 Fang Xiao
Fang Xiao Jixu Guo
Jixu Guo Stephen Tomlinson
Stephen Tomlinson Guandou Yuan
Guandou Yuan Songqing He
Songqing He- 1Division of Hepatobiliary Surgery, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
- 2Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, United States
- 3Key Laboratory of Early Prevention and Treatment for Regional High Frequency Tumor (Guangxi Medical University), Ministry of Education, Nanning, China
- 4Guangxi Key Laboratory of Immunology and Metabolism for Liver Diseases, Nanning, Guangxi, China
The complement system is one of the immune system’s oldest defense mechanisms and is historically regarded as a liver-derived and serum-active innate immune system that ‘complements’ cell-mediated and antibody-mediated immune responses against pathogens. However, the complement system is now recognized as a central component of both innate and adaptive immunity at both the systemic and local tissue levels. More findings have uncovered novel activities of an intracellularly active complement system—the complosome—that have shifted established functional paradigms in the field. The complosome has been shown to play a critical function in regulating T cell responses, cell physiology (such as metabolism), inflammatory disease processes, and cancer, which has amply proved its immense research potential and informed us that there is still much to learn about this system. Here, we summarize current understanding and discuss the emerging roles of the complosome in health and disease.
1 Introduction
The complement system is composed of more than 50 plasma and cell-bound components, and comprises a major arm of the immune system. From its identification over a hundred years ago, our understanding of the complement has completely changed from what was thought to be a blood-borne antimicrobial system to a global and local regulator of immunity and tissue homeostasis (1, 2). A long-held belief was that complement proteins are synthesized by various cell types, principally hepatocytes, and were operative only in the extracellular milieu. However, the discovery of functionally active complement proteins, such as C3 and C5, in the intracellular compartment of T cells initiated a new chapter for complement research (3–6). This intracellular complement system termed the ‘complosome’ has since been identified in a variety of cell populations, including T cells, monocytes, macrophages, neutrophils, tumor cells, epithelial cells, and other immune and non-immune cells. There is increasing recognition that the complosome contributes significantly to normal cell and organ development, homeostasis, and tissue repair (2, 7–9). The intracellular complement system also engages in crosstalk with intracellular innate sensor systems, such as the Pyrin domains-containing protein 3 (NLRP3) inflammasome in cell metabolic pathways (5, 10, 11). Our growing understanding of the complement system, particularly the complosome, in relation to health and disease is opening up new research avenues and potential therapeutic strategies.
2 Conventional complement cascades
Research into the complement system has undergone something of a renaissance over the past 15 years, principally due to the recognition that it plays a key role in many disease processes and, with it, the promise of anti-complement therapies. Complement proteins, in general, exist in inactive forms in body fluid, and are activated to produce proinflammatory mediators, opsonins, and cytolytic membrane attack complexes (MAC). Activation of complement normally occurs via one of three pathways (Figure 1): The classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP) (12, 13). All three pathways converge with the cleavage and activation of C3, leading to the generation of C3 and C5 convertase enzymes that amplify the activation cascade. Activation of the CP usually occurs following the binding of the C1 complex to Fc regions of complement-fixing antibodies, leading to cleavage of C2 and C4, and resulting in the formation of the CP C3 convertase (C4b2a) (14). The LP follows a similar activation mechanism, except that it employs a mannose-binding lectin/carbohydrate recognition system with different C2 and C4 cleaving enzymes (15). In the AP, soluble C3 is spontaneously hydrolyzed through a “C3 tick over” mechanism to form C3b(H2O), which results in exposure of a site that recognizes factor B and leads to the generation of the AP C3 convertase, C3bBb (16). However, it is initially activated, and the next step in the cascade occurs when either the CP or AP C3 convertase cleaves C3 into C3a and C3b. The larger C3b fragment can associate with additional C3 convertases to form CP and AP C5 convertases (C4b2a3b/C3bBb3b). These C5 convertases cleave C5 into C5a and C5b. The larger C5b fragment associates with C6 to form C5b6 which associates with cell membranes, followed by the sequential recruitment of C7, C8 and multiple C9’s to form the membrane penetrating and cytolytic MAC (17). There are, by necessity, several mechanisms in place to prevent uncontrolled activation of complement. In addition to the rate-limiting effect of the C3 and C5 convertase, there are also soluble regulatory proteins (i.e., C1INH, C4BP, and FH) and membrane regulatory proteins (i.e., CR1, MCP/CD46, and DAF/CD55) that regulate the activation of complement cascades (18). The intermediate products, C3a and C5a, which are anaphylatoxins, bind to corresponding transmembrane receptors and play an inflammatory role. As for C3b, it also can induce phagocytosis of opsonized targets as well as serves to initiate the amplification loop in the AP. The conventional complement regulatory cascades are shown in Figure 1. Activation of intracellular complement, the complosome, and its interaction with extracellular complement systems have received extensive focus since its discovery (3). The complosome not only affects how we interpret traditional complement cascades, but it also has the capacity to reconstruct the framework of complement cascades (19). Here, we aim to summarize current understanding and discuss the emerging roles of the complosome in health and disease.
