 Arthur Xuan Wang
Arthur Xuan Wang Xiao Jing Ong
Xiao Jing Ong Criselle D’Souza
Criselle D’Souza Paul J. Neeson
Paul J. Neeson Joe Jiang Zhu
Joe Jiang Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 06 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1140541
This article is part of the Research Topic Community Series in Combining Chemo/radio Therapy and Immunotherapy for Cancers—Perfect Mix of Old and New, volume II View all 31 articles
Chemotherapy has long been a standard treatment for a wide range of malignancies, where patients typically undergo multiple rounds of chemotherapy regimens to control tumor growth. In the clinic, the chemotherapy drugs cyclophosphamide and fludarabine are commonly used prior to Chimeric Antigen Receptor T (CAR-T) cell therapy to lymphodeplete and improve CAR-T cell engraftment. In this review, we discuss the use of chemotherapy in combination with CAR-T cell therapy. We also show that chemotherapy can deplete immunosuppressive cells, promote a pro-inflammatory tumor microenvironment, disrupt tumor stroma, and improve CAR-T cell recruitment to the tumor. Although the combination of chemotherapy plus CAR-T cell therapy is promising, certain aspects of chemotherapy also pose a challenge. In addition, the combined therapeutic effect may be heavily dependent on the dose and the treatment schedule. Thus, we also discussed the obstacles to effective clinical outcomes of the combination therapy.
Recently, a large number of cancer therapies have been developed to facilitate a patient’s immune system against cancers. One such therapy is called the Chimeric Antigen Receptor (CAR) T cell therapy, which involves the adoptive transfer of autologous T cells that have been genetically engineered with a CAR to target tumor cells (1). CAR-T cell therapy has induced remission in patients with relapsed/refractory B-cell malignancies (2–4). However, this success has not occurred in patients with solid tumors. The reasons for this poor outcome include tumor heterogeneity, an immunosuppressive tumor microenvironment (TME), insufficient T-cell trafficking to the tumor site, and poor T-cell persistence (5).
Chemotherapy has long been a standard-of-care treatment for many cancers, especially advanced solid tumors. The successful treatment of cancers requires a combination of different approaches. However, because chemotherapy can exert negative effects on the immune system, it is not clear whether chemotherapy can be combined with immunotherapy more broadly. Indeed, different classes of chemotherapeutic drugs such as alkylating agents (e.g., cyclophosphamide), platinum compounds (e.g., cisplatin, carboplatin, oxaliplatin), antimetabolites (e.g., methotrexate), anthracyclines, DNA methyltransferase inhibitors and spindle poisons (e.g., taxanes) have mixed effects on the immune system (6). Certain drugs can induce profound immunosuppression, while some drugs can enhance anti-tumor immunity. Some synergistic effects include relieving tumor-induced immunosuppression, augmenting the anti-tumor activity of cytotoxic immune cells, and improving immune cell trafficking to tumor sites. As a result, these positive effects of chemotherapy could overcome some of the roadblocks for CAR-T cell therapy in treating solid cancers. Hence, this review will summarise the main effects of chemotherapy when combined with CAR-T cells, including the promises and challenges of combination therapy for solid cancers.
CAR-T cell therapy have produced impressive clinical responses in relapsed/refractory B cell malignancies. However, there are numerous factors to be considered during the treatment, including the need for a bridging therapy or conditioning regimen prior to the infusion of CAR-T cell product. These regimens significantly impact the clinical outcomes.
The infusion of CAR-T cells must be performed in a timely manner to control disease progression (7). However, the period between apheresis and CAR-T cell infusion can be weeks to months, e.g. a maximum of 105 days (median time of 45 days) in the ELIANA trial (NCT02435849) (3). This time interval may lead to a treatment gap where some patients may experience disease progression and/or death. As a result, 7% of patients did not survive while awaiting the production of CAR-T cells (8, 9). Therefore, bridging therapy is critical in controlling disease burden prior to CAR-T cell treatment.
The choice of bridging therapy is highly variable, depending on the patient’s cancer type, disease stage, prior treatments, and disease burden. Chemotherapy is one of the most widely used bridging therapies, including kinase modulators, topoisomerase inhibitors, platinum-based agents, and drugs that interfere with DNA replication, synthesis and repair (10). These drugs can inhibit tumor growth to achieve disease control in the bridging period. As a bridging therapy, chemotherapy can also debulk the tumor before CAR-T cell infusion in some cases. This is an important consideration as high disease burdens have been associated with toxicities following CAR-T cell infusions, such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) (11).
Studies showed that lymphodepletion prior to adoptive T-cell transfer significantly enhanced their expansion, engraftment, and anti-tumor efficacy (12). This enhancement is likely due to reduced immunosuppressive cells (e.g., myeloid-derived suppressor cells and regulatory T cells) (12, 13), increased homeostatic cytokine production (14, 15), and the downregulation of indoleamine 2,3-dioxygenase (IDO) expression in the tumor (16). Together, these mechanisms may create an optimal environment for the anti-tumor function of infused CAR-T cells. Along with improved CAR-T cell engraftment and homeostatic expansion, these findings led to the introduction of lymphodepletion conditioning regimens with CAR-T cell therapy.
