 Leticia Muñoz
Leticia Muñoz Esther Caparrós
Esther Caparrós Agustín Albillos
Agustín Albillos Rubén Francés
Rubén Francés
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 April 2023
Sec. Mucosal Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1139554
Cirrhosis is the common end-stage of chronic liver diseases of different etiology. The altered bile acids metabolism in the cirrhotic liver and the increase in the blood-brain barrier permeability, along with the progressive dysbiosis of intestinal microbiota, contribute to gut immunity changes, from compromised antimicrobial host defense to pro-inflammatory adaptive responses. In turn, these changes elicit a disruption in the epithelial and gut vascular barriers, promoting the increased access of potential pathogenic microbial antigens to portal circulation, further aggravating liver disease. After summarizing the key aspects of gut immunity during homeostasis, this review is intended to update the contribution of liver and brain metabolites in shaping the intestinal immune status and, in turn, to understand how the loss of homeostasis in the gut-associated lymphoid tissue, as present in cirrhosis, cooperates in the advanced chronic liver disease progression. Finally, several therapeutic approaches targeting the intestinal homeostasis in cirrhosis are discussed.
Cirrhosis is an evolving liver disease that leads to portal hypertension and hepatic insufficiency. During cirrhosis, the composition of intestinal microbiota is dynamically altered. This microbiota state, known as dysbiosis, induces an increased intestinal permeability and gut barrier dysfunction. In these conditions, the homeostatic contribution of the liver and the brain to gut immunity that is established through the gut-liver-brain axis is then affected, accelerating disease decompensation and complications (1, 2). This review outlines intestinal immunity characteristics in the homeostatic condition and how metabolites derived from liver and brain during cirrhosis contribute to disturbing the gut tissue microenvironment, which further contributes to progression of advanced chronic disease.
Multiple barriers separate the commensal microbes in the gut from blood, avoiding bacteria entering the systemic circulation. From the lumen inwards, tissue arrangement comprises the mucus layer, the epithelial barrier, the immune system layer, and the gut vascular barrier (GVB) (3) (Figure 1).
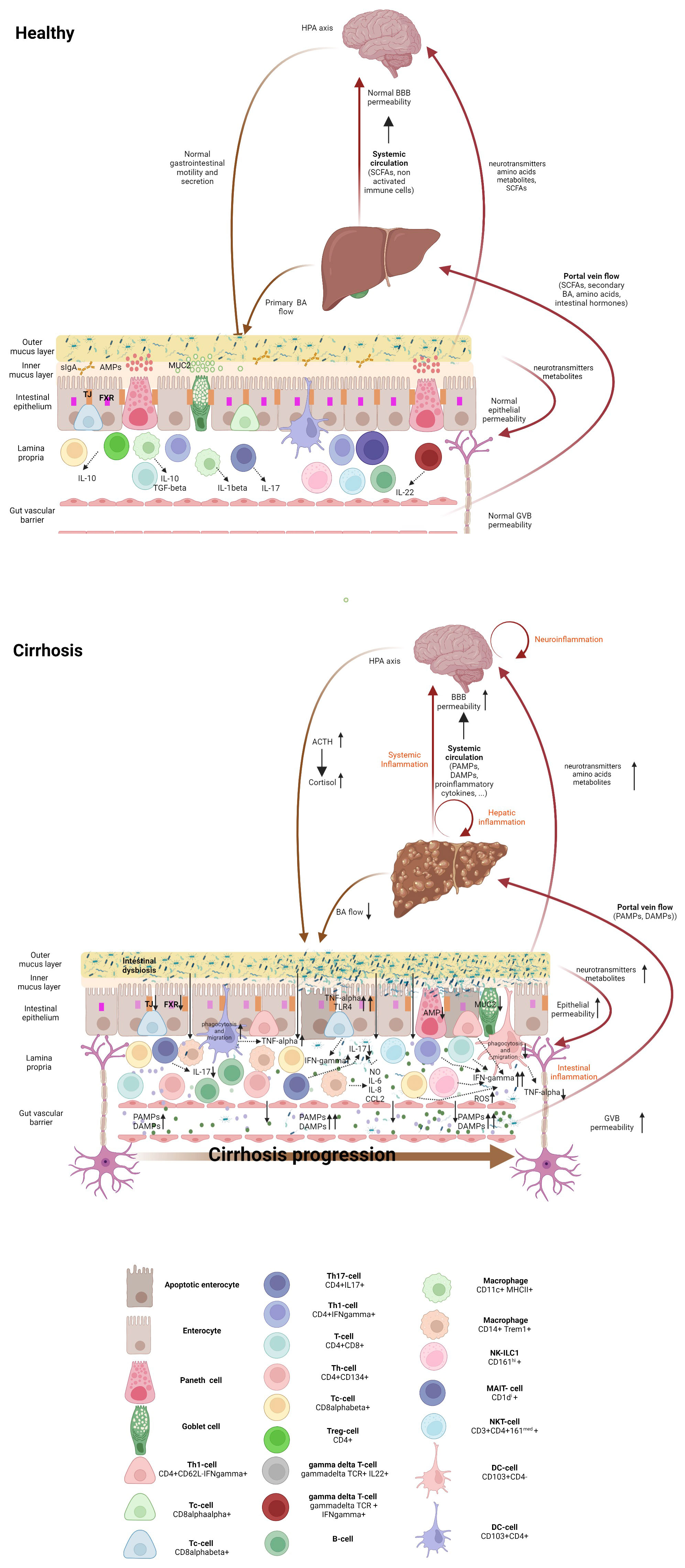
Figure 1 Gut-liver-brain axis in health and cirrhosis, highlighting the intestinal immune milieu. The intestinal epithelial barrier is a multi-layered structure that separates bacteria in the intestinal lumen from the systemic circulation. In healthy individuals, eubiosis, a tolerant immune system and an efficient innate antibacterial defense maintain the integrity of the epithelial and vascular barriers. Cirrhosis is associated with an altered microbiome which results from decreased bile flow, with deficient levels of primary but increased of secondary bile acids, together with intestinal hypomotility. Gut dysbiosis drives bacterial colonization of the inner mucous layer of the intestine, which facilitates the interaction of bacteria with the immune system, resulting in intestinal inflammation with recruitment and activation of immune system cells. Composition and severity of intestinal inflammation change with cirrhosis progression. Intestinal inflammation in the compensated stage is featured by activated innate cells, including DCs producing TNF-α and showing increased phagocytic and migratory abilities, along with the activation of the adaptive immune system, mainly located in the intraepithelial compartment. In the decompensated stage, intestinal immune system derangement is characterized by non-activated DCs, with lowered TNF-α secretion, and deficient phagocytosis and migration abilities, as well as expansion of activated macrophages and Th1 lymphocytes with concomitant Th17 reduction. Inflammation results in the release of proinflammatory cytokines and ROS, which worsens epithelial and the vascular barrier damage, hyperpermeability and access of bacterial products to the liver (via portal vein Flow) and to the systemic circulation. Elevated systemic levels of proinflammatory cytokines, PAMPs and DAMPs accelerate cirrhosis progression and increase the blood-brain-barrier permeability, inducing neuroinflammation (astrocyte swelling, activated microglia and immune system infiltration), which activates hypothalamic-pituitary-adrenal axis, and affects intestinal barrier integrity. Within the gut, the altered microbiota can produce neurotransmitters, amino acids and microbial metabolites. These metabolites can travel through portal circulation to interact with the host immune system, influence metabolism and/or affect local neuronal cells of the enteric nervous system and afferent pathways of the vagus nerve that signal directly to the brain. DC, dendritic cell; Treg, regulatory T cells; Tc, cytotoxic T cell; Th, helper T cell; ILC, innate lymphoid cell; NK cell, natural killer cell; NKT cell, natural killer T cell, MAIT cell, mucosal-associated invariant T cell, AMP, antimicrobial peptides; MUC-2, mucin-2; FXR, Farnesoid X receptor, TJ, Tight junction, BA, bile acid; NO, nitric oxide; IL-, interleukin; CCL2, C-C motif ligand 2; PAMPs, pathogen-associated molecular patterns; 318 DAMPs, damage-associated molecular patterns, ROS, radical oxygen species; TNF-γ, tumor necrosis factor-alpha; IFN-γ, interferon γ; ACTH, adrenocorticotrophic hormone; BBB, blood-brain barrier; HPA, hypothalamic-pituitary-adrenal.