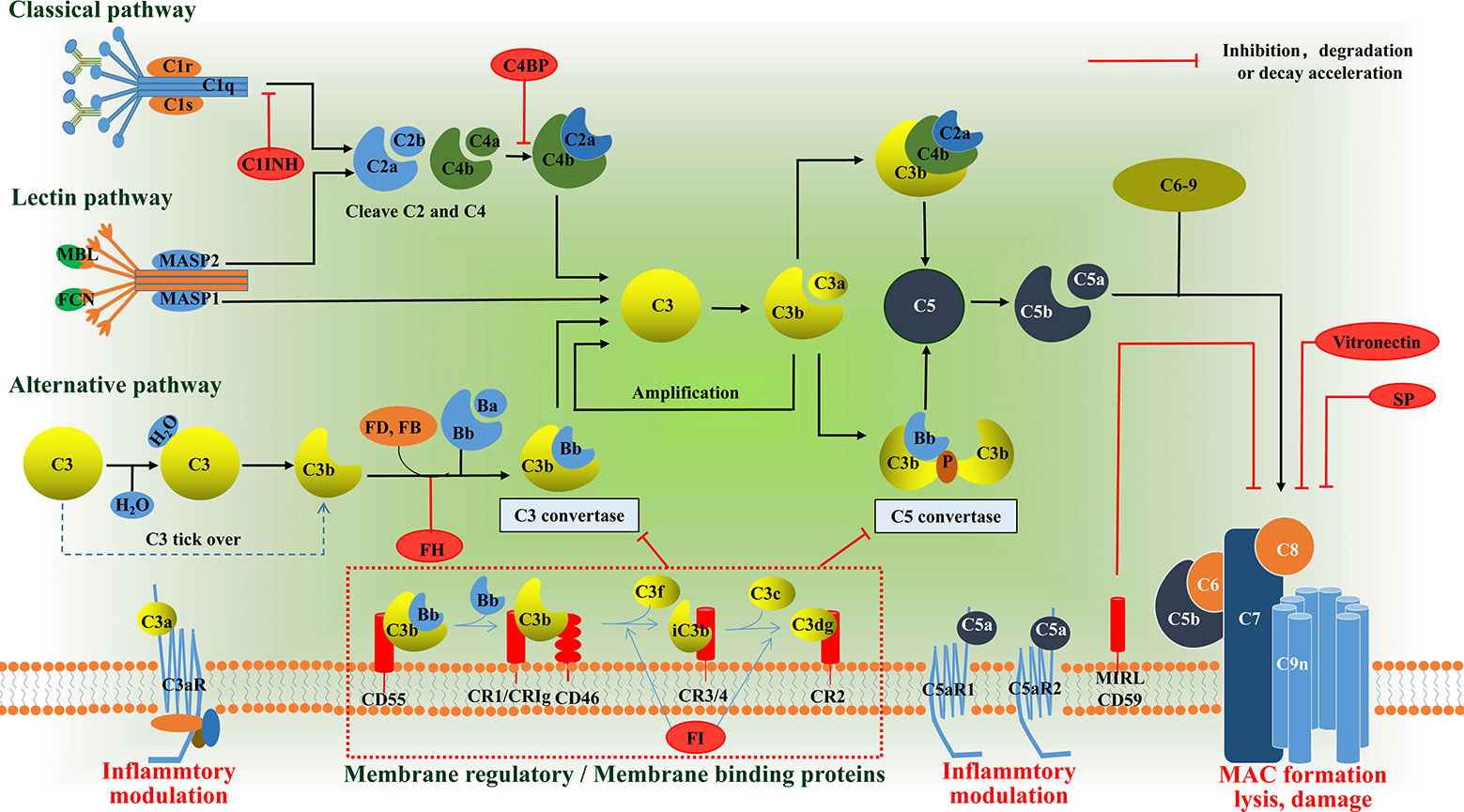
Figure 1 Schematic diagram of the complement system. The complement system can be activated via the classical pathway (CP), the lectin pathway (LP), or the alternative pathway (AP). These three pathways have their own pattern in forming C3 convertase: C4b2a or C3bBb. The C3a binds its anaphylatoxin receptor the C3aR, while C3b can induce phagocytosis of opsonized targets and its further degradation products, iC3b, C3c and C3d/C3dg are able to bind various complement receptors. C3b can also bind to the former C3 convertase, which then results in formation of the C5 convertases: C4bC2aC3b or C3bBbC3b. The C5 convertases cleaves C5 into the C5a and C5b. The C5a binds its anaphylatoxin receptor the C5aR1 and C5aR2, whereas C5b, C6, C7, C8 and up to 16 molecules of C9 bind together to form the MAC, which forms a channel on the surface of targeted cells and causes cells to swell and lysis. In addition to the rate-limiting effect of the C3 and C5 convertase, there are also soluble regulatory proteins (i.e., C1INH, C4BP, and FH) and membrane regulatory proteins (i.e., CR1, CD46, and CD55) that regulate the activation of complement cascades. The anaphylatoxins C3a and C5a mediate inflammatory mudulation through binding to their receptors, respectively. MBL, mannose binding lectin; MASP, MBL associated serine protease; FD, factor D; FB, factor B; FH, factor H; C1INH, C1 inhibitor; FI, factor I; C4BP, C4b-binding protein; CR, complement receptor; MAC, membrane attack complex, SP, S protein.
3 The history of the complosome
In 2013, a study on complement subverted our perception of complement activation and showed that complement is not confined to the extracellular space but can also occur intracellularly. This landmark study discovered that the intracellular reserves of C3, C3aR (located on lysosomes), and cathepsin L (CTSL) are present in human CD4+ T cells and are essential for maintaining T cell homeostasis and mediating effector differentiation. Furthermore, the intracellular C3 stores and ‘Tonic’ intracellular C3a generation have also been shown to occur in monocytes, neutrophils, CD8+ T cells, B cells, epithelial cells, endothelial cells, and fibroblasts, implying that intracellular complement activation might be of broad physiological significance (3). Inspired by this exciting discovery, the novel concept of ‘complosome’ was first introduced in 2014 by Claudia Kemper’s team, which was somewhat analogous to the inflammasome (19). Then other studies have shown that another important complement component, C5, also exists intracellularly in T cells and plays a role in regulating Th1 response, reactive oxygen species (ROS) generation, and NLRP3 inflammasome activation (5). Another important study demonstrated the movement of a portion of the intracellular C3 (also known as C3(H2O)) pools from the extracellular milieu (serum/blood) into T and B cells (20). The hydrolyzed form C3(H2O) was quickly taken up and mostly returned to the extracellular environment within 24 hours, and this mechanism was dubbed the recycling pathway for extracellular-derived C3 (20). In 2017, another complement component, Factor H (FH), has been found to be internalized (bound to nucleosomes) by apoptotic cells (Jurkat T cells), where it directly binds to CTSL rather than being degraded. The FH functions intracellularly as a cofactor for the CTSL-mediated cleavage of C3 (21). Emerging evidence supports that a variety of complement components, receptors, and regulators appear to be present inside T cells and other cells, just like the inflammasome, therefrom, the ‘complosome’ has ignited excitement in the field. An entire session at the XXVI International Complement Workshop in 2016 was dedicated to the complosome. Despite the robust evidence that has been accumulated to date, the veracity and relevance of the complosome are still being questioned. Even though there is controversy surrounding the complosome, scientists in the field still believe that this new area of research will remain mainstream for the near future, as it would help to understand the crosstalk between intracellular complement proteins and inflammasome molecules, as well as other intracellular proteins underlying in health and disease (22–24).