Chemotherapeutic drugs such as cyclophosphamide (Cy) and fludarabine (Flu) are the most commonly used lymphodepleting regimen in CAR-T cell therapy (250-500 mg/m2 cyclophosphamide, 25-30 mg/m2 fludarabine, 3-5 days before infusion) (17, 18). Compared to Cy alone, the combination of Cy/Flu significantly enhanced CAR-T cell expansion and was associated with better clinical responses (Cy/Flu = 50% CR, 72% ORR versus Cy alone = 8% CR, 50% ORR) (19). In the JULIET trial, patients with the Cy-resistant disease also received bendamustine (90 mg/m2, 2-11 days before infusion) in lieu of Cy/Flu, with similar clinical outcomes (20). However, there are risks associated with intense lymphodepletion, including prolonged neutropenia and toxicities. Therefore, further studies are needed to investigate the favourable dose and schedule of lymphodepletion with minimal side effects and enhanced clinical response to CAR-T therapy.
Although chemotherapy has not been conventionally utilized as a neoadjuvant or adjuvant treatment for CAR-T cell therapy, emerging evidence suggests that specific chemotherapy drugs may work in concert with CAR-T cells (6, 21) (Table 1). This novel combination strategy shows great potential to overcome some barriers in CAR-T cell therapy and provide synergistic effects in treating solid tumors.
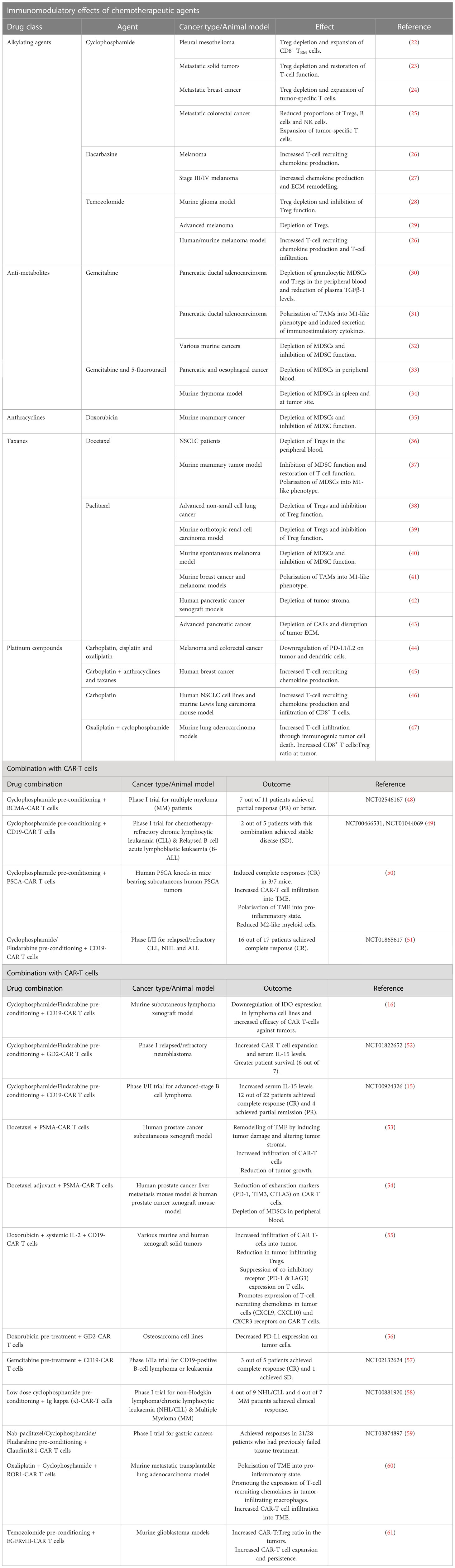
Table 1 Chemotherapeutic drugs that can be used as neoadjuvant or adjuvant regimen with CAR-T cell therapy - preclinical and clinical evidence.
The immunosuppressive tumor microenvironment (TME) in solid tumors is a significant obstacle to achieving clinical response to CAR-T cell therapy (62). Numerous immune suppressive cells infiltrate the TME, including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs). These cells promote immunosuppression and negatively regulate CAR-T cell effector function (63). In addition, cancer-associated fibroblasts (CAFs) produce extracellular matrix components in the TME (64), forming a physical barrier restricting CAR-T cell penetration. Together, these cells play a pro-tumorigenic role that interferes with CAR-T cell efficacy (65–67). This anti-inflammatory milieu would lead to poor CAR-T cell penetration, expansion, and persistence, rendering the therapy ineffective.
In this regard, chemotherapy is a promising approach for remodelling the TME, and could lead to the enhanced therapeutic efficacy of CAR-T cells in solid tumors. Chemotherapy drugs can modify the TME in four main ways: reducing immune suppressor cells, repolarising the anti-inflammatory immune microenvironment, disrupting the tumor stroma, and altering the chemokine profile for T cell trafficking. These mechanisms are discussed in the following sections of this review.
Chemotherapy has been shown to reduce the various immune suppressor cells in the TME. Cy/Flu conditioning regimen have enhanced CAR-T cell expansion and clinical responses in cancer patients (48, 49, 51, 52). Several preclinical and clinical studies have shown that metronomic, low-dose Cy regimen (12.5mg/kg, single dose) reduced the number and function of Tregs, thereby restoring tumor-specific T cell responses (22–25, 58, 68) (Figure 1A). Importantly, low-dose Cy does not affect tumor-responding T cells (24), suggesting that low-dose Cy could be used as an adjuvant treatment post-CAR-T cell infusion.