The mucus layer acts as a frontline barrier separating the intestinal lumen from underlying tissues. Mucins comprising the outer thick mucus layer, which help the adhesion of microorganisms and their products, are produced by goblet and epithelial cells, and facilitate the opsonizing activity of plasma cell-derived secretory Immunoglobulin A (sIgA) (Table 1). Mucins can be found either as membrane-attached, such as MUC1, MUC3, MUC4, MUC13, and MUC17, or as secreted gel-forming mucins, like MUC2, MUC5AC, MUC5B, and MUC6 (47). Particularly, MUC2 offers static protection and restricts the potential activation of dendritic cells (DC), conferring them a tolerogenic state through stimulation of interleukin 10 (IL-10) production and TNF-α signaling inhibition (48).
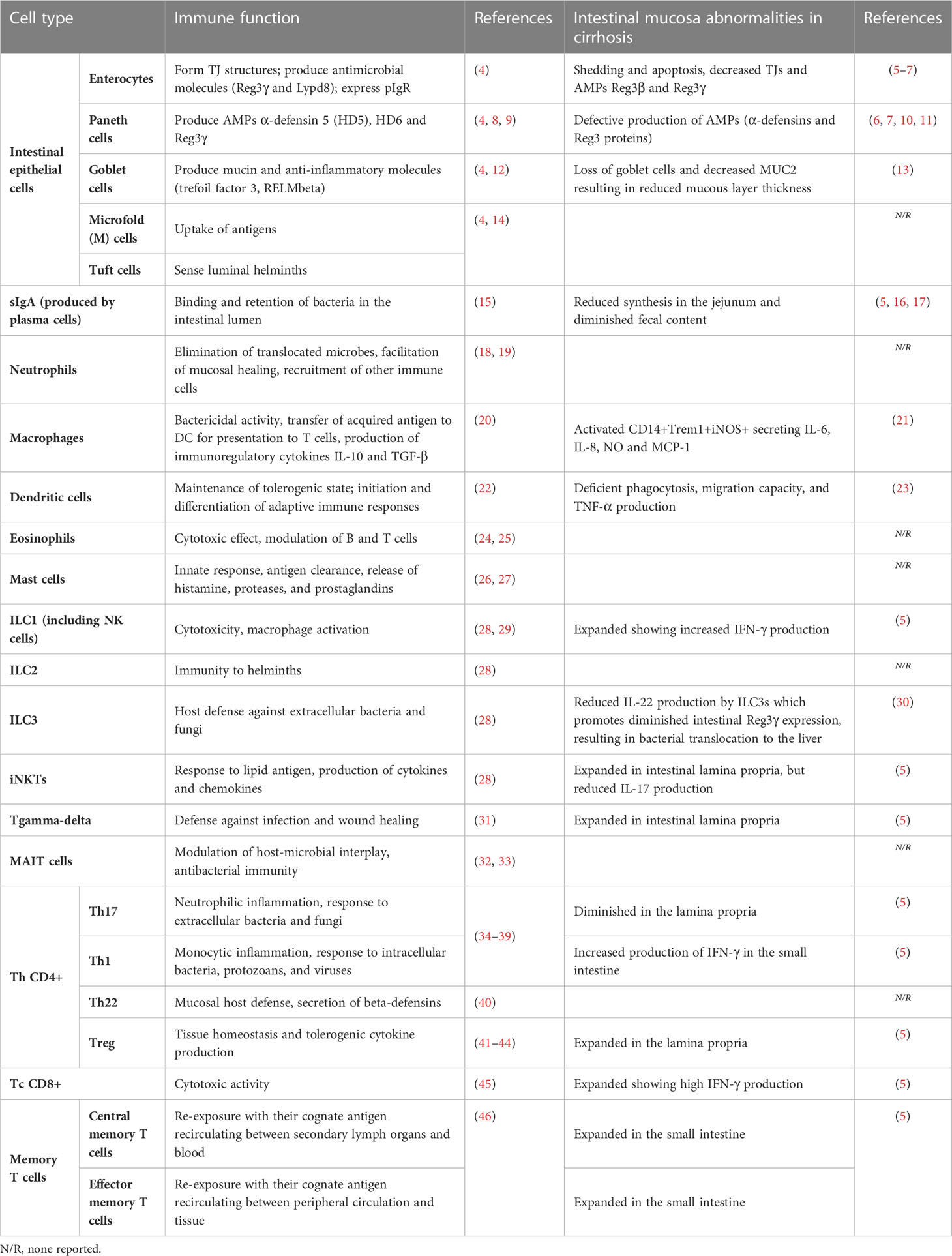
Table 1 Role of immune cells involved in gut homeostasis and abnormalities produced in cirrhosis.
The physical barrier integrated by mucins is reinforced by antimicrobial peptides (AMPs), that are produced by enterocytes and Paneth cells, and by microbial metabolites from commensal microbiota (49). AMPs act as efficient barrier against enteric pathogens (50). Cathelicidins, defensins and the regenerating islet-derived protein III-gamma (Reg3γ) prompt antimicrobial and anti-inflammatory functions, and induce immune cell recruitment, bacterial phagocytosis, and epithelial healing. The recently described lipopeptides, C14-R1 and C12-R2, have also shown AMP activity through the production of reactive oxygen species (ROS) (51). On the other hand, the barrier features of the mucus layer are completed with microbial metabolites produced by bacterial fermentation of dietary components, such as short-chain fatty acids (SCFAs). SCFAs regulate most intestinal epithelial cell functions, including cell turnover (52), mucus secretion by goblet cells (53), tight junction (TJ) proteins expression (54) and inflammasome- or hypoxia-inducible factor-mediated epithelial integrity (55, 56).
Below the mucus layer, the epithelial barrier maintains gut homeostasis and regulates immune responses (50). Epithelial cells are sealed by tight and adherens junctions (TJ and AJ, respectively), which are finely regulated by the influence of proinflammatory cytokines, and by signaling kinases and cytoskeleton, like myosin light chain kinases (5, 57). Intestinal epithelial cells possess a tightly regulated and specifically localized set of Pathogen-associated molecular pattern (PAMP) receptors and their signaling components for microbial detection. This innate immune-recognition equipment enables them to respond to microorganisms, thereby initiating the first steps in the host-pathogen interaction by regulating the immune response (58, 59). In addition, some studies unveil intestinal epithelial cells as non-professional antigen-presenting cells inducing inflammation or promoting tolerance (60–62). Following the detection of intestinal microbes through PAMP receptors, some specialized epithelial cells, such as goblet or Paneth cells (Table 1), respond generating molecules that protect mucosa from commensal microbes and invading pathogenic microorganisms, as mentioned above.
The Gut-associated lymphoid tissue (GALT) comprises both organized tissues such as lymphoid follicles, Peyer’s patches, and mesenteric lymph nodes (MLNs), where immune responses are induced, and scattered immune cells throughout the surface epithelium of the mucosa and in the underlying lamina propria, where effector functions are carried out (63).
In the steady state, neutrophils, located in the lamina propria, produce ROS and neutrophil extracellular traps (NETs) helping eliminating microbes translocated across the mucosal epithelium and facilitating tissue healing or immune cell recruitment (18). Eosinophils produce mediators including fibroblast growth factor (FGF)-2 and transforming growth factor (TGF)-β to promote tissue remodeling and repair (64, 65). In addition, mast cells, localizing along the gastrointestinal tract, differentially express PAMP receptors depending on their location and develop a fundamental regulatory and defensive function (26, 66). Intestinal macrophages remove senescent, apoptotic epithelial cells and promote epithelial integrity. They can also capture and destroy any bacteria that breach the barrier or send cellular processes across the epithelial barrier to sample luminal contents, which transfer to Dendritic cells (DCs) for presentation to T cells in the draining MLNs. Through their production of immunoregulatory cytokines, such as IL-10 and TGF-β, they maintain and facilitate secondary expansion of regulatory T cells (Tregs). In a similar manner, they support T helper (Th)17 cells and type 3 Innate lymphoid cells (ILC3) through their production of IL1-β (20). Also, differentially localized DC subsets can capture translocated IgA immune complexes (67) and continuously sample the lumen to establish a tolerogenic response to innocuous antigens (22). Finally, several innate lymphocytes like NK cells, invariant natural killer T cells (iNKT), and mucosal-associated invariant T (MAIT) cells have been identified as important in controlling gut immunity. Together with ILC3 and gamma delta (γδ)T, both iNKT and MAIT cells contribute to gut barrier integrity by producing IL-22, which is also important for promoting antibacterial defense (68–70) (Figure 1 and Table 1).
Th cells control the entry of translocated antigens at the gut (71). However, the sustained activation and proliferation of the Th response is an impeller of chronic inflammation. On the other hand, regulatory T (Treg) cells in the gut secrete TGF-β that negatively regulates T cell function (41). Both Th17 and Treg subsets show reciprocal phenotype plasticity based on the environmental milieu (72). Any imbalance between these populations in a tissue highly exposed to bacteria such as the gut barrier leads to a dysregulated mucosal immune response, with consequences for liver regulation and disease (73, 74) (Figure 1 and Table 1).
