 Vincent Gauthier1,2,3†
Vincent Gauthier1,2,3† Maria Kyriazi1,4†
Maria Kyriazi1,4† Meriam Nefla2,3Valentina Pucino2,3
Meriam Nefla2,3Valentina Pucino2,3 Karim Raza3,5
Karim Raza3,5 Christopher D. Buckley2,3*
Christopher D. Buckley2,3* Ghada Alsaleh1,2*
Ghada Alsaleh1,2*- 1Botnar Institute for Musculoskeletal Sciences, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences (NDORMS), University of Oxford, Oxford, United Kingdom
- 2The Kennedy Institute of Rheumatology, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences (NDORMS), University of Oxford, Oxford, United Kingdom
- 3Institute of Inflammation and Ageing, College of Medical and Dental Sciences, University of Birmingham, Birmingham, United Kingdom
- 4Faculty of Medical Sciences, Newcastle University, Newcastle-upon-Tyne, United Kingdom
- 5Department of Rheumatology, Sandwell and West, Birmingham Hospitals NHS Trust, Birmingham, United Kingdom
Fibroblasts, derived from the embryonic mesenchyme, are a diverse array of cells with roles in development, homeostasis, repair, and disease across tissues. In doing so, fibroblasts maintain micro-environmental homeostasis and create tissue niches by producing a complex extracellular matrix (ECM) including various structural proteins. Although long considered phenotypically homogenous and functionally identical, the emergence of novel technologies such as single cell transcriptomics has allowed the identification of different phenotypic and cellular states to be attributed to fibroblasts, highlighting their role in tissue regulation and inflammation. Therefore, fibroblasts are now recognised as central actors in many diseases, increasing the need to discover new therapies targeting those cells. Herein, we review the phenotypic heterogeneity and functionality of these cells and their roles in health and disease.
1 Introduction
Following their discovery in 1858 by Rudolf Virchow, and their description as fibroblasts by Ernst Ziegler in 1895, fibroblasts have been observed in anatomically diverse connective tissues, with a distinct spindle-shaped morphology, delineating them from the other structural cells (1–3). Their characterization has been historically driven by their distinct morphology and the absence of leucocyte, epithelial and vascular lineage markers (4). Identifying the functionality and prevalence of distinct populations of fibroblasts has led to an elucidation of their differential roles in pathological states and insights into how they may be therapeutically targeted. In the following sections, we summarize the origins, tissue-specific types, heterogeneity, and function of fibroblasts identified to date, with a primary focus on their roles in inflammation and ageing. We illustrate how impairments in fibroblast biology contribute to tissue and organismal ageing and examine how recent advances set the groundwork for therapeutically targeting these cells.
2 Origins and functional heterogeneity of fibroblasts across the tissues
2.1 Origins and tissue-specific types
