 Wenyuan Li1†
Wenyuan Li1† Yao Wang2*†
Yao Wang2*†- 1Department of Anesthesiology, Renmin Hospital of Wuhan University, Wuhan, Hubei, China
- 2Department of Infectious Diseases, Renmin Hospital of Wuhan University, Wuhan, Hubei, China
Eukaryotic cells are stimulated by external pressure such as that derived from heat shock, oxidative stress, nutrient deficiencies, or infections, which induce the formation of stress granules (SGs) that facilitates cellular adaptation to environmental pressures. As aggregated products of the translation initiation complex in the cytoplasm, SGs play important roles in cell gene expression and homeostasis. Infection induces SGs formation. Specifically, a pathogen that invades a host cell leverages the host cell translation machinery to complete the pathogen life cycle. In response, the host cell suspends translation, which leads to SGs formation, to resist pathogen invasion. This article reviews the production and function of SGs, the interaction between SGs and pathogens, and the relationship between SGs and pathogen-induced innate immunity to provide directions for further research into anti-infection and anti-inflammatory disease strategies.
1 Introduction
When cells are subjected to various degrees of damage, such as that induced by viral or bacterial infection, heat shock, oxidative stress, or nutrient starvation, membrane-less RNA/protein molecular condensates, known as stress granules (SGs), are formed in the cytoplasm (1). The generation of SGs is generally believed to be caused by abrogated translation that is induced to prevent abnormal protein synthesis, with the transcription and translation of translation-stalled nucleoproteins resuming in the absence of external stimuli or with SGs being captured during processing body (PB) disassembly (2). SGs are dynamic membrane-free cytoplasmic structures that are rapidly modified in response to changes in the external environment (3). SGs in the cytoplasm are complexes comprising protein and RNA, and the SGs composition varies according to different conditions (4).
Studies have shown that SGs are closely related to viral infections (5), genetic diseases (6), tumors (7) and other diseases. Because SGs are involved in innate immune regulatory processes, most of the studies on SGs have been focused on the field of virology. Viruses are characterized by their ability to hinder the growth of host cells in a way that is conducive to viral replication and spread, and many virus-infected cells show an increased SGs formation rate and blocked protein translation. Since a virus relies on the cellular translation mechanism for protein synthesis, SGs can hinder the replication of a virus in a cell while affecting the expression of cellular proteins. To escape host cell resistance, viruses have evolved various strategies to disrupt SGs formation and promote their own replication (8). The formation of SGs in most virus-infected cells is caused by the phosphorylation of eukaryotic translation initiation factor (TIF) 2α (eIF2α) through the activation of protein kinase R (PKR). SGs, as players downstream of the cellular stress response, may play a role in viral replication and may be promoters of cellular innate immune responses to viral infection.
An RNA virus or a DNA virus can induce or regulate the production of SGs in host cells during infection. Viruses play different regulatory roles on SGs formation throughout the replication cycle. Moreover, SGs act on viruses. Although research on the relationship between bacterial infection and SGs is in its infancy, bacteria are clearly closely related to SGs formation and change the level of intracellular p-eIF2α (9). Moreover, similar to cellular responses and viral infection, SGs and cellular inflammatory responses are related. For example, recent reports have shown that SGs attenuated macrophage-mediated cell inflammatory damage (10) and further regulated cell survival and pyroptosis by inhibiting inflammasome activation (10). In contrast, certain proinflammatory factors, such as IFN-γ and TNF-α, promote the formation of SGs through the phosphorylation of eIF2α (11). Despite reports indicating correlations between infective (viruses and bacteria) inflammatory factors and SGs, the molecular mechanisms underlying SGs formation remain unclear. Therefore, the correlations between infective and inflammatory factors and SGs deserves further exploration. This article reviews recent reports on the relationship among infective and inflammatory factors and SGs to identify ideas for finding new therapeutic targets for infection and inflammation.
2 SGs formation
2.1 SGs components
SGs are membraneless organelles produced by cells in response to stress-inducing external environmental stimuli (such as inflammatory stimuli, heat shock, oxidative stress, ischemia or viral infection) and temporarily abrogate the translation of most mRNAs. These untranslated mRNAs and related RNA-binding proteins (RBPs) form dynamic SGs in the cytoplasm. Studies have shown that the main components, which are shown in Figure 1; specifically, SGs comprise the following 7 components: (1) translation-blocked mRNAs that are protected from degradation (12); (2) mRNA-targeted TIFs, such as eukaryotic initiation factor 4E (eIF4E), elF3, poly(A)-binding protein 1 (PABPC1), phosphorylated elF2α (p-elF2α) and elF5a (13); (3) RBPs that regulate translation and protect mRNA stability, such as T-cell-restricted intracellular antigen-1 (TIA-1), TIA-related protein (TIAR), human antigen R/Elav-like RNA binding protein 1 (HuR/ELAVL1), fragment X intellectual disability protein (FMRP) and pumilio RNA-binding family member 1 (PUM1) (13); (4) mRNA metabolism-related proteins, such as Ras GTPase-activating protein-binding protein 1 (G3BP1), G3BP2, DEAD-box RNA helicase 6 (DDX6 or Rck/p54), plasma membrane-related Ca2+-ATPase 1 (PMR1), survival motor neuron protein (SMN), staufen1 (STAU1), DEAH (Asp-Glu-Ala-His) box polypeptide 36 (DHX36), cytoplasmic activation/proliferation-associated protein-1 (caprin-1), Z-DNA-binding protein 1 (ZBP1), histone deacetylase 6 (HDAC6) and adenosine deaminase acting on RNA1 (ADAR1) (14); (5) signaling proteins, such as mechanistic target of rapamycin (mTOR), receptor of activated protein kinase C1 (RACK1) and TNF receptor-associated factor 2 (TRAF2) (15); (6) expression products of interferon-stimulated genes (ISGs), such as PKR, RNA-sensing RIG-I-like receptor (RIG-I), melanoma differentiation-associated gene 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2), ribonuclease L (RNase L) and oligoadenylate synthetase (OAS) (16); and (7) regulatory proteins involved in SGs formation, such as apolipoprotein B mRNA-editing catalytic polypeptide enzyme-like 3G (APOBEC3G or A3G), argonaute 2 (Ago2), B-related factor 1 (BRF1), DDX3, Fas-activated serine/threonine phosphoprotein (FAST) and tristetraprolin (TTP) (17).
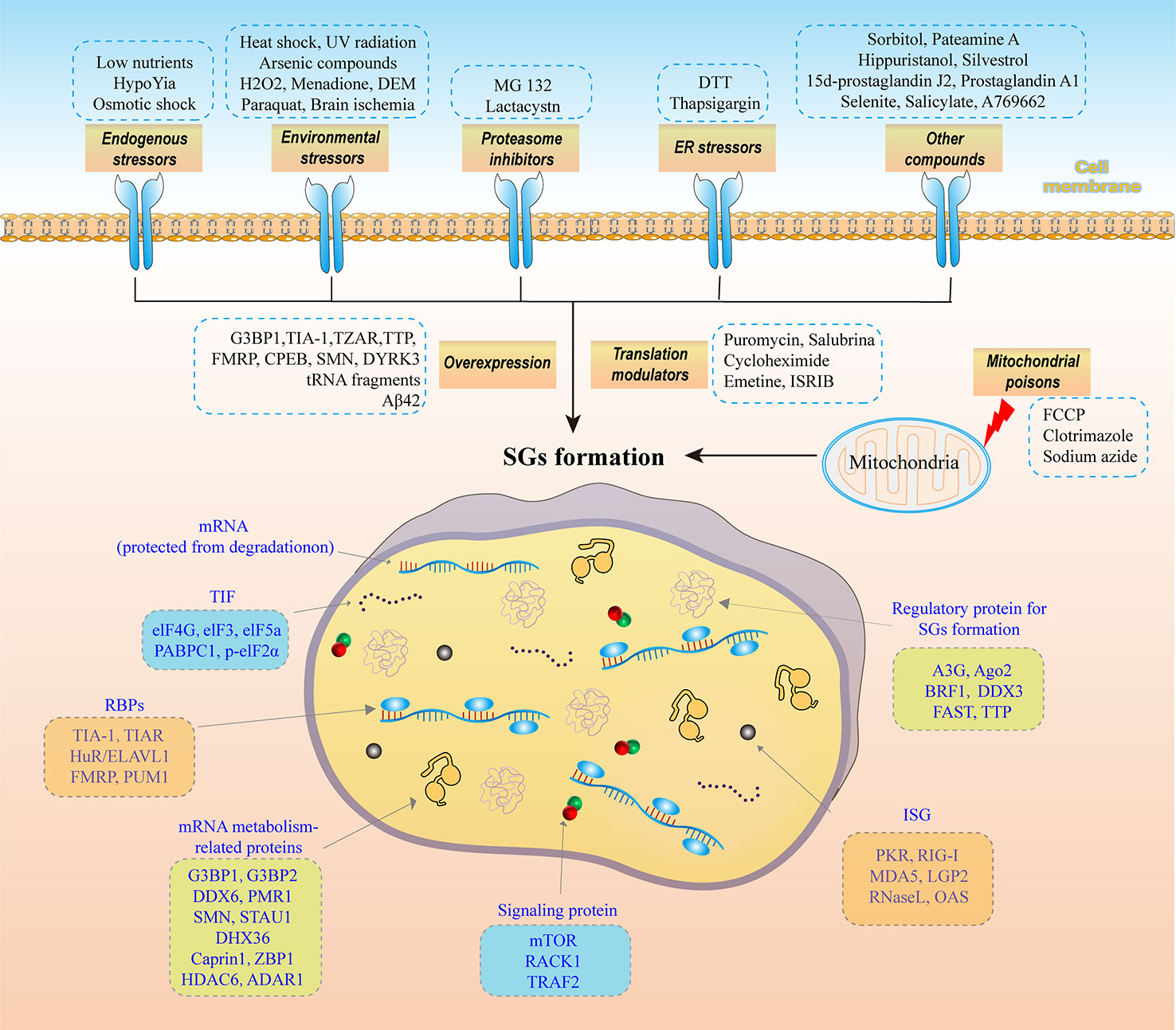
Figure 1 The main components of SGs and the factors that induce SGs formation. SGs comprise the following 7 components: (1) mRNAs that are protected from degradation. (2) TIFs targeting mRNAs, such as eIF4E, elF3, PABPC1, p-elF2α and elF5a; (3) RNA-binding proteins (RBPs) that regulate translation and protect mRNA stability, such as TIA-1, TIAR, HuR/ELAVL1, FMRP and PUM1; (4) mRNA metabolism-related proteins, such as G3BP1, G3BP2, DDX6, PMR1, SMN, STAU1, DHX36, caprin-1, ZBP1, HDAC6 and ADAR1; (5) signaling proteins, such as mTOR, RACK1 and TRAF; (6) expression products of interferon-stimulated genes (ISGs), such as PKR, RIG-I, MDA5 and LGP2, RNase L and OAS; and (7) regulatory proteins in SGs formation, such as APOBEC3G, Ago2, BRF1, DDX3, FAST and TTP. Eight factors induce SGs formation: (1) endogenous stressors, such as low-level nutrients, hypoxia, and osmotic shock; (2) environmental stressors, such as heat shock, UV radiation, arsenic compounds, H2O2, menadione, diethyl maleate (DEM), paraquat, and brain ischemia; (3) overexpressed G3BP1, TIA-1, TZAR, TTP, FMRP, CPEB, SMN, DYRK3, tRNA fragments, and Aβ42; (4) translation modulators, such as puromycin, salubrinal, cycloheximide, emetine, and ISRIB; (5) proteasome inhibitors, such as MG 132 and lactacystin; (6) ER stressors, such as DTT and thapsigargin; (7) mitochondrial poisons, such as FCCP, clotrimazole, and sodium azide; and (8) other compounds, such as sorbitol, pateamine A, hippuristanol, silvestrol, 15d-prostaglandin J2 (15d-PGJ2); prostaglandin A1, selenite, salicylate, and A769662.
Notably, G3BP1 is a phosphorylation-dependent endonuclease and a core protein that mediates SGs assembly. G3BP1 consists of an N-terminal nuclear transport factor 2 (NTF2) domain, a C-terminal receptor-binding domain (RBD) and an intermediate intrinsically disordered region (IDR) (18). The formation of SGs begins with the establishment of a core SGs network, which includes the G3BP1 protein, with interwoven protein-protein, protein-RNA, and RNA-RNA interacting pairs. In a mutually synergistic process, the G3BP1 complex induces phase separation, which leads to SGs assembly (19). Studies have shown that SGs assembly can be completely inhibited only after both G3BP1 and G3BP2 (G3BP2 and G3BP1 are highly similar) are knocked out (19). However, knocking out G3BP1, G3BP2, DDX3X, Caprin1 or another important SGs component alone does not completely inhibit SGs assembly. In addition, a G3BP1-mutant protein lacking the NTF2 or RBD domain failed to mediate SGs assembly, while G3BP1 with the both dual deletion of IDR1 and IDR2 mediated the SGs assembly process (20).