Just below the effector sites of the immune system layer, an additional cellular barrier, the GVB, controls entry into the portal circulation and access to the liver (75). Thus, if a molecule or a microorganism crosses the muco-epithelial barrier and escapes the local immune system, even to reach the systemic circulation the GVB must be disrupted. The GVB is characterized by the presence of TJ and AJ, which strictly control paracellular trafficking of solutes and fluids, together with other cell types, such as pericytes or fibroblasts, associated with the microvasculature and involved in the maintenance of the GVB, where they form a vascular unit.
Liver-derived molecules interact with the gut regional immune system defining tolerogenic or pathological interactions. In addition to metabolic functions, bile acids (BAs) are synthesized in the liver and incorporated by intestinal epithelial cells, exerting several immunomodulatory activities (76). Firstly, BAs induce anti-inflammatory responses through farnesoid X receptor (FXR) and Takeda G-protein-coupled receptor 5 (TGR5) on many tissues such as liver, intestine, or brain. Particularly, in macrophages, TGR5 activation inhibits nuclear translocation of nuclear factor κB (NF-kB), reducing the secretion of proinflammatory mediators such as TNF-α, IL-1β, IL-6, IFN-γ and nitric oxide (NO) (77, 78). Secondly, BAs interact with intestinal microbiota helping maintain its eubiotic state. In fact, a decrease in BAs levels may induce an overgrowth of pathogenic Gram-negative bacteria, with raised levels of lipopolysaccharide (LPS) (79) and subsequent induction of proinflammatory responses in the gut. Moreover, BAs such as deoxycholic acid have a direct impact on microbial membranes due to its hydrophobicity and detergent properties. Indirectly, BAs binding to FXR induces the control of gut microbiota through the production of intestinal antimicrobial molecules, such as angiogenin 1, that help control microbial overgrowth (80).
Together with BAs, IgA is the main isotype participating in microbial control in the gut-liver axis. Though main contribution on microbiota regulation is proportioned by sIgA produced by intestinal plasma cells, liver-derived IgA also contributes to the regulation of intestinal microbiota (81). Regardless the source, the sIgA patrols the intestinal mucus barrier, protecting epithelial cells and opsonizing luminal bacteria (15) without inducing a deleterious local inflammatory response (82). In fact, sIgA deficiency has been shown to disrupt the gut barrier via increased translocation of commensal bacteria (83).
The rest of metabolites and intermediaries drained by the liver into systemic circulation such as free fatty acids, choline and ethanol metabolites have also an impact on the physiological conditions of the gut, as they contribute to modulate the gut barrier interaction between the microbiome and the immune system (73).
Gut-brain axis involves a bidirectional communication route with a cellular and molecular interchange modulated by threatening inflammatory insults. Intestinal microbiota and its products can directly modulate the central nervous system (CNS) immune profile (84). Though specific immune responses coming from CNS and controlling the gut remains still elusive, different mechanisms have been recently uncovered (85). On this point, systemic, neuronal, and cellular intercommunication pathways have been described. First, systemic proinflammatory elements can alter blood brain barrier (BBB), as it happens with GVB, leading to increased permeability and passage of inflammatory products along the axis. Thus, neuroinflammatory triggering by such products could give rise to potential intestinal tract immune responses, as glucocorticoids release under hypothalamic-pituitary-function adrenal (HPA) activation, can modulate intestinal functions (84) (Figure 1). Moreover, neuronal connection of the CNS with the gut allows the signaling communication and the inhibition of proinflammatory macrophages, as well as M cell function to control pathogen irruption and microbiota homeostasis (Figure 1). Additionally, dysbiosis induced by stress and intestinal Th17 production of IL-17A, has been described to feedback this stress response (86).
In cirrhosis, the intestinal barrier is markedly disturbed, prompting the passage of live bacteria and bacterial components or metabolites to the internal milieu in experimental models and patients (5, 16, 87). This derangement increases in severity as cirrhosis progresses (Figure 1).
Gut barrier dysfunction affects both physical (muco-epithelial and GV barriers) and immunological layers, and results in hyperpermeability of the whole intestine (5, 13, 88). Disrupted mucosal barrier integrity plays a pivotal pathophysiological role not only because it increases the risk of severe complications such as spontaneous infections, but also and more importantly, because it affects the natural history of liver disease and patient survival.
Intestinal dysbiosis is the dominant player that sets the basis for epithelial barrier disruption. The gut microbiome in cirrhosis is characterized by reduced diversity and shows a significantly increased abundance of potentially pathogenic bacteria such as Enterococcaceae, Staphylococcaceae and Enterobacteriaceae, and a reduced relative abundance of potentially beneficial autochthonous bacteria such as Lachnospiraceae and Ruminococcaceae (89–91). Dysbiosis likely results from reduced bile flow with deficient levels of primary but increased levels of secondary BAs to the gut (92–94), which in turn results in reduced intestinal FXR signaling (10, 95), along with intestinal hypomotility. Intestinal dysbiosis and changes in BAs drive intestinal barrier functional abnormalities through changes in mucosal immunity and deficiencies in innate mechanisms of defense against bacteria (Figure 1).
The microbiome has been proposed to play a key role in mucus synthesis, release and barrier-function (96). In bile duct ligated (BDL) cirrhotic rats, there is lower mucus thickness, mucus weight per ileum length, and goblet cell numbers in the terminal ileum compared to healthy controls, predominantly with a reduction in mucin-filled goblet cells. This is associated with bacterial overgrowth in the inner most mucus layer (13). The closer proximity of bacteria to epithelial cells is one of the factors contributing to the passage of bacterial products in cirrhosis.
In humans and experimental models of cirrhosis, the observed structural changes in the small intestine include increased inter-enterocyte spacing with disorganization of TJ and AJ proteins, decreased intestinal mucosal proliferation and proliferation/apoptosis ratio, increased intestinal oxidative stress, edema of the lamina propria, infiltration by immune system cells, fibromuscular proliferation and a lowered villous/crypt ratio (5, 10, 21, 97–99). Diminished expression of zonula occludens-1 (ZO-1), occludin, claudin-1 and e-cadherin, and increased expression of claudin-2 have been reported in the small intestine of patients and experimental models of cirrhosis, especially in decompensated ones, with a concomitant increase produced in intestinal permeability, supporting the dynamic relationship between portal hypertension, gut bacterial translocation (GBT) and TJ and AJ expression in intestinal epithelial cells (5, 10, 21, 98). Portal hypertension results in intestinal mucosa hypoperfusion and hypoxia, which exacerbate oxidative damage in the gut mucosa (100). ROS can also enhance bacterial adhesion to epithelial cells and facilitate GBT across the mucosa (101). Increased cyclooxygenase-2 activity contributes to intestinal barrier disruption, as its inhibition increases ZO-1 and e-cadherin expression as well as intestinal permeability in rats with cirrhosis (102).
GVB is also profoundly altered in pre-clinical models of cirrhosis (13). This pathological endothelial permeability and accessibility in cirrhotic mice is associated with augmented expression of plasmalemma vesicle-associated protein (PV1), an integral membrane protein associated to the diaphragms of endothelial fenestrae, in intestinal vessels. Gut dysbiosis drives GVB disruption associated with liver disease, as mice receiving fecal microbiota transplantation (FMT) from high fat diet-fed mice, a pre-clinical model of non-alcoholic steatohepatitis, displayed increased PV1 expression in the small intestine compared to recipients receiving FMT from mice under control diet (103).
Besides damage to the physical barriers, cirrhosis is also associated with compromised antimicrobial host defense, which concerns certain specialized populations of epithelial cells having particularly important roles in innate immune defense of the intestine, such as Paneth cells, as well as innate immune cells. In this sense, the loss of Paneth cell α-5, α-6 and α-7-defensins, lysozyme and Reg3γ AMPs has been described in experimental models and patients with cirrhosis (6, 10, 11, 30, 104). Interestingly, some of these changes have been associated with GBT in CCl4-induced ascitic cirrhotic rats. Moreover, a diminished activity against E. coli and Enterococcus faecalis in the distal ileum has been found in rats with cirrhosis with GBT compared with non-GBT (11). Of note, a negative correlation between α-5, and α-7-defensins expression and circulating endotoxin levels has been described in cirrhotic patients (104). Therefore, a deficiency in Paneth cell AMPs likely decreases mucosal killing activity with a consequent shift of the luminal bacterial composition associated with intestinal bacterial overgrowth and increased GBT in liver cirrhosis. In this regard, angiogenin-1 is an AMP transcriptionally regulated by FXR (105), which displays a selective bactericidal activity against Gram negative bacteria and modulates intestinal inflammation (106). Reduced ileum expression of angiogenin-1 has been observed in rats with CCl4-induced cirrhosis with ascites suffering GBT (10).
