intramolecular cross-linking of normal TIMP3 upon oxidative stress or due to spontaneous misfolding. This alternate route to protein aggregation takes an extended time to form, which may explain why AMD has a later onset of symptoms than SFD (185). Additionally, TIMP3 may also play a role in DHRD/ML as it is a direct interaction partner of EFEMP1 (217). Curiously, EFEMP1 also interacts with CFH (218), although there are currently no hypotheses as to how a potential interactome of ECM modulators and complement components may participate in the maintenance of BrM homeostasis. Finally, experimental evidence also supports the role of TIMP3 in ameliorating the inflammation, angiogenesis and vascular damage associated with DR (78).
Observations of commonalities between different clinical pictures may provide a unique opportunity to provide not only an improved model of disease but also to understand retinal pathomechanisms, particularly in complex diseases such as AMD. In light of such a reasoning, targeting the ECM or its molecular constituents could provide an alternative but yet important avenue for therapeutic intervention in retinal diseases in addition to those currently being explored through isolated immune targets. Such an approach appears superior as a common pathway can be addressed regardless of many individual mutational events or risk predispositions.
KP, FB, PB, and BW were all involved in the concept, design, writing, and critical review of the content of this article. All authors agree to the accuracy and integrity of this work as presented. All authors contributed to the article and approved the submitted version.
This research was funded by the Australian National Health and Medical Research Council (NHMRC) Senior Research Fellowship grant number 1138585 to PB. The sponsor or funding organizations had no role in the design or conduct of this research.
Figure schematics and cartoons were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wiggs JL, Pierce EA. Genetic testing for inherited eye disease. JAMA Ophthalmol (2013) 131(10):1265. doi: 10.1001/jamaophthalmol.2013.4509
2. Lam BL, Leroy BP, Black G, Ong T, Yoon D, Trzupek K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J Rare Dis (2021) 16(1):514. doi: 10.1186/s13023-021-02145-0
3. Cremers F, Boon C, Bujakowska K, Zeitz C. Special issue introduction: Inherited retinal disease: Novel candidate genes, genotype–phenotype correlations, and inheritance models. Genes (Basel). (2018) 9(4):215. doi: 10.3390/genes9040215
4. Sorsby A, Mason MEJ, Gardener NA. FUNDUS DYSTROPHY WITH UNUSUAL FEATURES (Late onset and dominant inheritance of a central retinal lesion showing oedema, haemorrhage and exudates developing into generalised choroidal atrophy with massive pigment proliferation). Br J Ophthalmology. (1949) 33(2):67–97. doi: 10.1136/bjo.33.2.67
5. Weber BHF, Vogt G, Pruett RC, Stöhr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with sorsby’s fundus dystrophy. Nat Genet (1994) 8(4):352–6. doi: 10.1038/ng1294-352
6. Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet (2016) 48(2):134–43. doi: 10.1038/ng.3448
7. Murakami Y, Ishikawa K, Nakao S, Sonoda KH. Innate immune response in retinal homeostasis and inflammatory disorders. Prog Retin Eye Res (2020) 74:100778. doi: 10.1016/j.preteyeres.2019.100778
8. Takeda A, Yanai R, Murakami Y, Arima M, Sonoda KH. New insights into immunological therapy for retinal disorders. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01431
9. Hallmann R, Zhang X, di Russo J, Li L, Song J, Hannocks MJ, et al. The regulation of immune cell trafficking by the extracellular matrix. Curr Opin Cell Biol (2015) 36:54–61. doi: 10.1016/j.ceb.2015.06.006
10. Simon T, Bromberg JS. Regulation of the immune system by laminins. Trends Immunol (2017) 38(11):858–71. doi: 10.1016/j.it.2017.06.002
11. Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol (2011) 209(2):139–51. doi: 10.1530/JOE-10-0377
12. Papp A, Papp K, Uzonyi B, Cserhalmi M, Csincsi ÁI, Szabó Z, et al. Complement factor h-related proteins FHR1 and FHR5 interact with extracellular matrix ligands, reduce factor h regulatory activity and enhance complement activation. Front Immunol [Internet]. (2022) 13:845953. doi: 10.3389/fimmu.2022.845953
13. Reinhard J, Joachim SC, Faissner A. Extracellular matrix remodeling during retinal development. Exp Eye Res (2015) 133:132–40. doi: 10.1016/j.exer.2014.07.001
14. Curcio C, Johnson M. Structure, function, and pathology of bruch’s membrane. In: Retina fifth edition. 5th ed, vol. p. Amsterdam, Netherlands: Elsevier (2012). p. 465–81. doi: 10.1016/B978-1-4557-0737-9.00020-5
15. Moore DJ, Clover GM. The effect of age on the macromolecular permeability of human bruch’s membrane. Invest Ophthalmol Vis Sci (2001) 42(12):2970–5.
16. Sarma JV, Ward PA. The complement system. Cell Tissue Res (2011) 343(1):227–35. doi: 10.1007/s00441-010-1034-0
17. Clark SJ, Bishop PN. The eye as a complement dysregulation hotspot. Semin Immunopathol (2018) 40(1):65–74. doi: 10.1007/s00281-017-0649-6
18. Clark SJ, McHarg S, Tilakaratna V, Brace N, Bishop PN. Bruch’s membrane compartmentalizes complement regulation in the eye with implications for therapeutic design in age-related macular degeneration. Front Immunol (2017) 8:1778. doi: 10.3389/fimmu.2017.01778
19. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of factor h-like protein 1 as the predominant complement regulator in bruch’s membrane: Implications for age-related macular degeneration. J Immunol (2014) 193(10):4962–70. doi: 10.4049/jimmunol.1401613
20. Ferreira VP, Pangburn MK, Cortés C. Complement control protein factor h: the good, the bad, and the inadequate. Mol Immunol (2010) 47(13):2187–97. doi: 10.1016/j.molimm.2010.05.007
21. Kreuger J, Kjellén L. Heparan sulfate biosynthesis: Regulation and variability. J Histochem Cytochem (2012) 60(12):898–907. doi: 10.1369/0022155412464972
22. Avery VM, Gordon DL. Characterization of factor h binding to human polymorphonuclear leukocytes. J Immunol (1993) 151(10):5545–53. doi: 10.4049/jimmunol.151.10.5545
23. Malhotra R, Ward M, Sim RB, Bird MI. Identification of human complement factor h as a ligand for l-selectin. Biochem J (1999) 341(Pt 1):61–9. doi: 10.1042/bj3410061