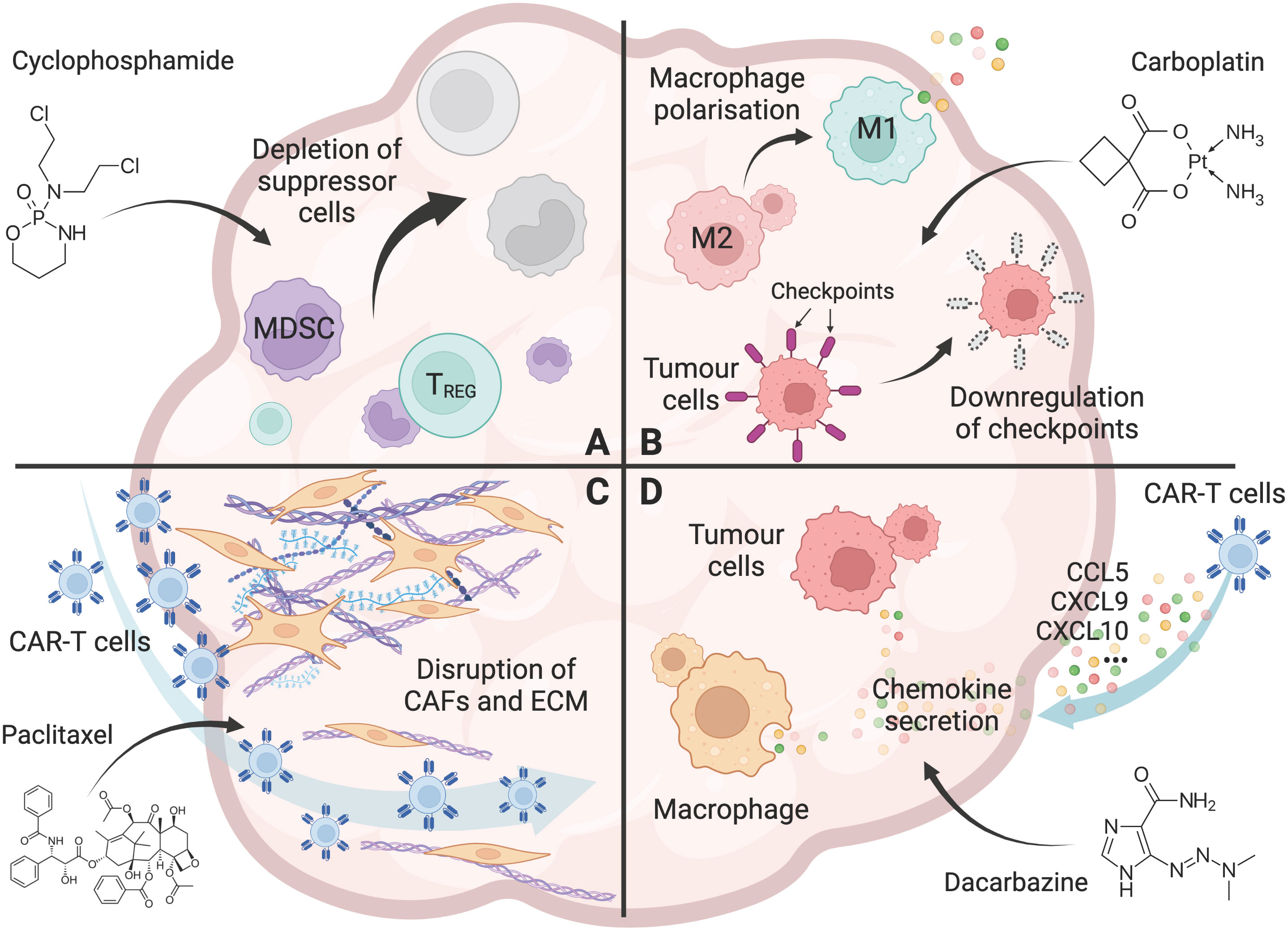
Figure 1 Chemotherapy overcomes the hurdles for CAR-T cells in solid tumors. (A) The effect of chemotherapy (Cyclophosphamide as an example) on the depletion of immunosuppressive cells, including Tregs, tumor associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment. (B) The effect of chemotherapy (Carboplatin as an example) on promoting a pro-inflammatory tumor microenvironment, including polarisation of macrophages from an M2 to an M1 phenotype, and downregulation of immune checkpoint molecules including PD-L1 and PD-L2 on tumor cells. (C) The effect of chemotherapy (Paclitaxel as an example) on the disruption of the tumor stroma, including removal of cancer-associated fibroblasts (CAFs) and their associated extracellular matrix (ECM) from the tumor barrier, allowing increased T-cell infiltration into the tumor. (D) The effect of chemotherapy (Dacarbazine as an example) on enhanced T-cell trafficking to the tumor, including the increased chemokine CCL5, CXCL9, and CXCL10 secretion by tumor cells and macrophages. Created with BioRender.com.
Temozolomide is another potential neoadjuvant treatment for patients with glioblastoma receiving CAR-T cells. Temozolomide reduced Tregs in both mouse models and in patients (28, 29). In murine glioblastoma models, temozolomide treatment also increased CAR-T cell expansion resulting in a greater CAR-T cell to Treg ratio (61).
Two taxane drugs (paclitaxel and docetaxel) reduced immune suppressor cells in the TME. Tregs in non-small cell lung cancer patients were reduced after four cycles of docetaxel (30 mg/m2) and cisplatin (75 mg/m2) (36). A similar reduction in Tregs and impairment of their inhibitory function were also observed with paclitaxel treatment (38, 39). In addition, when given as neoadjuvant treatment prior to CAR-T cell infusion, docetaxel was found to enhance the PSMA-CAR-T cell efficacy in prostate cancer mouse models, by reducing MDSCs in the tumor (54). Similarly, paclitaxel has also been found to reduce the tumor-infiltrating MDSCs and restore effector functions of CD8+ T cells (40). However, this impact was not consistently observed across other studies (34, 69). This discrepancy could be due to differences in drug dosage and treatment schedule.
