 Wenjie Zhang
Wenjie Zhang Yuan Chen3
Yuan Chen3 Hui Pei
Hui Pei- 1Department of Emergency Intensive Care Unit, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of General Practice, Xingyang Sishui Central Health Center, Zhengzhou, China
- 3Department of Neurology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
C1q is a crucial component of the complement system, which is activated through the classical pathway to perform non-specific immune functions, serving as the first line of defense against pathogens. C1q can also bind to specific receptors to carry out immune and other functions, playing a vital role in maintaining immune homeostasis and normal physiological functions. In the developing central nervous system (CNS), C1q functions in synapse formation and pruning, serving as a key player in the development and homeostasis of neuronal networks in the CNS. C1q has a close relationship with microglia and astrocytes, and under their influence, C1q may contribute to the development of CNS disorders. Furthermore, C1q can also have independent effects on neurological disorders, producing either beneficial or detrimental outcomes. Most of the evidence for these functions comes from animal models, with some also from human specimen studies. C1q is now emerging as a promising target for the treatment of a variety of diseases, and clinical trials are already underway for CNS disorders. This article highlights the role of C1q in CNS diseases, offering new directions for the diagnosis and treatment of these conditions.
1 Introduction
The complement system is a sophisticated protein response system consisting of over 30 components that are present in serum, tissue fluids, and cell surfaces. It possesses a complex regulatory mechanism (1). These components exist in a non-activated form in body fluids and are activated through a cascade of enzymatic reactions that produce various biological effects. There are three complement activation pathways, each with a common end-reaction process (2). The classical pathway involves a cascade of enzymatic reactions in which activators, mainly IgG and IgM bound to antigen, bind to C1q and sequentially activate C1r, C1s, C2, and C3, forming C3 convertase (C4b2a) and C5 convertase (C4b2a3b), ultimately leading to the formation of the C5b6789n complex, the membrane attack complex (MAC), at the cell membrane (Figure 1). The alternative pathway directly activates C3 by microorganisms or exogenous xenobiotics to form C3 convertase (C3bBbP) and C5 convertase (C3bBb3b), with the involvement of B-factor, D-factor, and P-factor. The lectin pathway, also known as the MBL pathway, involves a cascade of enzymatic reactions in which mannose-binding lectin (MBL) and ficolin (FCN) in plasma directly recognize the glycan structure on the surface of the pathogen, sequentially activating MBL-associated serine protease (MASP), C4, C2, and C3, ultimately forming the same C3 convertase and C5 convertase as in the classical pathway.
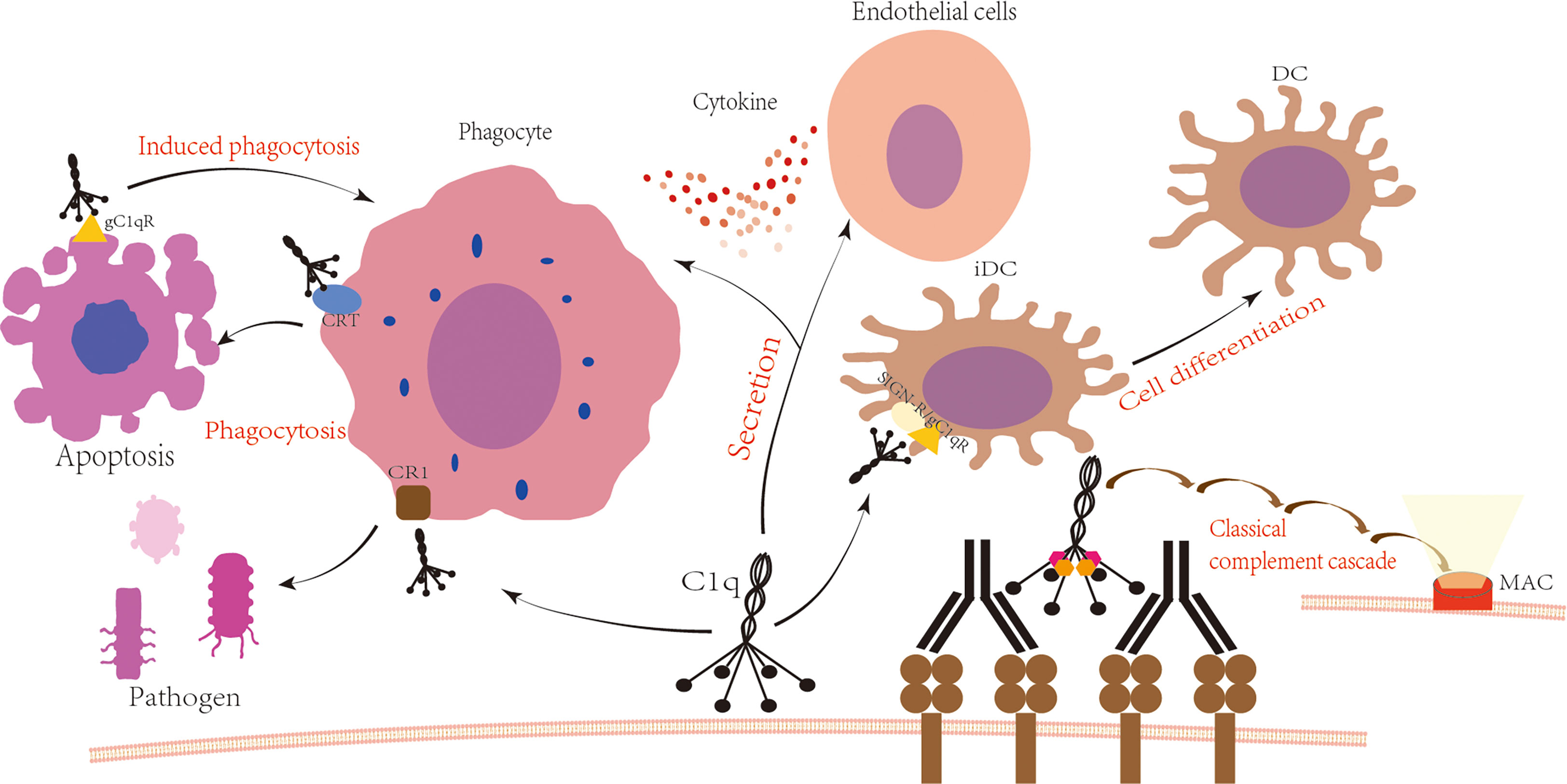
Figure 1 C1q is involved in the immune process. Upon antigen-antibody binding, C1q is activated and subsequently activates C1r and C1s, leading to the formation of C3 convertase C4b2a, C5 convertase C4b2a3b, and ultimately the formation of the membrane attack complex with the involvement of C6 to C9. The globular domain or collagen-like tail of C1q binds to cell surface receptors to produce various physiological effects. The globular domain of C1q binds to SIGN/gC1qR, which regulates the differentiation of immature dendritic cells (3, 4). The tail of C1q binds to the phagocyte surface receptor CR1, promoting the phagocytosis of pathogens (5). Additionally, the globular domain of C1q interacts with the phagocyte surface receptor CRT to promote phagocytosis of apoptotic cells, while binding to gC1qR on the surface of apoptotic cells induces phagocytosis (6, 7). Furthermore, C1q interacts with phagocytes and endothelial cells, increasing cytokine secretion (8, 9).
C1q is a crucial component of the complement system, produced mainly by myeloid cells such as monocytes, macrophages, and immature dendritic cells. It is composed of three genes (C1q A, B, and C) that encode proteins with a collagen-like region and a globular domain. The A, B, and C chains are assembled into trimers, and six trimers form a hexamer with a total of 18 polypeptide chains. The resulting complex molecule has multiple interaction sites on C1q. The crystallographic model revealed that the binding site of the immunoglobulin to C1q is located on the outer surface of the head, while the apoptotic cells bind on the inner surface of the sphere (10). C1q can act as a biomolecule recognized by pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) to perform immune functions (11). It has an essential role in removing foreign substances and pathogens, as well as maintaining internal stability. C1q is well known for its involvement in the classical activation pathway of complement. When the globular head of C1q binds to the Fc segment of IgM or IgG in the immune complex, the C1q conformation changes, leading to the sequential activation of C1r and C1s, initiating the classical activation pathway of complement. C1q has four main types of receptors, including Clq receptor for phagocytosis enhancement (ClqRp), complement receptor 1 (CR1), calreticulin (CRT), and C1q globular head binding protein (gClqbp) (12). By interacting with supracellular receptors, C1q stimulates variety of cells such as phagocytes and endothelial cells to secrete a variety of cytokines, exacerbating local inflammatory responses (8, 9, 11). Recent research has shown that the interaction of C-type lectin SIGN-R and C1q head can promote the maturation and differentiation of immature DCs(iDCs) cells (3). Additionally, the regulation of DC differentiation and function is through C1q binding to cell surface SIGN and gC1qR-specific molecules on the DC plasma membrane to form a trimolecular complex (4). In pneumococci, the SIGN-R1 receptor on macrophages binds directly to C1q replacing the Ig normally used in the classical pathway, forming C3 convertase, subsequently activating the rest of the pathway (13). CR1 binds to C1q and is involved in phagocytic activity (5), CR1 is widely expressed and is also found on CNS microglia (14). CRT binds to the C1q globular domain, enhances phagocytosis of apoptotic cells, and triggers an immunogenic response (6, 15). C1qRp binds to the C1q collagen tail and globular head sites and induces phagocytosis of apoptotic cells (7). C1q also has anti-cancer effects through immunosurveillance (16, 17), Antigen-presenting cancer-associated fibroblasts (apCAFs) in lung malignancies produce C1q, which binds to T cells C1qbp to inhibit T cell apoptosis and promote tumor killing by immune cells (18). The presence of C1r2s2 increases the stability of C1q-IgG and has a positive effect on the elimination of foreign substances and tumor cells (19). Apart from normal physiological immune responses such as the removal of foreign substances and abnormal cells, C1q also has an important impact on the development of other diseases. C1q can act in conjunction with ApoE to cause atherosclerotic plaque formation (20), can be present alone to participate in the early development of atherosclerosis (21). C1q complexes with vWF increase platelet rolling and adhesion and promote hemostasis (22). C1q binds to discoidal domain receptor 2 (DDR2) to regulate metalloproteinase expression for wound healing due to its specific collagen-like structure (23). The structure of the C1q collagen region binds to the sequence of fibronectin-binding protein B (PfbB) on the surface of Streptococcus pneumoniae and promotes the adhesion of Streptococcus pneumoniae to host cells and participates in the process of pneumococcal infection (24). Complement activation is well documented in the progression of autoimmune diseases and is especially important in the development, diagnosis and prognostic monitoring of lupus nephritis (25, 26). C1q blocks aberrant effector CD8+ T cell responses by recognizing activated CD8+ T cells through globular structural domains and affecting their mitochondrial metabolism, and such aberrant effector CD8+ T cells are known to be involved in the development of systemic lupus erythematosus (SLE) (27). A recent study by Zheng et al. in Behçet’s disease showed a significant pro-inflammatory effect of monocytes with high expression of C1q (C1qhi) in the cell membrane by single cell RNA sequencing (scRNA-seq) technology (28). This highlights the importance of understanding the role of C1q in autoimmune diseases and other inflammatory.