Both structural changes and compromised antimicrobial host defense in the small intestine allow for bacterial colonization of the inner mucous layer and further promote the interaction of an altered gut microbiota, including an augmented bacterial burden and abundance of potentially pathogenetic taxa, with mucosal immune system cells. The consequence of this interaction between pathobionts and GALT is a state of subclinical inflammation with activation of innate immune cells, leading to proinflammatory cytokine and chemokine release, oxidative stress, and further recruitment of lymphocytes (11, 13, 21, 23, 107, 108).
Experimental studies have shown the increased expression of Toll-like receptor 4 (TLR4) and the proinflammatory cytokine TNF-α in the terminal ileum of CCl4-cirrhotic rats, especially in those with GBT (10, 109). In addition, the TNF-α levels in both the serum and multiple organs are significantly increased in CCl4-cirrhotic rats with compared with those without GBT (109) Similarly, patients with advanced cirrhosis with ascites exhibit elevated blood TNF-α levels along with an increase in the local production of TNF-α in the MLNs, which correlates with GBT (110). These findings suggest that, in cirrhosis, bacterial overgrowth and endotoxins significantly stimulate the secretion of TNF-α in the small intestine through the activation of TLR4. In the gut, immune dysregulation with a functional proinflammatory switch contributes to perpetuate intestinal barrier failure and GBT, both to the MLNs, and to the liver through the portal-venous route, due to disruption of the GVB (5, 13, 23, 107, 109). Subsequently, clearance deficiency by the cirrhotic liver allows bacteria and PAMPs getting into the systemic circulation (5, 13, 23, 107–109, 111). Recirculation of gut activated effector immune system cells along with inflammatory mediators, such as TNF-α, PAMPs and damage-associated molecular patterns (DAMPs) spreads the systemic inflammatory response inducing circulatory and remote organ dysfunction (91, 107, 109–113) (Figure 1).
Regarding to innate immune cells, a deranged innate immune response has an important contribution to the gut barrier disruption that occurs in cirrhosis patients and experimental models and includes: i) activated CD14+Trem1+inducible nitric oxide synthase (iNOS)+ macrophages, secreting IL-8, CCL2, IL-6 and NO, which correlate with an increased expression of claudin-2 (21); ii) an increased number of activated CD103+ DCs producing TNF-α and showing higher phagocytic and migratory abilities, when GBT is not present, which switch to a non-activated phenotype, and relatively deficient function when cirrhosis progresses and GBT occurs, likely facilitating bacterial passage (23); iii) the expansion of IFN-γ-secreting γδT- and NK cells, that could be related with TJ disorganization (5); and iv) a reduced IL-22 production by ILC3s which promotes diminished intestinal Reg3γ expression, resulting in GBT to the liver (30) (Figure 1 and Table 1).
Changes in the small intestine’s adaptive immune response are also prominent especially as cirrhosis progresses to the ascites stage, and consist of: i) high infiltration of several activated Th and cytotoxic T (Tc) CD8αβ+ cell subsets (5, 109); ii) high proportion of Th1 and IFN-γ-producing Tc cells, which directly correlates with gut permeability that is probably due to endocytosis of TJ proteins (5, 114); iii) Th17 depletion, which also contributes to barrier damage as IL-17 play a role in intestinal epithelium homeostasis by inducing TJ function and AMPs production (115); iv) increased numbers of Tregs in a likely response to bacterial challenge, although they seem unable to offset the proinflammatory immune response; and v) an increased number of B cells, which contrasts with the reduced synthesis of IgA in the jejunum of rats with cirrhosis and the diminished fecal IgA content of the intestine in cirrhotic rats (5, 16, 17) (Figure 1 and Table 1).
Intestinal dysbiosis leads to a dysregulation of the innate and adaptive immune responses of the intestinal mucosa that further contribute to bacterial mucosal adhesion and colonization, as well as intestinal hyperpermeability. As cirrhosis progresses from the preascitic to the ascitic stage a dysregulated mucosal immune system contributes to the derangement of epithelial TJ, reduced secretion of AMPs and impaired phagocytic function of DCs, which facilitate the increased passage of bacteria and bacterial products to the systemic circulation (Figure 1 and Table 1).
The key pathogenetic role of bacteria is confirmed by the reversibility of most intestinal immune abnormalities and increased gut permeability provoked by antibiotic-induced microbiome reorganization (5, 23). Consistently, antibiotics reduce immune cell infiltration in the intestinal mucosa of cirrhotic animals. Of note, bowel decontamination normalizes the frequencies of proinflammatory IFN-γ and TNF-α cytokines, the phagocytic activity of DCs, and gut permeability, as revealed by a reduction in fecal albumin loss and by fully suppressed GBT in cirrhotic rats with ascites (5, 23).
A mild neurocognitive dysfunction is present in as much as 80% of patients with cirrhosis. This condition, named minimal or covert hepatic encephalopathy (HE) affects quality of life (116) and predicts the development of overt HE (117). This complication encompasses a wide range of cognitive, psychomotor and psychiatric disturbances that heralds a poor prognosis with negative impact on health-related quality of life, liver transplant priority and patient survival (118, 119).
From a neurologic point of view, hepatic encephalopathy in cirrhosis is primarily astroglial in nature, characterized by Alzheimer type 2 astrocytosis together with activation of microglia indicative of neuroinflammation (120, 121). Inflammation and hyperammonemia are associated with enhanced cognitive impairment in patients with cirrhosis (122). It has been proposed that inflammation synergistically acts with ammonia in driving nitrosation of brain proteins in BDL cirrhotic rats (123). Proinflammatory cytokine levels are also increased in patients with covert hepatic encephalopathy (124), and these inflammatory mediators have been correlated with serum ammonia in patients with extrahepatic portal venous obstruction (125). The relevance of the neutrophil response in the pathogenesis of hepatic encephalopathy has also been documented (126), and proinflammatory IL-1β and IL-6 have been correlated with neurocognitive scores and with health-related quality of live questionnaires in patients with cirrhosis (127) (Figure 1).
A link between altered flora (higher Veillonellaceae), poor cognition, endotoxemia, and inflammation (IL-6, TNF-α, IL-2, and IL-13) has been described in cirrhotic patients with compared with those without HE. In addition, in the cirrhosis group, Alcaligeneceae and Porphyromonadaceae positively correlated with cognitive impairment (128). An increased risk of cognitive impairment in patients with cirrhosis and bacterial infections has been reported, including those with subclinical cognitive alterations (129). In this latter group of patients, even bacterial antigen translocation, without the need of an over infection, is associated with increased serum ammonia and NOx levels, and a more severe neurocognitive condition (130).
Finally, the relationship between Th17 cells and cognitive impairment has also been documented. Th17 cells have been described to cross the BBB and promote brain injury in neurological diseases (131). Systemic inflammation associated with cirrhosis may therefore increase BBB permeability and facilitate brain T cell recruitment in the context of advanced liver disease (Figure 1).
Abnormalities in microbiota composition and bacterial overgrowth set the stage for the gut-liver axis disruption with intestinal barrier failure, which is distinctive of cirrhosis. These facts enable increased passage to the bloodstream of bacteria or their products, which promote systemic inflammation and bacterial infections, and worsen cirrhosis progression. The disrupted gut-liver axis observed in cirrhosis is a topic of active research, considering its contribution to disease progression. We thereby briefly summarize novel therapeutic approaches to target the gut-liver axis aimed to halt cirrhosis progression.
The effects of bowel decontamination with poorly absorbable antibiotics are the redistribution of microbiota composition, reduced intestinal permeability and GBT, and improved proinflammatory activation of mucosal and circulating immune cells along with circulatory dysfunction in human and experimental models (5, 112). Norfloxacin and ciprofloxacin are standard of care for primary and secondary prevention of spontaneous bacterial peritonitis, and norfloxacin reduces the risk of infection and 6-month mortality in patients with advanced cirrhosis (132, 133). Rifaximin is a broad-spectrum compound that improves and prevents overt hepatic encephalopathy and ameliorates systemic inflammation and endotoxinemia in patients with cirrhosis and encephalopathy (134, 135). Rather than promoting bowel decontamination, rifaximin exerts its effects by eliminating polarization of the gut microbiome via suppression of mucin-degrading species rich in sialidase and known to induce gut barrier damage while preserving beta-diversity, e.g. Streptococcus and Veillonella spp (135). Rifaximin associated with simvastatin to prevent Acute-on-Chronic Liver Failure and reduce complications in decompensated cirrhosis is being currently tested in clinical trials (NCT03780673). Alternatives to antibiotics are a topic of research as their long-term use has been linked to reduced bacterial diversity and multidrug-resistant microorganisms (136).