Gemcitabine can also deplete MDSCs in cancer patients. In a Phase I/IIa trial of CAR-T cell therapy, three out of five patients who received gemcitabine as neoadjuvant treatment achieved complete response (CR) following CAR-T infusion (57). Preclinical and clinical studies demonstrated that gemcitabine treatment reduced the amount of MDSCs (30, 33) and increased the cytotoxicity of anti-tumor CD8+ T cells and NK cells (32), this may contribute to enhanced responses to CAR-T cells.
Doxorubicin is another chemotherapeutic drug shown to reduce Tregs and MDSCs in the TME. When combined with CAR-T cells (55) or adoptively transferred T-helper cells (35), doxorubicin synergistically increased anti-tumor activity by diminishing the Tregs and MDSCs, leading to tumor suppression in both murine and human xenograft models.
In solid tumors, tumor cells can express high levels of immune checkpoint molecules, including PD-L1 and PD-L2, to inhibit the effector functions of tumor-infiltrating T cells and CAR-T cells (70). In this regard, doxorubicin has been shown to affect CAR-T cell activity by lowering the expression of the immune checkpoint PD-L1 on tumor cells in osteosarcomas (56). Platinum-based chemotherapeutic drugs such as carboplatin, cisplatin, and oxaliplatin have also been shown to reduce PD-L2 expression on tumor cells, resulting in increased tumor cell recognition, proliferation and cytokine secretion of tumor-specific T cells (44) (Figure 1B).
Studies also revealed that oxaliplatin ameliorates the anti-inflammatory TME and enhances the recruitment of ROR1-CAR-T cells through modulating the TME chemokine profile (60). This polarisation of anti-inflammatory TME into a favourable pro-inflammatory state was also observed in CAR-T cell therapy with prior Cy treatment (50). The change in TME polarisation into a more inflammatory milieu is most likely accomplished through the activation and differentiation of myeloid cells into a pro-inflammatory phenotype. This TME change produces chemokines that recruit CAR-T cells, transforming the “cold” tumor into a “hot” tumor. In addition, paclitaxel, docetaxel, and gemcitabine were also shown to polarise macrophages into an M1-like phenotype to facilitate anti-tumor immunity (31, 37, 41, 71). This further provides a rationale for the combination therapy of CAR-T cells with chemotherapeutic drugs to overcome the immunosuppression in solid tumors.
Besides the suppressor cells, the immunosuppressive tumor microenvironment is also enriched with stroma that could exclude CAR-T cell infiltration. The anti-microtubule agent, nab-paclitaxel, damaged the tumor stroma in primary tumors of advanced-stage prostate cancer patients (42, 43). Patients treated with nab-paclitaxel demonstrated less abundant fibrillar collagen matrices and lower CAF numbers, which were also confirmed using mice patient-derived xenograft (PDX) models (43). In a Phase I clinical trial of combination treatment of Claudin18.2-CAR-T cells with paclitaxel plus Cy, 21 out of 28 patients who failed prior taxane treatment achieved a significant clinical response to the combination treatment. The author suggested that this effect may be due to the accumulated nab-paclitaxel in the tumor stroma, which disrupted cancer-stromal interactions and helped with the CAR-T cell infiltration (59) (Figure 1C). Similarly, in a human xenograft mouse model, it was found that PSMA-CAR-T cells, in combination with low-dose, non-ablative docetaxel as a neoadjuvant regimen, eradicated large established tumors (53). Further analysis revealed that docetaxel could remodel the TME by altering the tumor stroma, which allowed the infiltration of CAR-T cells into the tumor site. These findings raise the possibility of using chemotherapy to overcome the physical barrier of TME, allowing CAR-T cell penetration.
T-cell trafficking to tumors remains a significant challenge for CAR-T cell efficacy. Successful T-cell trafficking depends on various chemokines including CCL5, CXCL9 and CXCL10 (72), and is critical for the response to CAR-T cell therapy. In cutaneous melanoma mouse models, temozolomide increased the expression of CCL5, CXCL9, and CXCL10, resulting in increased T-cell trafficking to the tumor (26). Furthermore, in patients treated with dacarbazine, CCL5, CXCL9, and CXCL10 expression was increased in chemotherapy-sensitive tumors, which was associated with increased T-cell infiltrate and enhanced patient survival (26). Other chemotherapeutic agents, such as dacarbazine, carboplatin, anthracycline and taxane, were also seen to increase the secretion of these chemokines and enhanced T cell trafficking to tumors (27, 45) (Figure 1D).
In preclinical mouse models, poor T-cell trafficking was associated with low levels of chemokine expression, such as ligands for CXCR3 and CXCR4 (73). Pre-treatment with oxaliplatin and cyclophosphamide induced a pro-inflammatory tumor microenvironment and enhanced secretion of CCL5, CXCL9, CXCL10, and CXCL16 by tumor-infiltrating macrophages, leading to enhanced ROR1-CAR-T cell infiltration (60). The combination was also able to increase CD8+ T cell infiltration and improved tumor control (47). This immunomodulatory effect was not restricted to oxaliplatin, other platinum-based drugs, such as carboplatin, have also increased tumor CD8+ T cell populations and enhanced CCL5 and CXCL10 mRNA levels in lung cancer cells (46).
Taken together, certain chemotherapeutic drugs can alter the immunosuppressive tumor microenvironment to facilitate CAR-T cell trafficking, infiltration, expansion, and anti-tumor efficacy in solid tumors. However, the evidence listed above also indicated the choice of chemotherapeutic drugs, the dosage and the treatment schedule must be taken into serious consideration when designed in combination with CAR-T cell therapy.