2 Neural development
A growing body of research has identified C1q as a key substance involved in neuronal network development and homeostasis within the central nervous system. Specifically, C1q serves as a pruning function during synapse development and synaptogenesis (29, 30). Microglia are extremely delicate and strict on synaptic pruning processes in the nervous system, and C1q and C3 may act as phagocytic signals for microglia-dependent phagocytosis of synapses (31, 32). Multiple EGF-like domains 10 (Megf10), a class F scavenger receptor expressed on astrocytes, binds to C1q with high affinity and induces phagocytosis of apoptotic cells (33). In developing mice, exposure to phosphatidylserine (PS) functions as a neuronal “eat-me” signal involved in C1q-mediated pruning with the microglia receptor TREM2 (34). PS is not exposed on the intact cell surface, but is exposed upon apoptosis (35). In a study of visual neurons in mice, C1qa was found to be critical in the formation of synapses between retinal ganglion neurons and dorsal lateral geniculate nucleus neurons in the visual thalamus, but C1qa did not affect synaptic density and microglia phagocytosis in the developing visual cortex (36). It indicates that different complement components are inconsistent at different developmental periods and regions, and their complex relationships need further study.
The role of the complement pathway in the brain is complex, and there is now evidence that complement plays an important role in neurological disorders in adulthood (37, 38). Complement activation may contribute to neurological disorders through the binding of C3b (iC3b)-CR3 to phagocytes for neuronal damage (39), or through the destruction of neurons by binding to specific allergenic toxin receptors on local glial cells (40). C1q on the one hand gives rise to the classical complement pathway and on the other hand is involved in several functions that may be independent of the complement cascade, including regulation of synaptic pruning (31, 32), protection against neurotoxicity (41) and promotion of angiogenesis (42). In neurological disorders, it may have many deleterious effects as well as some beneficial effects, while evidence for these effects primarily comes from animal models, there is also some evidence from human specimen studies. Overall, an in-depth understanding of the effects of C1q on the nervous system’s development and function can provide insight into the role of C1q in neurological disorders and may offer potential diagnostic and treatment strategies (Figure 2). This article revolves around a review of recent research advances on C1q in recent years, which are expected to provide new ideas for the diagnosis and treatment of neurological disorders.
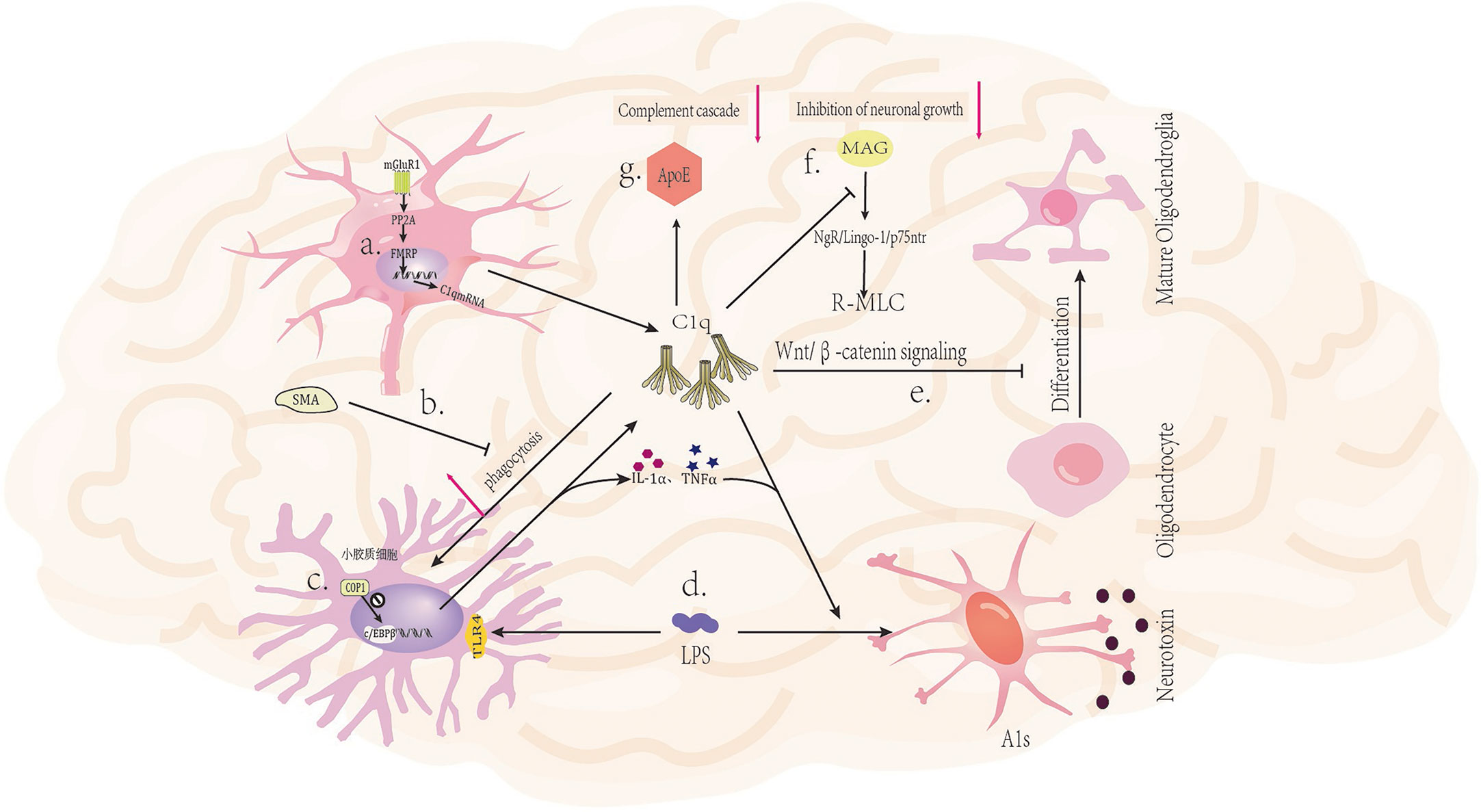
Figure 2 Mechanism of C1q in nervous system diseases. (A) Activation of glutamatergic neuronal mGluR1 receptors leads to increased expression of C1q mRNA (43). (B) The mGluR5 silent alteration modulator (SAM) inhibits C1q-mediated microglial phagocytosis (44). (C) The ubiquitin ligase COP1 regulates the transcription process of microglial inflammatory genes by controlling CCAAT/enhancer binding protein β (c/EBPβ) (45). (D) LPS (lipopolysaccharide) can bind to TLR4 receptors on microglia, triggering the secretion of C1q, Il-1α, and TNFα by microglia, and inducing A1 in astrocytes (46). (E) C1q prevents the differentiation of oligodendrocyte progenitor cells (OPCs) into mature oligodendrocytes by inhibiting the Wnt/β-catenin signaling pathway (47). (F) C1q interacts with MAG to reduce MAG inhibitory effect on neuronal signals, thereby promoting axonal growth (48). (G) The binding of ApoE to C1q inhibits the initiation of the classical complement cascade reaction (20).
3 Relationship between C1q and microglia and astrocytes
C1q, which is produced by microglia, astrocytes, and neurons, plays different roles in the CNS. Microglia and astrocytes are crucial cells in CNS disorders related to C1q. Astrocytes, as the most abundant glial cells, are a vital component of the blood-brain barrier and exhibit incredible complexity in different brain regions (49, 50). Microglia, as the brain’s resident macrophages, participate in various neuroimmune and non-immune processes (51). Microglia and astrocytes have important roles in neural repair, growth, homeostasis, and interneuronal signaling. Abnormalities in both will result in the development of a variety of diseases. During nervous system development, microglia interact with C1q and C3 to regulate synapse pruning. When stimulated by foreign C1q, microglia produce C1q to activate more microglia, creating a positive feedback loop and promoting inflammation (52). However, complement activation can also mediate pathological synaptic loss in microglia during aging, leading to degenerative diseases (31). Astrocytes can respond to various stimuli and undergo changes in gene expression, function, and morphology, resulting in “reactive astrocytes” that can have both protective and harmful effects on neurons (53). Inflammatory mediators secreted by microglia, such as IL-1α, C1q, and TNF, can induce astrocytes to become reactive astrocytes (A1s) (54). The pro-inflammatory factors TNF, IL1α and complement pathway component C1q are able to induce a deleterious response state in astrocytes that are distinct from healthy astrocytes (46). A1s are cells that have lost their typical astrocyte functions (promoting neuronal survival and growth, facilitating synapse formation, phagocytosis of synapses and myelin debris, etc.) (55–57). Studies with rodent and human astrocytes suggest that activated microglia are not sufficient to kill neurons, but rather induce A1s by inducing the secretion of IL-1α, TNFα, and C1q together, and that A1s are capable of secreting neurotoxins and releasing a variety of complement components that denature synapses, leading to neural deformation and death (46, 58). A1s can denature synapses, leading to neural deformation and death, and are implicated in several neurodegenerative diseases such as Alzheimer’s, Huntington’s, Parkinson’s, Amyotrophic Lateral Sclerosis, and Multiple sclerosis. Transcriptomic analysis reveals that the transcriptional profiles of reactive astrocytes (A1s) and inflammatory microglia (MIMS) overlap with those of other neurodegenerative glial cells. C1q has been identified as a crucial mediator of A1s and MIMS activation (59), which will be further analyzed in subsequent sections.
4 Relationship to degenerative diseases in the central nervous system
Neurodegenerative diseases are a group of chronic and progressive diseases that cause damage to tissues such as the central nervous system or peripheral nervous system, and their underlying mechanisms are not yet fully understood. These diseases include Alzheimer’s disease (AD), Amyotrophic Lateral Sclerosis (ALS), Parkinson’s disease (PD), among others. Recent studies utilizing single-cell RNA sequencing have revealed significant differences in gene expression in patient cells, and have identified the accumulation of complement C1q as a central factor underlying most proteotype-related functional synaptic dysfunction (60). C1q plays a crucial role in the development of degenerative diseases, and the following section provides a brief overview of the relationship between C1q and the aforementioned diseases in recent years (Table 1).