2.2 The driving force of SGs assembly: liquid–liquid phase separation
Hyman et al. first introduced the physicochemical concept of LLPS into biological research with the study of P granules in 2009 (21). Cell differentiation fate is determined through the asymmetric distribution of P granules as mediated by LLPS. LLPS in cells is initiated by the collision among different proteins and nucleic acid molecules, ultimately leading to aggregates that form droplet-like structures through multivalent weak interactions and ultimately specific cellular compartments. In addition to P granules, intracellular membraneless organelles such as SGs and paraspeckles are formed via LLPS (22). LLPS explains how specific molecules in cells aggregate, and how these aggregates are maintained in a state with specific fluidity without the involvement of membrane structures and physiologically function in specific areas within cells. Hence, LLPS is considered to be the main driver of membraneless organelle formation (23).
However, the fundamental criteria underlying the assembly and structure of SGs differ from those of similar P granule structures. Ribosomal machinery is constantly moving along the RNA assembly line, isolating genetic information from the environment and producing proteins necessary for the cell to survive. When external pressure disrupts the assembly line, the RNA is stripped away from ribosomes and aggregated into SGs and P granules. SGs protein assembly requires a specific set of building blocks that require a certain number of chemical linkers that grab RNA strands to bring them together. This linker, called an RNA-binding domain, is encoded in a variety of proteins. However, these linkers show different affinities that determine their biological functions. G3BP1 is the most abundant SGs protein with the greatest network affinities. Specifically, G3BP1 preferentially interacts with other proteins that also bind RNA. The reason that SGs and P granules are found together is that their networks overlap, exhibiting glue-like adhesiveness (18). In addition, membrane organelles affect the formation and function of SGs through LLPS; for example, the endoplasmic reticulum (ER) directly contacts SGs through ribonucleoprotein (RNP) bodies and thus regulates LLPS-mediated separation of SGs components (24). The membranous structure of the ER provides a platform for phase separation and actively participates in LLPS regulation. Conversely, LLPS affects the organization, storage, and release of membranous organelles.
2.3 Mechanism of SGs formation via p-eIF2α-dependent and -independent pathways
The evidence obtained thus far has suggested that the formation of SGs is achieved mainly by the phosphorylation of eIF2α and interference in the function of eIF4F, namely, via the p-eIF2α-dependent pathway and p-eIF2α-independent pathways (12). As shown on the left side of Figure 2, the phosphorylation of the α subunit of eIF2 is the most important initiator of SGs formation, as translation initiation is regulated by the phosphorylation of eIF2α. Four serine/threonine kinases can sense different types of stresses and phosphorylate the serine residue at position 51 of eIF2α: heme-regulated eIF2α kinase (HRI), general control nonderepressible 2 (GCN2), PKR and PKR-like ER kinase (PERK). GCN2 mainly senses the activation of intracellular stress caused by amino acid starvation (25). PERK is activated by endoplasmic reticulum stress (ERS) (26). PKR is a component of the interferon response mechanism and is activated by oxidative double-stranded RNA (dsRNA) in cells (27). HRI is activated after sensing changes in heme heavy metal levels, oxidative stress, thermal aggression, etc. (28).
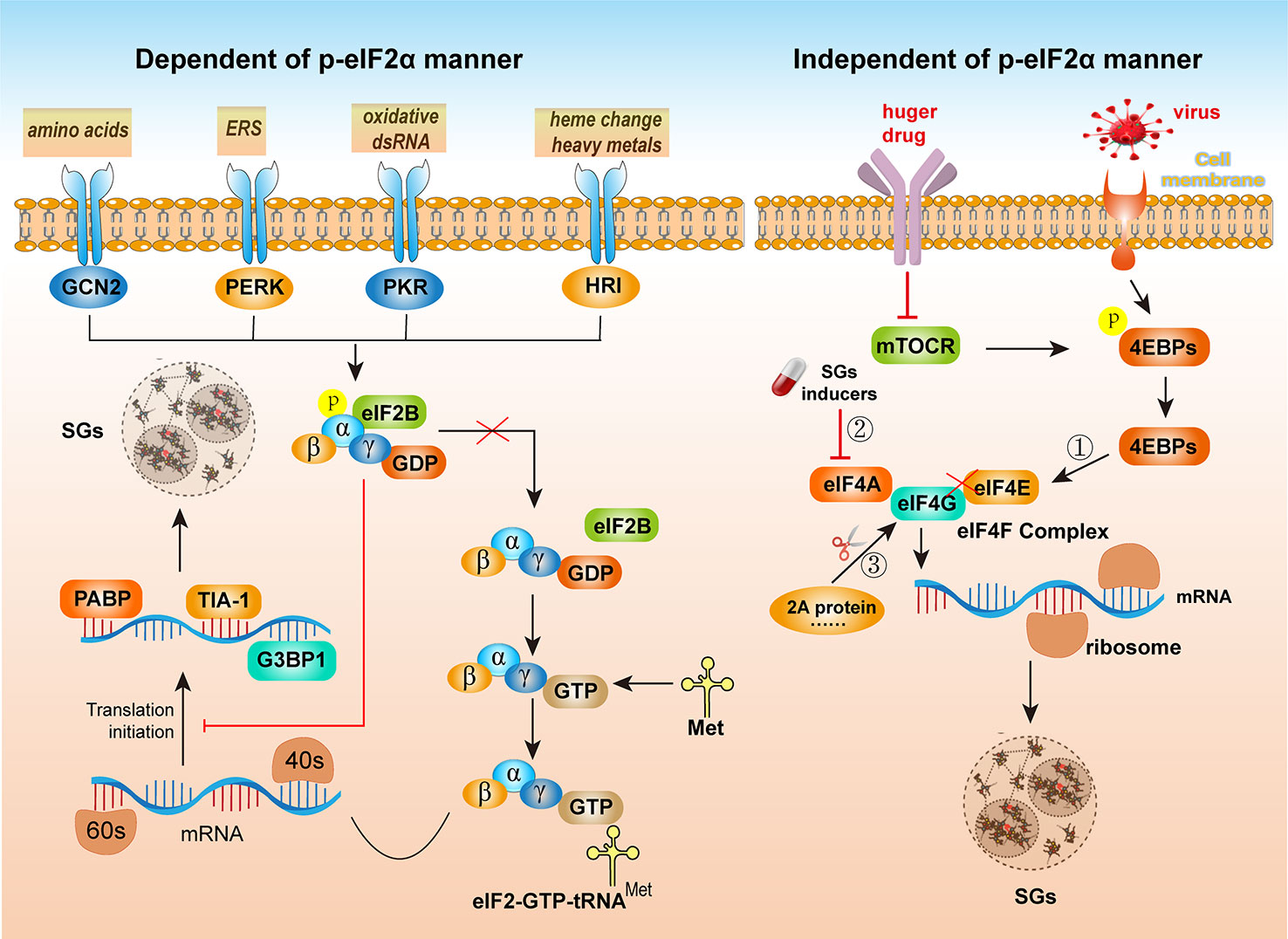
Figure 2 The formation of SGs is dependent or independent of eIF2α phosphorylation. (Left) Via different external stimuli, such as amino acids, ER stress (ERS), oxidative double-stranded RNA (dsRNA), and changes in heme heavy metal levels, four kinases, GCN2, PERK, PKR and HRI, can be activated. The α subunit of eIF2 is phosphorylated, preventing its separation from eIF2B. Subsequently, translation initiation is blocked. The mRNAs and certain proteins involved in the translation process aggregate to form SGs. Before recruiting the 60S ribosome to start translation, the eIF2-GTP-tRNAMet trimer complex is recruited to the mRNA/eIF4F complex bound to the 40S ribosomal subunit, presenting messenger RNA linked to the translation initiation amino acid to the 40S ribosomal subunit. However, when eIF2 and eIF2B are separated, the ternary complex of eIF2-GTP-tRNAMet is formed. The complex then binds to mRNA and 40S and 60S ribosomes and participates in translation initiation. Phosphorylated eIF2α interferes with the formation of the eIF2-GTP-tRNAMet complex, resulting in the retention of a large number of translation initiation complexes in the cytoplasm and ultimately induces SGs formation. (Left) The p-eIF2α-independent pathway formed by SGs requires disruption of the eIF4F complex to interfere with RNA helicase eIF4E activity and inhibit its interaction with eIF4G. Maintaining the structural and functional integrity of eIF4F is required for translation initiation and can induce SGs formation through the following three mechanisms: ① Inhibition of the interaction between eIF4E and eIF4G. The recruitment of eIF4F to mRNA may be regulated by mTOR. When cells are starved or damaged by drugs, mTOR is inactivated, resulting in the accumulation of unphosphorylated 4EBP in cells. Unphosphorylated 4EBP competes with eIF4F to bind eIF4E, preventing the formation of eIF4F. Therefore, inhibiting the initiation of translation results in a reduction in the number of polysomes, and translationally stopped preinitiation complexes (PICs) accumulate in the cell and are thus assembled into SGs; ② Inhibition of the interaction between eIF4A and eIF4G or inhibition of the RNA helicase activity of eIF4A. ③ Destruction of the eIF4G structure. For example, after CVB infection, the 2A protein cleaves eIF4G.
eIF2B is an important protein involved in eukaryotic translation initiation. During translation initiation, eIF2-bound GDP (eIF2-GDP, the inactive state of eIF2) can be converted to eIF2-bound GTP (eIF2-GTP, the active state of eIF2). Therefore, when present in a sufficient amount, eIF2-GTP participates in translation initiation (29). When GCN2, PERK, PKR, and HRI kinases are activated, the α subunit of eIF2 is phosphorylated, preventing its separation from eIF2B. Subsequently, translation initiation is blocked, and mRNAs and certain proteins involved in the translation process aggregate to form SGs. In these cases, dsRNA formation or ERS induced by viral infection activates PKR and PERK, induces eIF2α phosphorylation, and leads to SGs formation. The formation of the eIF2-GTP-tRNAMet trimer complex is also a key step in the cellular regulation of translation. Before recruiting the 60S ribosome to start translation, the eIF2-GTP-tRNAMet trimer complex is recruited to the mRNA/eIF4F complex bound to the 40S ribosomal subunit, thereby presenting the messenger RNA linked to the translation initiation amino acid to 40S of the ribosomal subunit. However, when eIF2 and eIF2B are separated, a ternary complex with tRNAMet, eIF2-GTP-tRNAMet, is formed. This ternary complex binds to mRNA and the 40S and 60S ribosome subunits and thus participate in the translation initiation process. Phosphorylated eIF2α interferes with the formation of the eIF2-GTP-tRNAMet complex, resulting in the retention of a large number of translation initiation complexes in the cytoplasm and ultimately inducing SGs formation (30). Therefore, when eIF2α is phosphorylated, the efficiency of the eIF2-GDP-to-EIF2-GTP conversion is inhibited, resulting in the inability of tRNAMet to bind to eIF2, inhibiting the formation of the eIF2-GTP-tRNAMet trimer complex and thereby inhibiting translation initiation. Because eIF2α phosphorylation does not affect translation elongation, ribosomes that have already begun the translation process are detached from mRNA, resulting in the polysome depolymerization, and cells accumulate a large number of 48S units lacking pretranslational eIF2, eIF5, or tRNAMet and show no translation initiation function, called preinitiation complexes (PICs), which are assembled into SGs (31).