24. Mnjoyan Z, Li J, Afshar-Kharghan V. Factor h binds to platelet integrin αIIbβ3. Platelets. (2008) 19(7):512–9. doi: 10.1080/09537100802238494
25. Losse J, Zipfel PF, Józsi M. Factor h and factor h-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans , and enhance neutrophil antimicrobial activity. J Immunol (2010) 184(2):912–21. doi: 10.4049/jimmunol.0901702
26. Schneider AE, Sándor N, Kárpáti É, Józsi M. Complement factor h modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol (2016) 72:37–48. doi: 10.1016/j.molimm.2016.02.011
27. Proudfoot AEI, Johnson Z, Bonvin P, Handel TM. Glycosaminoglycan interactions with chemokines add complexity to a complex system. Pharm (Basel). (2017) 10(3):70. doi: 10.3390/ph10030070
28. Mulloy B, Rider CC. Cytokines and proteoglycans: An introductory overview. Biochem Soc Trans (2006) 34(Pt 3):409–13. doi: 10.1042/BST0340409
29. Clark SJ, Keenan TDL, Fielder HL, Collinson LJ, Holley RJ, Merry CLR, et al. Mapping the differential distribution of glycosaminoglycans in the adult human retina, choroid, and sclera. Invest Ophthalmol Vis Sci (2011) 52(9):6511–21. doi: 10.1167/iovs.11-7909
30. Puré E, Cuff CA. A crucial role for CD44 in inflammation. Trends Mol Med (2001) 7(5):213–21. doi: 10.1016/S1471-4914(01)01963-3
31. Lebbink RJ, de Ruiter T, Adelmeijer J, Brenkman AB, van Helvoort JM, Koch M, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med (2006) 203(6):1419–25. doi: 10.1084/jem.20052554
32. Larsen AMH, Kuczek DE, Kalvisa A, Siersbæk MS, Thorseth ML, Johansen AZ, et al. Collagen density modulates the immunosuppressive functions of macrophages. J Immunol (2020) 205(5):1461–72. doi: 10.4049/jimmunol.1900789
33. Shields KJ, Stolz D, Watkins SC, Ahearn JM. Complement proteins C3 and C4 bind to collagen and elastin in the vascular wall: A potential role in vascular stiffness and atherosclerosis. Clin Transl Sci (2011) 4(3):146–52. doi: 10.1111/j.1752-8062.2011.00304.x
34. Fernandez-Godino R, Bujakowska KM, Pierce EA. Changes in extracellular matrix cause RPE cells to make basal deposits and activate the alternative complement pathway. Hum Mol Genet (2018) 27(1):147–59. doi: 10.1093/hmg/ddx392
35. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol (2011) 3(12):a005058. doi: 10.1101/cshperspect.a005058
36. de Almeida LGN, Thode H, Eslambolchi Y, Chopra S, Young D, Gill S, et al. Matrix metalloproteinases: From molecular mechanisms to physiology, pathophysiology, and pharmacology. Pharmacol Rev (2022) 74(3):712–68. doi: 10.1124/pharmrev.121.000349
37. Sivak JM, Fini ME. MMPs in the eye: emerging roles for matrix metalloproteinases in ocular physiology. Prog Retin Eye Res (2002) 21(1):1–14. doi: 10.1016/S1350-9462(01)00015-5
38. Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJG. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog (2012) 8(4):e1002641. doi: 10.1371/journal.ppat.1002641
39. Murray MY, Birkland TP, Howe JD, Rowan AD, Fidock M, Parks WC, et al. Macrophage migration and invasion is regulated by MMP10 expression. PLoS One (2013) 8(5):e63555. doi: 10.1371/journal.pone.0063555
40. van den Steen PE, Proost P, Wuyts A, van Damme J, Opdenakker G. Neutrophil gelatinase b potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. (2000) 96(8):2673–81. doi: 10.1182/blood.V96.8.2673
41. Starr AE, Dufour A, Maier J, Overall CM. Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J Biol Chem (2012) 287(8):5848–60. doi: 10.1074/jbc.M111.314609
42. Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. (2002) 111(5):635–46. doi: 10.1016/S0092-8674(02)01079-6
43. Pruijt JF, Fibbe WE, Laterveer L, Pieters RA, Lindley IJ, Paemen L, et al. Prevention of interleukin-8-induced mobilization of hematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against the metalloproteinase gelatinase b (MMP-9). Proc Natl Acad Sci U S A. (1999) 96(19):10863–8. doi: 10.1073/pnas.96.19.10863
44. McMahon M, Ye S, Pedrina J, Dlugolenski D, Stambas J. Extracellular matrix enzymes and immune cell biology. Front Mol Biosci (2021) 8:703868. doi: 10.3389/fmolb.2021.703868
45. Caban M, Owczarek K, Lewandowska U. The role of metalloproteinases and their tissue inhibitors on ocular diseases: Focusing on potential mechanisms. Int J Mol Sci (2022) 23(8):4256. doi: 10.3390/ijms23084256
46. Padgett LC, Lui GM, Werb Z, LaVail MM. Matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-1 in the retinal pigment epithelium and interphotoreceptor matrix: vectorial secretion and regulation. Exp Eye Res (1997) 64(6):927–38. doi: 10.1006/exer.1997.0287
47. Guo L, Hussain AA, Limb GA, Marshall J. Age-dependent variation in metalloproteinase activity of isolated human bruch’s membrane and choroid. Invest Ophthalmol Vis Sci (1999) 40(11):2676–82.
48. van Hove I, Lefevere E, de Groef L, Sergeys J, Salinas-Navarro M, Libert C, et al. MMP-3 deficiency alleviates endotoxin-induced acute inflammation in the posterior eye segment. Int J Mol Sci (2016) 17(11):1825. doi: 10.3390/ijms17111825
49. Opdenakker G, van den Steen PE, van Damme J. Gelatinase b: a tuner and amplifier of immune functions. Trends Immunol (2001) 22(10):571–9. doi: 10.1016/S1471-4906(01)02023-3
50. Goverdhan S v, Ennis S, Hannan SR, Madhusudhana KC, Cree AJ, Luff AJ, et al. Interleukin-8 promoter polymorphism -251A/T is a risk factor for age-related macular degeneration. Br J Ophthalmology. (2008) 92(4):537–40. doi: 10.1136/bjo.2007.123190
51. Ulhaq ZS, Soraya GV. Roles of IL-8 –251A/T and +781C/T polymorphisms, IL-8 level, and the risk of age-related macular degeneration. Archivos la Sociedad Española Oftalmología (English Edition). (2021) 96(9):476–87. doi: 10.1016/j.oftale.2020.02.017
52. Jonas JB, Jonas RA, Neumaier M, Findeisen P. Cytokine concentration in aqueous humor of eyes with diabetic macular edema. Retina. (2012) 32(10):2150–7. doi: 10.1097/IAE.0b013e3182576d07
53. Ecker SM, Pfahler SM, Hines JC, Lovelace AS, Glaser BM. Sequential in-office vitreous aspirates demonstrate vitreous matrix metalloproteinase 9 levels correlate with the amount of subretinal fluid in eyes with wet age-related macular degeneration. Mol Vis (2012) 18:1658–67.