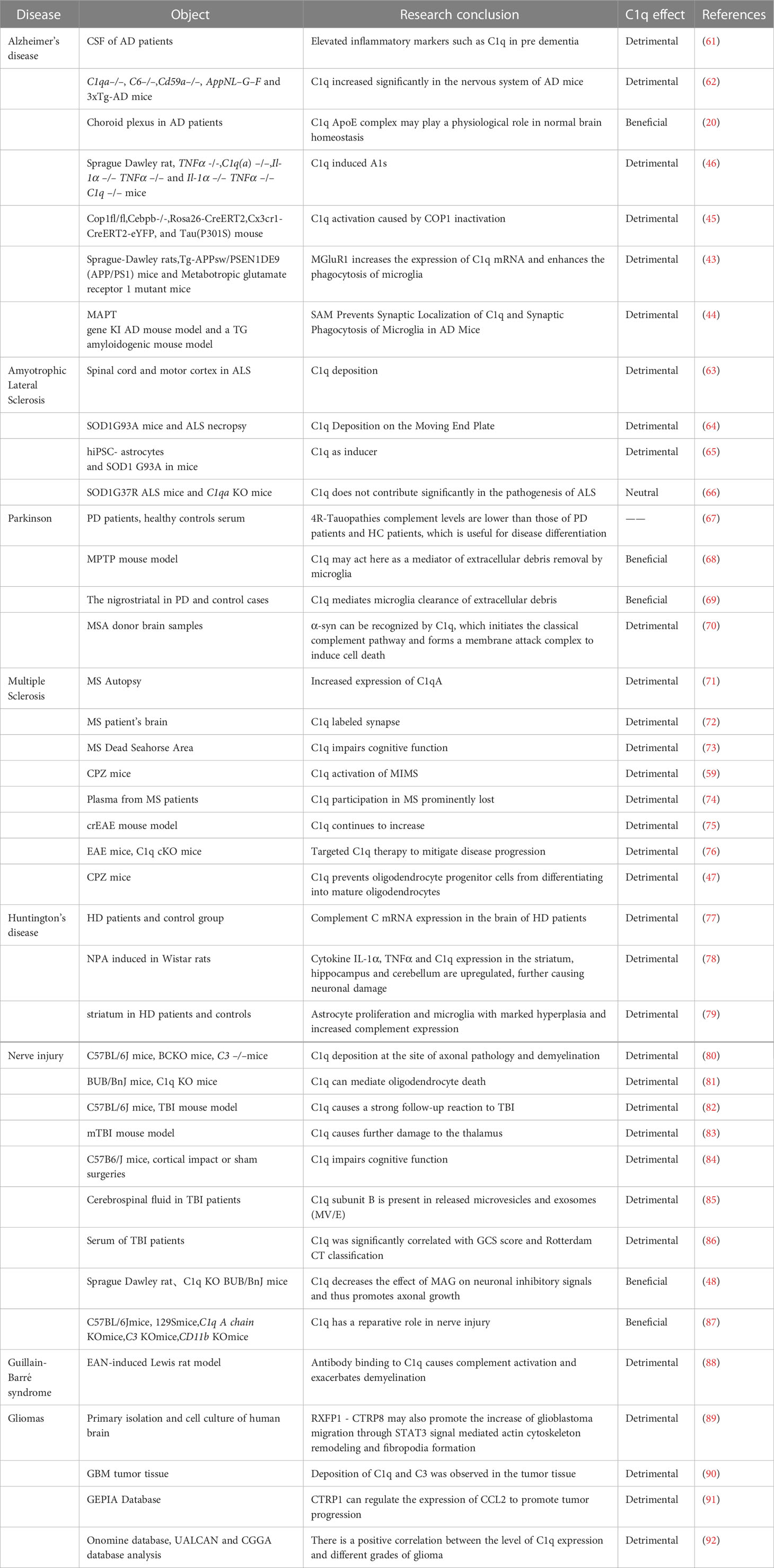
Table 1 The effect of C1q in neurological disorders.
4.1 Alzheimer’s disease (AD)
Alzheimer’s disease (AD) is a common neurodegenerative disorder that affects the elderly, but its underlying mechanisms are still not fully understood. The cognitive impairment observed in AD is associated with synaptic loss and morphological changes in the brain (93). A study of cerebrospinal fluid from patients with AD pathology (beta-amyloid (Ab) and tau subtypes) but not yet symptomatic or in pre-dementia found elevated YKL-40, sTREM2, sAXL, sTyro3, MIF, complement factors C1q, C4 and H, ferritin, and ApoE inflammatory markers (61). C1q has been found to induce excessive phagocytosis and synapse clearance by microglia, leading to synapse loss in AD patients (93, 94). Carpanini et al. conducted experiments on AD model mice and discovered complement dysregulation, with C1q significantly increased in the nervous system of AD mice (62). Yin et al. studied the inflammatory response in the choroid plexus (ChP) of AD patients and found a specific interaction between ApoE and C1q. They observed that the high-affinity binding of ApoE to C1q inhibits the initiation phase of the classical complement cascade reaction (CCC), with the ApoE isoform binding to the activated form of C1q in a Ca2+-dependent manner (20). These findings suggest a close relationship between C1q and neurodegenerative lesions in AD. Liddelow et al. used a mouse model of AD and found that reactive astrocyte A1 (A1s) plays an important role in AD progression (46). A1s-reactive astrocytes can also cause neurological damage via astrocyte-derived exosomes (ADE) and deliver them to other CNS cells via ADE (95). Another study found that the transcription factor CCAAT/enhancer binding protein β (c/EBPβ) promotes inflammatory gene transcription in microglia, and that microglia c/EBPβ expression can be regulated by the ubiquitin ligase COP1. Microglia knocked out of COP1 produce c/EBPβ-dependent neurotoxicity mainly through C1q activation of the classical complement pathway. This suggests that the activation of C1q caused by COP1 inactivation plays an important role in AD progression (45). Previous reports have found a strong link between glutamate and the development of AD, so how is C1q produced and regulated in the brain, and what is the link between it and glutamate? Mouse astrocyte glutamate transporter 1 (GLT1) maintains intersynaptic glutamate concentration homeostasis by removing glutamate from the synaptic gap via active transport via H+-K+ or Na+-K+ pumps or by synthesizing glutamine for reuse via glutamine synthetase. This suggests that abnormal function or reduced number of the astrocyte glutamate transporter GLT1 may be responsible for the occurrence of AD (96). Bie et al. (43) concluded from their mouse model studies that metabotropic glutamate receptor 1 (mGluR1), when induced by amyloid, activates mGluR protein phosphatase 2A (PP2A), which causes dephosphorylation of fragile X messenger ribonucleoprotein (FMRP), resulting in increased expression of C1qmRNA and enhanced phagocytosis of microglia. Spurrier et al. confirmed this mechanism in the treatment of aged AD mice with metabotropic glutamate receptor 5 (mGluR5) silent alteration modulator (SAM). SAM restored synaptic density and prevented synaptic localization of C1q and synaptic phagocytosis of AD mice by microglia. It also prevented abnormal synaptic signaling induced by β-amyloid oligomers, while maintaining physiological glutamate responses and reducing the accumulation of phospho-TAU. This suggests the possibility of treating AD specifically by targeting mGluR5 for modulation (44).
4.2 Amyotrophic lateral sclerosis (ALS)
ALS is the most common adult motor neuron disease, characterized by the progressive loss of upper and lower motor neurons leading to muscle atrophy and eventual death (97). The mechanism underlying the development of ALS is still unclear, and mutations in approximately 30 genes have been identified as causative factors of ALS, including C9orf72, SOD1, FUS, TARDBP, and VCP (98). While most of ALS cases are sporadic, familial cases are also present, with mutations in the gene encoding copper-zinc superoxide dismutase-1 (SOD-1) being more common. As such, many studies on ALS have been conducted on mice with SOD1 mutations. Ferraiuolo et al. used laser capture microdissection and microarray to analyze motor neuron changes in SOD1 G93A mice, revealing significant transcriptional repression, metabolic function decline, upregulation of complement components, and increased expression of cell cycle proteins involved in the cell cycle during the late stage of the disease, indicating the crucial role of complement components in ALS development (99). In ALS, complement components have been found at multiple nerve sites, with deposition of C1q and C4 observed in both the spinal cord and motor cortex of ALS patients, along with higher levels of inflammatory response and microglia activation in patients with rapidly developing ALS (63, 99). Bahia El Idrissi et al. studied immunofluorescence staining of SOD1G93A gastrocnemius and ALS donor intercostal muscle tissue and found that deposition of complement activation products C3/C3b and C1q in the motor endplates during the early stages of ALS symptoms. C1q deposition on the motor endplates of ALS donor intercostal muscles in the SOD1G93A mouse model was detected before the appearance of clinical signs, indicating that complement activation is an early event. C1q immunoreactivity is present in most intercostal muscle tissues of ALS donors, in addition to C1q deposition on motor nerve terminals and terminal Schwann cells of ALS donor intercostal muscles. The study also explored regulatory factors such as CD55 and CD59, which protect tissues from complement system attack and could provide new therapeutic approaches for ALS (64). C1q not only destroys neurons via the classical complement system, but also acts as an inducer of reactive astrocytes, as has been briefly suggested above in AD patients. Among the damaged astrocyte mechanisms, damaged astrocyte glutamate uptake was found to cause excitotoxicity and has been suggested to play an important role in motor neuron hyperexcitability and death in ALS (100). In the ALS mouse model astrocytes were compared to A1s astrocytes with respect to glutamate uptake and subsequent toxic response and there was a great similarity. As seen by gene expression analysis of ALS astrocytes versus protective astrocytes, multiple genes appear opposite (65), signaling pathway analysis revealed that inflammatory pathways such as JAK-STAT, NF-kB and TNF are significantly increased in mouse ALS models, but C1q is able to inhibit the activation of these inflammatory pathways in phagocytes (101–103). Peng showed that TDP-43 is required to maintain the protective properties of astrocytes, and TDP-43-deficient astrocytes exhibited increased immunoreactivity but did not affect the proliferation of astrocytes or microglia. At the transcriptome level, TDP-43-deficient astrocytes resembled A1-responsive astrocytes and induced increased C1q expression in microglia (104). Interestingly, Lobsiger et al. found that the induction of complement pathway activation by C1q did not significantly contribute to the ALS pathogenesis in SOD1 G37R mutant mice (66). The role of C1q in ALS has been recognized by most researchers, and although some studies are inconsistent, we still cannot deny the important role of C1q in ALS, a disease that requires continued efforts to explore and study to find more treatments. Clinical trials about C1q inhibitors in ALS have emerged(NCT04569435)(EUCTR2021-000325-26-FR) (Table 2).
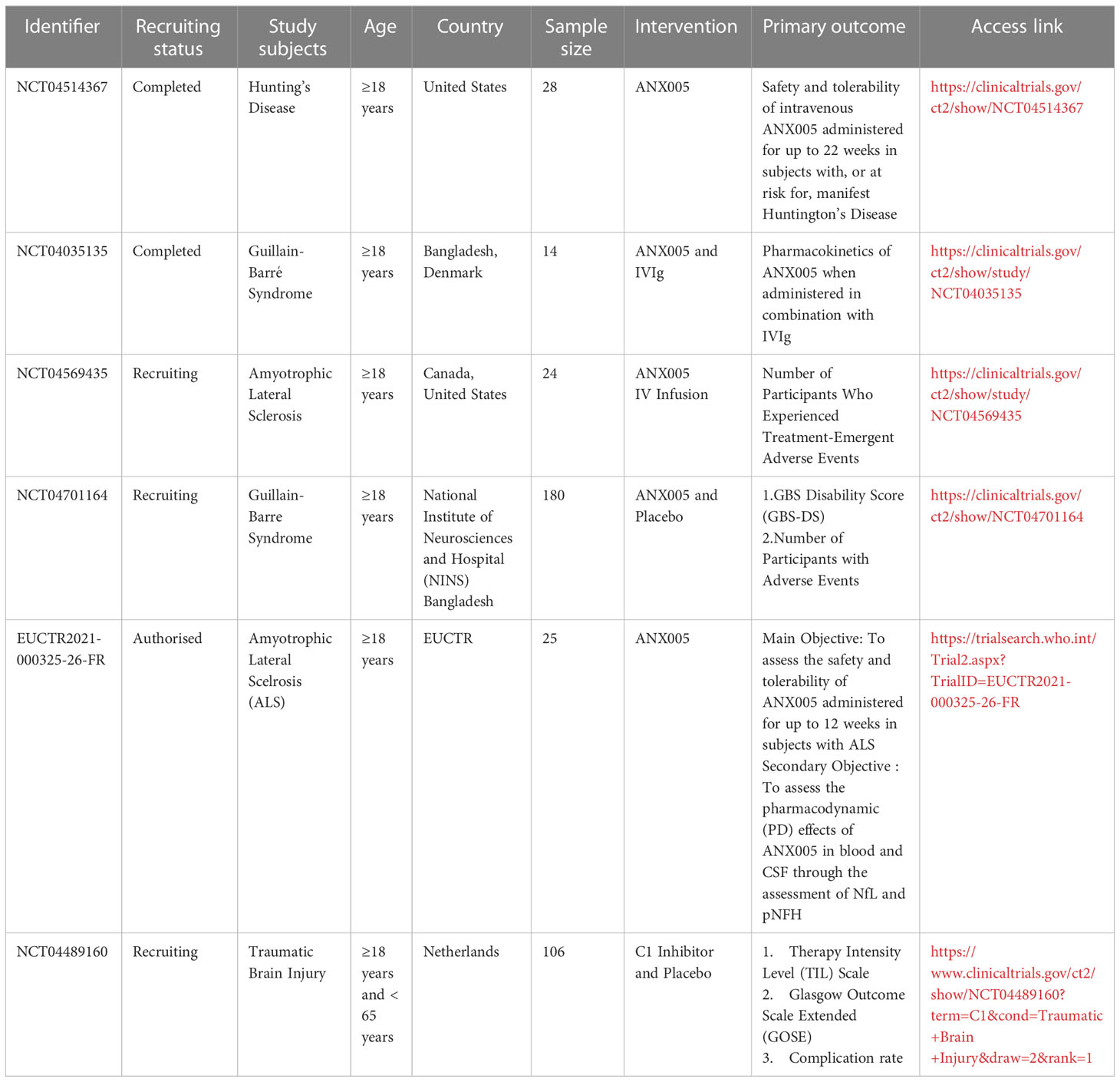
Table 2 Anti-C1q clinical trials on neurological disorders.