As shown in the right side of Figure 2, the eIF2α phosphorylation-independent pathway formation of SGs requires disruption of the eIF4F complex and interference with RNA helicase eIF4E activity to inhibit its interaction with eIF4G. During translation initiation, eIF4F is recruited by the vast majority of mRNAs in the initial step of translation. eIF4F is a heterotrimer consisting of eIF4A, eIF4E and eIF4G. eIF4A is an ATPase activity-dependent RNA helicase that opens the secondary structure of RNA and facilitates translation initiation complex scanning of RNA to recognize the AUG start codon. eIF4E binds to the cap structure at the 5’ end of mRNA, and the interaction between the eIF4E and mRNA mediates the recruitment of eIF4F via mRNA. The N-terminus of eIF4G interacts with eIF4E, and the C-terminus of eIF4G interacts with eIF4A, which forms a bridge to eIF4F. Maintaining the structural and functional integrity of eIF4F is required for translation initiation and can induce the formation of SGs through the following three mechanisms: (1) inhibition of the interaction between eIF4E and eIF4G. The recruitment of eIF4F to mRNA is regulated by mTOR, which is a serine/threonine kinase that controls cellular metabolism by regulating cellular protein synthesis (32). Under normal growth conditions, mTOR is activated. Phosphorylated eIF4E-binding proteins (4EBPs), however, cannot bind eIF4E. Therefore, eIF4F is formed, and translation proceeds normally. When cells are starved and damaged by drugs, mTOR is inactive. The accumulation of the unphosphorylated form of 4EBP in cells competes with eIF4F for binding to eIF4E, preventing the formation of eIF4F-mRNA complexes, thereby inhibiting the initiation of translation. This abrogated translation results in a reduction in the number of polysomes, and the PICs that accumulate in cells lacking key initiation factors are assembled into SGs. Similarly, tRNA-derived stress-induced RNAs (tiRNAs), produced by angiogenin-induced cleavage of mature messenger RNA, can dissociate eIF4G/eIF4A from eIF4E bound to the mRNA cap structure to generate PICs. These PICs are assembled into SGs facilitated by the RNA chaperone Y box-binding protein-1 (YB-1) (33). (2) Inhibition of the interaction between eIF4A and eIF4G or inhibition of the RNA helicase activity of eIF4A is a mechanism by which most instances of SGs assembly are induced. 15-Deoxy-delta 12,14-prostaglandinJ2 (15d-PGJ2) is a natural lipid inflammation mediator belonging to the prostaglandin family that can bind eIF4A and thus prevent the interaction between eIF4A and eIF4G (34). Pateamine A (PatA) and hippuristanol disrupts the function of eIF4F by inhibiting the helicase, ATPase activity or RNA-binding ability of eIF4A (35). (3) Destruction of the eIF4G structure. For example, in coxsackievirus B (CVB) infection, the 2A protein cleaves eIF4G (36).
2.4 Factors inducing SGs formation
In the body, the amount of mRNA in a polysome is regulated mainly via the initiation of translation. When translation initiation is successful, mRNA is located within a polysome. When translation initiation is blocked, mRNA is in a free state and is diverted to form a SGs. Therefore, any factor that blocks translation initiation can promote SGs formation. Numerous stresses and agents can also induce SGs formation (37).
Low nutrient levels, hypoxia, and osmotic shock are endogenous stressors; arsenic compounds, diethyl maleate (DEM), and paraquat are environmental stressors; overexpressed amyloid-beta 42 (Aβ42), MG132 and lactacystin are proteasome inhibitors; dithiothreitol (DTT) and thapsigargin are ER stressors, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and clotrimazole are mitochondrial poisons, and sorbitol (38), among other compounds, promote SGs formation in a p-eIF2α-dependent manner. Furthermore, when combined with stress, puromycin (39) and salubrinal, translation-modulating compounds stimulate SGs production in a p-eIF2α-dependent manner.
Ultraviolet (UV) radiation and brain ischemia are environmental stressors, overexpression of G3BP1, TIA-1, TIAR, TTP, FMRP, cytoplasmic polyadenylation element-binding protein (CPEB), SMN, dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 3 (DYRK3), tRNA fragments, and sodium azide are mitochondrial poisons; pateamine A, hippuristanol, silvestrol, 15d-PGJ2, prostaglandin A1 (PGA1), selenite and other compounds promote SGs formation in a p-eIF2α-dependent manner.
For certain novel molecules, such as menadione, among environmental stressors, and salicylate and 6,7-dihydro-4-hydroxy-3-(2′-hydroxy[1,1′-biphenyl]-4-yl)-6-oxo-thieno[2,3-b]pyridine-5-carbonitrile (A769662), among other compounds, combine to promote SGs formation. However, whether these novel SGs inducers dependent on p-eIF2α remains to be determined. Heat shock, among environmental stressors, induces SGs production in a p-eIF2α-dependent manner in mammalian cells (40), but its effects are p-eIF2α independent in Drosophila, C. elegans, and yeast. H2O2 can inhibit or promote SGs assembly in a p-eIF2α-independent manner, depending on the cell species and H2O2 concentration. Cycloheximide and emetine, which are translation modulators, induce SGs formation in a p-eIF2α-independent manner, and ISRIB, a translation modulator, first promotes SGs assembly and then promotes SGs disassembly in a p-eIF2α-dependent manner.
In conclusion, SGs formation is closely related to the depolymerization of translation polysomes, and the increase in untranslated mRNA levels in cells is conducive to SGs assembly. The disassembly and homeostasis of SGs and polysomes are regulated via translation initiation mechanisms (12). Therefore, pharmacological interventions that disrupt homeostatic translation can exert a marked impact on SGs formation. For example, emetine traps mRNA in polysomes, promotes SGs component depolymerization and inhibits SGs assembly. Puromycin promotes ribosome detachment from translated mRNA and releases the cleaved mRNA from polysomes, which significantly increases SGs formation.
3 Biological properties of SGs
3.1 Dispelling traditional concepts of mRNA translation with respect to SGs
It was previously believed that the formation of SGs was accompanied by the stagnation of mRNA translation. However, the formation of SGs in cells induced by sodium arsenite treatment favored subsequent reovirus replication: sodium arsenite pretreated cells had higher levels of viral protein synthesis and viral titers. Meanwhile, viral core particles were located in SGs. Therefore, the formation of SGs did not inhibit the translation of reovirus mRNA in the early postinfection period (41). Whether it means that mRNA not only did not degrade in SGs, but would further be translated. Since scientists had not conducted in-depth studies on the mRNAs in SGs, it is unclear whether the translation mRNA sequestered into SGs is inhibited.
In 2020, Mateju et al. made a major discovery showing that SGs do not directly inhibit any step of mRNA translation and that the translation of mRNAs located in SGs is not blocked (42). Using live-cell single-molecule imaging, the authors of this study found that untranslated mRNAs were preferentially recruited to SGs and that mRNAs localized to SGs could be translated. The translational state of the mRNA that was translocated between the cytosol and SGs was unchanged. By quantifying the correlation between translation status and localization of SGs, the authors found that 30% of translated mRNAs colocalized with SGs signals, suggesting that SGs-associated transcripts are commonly translated. When the distribution of mRNAs in SGs and cytoplasm was evaluated according to their translation status, only a small fraction of translated (12%) and untranslated (19%) transcripts were found to be located in SGs, with most transcripts retained in the cytoplasm. In examining the relationship between mRNA translation status and mRNA location in SGs, most translated (76%) and untranslated (65%) mRNAs in SGs were found to be located within 0.3 μm of the detected SGs boundaries, with translated mRNAs showing a slight propensity to be located close to the surface (mean distance ± SD: 0.22 ± 0.16 μm vs. 0.27 ± 0.18 μm), but translated mRNA was also detected deep inside the SGs, in large SGs and 1 μm or farther from the SGs boundary. The distance of mRNA from the SGs border was independent of the intensity of the translated mRNA signal. These findings suggest that the position of the mRNA in SGs is not changed via translation (42).
3.2 The regulatory effects of SGs on innate immunity
Viral infection can activate innate immune signaling pathways. The innate immune system includes pattern recognition receptors (PRRs), which sense viral RNA and present it sequentially in the innate immune signaling pathway, eventually inducing the production of a large number of IFNs and inhibiting the replication and spread of the virus (43). Viral dsRNA activates PKR phosphorylation, which ultimately leads to inhibited mRNA translation, RNase L-mediated RNA degradation, etc. (44). In addition, viral nucleic acid molecules of RNA and DNA viruses activate the innate immune signaling pathway. The RNA and DNA produced during viral replication are mediated by cellular Toll-like receptors (TLRs), retinoic acid inducible gene-1 (RIG-1)-like receptors (RLRs), DNA-mediated IFN regulators (DAIs), activator of IFN (STING/MITA/ERIS/MPYS), DEAD box polypeptide 41 (DDX41) and cGMP/CAMP synthase (cGAS) (45). Among these nucleic acid recognition molecules, RLRs carry three RNA helicases, RIG-5, MDA5 and DHX58 (LGP2), which sense mainly RNA molecules in the cytoplasm. These RLRs distinguish viral RNA molecules from host cell RNA molecules. For example, RNA molecules synthesized by viruses usually carry a 5’-ppp modification at the 5’ end (5’-PPP-containing single stranded RNA), whereas RNA molecules synthesized by host cells do not carry this modification (46). In addition, RLRs also mediate the activation of innate immune signaling pathways. RIG-1 generally recognizes viral single-stranded RNA (ssRNA) and short dsRNA, and MDA5 generally recognizes long dsRNA produced during viral replication. After recognizing viral RNA, RIG-1 and MDA5 undergo a conformational change, bind to receptor proteins called virus-induced signaling adapters (VISAs), such as mitochondrial antiviral signaling protein (MAVS), IPS-1, and Cardif on the mitochondrial outer membrane, and subsequently recruit TRAFs and activate TBK1/IKKi and IKKa/IKKp complexes, which in turn activate IRFs and NF-κB; ultimately, activated IRF3, IRF7 and NF-κB enter the nucleus to activate the gene transcription and expression of IFN and inflammatory factors (47).
SGs regulate innate immune responses. In recent years, certain sensors and elements involved in these signaling pathways, namely, RIG-1, MDA5, LGP2, and various sensors, such as PKR, RNase L, 2’-5’-oligoadenylate synthetase (OAS), have been found to be recruited to SGs, and multiple marker proteins of SGs have been found to be colocalized (48), suggesting a tight connection between SGs the formation and innate immune responses. Among the aforementioned sensors, OAS/RNase L cleaves viral RNA, and the cleaved viral RNA is recognized by pattern recognition molecules, which further activate the antiviral immune pathway. OASs are interferon-induced antiviral enzymes, and the human OAS family consists of four members: OAS1, OAS2, OAS3, and OAS-like protein (OASL). The profiles of SGs with assembly induced by viral infection and the innate immunity responses are shown in Figure 3. The interferon response protein molecule PKR recognizes viral dsRNA and 5’ppp RNA, senses the presence of viral nucleic acids in cells, and mediates downstream interferon responses. In addition, the activation of PKR induces the phosphorylation of eIF2α, which blocks the translational process in cells and leads to SGs formation (49). Cytoplasmic dsRNA and 5’ppp-RNA are also recognized by the pattern recognition molecules MDA5 and RIG-I, which are SGs components (50). After viral RNA is recognized by MDA5, RIG-I or LGP2, a signal is transmitted to MAVS (on the outer mitochondrial membrane. Subsequently, TBK1/IKK are phosphorylated, allowing the transcription factors IRF3/7 and NF-κB to enter the nucleus and activate the expression and secretion of type I interferons and certain inflammatory factors (51). In addition, after autocrine or paracrine IFN-α and IFN-β bind with IFN receptors on the cell membrane, leading to STAT1/2 phosphorylation in the cell. Phosphorylated STAT1/2 enters the nucleus together with IRF9, recognizes internal ribosome entry site (IRSE) regions, and activates the expression of more ISGs, producing antiviral effects.