Recent technological advances have allowed a better understanding of the origin of fibroblast (summarised in Figure 1). During early embryonic development, gastrulation forms three primary layers, so-called ectoderm, mesoderm, and endoderm. These layers give rise to specific tissues and organs in the developing embryo. During the process of gastrulation, the epiblast gives rise to two main cell types: the primary mesenchyme and the ectoderm. The primary mesenchyme is made up of primary fibroblasts, and further differentiation of these cells leads to the development of the endoderm and mesoderm. The mesoderm gives rise to various cell types, including endothelial cells, pericytes, adipocytes, and mesenchyme. Mesenchymal stromal cells may also develop from the mesoderm and help create a supportive environment in certain tissues. In adults, the mesenchyme becomes quiescent fibroblasts known as resident quiescent fibroblasts (RQF). Together, epithelial cells, endothelial cells, perivascular cells, adipocytes, and fibrocytes from the bone marrow, participate in the generation of fibroblast-like cells during injury through processes known as Type 2 and Type 3 epithelial-to-mesenchymal transitions (EMT) (5). In adults the mesenchyme comprises resident fibroblasts that support ECM formation and remain quiescent in the absence of stimulation. Thus, fibroblasts are responsible for tissue homeostasis and contribute to extrinsic tissue ageing (5). Transcriptional regulation determines the localization of resident fibroblasts and defines their distribution through development. Additionally, fibroblasts are imprinted with positional identities laid down during development and maintained by the epigenetic regulation of homeobox transcription factors, so-called HOX genes, responsible for positional patterning during development (6, 7). Although all fibroblast subsets share a mesenchymal origin, their differences rely upon their localization, functionality, and cellular state. The different fibroblast types defined by their tissue specific gene enrichment and marker expression allows the depiction of fibroblast subtypes in tissues including the heart, gut, lungs, colon, skin, and joints. Cardiac/Myocardial fibroblasts express atrial and ventricular markers (8). Fibroblasts found in the lungs comprise lipofibroblasts (LipFB), myofibroblasts (MyoFB), alveolar (AlvFB) and adventitial fibroblasts (AdvFB) (9). The skin includes three fibroblast subtypes, type A (FB-A), responsible for dermal cell and ECM homeostasis, type B (FB-B), regulating immune surveillance and inflammation and type C (FB-C), comprising specialized subpopulations (10). Recent evidence also suggests the expression of myofibroblasts and pericytes, as well as a distinct population of fibroblast-like cells expressed in the colon that are associated with health status or with the development of inflammatory bowel disease (IBD) and colitis (11). Gut fibroblasts, including interstitial fibroblasts, are characterized by the expression of differential markers depending on their localization within the gut (e.g., villus, crypt). In healthy adult joints, fibroblast-like synoviocytes here termed synovial fibroblasts (SFs) are observed in both the synovial lining layer (LL) and the sub-lining layer (SL), expressing location-specific proteins and cytokines, and associated with either inflammatory or destructive diseases. Therefore, one could logically think that different fibroblast subsets exist in both healthy and diseased states. Indeed, recent data published by Buechler et al. indicate that universal and specialized fibroblast subsets, so-called steady state subsets, can exist together with activated, perturbed state subsets, within the same tissue (12). Understanding the different fibroblast markers observed in steady and perturbed states is thus likely to further our understanding of the mechanistic roles of these cells in the co-existing microenvironment.