54. van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol (2007) 82(6):1375–81. doi: 10.1189/jlb.0607338
55. McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. (2002) 100(4):1160–7. doi: 10.1182/blood.V100.4.1160.h81602001160_1160_1167
56. Eichler W, Friedrichs U, Thies A, Tratz C, Wiedemann P. Modulation of matrix metalloproteinase and TIMP-1 expression by cytokines in human RPE cells. Invest Ophthalmol Vis Sci (2002) 43(8):2767–73.
57. Garcia C, Bartsch DU, Rivero ME, Hagedorn M, McDermott CD, Bergeron-Lynn G, et al. Efficacy of prinomastat) (AG3340), a matrix metalloprotease inhibitor, in treatment of retinal neovascularization. Curr Eye Res (2002) 24(1):33–8. doi: 10.1076/ceyr.24.1.33.5429
58. Bhatt LK, Addepalli V. Attenuation of diabetic retinopathy by enhanced inhibition of MMP-2 and MMP-9 using aspirin and minocycline in streptozotocin-diabetic rats. Am J Transl Res (2010) 2(2):181–9.
59. Becerra E M, Morescalchi F, Gandolfo F, Danzi P, Nascimbeni G, Arcidiacono B, et al. Clinical evidence of intravitreal triamcinolone acetonide in the management of age-related macular degeneration. Curr Drug Targets. (2011) 12(2):149–72. doi: 10.2174/138945011794182746
60. Webb AH, Gao BT, Goldsmith ZK, Irvine AS, Saleh N, Lee RP, et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer. (2017) 17(1):434. doi: 10.1186/s12885-017-3418-y
61. Plantner JJ, Jiang C, Smine A. Increase in interphotoreceptor matrix gelatinase a (MMP-2) associated with age-related macular degeneration. Exp Eye Res (1998) 67(6):637–45. doi: 10.1006/exer.1998.0552
62. Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. (2005) 85(5):597–607. doi: 10.1038/labinvest.3700251
63. Chau KY, Sivaprasad S, Patel N, Donaldson TA, Luthert PJ, Chong N v. Plasma levels of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9) in age-related macular degeneration. Eye (Lond). (2007) 21(12):1511–5. doi: 10.1038/sj.eye.6702722
64. Kim J, Kim JH, Do JY, Lee JY, Yanai R, Lee Ik, et al. Key role of microglial matrix metalloproteinases in choroidal neovascularization. Front Cell Neurosci (2021) 15. doi: 10.3389/fncel.2021.638098
65. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta (2010) 1803(1):55–71. doi: 10.1016/j.bbamcr.2010.01.003
66. Leco KJ, Khokha R, Pavloff N, Hawkes SP, Edwards DR. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J Biol Chem (1994) 269(12):9352–60. doi: 10.1016/S0021-9258(17)37115-6
67. Ruiz A, Brett P, Bok D. TIMP-3 is expressed in the human retinal pigment epithelium. Biochem Biophys Res Commun (1996) 226(2):467–74. doi: 10.1006/bbrc.1996.1379
68. Fariss RN, Apte SS, Olsen BR, Iwata K, Milam AH. Tissue inhibitor of metalloproteinases-3 is a component of bruch’s membrane of the eye. Am J Pathol (1997) 150(1):323–8.
69. Mohammed FF, Smookler DS, Taylor SEM, Fingleton B, Kassiri Z, Sanchez OH, et al. Abnormal TNF activity in Timp3-/- mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet (2004) 36(9):969–77. doi: 10.1038/ng1413
70. Hojilla C v, Jackson HW, Khokha R. TIMP3 regulates mammary epithelial apoptosis with immune cell recruitment through differential TNF dependence. PLoS One (2011) 6(10):e26718. doi: 10.1371/journal.pone.0026718
71. Monteleone I, Federici M, Sarra M, Franzè E, Casagrande V, Zorzi F, et al. Tissue inhibitor of metalloproteinase-3 regulates inflammation in human and mouse intestine. Gastroenterology. (2012) 143(5):1277–1287.e4. doi: 10.1053/j.gastro.2012.07.016
72. Gill SE, Huizar I, Bench EM, Sussman SW, Wang Y, Khokha R, et al. Tissue inhibitor of metalloproteinases 3 regulates resolution of inflammation following acute lung injury. Am J Pathol (2010) 176(1):64–73. doi: 10.2353/ajpath.2010.090158
73. Gill SE, Gharib SA, Bench EM, Sussman SW, Wang RT, Rims C, et al. Tissue inhibitor of metalloproteinases-3 moderates the proinflammatory status of macrophages. Am J Respir Cell Mol Biol (2013) 49(5):768–77. doi: 10.1165/rcmb.2012-0377OC
74. Chen G, Goeddel D v. TNF-R1 signaling: a beautiful pathway. Science. (2002) 296(5573):1634–5. doi: 10.1126/science.1071924
75. Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett (1998) 435(1):39–44. doi: 10.1016/S0014-5793(98)01031-X