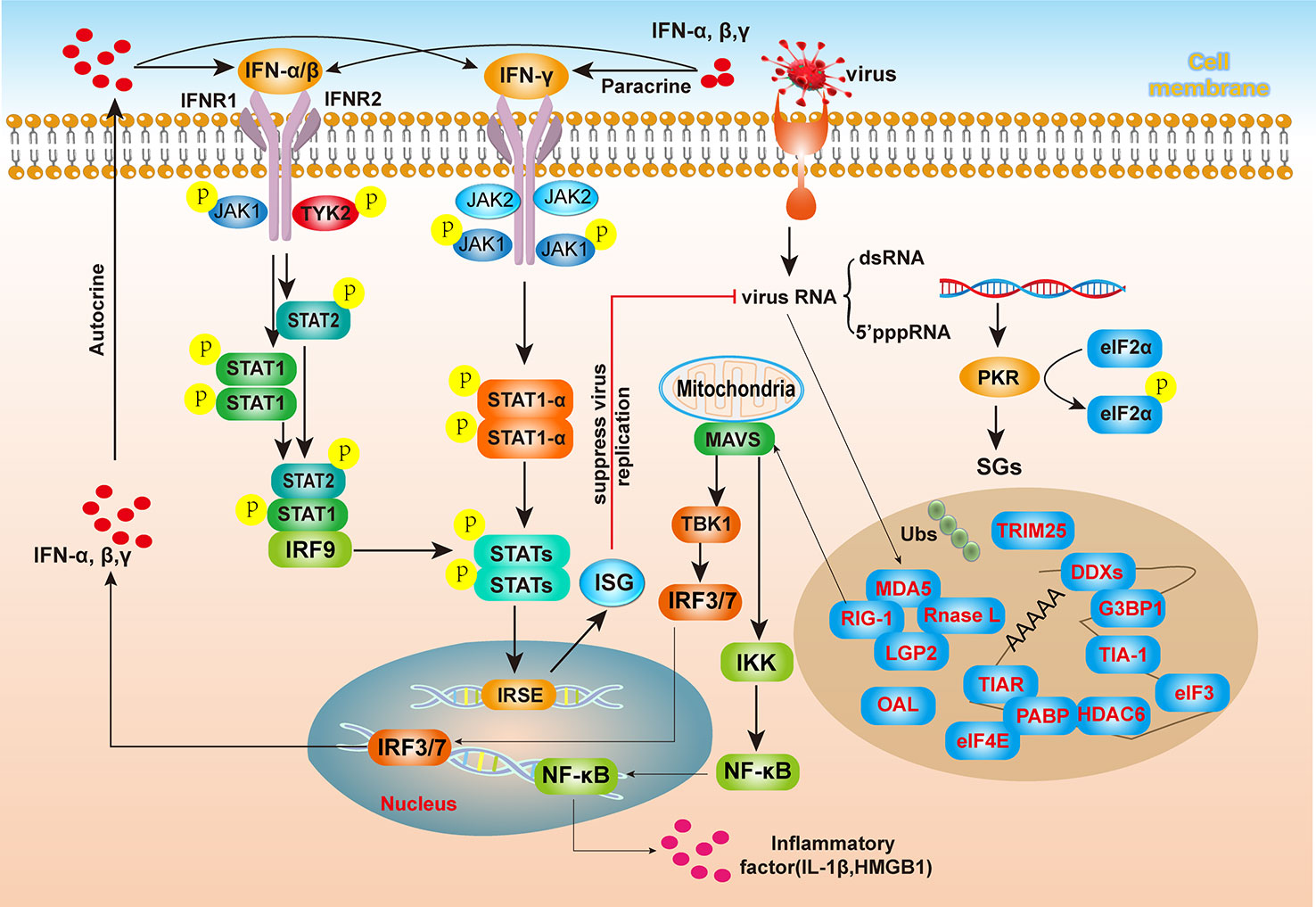
Figure 3 The relationship between SGs and antiviral innate immunity. After viral invasion into host cells, viral RNA (dsRNA or 5’pppRNA) activates the PKR kinase, resulting in eIF2α phosphorylation and SGs assembly. SGs contain the RIG-I-like receptors MDA5 and RIG-I, which can interact with MAVS on the mitochondrial membrane to activate the IFN signaling pathway and promote IFN production. After binding to IFN receptors on the cell membrane, interferon-stimulated genes (ISGs) induce the inhibition of viral RNA replication through the JAK/STAT pathway. After viral infection of cells, viral genomic RNA activates PKR, phosphorylates eIF2α, and inhibits the initiation of translation, thereby inhibiting viral protein synthesis and inducing SGs formation. SGs are composed of diverse components, including viral RNA, RNA-binding proteins (RBPs), and translation initiation factors, and especially, many innate immune pattern recognition molecules, such as MDA5, RIG-I, LDP2, and RNase L. These pattern recognition molecules bind viral RNA, which in turn transmits signals to MAVS on the outer mitochondrial membrane, activates TBK1/IKK phosphorylation, and allows transcription factors IRF3/7 and NF-κB to enter the nucleus, where they activate the expression and secretion of type I interferons and certain inflammatory factors. IFN activates intracellular STAT1/2 phosphorylation after autocrine or paracrine binding to cell membrane interferon receptors. Subsequently, p-STAT1/2 enters the nucleus together with IRF9, recognizes internal ribosome entry site (IRSE) regions, activates the expression of various ISGs, and produces antiviral effects.
Notably, many proteins encoded by ISGs that induce IFN signaling pathway activation are recruited to SGs and regulate SGs formation, and SGs also create platforms for a host antiviral response (52). OASs induced by IFN recognize dsRNA and then catalyze the production of 2-5A from ATP, thereby inducing dsRNA degradation by RNase L, an SGs component. ADAR1, another SGs protein, is also activated by IFN to convert A to I in dsRNA, thereby altering viral dsRNA and interfering with viral replication (53). SGs formation induced by which virus is not a result of IFN signaling pathway activation, but when G3BP1 is silenced, the ability of influenza A virus (IAV) infection to induce SGs formation in cells is significantly weakened, and the IFN activity level is also significantly reduced (54).
Specific viruses exhibit unique regulatory programs. For example, the viral protein NS1 in IAV strongly inhibits SGs formation initially induced by IAV and activates innate immunity mediated by RIG-1. Proteins that engage in signaling mediated through IFN receptor 1 (IFNAR1) and are encoded by ISGs establish an antiviral state in both infected and uninfected cells (55). In addition to the aforementioned molecular mechanisms, PKR activates inflammasome and macrophage responses during bacterial and dsRNA virus infection, resulting in the release of the cytokines IL-1β and HMGB1 (56).
In summary, virus-induced SGs formation generates intracellular platforms through which pattern recognition molecules regulate downstream signaling (57). The specific regulatory mechanisms are relatively complex, and research in recent years has revealed that interactions, such as the interaction between OASL and MDA5, enhance MDA5-mediated type I interferon signaling pathway activation (58). TRIM25 can also be recruited to SGs and is required for full RIG-I activation (59). Two types of enzymes are encoded by ISGs: one type constitutes dsRNA-binding proteins, such as the protein kinase PKR, which is regulated by dsRNA, and the other type comprises adenosine deaminase ADAR1, which acts on dsRNA (60). G3BP1 contributes to the induction of antiviral cytokine IFN-β production induced by RIG-1 (61). Studies are gradually revealing the connections among various innate immune signaling pathways in cells, SGs and various functional interactions.
4 SGs and viruses
4.1 Viruses and SGs coexist
After a virus infects a cell, it causes a series of changes in the intracellular environment, and after these changes are sensed by cells, the series of stress responses are initiated to resist viral infection and promote cell survival. However, a virus may evade or even exploit the stress responses of host cells in a number of ways to promote their own replication. SGs formation is a stress response in cells that can inhibit translation; however, viruses can regulate and change SGs assembly in various ways. Therefore, the relationships between viruses and SGs are diverse. Generally, the relationships can be roughly classified into three categories: viral infection is induced first and then SGs formation is inhibited; viral infection inhibits SGs formation; and viruses and SGs coexist. Therefore, as shown in Table 1, various modes of viral infection of host cells affects SGs: After viral infections, SGs are quickly disassembled after formation, stable SGs are formed, or SGs are not formed. After the initial synthesis of viral proteins, positive-strand RNA viruses switch individual genome recruitment of ribosomes to translational repression to clear ribosomes from templates and facilitate viral RNA replication (114). Therefore, a large number of RNA regulatory proteins are involved in viral replication. The key SGs protein G3BP1 interacts with nonstructural proteins (NSPs) of the virus to form complexes that inhibit viral replication. Studies have shown that G3BP1 forms a complex with the viral polymerases nsp3, nsp2 and nsp4 (115).
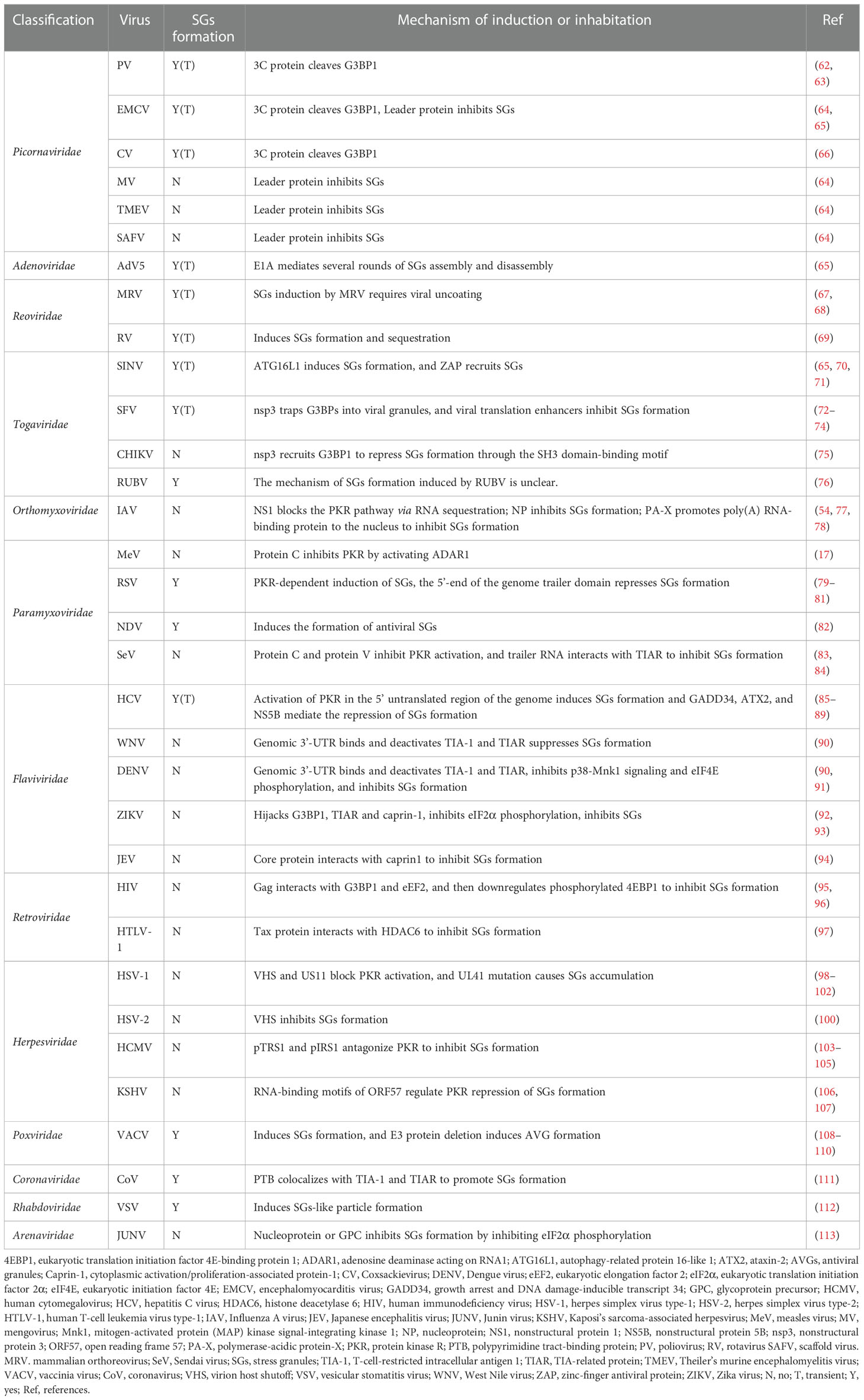
Table 1 Viral infection and SGs formation.
4.1.1 Viral infection induces and then suppresses SGs formation or leads to SGs disassembly
Viruses, especially RNA viruses can cause the formation of SGs-like bodies in the cytoplasm. Some viruses induce SGs formation by activating the eIF2α kinases PKR and GCN2. When cells detect viral RNA in the cytoplasm, these two factors are activated and play specific roles. Viral infection produces a unique type of cellular stress response and often induces the formation of viral-type SGs (V-SGs), and some V-SGs specifically contain the RBP SRC associated in mitosis of 68 kd (Sam68) (116). As shown in Table 1, during infection with viruses of the Picornavirus family, such as poliovirus (PV) (63), CVs (66), encephalomyocarditis virus (EMCV) (65), and Theiler’s murine encephalomyelitis virus (TMEV) (64), SGs formation is transient and closely related to the viral protease 3C. However, in the later stages of infection, the breakdown of the G3BP1 or L protein in the cell leads to the disintegration of SGs and inhibits the formation of SGs (62–64). The Flaviviridae family member Hepatitis C virus (HCV) induces the formation of SGs which are dissembled in later infection stages (85, 88, 89) (86). The Adenoviridae family member Adenovirus type 5 (AdV5) can induce several rounds of SGs formation and degradation (65). The Reoviridae family member Mammalian orthoreovirus (MRV) induces the SGs formed in the early stage, this in turn promote the replication of the virus (67) (117–119). Rotavirus (RV) infection can lead to eIF2α phosphorylation but inhibit SGs formation. However, RV can induce SGs formation and SGs sequestration by viroplasms (VMs) to promote viral replication in early stage (69).