4 Complosome in immune cells
4.1 T cells
To date, Claudia Kemper’s team has taken the lead in conducting most of the research on the complosome and have mostly described it in human T cells, highlighting its connection to inflammasome activation and immunometabolism (Figure 2A). As mentioned above, both intracellular C3 and C5 system, together with their interaction with the inflammasome, are indeed required for effective Th1 immunity (3, 5). The complosome is required to induce metabolic reprogramming of immune cells, including increased glycolytic flux and oxidative phosphorylation (OXPHOS), which promote the secretion of the proinflammatory cytokine IFN-γ (25). In resting T cells, CTSL constantly cleaves intracellular C3 into bioactive C3a and C3b, and the intracellular C3a/C3aR engagement maintain T cell survival via low-level mTOR induction (3). Upon toll like receptor activation, the entire intracellular C3 system translocates rapidly to the cell surface, where C3a and C3b signal in an autocrine manner via their receptors, C3aR and CD46, respectively, to induce Th1 immunity and IFN-γ production. Moreover, during T cell activation, CD46-mediated signals permit nutrient influx via expression of the large neutral amino acid transporter 1 (LAT1) and glucose transporter 1 (GLUT1). Autocrine activation of CD46 simultaneously drives induction of late endosomal or lysosomal adaptor and MAPK and mTOR activator 5 (LAMTOR5), which facilitates the assembly of the Amino Acid-sensing Ragulator-Rag-mTORC1 complex and promotes OXPHOS and glycolysis, two processes necessary for IFN-γ production and Th1 lineage commitment (4). This demonstrates a crucial connection between the complement system and immune metabolism guiding human CD4+ T cell effector function. Similarly, upon TCR and CD46 co-stimulation, the intracellular C5a interacts with the mitochondrially expressed C5aR1, resulting in mitochondrial ROS production, intrinsic NLRP3 inflammasome assembly and activation, maintenance of IFN-γ secretion, and subsequent mature IL-1β secretion during T cell migration into inflamed tissues. Additionally, CD4+ T cells express C5aR2 on their surface and intracellularly, and both secreted C5a and the desarginated version of C5a (C5a-desArg) interact with C5aR2 in an autocrine manner. C5aR2 negatively regulates the C5aR1-driven NLRP3 inflammasome activity, which results in a reduction in IFN-γ production, a switch to IL-10 secretion, and a suppression of Th1 responses (5). Moreover, the complosome also participates in the contraction phase of T cell responses. Following effective Th1 induction, CD46-mediated signals interacts with the interleukin (IL)-2 receptor, reduces glycolysis and OXPHOS, promotes IL-10 production in these cells, which induces them to enter a self-regulative contraction phase (8). Moreover, human cytotoxic CD8+ T cells (CTLs) also harbor a complosome system. The TCR and autocrine CD46 costimulation drives IFN-γ production, nutrient influx, and cytotoxic activity in these cells. Interestingly, in CTLs, aside from a strong OXPHOS induction, CD46 is also a potent inducer of fatty acid synthesis. Although CD8+ T cells express NLRP3, the inflammasome is not required for normal IFN-γ secretion or cytotoxicity in CTLs (26, 27). The activated CD8+T cell can also take up C1q, another complement protein, and restrains the response to self-antigens by modulating the mitochondrial metabolism via gC1qR and a yet unknown mechanism (28). Given what we presently know about the complosome in T cells, it is possible that the complosome also plays a role in the development of T cell memory and/or tissue residency, these subjects are being explored currently (29).