76. Tezel G, Li LY, Patil R v, Wax MB. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci (2001) 42(8):1787–94.
77. Costagliola C, Romano V, de Tollis M, Aceto F, dell’Omo R, Romano MR, et al. TNF-alpha levels in tears: a novel biomarker to assess the degree of diabetic retinopathy. Mediators Inflamm (2013) 2013:629529. doi: 10.1155/2013/629529
78. Abu El-Asrar AM, Ahmad A, Nawaz MI, Siddiquei MM, de Zutter A, Vanbrabant L, et al. Tissue inhibitor of metalloproteinase-3 ameliorates diabetes-induced retinal inflammation. Front Physiol (2021) 12:807747. doi: 10.3389/fphys.2021.807747
79. Qi JH, Ebrahem Q, Ali M, Cutler A, Bell B, Prayson N, et al. Tissue inhibitor of metalloproteinases-3 peptides inhibit angiogenesis and choroidal neovascularization in mice. PLoS One (2013) 8(3):e55667. doi: 10.1371/journal.pone.0055667
80. Cruz-Muñoz W, Kim I, Khokha R. TIMP-3 deficiency in the host, but not in the tumor, enhances tumor growth and angiogenesis. Oncogene. (2006) 25(4):650–5. doi: 10.1038/sj.onc.1209104
81. Wells JM, Gaggar A, Blalock JE. MMP generated matrikines. Matrix Biol (2015) 44–46:122–9. doi: 10.1016/j.matbio.2015.01.016
82. Gaggar A, Weathington N. Bioactive extracellular matrix fragments in lung health and disease. J Clin Invest. (2016) 126(9):3176–84. doi: 10.1172/JCI83147
83. Bhutto IA, Kim SY, McLeod DS, Merges C, Fukai N, Olsen BR, et al. Localization of collagen XVIII and the endostatin portion of collagen XVIII in aged human control eyes and eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci (2004) 45(5):1544–52. doi: 10.1167/iovs.03-0862
84. Bonet A, Valença A, Mendes-Jorge L, Casellas A, Rodríguez-Baeza A, Nacher V, et al. Decreased endostatin in db/db retinas is associated with optic disc intravitreal vascularization. Exp Eye Res (2021) 212:108801. doi: 10.1016/j.exer.2021.108801
85. Yuan Y, Kong W, Liu XM, Shi GH. A hypoxia-regulated retinal pigment epithelium-specific gene therapy vector reduces choroidal neovascularization in a mouse model. Curr Gene Ther (2022) 22(5):417–26. doi: 10.2174/1566523222666220405135135
86. Yamaguchi N, Anand-Apte B, Lee M, Sasaki T, Fukai N, Shapiro R, et al. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J (1999) 18(16):4414–23. doi: 10.1093/emboj/18.16.4414
87. Skovseth DK, Veuger MJT, Sorensen DR, de Angelis PM, Haraldsen G. Endostatin dramatically inhibits endothelial cell migration, vascular morphogenesis, and perivascular cell recruitment in vivo. Blood. (2005) 105(3):1044–51. doi: 10.1182/blood-2004-03-1164
88. Halfter W, Dong S, Schurer B, Cole GJ. Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem (1998) 273(39):25404–12. doi: 10.1074/jbc.273.39.25404
89. Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, et al. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J (2002) 21(7):1535–44. doi: 10.1093/emboj/21.7.1535
90. Marneros AG, Keene DR, Hansen U, Fukai N, Moulton K, Goletz PL, et al. Collagen XVIII/endostatin is essential for vision and retinal pigment epithelial function. EMBO J (2004) 23(1):89–99. doi: 10.1038/sj.emboj.7600014
91. Liu X, Nie W, Xie Q, Chen G, Li X, Jia Y, et al. Endostatin reverses immunosuppression of the tumor microenvironment in lung carcinoma. Oncol Lett (2018) 15(2):1874–80. doi: 10.3892/ol.2017.7455
92. Mydel P, Shipley JM, Adair-Kirk TL, Kelley DG, Broekelmann TJ, Mecham RP, et al. Neutrophil elastase cleaves laminin-332 (laminin-5) generating peptides that are chemotactic for neutrophils. J Biol Chem (2008) 283(15):9513–22. doi: 10.1074/jbc.M706239200
93. Akthar S, Patel DF, Beale RC, Peiró T, Xu X, Gaggar A, et al. Matrikines are key regulators in modulating the amplitude of lung inflammation in acute pulmonary infection. Nat Commun (2015) 6:8423. doi: 10.1038/ncomms9423
94. Hope C, Emmerich PB, Papadas A, Pagenkopf A, Matkowskyj KA, van de Hey DR, et al. Versican-derived matrikines regulate Batf3-dendritic cell differentiation and promote T cell infiltration in colorectal cancer. J Immunol (2017) 199(5):1933–41. doi: 10.4049/jimmunol.1700529
95. Robison SW, Li J, Viera L, Blackburn JP, Patel RP, Blalock JE, et al. A mechanism for matrikine regulation in acute inflammatory lung injury. JCI Insight (2021) 6(7):e140750. doi: 10.1172/jci.insight.140750
96. Austin BA, Liu B, Li Z, Nussenblatt RB. Biologically active fibronectin fragments stimulate release of MCP-1 and catabolic cytokines from murine retinal pigment epithelium. Invest Opthalmology Visual Science. (2009) 50(6):2896. doi: 10.1167/iovs.08-2495
97. An E, Lu X, Flippin J, Devaney JM, Halligan B, Hoffman EP, et al. Secreted proteome profiling in human RPE cell cultures derived from donors with age related macular degeneration and age matched healthy donors. J Proteome Res (2006) 5(10):2599–610. doi: 10.1021/pr060121j
98. Sivaprasad S, Chong NV, Bailey TA. Serum elastin-derived peptides in age-related macular degeneration. Invest Ophthalmol Vis Sci (2005) 46(9):3046–51. doi: 10.1167/iovs.04-1277
99. Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol (2014) 37:1–14. doi: 10.1016/j.matbio.2014.07.005
100. Leavesley DI, Kashyap AS, Croll T, Sivaramakrishnan M, Shokoohmand A, Hollier BG, et al. Vitronectin–master controller or micromanager? IUBMB Life (2013) 65(10):807–18. doi: 10.1002/iub.1203
101. Preissner KT. Structure and biological role of vitronectin. Annu Rev Cell Biol (1991) 7:275–310. doi: 10.1146/annurev.cb.07.110191.001423
102. Stoop AA, Lupu F, Pannekoek H. Colocalization of thrombin, PAI-1, and vitronectin in the atherosclerotic vessel wall: A potential regulatory mechanism of thrombin activity by PAI-1/vitronectin complexes. Arterioscler Thromb Vasc Biol (2000) 20(4):1143–9. doi: 10.1161/01.ATV.20.4.1143
103. Dufourcq P, Couffinhal T, Alzieu P, Daret D, Moreau C, Duplàa C, et al. Vitronectin is up-regulated after vascular injury and vitronectin blockade prevents neointima formation. Cardiovasc Res (2002) 53(4):952–62. doi: 10.1016/S0008-6363(01)00547-8
104. Singh B, Janardhan KS, Kanthan R. Expression of angiostatin, integrin alphavbeta3, and vitronectin in human lungs in sepsis. Exp Lung Res (2005) 31(8):771–82. doi: 10.1080/01902140500324901