The Togaviridae family member Sindbis virus (SINV) induces SGs formation in the early stages of infection, a process requiring autophagy-related protein 16-like 1 (ATG16L1), but then, the SGs disappear in the late stages of infection (65, 70). Rubella virus (RUBV) infection promotes SGs assembly, but the number of SGs decreases in the late stages of infection, and the specific molecular mechanisms involved in these changes are unknown (76). Replication of Semliki forest virus (SFV) RNA induces SGs formation that depends on the level of eIF2α phosphorylation. Throughout viral translation, viral nonstructural protein 3 (nsp3) traps G3BP1 into viral granules, thereby inhibiting the formation of SGs and promoting viral replication (74). In addition, the viral translation enhancer near the initiation codon of SFV viral RNA, AUG promotes SG disassembly during infection (73).
4.1.2 Viral infections that inhibit SGs formation
During the life cycle of many viruses, SGs formation is not observed in cells. In some cases, SGs failed to form despite high p-eIF2α levels in infected cells or exposure to new SGs-induced stress. These outcomes indicate that some viral infections inhibit SGs formation. As shown in Table 1, the Picornaviridae family Mengovirus (MV), EMCV, TMEV and Saffold virus (SAFV) members inhibit the formation of SGs through L protein and interferon (IFN) (64). The Retroviridae family members human immunodeficiency virus (HIV) and human T-cell leukemia virus type-1 (HTLV-1) inhibit SGs formation through the Gag protein and Tax protein (95–97). The Paramyxoviridae family member Measles virus (MeV) and Sendai virus (SeV) inhibit SGs formation through protein C and protein V (17, 83, 84). The Orthomyxoviridae family member IAV inhibit SGs formation through NS1 protein, nucleoprotein (NP) (54, 78, 120). The Flavivirus family members West Nile virus (WNV) (121), Dengue virus (DENV) (91), ZIKA virus (ZIKV) (93) and Japanese encephalitis virus (JEV) (122) inhibit SGs assembly by PKR, genomic 3′-UTR, p38/mitogen-activated protein (MAP) kinase signal-integrating kinase 1 (Mnk1) signaling, or Caprin1 (90) (91–94, 123). The Herpesviridae family member herpes simplex virus type-1 (HSV-1), herpes simplex virus type-2 (HSV-2), Human cytomegalovirus (HCMV), Kaposi’s sarcoma-associated herpesvirus (KSHV), Pseudorabies virus (PRV) inhibit SGs formation through virion host shutoff (VHS) protein, US11 protein, unfolded protein response (UPR), p-TRS1 and p-IRS1 proteins, open reading frame 57 (ORF57), growth arrest and DNA damage-inducible protein 34 (GADD34), protein phosphatase 1 (PP1) (98, 100–102, 104, 105, 107, 124, 125). The Togaviridae family member Chikungunya virus (CHIKV) inhibits SGs formation by Nsp3 (75). The Arenaviridae family member Junin virus (JUNV) inhibits SGs formation through the nucleoprotein (N) or glycoprotein precursor (GPC) (113).
4.1.3 The coexistence of viral infection and SGs
As shown in Table 1, the Poxviridae family is a group of enveloped dsDNA viruses that cause smallpox, monkeypox, and vaccinia in humans. Most research on poxviridae and SGs has focused on vaccinia virus (VACV). VACV leverages different SGs proteins at different replication stages to facilitate the completion of each replication cycle (126). Some VACV mutant-infected cells form SGs containing components such as TIA-1, and these structures are called antiviral stress granules (avSGs) due to their ability to inhibit viral replication (108–110). It is worth noting that avSGs is a type of SGs. The avSGs are not formed in all virus-infected cells. Although some cells infected with the virus form SGs, these SGs do not have antiviral ability and instead disappear in the later stages of infection. The presence of SGs facilitates both viral proliferation and host growth. The aforementioned studies have shown that SGs and VACV coexist, and the components of SGs can promote the transcription and translation of VACV. The Paramyxoviridae family member respiratory syncytial virus (RSV) can induce SGs formation in some cells after infection and can form large inclusion bodies in cells with SGs formation depending on the PKR-mediated eIF2α phosphorylation pathway (81). The SGs formation may facilitate the translation and replication of this virus by inhibiting the translation of host cells (80). However, only RSV with mutations in the 5’-end of the genome have been reported to induce SGs formation, even though the 5’-end of the viral genome is required for the transcription of RSV (79). Newcastle disease virus (NDV), another Paramyxoviridae virus, can form antiviral SGs similar to those induced by VACV in infected cells. When the formation of antiviral SGs was inhibited, NDV-induced interferon levels were greatly reduced (82). The Coronaviridae family menber coronavirus (CoV) colocalizes TIA-1 and TIAR with polypyrimidine tract-binding protein (PTB) to promote SGs formation (111). The Rhabdoviridae family member vesicular stomatitis virus (VSV) induces the production of particles that contain PCBP2, TIA1, and TIA1 proteins. These particles are exclusively located in SGs-like structures (112).
4.2 SGs inducers are expected to become novel antiviral drugs
Drug-induced PKR activation of SGs formation generated by p-elF2α is thought to control viral infection. TIA-1 and its homolog TIAR constitute a class of proteins that specifically bind to the 3’-non-coding region of TNF-α and matrix metalloproteinase-13 (MMP-13) mRNA and exert a translational silencing effect. Both proteins carry three RNA recognition motifs that bind with high affinity to U- and A+U-rich RNAs. Sustained TIA1 or TIAR expression inhibits cell proliferation, favors cell cycle arrest in the G1/S phase, and triggers cell death through slow caspase-dependent apoptosis and late autophagy (127). Therefore, viral spread can be inhibited by inducing the aggregation of TIA-1 (127). In vitro studies have shown that eIF4A helicase inhibitors exert antiviral effects, pateamine A inhibits replication of IAV and cytomegalovirus (128), and silvestrol effectively inhibits Ebola virus (EBOV) replication (129). eIF4A helicase inhibitors have been shown to exhibit antiviral activity at concentrations that do not lead to significant cytotoxicity. Therefore, it is of great value to develop relevant antiviral agents for determining the interaction of different viruses with SGs.
5 SGs and bacteria and other microorganisms
Some scholars have proposed that bacteria affect the level of p-eIF2α and maintain a close relationship with SGs (9). Gram-negative bacteria such as Shiga toxin-producing Escherichia coli (STEC) (130), Aeromonas hydrophila (131), Vibrio vulnificus (V. vulnificus) (132), Pseudomonas aeruginosa (PA) (133), and Yersinia pseudotuberculosis (134) significantly increased p-eIF2α levels in cell lines in vitro. In a study of the formation and kinetics of SGs in bacteria, it the ability of the SGs markers eIF3 and eIF4B to move to SGs was found to be inhibited in Shigella flexneri-infected cells, and the aggregation of SGs containing G3BP1 and eIF4G was also inhibited. Further studies have shown that invasive bacteria exhibit this inhibitory effect based on conditions that promote eIF2α phosphorylation or inhibit eIF4A helicase activity (135). The assembly of SGs requires the involvement of dyneins, which that regulate microtubule movement. During the inhibition of SGs formation mediated by S. flexneri, the acetylation level of alpha-tubulin was significantly increased, and the number of alpha-tubulin-mediated microtubule bundles, microtubule stability, and microtubule-based transport and regulatory functions were subsequently affected (135).
Gram-positive bacteria and Gram-negative bacteria induce elevated p-eIF2α levels. However, the species found to cause elevated p-eIF2α levels were members of the Firmicutes phylum, namely, Bacilli (Listeria monocytogenes, Streptococcus pneumoniae, and Staphylococcus aureus) or Clostridia (Clostridium difficile) (9). Listeria monocytogenes, one of the gram-positive bacteria, was the most studied p-eIF2α-inducing species (9). Mycobacteria belong to the gram-positive Actinobacteria phylum and are characterized by their “acid resistance”, which prevents gram-positive staining due to lipids attached to the cell wall. However, the ability of other mycobacteria to induce p-eIF2α in host cells appeared to be no different. Notably, Mycobacterium avium (136), Mycobacterium ulcerans (137), and Mycobacterium tuberculosis (138, 139) can activate the PERK/eIF2α/CHOP pathway.
In addition, other pathogens, such as Mycoplasma hyopneumoniae, can activate the PERK/eIF2α/CHOP pathway (140). Studies have reported that, three protozoan species, Leishmania amazonensis (141), Plasmodium berghei (142), and Toxoplasma gondii (143), and one fungal species, Histoplasma capsulatum (144), can increase p-eIF2α levels, after an organism infects the cell, the host p-eIF2α level increases. However, for some microorganisms, the change in p-eIF2α level does not involve a constant increase with the change in infection time, and therefore, more research in the future is needed to confirm the patterns of p-eIF2α level changes and microorganism infection (9).
6 SGs and inflammation
Similar to their relationship after viral infection, SGs and inflammatory factors can interact. Studies on the effect of SGs on inflammation have reported that in primary bone marrow-derived macrophages stimulated by endotoxin, SGs can reduce the degree of cellular inflammatory damage (10). SGs can also inhibit apoptosis by reducing the production of reactive oxygen species (145). An important component of SGs, DDX3X, competitively binds to NLRP3, thereby inhibiting inflammasome activation and regulating the “survival-related pyroptosis” of cells (10). The core protein HuR in SGs can bind to the 3′-UTR of COX-2 to stabilize its mRNA (146).
In contrast, certain inflammatory factors are directly or indirectly related to SGs formation. In mucosal inflammation, the proinflammatory factors IFN-γ and TNF-α cause the phosphorylation of eIF2α to induce SGs formation, and HSP70 mRNA is encapsulated into SGs, reducing the expression of HSP70 (11). SGs generated after heat shock can recruit TRAF2 and inhibit TNF-α-mediated activation of NF-κB through its interaction with eIF4G (147). In the immune response, eIF2α in T cells is phosphorylated after the first antigen is presented to form SGs, which encapsulate cytokine mRNA. When the antigen is re-presented by T cells, a mechanism of SGs disassembly is initiated, and mRNA is released for the translation and secretion of cytokines (148). Atherosclerotic inflammation promotes the formation of SGs that accumulate in endometrial macrophages and vascular smooth muscle cells (VSMCs) as the disease progresses, sequester mRNA transcripts and halt translation. Knocking down G3BP1 in VSMCs inhibits SGs formation while altering the inflammatory gene expression profile (149), and IL-19 can reduce vascular inflammation by reducing the rate of SGs formation and alleviate atherosclerosis (149).
Currently, the impact of SGs on the inflammatory response has not been determined. Different diseases exhibit different anti-inflammatory or proinflammatory effects of SGs or their key components. In the process of atherosclerosis, SGs can be formed in vascular smooth muscle cells (VSMCs). Decreasing the expression of G3BP2, a key component of SGs, can reduce oxidized low-density lipoprotein (ox-LDL) - induced inflammation in the atherosclerotic lesions. This study only explored the effect of G3BP2 on the atherosclerotic lesions (150). However, it remains unclear whether inhibiting the expression of G3BP2 leads to a decrease in the formation of SGs, and the subsequent reduction in vascular inflammation levels during the process of atherosclerotic lesions. The anti-inflammatory drug cortisone can induce the production of SGs (151). Whether the powerful anti-inflammatory effects of cortisone are mediated by SGs will be a very interesting issue. The signaling -lipid molecule 15-deoxy-delta 12,14-prostaglandin J2 (15d-PGJ2) inhibits translation leading to an increase in SGs production and TRAF2 is sequestered in these SGs. The sequestration of TRAF2 contributes to the anti-inflammatory activity of 15d-PGJ2 (34). Carbon monoxide (CO) can play an anti-inflammatory role by inducing the formation of SGs (152). SGs could protect hepatocytes from hypoxia-induced damage during acute liver failure by reducing endoplasmic reticulum stress (ERS)-mediated apoptosis (153). ERS is a key process in mediating inflammatory responses, so molecules related to the ERS pathway can serve as potential anti-inflammatory targets for SGs.
7 Discussion
Cells are often exposed to potentially fluctuating adverse environmental conditions, and SGs formation enables cells to adapt to different environmental changes as SGs provide protection for important cellular components. SGs can transduce signals in response to cellular stress and show protective functions. The formation and function of SGs are tightly regulated dynamic processes. When the translation initiation mechanism is blocked, mRNA and many translation-related proteins aggregate into SGs and are temporarily stored in SGs to prevent the mediation of degradation mechanisms. Moreover, mRNAs can also be translocated into SGs from P granules or cytoplasmic mRNAs. During an integrated stress response, cells trigger phosphorylation of the TIF eIF2α, resulting in the inhibited translation of most mRNAs. This inhibitory state allows cells to conserve energy and use their resources to restore homeostasis. During stress, SGs temporarily store mRNAs and proteins and protect both types of molecules from autophagy and proteasome degradation, with cells rapidly restarting translation and activating other signaling pathways after recovery from stress. The aggregation and depolymerization in SGs partly depend on phase separation, interactions between microtubules and dyneins, and RBPs.