4.3 Parkinson’s disease (PD)
PD is the most prevalent movement disorder of the nervous system and the second most common neurodegenerative disease after AD. The lesions are found in the substantia nigra-striatal area and are characterized by the loss of nigrostriatal neurons, striatal dopaminergic deficiency, and the buildup of alpha-synuclein (α-syn) in intraneural inclusions (105). The incidence of PD tends to increase with age, which is significantly associated with age (106). Initially, PD was thought to be a movement disorder without dementia, with major symptoms in the motor system such as bradykinesia (slow movement), rigidity, and resting tremor. However, with the growing awareness of Parkinson’s disease, PD also affects other extrapyramidal dopaminergic, cholinergic, and serotonergic bundles, leading to non-motor symptoms, including loss of smell, sleep disturbances, and constipation, as well as cognitive and psychiatric symptoms, such as dementia and depression (107). Cognitive impairment is six times more prevalent in individuals with PD than in the healthy population (108). It is one of the most significant non-motor manifestations of PD, and cognitive impairment can significantly impair the quality of life and function of PD patients, with the majority of patients developing dementia within 20 years of diagnosis (109). The pathogenesis of PD is still unknown, and clinical data suggest that PD has a genetic origin, with mutated genes including those encoding alpha-synuclein, DJ-1, PINK, LRRK2, and others (110). Numerous studies have highlighted the involvement of both innate and adaptive immune systems in the development of PD (111, 112). Clinical studies have demonstrated that non-steroidal anti-inflammatory drugs (NSAIDs), including ibuprofen (non-aspirin), may reduce the risk of PD and have a protective effect, particularly in long-term, regular users (113, 114). The difficulty in distinguishing early PD from some Four-repeat (4R-) Tauopathies arises from the lack of specificity in clinical presentation. To address this issue, Khosousi investigated serum C1q and C3 levels and found that 4R-Tauopathies had lower levels of complement compared to PD patients and healthy controls (67). Mice models of nigrostriatal pathway injury induced by MPTP have confirmed microglia activation and increased expression of C1q in the nigrostriatal system. However, in the subchronic MPTP model, C1q may act as a mediator of extracellular debris removal by microglia and did not affect nigrostriatal injury (68). These findings were also confirmed in the nigrostriatal SNc in the Depboylu study of PD and control cases (69). PD is characterized by the accumulation of alpha-synuclein (α-syn) in intracellular Lewy bodies (115), α-syn can activate the classical complement pathway by acting at an early step of the complement cascade. α-syn is secreted extracellularly through the formation of transmembrane pores or released as vesicles to bind to cell surface receptors (115, 116). α-syn can be recognized by C1q, which initiates the classical complement pathway and forms a membrane attack complex to induce cell death (70). The relationship between α-syn and the innate immune system could open new avenues for PD treatment.
4.4 Multiple sclerosis (MS)
MS is a chronic autoimmune demyelinating disease of the central nervous system, characterized by lesions in the white matter, gray matter, brainstem, spinal cord, and optic nerve (117). Pathologically, the disease is associated with increased demyelination of white matter, inflammatory response, and gliosis (117, 118). Demyelination is not unique to the white matter, but also involves the gray matter (119, 120). The disease is characterized by recurrent episodes of inflammatory demyelination, which can lead to neurodegeneration, accompanied by a relapse-remission process (121). MS not only leads to physical disability, but also cognitive impairment and a decline in quality of life (122), and an in-depth understanding of the pathogenesis of MS can provide new directions and ideas for its treatment. Several genetic variants, including single-nucleotide polymorphisms and mutations have been identified in complement genes (123). Previous studies have found that genetic abnormalities in the complement pathway are more likely to cause retinal neurodegeneration or increased susceptibility to visual loss in MS patients (124). A study by Vilariño-Güell analyzed the DNA of 132 MS patients and identified 12 genetic variants in genes associated with the immune pathway, providing further evidence that immunity drives MS development (125). Ingram shows that inflammation progression in the MS CNS is not dependent on infiltrating cells; inflammation can be driven by innate immune mechanisms such as complement (126). Complement activation occurs universally in MS, where complement proteins (C1q, C3) persist in MS plaques, and C1q is present in all MS plaques and plays a dominant role in the classical cascade response (126). Watkins analyzed complement expression and activation in the MS deceased organization and found C1qA expression in neurons and glial cells in the MS cortex and deep gray matter, and an increase in the upregulation and number of microglia allergenic toxin receptors in the damaged cortical gray matter area (71). C1q and C3 are deposited at synapses in the MS brain, and microglia phagocytosis of labeled synapses leads to a significant decrease in synaptic density (72). Human cadaveric samples have shown the accumulation of early complement components, including C1q, C4d, Bb, C3b-iC3b, C3d, and MAC in the cortical, hippocampal, and thalamic gray matter of MS patients (72, 127). Ramaglia et al. found a significant increase in C1q expression in the CA2 region of the hippocampus, suggesting a link between cognitive dysfunction and C1q deposition in hippocampal CA2 in MS patients (73). Interestingly, in a study by Hammond et al. using MOG 35-55 induced experimental autoimmune encephalomyelitis (EAE), they found that C1q was not significant in causing hippocampal synapse loss and microglia activation (128). Inflammatory microglia (MIMS) and reactive astrocytes were also found in the MS nervous system. Extracellular vesicles (EVs) of astrocytes containing high levels of C1q, C3, and other complement proteins have been detected in the plasma of MS patients, which are involved in the synaptic loss process in MS (74). A positive correlation has been observed between the number density of C1q+ cells and tissue damage (129). In an experimental analysis of the spinal cord of a mouse model of chronic recurrent experimental autoimmune encephalomyelitis (crEAE), C1q expression increased throughout the disease progression phase, but complement expression decreased during the early and remission phases (75). By knocking out C1q receptors on microglia in a mouse model of MS, Absinta’s team showed that microglia proliferation indicators were largely attenuated, suggesting that C1q inhibition may be a potential way to treat MS (76). However, the Vanguri study recognized that damage to myelin phospholipids can also occur in the absence of antibodies (130). Gao demonstrated in a demyelinating mouse model that C1q may be involved in demyelination by preventing the differentiation of oligodendrocyte progenitors into mature oligodendrocytes through Wnt/β-catenin signaling activation (47).
4.5 Huntington’s disease (HD)
Huntington’s chorea (HD) is an autosomal dominant inherited neurodegenerative disorder (131). It is caused by an abnormally expanded CAG repeat near the N terminus of the Huntington protein gene (HTT), which produces mutant Huntington proteins upon translation(mHTT) (132). HD patients are indistinguishable from normal individuals before the onset of clinical symptoms (133). which usually start with mental changes such as personality, cognition, irritability, forgetfulness, and anxiety. As the disease progresses, motor limb incoordination appears (131, 134). HD patients are often also suicidal (135), and there is a pressing need for more tools and methods to treat this disease. Early recognition of potential suicidal ideation and symptom improvement are imperative. Complement C expression was higher in the brain of HD patients with early disease compared to controls (77), especially in the primary lesion area (136). Francis et al. suggested that C1q may co-mediate with Huntington’s protein to cause HD by inducing apoptosis in the caudate nucleus (136). Lopez-Sanchez et al. injected 3-Nitropropionic acid (NPA) into adult Wistar rats to induce an animal model of HD (137). Protein blotting and immunohistochemical analysis of brain sections from these rats showed an increase in the C3α subunit, a marker of neurotoxic A1 astrocytes, and an upregulation of cytokine IL-1α, TNFα and C1q expression in the striatum, hippocampus and cerebellum, leading to neuronal damage (78). Singhrao analyzed the complement profile of striatum in HD patients versus normal subjects and observed significant astrocyte and microglia proliferation in the caudate nucleus and internal capsule of HD, with increased complement expression, further confirmed by the increased inflammatory response in HD (79). ANX005 is a monoclonal antibody that inhibits C1q, and the safety and tolerability of ANX005 are currently being risk assessed in subjects with significant HD (NCT04514367) (134) (Table 2).
5 Relationship to inflammatory diseases in the nervous system
C1q acts as an ancient substance in the innate immune response and is naturally indispensable for its value in inflammatory diseases of the nervous system. In many diseases of the nervous system, the inflammatory response plays an important role in the pathology of neurological lesions.
5.1 Nerve injury
Injuries to the central nervous system, such as brain and spinal cord, trigger a robust inflammatory response, with non-adaptive immunity being the first line of defense. Numerous studies have emerged with the goal of understanding the mechanisms of nerve injury, reducing injury, improving patient outcomes, and enhancing their quality of life. Research on spinal cord injury (SCI) patients have identified polymorphonuclear leukocytes (PMNs) as the first immune cells to infiltrate the CNS following SCI, and these cells are believed to contribute to subsequent damage after CNS trauma (138). Studies have detected the expression of mRNAs encoding C1q, C3, and C4, as well as the complement proteins C1q and C3, in PMN cells in a rat model of SCI (139). Deposition of C1q was also found at the site of axon pathology and demyelination in SCI model mice (80). Moreover, C1q has been shown to mediate oligodendrocyte death, leading to demyelination and axonal loss (81). Peterson et al. found in myelin isolated from Sprague Dawley rat brain that C1q interacts with myelin-associated glycoprotein (MAG) to reduce MAG’s inhibitory effect on neurons and promote axonal growth (48). MAG is a protein associated with myelin that is produced after nerve injury and can inhibit nerve growth by binding to a common receptor complex composed of NgR, LINGO-1, and p75 ntr (140).