105. Milner R, Crocker SJ, Hung S, Wang X, Frausto RF, del Zoppo GJ. Fibronectin- and vitronectin-induced microglial activation and matrix metalloproteinase-9 expression is mediated by integrins alpha5beta1 and alphavbeta5. J Immunol (2007) 178(12):8158–67. doi: 10.4049/jimmunol.178.12.8158
106. Tsuruta Y, Park YJ, Siegal GP, Liu G, Abraham E. Involvement of vitronectin in lipopolysaccaride-induced acute lung injury. J Immunol (2007) 179(10):7079–86. doi: 10.4049/jimmunol.179.10.7079
107. Martins C de O, Demarchi L, Ferreira FM, Pomerantzeff PMA, Brandao C, Sampaio RO, et al. Rheumatic heart disease and myxomatous degeneration: Differences and similarities of valve damage resulting from autoimmune reactions and matrix disorganization. PLoS One (2017) 12(1):e0170191. doi: 10.1371/journal.pone.0170191
108. Zuchtriegel G, Uhl B, Pick R, Ramsauer M, Dominik J, Mittmann LA, et al. Vitronectin stabilizes intravascular adhesion of neutrophils by coordinating β2 integrin clustering. Haematologica. (2021) 106(10):2641–53. doi: 10.3324/haematol.2019.226241
109. Ho TC, Yeh SI, Chen SL, Tsao YP. Integrin αv and vitronectin prime macrophage-related inflammation and contribute the development of dry eye disease. Int J Mol Sci (2021) 22(16):8410. doi: 10.3390/ijms22168410
110. Podack ER, Müller-Eberhard HJ. Isolation of human s-protein, an inhibitor of the membrane attack complex of complement. J Biol Chem (1979) 254(19):9808–14. doi: 10.1016/S0021-9258(19)83603-7
111. Kanse SM, Matz RL, Preissner KT, Peter K. Promotion of leukocyte adhesion by a novel interaction between vitronectin and the beta2 integrin mac-1 (alphaMbeta2, CD11b/CD18). Arterioscler Thromb Vasc Biol (2004) 24(12):2251–6. doi: 10.1161/01.ATV.0000146529.68729.8b
112. Li R, Ren M, Chen N, Luo M, Zhang Z, Wu J. Vitronectin increases vascular permeability by promoting VE-cadherin internalization at cell junctions. PLoS One (2012) 7(5):e37195. doi: 10.1371/journal.pone.0037195
113. Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun (2010) 2(3):216–27. doi: 10.1159/000284367
114. Bae HB, Tadie JM, Jiang S, Park DW, Bell CP, Thompson LC, et al. Vitronectin inhibits efferocytosis through interactions with apoptotic cells as well as with macrophages. J Immunol (2013) 190(5):2273–81. doi: 10.4049/jimmunol.1200625
115. Keasey MP, Jia C, Pimentel LF, Sante RR, Lovins C, Hagg T. Blood vitronectin is a major activator of LIF and IL-6 in the brain through integrin-FAK and uPAR signaling. J Cell Sci (2018) 131(3):jcs202580. doi: 10.1242/jcs.202580
116. Chakravarty D, Ray AG, Chander V, Mabalirajan U, Mondal PC, Siddiqui KN, et al. Systemic deficiency of vitronectin is associated with aortic inflammation and plaque progression in ApoE-knockout mice. FASEB Bioadv. (2022) 4(2):121–37. doi: 10.1096/fba.2021-00108
117. Anderson DH, Hageman GS, Mullins RF, Neitz M, Neitz J, Ozaki S, et al. Vitronectin gene expression in the adult human retina. Invest Ophthalmol Vis Sci (1999) 40(13):3305–15.
118. Li M, Jia C, Kazmierkiewicz KL, Bowman AS, Tian L, Liu Y, et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum Mol Genet (2014) 23(15):4001–14. doi: 10.1093/hmg/ddu114
119. Wasmuth S, Lueck K, Baehler H, Lommatzsch A, Pauleikhoff D. Increased vitronectin production by complement-stimulated human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci (2009) 50(11):5304–9. doi: 10.1167/iovs.08-3326
120. Lueck K, Wasmuth S, Williams J, Hughes TR, Morgan BP, Lommatzsch A, et al. Sub-Lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye (Lond). (2011) 25(8):1074–82. doi: 10.1038/eye.2011.109
121. Chen M, Xu H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukoc Biol (2015) 98(5):713–25. doi: 10.1189/jlb.3RI0615-239R
122. Hageman GS, Mullins RF, Russell SR, v. JL, Anderson DH. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J (1999) 13(3):477–84. doi: 10.1096/fasebj.13.3.477
123. Wang L, Clark ME, Crossman DK, Kojima K, Messinger JD, Mobley JA, et al. Abundant lipid and protein components of drusen. PLoS One (2010) 5(4):e10329. doi: 10.1371/journal.pone.0010329
124. Biasella F, Plössl K, Karl C, Weber BHF, Friedrich U. Altered protein function caused by AMD-associated variant rs704 links vitronectin to disease pathology. Invest Ophthalmol Vis Sci (2020) 61(14):2. doi: 10.1167/iovs.61.14.2
125. Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. (2018) 558(7708):73–9. doi: 10.1038/s41586-018-0175-2
126. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol (2013) 13(6):438–51. doi: 10.1038/nri3459
127. Marzeda AM, Midwood KS. Internal affairs: Tenascin-c as a clinically relevant, endogenous driver of innate immunity. J Histochem Cytochem (2018) 66(4):289–304. doi: 10.1369/0022155418757443
128. Wiemann S, Reinhard J, Faissner A. Immunomodulatory role of the extracellular matrix protein tenascin-c in neuroinflammation. Biochem Soc Trans (2019) 47(6):1651–60. doi: 10.1042/BST20190081
129. Manrique-Castano D, Dzyubenko E, Borbor M, Vasileiadou P, Kleinschnitz C, Roll L, et al. Tenascin-c preserves microglia surveillance and restricts leukocyte and, more specifically, T cell infiltration of the ischemic brain. Brain Behav Immun (2021) 91:639–48. doi: 10.1016/j.bbi.2020.10.016
130. Bartsch U, Pesheva P, Raff M, Schachner M. Expression of janusin (J1-160/180) in the retina and optic nerve of the developing and adult mouse. Glia. (1993) 9(1):57–69. doi: 10.1002/glia.440090108
131. D’Alessandri L, Ranscht B, Winterhalter KH, Vaughan L. Contactin/F11 and tenascin-c co-expression in the chick retina correlates with formation of the synaptic plexiform layers. Curr Eye Res (1995) 14(10):911–26. doi: 10.3109/02713689508995131
132. Sánchez-López A, Cuadros MA, Calvente R, Tassi M, Marín-Teva JL, Navascués J. Radial migration of developing microglial cells in quail retina: a confocal microscopy study. Glia. (2004) 46(3):261–73. doi: 10.1002/glia.20007
133. Stein JD, Khawaja AP, Weizer JS. Glaucoma in adults-screening, diagnosis, and management: A review. JAMA. (2021) 325(2):164–74. doi: 10.1001/jama.2020.21899