In addition to protecting mRNA from degradation, promoting mRNA transcription in SGs, and regulating innate immunity, SGs have also been implicated in apoptosis and other intranuclear processes (154). In addition, the key molecules formed by SGs can interact with some molecules in pathogens. Based on the aforementioned molecular mechanisms, SGs have been shown to be involved in viral infections; bacterial infections; novel pathogen infections; inflammatory diseases; cancers; various neurological diseases, including Fragile X syndrome, spinal muscular atrophy, spinocerebellar ataxia, myotonia; and pathological processes such as malnutrition responses. However, the functions of SGs differ depending on the virus, bacteria, inflammatory response and host cell involvement (80, 155).
Recently, research on SGs has been focused mainly on infectious diseases, especially viral infections. Three modes of viral infection and SGs formation have been described. Some viruses induce SGs production in the early stage of infection. However, most viruses inhibit SGs formation during a certain period in the infection cycle. When the viral gene expression level is high, SGs coexist in virus-infected cells, and some viruses leverage SGs to facilitate their own replication. In the process of virus infection, SGs formation is induced in various ways. Among the SGs formation processes, dsRNA or RNA with novel structures produced via virus replication is recognized by PKR, which is the most common way in which viruses induce SGs formation. The shutdown of host cell translation by viral components is another mechanism by which viruses induce SGs formation. SGs are the products of the mutual gamesmanship observed between viruses and cells. Viral infection of cells can cause eIF2α phosphorylation, but p-eIF2α does not necessarily lead to SGs formation. SGs not only directly inhibit viral translation but also promote cells survival by inducing innate immunity to suppress viral replication. For example, SGs formed by RSV infection show enhanced replication, while SGs formed by MRV infection show inhibited replication. The SGs that inhibit viral replication are attacked by viruses at a later stage of formation, resulting in no prolonged assembly or even the disassembly of SGs. The constituent proteins in SGs also exert effects on viral replication, and the replication efficiency of MRV is significantly increased in G3BP1-knockout cells. In conclusion, when host cells are infected by viruses, they protect themselves by regulating SGs formation, and a virus also targets SGs to benefit itself. Therefore, the interactions between different viruses and different cells are not the same, and the mechanisms underlying these interactions may also be dynamic developmental processes.
Compared with that of RNA viruses, the regulatory mechanism of DNA virus on SGs formation has been less frequently reported. Whether SGs formation contributes to the antiviral effects of these compounds is unclear, and inhibition of viral protein synthesis is likely sufficient to disrupt viral replication. The speculation that SGs compete with bacteria and other pathogens has been based on a large number of related studies of SGs and viruses, which have provided great references for future research directions. We believe that in the future, more studies on SGs related to bacterial infection and the inflammatory response will be reported, and some valuable targets for antiviral, antibacterial, and anti-inflammatory treatment will be found.
However, many limitations in the research on SGs are evident. Under different stimuli, the components of SGs differ, and the specific composition of SGs is not fully understood. With in-depth related research, some novel proteins in SGs have been gradually identified. Recently, many reports on the interaction between SGs and various viruses have become available, but the specific molecular mechanisms are still unclear, and detailed research is lacking. Because SGs lack membranes, SGs components cannot be directly extracted or identified, and SGs are aggregates with dynamic formation and disaggregation kinetics, which makes studying their nature difficult. No effective method to detect SGs in tissues has been established; therefore, the study of SGs has remained at the cellular level, which has resulted in an in-depth study of the relationship between SGs and diseases. Therefore, drugs that activate p-elF2α through PKR to promote SGs formation are expected to be a developed to control viral infection, but it is unclear whether SGs formation contributes to the antiviral effects of these compounds, and they likely exert an inhibitory effect on viral protein synthesis. Whether SGs can be targeted in effective strategies for inducing bacteriostasis or anti-inflammation effects or inhibiting tumor growth remains unknown due a lack of relevant research.
However, with increasingly in-depth research, especially in recent years, it has been revealed that the translation of mRNA in SGs is not suspended but is independent of the type of cell and is mediated by a complete set of transcriptional and translational regulatory systems. Many problems identified in previous studies, such as how polyadenylation and the RNA-induced silencing complex (RISC) affect the relationship between SGs and mRNA and how viral RNA escapes from SGs and is translated in the presence of high concentrations of phosphorylated eIF2α, have been partially resolved. Moreover, translationally blocked mRNA promotes SGs formation, and with an increase in the mRNA levels, the number of SGs formed is increased. SGs contain mRNAs that are protected from degradation and for which translation has been initially blocked, but these mRNAs can be replicated in SGs. Moreover, in-depth study into SGs and infectious and inflammatory diseases will help to identify potential therapeutic targets for diseases.
Author contributions
WL and YW wrote the manuscript, completed the review process, contributed to the conception and design of manuscript. YW contributed to the conception and design of the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82100630, 82100894) and by Hubei Provincial Natural Science Foundation of China (2022CFB118).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol (2005) 169(6):871–84. doi: 10.1083/jcb.200502088
2. Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans (2002) 30(Pt 6):963–9. doi: 10.1042/bst0300963
3. Ivanov P, Kedersha N, Anderson P. Stress granules and processing bodies in translational control. Cold Spring Harbor Perspect Biol (2019) 11(5):a032813. doi: 10.1101/cshperspect.a032813
4. Emara MM, Fujimura K, Sciaranghella D, Ivanova V, Ivanov P, Anderson P. Hydrogen peroxide induces stress granule formation independent of eIF2alpha phosphorylation. Biochem Biophys Res Commun (2012) 423(4):763–9. doi: 10.1016/j.bbrc.2012.06.033
5. Fernandez-Carrillo C, Perez-Vilaro G, Diez J, Perez-Del-Pulgar S. Hepatitis c virus plays with fire and yet avoids getting burned. a review for clinicians on processing bodies and stress granules. Liver Int (2018) 38(3):388–98. doi: 10.1111/liv.13541
6. Wang B, Maxwell BA, Joo JH, Gwon Y, Messing J, Mishra A, et al. ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol Cell (2019) 74(4):742–757 e748. doi: 10.1016/j.molcel.2019.03.027
7. Gao X, Jiang L, Gong Y, Chen X, Ying M, Zhu H, et al. Stress granule: a promising target for cancer treatment. Br J Pharmacol (2019) 176(23):4421–33. doi: 10.1111/bph.14790
8. Basu M, Courtney SC, Brinton MA. Arsenite-induced stress granule formation is inhibited by elevated levels of reduced glutathione in West Nile virus-infected cells. PloS Pathog (2017) 13(2):e1006240. doi: 10.1371/journal.ppat.1006240
9. Tweedie A, Nissan T. Hiding in plain sight: formation and function of stress granules during microbial infection of mammalian cells. Front Mol Biosci (2021) 8:647884. doi: 10.3389/fmolb.2021.647884
10. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature (2019) 573(7775):590–4. doi: 10.1038/s41586-019-1551-2
11. Hu S, Claud EC, Musch MW, Chang EB. Stress granule formation mediates the inhibition of colonic Hsp70 translation by interferon-gamma and tumor necrosis factor-alpha. American journal of physiology. Gastrointestinal liver Physiol (2010) 298(4):G481–492. doi: 10.1152/ajpgi.00234.2009
12. Panas MD, Ivanov P, Anderson P. Mechanistic insights into mammalian stress granule dynamics. J Of Cell Biol (2016) 215(3):313–23. doi: 10.1083/jcb.201609081
13. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol (2010) 11(2):113–27. doi: 10.1038/nrm2838
14. Thomas MG, Martinez Tosar LJ, Desbats MA, Leishman CC, Boccaccio GL. Mammalian staufen 1 is recruited to stress granules and impairs their assembly. J Cell Sci (2009) 122(Pt 4):563–73. doi: 10.1242/jcs.038208
15. Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell (2013) 152(4):791–805. doi: 10.1016/j.cell.2013.01.033
16. Gabriel L, Srinivasan B, Kus K, Mata JF, Joao Amorim M, Jansen LET, et al. Enrichment of zalpha domains at cytoplasmic stress granules is due to their innate ability to bind to nucleic acids. J Cell Sci (2021) 134(10):jcs258446. doi: 10.1242/jcs.258446
17. Okonski KM, Samuel CE. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J Virol (2013) 87(2):756–66. doi: 10.1128/JVI.02270-12
18. Sanders DW, Kedersha N, Lee DSW, Strom AR, Drake V, Riback JA, et al. Competing protein-RNA interaction networks control multiphase intracellular organization. Cell (2020) 181(2):306–324 e328. doi: 10.1016/j.cell.2020.03.050
19. Yang P, Mathieu C, Kolaitis RM, Zhang P, Messing J, Yurtsever U, et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell (2020) 181(2):325–345 e328. doi: 10.1016/j.cell.2020.03.046
20. Guillen-Boixet J, Kopach A, Holehouse AS, Wittmann S, Jahnel M, Schlussler R, et al. RNA-Induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell (2020) 181(2):346–361 e317. doi: 10.1016/j.cell.2020.03.049
21. Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, et al. Germline p granules are liquid droplets that localize by controlled dissolution/condensation. Science (2009) 324(5935):1729–32. doi: 10.1126/science.1172046
23. Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science (2017) 357(6357):eaaf4382. doi: 10.1126/science.aaf4382
24. Lee JE, Cathey PI, Wu H, Parker R, Voeltz GK. Endoplasmic reticulum contact sites regulate the dynamics of membraneless organelles. Science (2020) 367(6477):eaay7108. doi: 10.1126/science.aay7108
25. Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol (1995) 15(8):4497–506. doi: 10.1128/MCB.15.8.4497
26. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell (2000) 5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5
27. Srivastava SP, Kumar KU, Kaufman RJ. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem (1998) 273(4):2416–23. doi: 10.1074/jbc.273.4.2416
28. McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, et al. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem (2005) 280(17):16925–33. doi: 10.1074/jbc.M412882200
29. Stolboushkina EA, Garber MB. Eukaryotic type translation initiation factor 2: structure-functional aspects. Biochem Biokhimiia (2011) 76(3):283–94. doi: 10.1134/s0006297911030011
30. Nakagawa K, Narayanan K, Wada M, Makino S. Inhibition of stress granule formation by middle East respiratory syndrome coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J Virol (2018) 92(20):e00902–18. doi: 10.1128/JVI.00902-18
31. Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, et al. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell (2002) 13(1):195–210. doi: 10.1091/mbc.01-05-0221
32. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell (2009) 136(4):731–45. doi: 10.1016/j.cell.2009.01.042
33. Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell (2011) 43(4):613–23. doi: 10.1016/j.molcel.2011.06.022
34. Kim WJ, Kim JH, Jang SK. Anti-inflammatory lipid mediator 15d-PGJ2 inhibits translation through inactivation of eIF4A. EMBO J (2007) 26(24):5020–32. doi: 10.1038/sj.emboj.7601920
35. Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, et al. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol (2006) 2(4):213–20. doi: 10.1038/nchembio776