Traumatic brain injury (TBI) is a mechanical injury that results in rupture of brain parenchyma and blood vessels, which can be divided into primary and secondary injuries (141). The complement system promotes secondary injury in TBI, and circulating complement components can enter the brain through the injured blood-brain barrier (BBB), locally reactive microglia can activate complement-producing neurons in response to injury (142). At the site of brain injury, C1q accumulates in microglia/macrophages and neurons, and the brain can also produce a strong subsequent response to TBI by activating the local synthesis of classical and lectin complement pathway activators (82). In the TBI mouse model, it was found that the extensive connection between the thalamus and the cerebral cortex can cause secondary brain damage to the thalamus, leading to inflammation, chronic neurodegeneration, disruption of sleep spindle waves, and the occurrence of epileptic brain waves. C1q was found to accumulate around the thalamus, causing further damage through cascade reactions (83), instead, anti-C1q antibodies were used to treat TBI mice and improve their prognosis. TBI not only presents with physical motor and sensory impairment, but also has a strong correlation with the risk of dementia, especially as age increases and the degree of brain damage becomes more severe (143). Study of long-term memory deficits in aged mice with TBI may depend on the accumulation of early complement cascade components (C1q, C3, and CR3) in the brain, and inhibition of complement responses reduces cognitive impairment, and the presence of these complements may be a potential modifier of cognitive decline in the aged damaged brain (84). Manek et al. found an increased number of released microvesicles and exosome (MV/E) from human TBI cerebrospinal fluid, which is rich in cytoskeletal proteins, synaptophysin, C1q subunit B, etc. This specific MV/E causes complement activation, axonal damage, cell death and other processes (85). The level of serum C1q correlates significantly with the severity of trauma as indicated by the GCS score and the Rotterdam CT classification, and serum C1q may be a biomarker for predicting the prognosis of TBI (86). Clinical trials for complement inhibitors in TBI treatment are ongoing (NCT04489160), with promising potential for future therapies in this area (Table 2).
In cases of neurological injury, the complement system activation triggers an inflammatory response that worsens the injury, but complement can also attract phagocytes to promote repair and regeneration effects after CNS injury (144, 145). In a mouse model of optic nerve injury, C1q was found to play a role in nerve injury repair by interacting with microglia to phagocytose the myelin sheath of the injured optic nerve, thereby facilitating repair (87). Clinical trials examining the use of C1 inhibitors for traumatic brain injury are currently underway (NCT04489160). There are conflicting opinions on the role of C1q in nerve injury, and more research is necessary to determine how to regulate C1q in a way that benefits us.
5.2 Guillain-Barré syndrome
Guillain-Barré syndrome (GBS) is an acute autoimmune disease involving peripheral nerves, characterized by symmetrical progressive flaccid paralysis of the extremities, diminished neurological reflexes, and sensory abnormalities (146). GBS is mainly divided into acute inflammatory demyelinating polyneuropathy (AIDP) (which is the most common type of Grinbarism), acute motor axonal neuropathy (AMAN) and Miller-Fischer syndrome (MFS), depending on the site of onset. The etiology of the disease is still unclear, and the more authoritative factors are molecular mimicry and Campylobacter jejuni infection-related (147). Infection produces cross-reactive antibodies to human peripheral gangliosides, and complement plays important role in its pathogenesis (148). Human ganglioside GM1 antibodies accumulate mainly at the node of ranvier nerve fibers and cause their destruction. Studies on the mechanism of GM1 antibodies in model membranes have revealed that GM1 antibodies form a hexameric ring in the membrane, which can be inhibited by staphylococcal protein A binding between CH2 and CH3, and that binding of the GM1 antibody hexameric CH2 structural domain to each spherical head of C1q causes complement activation, indicating that complement is involved in an important step in the development of Grimballi syndrome disease (149). Experiments using anti-C1q antibodies in a mouse model have shown a reduction in demyelination (88). Treatment of AMAN model mice with C1q antibody can reduce the level of serum C1q and also prevent progressive development of respiratory function and neurological damage in MFS mouse model and prevent further deterioration of the disease (150). The use of anti-C1q antibodies in the treatment of GBS patients is currently being studied in clinical trials (NCT04035135)(NCT04701164) (151) (Table 2).
6 Relationship to glioma
Glioma is the most common type of brain cancer (152), glioblastoma(GBM) is the most malignant form. Currently, the standard treatment for glioblastoma involves surgical resection followed by radiotherapy and temozolomide chemotherapy (153). However, due to its rapid growth rate and poor treatment outcomes, effective management of the disease remains a challenge for clinicians seeking to improve survival time and quality of life for glioma patients. With increasing understanding of the tumor microenvironment, there is growing recognition of the role played by surface-associated proteins in tumor signaling, both between tumor cells and between tumor cells and non-tumor cells. One such protein is complement C1q, which is expressed in the human tumor microenvironment and appears to play a variety of biological roles. Recent studies have identified that complement C1q appears as an immune tolerance and immunosuppressive marker in cells of macrophage populations of healthy and tumor tissues, called tumor-associated macrophages (TAM), which can suppress cellular immunity and promote tumor growth (154, 155). Deposition of C1q and C3 was observed in tumor tissues, suggesting a role for complement in the pathogenesis of GBM (90). Complement activation has been shown to promote carcinogenesis and support the basic needs of malignant cells by maintaining proliferative signaling, angiogenesis, and anti-apoptosis, while also regulating anti-tumor immunity and promoting invasion and migration (156). There is also emerging evidence of the important role played by C1q/TNF-related proteins (CTRPs) in glioblastoma. CTRPs have been identified because they all contain a C1q globular domain, also known as the C1q/TNF superfamily, and the family includes 16 members that have been shown to have a wide range of effects on metabolism, food intake, tumor metastasis, apoptosis, vascular disease, ischemic injury, inflammation (157). Binding of the leucine-rich G protein-coupled relaxin receptor RXFP1 to C1q-tumor necrosis factor-related protein 8 (CTRP8) ligand mediates increased GBM cell migration, protein kinase C pathway activation, and lysosomal protease cathepsin B production in glioblastoma progenitor cells (158–160). Additionally, RXFP1-CTRP8 promotes actin cytoskeletal remodeling and filopodia formation through STAT3 signaling, further enhancing glioblastoma migration (89). C1q/TNF-related protein 1 (CTRP1), a member of the CTRP family, is strongly correlated with glioblastoma multiforme (GBM) and can regulate CCL2 expression, promoting tumor progression (91). Moreover, the expression level of C1q is positively correlated with different grades of glioma, and its expression may serve as a prognostic indicator. Large deposits of C1q were found in the stroma around tumor vessels, indicating that C1q may promote angiogenesis and nutrient supply to tumor cells, facilitating tumor growth (92). C1q plays a role in promoting glioma disease progression, especially through its membrane-bound form, which provides a new idea that C1q can be used as a potential target for the treatment of glioma.
7 Summary
C1q, as the first initiator of the classical complement response, plays an important role in the immune process in the normal physiological state, and is essential for our organism to face microbial invasion, tumor cell elimination and abnormal cell apoptosis, etc. However, if the complement regulation balance is disrupted, uncontrolled activation of C1q can lead to inflammatory and progressive damage to the host organ, generating new pathological effects, and causing various diseases. C1q plays a crucial role in neurodevelopment and neurological diseases, contributing to the development of disease progression and cognitive impairment. There are already C1q antibodies used in animal testing, and there are various types of antibody designs, and continued research is needed on how to produce the best antibodies. Additionally, regulating the activation of microglia may also provide a direction as it has been mentioned that microglia and C1q work together to produce an inflammatory effect. However, a few studies have suggested that C1q plays an active role in the repair of neurological damage, so we still need to further understand the role of C1q in the development of the disease and target interventions to fully utilize the role of C1q. In-depth exploration of the relationship between C1q and the nervous system has shown that C1q has a high potential clinical value in the diagnosis of more neurological diseases, and the amount of complement markers may be a valuable evaluation indicator for the severity and activity of the disease. As research on neurological tumors progresses, the relationship between C1q and neurological tumors continues to be confirmed, providing new directions for the treatment of neurological tumors. Although most of the current studies on the mechanism of C1q and its role in the CNS are performed in animal models, they provide many clues to the understanding of the relationship between C1q and neurological diseases, laying the foundation for the physiological functions played by C1q in the CNS and providing new ideas for the direction of treatment of related diseases. However, there is still a lack of clinical studies on C1q, its potential as a therapeutic target for neurological diseases, and the safety and efficacy of C1q inhibitors in clinical trials. C1q as a new target provides new directions for doctors and pharmaceutical companies to explore new treatment modalities, but more experiments are needed to investigate how C1q can be better applied in the clinic.
Author contributions
WZ and YC wrote this manuscript. YC and HP revised this manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank HP and YC for the critical reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pouw RB, Ricklin D. Tipping the balance: Intricate roles of the complement system in disease and therapy. Semin Immunopathol (2021) 43(6):757–71. doi: 10.1007/s00281-021-00892-7
2. Elieh Ali Komi D, Shafaghat F, Kovanen PT, Meri S. Mast cells and complement system: Ancient interactions between components of innate immunity. Allergy (2020) 75(11):2818–28. doi: 10.1111/all.14413
3. Hosszu KK, Valentino A, Vinayagasundaram U, Vinayagasundaram R, Joyce MG, Ji Y, et al. DC-SIGN, C1q, and gC1qR form a trimolecular receptor complex on the surface of monocyte-derived immature dendritic cells. Blood (2012) 120(6):1228–36. doi: 10.1182/blood-2011-07-369728
4. Son M, Diamond B, Santiago-Schwarz F. Fundamental role of C1q in autoimmunity and inflammation. Immunol Res (2015) 63(1-3):101–6. doi: 10.1007/s12026-015-8705-6
5. Nicholson-Weller A, Klickstein LB. C1q-binding proteins and C1q receptors. Curr Opin Immunol (1999) 11(1):42–6. doi: 10.1016/S0952-7915(99)80008-9
6. Cockram TOJ, Dundee JM, Popescu AS, Brown GC. The phagocytic code regulating phagocytosis of mammalian cells. Front Immunol (2021) 12:629979. doi: 10.3389/fimmu.2021.629979
7. Baldwin WM, Valujskikh A, Fairchild RL. C1q as a potential tolerogenic therapeutic in transplantation. Am J Transplant (2021) 21(11):3519–23. doi: 10.1111/ajt.16705
8. Guo WX, Ghebrehiwet B, Weksler B, Schweitzer K, Peerschke EI. Up-regulation of endothelial cell binding proteins/receptors for complement component C1q by inflammatory cytokines. J Lab Clin Med (1999) 133(6):541–50. doi: 10.1016/S0022-2143(99)90183-X
9. Ghebrehiwet B, Peerschke EIB. cC1q-r (calreticulin) and gC1q-R/p33: Ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol Immunol (2004) 41(2-3):173–83. doi: 10.1016/j.molimm.2004.03.014
10. Gaboriaud C, Frachet P, Thielens NM, Arlaud GJ. The human c1q globular domain: structure and recognition of non-immune self ligands. Front Immunol (2011) 2:92. doi: 10.3389/fimmu.2011.00092
11. Ghebrehiwet B, Hosszu KK, Valentino A, Peerschke EIB. The C1q family of proteins: insights into the emerging non-traditional functions. Front Immunol (2012) 3. doi: 10.3389/fimmu.2012.00052
12. McGreal E, Gasque P. Structure-function studies of the receptors for complement C1q. Biochem Soc Trans (2002) 30(Pt 6):1010–4. doi: 10.1042/bst0301010
13. Kang Y-S, Do Y, Lee H-K, Park SH, Cheong C, Lynch RM, et al. A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell (2006) 125(1):47–58. doi: 10.1016/j.cell.2006.01.046
14. Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P. Differential expression of individual complement regulators in the brain and choroid plexus. Lab Invest (1999) 79(10):1247–59.