Chemotherapy is well known for its cytotoxic effects, which may also have a deleterious effect on CAR-T cell viability and function, inducing apoptosis of immune cells, and often leading to lymphopenia (6). The cytotoxic effect of chemotherapy is often mediated via mechanisms such as DNA damage and cell cycle arrest, and preferentially targets rapidly dividing cells such as tumour cells and bone marrow stem cells. The effect is drug and dosage dependent. Some drugs, such as cyclophosphamide and fludarabine, are considered as non-myeloablative but lymphodepleting. They may affect the transferred CAR-T cells both in circulation and at tumor site. Thus, the systemic delivery of chemotherapy may not only affect the endogenous T cell population generated at bone marrow, but also induces cytotoxicity on transferred CAR-T cells directly. Because CAR-T cells are usually given in one dose or several doses in a short period of time, the long persistence of the CAR-T cells and sustained anti-tumor function are the keys to therapeutic success. Thus, any adjuvant treatment post-CAR-T infusion may have an impact on long-term CAR-T cell efficacy.
After chemotherapy regimens, T cell numbers decrease, which gradually recovers over time (74). Notably, chemotherapy may preferentially deplete certain T-cell populations over others. In glioblastoma patients treated with combined radiotherapy and temozolomide, an increased effector memory T-cell (TEM) population and a decreased CD45RA-expressing effector memory T-cell population (TEMRA) population was observed (75). Additionally, cyclophosphamide treatment was found to decrease the mitochondrial function of naïve T-cells significantly more than central memory (TCM) and effector memory (TEM) T cells (76). The evidence indicated that certain chemotherapeutic drugs might significantly impair the CAR-T cell function when used in combination.
Chemotherapy may also negatively regulate the CD4+ T-cell population. Compared with CD8+ T cells, naïve CD4+ T cells did not recover to pre-treatment numbers in breast cancer patients following chemotherapy (77). Another study found that naïve CD4+ T cells were preferentially depleted after chemotherapy, with memory CD4+ T cells forming the majority of the CD4+ T cell population instead (78). This preferential depletion of naïve CD4+ T cells may negatively affect CAR-T cell efficacy, given the importance of CD4+ T cells in achieving long-term remission (79).
In CAR-T cell therapy, less differentiated T-cell subsets, including naïve T-cells and TCM cells, have shown greater efficacy compared to more differentiated effector T-cell and TEM cells (80). Thus, the susceptibility of naïve T-cells towards chemotherapy may decrease CAR-T cell efficacy when the chemotherapy is used as an adjuvant treatment.
Besides the effect on T-cell subsets, chemotherapy, such as carboplatin and taxane, may also induce the expression of immune checkpoint molecules on T cells, including PD-1 and CTLA4 in breast cancer patients, as a feedback immunosuppressive pathway following immune activation (45). This effect was also seen in other drugs, such as the oxaliplatin plus cyclophosphamide combination (47). Although combining with checkpoint blockades such as anti-PD1 or anti-CTLA4 may overcome this issue, the potential chemotherapy-induced early T-cell exhaustion should be considered for combination treatments.
Despite these challenges, the heterogenous response between studies highlights the multitude of factors affecting chemotherapy efficacy in combination with CAR-T therapy, including the type of drugs, treatment dose, regimen schedule, and cancer types. Thus, a successful combination of chemotherapy and CAR-T cell therapy relies greatly on the design of the treatment. To achieve this, the treatment plan must be optimised to allow long-term CAR-T cell persistence and anti-tumor function to provide the best clinical outcomes. In addition, CAR-T cells can also be engineered to resist these drugs, making it easier to optimise this combination. Such engineering may take inspirations from the chemo-resistance mechanisms observed in cancers, such as the upregulation of detoxifying molecules in response to platinum-based chemotherapies (81). The engineered expression chemo-resistance proteins such as platinum transporters and chelators may confer CAR-T cell resistance against platinum-based drugs. The CRISPR drug screening technology can also be used to select potential targets for drug resistance in CAR-T cells. Such candidates can then be conditionally expressed in CAR-T cells to gain chemo-resistance. In addition, chemotherapeutic drugs targeting DNA damage and the cell cycle also induce apoptosis in CAR-T cells. Thus, engineering mutated apoptotic machinery, such as the BCL-2 family proteins, may facilitate resistance to this drug-induced apoptosis in CAR-T cells. However, engineering CAR-T cells to make them resistant to apoptosis should be viewed with caution to avoid unexpected uncontrolled CAR-T cell proliferation.
Moreover, clinical strategies for combining chemotherapy with CAR-T cell therapy need to be evaluated. This combination therapy could be performed via two approaches. Chemotherapy’s anti-tumor effect and immunomodulatory properties are traditionally studied in the context of specific tumor types. Thus, chemotherapeutic drugs could be selected based on the patient’s tumor type. However, given the significant heterogeneity between tumor subtypes, such as in breast cancer, different drugs may be needed for different patients with the same cancer (82). Alternatively, a more personalised approach where drug choices are tailored to the patient’s TME may be used. By testing patient samples, the TME components can be revealed. This information can be used to select the best chemotherapeutic drugs to facilitate the CAR-T cell attack. For instance, if poor T cell trafficking is due to immunosuppressive cells such as MDSCs and Tregs, cyclophosphamide may be used. If the immune exclusion is caused by CAFs, paclitaxel could be used instead. In addition, it should be noted that many chemotherapeutic drugs, including paclitaxel, have a wide range of effects on the TME, e.g., depleting MDSCs, Tregs and CAFs at the same time, as outlined in Table 1. These personalised strategies focus more on the challenges for CAR-T cells in each individual patient’s tumor microenvironment and will be more likely to achieve better clinical outcomes for the ‘chemo plus CAR-T’ combination therapy.