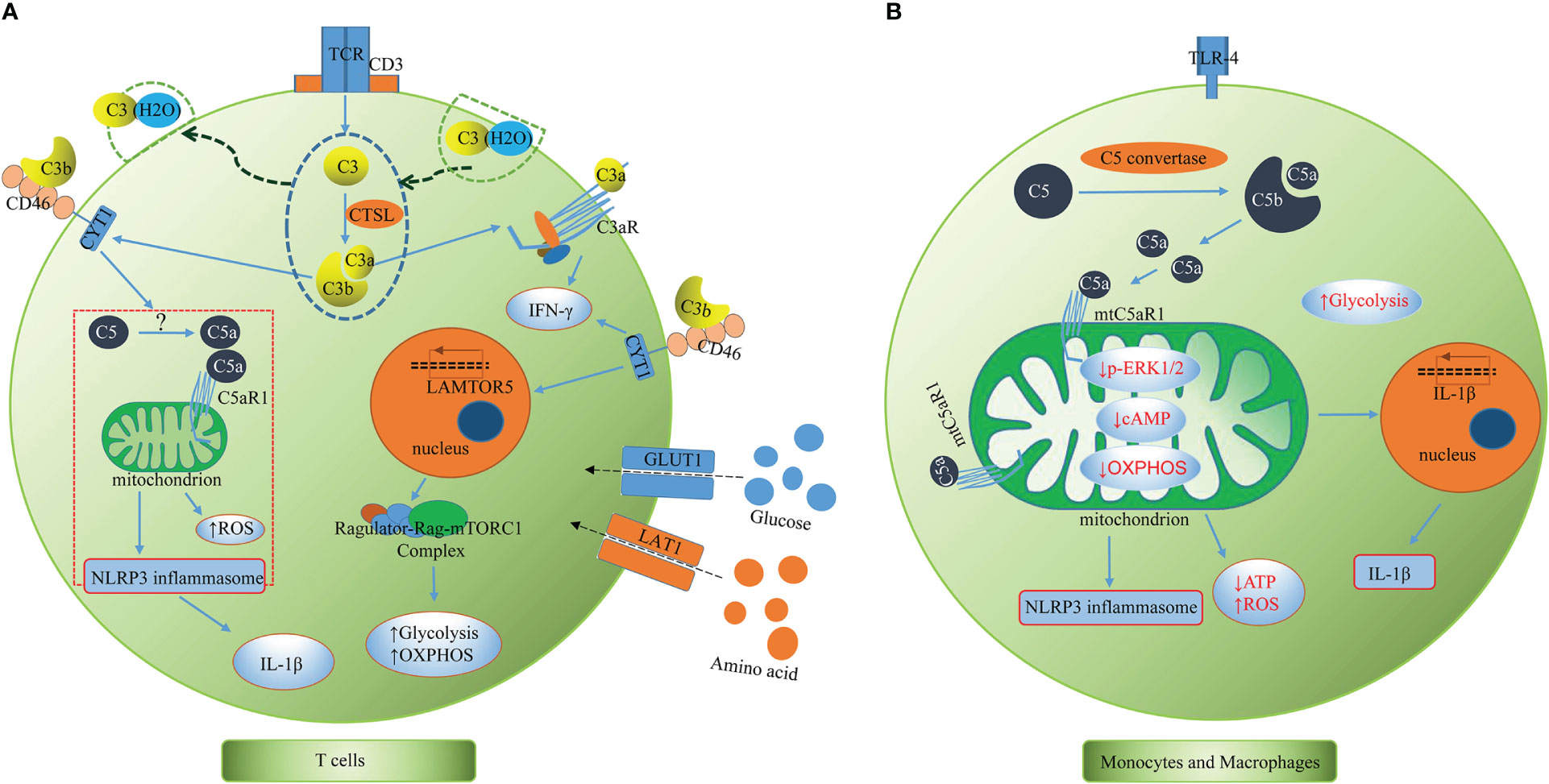
Figure 2 The complosome in T cells and monocytes and macrophages. (A) T cells. T cells have intracellular store of C3 (partly taken up from serum as C3(H2O)), C5 and CTSL. CTSL continuously cleaves intracellular C3 into bioactive C3a and C3b. Upon TCR stimulation, the intracellular C3 and activation fragments C3a, C3b and CTSL translocate to the cell surface where C3a and C3b signal in an autocrine activation though C3aR and CD46, respectively, to to induce Th1 immunity and IFN-γ production. The CD46 activation drives expression of LAMTOR5, LAT1 and GLUT1, which promotes mTORC1 activation, nutrient influx, glycolysis, and OXPHOS. CD46 also induces intracellular C5 activation and C5a generation via yet unknown mechanisms. The intracellular C5a interacts with the mitochondrially expressed C5aR1, resulting in mitochondrial ROS production, intrinsic NLRP3 inflammasome assembly and activation, maintenance of IFN-γ secretion, and subsequent mature IL-1β secretion during T cell migration into inflamed tissues. CTSL, cathepsin L; TCR, T cell receptor; LAMTOR5, late endosomal/lysosomal adaptor and MAPK and mTOR activator 5; GLUT1, glucose transporter; LAT1, large neutral amino acid transporter 1; mTOR, mammalian target of rapamycin; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species. (B) Monocytes and macrophages. Intracellular complement C5 is cleaved and generates C5a via intracellular C5 convertase (C3bBbC3b). C5a/C5aR1 signaling on mitochondrial membranes alters mitochondrial activity, reduces mitochondrial ERK1/2 phosphorylation, cAMP formation and shifts ATP production via reverse electron chain flux toward ROS production and aerobic glycolysis, promoting IL-1β gene expression and processing of bioactive Il-1β upon DAMP sensing. cAMP, Cyclic Adenosine monophosphate; ATP, adenosine triphosphate; ROS, reactive oxygen species; DAMP, damage-associated molecular pattern.
This novel pathway of complosome in T cells contributes greatly to disease. For example, Juvenile idiopathic arthritis (JIA) patients’ T cells have increased intracellular C3. In vitro treatment with a cell permeable CTSL inhibitor can normalize the hyper-active intracellular C3 activation and decrease IFN-γ production in patients’ T cells (3). Intracellular C3 system dysregulation contributes to human autoimmune diseases such as scleroderma, systemic lupus erythematosus (SLE), RA, and multiple sclerosis, as pathological intracellular C3 hyperactivation contributes to Th1 hyperactivity in these autoimmune conditions (4, 30). Individuals lacking CD46 expression or C3 secretion by T cells have diminished Th1 immunity (but normal Th2 and TH17 responses) and suffer from recurrent infections (31, 32). Patients with leukocyte adhesion deficiency type 1 (LAD-1) have reduced C3 transcripts and diminished effector activities in immune cells, which have been shown to be rescued proportionally by intracellular C3 provision. It was also shown that C3 transcription is LFA-1 (lymphocyte function-associated antigen 1)-dependent, and perturbations in the LFA-1-C3-axis contribute to primary immunodeficiency (33).