15. Verneret M, Tacnet-Delorme P, Osman R, Awad R, Grichine A, Kleman J-P, et al. Relative contribution of c1q and apoptotic cell-surface calreticulin to macrophage phagocytosis. J Innate Immun (2014) 6(4):426–34. doi: 10.1159/000358834
16. Kavvadas E. Autoantibodies specific for C1q, C3b, β2-glycoprotein 1 and annexins may amplify complement activity and reduce apoptosis-mediated immune suppression. Med Hypotheses (2020) 144:110286. doi: 10.1016/j.mehy.2020.110286
17. Cho K. Emerging roles of complement protein C1q in neurodegeneration. Aging Dis (2019) 10(3):652–63. doi: 10.14336/AD.2019.0118
18. Kerdidani D, Aerakis E, Verrou K-M, Angelidis I, Douka K, Maniou M-A, et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J Exp Med (2022) 219(2):e20210815. doi: 10.1084/jem.20210815
19. Zwarthoff SA, Widmer K, Kuipers A, Strasser J, Ruyken M, Aerts PC, et al. C1q binding to surface-bound IgG is stabilized by C1rs proteases. Proc Natl Acad Sci USA (2021) 118(26):e2102787118. doi: 10.1073/pnas.2102787118
20. Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med (2019) 25(3):496–506. doi: 10.1038/s41591-018-0336-8
21. Sasaki S, Nishihira K, Yamashita A, Fujii T, Onoue K, Saito Y, et al. Involvement of enhanced expression of classical complement C1q in atherosclerosis progression and plaque instability: C1q as an indicator of clinical outcome. PloS One (2022) 17(1):e0262413. doi: 10.1371/journal.pone.0262413
22. Donat C, Kölm R, Csorba K, Tuncer E, Tsakiris DA, Trendelenburg M. Complement C1q enhances primary hemostasis. Front Immunol (2020) 11:1522. doi: 10.3389/fimmu.2020.01522
23. Hayuningtyas RA, Han M, Choi S, Kwak MS, Park IH, Lee J-H, et al. The collagen structure of C1q induces wound healing by engaging discoidin domain receptor 2. Mol Med (2021) 27(1):125. doi: 10.1186/s10020-021-00388-y
24. De Gaetano GV, Coppolino F, Lentini G, Famà A, Cullotta C, Raffaele I, et al. Streptococcus pneumoniae binds collagens and C1q via the SSURE repeats of the PfbB adhesin. Mol Microbiol (2022) 117(6):1479–92. doi: 10.1111/mmi.14920
25. Jiao Y, Jiang S, Wang Y, Yu T, Zou G, Zhuo L, et al. Activation of complement C1q and C3 in glomeruli might accelerate the progression of diabetic nephropathy: Evidence from transcriptomic data and renal histopathology. J Diabetes Investig (2022) 13(5):839–49. doi: 10.1111/jdi.13739
26. Kallenberg CGM. Anti-C1q autoantibodies. Autoimmun Rev (2008) 7(8):612–5. doi: 10.1016/j.autrev.2008.06.006
27. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, et al. C1q restrains autoimmunity and viral infection by regulating CD8 T cell metabolism. Science (2018) 360(6388):558–63. doi: 10.1126/science.aao4555
28. Zheng W, Wang X, Liu J, Yu X, Li L, Wang H, et al. Single-cell analyses highlight the proinflammatory contribution of C1q-high monocytes to behçet’s disease. Proc Natl Acad Sci USA (2022) 119(26):e2204289119. doi: 10.1073/pnas.2204289119
29. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74(4):691–705. doi: 10.1016/j.neuron.2012.03.026
30. Perry VH, O’Connor V. C1q: the perfect complement for a synaptic feast? Nat Rev Neurosci (2008) 9(11):807–11. doi: 10.1038/nrn2394
31. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell (2016) 165(4):921–35. doi: 10.1016/j.cell.2016.04.001
32. Kovács RÁ., Vadászi H, Bulyáki É, Török G, Tóth V, Mátyás D, et al. Identification of neuronal pentraxins as synaptic binding partners of C1q and the involvement of NP1 in synaptic pruning in adult mice. Front Immunol (2020) 11:599771. doi: 10.3389/fimmu.2020.599771
33. Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, et al. Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci (2016) 36(19):5185–92. doi: 10.1523/JNEUROSCI.3850-15.2016
34. Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J (2020) 39(16):e105380. doi: 10.15252/embj.2020105380
35. Shirotani K, Hori Y, Yoshizaki R, Higuchi E, Colonna M, Saito T, et al. Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci Rep (2019) 9(1):7508. doi: 10.1038/s41598-019-43535-6
36. Cong Q, Soteros BM, Huo A, Li Y, Tenner AJ, Sia GM. C1q and SRPX2 regulate microglia mediated synapse elimination during early development in the visual thalamus but not the visual cortex. Glia (2022) 70(3):451–65. doi: 10.1002/glia.24114
37. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131(6):1164–78. doi: 10.1016/j.cell.2007.10.036
38. Datta D, Leslie SN, Morozov YM, Duque A, Rakic P, van Dyck CH, et al. Classical complement cascade initiating C1q protein within neurons in the aged rhesus macaque dorsolateral prefrontal cortex. J Neuroinflamm (2020) 17(1):8. doi: 10.1186/s12974-019-1683-1
39. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci (2014) 15(4):209–16. doi: 10.1038/nrn3710
40. Lian H, Yang L, Cole A, Sun L, Chiang ACA, Fowler SW, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with alzheimer’s disease. Neuron (2015) 85(1):101–15. doi: 10.1016/j.neuron.2014.11.018
41. Benoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J Biol Chem (2013) 288(1):654–65. doi: 10.1074/jbc.M112.400168
42. Fan G, Li Q, Qian J. C1q contributes to post-stroke angiogenesis via LAIR1-HIF1α-VEGF pathway. Front Biosci (Landmark Ed) (2019) 24(6):1050–9. doi: 10.2741/4767
43. Bie B, Wu J, Foss JF, Naguib M. Activation of mGluR1 mediates C1q-dependent microglial phagocytosis of glutamatergic synapses in alzheimer’s rodent models. Mol Neurobiol (2019) 56(8):5568–85. doi: 10.1007/s12035-019-1467-8
44. Spurrier J, Nicholson L, Fang XT, Stoner AJ, Toyonaga T, Holden D, et al. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci Transl Med (2022) 14(647):eabi8593. doi: 10.1126/scitranslmed.abi8593
45. Ndoja A, Reja R, Lee S-H, Webster JD, Ngu H, Rose CM, et al. Ubiquitin ligase COP1 suppresses neuroinflammation by degrading c/EBPβ in microglia. Cell (2020) 182(5):1156–69.e12. doi: 10.1016/j.cell.2020.07.011
46. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541(7638):481–7. doi: 10.1038/nature21029
47. Gao Z, Zhang C, Feng Z, Liu Z, Yang Y, Yang K, et al. C1q inhibits differentiation of oligodendrocyte progenitor cells via wnt/β-catenin signaling activation in a cuprizone-induced mouse model of multiple sclerosis. Exp Neurol (2022) 348:113947. doi: 10.1016/j.expneurol.2021.113947
48. Peterson SL, Nguyen HX, Mendez OA, Anderson AJ. Complement protein C1q modulates neurite outgrowth in vitro and spinal cord axon regeneration in vivo. J Neurosci (2015) 35(10):4332–49. doi: 10.1523/JNEUROSCI.4473-12.2015
49. Endo F, Kasai A, Soto JS, Yu X, Qu Z, Hashimoto H, et al. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science (2022) 378(6619):eadc9020. doi: 10.1126/science.adc9020
50. Zhou B, Zuo Y-X, Jiang R-T. Astrocyte morphology: Diversity, plasticity, and role in neurological diseases. CNS Neurosci Ther (2019) 25(6):665–73. doi: 10.1111/cns.13123
51. Borst K, Dumas AA, Prinz M. Microglia: Immune and non-immune functions. Immunity (2021) 54(10):2194–208. doi: 10.1016/j.immuni.2021.09.014
52. Färber K, Cheung G, Mitchell D, Wallis R, Weihe E, Schwaeble W, et al. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res (2009) 87(3):644–52. doi: 10.1002/jnr.21875
53. Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci (2021) 24(3):312–25. doi: 10.1038/s41593-020-00783-4
54. Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA (2018) 115(8):E1896–905. doi: 10.1073/pnas.1800165115
55. Li K, Li J, Zheng J, Qin S. Reactive astrocytes in neurodegenerative diseases. Aging Dis (2019) 10(3):664–75. doi: 10.14336/AD.2018.0720
56. Ding Z-B, Song L-J, Wang Q, Kumar G, Yan Y-Q, Ma C-G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regener Res (2021) 16(9):1702–10. doi: 10.4103/1673-5374.306064
57. Hinkle JT, Dawson VL, Dawson TM. The A1 astrocyte paradigm: New avenues for pharmacological intervention in neurodegeneration. Mov Disord (2019) 34(7):959–69. doi: 10.1002/mds.27718
58. Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature (2021) 599(7883):102–7. doi: 10.1038/s41586-021-03960-y
59. Zha Z, Liu Y-J, Liu S-S, Zhang N, Li J-L, Qi F, et al. Bu shen yi sui capsule promotes myelin repair by modulating the transformation of A1/A2 reactive astrocytes and. Oxid Med Cell Longev (2022) 2022:3800004. doi: 10.1155/2022/3800004
60. Haure-Mirande J-V, Audrain M, Ehrlich ME, Gandy S. Microglial TYROBP/DAP12 in alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol Neurodegener (2022) 17(1):55. doi: 10.1186/s13024-022-00552-w
61. Brosseron F, Maass A, Kleineidam L, Ravichandran KA, González PG, McManus RM, et al. Soluble TAM receptors sAXL and sTyro3 predict structural and functional protection in alzheimer’s disease. Neuron (2022) 110(6):1009–22.e4. doi: 10.1016/j.neuron.2021.12.016
62. Carpanini SM, Torvell M, Bevan RJ, Byrne RAJ, Daskoulidou N, Saito T, et al. Terminal complement pathway activation drives synaptic loss in alzheimer’s disease models. Acta Neuropathol Commun (2022) 10(1):99. doi: 10.1186/s40478-022-01404-w
63. Sta M, Sylva-Steenland RMR, Casula M, de Jong JMBV, Troost D, Aronica E, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis (2011) 42(3):211–20. doi: 10.1016/j.nbd.2011.01.002
64. Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflamm (2016) 13(1):72. doi: 10.1186/s12974-016-0538-2
65. Ziff OJ, Clarke BE, Taha DM, Crerar H, Luscombe NM, Patani R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res (2022) 32(1):71–84. doi: 10.1101/gr.275939.121
66. Lobsiger CS, Boillée S, Pozniak C, Khan AM, McAlonis-Downes M, Lewcock JW, et al. C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci USA (2013) 110(46):E4385–92. doi: 10.1073/pnas.1318309110
67. Khosousi S, Hye A, Velayudhan L, Bloth B, Tsitsi P, Markaki I, et al. Complement system changes in blood in parkinson’s disease and progressive supranuclear Palsy/Corticobasal syndrome. Parkinsonism Relat Disord (2023) 108:105313. doi: 10.1016/j.parkreldis.2023.105313