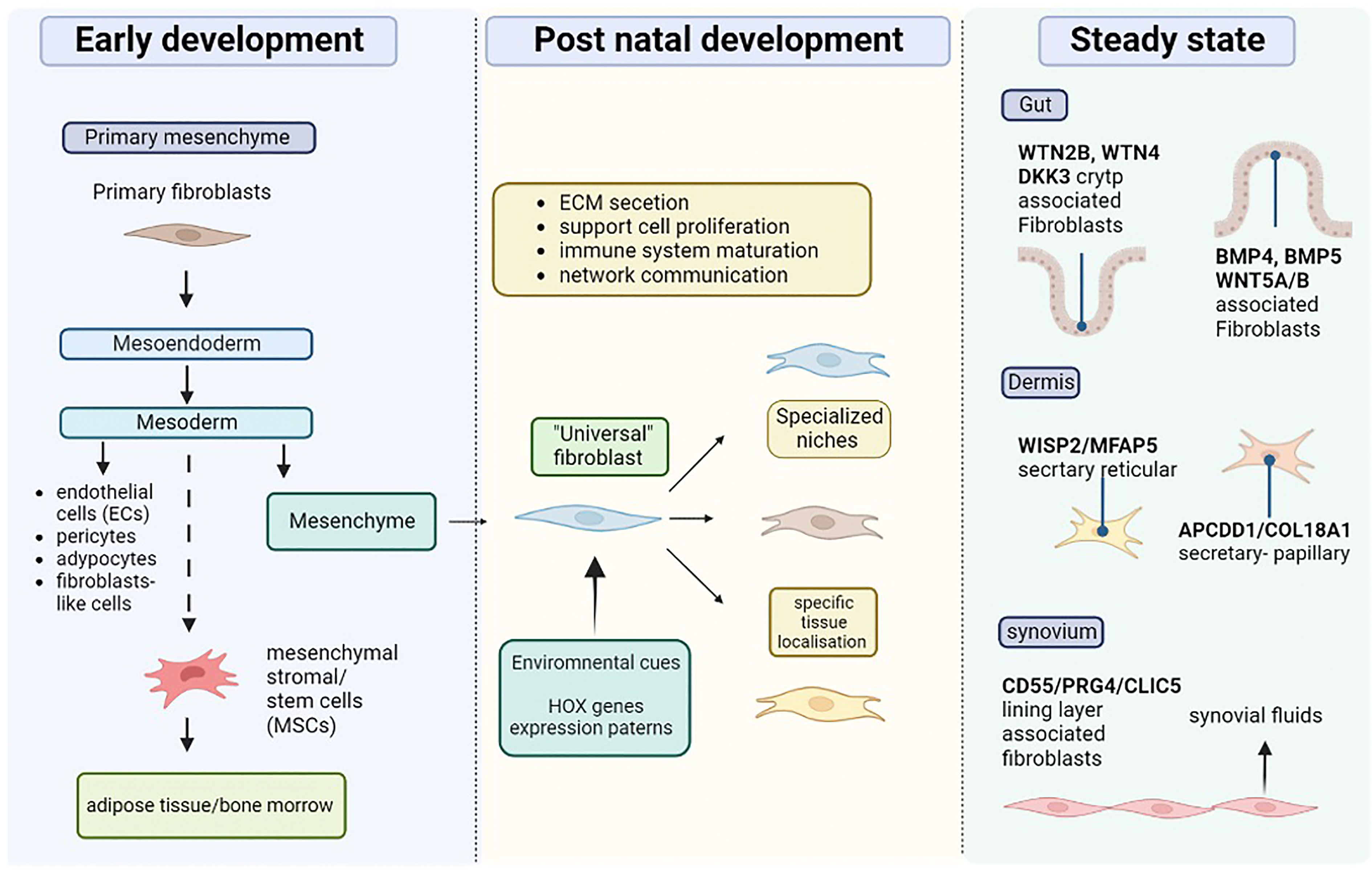
Figure 1 Origin and diversity of tissue resident fibroblast. During the early stages of development, the primary mesenchyme is mad up of primary fibro plats, which eventually give rise to the mesoendoderm. The mesoderm also gives rise to mesenchyme stromal or stem cells (MSCs), which can populate specific areas such as the bone marrow and adipose tissue. In addition, the mesoderm produces endothelial cells (ECs), adipocytes, and mesenchyme, which is where resident fibroblasts originate. Environmental cues and transcription of HOX genes give tissue localization instruction to resident fibroblasts which leads to their specific roles. Fibroblasts populate specific niches in different tissue supporting roles including cell proliferation, ECM secretion, immune cells, and maturation. According to the tissue and environmental needs fibroblasts develop specific function, (e.g., synovial fluid secretion).
2.2 Heterogeneity across the tissues
Fibroblasts are characterized by their secretion of ECM molecules including elastin, fibronectin, periostin, laminins, proteoglycans, microfibrillar proteins, and fiber- or sheet-forming collagens, creating the “matrisome” and which in turn influences their function. Since no totally lineage specific markers have been identified, fibroblasts are captured using archetypal mesenchymal markers, including vimentin, PDGFRα/β (platelet-derived growth factor receptor-alpha and -beta), PDPN (podoplanin), CD90 (THY-1), FAP (fibroblast activation protein) marking activated fibroblasts, and αSMA (α-smooth muscle actin) typically expressed in myofibroblasts, the fibroblast type that is activated in response to injury and is responsible for the tissue repair (5, 13–15). However, these markers are not ubiquitously expressed upon all the fibroblasts, diminishing our understanding of the heterogeneity of this cell type.
Recent technological advances, including single cell RNA sequencing (scRNA-seq) have begun to reveal fibroblast heterogeneity and provide insights into their roles in disease (Table 1). However, whether subtype heterogeneity emerges from broad transcriptional programming (intrinsic triggers), or from other cell types in an extrinsic context dependant manner still remains to be established. A recent study in a murine model suggests the existence of dpt+ universal fibroblasts providing functional plasticity to activated fibroblasts across tissues (12). This supports the idea that fibroblast phenotype depends on their activation state in addition to potential tissue-specific cues. The authors propose that fibroblasts exist in a universal and specialized state, called steady state, or an activated state during perturbation (inflammation, wound healing, fibrosis, or cancer). Consequently, fibroblast identity changes depending on the surrounding environment.

Table 1 Fibroblasts subsets identified in health or disease and their localization in the tissues.