Probiotics displace resident dysbiotic bacteria and reconstitute a healthy microbiome. Bifidobacterium pseudocatenulatum CECT7765 induces a shift towards an TLR2-mediated anti-inflammatory cytokine profile in intestinal lymphocytes and macrophages, improves gut barrier integrity, reduces intestinal permeability, and decreases bacterial DNA translocation, serum endotoxin levels and burden of bacterial antigens in the liver in cirrhotic patients and experimental models (137–139). Lactobacillus rhamnosus GG supernatant therapy also increases mRNA expression of TJ proteins and reduces intestinal permeability in a mouse model of chronic-binge alcohol feeding. This therapy led to a decrease of GBT to the liver and an overall balance restored of Tregs, Th17s, and IL-17 (140, 141). Furthermore, probiotic supplementation has shown significantly increase serum neopterin levels and the production of ROS by neutrophils (142). These findings might explain the beneficial effects of probiotics on immune function, as well as in ameliorating gut barrier dysfunction in cirrhosis. However, studies on the impact of probiotics on cirrhosis complications and progression, including hepatic encephalopathy, have severe methodological limitations and patient numbers have been too small to draw valid conclusions. In parallel, an international study has shown that a diet rich in coffee, tea, fresh vegetables and fermented milk is associated with an increased diversity of microbiota species and reduced number of hospitalizations in patients with cirrhosis, suggesting the benefits of dietary modulation of the gut microbiome in favor of liver health (143).
Another therapeutic approach is to reduce the amount of bacterial toxin being absorbed from the intestine in patients and experimental models with chronic liver disease. In contrast to the strategies based on the prolonged use of antibiotics that reduce intestinal bacterial populations, use of scavengers of bacterial toxins has the advantage of avoiding the risk of establishing antimicrobial drug resistant intestinal bacteria. Yaq-001 is a non-absorbable activated charcoal with a high adsorptive capacity for endotoxins and other products of bacterial metabolism preventing their absorption into the bloodstream. Evidence in BDL rats showed that Yaq-001 was associated with shifts in microbiome composition, attenuated the LPS-induced production of ROS by monocytes, and reductions in liver injury and portal pressure (144). A phase-II clinical study in patients with cirrhosis and diuretic-sensitive ascites treated with Yaq-001 or placebo for 12 weeks suggested proof of a mechanism that Yak-001 modulated systemic endotoxinemia and inflammation by improving gut inflammation and its permeability (NCT03202498).
FMT restores a healthy gut microbial environment and physiological colonization via different routes of administration. Microbiota transplantation in patients with recurrent hepatic encephalopathy proved safe in the long-term and has shown to rescue antibiotic-associated disruption in microbial diversity, improve cognitive function, and reduce encephalopathy recurrence and hospitalizations (145). These studies have involved a limited number of patients and efficacy trials are pending. In this regard, FMT is currently being tested in a randomized controlled trial in patients with decompensated cirrhosis to address survival and liver related outcomes (NCT04932577).
Diminished luminal BA availability in cirrhosis provokes a reduction in intestinal FXR signaling which seems, at least partly, to mediate the gut barrier disruption in cirrhosis, as FXR-agonists reduce GBT via the portal-venous route to the liver (13). In experimental cirrhosis, FXR activation by obeticholic acid (OCA) increases ileal protein expression of the main TJ proteins and AMP secretion (10, 13), influences intestinal epithelial cell proliferation and apoptosis (146), and exerts potent antiinflammatory actions in the intestine, stabilizing epithelial integrity (10). Herein, OCA impacts the mucous machinery by increasing ileal goblet cell numbers in cirrhotic animals (13). On the vascular side, the FXR agonist OCA stabilizes the dysfunctional GVB in experimental cirrhosis (13).
IL-22, a cytokine member in IL-10 family, is mainly secreted by various immune cells, such as ILCs and MAIT cells, which targets epithelial cells in several organs, including the intestine and the liver. IL-22 promotes survival and proliferation of hepatocytes and liver progenitor cells, thereby promoting liver repair (147). Additionally, IL-22 increases the expression of intestinal Reg3 lectins, which maintain low bacterial colonization of the inner mucus layer and reduce GBT to the liver. In a mouse model of ethanol-induced liver disease, intestinal IL-22 has the beneficial effect of reducing GBT in the intestine by increasing the expression of Reg3γ. Bacteria engineered to produce IL-22 in the intestine (without increasing systemic IL-22) ameliorate experimental ethanol-induced steatohepatitis via induction of Reg3γ (30). Importantly, due to this combination of protective effects in the liver and in the intestinal barrier IL-22 is a promising drug for the treatment of alcoholic hepatitis.
While the gut immunity during homeostasis plays a crucial role in liver and brain functions, mainly through its interaction with symbiotic microbiota, the progression of advanced chronic liver disease significantly compromises the immunological behavior at the gut and brain barriers of cirrhotic patients. Therefore, therapeutic options aimed at restoring gut immune homeostasis in these patients are of relevance to prevent cirrhosis-derived complications. However, despite intense experimental research, its translation into clinic remains a lengthy goal.
LM, EC, AA, RF: manuscript design, writing and final version approval.
This work was supported by grants from the Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III PI20/01302 awarded to AA, and Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III PID2019-107036RB-I00 and PMP21/0082, and Generalitat Valenciana Prometeo 2021/033 awarded to RF. CIBEREHD is funded by the Instituto de Salud Carlos III using grants cofinanced by the European Development Regional Fund “A way to achieve Europe” (EDRF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Brescia P, Rescigno M. The gut vascular barrier: a new player in the gut-liver-brain axis. Trends Mol Med (2021) 27:844–55. doi: 10.1016/j.molmed.2021.06.007
2. Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet (2021) 398:1359–76. doi: 10.1016/S0140-6736(21)01374-X
3. Okumura R, Takeda K. Maintenance of intestinal homeostasis by mucosal barriers. Inflammation Regener (2018) 38:5. doi: 10.1186/s41232-018-0063-z
4. Goto Y. Epithelial cells as a transmitter of signals from commensal bacteria and host immune cells. Front Immunol (2019) 10:2057. doi: 10.3389/fimmu.2019.02057
5. Muñoz L, Borrero MJ, Úbeda M, Conde E, Del Campo R, Rodríguez-Serrano M, et al. Intestinal immune dysregulation driven by dysbiosis promotes barrier disruption and bacterial translocation in rats with cirrhosis. Hepatology (2019) 70(3):925–38. doi: 10.1002/hep.30349
6. Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology (2011) 53:96–105. doi: 10.1002/hep.24018
7. Schnabl B. Linking intestinal homeostasis and liver disease. Curr Opin Gastroenterol (2013) 29:264–70. doi: 10.1097/MOG.0b013e32835ff948
8. Ouellette AJ, Bevins CL. Paneth cell defensins and innate immunity of the small bowel. Inflammation Bowel Dis (2001) 7:43–50. doi: 10.1097/00054725-200102000-00007
9. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol (2013) 75:289–311. doi: 10.1146/annurev-physiol-030212-183744
10. Ubeda M, Lario M, Munoz L, Borrero MJ, Rodríguez-Serrano M, Sánchez-Díaz AM, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol (2016) 64:1049–57. doi: 10.1016/j.jhep.2015.12.010
11. Teltschik Z, Wiest R, Beisner J, Nuding S, Hofmann C, Schoelmerich J, et al. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology (2012) 55(4):1154–63. doi: 10.1002/hep.24789
12. Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep (2010) 12:319–30. doi: 10.1007/s11894-010-0131-2