68. Depboylu C, Schorlemmer K, Klietz M, Oertel WH, Weihe E, Höglinger GU, et al. Upregulation of microglial C1q expression has no effects on nigrostriatal dopaminergic injury in the MPTP mouse model of Parkinson disease. J Neuroimmunol (2011) 236(1-2):39–46. doi: 10.1016/j.jneuroim.2011.05.006
69. Depboylu C, Schäfer MKH, Arias-Carrión O, Oertel WH, Weihe E, Höglinger GU. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J Neuropathol Exp Neurol (2011) 70(2):125–32. doi: 10.1097/NEN.0b013e31820805b9
70. Gregersen E, Betzer C, Kim WS, Kovacs G, Reimer L, Halliday GM, et al. Alpha-synuclein activates the classical complement pathway and mediates complement-dependent cell toxicity. J Neuroinflamm (2021) 18(1):177. doi: 10.1186/s12974-021-02225-9
71. Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, Rees MI, et al. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflamm (2016) 13(1):161. doi: 10.1186/s12974-016-0611-x
72. Michailidou I, Willems JGP, Kooi E-J, van Eden C, Gold SM, Geurts JJG, et al. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol (2015) 77(6):1007–26. doi: 10.1002/ana.24398
73. Ramaglia V, Dubey M, Malpede MA, Petersen N, de Vries SI, Ahmed SM, et al. Complement-associated loss of CA2 inhibitory synapses in the demyelinated hippocampus impairs memory. Acta Neuropathol (2021) 142(4):643–67. doi: 10.1007/s00401-021-02338-8
74. Bhargava P, Nogueras-Ortiz C, Kim S, Delgado-Peraza F, Calabresi PA, Kapogiannis D. Synaptic and complement markers in extracellular vesicles in multiple sclerosis. Mult Scler (2021) 27(4):509–18. doi: 10.1177/1352458520924590
75. Ramaglia V, Jackson SJ, Hughes TR, Neal JW, Baker D, Morgan BP. Complement activation and expression during chronic relapsing experimental autoimmune encephalomyelitis in the biozzi ABH mouse. Clin Exp Immunol (2015) 180(3):432–41. doi: 10.1111/cei.12595
76. Absinta M, Maric D, Gharagozloo M, Garton T, Smith MD, Jin J, et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature (2021) 597(7878):709–14. doi: 10.1038/s41586-021-03892-7
77. Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology (2000) 49(1-2):171–86. doi: 10.1016/S0162-3109(00)80302-1
78. Lopez-Sanchez C, Garcia-Martinez V, Poejo J, Garcia-Lopez V, Salazar J, Gutierrez-Merino C. Early reactive A1 astrocytes induction by the neurotoxin 3-nitropropionic acid in rat brain. Int J Mol Sci (2020) 21(10):3609. doi: 10.3390/ijms21103609
79. Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in huntington’s disease. Exp Neurol (1999) 159(2):362–76. doi: 10.1006/exnr.1999.7170
80. Ankeny DP, Guan Z, Popovich PG. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J Clin Invest (2009) 119(10):2990–9. doi: 10.1172/JCI39780
81. Galvan MD, Luchetti S, Burgos AM, Nguyen HX, Hooshmand MJ, Hamers FPT, et al. Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. J Neurosci (2008) 28(51):13876–88. doi: 10.1523/JNEUROSCI.2823-08.2008
82. Ciechanowska A, Ciapała K, Pawlik K, Oggioni M, Mercurio D, De Simoni M-G, et al. Initiators of classical and lectin complement pathways are differently engaged after traumatic brain injury-Time-Dependent changes in the cortex, striatum, thalamus and hippocampus in a mouse model. Int J Mol Sci (2020) 22(1):45. doi: 10.3390/ijms22010045
83. Holden SS, Grandi FC, Aboubakr O, Higashikubo B, Cho FS, Chang AH, et al. Complement factor C1q mediates sleep spindle loss and epileptic spikes after mild brain injury. Science (2021) 373(6560):eabj2685. doi: 10.1126/science.abj2685
84. Krukowski K, Chou A, Feng X, Tiret B, Paladini M-S, Riparip L-K, et al. Traumatic brain injury in aged mice induces chronic microglia activation, synapse loss, and complement-dependent memory deficits. Int J Mol Sci (2018) 19(12):3753. doi: 10.3390/ijms19123753
85. Manek R, Moghieb A, Yang Z, Kumar D, Kobessiy F, Sarkis GA, et al. Protein biomarkers and neuroproteomics characterization of Microvesicles/Exosomes from human cerebrospinal fluid following traumatic brain injury. Mol Neurobiol (2018) 55(7):6112–28. doi: 10.1007/s12035-017-0821-y
86. Yan X-J, Li Y-B, Liu W, Wu H-Y, Yu G-F. Elevated serum complement C1q levels after traumatic brain injury and its association with poor prognosis. Neuropsychiatr Dis Treat (2022) 18:47–55. doi: 10.2147/NDT.S348682
87. Peterson SL, Li Y, Sun CJ, Wong KA, Leung KS, de Lima S, et al. Retinal ganglion cell axon regeneration requires complement and myeloid cell activity within the optic nerve. J Neurosci (2021) 41(41):8508–31. doi: 10.1523/JNEUROSCI.0555-21.2021
88. Motamed-Gorji N, Matin N, Tabatabaie O, Pavone P, Romano C, Falsaperla R, et al. Biological drugs in Guillain-Barré syndrome: An update. Curr Neuropharmacol (2017) 15(7):938–50. doi: 10.2174/1570159X14666161213114904
89. Glogowska A, Thanasupawat T, Beiko J, Pitz M, Hombach-Klonisch S, Klonisch T. Novel CTRP8-RXFP1-JAK3-STAT3 axis promotes Cdc42-dependent actin remodeling for enhanced filopodia formation and motility in human glioblastoma cells. Mol Oncol (2022) 16(2):368–87. doi: 10.1002/1878-0261.12981
90. Bouwens TAM, Trouw LA, Veerhuis R, Dirven CMF, Lamfers MLM, Al-Khawaja H. Complement activation in glioblastoma multiforme pathophysiology: evidence from serum levels and presence of complement activation products in tumor tissue. J Neuroimmunol (2015) 278:271–6. doi: 10.1016/j.jneuroim.2014.11.016
91. Chen L, Su G. Identification of CTRP1 as a prognostic biomarker and oncogene in human glioblastoma. BioMed Res Int (2019) 2019:2582416. doi: 10.1155/2019/2582416
92. Mangogna A, Belmonte B, Agostinis C, Zacchi P, Iacopino DG, Martorana A, et al. Prognostic implications of the complement protein C1q in gliomas. Front Immunol (2019) 10:2366. doi: 10.3389/fimmu.2019.02366
93. Qin Q, Wang M, Yin Y, Tang Y. The specific mechanism of TREM2 regulation of synaptic clearance in alzheimer’s disease. Front Immunol (2022) 13:845897. doi: 10.3389/fimmu.2022.845897
94. Hansen DV, Hanson JE, Sheng M. Microglia in alzheimer’s disease. J Cell Biol (2018) 217(2):459–72. doi: 10.1083/jcb.201709069
95. Goetzl EJ, Schwartz JB, Abner EL, Jicha GA, Kapogiannis D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol (2018) 83(3):544–52. doi: 10.1002/ana.25172
96. Wu J, Bie B, Foss JF, Naguib M. Amyloid fibril-induced astrocytic glutamate transporter disruption contributes to complement C1q-mediated microglial pruning of glutamatergic synapses. Mol Neurobiol (2020) 57(5):2290–300. doi: 10.1007/s12035-020-01885-7
97. Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet (2007) 369(9578):2031–41. doi: 10.1016/S0140-6736(07)60944-1
98. Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol (2017) 13(2):96–104. doi: 10.1038/nrneurol.2016.182
99. Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci (2007) 27(34):9201–19. doi: 10.1523/JNEUROSCI.1470-07.2007
100. Dafinca R, Barbagallo P, Farrimond L, Candalija A, Scaber J, Ababneh N, et al. Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep (2020) 14(5):892–908. doi: 10.1016/j.stemcr.2020.03.023
101. Ho M-M, Manughian-Peter A, Spivia WR, Taylor A, Fraser DA. Macrophage molecular signaling and inflammatory responses during ingestion of atherogenic lipoproteins are modulated by complement protein C1q. Atherosclerosis (2016) 253:38–46. doi: 10.1016/j.atherosclerosis.2016.08.019
102. Ye L, Jia G, Li Y, Wang Y, Chen H, Yu L, et al. C1q/TNF-related protein 4 restores leptin sensitivity by downregulating NF-κB signaling and microglial activation. J Neuroinflamm (2021) 18(1):159. doi: 10.1186/s12974-021-02167-2
103. Zheng S, Ren J, Gong S, Qiao F, He J. CTRP9 protects against MIA-induced inflammation and knee cartilage damage by deactivating the MAPK/NF-κB pathway in rats with osteoarthritis. Open Life Sci (2020) 15(1):971–80. doi: 10.1515/biol-2020-0105
104. Peng AYT, Agrawal I, Ho WY, Yen Y-C, Pinter AJ, Liu J, et al. Loss of TDP-43 in astrocytes leads to motor deficits by triggering A1-like reactive phenotype and triglial dysfunction. Proc Natl Acad Sci USA (2020) 117(46):29101–12. doi: 10.1073/pnas.2007806117
105. Ahn EH, Kang SS, Liu X, Chen G, Zhang Z, Chandrasekharan B, et al. Initiation of parkinson’s disease from gut to brain by δ-secretase. Cell Res (2020) 30(1):70–87. doi: 10.1038/s41422-019-0241-9
106. de Lau LML, Breteler MMB. Epidemiology of parkinson’s disease. Lancet Neurol (2006) 5(6):525–35. doi: 10.1016/S1474-4422(06)70471-9
107. Jankovic J, Tan EK. Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry (2020) 91(8):795–808. doi: 10.1136/jnnp-2019-322338
108. Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sørensen P. Risk of dementia in parkinson’s disease: A community-based, prospective study. Neurology (2001) 56(6):730–6. doi: 10.1212/WNL.56.6.730
109. Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Ray Chaudhuri K, et al. Parkinson Disease-associated cognitive impairment. Nat Rev Dis Primers (2021) 7(1):47. doi: 10.1038/s41572-021-00280-3
110. Watanabe R, Buschauer R, Böhning J, Audagnotto M, Lasker K, Lu T-W, et al. The in situ structure of parkinson’s disease-linked LRRK2. Cell (2020) 182(6):1508–18.e16. doi: 10.1016/j.cell.2020.08.004
111. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest (2009) 119(1):182–92. doi: 10.1172/JCI36470
112. Sabatino JJ, Pröbstel A-K, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci (2019) 20(12):728–45. doi: 10.1038/s41583-019-0233-2
113. Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: A meta-analysis. Neurology (2010) 74(12):995–1002. doi: 10.1212/WNL.0b013e3181d5a4a3
114. Poly TN, Islam MMR, Yang H-C, Li Y-CJ. Non-steroidal anti-inflammatory drugs and risk of parkinson’s disease in the elderly population: A meta-analysis. Eur J Clin Pharmacol (2019) 75(1):99–108. doi: 10.1007/s00228-018-2561-y