Although a limited number of clinical trials have been performed, the combination treatment of chemotherapy and CAR-T cell therapy has significant capacity to improve the current clinical outcomes. It is a promising option for patients with advanced solid tumors, and further studies on the dose, treatment schedule, immune context, tumor types, and CAR-T cell engineering should be investigated to achieve best clinical outcomes. In addition, more mechanistic studies are still needed to understand how these therapies will best work together. Our increased understanding of the immunomodulatory effects of chemotherapy, together with the engineering of novel CAR-T cells, will further facilitate this combination strategy in the clinic and benefit more cancer patients.
Writing-original draft preparation, AW, XO. Writing-review and editing, CD’S, PN, JZ. Supervision, PN. AW and XO share first authorship. JZ, PN and CD’S share senior authorship. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ALL, acute lymphoblastic leukaemia; BCMA, B-cell maturation antigen; CAFs, cancer associated fibroblast; CAIX, carbonic anhydrase IX; CCL, C-C motif ligand; CLL, chronic lymphocytic leukaemia; CR, complete response; CXCL, C-X-C motif chemokine ligand; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; IDO, indolamine-2,3-dioxygenase 1; MDSCs, myeloid-derived suppressor cells; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; NK cells, natural killer cells; NSCLC, non-small cell lung cancer; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; PD-L2, programmed cell death ligand 2; PR, partial response; PSCA, prostate stem cell antigen; ROR1, tyrosine kinase-like orphan receptor 1; SD, stable disease; TAMs, tumor-associated macrophages; TGFβ, transforming growth factor beta; TILs, tumor-infiltrating lymphocytes; TME, tumor microenvironment; Tregs, regulatory T cells; TEM T effector memory; CAR, Chimeric Antigen Receptor; CRS, Cytokine Release Syndrome; ICANS, Immune effector cell-associated neurotoxicity syndrome.
1. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMra1706169
2. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a b-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet (2021) 398(10297):314–24. doi: 10.1016/S0140-6736(21)00933-8
3. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
4. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large b-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
5. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther (2021) 12(1):81. doi: 10.1186/s13287-020-02128-1
6. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi: 10.1038/nri2216
7. Johnson PC, Abramson JS. Patient selection for chimeric antigen receptor (CAR) T-cell therapy for aggressive b-cell non-Hodgkin lymphomas. Leuk Lymphoma (2020) 61(11):2561–7. doi: 10.1080/10428194.2020.1786563
8. Jain MD, Jacobs MT, Nastoupil LJ, Spiegel JY, Feng G, Lin Y, et al. Characteristics and outcomes of patients receiving bridging therapy while awaiting manufacture of standard of care axicabtagene ciloleucel CD19 chimeric antigen receptor (CAR) T-cell therapy for Relapsed/Refractory Large b-cell lymphoma: Results from the US lymphoma CAR-T consortium. Blood (2019) 134(Supplement_1):245–5. doi: 10.1182/blood-2019-129624
9. Nastoupil LJ, Jain MD, Spiegel JY, Ghobadi A, Lin Y, Dahiya S, et al. Axicabtagene ciloleucel (Axi-cel) CD19 chimeric antigen receptor (CAR) T-cell therapy for Relapsed/Refractory Large b-cell lymphoma: Real world experience. Blood (2018) 132(Supplement 1):91–1. doi: 10.1182/blood-2018-99-114152
10. Amini L, Silbert SK, Maude SL, Nastoupil LJ, Ramos CA, Brentjens RJ, et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol (2022) 19(5):342–55. doi: 10.1038/s41571-022-00607-3
11. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with b-cell acute lymphoblastic leukemia. Cancer Discovery (2018) 8(8):958–71. doi: 10.1158/2159-8290.CD-17-1319
12. Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, et al. Increased intensity lymphodepletion and adoptive immunotherapy–how far can we go? Nat Clin Pract Oncol (2006) 3(12):668–81. doi: 10.1038/ncponc0666
13. Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother (2010) 33(1):1–7. doi: 10.1097/CJI.0b013e3181b88ffc
14. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med (2005) 202(7):907–12. doi: 10.1084/jem.20050732
15. Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol (2017) 35(16):1803–13. doi: 10.1200/JCO.2016.71.3024
16. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood (2015) 125(25):3905–16. doi: 10.1182/blood-2015-01-621474
17. Sesques P, Ferrant E, Safar V, Wallet F, Tordo J, Dhomps A, et al. Commercial anti-CD19 CAR T cell therapy for patients with relapsed/refractory aggressive b cell lymphoma in a European center. Am J Hematol (2020) 95(11):1324–33. doi: 10.1002/ajh.25951
18. Grigor EJM, Fergusson D, Kekre N, Montroy J, Atkins H, Seftel MD, et al. Risks and benefits of chimeric antigen receptor T-cell (CAR-T) therapy in cancer: A systematic review and meta-analysis. Transfus Med Rev (2019) 33(2):98–110. doi: 10.1016/j.tmrv.2019.01.005
19. Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med (2016) 8(355):355ra116. doi: 10.1126/scitranslmed.aaf8621
20. Andreadis C, Tam CS, Borchmann P, Jaeger U, McGuirk JP, Holte H, et al. Correlation of bridging and lymphodepleting chemotherapy with clinical outcomes in patients with relapsed/refractory diffuse large b-cell lymphoma treated with tisagenlecleucel. Blood (2019) 134(Supplement_1):2883–3. doi: 10.1182/blood-2019-124492
21. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ (2014) 21(1):15–25. doi: 10.1038/cdd.2013.67