4.2 Monocytes and macrophages
The complosome plays a crucial orchestrating role in cell metabolic processes that regulate T cell effector responses, but the role of the complosome in myeloid immune cells was unknown for several years. There was preliminary evidence showing human monocytes produce IL-1β during infections by relying on intrinsic C3a activity (10). Macrophage populations express C3a receptor (C3aR) intracellularly (34). M2 macrophages highly express intracellular C3/C3b, which modulates the proinflammatory profile during endoplasmic reticulum (ER) stress (35). In general, studies regarding this topic were relatively scarce until recently, when Kemper’s team published a remarkable study that revealed that human monocytes and macrophages continuously synthesized C5 and produced C5a intracellularly via an intrinsic intracellular C5 convertase (Figure 2B). C5a/C5aR1 signaling on mitochondrial membranes was shown to alter mitochondrial activity and shift adenosine triphosphate (ATP) production via reverse electron chain flux toward ROS production and aerobic glycolysis, which not only promoted IL-1β gene expression, but also processing of bioactive Il-1β upon damage-associated molecular pattern (DAMP) sensing in monocytes and macrophages (36). These results further elaborated previous findings that C5a augments physiologic inflammasome responses (11, 37) from the perspective of intracellular complement. Unlike the previous conclusions that cell surface anaphylatoxin receptors are indirect regulators of mitochondrial activity (38), evidence has demonstrated that cellular mitochondrial C5aR1 (mtC5aR1) activation can directly reduce mitochondrial ERK1/2 phosphorylation, as well as ATP and cAMP formation and OXPHOS, simultaneously increasing ROS production and glycolysis, thereby establishing a direct link between cellular C5a/C5aR1 signaling and cell metabolic pathways located within the mitochondria. Upon mtC5aR1 blockade, IL-1β suppression was only observed in M2 macrophages, but not M1 macrophages. Conversely, mtC5aR1 inhibition reduced IL-10 production in differentiated M1 macrophages and not M2 macrophages, suggesting that there is still much to learn regarding the interaction between the complosome and macrophages. Furthermore, intracellular factor B was proposed to play a role in this process in macrophages. In diseased states, mice benefited from conditional knockout of C5ar1 in myeloid cells in models of crystal-triggered sterile inflammation (folic acid–induced kidney injury and atherosclerotic cardiovascular disease (ACVD) (36). This research has greatly increased our current knowledge on the complosome in myeloid immune cells and it is of great significance to the follow-up research.
4.3 Other immune cells
There is growing evidence that the complosome exists and plays a crucial role in other immune cells, even though the majority of research on the topic focuses on T cells and macrophages/monocytes. Human neutrophils, for instance, have intracellular stores of C3, CR1, and FB (2). C3 and FB are released from stores to trigger a local inflammatory process upon neutrophil activation. It is not yet known whether this process involves a traditional cascade reaction or simple proteolytic cleavage to release functional fragments, such as C3a or Ba (24, 39). M-ficolin/Ficolin-1, an activator of the LP pathway, was also found to be localized in neutrophils secretory granules (40).Neutrophil stimulation with C5a resulted in a quick release of the complement proteins FP, C3, and FB, which are critical for the assembly of the AP convertase C3bBb, linking a positive feedback loop between neutrophil and complement activation (41, 42). B cells also harbor a so-called B cell complosome (3), as mentioned above, B cells can transport C3(H2O) from the extracellular milieu into the cell and process it intracellularly via CTSL into C3b and C3a, however the biological implication of this observation remains to be established (20). The endogenous C3 expression is relatively low, and the serum is the main source of intracellular C3 in human B cells. According to research, both serum derived and pure C3 may enter the nucleus of live B cells, and its strong interaction with histone proteins and potential capacity to induce chromatin rearrangement implies that C3 might regulate DNA transcription via chromatin remodeling (43, 44). Natural killer cells (NK cells) express several complement receptors such as C3aR, CR3, CR4, C5aR1 and C5aR2, while C5aR1 and C5aR2 proteins were reported to be only expressed inside NK cells upon permeabilization. The intracellular expression of C5aR1 and C5aR2 was down-regulated in response to Poly (I: C) (a synthetic analogue of double-stranded RNA) or TNF-α (45). A similar pattern of intracellular C5aR1 and C5aR2 expression was also reported in CD3+CD56+ NKT-like cells (46). However, it is uncertain if this intracellular expression regulates NK cell activity or whether complement components such as C3 or C5 can be generated and secreted by the NK cell.
5 Complosome in non-immune cells
5.1 Cancer cells
Extensive studies have highlighted the importance of tumor cell-derived complosome (particularly C3/C3a) in cancer progression (47, 48). Intracellular activation of tumor cell-derived C3 inhibited CD8+ T cell infiltration and function by driving the accumulation and immune-suppressive activity of tumor-associated macrophages (TAMs) in a C3a-C3aR-dependent manner through the C3a-C3aR-PI3Kγ signaling pathway (49). More importantly, deletion of C3 in tumor cells that had high C3 expression enhanced the efficacy of anti–PD-L1 treatment (49). C3 downregulation by siRNA in ovarian cancer cells SKOV3ip1 inhibits tumor cell growth and migration (47). It has been shown that intracellular C3 in human colon carcinoma Caco2 cells can cleave cathepsin L and B and release C3a, just like CTSL can cleave C3 in CD4+ T cells in a non-C3 convertase manner, and C3a secretion can be downregulated by treatment with cathepsin L and B inhibitors in these cells (50). Other tumor cells, such as leukemia cells, express C3a and C5a receptors and respond to C3a and C5a stimulation by phosphorylation of p44/42 MAPK and AKT (51). Additionally, tumor cell derived-FB was found to be upregulated in pancreatic ductal adenocarcinoma (PDAC), promoting cancer cell proliferation by preventing cellular senescence and associated with myeloid-derived suppressor cells (MDSCs), immunosuppressive regulatory T-cells (Tregs), and TAMs in immunological tumor promotion (52). As previously stated, the complosme in CD4+ T cells and monocytes regulates basic metabolic processes and mTOR during T cell activation. Although evidence is lacking, it is very likely that the complosme functions similarly in tumor cells, which rely on aerobic glycolysis for cell survival, proliferation, and metastasis (48).