36. Wu S, Wang Y, Lin L, Si X, Wang T, Zhong X, et al. Protease 2A induces stress granule formation during coxsackievirus B3 and enterovirus 71 infections. Virol J (2014) 11:192. doi: 10.1186/s12985-014-0192-1
37. Mahboubi H, Stochaj U. Cytoplasmic stress granules: dynamic modulators of cell signaling and disease. Biochim Biophys Acta Mol basis Dis (2017) 1863(4):884–95. doi: 10.1016/j.bbadis.2016.12.022
38. Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P 3rd, Good SK, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol (2011) 31(5):1098–108. doi: 10.1128/MCB.01279-10
39. Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, et al. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol (2000) 151(6):1257–68. doi: 10.1083/jcb.151.6.1257
40. Mahboubi H, Kodiha M, Stochaj U. Automated detection and quantification of granular cell compartments. Microscopy microanalysis (2013) 19(3):617–28. doi: 10.1017/S1431927613000159
41. Lutz M, Worth MP, Hinchman MM, Parker JSL, Ledgerwood ED. Mammalian orthoreovirus infection is enhanced in cells pre-treated with sodium arsenite. Viruses (2019) 11(6):563. doi: 10.3390/v11060563
42. Mateju D, Eichenberger B, Voigt F, Eglinger J, Roth G, Chao JA. Single-molecule imaging reveals translation of mRNAs localized to stress granules. Cell (2020) 183(7):1801–1812 e1813. doi: 10.1016/j.cell.2020.11.010
43. Gurtler C, Bowie AG. Innate immune detection of microbial nucleic acids. Trends Microbiol (2013) 21(8):413–20. doi: 10.1016/j.tim.2013.04.004
44. Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8(7):559–68. doi: 10.1038/nri2314
45. Carty M, Guy C, Bowie AG. Detection of viral infections by innate immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
46. Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, et al. 5’-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci USA (2009) 106(29):12067–72. doi: 10.1073/pnas.0900971106
47. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25(3):349–60. doi: 10.1016/j.immuni.2006.08.009
48. Langereis MA, Feng Q, van Kuppeveld FJ. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol (2013) 87(11):6314–25. doi: 10.1128/JVI.03213-12
49. Taghavi N, Samuel CE. Protein kinase PKR catalytic activity is required for the PKR-dependent activation of mitogen-activated protein kinases and amplification of interferon beta induction following virus infection. Virology (2012) 427(2):208–16. doi: 10.1016/j.virol.2012.01.029
50. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol (2014) 5:342. doi: 10.3389/fimmu.2014.00342
51. Dixit E, Kagan JC. Intracellular pathogen detection by RIG-i-like receptors. Adv Immunol (2013) 117:99–125. doi: 10.1016/B978-0-12-410524-9.00004-9
52. George CX, Ramaswami G, Li JB, Samuel CE. Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J Biol Chem (2016) 291(12):6158–68. doi: 10.1074/jbc.M115.709014
53. Silverman RH. Viral encounters with 2’,5’-oligoadenylate synthetase and RNase l during the interferon antiviral response. J Virol (2007) 81(23):12720–9. doi: 10.1128/JVI.01471-07
54. Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PloS One (2012) 7(8):e43031. doi: 10.1371/journal.pone.0043031
55. Teijaro JR. Type I interferons in viral control and immune regulation. Curr Opin Virol (2016) 16:31–40. doi: 10.1016/j.coviro.2016.01.001
56. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature (2012) 488(7413):670–4. doi: 10.1038/nature11290
57. Wu B, Hur S. How RIG-I like receptors activate MAVS. Curr Opin Virol (2015) 12:91–8. doi: 10.1016/j.coviro.2015.04.004
58. Li LF, Yu J, Zhang Y, Yang Q, Li Y, Zhang L, et al. Interferon-inducible oligoadenylate synthetase-like protein acts as an antiviral effector against classical swine fever virus via the MDA5-mediated type I interferon-signaling pathway. J Virol (2017) 91(11):e01514–16. doi: 10.1128/JVI.01514-16
59. Sanchez-Aparicio MT, Ayllon J, Leo-Macias A, Wolff T, Garcia-Sastre A. Subcellular localizations of RIG-I, TRIM25, and MAVS complexes. J Virol (2017) 91(2):e01155–16. doi: 10.1128/JVI.01155-16
60. Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol (2008) 89(Pt 1):1–47. doi: 10.1099/vir.0.83391-0
61. Kim SS, Sze L, Liu C, Lam KP. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-beta response. J Biol Chem (2019) 294(16):6430–8. doi: 10.1074/jbc.RA118.005868
62. Mazroui R, Sukarieh R, Bordeleau ME, Kaufman RJ, Northcote P, Tanaka J, et al. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol Biol Cell (2006) 17(10):4212–9. doi: 10.1091/mbc.e06-04-0318
63. White JP, Cardenas AM, Marissen WE, Lloyd RE. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe (2007) 2(5):295–305. doi: 10.1016/j.chom.2007.08.006
64. Borghese F, Michiels T. The leader protein of cardioviruses inhibits stress granule assembly. J Virol (2011) 85(18):9614–22. doi: 10.1128/JVI.00480-11
65. Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, et al. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol (2013) 87(17):9511–22. doi: 10.1128/JVI.03248-12
66. Xue YC, Ng CS, Mohamud Y, Fung G, Liu H, Bahreyni A, et al. FUS/TLS suppresses enterovirus replication and promotes antiviral innate immune responses. J Virol (2021) 95(12):e00304–21. doi: 10.1128/JVI.00304-21
67. Qin Q, Hastings C, Miller CL. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J Virol (2009) 83(21):11090–101. doi: 10.1128/JVI.01239-09
68. Qin Q, Carroll K, Hastings C, Miller CL. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2alpha phosphorylation and PKR. J Virol (2011) 85(17):8798–810. doi: 10.1128/JVI.01831-10
69. Montero H, Rojas M, Arias CF, Lopez S. Rotavirus infection induces the phosphorylation of eIF2alpha but prevents the formation of stress granules. J Virol (2008) 82(3):1496–504. doi: 10.1128/JVI.01779-07
70. Jefferson M, Bone B, Buck JL, Powell PP. The autophagy protein ATG16L1 is required for sindbis virus-induced eIF2alpha phosphorylation and stress granule formation. Viruses (2019) 12(1):39. doi: 10.3390/v12010039
71. Law LMJ, Razooky BS, Li MMH, You S, Jurado A, Rice CM, et al. ZAP’s stress granule localization is correlated with its antiviral activity and induced by virus replication. PloS Pathog (2019) 15(5):e1007798. doi: 10.1371/journal.ppat.1007798
72. McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljestrom P. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol Biol Cell (2005) 16(8):3753–63. doi: 10.1091/mbc.e05-02-0124
73. Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, et al. Sequestration of G3BP coupled with efficient translation inhibits stress granules in semliki forest virus infection. Mol Biol Cell (2012) 23(24):4701–12. doi: 10.1091/mbc.E12-08-0619
74. Nowee G, Bakker JW, Geertsema C, Ros VID, Goertz GP, Fros JJ, et al. A tale of 20 alphaviruses; inter-species diversity and conserved interactions between viral non-structural protein 3 and stress granule proteins. Front Cell Dev Biol (2021) 9:625711. doi: 10.3389/fcell.2021.625711
75. Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, et al. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol (2012) 86(19):10873–9. doi: 10.1128/JVI.01506-12
76. Matthews JD, Frey TK. Analysis of subcellular G3BP redistribution during rubella virus infection. J Gen Virol (2012) 93(Pt 2):267–74. doi: 10.1099/vir.0.036780-0
77. Khaperskyy DA, Hatchette TF, McCormick C. Influenza a virus inhibits cytoplasmic stress granule formation. FASEB J (2012) 26(4):1629–39. doi: 10.1096/fj.11-196915
78. Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. Influenza a virus host shutoff disables antiviral stress-induced translation arrest. PloS Pathog (2014) 10(7):e1004217. doi: 10.1371/journal.ppat.1004217
79. Hanley LL, McGivern DR, Teng MN, Djang R, Collins PL, Fearns R. Roles of the respiratory syncytial virus trailer region: effects of mutations on genome production and stress granule formation. Virology (2010) 406(2):241–52. doi: 10.1016/j.virol.2010.07.006
80. Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE Jr. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol (2010) 84(23):12274–84. doi: 10.1128/JVI.00260-10
81. Lindquist ME, Mainou BA, Dermody TS, Crowe JE Jr. Activation of protein kinase r is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology (2011) 413(1):103–10. doi: 10.1016/j.virol.2011.02.009
82. Oh SW, Onomoto K, Wakimoto M, Onoguchi K, Ishidate F, Fujiwara T, et al. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PloS Pathog (2016) 12(2):e1005444. doi: 10.1371/journal.ppat.1005444
83. Iseni F, Garcin D, Nishio M, Kedersha N, Anderson P, Kolakofsky D. Sendai Virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J (2002) 21(19):5141–50. doi: 10.1093/emboj/cdf513
84. Takeuchi K, Komatsu T, Kitagawa Y, Sada K, Gotoh B. Sendai Virus c protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J Virol (2008) 82(20):10102–10. doi: 10.1128/JVI.00599-08
85. Samuel CE, Duncan R, Knutson GS, Hershey JW. Mechanism of interferon action. increased phosphorylation of protein synthesis initiation factor eIF-2 alpha in interferon-treated, reovirus-infected mouse L929 fibroblasts. Vitro vivo. J Biol Chem (1984) 259(21):13451–7. doi: 10.1016/S0021-9258(18)90715-5
86. Yi Z, Fang C, Pan T, Wang J, Yang P, Yuan Z. Subproteomic study of hepatitis c virus replicon reveals ras-GTPase-activating protein binding protein 1 as potential HCV RC component. Biochem Biophys Res Commun (2006) 350(1):174–8. doi: 10.1016/j.bbrc.2006.09.027
87. Toroney R, Nallagatla SR, Boyer JA, Cameron CE, Bevilacqua PC. Regulation of PKR by HCV IRES RNA: importance of domain II and NS5A. J Mol Biol (2010) 400(3):393–412. doi: 10.1016/j.jmb.2010.04.059
88. Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, et al. Hepatitis c virus hijacks p-body and stress granule components around lipid droplets. J Virol (2011) 85(14):6882–92. doi: 10.1128/JVI.02418-10
89. Garaigorta U, Heim MH, Boyd B, Wieland S, Chisari FV. Hepatitis c virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J Virol (2012) 86(20):11043–56. doi: 10.1128/JVI.07101-11
90. Emara MM, Brinton MA. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci United States America (2007) 104(21):9041–6. doi: 10.1073/pnas.0703348104
91. Roth H, Magg V, Uch F, Mutz P, Klein P, Haneke K, et al. Flavivirus infection uncouples translation suppression from cellular stress responses. mBio (2017) 8(1):e00488–17. doi: 10.1128/mBio.02150-16
92. Amorim R, Temzi A, Griffin BD, Mouland AJ. Zika virus inhibits eIF2alpha-dependent stress granule assembly. PloS Negl Trop Dis (2017) 11(7):e0005775. doi: 10.1371/journal.pntd.0005775
93. Hou S, Kumar A, Xu Z, Airo AM, Stryapunina I, Wong CP, et al. Zika virus hijacks stress granule proteins and modulates the host stress response. J Virol (2017) 91(16):e00474–17. doi: 10.1128/JVI.00474-17
94. Katoh H, Okamoto T, Fukuhara T, Kambara H, Morita E, Mori Y, et al. Japanese Encephalitis virus core protein inhibits stress granule formation through an interaction with caprin-1 and facilitates viral propagation. J Virol (2013) 87(1):489–502. doi: 10.1128/JVI.02186-12
95. Valiente-Echeverria F, Melnychuk L, Vyboh K, Ajamian L, Gallouzi IE, Bernard N, et al. eEF2 and ras-GAP SH3 domain-binding protein (G3BP1) modulate stress granule assembly during HIV-1 infection. Nat Commun (2014) 5:4819. doi: 10.1038/ncomms5819
96. Cinti A, Le Sage V, Ghanem M, Mouland AJ. HIV-1 gag blocks selenite-induced stress granule assembly by altering the mRNA cap-binding complex. mBio (2016) 7(2):e00329. doi: 10.1128/mBio.00329-16
97. Legros S, Boxus M, Gatot JS, Van Lint C, Kruys V, Kettmann R, et al. The HTLV-1 tax protein inhibits formation of stress granules by interacting with histone deacetylase 6. Oncogene (2011) 30(38):4050–62. doi: 10.1038/onc.2011.120
98. Cassady KA, Gross M. The herpes simplex virus type 1 U(S)11 protein interacts with protein kinase r in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J Virol (2002) 76(5):2029–35. doi: 10.1128/jvi.76.5.2029-2035.2002
99. Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase r in cells infected with herpes simplex virus 1. J Virol (2013) 87(6):3271–6. doi: 10.1128/JVI.03049-12
100. Finnen RL, Hay TJ, Dauber B, Smiley JR, Banfield BW. The herpes simplex virus 2 virion-associated ribonuclease vhs interferes with stress granule formation. J Virol (2014) 88(21):12727–39. doi: 10.1128/JVI.01554-14