22. Noordam L, Kaijen MEH, Bezemer K, Cornelissen R, Maat LAPWM, Hoogsteden HC, et al. Low-dose cyclophosphamide depletes circulating naïve and activated regulatory T cells in malignant pleural mesothelioma patients synergistically treated with dendritic cell-based immunotherapy. Oncoimmunology (2018) 7(12):e1474318. doi: 10.1080/2162402X.2018.1474318
23. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother (2007) 56(5):641–8. doi: 10.1007/s00262-006-0225-8
24. Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother (2012) 61(3):353–62. doi: 10.1007/s00262-011-1106-3
25. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, et al. Low-dose cyclophosphamide induces antitumor T-cell responses, which associate with survival in metastatic colorectal cancer. Clin Cancer Res (2017) 23(22):6771–80. doi: 10.1158/1078-0432.CCR-17-0895
26. Hong M, Puaux A-L, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res (2011) 71(22):6997–7009. doi: 10.1158/0008-5472.CAN-11-1466
27. Nardin A, Wong W-C, Tow C, Molina TJ, Tissier F, Audebourg A, et al. Dacarbazine promotes stromal remodeling and lymphocyte infiltration in cutaneous melanoma lesions. J Invest Dermatol (2011) 131(9):1896–905. doi: 10.1038/jid.2011.128
28. Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother (2009) 58(10):1627–34. doi: 10.1007/s00262-009-0671-1
29. Ridolfi L, Petrini M, Granato AM, Gentilcore G, Simeone E, Ascierto PA, et al. Low-dose temozolomide before dendritic-cell vaccination reduces (specifically) CD4+CD25++Foxp3+ regulatory T-cells in advanced melanoma patients. J Transl Med (2013) 11(1):135. doi: 10.1186/1479-5876-11-135
30. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med (2016) 14(1):282. doi: 10.1186/s12967-016-1037-z
31. Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut (2016) 65(10):1710–20. doi: 10.1136/gutjnl-2015-309193
32. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11(18):6713–21. doi: 10.1158/1078-0432.CCR-05-0883
33. Gabitass RF, Annels NE, Crawshaw J, Pandha HS, Middleton GW. Use of gemcitabine- (Gem) and fluropyrimidine (FP)-based chemotherapy to reduce myeloid-derived suppressor cells (MDSCs) in pancreatic (PC) and esophagogastric cancer (EGC). J Clin Orthod (2011) 29(15_suppl):2588–8. doi: 10.1200/jco.2011.29.15_suppl.2588
34. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70(8):3052–61. doi: 10.1158/0008-5472.CAN-09-3690
35. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res (2014) 74(1):104–18. doi: 10.1158/0008-5472.CAN-13-1545
36. Li J-Y, Duan X-F, Wang L-P, Xu Y-J, Huang L, Zhang T-F, et al. Selective depletion of regulatory T cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J Immunol Res (2014) 2014:286170. doi: 10.1155/2014/286170
37. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16(18):4583–94. doi: 10.1158/1078-0432.CCR-10-0733
38. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol (2008) 129(2):219–29. doi: 10.1016/j.clim.2008.07.013
39. Vicari AP, Luu R, Zhang N, Patel S, Makinen SR, Hanson DC, et al. Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother (2009) 58(4):615–28. doi: 10.1007/s00262-008-0586-2
40. Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol (2013) 190(5):2464–71. doi: 10.4049/jimmunol.1202781
41. Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res (2018) 78(20):5891–900. doi: 10.1158/0008-5472.CAN-17-3480
42. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol (2011) 29(34):4548–54. doi: 10.1200/JCO.2011.36.5742
43. Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer (2013) 109(4):926–33. doi: 10.1038/bjc.2013.415
44. Lesterhuis WJ, Punt CJA, Hato SV, Eleveld-Trancikova D, Jansen BJH, Nierkens S, et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Invest (2011) 121(8):3100–8. doi: 10.1172/JCI43656
45. Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol (2015) 33(9):983–91. doi: 10.1200/JCO.2014.58.1967
46. Zhou L, Xu Q, Huang L, Jin J, Zuo X, Zhang Q, et al. Low-dose carboplatin reprograms tumor immune microenvironment through STING signaling pathway and synergizes with PD-1 inhibitors in lung cancer. Cancer Lett (2021) 500:163–71. doi: 10.1016/j.canlet.2020.11.049
47. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity (2016) 44(2):343–54. doi: 10.1016/j.immuni.2015.11.024
48. Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest (2019) 129(6):2210–21. doi: 10.1172/JCI126397
49. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory b-cell leukemias. Blood (2011) 118(18):4817–28. doi: 10.1182/blood-2011-04-348540
50. Murad JP, Tilakawardane D, Park AK, Lopez LS, Young CA, Gibson J, et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther (2021) 29(7):2335–49. doi: 10.1016/j.ymthe.2021.02.024
51. Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult b cell ALL patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
52. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther (2017) 25(9):2214–24. doi: 10.1016/j.ymthe.2017.05.012
53. Alzubi J, Dettmer-Monaco V, Kuehle J, Thorausch N, Seidl M, Taromi S, et al. PSMA-directed CAR T cells combined with low-dose docetaxel treatment induce tumor regression in a prostate cancer xenograft model. Mol Ther Oncolytics (2020) 18:226–35. doi: 10.1016/j.omto.2020.06.014
54. Zhang X, Sun S, Miao Y, Yuan Y, Zhao W, Li H, et al. Docetaxel enhances the therapeutic efficacy of PSMA-specific CAR-T cells against prostate cancer models by suppressing MDSCs. J Cancer Res Clin Oncol (2022) 148(12):3511–20. doi: 10.1007/s00432-022-04248-y
55. Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, et al. T-Cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res (2018) 24(12):2920–34. doi: 10.1158/1078-0432.CCR-17-1365
56. Chulanetra M, Morchang A, Sayour E, Eldjerou L, Milner R, Lagmay J, et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am J Cancer Res (2020) 10(2):674–87.