134. Reinhard J, Roll L, Faissner A. Tenascins in retinal and optic nerve neurodegeneration. Front Integr Neurosci (2017) 11:30. doi: 10.3389/fnint.2017.00030
135. Wax MB. The case for autoimmunity in glaucoma. Exp Eye Res (2011) 93(2):187–90. doi: 10.1016/j.exer.2010.08.016
136. Geyer O, Levo Y. Glaucoma is an autoimmune disease. Autoimmun Rev (2020) 19(6):102535. doi: 10.1016/j.autrev.2020.102535
137. Wiemann S, Reinhard J, Reinehr S, Cibir Z, Joachim SC, Faissner A. Loss of the extracellular matrix molecule tenascin-c leads to absence of reactive gliosis and promotes anti-inflammatory cytokine expression in an autoimmune glaucoma mouse model. Front Immunol (2020) 11:566279. doi: 10.3389/fimmu.2020.566279
138. Pena JD, Varela HJ, Ricard CS, Hernandez MR. Enhanced tenascin expression associated with reactive astrocytes in human optic nerve heads with primary open angle glaucoma. Exp Eye Res (1999) 68(1):29–40. doi: 10.1006/exer.1998.0577
139. Johnson EC, Jia L, Cepurna WO, Doser TA, Morrison JC. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci (2007) 48(7):3161–77. doi: 10.1167/iovs.06-1282
140. Reinehr S, Reinhard J, Wiemann S, Stute G, Kuehn S, Woestmann J, et al. Early remodelling of the extracellular matrix proteins tenascin-c and phosphacan in retina and optic nerve of an experimental autoimmune glaucoma model. J Cell Mol Med (2016) 20(11):2122–37. doi: 10.1111/jcmm.12909
141. Reinhard J, Wiemann S, Hildebrandt S, Faissner A. Extracellular matrix remodeling in the retina and optic nerve of a novel glaucoma mouse model. Biol (Basel). (2021) 10(3):169. doi: 10.3390/biology10030169
142. Wiemann S, Yousf A, Joachim SC, Peters C, Mueller-Buehl AM, Wagner N, et al. Knock-out of tenascin-c ameliorates ischemia-induced rod-photoreceptor degeneration and retinal dysfunction. Front Neurosci (2021) 15:642176. doi: 10.3389/fnins.2021.642176
143. Kubo Y, Ishikawa K, Mori K, Kobayashi Y, Nakama T, Arima M, et al. Periostin and tenascin-c interaction promotes angiogenesis in ischemic proliferative retinopathy. Sci Rep (2020) 10(1):9299. doi: 10.1038/s41598-020-66278-1
144. Mitamura Y, Takeuchi S, Ohtsuka K, Matsuda A, Hiraiwa N, Kusakabe M. Tenascin-c levels in the vitreous of patients with proliferative diabetic retinopathy. Diabetes Care United States; (2002) 25(10):1899. doi: 10.2337/diacare.25.10.1899
145. Castellon R, Caballero S, Hamdi HK, Atilano SR, Aoki AM, Tarnuzzer RW, et al. Effects of tenascin-c on normal and diabetic retinal endothelial cells in culture. Invest Ophthalmol Vis Sci (2002) 43(8):2758–66.
146. Palenski TL, Sorenson CM, Sheibani N. Inflammatory cytokine-specific alterations in retinal endothelial cell function. Microvasc Res (2013), 89:57–69. doi: 10.1016/j.mvr.2013.06.007
147. Kobayashi Y, Yoshida S, Zhou Y, Nakama T, Ishikawa K, Arima M, et al. Tenascin-c promotes angiogenesis in fibrovascular membranes in eyes with proliferative diabetic retinopathy. Mol Vis (2016) 22:436–45.
148. Antonetti DA, Silva PS, Stitt AW. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol (2021) 17(4):195–206. doi: 10.1038/s41574-020-00451-4
149. Nicolò M, Piccolino FC, Zardi L, Giovannini A, Mariotti C. Detection of tenascin-c in surgically excised choroidal neovascular membranes. Graefes Arch Clin Exp Ophthalmol (2000) 238(2):107–11. doi: 10.1007/PL00021257
150. Fasler-Kan E, Wunderlich K, Hildebrand P, Flammer J, Meyer P. Activated STAT 3 in choroidal neovascular membranes of patients with age-related macular degeneration. Ophthalmologica. (2005) 219(4):214–21. doi: 10.1159/000085730
151. Kobayashi Y, Yoshida S, Zhou Y, Nakama T, Ishikawa K, Kubo Y, et al. Tenascin-c secreted by transdifferentiated retinal pigment epithelial cells promotes choroidal neovascularization via integrin αV. Lab Invest. (2016) 96(11):1178–88. doi: 10.1038/labinvest.2016.99
152. Afshari FT, Kwok JC, Andrews MR, Blits B, Martin KR, Faissner A, et al. Integrin activation or alpha 9 expression allows retinal pigmented epithelial cell adhesion on bruch’s membrane in wet age-related macular degeneration. Brain. (2010) 133(Pt 2):448–64. doi: 10.1093/brain/awp319
153. Moreau JF, Pradeu T, Grignolio A, Nardini C, Castiglione F, Tieri P, et al. The emerging role of ECM crosslinking in T cell mobility as a hallmark of immunosenescence in humans. Ageing Res Rev (2017) 35:322–35. doi: 10.1016/j.arr.2016.11.005
154. Abaricia JO, Shah AH, Olivares-Navarrete R. Substrate stiffness induces neutrophil extracellular trap (NET) formation through focal adhesion kinase activation. Biomaterials. (2021) 271:120715. doi: 10.1016/j.biomaterials.2021.120715
155. Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J BioMed Mater Res A. (2012) 100(6):1375–86. doi: 10.1002/jbm.a.34104
156. Friedemann M, Kalbitzer L, Franz S, Moeller S, Schnabelrauch M, Simon JC, et al. Instructing human macrophage polarization by stiffness and glycosaminoglycan functionalization in 3D collagen networks. Adv Healthc Mater (2017) 6(7). doi: 10.1002/adhm.201600967
157. Gruber E, Heyward C, Cameron J, Leifer C. Toll-like receptor signaling in macrophages is regulated by extracellular substrate stiffness and rho-associated coiled-coil kinase (ROCK1/2). Int Immunol (2018) 30(6):267–78. doi: 10.1093/intimm/dxy027
158. Sridharan R, Cavanagh B, Cameron AR, Kelly DJ, O’Brien FJ. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. (2019) 89:47–59. doi: 10.1016/j.actbio.2019.02.048
159. Hsieh JY, Keating MT, Smith TD, Meli VS, Botvinick EL, Liu WF. Matrix crosslinking enhances macrophage adhesion, migration, and inflammatory activation. APL Bioeng. (2019) 3(1):16103. doi: 10.1063/1.5067301
160. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol (2013) 229(2):176–85. doi: 10.1002/path.4133
161. Wong SW, Lenzini S, Cooper MH, Mooney DJ, Shin JW. Soft extracellular matrix enhances inflammatory activation of mesenchymal stromal cells to induce monocyte production and trafficking. Sci Adv (2020) 6(15):eaaw0158. doi: 10.1126/sciadv.aaw0158.
162. Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, et al. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med (2011) 3(112):112ra122. doi: 10.1126/scitranslmed.3002761
163. Chen W, Tian B, Liang J, Yu S, Zhou Y, Li S. Matrix stiffness regulates the interactions between endothelial cells and monocytes. Biomaterials. (2019) 221:119362. doi: 10.1016/j.biomaterials.2019.119362
164. Fields MA, Bowrey HE, Gong J, Moreira EF, Cai H, del Priore L v. Extracellular matrix nitration alters growth factor release and activates bioactive complement in human retinal pigment epithelial cells. PLoS One (2017) 12(5):e0177763. doi: 10.1371/journal.pone.0177763
165. Gong J, Cai H, Noggle S, Paull D, Rizzolo LJ, del Priore L v, et al. Stem cell-derived retinal pigment epithelium from patients with age-related macular degeneration exhibit reduced metabolism and matrix interactions. Stem Cells Transl Med (2020) 9(3):364–76. doi: 10.1002/sctm.19-0321
166. Keenan TDL, Pickford CE, Holley RJ, Clark SJ, Lin W, Dowsey AW, et al. Age-dependent changes in heparan sulfate in human bruch’s membrane: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci (2014) 55(8):5370–9. doi: 10.1167/iovs.14-14126
167. Kumar A, El-Osta A, Hussain AA, Marshall J. Increased sequestration of matrix metalloproteinases in ageing human bruch’s membrane: Implications for ECM turnover. Invest Ophthalmol Vis Sci (2010) 51(5):2664–70. doi: 10.1167/iovs.09-4195
168. Biasella F, Strunz T, Kiel C, Weber BHF, Friedrich U. Vitronectin and its interaction with PAI-1 suggests a functional link to vascular changes in AMD pathobiology. Cells. (2022) 11(11):1766. doi: 10.3390/cells11111766
169. Hejtmancik JF, Daiger SP. Understanding the genetic architecture of human retinal degenerations. Proc Natl Acad Sci (2020) 117(8):3904–6. doi: 10.1073/pnas.1922925117
170. Resnikoff S, Pascolini D, Etya’ale D, Kocur I, Pararajasegaram R, Pokharel G, et al. Global data on visual impairment in the year 2002. Bull World Health Organ. (2004) 82(11):844–51.
171. Pascolini D, Mariotti SP. Global estimates of visual impairment: 2010. Br J Ophthalmology. (2012) 96(5):614–8. doi: 10.1136/bjophthalmol-2011-300539
172. Stone EM, Lotery AJ, Munier FL, Héon E, Piguet B, Guymer RH, et al. A single EFEMP1 mutation associated with both malattia leventinese and doyne honeycomb retinal dystrophy. Nat Genet (1999) 22(2):199–202. doi: 10.1038/9722
173. Roybal CN, Marmorstein LY, Vander Jagt DL, Abcouwer SF. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest Ophthalmol Vis Sci (2005) 46(11):3973–9. doi: 10.1167/iovs.05-0070
174. Hulleman JD, Kaushal S, Balch WE, Kelly JW. Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol Biol Cell (2011) 22(24):4765–75. doi: 10.1091/mbc.e11-08-0695
175. Garland DL, Fernandez-Godino R, Kaur I, Speicher KD, Harnly JM, Lambris JD, et al. Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Hum Mol Genet (2014) 23(1):52–68. doi: 10.1093/hmg/ddt395
176. Sohn EH, Wang K, Thompson S, Riker MJ, Hoffmann JM, Stone EM, et al. Comparison of drusen and modifying genes in autosomal dominant radial drusen and age-related macular degeneration. Retina. (2015) 35(1):48–57. doi: 10.1097/IAE.0000000000000263
177. Crowley MA, Garland DL, Sellner H, Banks A, Fan L, Rejtar T, et al. Complement factor B is critical for sub-RPE deposit accumulation in a model of Doyne honeycomb retinal dystrophy with features of age-related macular degeneration. Hum Mol Genet (2023) 32(2):204–17. doi: 10.1093/hmg/ddac187
178. Stanton JB, Marmorstein AD, Zhang Y, Marmorstein LY. Deletion of Efemp1 is protective against the development of sub-rpe deposits in mouse eyes. Invest Ophthalmol Vis Sci (2017) 58(3):1455–61. doi: 10.1167/iovs.16-20955
179. Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E, et al. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet (2007) 16(20):2411–22. doi: 10.1093/hmg/ddm198
180. Garland DL, Pierce EA, Fernandez-Godino R. Complement C5 is not critical for the formation of sub-RPE deposits in Efemp1 mutant mice. Sci Rep [Internet]. (2021) 11(1):1–11. doi: 10.1038/s41598-021-89978-8
181. Tan PL, Bowes Rickman C, Katsanis N. AMD And the alternative complement pathway: genetics and functional implications. Hum Genomics (2016) 10(1):23. doi: 10.1186/s40246-016-0079-x
182. Thakkinstian A, McEvoy M, Chakravarthy U, Chakrabarti S, McKay GJ, Ryu E, et al. The association between complement component 2/complement factor b polymorphisms and age-related macular degeneration: a HuGE review and meta-analysis. Am J Epidemiol. (2012) 176(5):361–72. doi: 10.1093/aje/kws031
183. Galloway CA, Dalvi S, Hung SSC, MacDonald LA, Latchney LR, Wong RCB, et al. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc Natl Acad Sci U S A. (2017) 114(39):E8214–23. doi: 10.1073/pnas.1710430114
184. Anand-Apte B, Chao JR, Singh R, Stöhr H. Sorsby fundus dystrophy: Insights from the past and looking to the future. J Neurosci Res (2019) 97(1):88–97. doi: 10.1002/jnr.24317
185. Christensen DRG, Brown FE, Cree AJ, Ratnayaka JA, Lotery AJ. Sorsby fundus dystrophy – a review of pathology and disease mechanisms. Exp Eye Res (2017) 165:35–46. doi: 10.1016/j.exer.2017.08.014
186. Arris CE, Bevitt DJ, Mohamed J, Li Z, Langton KP, Barker MD, et al. Expression of mutant and wild-type TIMP3 in primary gingival fibroblasts from sorsby’s fundus dystrophy patients. Biochim Biophys Acta (2003) 1638(1):20–8. doi: 10.1016/S0925-4439(03)00036-X
187. Langton KP, McKie N, Smith BM, Brown NJ, Barker MD. Sorsby’s fundus dystrophy mutations impair turnover of TIMP-3 by retinal pigment epithelial cells†. Hum Mol Genet [Internet]. (2005) 14(23):3579–86. doi: 10.1093/hmg/ddi385
188. Langton KP, McKie N, Curtis A, Goodship JA, Bond PM, Barker MD, et al. A novel tissue inhibitor of metalloproteinases-3 mutation reveals a common molecular phenotype in sorsby’s fundus dystrophy. J Biol Chem (2000) 275(35):27027–31. doi: 10.1016/S0021-9258(19)61475-4
189. Weber BHF, Lin B, White K, Kohler K, Soboleva G, Herterich S, et al. A mouse model for sorsby fundus dystrophy. Invest Ophthalmol Vis Sci (2002) 43(8):2732–40.