101. Dauber B, Poon D, Dos Santos T, Duguay BA, Mehta N, Saffran HA, et al. The herpes simplex virus virion host shutoff protein enhances translation of viral true late mRNAs independently of suppressing protein kinase r and stress granule formation. J Virol (2016) 90(13):6049–57. doi: 10.1128/JVI.03180-15
102. Finnen RL, Zhu MZ, Li J, Romo D, Banfield BW. Herpes simplex virus 2 virion host shutoff endoribonuclease activity is required to disrupt stress granule formation. J Virol (2016) 90(17):7943–55. doi: 10.1128/Jvi.00947-16
103. Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol (2005) 79(11):6890–9. doi: 10.1128/JVI.79.11.6890-6899.2005
104. Li S, Peters GA, Ding K, Zhang X, Qin J, Sen GC. Molecular basis for PKR activation by PACT or dsRNA. Proc Natl Acad Sci United States America (2006) 103(26):10005–10. doi: 10.1073/pnas.0602317103
105. Ziehr B, Vincent HA, Moorman NJ. Human cytomegalovirus pTRS1 and pIRS1 antagonize protein kinase r to facilitate virus replication. J Virol (2016) 90(8):3839–48. doi: 10.1128/JVI.02714-15
106. Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem (2000) 275(48):37993–8. doi: 10.1074/jbc.M004762200
107. Sharma NR, Majerciak V, Kruhlak MJ, Zheng ZM. KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PloS Pathog (2017) 13(10):e1006677. doi: 10.1371/journal.ppat.1006677
108. Katsafanas GC, Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe (2007) 2(4):221–8. doi: 10.1016/j.chom.2007.08.005
109. Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, et al. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J Virol (2011) 85(4):1581–93. doi: 10.1128/JVI.02247-10
110. Rozelle DK, Filone CM, Kedersha N, Connor JH. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol Cell Biol (2014) 34(11):2003–16. doi: 10.1128/MCB.01630-13
111. Sola I, Galan C, Mateos-Gomez PA, Palacio L, Zuniga S, Cruz JL, et al. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J Virol (2011) 85(10):5136–49. doi: 10.1128/JVI.00195-11
112. Dinh PX, Beura LK, Das PB, Panda D, Das A, Pattnaik AK. Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J Virol (2013) 87(1):372–83. doi: 10.1128/JVI.02305-12
113. Linero FN, Thomas MG, Boccaccio GL, Scolaro LA. Junin virus infection impairs stress-granule formation in vero cells treated with arsenite via inhibition of eIF2alpha phosphorylation. J Gen Virol (2011) 92(Pt 12):2889–99. doi: 10.1099/vir.0.033407-0
114. Lloyd RE. Regulation of stress granules and p-bodies during RNA virus infection. Wiley Interdiscip Rev RNA (2013) 4(3):317–31. doi: 10.1002/wrna.1162
115. Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, et al. Host factors associated with the sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J Virol (2010) 84(13):6720–32. doi: 10.1128/JVI.01983-09
116. Piotrowska J, Hansen SJ, Park N, Jamka K, Sarnow P, Gustin KE. Stable formation of compositionally unique stress granules in virus-infected cells. J Virol (2010) 84(7):3654–65. doi: 10.1128/JVI.01320-09
117. Preiss T, W Hentze M. Starting the protein synthesis machine: eukaryotic translation initiation. BioEssays (2003) 25(12):1201–11. doi: 10.1002/bies.10362
118. Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, et al. Reovirus induces and benefits from an integrated cellular stress response. J Virol (2006) 80(4):2019–33. doi: 10.1128/JVI.80.4.2019-2033.2006
119. Carroll K, Hastings C, Miller CL. Amino acids 78 and 79 of mammalian orthoreovirus protein microNS are necessary for stress granule localization, core protein lambda2 interaction, and. novo Virus replication. Virol (2014) 448:133–45. doi: 10.1016/j.virol.2013.10.009
120. Thulasi Raman SN, Liu G, Pyo HM, Cui YC, Xu F, Ayalew LE, et al. DDX3 interacts with influenza a virus NS1 and NP proteins and exerts antiviral function through regulation of stress granule formation. J Virol (2016) 90(7):3661–75. doi: 10.1128/JVI.03010-15
121. Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BR, et al. PKR and RNase l contribute to protection against lethal West Nile virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol (2006) 80(14):7009–19. doi: 10.1128/JVI.00489-06
122. Tu YC, Yu CY, Liang JJ, Lin E, Liao CL, Lin YL. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J Virol (2012) 86(19):10347–58. doi: 10.1128/JVI.00525-12
123. Bidet K, Dadlani D, Garcia-Blanco MA. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PloS Pathog (2014) 10(7):e1004242. doi: 10.1371/journal.ppat.1004242
124. Xu S, Chen D, Chen D, Hu Q, Zhou L, Ge X, et al. Pseudorabies virus infection inhibits stress granules formation via dephosphorylating eIF2alpha. Veterinary Microbiol (2020) 247:108786. doi: 10.1016/j.vetmic.2020.108786
125. Zhu T, Jiang X, Xin H, Zheng X, Xue X, Chen JL, et al. GADD34-mediated dephosphorylation of eIF2alpha facilitates pseudorabies virus replication by maintaining de novo protein synthesis. Vet Res (2021) 52(1):148. doi: 10.1186/s13567-021-01018-5
126. Katsafanas GC, Moss B. Vaccinia virus intermediate stage transcription is complemented by ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem (2004) 279(50):52210–7. doi: 10.1074/jbc.M411033200
127. Sanchez-Jimenez C, Ludena MD, Izquierdo JM. T-Cell intracellular antigens function as tumor suppressor genes. Cell Death Dis (2015) 6:e1669. doi: 10.1038/Cddis.2015.43
128. Ziehr B, Lenarcic E, Cecil C, Moorman NJ. The eIF4AIII RNA helicase is a critical determinant of human cytomegalovirus replication. Virology (2016) 489:194–201. doi: 10.1016/j.virol.2015.12.009
129. Biedenkopf N, Lange-Grunweller K, Schulte FW, Weisser A, Muller C, Becker D, et al. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antiviral Res (2017) 137:76–81. doi: 10.1016/j.antiviral.2016.11.011
130. Morinaga N, Yahiro K, Matsuura G, Moss J, Noda M. Subtilase cytotoxin, produced by shiga-toxigenic escherichia coli, transiently inhibits protein synthesis of vero cells via degradation of BiP and induces cell cycle arrest at G1 by downregulation of cyclin D1. Cell Microbiol (2008) 10(4):921–9. doi: 10.1111/j.1462-5822.2007.01094.x
131. Gonzalez MR, Bischofberger M, Freche B, Ho S, Parton RG, van der Goot FG. Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol (2011) 13(7):1026–43. doi: 10.1111/j.1462-5822.2011.01600.x
132. Song EJ, Lee SJ, Lim HS, Kim JS, Jang KK, Choi SH, et al. Vibrio vulnificus VvhA induces autophagy-related cell death through the lipid raft-dependent c-Src/NOX signaling pathway. Sci Rep (2016) 6:27080. doi: 10.1038/srep27080
133. van ‘t Wout EF, van Schadewijk A, van Boxtel R, Dalton LE, Clarke HJ, Tommassen J, et al. Virulence factors of pseudomonas aeruginosa induce both the unfolded protein and integrated stress responses in airway epithelial cells. PloS Pathog (2015) 11(6):e1004946. doi: 10.1371/journal.ppat.1004946
134. Shrestha N, Bahnan W, Wiley DJ, Barber G, Fields KA, Schesser K. Eukaryotic initiation factor 2 (eIF2) signaling regulates proinflammatory cytokine expression and bacterial invasion. J Biol Chem (2012) 287(34):28738–44. doi: 10.1074/jbc.M112.375915
135. Vonaesch P, Campbell-Valois FX, Dufour A, Sansonetti PJ, Schnupf P. Shigella flexneri modulates stress granule composition and inhibits stress granule aggregation. Cell Microbiol (2016) 18(7):982–97. doi: 10.1111/cmi.12561
136. Go D, Lee J, Choi JA, Cho SN, Kim SH, Son SH, et al. Reactive oxygen species-mediated endoplasmic reticulum stress response induces apoptosis of mycobacterium avium-infected macrophages by activating regulated IRE1-dependent decay pathway. Cell Microbiol (2019) 21(12):e13094. doi: 10.1111/cmi.13094
137. Ogbechi J, Hall BS, Sbarrato T, Taunton J, Willis AE, Wek RC, et al. Inhibition of Sec61-dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis (2018) 9(3):397. doi: 10.1038/s41419-018-0427-y
138. Choi HH, Shin DM, Kang G, Kim KH, Park JB, Hur GM, et al. Endoplasmic reticulum stress response is involved in mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett (2010) 584(11):2445–54. doi: 10.1016/j.febslet.2010.04.050
139. Lim YJ, Choi JA, Choi HH, Cho SN, Kim HJ, Jo EK, et al. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of mycobacterium tuberculosis. PloS One (2011) 6(12):e28531. doi: 10.1371/journal.pone.0028531
140. Pan Q, Wang X, Liu T, Yu Y, Li L, Zhou R, et al. Mycoplasma hyopneumoniae inhibits porcine beta-defensin 2 production by blocking the unfolded protein response to facilitate epithelial adhesion and infection. Infection Immun (2020) 88(7):e00164–20. doi: 10.1128/IAI.00164-20
141. Pereira RM, Teixeira KL, Barreto-de-Souza V, Calegari-Silva TC, De-Melo LD, Soares DC, et al. Novel role for the double-stranded RNA-activated protein kinase PKR: modulation of macrophage infection by the protozoan parasite leishmania. FASEB J (2010) 24(2):617–26. doi: 10.1096/fj.09-140053
142. Anand SS, Babu PP. Endoplasmic reticulum stress and neurodegeneration in experimental cerebral malaria. Neurosignals (2013) 21(1-2):99–111. doi: 10.1159/000336970
143. Ietta F, Maioli E, Daveri E, Gonzaga Oliveira J, da Silva RJ, Romagnoli R, et al. Rottlerin-mediated inhibition of toxoplasma gondii growth in BeWo trophoblast-like cells. Sci Rep (2017) 7(1):1279. doi: 10.1038/s41598-017-01525-6
144. English BC, Van Prooyen N, Ord T, Ord T, Sil A. The transcription factor CHOP, an effector of the integrated stress response, is required for host sensitivity to the fungal intracellular pathogen histoplasma capsulatum. PloS Pathog (2017) 13(9):e1006589. doi: 10.1371/journal.ppat.1006589
145. Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, et al. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol Cell Biol (2013) 33(4):815–29. doi: 10.1128/MCB.00763-12
146. Feng S, Chen W, Cao D, Bian J, Gong FY, Cheng W, et al. Involvement of na(+), K (+)-ATPase and its inhibitors in HuR-mediated cytokine mRNA stabilization in lung epithelial cells. Cell Mol Life sciences: CMLS (2011) 68(1):109–24. doi: 10.1007/s00018-010-0444-1
147. Kim WJ, Back SH, Kim V, Ryu I, Jang SK. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol (2005) 25(6):2450–62. doi: 10.1128/MCB.25.6.2450-2462.2005
148. Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol (2006) 7(6):644–51. doi: 10.1038/ni1338
149. Herman AB, Silva Afonso M, Kelemen SE, Ray M, Vrakas CN, Burke AC, et al. Regulation of stress granule formation by inflammation, vascular injury, and atherosclerosis. Arteriosclerosis thrombosis Vasc Biol (2019) 39(10):2014–27. doi: 10.1161/ATVBAHA.119.313034
150. Li T, Safitri M, Zhang K, Wang Y, Huang L, Zhu Y, et al. Downregulation of G3BP2 reduces atherosclerotic lesions in ApoE(-/-) mice. Atherosclerosis (2020) 310:64–74. doi: 10.1016/j.atherosclerosis.2020.08.003
151. Schwed-Gross A, Hamiel H, Faber GP, Angel M, Ben-Yishay R, Benichou JIC, et al. Glucocorticoids enhance chemotherapy-driven stress granule assembly and impair granule dynamics, leading to cell death. J Cell Sci (2022) 135(14):jcs259629. doi: 10.1242/jcs.259629
152. Chen Y, Joe Y, Park J, Song HC, Kim UH, Chung HT. Carbon monoxide induces the assembly of stress granule through the integrated stress response. Biochem Biophys Res Commun (2019) 512(2):289–94. doi: 10.1016/j.bbrc.2019.03.017
153. Li WY, Yang F, Li X, Wang LW, Wang Y. Stress granules inhibit endoplasmic reticulum stress-mediated apoptosis during hypoxia-induced injury in acute liver failure. World J Gastroenterol (2023) 29(8):1315–29. doi: 10.3748/wjg.v29.i8.1315
154. Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell (2009) 36(6):932–41. doi: 10.1016/j.molcel.2009.11.020
Keywords: stress granules, infection, inflammation, innate immunity, therapeutic targets
Citation: Li W and Wang Y (2023) Stress granules: potential therapeutic targets for infectious and inflammatory diseases. Front. Immunol. 14:1145346. doi: 10.3389/fimmu.2023.1145346
Received: 16 January 2023; Accepted: 12 April 2023;
Published: 02 May 2023.
Edited by:
Belinda Hall, University of Surrey, United KingdomReviewed by:
Szu Ting Jessica Chen, National Yang-Ming University, TaiwanBéatrice Nal, INSERM U1104 Centre d’immunologie de Marseille-Luminy (CIML), France
Copyright © 2023 Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yao Wang, cm0wMDM3NDNAd2h1LmVkdS5jbg==
†ORCID: Wenyuan Li, orcid.org/0000-0001-9385-3069
Yao Wang, orcid.org/0000-0001-7701-5313