5.2 Other non-immune cells
There is also a growing number of reports about intracellular complement expression from stromal cells. For example, it have been demonstrated to regulate autophagy, energy management, and insulin production by pancreatic β cells (53, 54). Human pancreatic islets highly express intracellular C3, and C3 binds autophagy-related protein 16-1 (ATG16L1), thus regulating autophagy and contributing to β cell survival in human islet inflammation and diabetes (54). The C5b-C9 formation inhibitor protein, CD59, was demonstrated to be localized intracellularly in insulin granules in pancreatic β cells, thereby regulating insulin secretion by exocytosis (55, 56). Adipocytes can also produce complement components such as C3, C1q, properdin, and Components B and D, which have been linked to abnormal adipocyte function, immune cell infiltration, obesity, and insulin resistance (56, 57). Similarly to pancreatic β cells, intracellular C3 also protects human airway epithelial cells from stress-induced cell death (58). Intracellular C3 regulates Paneth cell turnover during repair of intestinal epithelial cells after injury (59). During mesenteric ischemia, C3 activation in a cathepsin-dependent manner in intestinal epithelial cells contribute greatly to intestinal tissue damage (50), confirming the pro-inflammatory role of intracellular C3. During SARS-CoV-2 infection, the complosome also plays an important role in lung epithelial cells by activating intracellular C3a, which can be blocked by a cell-permeable FB inhibitor, confirming the presence of an inducible cell-intrinsic C3 convertase in lung epithelial cells. Mechanically, complement gene transcription in lung epithelial cells was shown to be JAK1/2-STAT1–dependent (60). In retinal pigment epithelial (RPE) cells, intracellular expression of C3, C3a, C3aR, CR3, and FB was reported to be induced by internalized complement factor H-related 3 (FHR-3), in addition to proinflammatory cytokine secretion and inflammasome NLRP3 activation (61).
6 Conclusions and perspectives
Today, the complement system is defined as a complex network of fluid-phase, cell-surface-associated and intracellular proteins that act as pattern-recognition molecules, regulators, convertases, proteases, and signaling receptors that collectively mediate immunosurveillance and tissue homeostasis (62). The discovery of the complosome and its significant roles in metabolism, inflammatory response, and cancer progression (4, 63) in the last decade have brought fresh vitality into this field, but it has also generated more questions than answers. Several questions that need to be answered include: Where and how is the complosome activated and regulated/dysregulated? Is this cell specific? How does the complosome intersect with extracellular complement? What is the mechanism underlying the location–function of complement? How can the complosome be targeted therapeutically? Finding the answers to these questions will not only deepen our understanding of the complosome but also open up new insights into the development of complosome-targeted therapeutics.
Author contributions
GY and FX conceived this study, SH and GY directed the study. FX, JG, and GY performed literature search. FX and JG drafted the manuscript. SH, ST, and GY provided critical intellectual revision. SH and GY provided financial support. All authors contributed to the article and approved the submitted version.
Fundings
This work was supported in part by National Key Research and Development Program (2022YFE0131600); National Natural Science Foundation of China (82160500); The 111 Project (D17011); Special project of central government guiding local science and technology development (ZY20198011); Guangxi science and technology base and talent project (GuikeAA21220002); Natural Science Foundation of Guangxi (2022GXNSFAA035642).
Acknowledgments
We thank LetPub (www.letpub.com) for linguistic assistance and pre-submission expert review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol (2017) 18(12):1288–98. doi: 10.1038/ni.3858
2. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol (2017) 188(2):183–94. doi: 10.1111/cei.12952
3. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity (2013) 39(6):1143–57. doi: 10.1016/j.immuni.2013.10.018
4. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity (2015) 42(6):1033–47. doi: 10.1016/j.immuni.2015.05.024
5. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T Helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science (2016) 352(6292):aad1210. doi: 10.1126/science.aad1210
6. Reichhardt MP, Meri S. Intracellular complement activation-an alarm raising mechanism? Semin Immunol (2018) 38:54–62. doi: 10.1016/j.smim.2018.03.003
7. Hansen CB, Willer A, Bayarri-Olmos R, Kemper C, Garred P. Expression of complement C3, C5, C3aR and C5aR1 genes in resting and activated CD4(+) T cells. Immunobiology (2019) 224(2):307–15. doi: 10.1016/j.imbio.2018.12.004
8. Liszewski MK, Kemper C. Complement in motion: The evolution of CD46 from a complement regulator to an orchestrator of normal cell physiology. J Immunol (2019) 203(1):3–5. doi: 10.4049/jimmunol.1900527
9. Kunz N, Kemper C. Complement has brains-do intracellular complement and immunometabolism cooperate in tissue homeostasis and behavior? Front Immunol (2021) 12:629986. doi: 10.3389/fimmu.2021.629986
10. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Köhl J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122(20):3473–81. doi: 10.1182/blood-2013-05-502229
11. Haggadone MD, Grailer JJ, Fattahi F, Zetoune FS, Ward PA. Bidirectional crosstalk between C5a receptors and the NLRP3 inflammasome in macrophages and monocytes. Mediators Inflamm (2016) 2016:1340156. doi: 10.1155/2016/1340156
12. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: A key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
13. Guo Z, Fan X, Yao J, Tomlinson S, Yuan G, He S. The role of complement in nonalcoholic fatty liver disease. Front Immunol (2022) 13:1017467. doi: 10.3389/fimmu.2022.1017467
14. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res (2010) 20(1):34–50. doi: 10.1038/cr.2009.139
15. Rawal N, Rajagopalan R, Salvi VP. Activation of complement component C5: Comparison of C5 convertases of the lectin pathway and the classical pathway of complement. J Biol Chem (2008) 283(12):7853–63. doi: 10.1074/jbc.M707591200
16. Pangburn MK. Initiation of the alternative pathway of complement and the history of "tickover". Immunol Rev (2023) 313(1):64–70. doi: 10.1111/imr.13130
17. Xie CB, Jane-Wit D, Pober JS. Complement membrane attack complex: New roles, mechanisms of action, and therapeutic targets. Am J Pathol (2020) 190(6):1138–50. doi: 10.1016/j.ajpath.2020.02.006
18. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9(10):729–40. doi: 10.1038/nri2620
19. Kolev M, Le Friec G, Kemper C. Complement–tapping into new sites and effector systems. Nat Rev Immunol (2014) 14(12):811–20. doi: 10.1038/nri3761
20. Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Invest (2017) 127(3):970–81. doi: 10.1172/JCI89412
21. Martin M, Leffler J, Smoląg KI, Mytych J, Björk A, Chaves LD, et al. Factor h uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ (2016) 23(5):903–11. doi: 10.1038/cdd.2015.164
22. Arbore G, Kemper C, Kolev M. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
23. Dutta K, Friscic J, Hoffmann MH. Targeting the tissue-complosome for curbing inflammatory disease. Semin Immunol (2022) 60:101644. doi: 10.1016/j.smim.2022.101644
24. Ghebrehiwet B. Complement proteins in unexpected places: Why we should be excited, not concerned! F1000Res (2020) 9. doi: 10.1126/sciimmunol.abf2489
25. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
26. Arbore G, West EE, Rahman J, Le Friec G, Niyonzima N, Pirooznia M, et al. Complement receptor CD46 co-stimulates optimal human CD8(+) T cell effector function via fatty acid metabolism. Nat Commun (2018) 9(1):4186. doi: 10.1038/s41467-018-06706-z
27. Hansen AS, Slater J, Biltoft M, Bundgaard BB, Moller BK, Hollsberg P. CD46 is a potent co-stimulatory receptor for expansion of human IFN-gamma-producing CD8(+) T cells. Immunol Lett (2018) 200:26–32. doi: 10.1016/j.imlet.2018.06.003
28. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, et al. C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science (2018) 360(6388):558–63. doi: 10.1126/science.aao4555
29. West EE, Kunz N, Kemper C. Complement and human T cell metabolism: Location, location, location. Immunol Rev (2020) 295(1):68–81. doi: 10.1111/imr.12852
30. Arbore G, Ong VH, Costantini B, Denton CP, Abraham D, Placais L, et al. Deep phenotyping detects a pathological CD4(+) T-cell complosome signature in systemic sclerosis. Cell Mol Immunol (2020) 17(9):1010–3. doi: 10.1038/s41423-019-0360-8
31. Ellinghaus U, Cortini A, Pinder CL, Le Friec G, Kemper C, Vyse TJ. Dysregulated CD46 shedding interferes with Th1-contraction in systemic lupus erythematosus. Eur J Immunol (2017) 47(7):1200–10. doi: 10.1002/eji.201646822
32. Liszewski MK, Atkinson JP. Membrane cofactor protein (MCP; CD46): deficiency states and pathogen connections. Curr Opin Immunol (2021) 72:126–34. doi: 10.1016/j.coi.2021.04.005
33. Kolev M, West EE, Kunz N, Chauss D, Moseman EA, Rahman J, et al. Diapedesis-induced integrin signaling via LFA-1 facilitates tissue immunity by inducing intrinsic complement C3 expression in immune cells. Immunity (2020) 52(3):513–27.e8. doi: 10.1016/j.immuni.2020.02.006
34. Quell KM, Karsten CM, Kordowski A, Almeida LN, Briukhovetska D, Wiese AV, et al. Monitoring C3aR expression using a floxed tdTomato-C3aR reporter knock-in mouse. J Immunol (2017) 199(2):688–706. doi: 10.4049/jimmunol.1700318
35. Chaumonnot K, Masson S, Sikner H, Bouchard A, Baverel V, Bellaye PS, et al. The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death Dis (2021) 12(1):114. doi: 10.1038/s41419-020-03288-x
36. Niyonzima N, Rahman J, Kunz N, West EE, Freiwald T, Desai JV, et al. Mitochondrial C5aR1 activity in macrophages controls IL-1β production underlying sterile inflammation. Sci Immunol (2021) 6(66):eabf2489. doi: 10.1126/sciimmunol.abf2489
37. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol (2014) 192(6):2837–45. doi: 10.4049/jimmunol.1302484
38. Rahman J, Singh P, Merle NS, Niyonzima N, Kemper C. Complement's favourite organelle-mitochondria? Br J Pharmacol (2021) 178(14):2771–85. doi: 10.1111/bph.15238
39. Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int (2002) 61(2):456–63. doi: 10.1046/j.1523-1755.2002.00139.x
40. Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, et al. Human m-ficolin is a secretory protein that activates the lectin complement pathway. J Immunol (2005) 175(5):3150–6. doi: 10.4049/jimmunol.175.5.3150
41. Yuen J, Pluthero FG, Douda DN, Riedl M, Cherry A, Ulanova M, et al. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front Immunol (2016) 7:137. doi: 10.3389/fimmu.2016.00137
42. Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux-Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood (2011) 117(4):1340–9. doi: 10.1182/blood-2010-05-283564
43. Jimenez-Reinoso A, Marin AV, Subias M, Lopez-Lera A, Roman-Ortiz E, Payne K, et al. Human plasma C3 is essential for the development of memory b, but not T, lymphocytes. J Allergy Clin Immunol (2018) 141(3):1151–4 e14. doi: 10.1016/j.jaci.2017.09.037
44. Kremlitzka M, Nowacka AA, Mohlin FC, Bompada P, De Marinis Y, Blom AM. Interaction of serum-derived and internalized C3 with DNA in human b cells-a potential involvement in regulation of gene transcription. Front Immunol (2019) 10:493. doi: 10.3389/fimmu.2019.00493
45. Min X, Liu C, Wei Y, Wang N, Yuan G, Liu D, et al. Expression and regulation of complement receptors by human natural killer cells. Immunobiology (2014) 219(9):671–9. doi: 10.1016/j.imbio.2014.03.018
46. Min XY, Liu CF, Cao B, Zhang T, Yang X, Ma N, et al. Human CD3(+)CD56(+)NKT-like cells express a range of complement receptors and C3 activation has negative effects on these cell activity and effector function. Hum Immunol (2021) 82(9):625–33. doi: 10.1016/j.humimm.2021.06.001
47. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep (2014) 6(6):1085–95. doi: 10.1016/j.celrep.2014.02.014
48. Kolev M, Das M, Gerber M, Baver S, Deschatelets P, Markiewski MM. Inside-out of complement in cancer. Front Immunol (2022) 13:931273. doi: 10.3389/fimmu.2022.931273
49. Zha H, Wang X, Zhu Y, Chen D, Han X, Yang F, et al. Intracellular activation of complement C3 leads to PD-L1 antibody treatment resistance by modulating tumor-associated macrophages. Cancer Immunol Res (2019) 7(2):193–207. doi: 10.1158/2326-6066.CIR-18-0272
50. Satyam A, Kannan L, Matsumoto N, Geha M, Lapchak PH, Bosse R, et al. Intracellular activation of complement 3 is responsible for intestinal tissue damage during mesenteric ischemia. J Immunol (2017) 198(2):788–97. doi: 10.4049/jimmunol.1502287
51. Lenkiewicz A, Bujko K, Brzezniakiewicz-Janus K, Xu B, Ratajczak MZ. The complement cascade as a mediator of human malignant hematopoietic cell trafficking. Front Immunol (2019) 10:1292. doi: 10.3389/fimmu.2019.01292
52. Shimazaki R, Takano S, Satoh M, Takada M, Miyahara Y, Sasaki K, et al. Complement factor b regulates cellular senescence and is associated with poor prognosis in pancreatic cancer. Cell Oncol (Dordr) (2021) 44(4):937–50. doi: 10.1007/s13402-021-00614-z
53. Golec E, Ekström A, Noga M, Omar-Hmeadi M, Lund PE, Villoutreix BO, et al. Alternative splicing encodes functional intracellular CD59 isoforms that mediate insulin secretion and are down-regulated in diabetic islets. Proc Natl Acad Sci USA (2022) 119(24):e2120083119. doi: 10.1073/pnas.2120083119
54. King BC, Kulak K, Krus U, Rosberg R, Golec E, Wozniak K, et al. Complement component C3 is highly expressed in human pancreatic islets and prevents beta cell death via ATG16L1 interaction and autophagy regulation. Cell Metab (2019) 29(1):202–10.e6. doi: 10.1016/j.cmet.2018.09.009
55. Krus U, King BC, Nagaraj V, Gandasi NR, Sjolander J, Buda P, et al. The complement inhibitor CD59 regulates insulin secretion by modulating exocytotic events. Cell Metab (2014) 19(5):883–90. doi: 10.1016/j.cmet.2014.03.001
56. Vlaicu SI, Tatomir A, Boodhoo D, Vesa S, Mircea PA, Rus H. The role of complement system in adipose tissue-related inflammation. Immunol Res (2016) 64(3):653–64. doi: 10.1007/s12026-015-8783-5
57. Gauvreau D, Roy C, Tom FQ, Lu H, Miegueu P, Richard D, et al. A new effector of lipid metabolism: complement factor properdin. Mol Immunol (2012) 51(1):73–81. doi: 10.1016/j.molimm.2012.02.110
58. Kulkarni HS, Elvington ML, Perng YC, Liszewski MK, Byers DE, Farkouh C, et al. Intracellular C3 protects human airway epithelial cells from stress-associated cell death. Am J Respir Cell Mol Biol (2019) 60(2):144–57. doi: 10.1165/rcmb.2017-0405OC
59. Zhang J, Ye J, Ren Y, Zuo J, Dai W, He Y, et al. Intracellular activation of complement C3 in paneth cells improves repair of intestinal epithelia during acute injury. Immunotherapy (2018) 10(15):1325–36. doi: 10.2217/imt-2018-0122
60. Yan B, Freiwald T, Chauss D, Wang L, West E, Mirabelli C, et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci Immunol (2021) 6(58):eabg0833. doi: 10.1126/sciimmunol.abg0833
61. Schafer N, Rasras A, Ormenisan DM, Amslinger S, Enzmann V, Jagle H, et al. Complement factor h-related 3 enhanced inflammation and complement activation in human RPE cells. Front Immunol (2021) 12:769242. doi: 10.3389/fimmu.2021.769242
62. Kemper C, Kohl J. Back to the future - non-canonical functions of complement. Semin Immunol (2018) 37:1–3. doi: 10.1016/j.smim.2018.05.002
Keywords: complement, complosome, T cells, monocytes, tumor cells
Citation: Xiao F, Guo J, Tomlinson S, Yuan G and He S (2023) The role of the complosome in health and disease. Front. Immunol. 14:1146167. doi: 10.3389/fimmu.2023.1146167
Received: 17 January 2023; Accepted: 28 February 2023;
Published: 10 March 2023.
Edited by:
Peter Epeh Kima, University of Florida, United StatesReviewed by:
Kalyani Pyaram, Kansas State University, United StatesCopyright © 2023 Xiao, Guo, Tomlinson, Yuan and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guandou Yuan, ZHJfeXVhbmdkQGd4bXUuZWR1LmNu; Songqing He, ZHJfaGVzb25ncWluZ0AxNjMuY29t
†These authors have contributed equally to this work