190. Terman A, Brunk UT. Lipofuscin. Int J Biochem Cell Biol (2004) 36(8):1400–4. doi: 10.1016/j.biocel.2003.08.009
191. Anderson OA, Finkelstein A, Shima DT. A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS One (2013) 8(6):e67263. doi: 10.1371/journal.pone.0067263
192. Radu RA, Hu J, Yuan Q, Welch DL, Makshanoff J, Lloyd M, et al. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for stargardt macular degeneration. J Biol Chem (2011) 286(21):18593–601. doi: 10.1074/jbc.M110.191866
193. Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci (2006) 103(44):16182–7. doi: 10.1073/pnas.0604255103
194. Lee MH, Verma V, Maskos K, Becherer JD, Knäuper V, Dodds P, et al. The c-terminal domains of TACE weaken the inhibitory action of n-TIMP-3. FEBS Lett (2002) 520(1–3):102–6. doi: 10.1016/S0014-5793(02)02776-X
195. Dewing JM, Carare RO, Lotery AJ, Ratnayaka JA. The diverse roles of TIMP-3: Insights into degenerative diseases of the senescent retina and brain. Cells. (2019) 9(1):39. doi: 10.3390/cells9010039
196. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J (2000) 14(7):835–46. doi: 10.1096/fasebj.14.7.835
197. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res (2001) 73(6):887–96. doi: 10.1006/exer.2001.1094
198. Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res (2000) 70(4):441–9. doi: 10.1006/exer.1999.0798
199. de Jong S, Gagliardi G, Garanto A, de Breuk A, Lechanteur YTE, Katti S, et al. Implications of genetic variation in the complement system in age-related macular degeneration. Prog Retin Eye Res (2021) 84:100952. doi: 10.1016/j.preteyeres.2021.100952
200. Armento A, Ueffing M, Clark SJ. The complement system in age-related macular degeneration. Cell Mol Life Sci (2021) 78(10):4487–505. doi: 10.1007/s00018-021-03796-9
201. Wang Z, Paik DC, del Priore L v., Burch RL III Scholar, Gaillard ER. Nitrite-modified extracellular matrix proteins deleteriously affect retinal pigment epithelial cell function and viability: A comparison study with nonenzymatic glycation mechanisms. Curr Eye Res (2005) 30(8):691–702. doi: 10.1080/02713680590968259
202. Fields MA, Cai H, Bowrey HE, Moreira EF, Beck Gooz M, Kunchithapautham K, et al. Nitrite modification of extracellular matrix alters CD46 expression and VEGF release in human retinal pigment epithelium. Invest Ophthalmol Vis Sci (2015) 56(8):4231–8. doi: 10.1167/iovs.15-16438
203. Lee J, Cooke JP. Nicotine and pathological angiogenesis. Life Sci (2012) 91(21–22):1058–64. doi: 10.1016/j.lfs.2012.06.032
204. Vogt SD, Curcio CA, Wang L, Li CM, McGwin GJ, Medeiros NE, et al. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp Eye Res (2011) 93(4):413–23. doi: 10.1016/j.exer.2011.06.002
205. Lyzogubov V, Wu X, Jha P, Tytarenko R, Triebwasser M, Kolar G, et al. Complement regulatory protein CD46 protects against choroidal neovascularization in mice. Am J Pathol (2014) 184(9):2537–48. doi: 10.1016/j.ajpath.2014.06.001
206. Chinchilla B, Foltopoulou P, Fernandez-Godino R. Tick-over-mediated complement activation is sufficient to cause basal deposit formation in cell-based models of macular degeneration. J Pathology. (2021) 255(2):120–31. doi: 10.1002/path.5747
207. Lin MK, Yang J, Hsu CW, Gore A, Bassuk AG, Brown LM, et al. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell (2018) 17(4):e12710. doi: 10.1111/acel.12710
208. Anderson DH, Mullins RF, Hageman GS, Johnson L v. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol (2002) 134(3):411–31. doi: 10.1016/S0002-9394(02)01624-0
209. Graue-Hernandez EO, Rivera-De-La-Parra D, Hernandez-Jimenez S, Aguilar-Salinas CA, Kershenobich-Stalnikowitz D, Jimenez-Corona A. Prevalence and associated risk factors of diabetic retinopathy and macular oedema in patients recently diagnosed with type 2 diabetes. BMJ Open Ophthalmol (2020) 5(1):e000304. doi: 10.1136/bmjophth-2019-000304
210. Liu Y, Wang Y, Qin S, Jin X, Jin L, Gu W, et al. Insights into genome-wide association study for diabetes: A bibliometric and visual analysis from 2001 to 2021. Front Endocrinol (Lausanne). (2022) 13. doi: 10.3389/fendo.2022.817620
211. Tonade D, Kern TS. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog Retin Eye Res (2021) 83:100919. doi: 10.1016/j.preteyeres.2020.100919
212. Gomułka K, Ruta M. The role of inflammation and therapeutic concepts in diabetic retinopathy–a short review. Int J Mol Sci (2023) 24(2):1024. doi: 10.3390/ijms24021024
213. Resnikoff HA, Miller CG, Schwarzbauer JE. Implications of fibrotic extracellular matrix in diabetic retinopathy. Exp Biol Med (2022) 247(13):1093–102. doi: 10.1177/15353702221087175
214. Coughlin BA, Feenstra DJ, Mohr S. Müller cells and diabetic retinopathy. Vision Res (2017) 139:93–100. doi: 10.1016/j.visres.2017.03.013
215. Sagmeister S, Merl-Pham J, Petrera A, Deeg CA, Hauck SM. High glucose treatment promotes extracellular matrix proteome remodeling in möller glial cells. PeerJ. (2021) 9:1–23. doi: 10.7717/peerj.11316
216. Giblin MJ, Ontko CD, Penn JS. Effect of cytokine-induced alterations in extracellular matrix composition on diabetic retinopathy-relevant endothelial cell behaviors. Sci Rep (2022) 12(1):12955. doi: 10.1038/s41598-022-12683-7
217. Klenotic PA, Munier FL, Marmorstein LY, Anand-Apte B. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1): Implications for macular degenerations. J Biol Chem (2004) 279(29):30469–73. doi: 10.1074/jbc.M403026200
Keywords: extracellular matrix, Bruch’s membrane, matricellular proteins, inflammation, retinal disease, age-related macular degeneration, Sorsby fundus dystrophy, TIMP3
Citation: Biasella F, Plössl K, Baird PN and Weber BHF (2023) The extracellular microenvironment in immune dysregulation and inflammation in retinal disorders. Front. Immunol. 14:1147037. doi: 10.3389/fimmu.2023.1147037
Received: 18 January 2023; Accepted: 15 February 2023;
Published: 01 March 2023.
Edited by:
Bikash Pattnaik, University of Wisconsin-Madison, United StatesReviewed by:
Antonio Inforzato, Humanitas University, ItalyCopyright © 2023 Biasella, Plössl, Baird and Weber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul N. Baird, cGJhaXJkQHVuaW1lbGIuZWR1LmF1; Bernhard H. F. Weber, YndlYkBrbGluaWsudW5pLXJlZ2Vuc2J1cmcuZGU=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.