13. Sorribas M, Jakob MO, Yilmaz B, Li H, Stutz D, Noser Y, et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol (2019) 71:1126–40. doi: 10.1016/j.jhep.2019.06.017
14. Schneider C, O'Leary CE, Locksley RM. Regulation of immune responses by tuft cells. Nat Rev Immunol (2019) 19:584–93. doi: 10.1038/s41577-019-0176-x
15. Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol (2008) 1:11–22. doi: 10.1038/mi.2007.6
16. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol (2014) 60:197–209. doi: 10.1016/j.jhep.2013.07.044
17. Pelletier G, Briantais MJ, Buffet C, Pillot J, Etienne JP. Serum and intestinal secretory IgA in alcoholic cirrhosis of the liver. Gut (1982) 23:475–80. doi: 10.1136/gut.23.6.475
18. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5:354–66. doi: 10.1038/mi.2012.24
19. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol (2007) 5:577–82. doi: 10.1038/nrmicro1710
20. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
21. Du Plessis J, Vanheel H, Janssen CE, Roos L, Slavik T, Stivaktas PI, et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J Hepatol (2013) 58:1125–32. doi: 10.1016/j.jhep.2013.01.038
22. Stagg AJ. Intestinal dendritic cells in health and gut inflammation. Front Immunol (2018) 9:2883. doi: 10.3389/fimmu.2018.02883
23. Munoz L, Jose Borrero M, Ubeda M, Lario M, Díaz D, Francés R, et al. Interaction between intestinal dendritic cells and bacteria translocated from the gut in rats with cirrhosis. Hepatology (2012) 56:1861–9. doi: 10.1002/hep.25854
24. Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol (2013) 13:9–22. doi: 10.1038/nri3341
25. Yantiss RK. Eosinophils in the GI tract: how many is too many and what do they mean? Mod Pathol (2015) 28 Suppl 1:S7–21. doi: 10.1038/modpathol.2014.132
26. Albert-Bayo M, Paracuellos I, Gonzalez-Castro AM, Rodríguez-Urrutia A, Rodríguez-Lagunas MJ, Alonso-Cotoner C, et al. Intestinal mucosal mast cells: Key modulators of barrier function and homeostasis. Cells (2019) 8(2):135. doi: 10.3390/cells8020135
27. Frossi B, Mion F, Sibilano R, Danelli L, Pucillo CEM. Is it time for a new classification of mast cells? what do we know about mast cell heterogeneity? Immunol Rev (2018) 282:35–46. doi: 10.1111/imr.12636
28. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. innate lymphoid cells: a new paradigm in immunology. Science (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
29. Poggi A, Benelli R, Vene R, Costa D, Ferrari N, Tosetti F, et al. Human gut-associated natural killer cells in health and disease. Front Immunol (2019) 10:961. doi: 10.3389/fimmu.2019.00961
30. Hendrikx T, Duan Y, Wang Y, Oh JH, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut (2019) 68:1504–15. doi: 10.1136/gutjnl-2018-317232
31. Nielsen MM, Witherden DA, Havran WL. Gammadelta T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol (2017) 17:733–45. doi: 10.1038/nri.2017.101
32. Mortier C, Govindarajan S, Venken K, Elewaut D. It takes "Guts" to cause joint inflammation: Role of innate-like T cells. Front Immunol (2018) 9:1489. doi: 10.3389/fimmu.2018.01489
33. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature (2003) 422:164–9. doi: 10.1038/nature01433
34. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol (2013) 8:477–512. doi: 10.1146/annurev-pathol-011110-130318
35. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol (2010) 11:674–80. doi: 10.1038/ni.1899
36. Shaw MH, Kamada N, Kim YG, Núñez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med (2012) 209:251–8. doi: 10.1084/jem.20111703
37. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
38. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe (2008) 4:337–49. doi: 10.1016/j.chom.2008.09.009
39. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature (2008) 455:808–12. doi: 10.1038/nature07240
40. Zhang N, Pan HF, Ye DQ. Th22 in inflammatory and autoimmune disease: prospects for therapeutic intervention. Mol Cell Biochem (2011) 353:41–6. doi: 10.1007/s11010-011-0772-y
41. O'Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med (2004) 10:801–5. doi: 10.1038/nm0804-801
42. Neumann C, Blume J, Roy U, Teh PP, Vasanthakumar A, Beller A, et al. C-maf-dependent treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat Immunol (2019) 20:471–81. doi: 10.1038/s41590-019-0316-2
43. Ueno A, Jeffery L, Kobayashi T, Hibi T, Ghosh S, Jijon H. Th17 plasticity and its relevance to inflammatory bowel disease. J Autoimmun (2018) 87:38–49. doi: 10.1016/j.jaut.2017.12.004
44. Sorini C, Cardoso RF, Gagliani N, Villablanca EJ. Commensal bacteria-specific CD4(+) T cell responses in health and disease. Front Immunol (2018) 9:2667. doi: 10.3389/fimmu.2018.02667
45. Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, et al. Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci Rep (2018) 8:14430. doi: 10.1038/s41598-018-32860-x
46. Zundler S, Becker E, Schulze LL, Neurath MF. Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances. Gut (2019) 68:1688–700. doi: 10.1136/gutjnl-2018-317977
47. Tailford LE, Crost EH, Kavanaugh D, Juge N. Mucin glycan foraging in the human gut microbiome. Front Genet (2015) 6:81. doi: 10.3389/fgene.2015.00081
48. Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science (2013) 342:447–53. doi: 10.1126/science.1237910
49. Lievin-Le Moal V, Servin AL. The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev (2006) 19:315–37. doi: 10.1128/CMR.19.2.315-337.2006
50. Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med (2017) 49:e338. doi: 10.1038/emm.2017.20
51. Zhong C, Zhang F, Zhu N, Zhu Y, Yao J, Gou S, et al. Ultra-short lipopeptides against gram-positive bacteria while alleviating antimicrobial resistance. Eur J Med Chem (2021) 212:113138. doi: 10.1016/j.ejmech.2020.113138
52. Park JH, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of intestinal epithelial cell turnover by commensal bacteria: Role of short-chain fatty acids. PloS One (2016) 11:e0156334. doi: 10.1371/journal.pone.0156334
53. Burger-van Paassen N, Vincent A, Puiman PJ, van der Sluis M, Bouma J, Boehm G, et al. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: implications for epithelial protection. Biochem J (2009) 420:211–9. doi: 10.1042/BJ20082222
54. Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, et al. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J Immunol (2017) 199:2976–84. doi: 10.4049/jimmunol.1700105
55. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun (2015) 6:6734. doi: 10.1038/ncomms7734
56. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
57. Du L, Kim JJ, Shen J, Dai N. Crosstalk between inflammation and ROCK/MLCK signaling pathways in gastrointestinal disorders with intestinal hyperpermeability. Gastroenterol Res Pract (2016) 2016:7374197. doi: 10.1155/2016/7374197
58. Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL, et al. A map of toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity (2018) 49:560–575 e6. doi: 10.1016/j.immuni.2018.07.016
59. McClure R, Massari P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol (2014) 5:386. doi: 10.3389/fimmu.2014.00386
60. Buning J, Hundorfean G, Schmitz M, Zimmer KP, Strobel S, Gebert A, et al. Antigen targeting to MHC class II-enriched late endosomes in colonic epithelial cells: trafficking of luminal antigens studied in vivo in crohn's colitis patients. FASEB J (2006) 20:359–61. doi: 10.1096/fj.05-4807fje
61. Dahan S, Roth-Walter F, Arnaboldi P, Agarwal S, Mayer L. Epithelia: lymphocyte interactions in the gut. Immunol Rev (2007) 215:243–53. doi: 10.1111/j.1600-065X.2006.00484.x
62. Danese S. Nonimmune cells in inflammatory bowel disease: from victim to villain. Trends Immunol (2008) 29:555–64. doi: 10.1016/j.it.2008.07.009
63. Morbe UM, Jorgensen PB, Fenton TM, von Burg N, Riis LB, Spencer J, et al. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol (2021) 14:793–802. doi: 10.1038/s41385-021-00389-4
64. Shamri R, Xenakis JJ, Spencer LA. Eosinophils in innate immunity: an evolving story. Cell Tissue Res (2011) 343:57–83. doi: 10.1007/s00441-010-1049-6
65. Chu VT, Beller A, Rausch S, Strandmark J, Zänker M, Arbach O, et al. Eosinophils promote generation and maintenance of immunoglobulin-a-expressing plasma cells and contribute to gut immune homeostasis. Immunity (2014) 40:582–93. doi: 10.1016/j.immuni.2014.02.014