57. Enblad G, Karlsson H, Gammelgård G, Wenthe J, Lövgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res (2018) 24(24):6185–94. doi: 10.1158/1078-0432.CCR-18-0426
58. Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J Clin Invest (2016) 126(7):2588–96. doi: 10.1172/JCI86000
59. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat Med (2022) 28(6):1189–98. doi: 10.1038/s41591-022-01800-8
60. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell (2021) 39(2):193–208.e10. doi: 10.1016/j.ccell.2020.11.005
61. Suryadevara CM, Desai R, Abel ML, Riccione KA, Batich KA, Shen SH, et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology (2018) 7(6):e1434464. doi: 10.1080/2162402X.2018.1434464
62. Zhao Z, Xiao X, Saw PE, Wu W, Huang H, Chen J, et al. Chimeric antigen receptor T cells in solid tumors: a war against the tumor microenvironment. Sci China Life Sci (2020) 63(2):180–205. doi: 10.1007/s11427-019-9665-8
63. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
64. Santi A, Kugeratski FG, Zanivan S. Cancer associated fibroblasts: The architects of stroma remodeling. Proteomics (2018) 18(5–6):e1700167. doi: 10.1002/pmic.201700167
65. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
66. Guo F, Cui J. CAR-T in solid tumors: Blazing a new trail through the brambles. Life Sci (2020) 260(118300):118300. doi: 10.1016/j.lfs.2020.118300
67. Yin Y, Boesteanu AC, Binder ZA, Xu C, Reid RA, Rodriguez JL, et al. Checkpoint blockade reverses anergy in IL-13Rα2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics (2018) 11:20–38. doi: 10.1016/j.omto.2018.08.002
68. Lutsiak MEC, Semnani RT, De Pascalis R, Kashmiri SVS, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood (2005) 105(7):2862–8. doi: 10.1182/blood-2004-06-2410
69. Cattin S, Fellay B, Pradervand S, Trojan A, Ruhstaller T, Rüegg C, et al. Bevacizumab specifically decreases elevated levels of circulating KIT+CD11b+ cells and IL-10 in metastatic breast cancer patients. Oncotarget (2016) 7(10):11137–50. doi: 10.18632/oncotarget.7097
70. Seliger B, Quandt D. The expression, function, and clinical relevance of B7 family members in cancer. Cancer Immunol Immunother (2012) 61(8):1327–41. doi: 10.1007/s00262-012-1293-6
71. Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol (2012) 9(3):292–300. doi: 10.3109/1547691X.2011.642418
72. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. doi: 10.1158/0008-5472.CAN-08-2281
73. Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther (2022) 29(1):10–21. doi: 10.1038/s41417-021-00303-x
74. Truong NTH, Gargett T, Brown MP, Ebert LM. Effects of chemotherapy agents on circulating leukocyte populations: Potential implications for the success of CAR-T cell therapies. Cancers (Basel) (2021) 13(9):2225. doi: 10.3390/cancers13092225
75. Fadul CE, Fisher JL, Gui J, Hampton TH, Côté AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro-Oncology (2011) 13(4):393–400. doi: 10.1093/neuonc/noq204
76. Das RK, O’Connor RS, Grupp SA, Barrett DM. Lingering effects of chemotherapy on mature T cells impair proliferation. Blood Advances (2020) 4(19):4653–64. doi: 10.1182/bloodadvances.2020001797
77. Fagnoni FF, Lozza L, Zibera C, Zambelli A, Ponchio L, Gibelli N, et al. T-Cell dynamics after high-dose chemotherapy in adults: Elucidation of the elusive CD8+ subset reveals multiple homeostatic T-cell compartments with distinct implications for immune competence. Immunology (2002) 106(1):27–37. doi: 10.1046/j.1365-2567.2002.01400.x
78. Verma R, Foster RE, Horgan K, Mounsey K, Nixon H, Smalle N, et al. Lymphocyte depletion and repopulation after chemotherapy for primary breast cancer. Breast Cancer Res (2016) 18(1):10. doi: 10.1186/s13058-015-0669-x
79. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature (2022) 602(7897):503–9. doi: 10.1038/s41586-021-04390-6
80. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol (2019) 97(7):664–74. doi: 10.1111/imcb.12254
81. Zhou J, Kang Y, Chen L, Wang H, Liu J, Zeng S, et al. The drug-resistance mechanisms of five platinum-based antitumor agents. Front Pharmacol (2020) 11:343. doi: 10.3389/fphar.2020.00343
Keywords: chemotherapy, Chimeric Antigen Receptor T cell (CAR-T), solid tumor, tumor microenvironment (TME), personalized combination
Citation: Wang AX, Ong XJ, D’Souza C, Neeson PJ and Zhu JJ (2023) Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front. Immunol. 14:1140541. doi: 10.3389/fimmu.2023.1140541
Received: 09 January 2023; Accepted: 22 February 2023;
Published: 06 March 2023.
Edited by:
Jian Zhang, Southern Medical University, ChinaReviewed by:
Marta Barisa, University College London, United KingdomCopyright © 2023 Wang, Ong, D’Souza, Neeson and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joe Jiang Zhu, am9lLnpodUBwZXRlcm1hYy5vcmc=
†These authors share first authorship
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.