115. Ferreira DG, Temido-Ferreira M, Vicente Miranda H, Batalha VL, Coelho JE, Szegö ÉM, et al. α-synuclein interacts with PrP to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci (2017) 20(11):1569–79. doi: 10.1038/nn.4648
116. Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, et al. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc Natl Acad Sci USA (2005) 102(30):10427–32. doi: 10.1073/pnas.0502066102
117. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med (2018) 378(2):169–80. doi: 10.1056/NEJMra1401483
119. Bø L, Vedeler CA, Nyland HI, Trapp BD, Mørk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol (2003) 62(7):723–32. doi: 10.1093/jnen/62.7.723
120. Lucchinetti CF, Popescu BFG, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med (2011) 365(23):2188–97. doi: 10.1056/NEJMoa1100648
121. Amato MP, Portaccio E, Goretti B, Zipoli V, Hakiki B, Giannini M, et al. Cognitive impairment in early stages of multiple sclerosis. Neurol Sci (2010) 31(Suppl 2):S211–4. doi: 10.1007/s10072-010-0376-4
122. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: A review. JAMA (2021) 325(8):765–79. doi: 10.1001/jama.2020.26858
123. Hecker M, Fitzner B, Putscher E, Schwartz M, Winkelmann A, Meister S, et al. Implication of genetic variants in primary microRNA processing sites in the risk of multiple sclerosis. EBioMedicine (2022) 80:104052. doi: 10.1016/j.ebiom.2022.104052
124. Fitzgerald KC, Kim K, Smith MD, Aston SA, Fioravante N, Rothman AM, et al. Early complement genes are associated with visual system degeneration in multiple sclerosis. Brain (2019) 142(9):2722–36. doi: 10.1093/brain/awz188
125. Vilariño-Güell C, Zimprich A, Martinelli-Boneschi F, Herculano B, Wang Z, Matesanz F, et al. Exome sequencing in multiple sclerosis families identifies 12 candidate genes and nominates biological pathways for the genesis of disease. PloS Genet (2019) 15(6):e1008180. doi: 10.1371/journal.pgen.1008180
126. Ingram G, Loveless S, Howell OW, Hakobyan S, Dancey B, Harris CL, et al. Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol Commun (2014) 2:53. doi: 10.1186/2051-5960-2-53
127. Cooze BJ, Dickerson M, Loganathan R, Watkins LM, Grounds E, Pearson BR, et al. The association between neurodegeneration and local complement activation in the thalamus to progressive multiple sclerosis outcome. Brain Pathol (2022) 32(5):e13054. doi: 10.1111/bpa.13054
128. Hammond JW, Bellizzi MJ, Ware C, Qiu WQ, Saminathan P, Li H, et al. Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain Behav Immun (2020) 87:739–50. doi: 10.1016/j.bbi.2020.03.004
129. Loveless S, Neal JW, Howell OW, Harding KE, Sarkies P, Evans R, et al. Tissue microarray methodology identifies complement pathway activation and dysregulation in progressive multiple sclerosis. Brain Pathol (2018) 28(4):507–20. doi: 10.1111/bpa.12546
130. Vanguri P, Koski CL, Silverman B, Shin ML. Complement activation by isolated myelin: activation of the classical pathway in the absence of myelin-specific antibodies. Proc Natl Acad Sci USA (1982) 79(10):3290–4. doi: 10.1073/pnas.79.10.3290
131. Walker FO. Huntington’s disease. Lancet (2007) 369(9557):218–28. doi: 10.1016/S0140-6736(07)60111-1
132. Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in huntington’s disease. Neuron (2019) 101(5):801–19. doi: 10.1016/j.neuron.2019.01.039
133. Myers RH. Huntington’s disease genetics. NeuroRx (2004) 1(2):255–62. doi: 10.1602/neurorx.1.2.255
134. Kim A, Lalonde K, Truesdell A, Gomes Welter P, Brocardo PS, Rosenstock TR, et al. New avenues for the treatment of huntington’s disease. Int J Mol Sci (2021) 22(16):8363. doi: 10.3390/ijms22168363
135. Paulsen JS, Hoth KF, Nehl C, Stierman L. Critical periods of suicide risk in huntington’s disease. Am J Psychiatry (2005) 162(4):725–31. doi: 10.1176/appi.ajp.162.4.725
136. Francis K, van Beek J, Canova C, Neal JW, Gasque P. Innate immunity and brain inflammation: the key role of complement. Expert Rev Mol Med (2003) 5(15):1–19. doi: 10.1017/S1462399403006252
137. Lopez-Sanchez C, Poejo J, Garcia-Lopez V, Salazar J, Garcia-Martinez V, Gutierrez-Merino C. Kaempferol prevents the activation of complement C3 protein and the generation of reactive A1 astrocytes that mediate rat brain degeneration induced by 3-nitropropionic acid. Food Chem Toxicol (2022) 164:113017. doi: 10.1016/j.fct.2022.113017
138. Saville LR, Pospisil CH, Mawhinney LA, Bao F, Simedrea FC, Peters AA, et al. A monoclonal antibody to CD11d reduces the inflammatory infiltrate into the injured spinal cord: A potential neuroprotective treatment. J Neuroimmunol (2004) 156(1-2):42–57. doi: 10.1016/j.jneuroim.2004.07.002
139. Nguyen HX, Galvan MD, Anderson AJ. Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes in vitro and in vivo after spinal cord injury. J Neuroinflamm (2008) 5:26. doi: 10.1186/1742-2094-5-26
140. Chaudhry N, Filbin MT. Myelin-associated inhibitory signaling and strategies to overcome inhibition. J Cereb Blood Flow Metab (2007) 27(6):1096–107. doi: 10.1038/sj.jcbfm.9600407
141. Al Nimer F, Lindblom R, Ström M, Guerreiro-Cacais AO, Parsa R, Aeinehband S, et al. Strain influences on inflammatory pathway activation, cell infiltration and complement cascade after traumatic brain injury in the rat. Brain Behav Immun (2013) 27(1):109–22. doi: 10.1016/j.bbi.2012.10.002
142. Bellander B-M, Bendel O, Von Euler G, Ohlsson M, Svensson M. Activation of microglial cells and complement following traumatic injury in rat entorhinal-hippocampal slice cultures. J Neurotrauma (2004) 21(5):605–15. doi: 10.1089/089771504774129937
143. Schönberger M, Ponsford J, Reutens D, Beare R, O’Sullivan R. The relationship between age, injury severity, and MRI findings after traumatic brain injury. J Neurotrauma (2009) 26(12):2157–67. doi: 10.1089/neu.2009.0939
144. Hammad A, Westacott L, Zaben M. The role of the complement system in traumatic brain injury: A review. J Neuroinflamm (2018) 15(1):24. doi: 10.1186/s12974-018-1066-z
145. Jones TB, McDaniel EE, Popovich PG. Inflammatory-mediated injury and repair in the traumatically injured spinal cord. Curr Pharm Des (2005) 11(10):1223–36. doi: 10.2174/1381612053507468
146. van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barré Syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol (2014) 10(8):469–82. doi: 10.1038/nrneurol.2014.121
147. Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-Barré Syndrome. Lancet (2021) 397(10280):1214–28. doi: 10.1016/S0140-6736(21)00517-1
148. Laman JD, Huizinga R, Boons G-J, Jacobs BC. Guillain-Barré Syndrome: Expanding the concept of molecular mimicry. Trends Immunol (2022) 43(4):296–308. doi: 10.1016/j.it.2022.02.003
149. Yanaka S, Yogo R, Watanabe H, Taniguchi Y, Satoh T, Komura N, et al. On-membrane dynamic interplay between anti-GM1 IgG antibodies and complement component C1q. Int J Mol Sci (2019) 21(1):147. doi: 10.3390/ijms21010147
150. McGonigal R, Cunningham ME, Yao D, Barrie JA, Sankaranarayanan S, Fewou SN, et al. C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy. Acta Neuropathol Commun (2016) 4:23. doi: 10.1186/s40478-016-0291-x
151. Rajabally YA. Immunoglobulin and monoclonal antibody therapies in Guillain-Barré syndrome. Neurotherapeutics (2022) 19(3):885–96. doi: 10.1007/s13311-022-01253-4
152. Gusyatiner O, Hegi ME. Glioma epigenetics: From subclassification to novel treatment options. Semin Cancer Biol (2018) 51:50–8. doi: 10.1016/j.semcancer.2017.11.010
153. Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang Z, et al. Glioma targeted therapy: insight into future of molecular approaches. Mol Cancer (2022) 21(1):39. doi: 10.1186/s12943-022-01513-z
154. Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell (2021) 184(11):2988–3005.e16. doi: 10.1016/j.cell.2021.04.038
155. Khaliq AM, Erdogan C, Kurt Z, Turgut SS, Grunvald MW, Rand T, et al. Refining colorectal cancer classification and clinical stratification through a single-cell atlas. Genome Biol (2022) 23(1):113. doi: 10.1186/s13059-022-02677-z
156. Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res (2010) 8(11):1453–65. doi: 10.1158/1541-7786.MCR-10-0225
157. Li Y, Wright GL, Peterson JM. C1q/TNF-related protein 3 (CTRP3) function and regulation. Compr Physiol (2017) 7(3):863–78. doi: 10.1002/cphy.c160044
158. Glogowska A, Kunanuvat U, Stetefeld J, Patel TR, Thanasupawat T, Krcek J, et al. C1q-tumour necrosis factor-related protein 8 (CTRP8) is a novel interaction partner of relaxin receptor RXFP1 in human brain cancer cells. J Pathol (2013) 231(4):466–79. doi: 10.1002/path.4257
159. Thanasupawat T, Glogowska A, Burg M, Krcek J, Beiko J, Pitz M, et al. C1q/TNF-related peptide 8 (CTRP8) promotes temozolomide resistance in human glioblastoma. Mol Oncol (2018) 12(9):1464–79. doi: 10.1002/1878-0261.12349
160. Klonisch T, Glogowska A, Thanasupawat T, Burg M, Krcek J, Pitz M, et al. Structural commonality of C1q TNF-related proteins and their potential to activate relaxin/insulin-like family peptide receptor 1 signalling pathways in cancer cells. Br J Pharmacol (2017) 174(10):1025–33. doi: 10.1111/bph.13559
Keywords: c1q, central nervous system, neurodevelopment, degenerative diseases, inflammatory diseases, glioma
Citation: Zhang W, Chen Y and Pei H (2023) C1q and central nervous system disorders. Front. Immunol. 14:1145649. doi: 10.3389/fimmu.2023.1145649
Received: 16 January 2023; Accepted: 07 March 2023;
Published: 23 March 2023.
Edited by:
Heela Sarlus, Karolinska Institutet (KI), SwedenReviewed by:
Rasmus Berglund, Karolinska Institutet (KI), SwedenDavis M. Borucki, Medical University of South Carolina, United States
Copyright © 2023 Zhang, Chen and Pei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Pei, cGVpaHVpQHp6dS5lZHUuY24=