66. Wu L, Feng BS, He SH, Zheng PY, Croitoru K, Yang PC. Bacterial peptidoglycan breaks down intestinal tolerance via mast cell activation: the role of TLR2 and NOD2. Immunol Cell Biol (2007) 85:538–45. doi: 10.1038/sj.icb.7100079
67. Kadaoui KA, Corthesy B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse peyer's patches with restriction to mucosal compartment. J Immunol (2007) 179:7751–7. doi: 10.4049/jimmunol.179.11.7751
68. Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol (2019) 20:1110–28. doi: 10.1038/s41590-019-0444-8
69. Victor AR, Nalin AP, Dong W, McClory S, Wei M, Mao C, et al. IL-18 drives ILC3 proliferation and promotes IL-22 production via NF-kappaB. J Immunol (2017) 199:2333–42. doi: 10.4049/jimmunol.1601554
70. Middendorp S, Nieuwenhuis EE. NKT cells in mucosal immunity. Mucosal Immunol (2009) 2:393–402. doi: 10.1038/mi.2009.99
71. Edelblum KL, Sharon G, Singh G, Odenwald MA, Sailer A, Cao S, et al. The microbiome activates CD4 T-cell-mediated immunity to compensate for increased intestinal permeability. Cell Mol Gastroenterol Hepatol (2017) 4:285–97. doi: 10.1016/j.jcmgh.2017.06.001
72. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
73. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
74. Yang X, Lu D, Zhuo J, Lin Z, Yang M, Xu X. The gut-liver axis in immune remodeling: New insight into liver diseases. Int J Biol Sci (2020) 16:2357–66. doi: 10.7150/ijbs.46405
75. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science (2015) 350:830–4. doi: 10.1126/science.aad0135
76. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol (2018) 15:397–411. doi: 10.1038/s41575-018-0011-z
77. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol (2014) 11:55–67. doi: 10.1038/nrgastro.2013.151
78. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated b cells (NF-kappaB) in mice. Hepatology (2011) 54:1421–32. doi: 10.1002/hep.24525
79. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol (2014) 30:332–8. doi: 10.1097/MOG.0000000000000057
80. Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev (2005) 29:625–51. doi: 10.1016/j.femsre.2004.09.003
81. Inamine T, Schnabl B. Immunoglobulin a and liver diseases. J Gastroenterol (2018) 53:691–700. doi: 10.1007/s00535-017-1400-8
82. Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev (2014) 260:76–85. doi: 10.1111/imr.12189
83. Suzuki K, Meek B, Doi Y, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci U.S.A. (2004) 101:1981–6. doi: 10.1073/pnas.0307317101
84. Cryan JF, O'Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-Gut-Brain axis. Physiol Rev (2019) 99:1877–2013. doi: 10.1152/physrev.00018.2018
85. Agirman G, Yu KB, Hsiao EY. Signaling inflammation across the gut-brain axis. Science (2021) 374:1087–92. doi: 10.1126/science.abi6087
86. Xu C, Lee SK, Zhang D, Frenette PS. The gut microbiome regulates psychological-Stress-Induced inflammation. Immunity (2020) 53:417–428 e4. doi: 10.1016/j.immuni.2020.06.025
87. Albillos A, de la HA. Multifactorial gut barrier failure in cirrhosis and bacterial translocation: working out the role of probiotics and antioxidants. J.Hepatol (2002) 37:523. doi: 10.1016/S0168-8278(02)00265-9
88. Perez-Paramo M, Munoz J, Albillos A, Freile I, Portero F, Santos M, et al. Effect of propranolol on the factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology (2000) 31:43–8. doi: 10.1002/hep.510310109
89. Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology (2011) 54:562–72. doi: 10.1002/hep.24423
90. Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, et al. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol (2012) 303:G675–85. doi: 10.1152/ajpgi.00152.2012
91. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol (2014) 60:940–7. doi: 10.1016/j.jhep.2013.12.019
92. Gunnarsdottir SA, Sadik R, Shev S, Simrén M, Sjövall H, Stotzer PO, et al. Small intestinal motility disturbances and bacterial overgrowth in patients with liver cirrhosis and portal hypertension. Am J Gastroenterol (2003) 98:1362–70. doi: 10.1111/j.1572-0241.2003.07475.x
93. Lorenzo-Zuniga V, Bartoli R, Planas R, Hofmann AF, Viñado B, Hagey LR, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology (2003) 37:551–7. doi: 10.1053/jhep.2003.50116
94. Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol (2014) 306:G929–37. doi: 10.1152/ajpgi.00315.2013
95. Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol (2013) 58:949–55. doi: 10.1016/j.jhep.2013.01.003
96. Jakobsson HE, Rodriguez-Pineiro AM, Schutte A, Ermund A, Boysen P, Bemark M, et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep (2015) 16:164–77. doi: 10.15252/embr.201439263
97. Such J, Guardiola JV, de Juan J, Casellas JA, Pascual S, Aparicio JR, et al. Ultrastructural characteristics of distal duodenum mucosa in patients with cirrhosis. Eur.J Gastroenterol Hepatol (2002) 14:371–6. doi: 10.1097/00042737-200204000-00006
98. Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Triantos C, Vagianos CE, et al. Altered intestinal tight junctions' expression in patients with liver cirrhosis: a pathogenetic mechanism of intestinal hyperpermeability. Eur J Clin Invest (2012) 42:439–46. doi: 10.1111/j.1365-2362.2011.02609.x
99. Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Zisimopoulos D, Maroulis I, et al. Intestinal mucosal proliferation, apoptosis and oxidative stress in patients with liver cirrhosis. Ann Hepatol (2013) 12:301–7. doi: 10.1016/S1665-2681(19)31369-9
100. Ramachandran A, Prabhu R, Thomas S, Reddy JB, Pulimood A, Balasubramanian KA. Intestinal mucosal alterations in experimental cirrhosis in the rat: role of oxygen free radicals. Hepatology (2002) 35:622–9. doi: 10.1053/jhep.2002.31656
101. Natarajan SK, Ramamoorthy P, Thomas S, Basivireddy J, Kang G, Ramachandran A, et al. Intestinal mucosal alterations in rats with carbon tetrachloride-induced cirrhosis: changes in glycosylation and luminal bacteria. Hepatology (2006) 43:837–46. doi: 10.1002/hep.21097
102. Gao JH, Wen SL, Tong H, Wang CH, Yang WJ, Tang SH, et al. Inhibition of cyclooxygenase-2 alleviates liver cirrhosis via improvement of the dysfunctional gut-liver axis in rats. Am J Physiol Gastrointest Liver Physiol (2016) 310:G962–72. doi: 10.1152/ajpgi.00428.2015
103. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol (2019) 71:1216–28. doi: 10.1016/j.jhep.2019.08.005
104. Tsiaoussis GI, Papaioannou EC, Kourea EP, Assimakopoulos SF, Theocharis GI, Petropoulos M, et al. Expression of alpha-defensins, CD20+ b-lymphocytes, and intraepithelial CD3+ T-lymphocytes in the intestinal mucosa of patients with liver cirrhosis: Emerging mediators of intestinal barrier function. Dig Dis Sci (2018) 63:2582–92. doi: 10.1007/s10620-018-5146-9
105. Urdaneta V, Casadesus J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med (Lausanne) (2017) 4:163. doi: 10.3389/fmed.2017.00163
106. Sun D, Bai R, Zhou W, Yao Z, Liu Y, Tang S, et al. Angiogenin maintains gut microbe homeostasis by balancing alpha-proteobacteria and lachnospiraceae. Gut (2021) 70:666–76. doi: 10.1136/gutjnl-2019-320135
107. Munoz L, Albillos A, Nieto M, Reyes E, Lledó L, Monserrat J, et al. Mesenteric Th1 polarization and monocyte TNF-α production: first steps to systemic inflammation in rats with cirrhosis. Hepatology (2005) 42:411–9. doi: 10.1002/hep.20799
108. Ubeda M, Munoz L, Borrero MJ, Díaz D, Francés R, Monserrat J, et al. Critical role of the liver in the induction of systemic inflammation in rats with preascitic cirrhosis. Hepatology (2010) 52:2086–95. doi: 10.1002/hep.23961
109. Liu L, Zhang C, Hu Y, Zhou L, Tan Q. Changes in gut toll-like receptor-4 and nod-like receptor family pyrin domain containing-3 innate pathways in liver cirrhosis rats with bacterial translocation. Clin Res Hepatol Gastroenterol (2016) 40:575–83. doi: 10.1016/j.clinre.2016.02.008
110. Genesca J, Marti R, Rojo F, Campos F, Peribáñez V, Gónzalez A, et al. Increased tumour necrosis factor alpha production in mesenteric lymph nodes of cirrhotic patients with ascites. Gut (2003) 52:1054–9. doi: 10.1136/gut.52.7.1054
111. Albillos A, Martin-Mateos R, van der Merwe S, Wiest R, Jalan R, Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol (2022) 19:112–34. doi: 10.1038/s41575-021-00520-7
112. Albillos A, de la HA, Gonzalez M, Moya JL, Calleja JL, Monserrat J, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology (2003) 37:208–17. doi: 10.1053/jhep.2003.50038
113. Schierwagen R, Alvarez-Silva C, Madsen MSA, Kolbe CC, Meyer C, Thomas D, et al. Circulating microbiome in blood of different circulatory compartments. Gut (2019) 68:578–80. doi: 10.1136/gutjnl-2018-316227
114. Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, et al. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell (2005) 16:5040–52. doi: 10.1091/mbc.e05-03-0193
115. Guglani L, Khader SA. Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS (2010) 5:120–7. doi: 10.1097/COH.0b013e328335c2f6
116. Bajaj JS, Hafeezullah M, Hoffmann RG, Varma RR, Franco J, Binion DG, et al. Navigation skill impairment: Another dimension of the driving difficulties in minimal hepatic encephalopathy. Hepatology (2008) 47:596–604. doi: 10.1002/hep.22032
117. Romero-Gomez M, Boza F, Garcia-Valdecasas MS, García E, Aguilar-Reina J. Subclinical hepatic encephalopathy predicts the development of overt hepatic encephalopathy. Am J Gastroenterol (2001) 96:2718–23. doi: 10.1016/S0002-9270(01)02692-2
118. Butterworth RF. Hepatic encephalopathy in cirrhosis: Pathology and pathophysiology. Drugs (2019) 79:17–21. doi: 10.1007/s40265-018-1017-0
119. Ezaz G, Murphy SL, Mellinger J, Tapper EB. Increased morbidity and mortality associated with falls among patients with cirrhosis. Am J Med (2018) 131:645–650 e2. doi: 10.1016/j.amjmed.2018.01.026
120. Butterworth RF, Giguere JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol (1987) 6:1–12. doi: 10.1007/BF02833598
121. Butterworth RF. Hepatic encephalopathy: a central neuroinflammatory disorder? Hepatology (2011) 53:1372–6. doi: 10.1002/hep.24228
122. Shawcross DL, Davies NA, Williams R, Jalan R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol (2004) 40:247–54. doi: 10.1016/j.jhep.2003.10.016
123. Wright G, Davies NA, Shawcross DL, Hodges SJ, Zwingmann C, Brooks HF. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology (2007) 45:1517–26. doi: 10.1002/hep.21599
124. Jain L, Sharma BC, Srivastava S, Puri SK, Sharma P, Sarin S. Serum endotoxin, inflammatory mediators, and magnetic resonance spectroscopy before and after treatment in patients with minimal hepatic encephalopathy. J Gastroenterol Hepatol (2013) 28:1187–93. doi: 10.1111/jgh.12160
125. Srivastava A, Yadav SK, Yachha SK, Thomas MA, Saraswat VA, Gupta RK. Pro-inflammatory cytokines are raised in extrahepatic portal venous obstruction, with minimal hepatic encephalopathy. J Gastroenterol Hepatol (2011) 26:979–86. doi: 10.1111/j.1440-1746.2011.06706.x
126. Shawcross DL, Shabbir SS, Taylor NJ, Hughes RD. Ammonia and the neutrophil in the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology (2010) 51:1062–9. doi: 10.1002/hep.23367
127. Wunsch E, Koziarska D, Milkiewicz M, Naprawa G, Nowacki P, Hartleb M, et al. In patients with liver cirrhosis, proinflammatory interleukins correlate with health-related quality of life irrespective of minimal hepatic encephalopathy. Eur J Gastroenterol Hepatol (2013) 25:1402–7. doi: 10.1097/MEG.0b013e328365a447
128. Bajaj JS, Ridlon JM, Hylemon PB, Thacker LR, Heuman DM, Smith S, et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointest Liver Physiol (2012) 302:G168–75. doi: 10.1152/ajpgi.00190.2011
129. Merli M, Lucidi C, Pentassuglio I, Giannelli V, Giusto M, Di Gregorio V, et al. Increased risk of cognitive impairment in cirrhotic patients with bacterial infections. J Hepatol (2013) 59:243–50. doi: 10.1016/j.jhep.2013.03.012
130. Moratalla A, Ampuero J, Bellot P, Gallego-Durán R, Zapater P, Roger M, et al. Lactulose reduces bacterial DNA translocation, which worsens neurocognitive shape in cirrhotic patients with minimal hepatic encephalopathy. Liver Int (2017) 37:212–23. doi: 10.1111/liv.13200
131. Cipollini V, Anrather J, Orzi F, Iadecola C. Th17 and cognitive impairment: Possible mechanisms of action. Front Neuroanat (2019) 13:95. doi: 10.3389/fnana.2019.00095
132. Moreau R, Elkrief L, Bureau C, Perarnau JM, Thévenot T, Saliba F, et al. Effects of long-term norfloxacin therapy in patients with advanced cirrhosis. Gastroenterology (2018) 155:1816–1827 e9. doi: 10.1053/j.gastro.2018.08.026
133. European Association for the Study of the Liver. Electronic address eee, European association for the study of the l. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol (2018) 69:406–60. doi: 10.1016/j.jhep.2018.03.024
134. Bajaj JS, Heuman DM, Sanyal AJ, Hylemon PB, Sterling RK, Stravitz RT, et al. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PloS One (2013) 8:e60042. doi: 10.1371/journal.pone.0060042
135. Patel VC, Lee S, McPhail MJW, Da Silva K, Guilly S, Zamalloa A, et al. Rifaximin-alpha reduces gut-derived inflammation and mucin degradation in cirrhosis and encephalopathy: RIFSYS randomised controlled trial. J Hepatol (2022) 76:332–42. doi: 10.1016/j.jhep.2021.09.010
136. Fernandez J, Piano S, Bartoletti M, Wey EQ. Management of bacterial and fungal infections in cirrhosis: The MDRO challenge. J Hepatol (2021) 75(Suppl 1):S101–17. doi: 10.1016/j.jhep.2020.11.010
137. Moratalla A, Gomez-Hurtado I, Santacruz A, Moya Á, Peiró G, Zapater P, et al. Protective effect of bifidobacterium pseudocatenulatum CECT7765 against induced bacterial antigen translocation in experimental cirrhosis. Liver Int (2014) 34:850–8. doi: 10.1111/liv.12380
138. Moratalla A, Gomez-Hurtado I, Moya-Perez A, Zapater P, Peiró G, González-Navajas JM, et al. Bifidobacterium pseudocatenulatum CECT7765 promotes a TLR2-dependent anti-inflammatory response in intestinal lymphocytes from mice with cirrhosis. Eur J Nutr (2016) 55:197–206. doi: 10.1007/s00394-015-0837-x
139. Moratalla A, Caparros E, Juanola O, Portune K, Puig-Kröger A, Estrada-Capetillo L, et al. Bifidobacterium pseudocatenulatum CECT7765 induces an M2 anti-inflammatory transition in macrophages from patients with cirrhosis. J Hepatol (2016) 64:135–45. doi: 10.1016/j.jhep.2015.08.020
140. Chen RC, Xu LM, Du SJ, Huang SS, Wu H, Dong JJ, et al. Lactobacillus rhamnosus GG supernatant promotes intestinal barrier function, balances treg and TH17 cells and ameliorates hepatic injury in a mouse model of chronic-binge alcohol feeding. Toxicol Lett (2016) 241:103–10. doi: 10.1016/j.toxlet.2015.11.019
141. Bull-Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of lactobacillus rhamnosus GG treatment. PloS One (2013) 8:e53028. doi: 10.1371/journal.pone.0053028
142. Horvath A, Leber B, Schmerboeck B, Tawdrous M, Zettel G, Hartl A, et al. Randomised clinical trial: the effects of a multispecies probiotic vs. placebo on innate immune function, bacterial translocation and gut permeability in patients with cirrhosis. Aliment Pharmacol Ther (2016) 44:926–35. doi: 10.1111/apt.13788
143. Bajaj JS, Idilman R, Mabudian L, Hood M, Fagan A, Turan D, et al. Diet affects gut microbiota and modulates hospitalization risk differentially in an international cirrhosis cohort. Hepatology (2018) 68:234–47. doi: 10.1002/hep.29791
144. Macnaughtan J, Albillos A, Kerbert A, Vargas V, Durand F, Gines P, et al. O09 a double blind, randomised, placebo-controlled study to assess safety and tolerability of oral enterosorbent carbalive (Yaq-001) in cirrhotic patients. Gut (2021) 70:A5–6. doi: 10.1136/gutjnl-2021-BASL.9
145. Bajaj JS, Khoruts A. Microbiota changes and intestinal microbiota transplantation in liver diseases and cirrhosis. J Hepatol (2020) 72:1003–27. doi: 10.1016/j.jhep.2020.01.017
146. Maran RR, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther (2009) 328:469–77. doi: 10.1124/jpet.108.145409
Keywords: cirrhosis, gut permeability, inflammation, bacterial translocation, microbiota
Citation: Muñoz L, Caparrós E, Albillos A and Francés R (2023) The shaping of gut immunity in cirrhosis. Front. Immunol. 14:1139554. doi: 10.3389/fimmu.2023.1139554
Received: 07 January 2023; Accepted: 27 March 2023;
Published: 14 April 2023.
Edited by:
Javier Leceta, Complutense University of Madrid, SpainReviewed by:
Ian Cartwright, University of Colorado Anschutz Medical Campus, United StatesCopyright © 2023 Muñoz, Caparrós, Albillos and Francés. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agustín Albillos, YWd1c3Rpbi5hbGJpbGxvc0B1YWguZXM=; Rubén Frances, cmZyYW5jZXNAdW1oLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.