2.2.1 Gut
Differential expression of the WNT/BMP pathway is observed in gut fibroblasts, providing them with location-specific properties. Indeed, WTN2B, WTN4 and DKK3 enriched fibroblasts are associated with the crypt, while BMP4, BMP5 and WNT5A/B enriched fibroblasts reside in the villus (21). These transcriptional and positional differences underline the role of fibroblasts in supporting other cell types in specific niches. In this context, a POSTN fibroblast population has been identified to support epithelium by expressing factors related to epithelial cell proliferation and maintenance (11). Similarly, within the WNT2B population, a RSPON3+ (R-spondin-3) subset supports the intestinal stem cell niche through its interaction with the LCR5 receptor, highlighting the role of fibroblasts heterogeneity in maintaining sub-compartmental function within the tissue.
2.2.2 Skin and dermis
The dermis is an important regulatory layer of the skin underlining the epidermis. It principally consists of ECM secreted by numerous fibroblasts and is divided in two regions: the papillary dermis (PD) and the reticular dermis (RD) (15). As in the gut, different fibroblast populations are localized in the distinct structures of the dermis. PD fibroblasts exhibit morphological differences compared to RD fibroblasts. While PD fibroblasts are thin and spindle-shaped, RD fibroblasts have a squarer shape (19). ScRNA-seq studies on skin biopsies identified 4 clusters of dermal fibroblasts and two subpopulations with important structural and ECM organization roles. The first, in the RD (so-called secretary-reticular), is enriched with WISP2 or MFAP5. The second sub-population (so-called secretary-papillary) is found in the PD region and expresses APCDD1 or COL18A1, previously described as papillary markers. The third population expressing Asporin (ASPN) and Periostin (POSTN) is implicated in mesenchymal regulation. The final sub-population represents a pro-inflammatory state, enriched with CCL19, APOE, or CXCL2 expression (24). However, poor overlap of adult dermal fibroblasts subpopulations has been observed in other scRNA-seq datasets (20, 25–27). A comparative study on all published datasets on human skin, identified 3 mains dermal fibroblast types, type A fibroblasts implicated in ECM homeostasis and potentially in fibrotic linage, type B fibroblasts involved in inflammation and immune surveillance and type C corresponding to a specialized skin specific subpopulation such as papillary dermis fibroblasts. While disparities between datasets can be explained by variation in experimental processes, once clustered together those datasets reveal biological similarities between them. Thus, these discoveries are improving our knowledge on the heterogeneity in skin fibroblasts populations paving the way to understand potential dysregulation in diseases.
2.2.3 Oral mucosa
The oral mucosa is the first site of encountered for food, airborne antigens, and commensal microbiome. The oral cavity mucosal tissue comprises multilayer squamous epithelium divided in three main regions; the masticatory mucosa (gingiva, hard palate and dorsum of the tongue), the specialized mucosa (taste buds associated), and the lining mucosa (inside the cheeks, floor of the mouth) (28, 29). Despite the constant exposure to antigens, commensals and microdamage, the oral mucosa display efficient wound healing and minimal scar formation without aberrant inflammation. This suggests meticulous regulation between the stromal compartment and the immune cells at those sites. Recently, the transcriptomic profile of gingival/oral fibroblasts reveals 5 sub populations in adult healthy gingiva and lining mucosa (22). One of the fibroblast cluster display collagen synthesis and ECM remodelling functions (COL1A1, COL1A2, MMP2). A second cluster expressing SPARCL1 display an active translation status. Finally, 3 clusters are associated with inflammatory signature genes: ANXA1+IGBP2+ enriched fibroblasts associated with leucocytes proliferation, CXCL1+CXCL2+CXCL8+ fibroblasts involved in granulocyte recruitment, and C3+CDF+ fibroblasts for the complement activation (22).
2.2.4 Heart
The heart is another fibroblast-enriched organ. Cardiac fibroblasts are responsible for the ECM remodelling, which is critical for electrical conductivity and heartbeat rhythm (30). Adult cardiac fibroblasts comprise myocardial, pericardial and epicardium layers, containing specialized adipose tissue and endothelial cells, respectively (30). These populations, revealed by single-cell transcriptomic studies in mouse, comprise two main groups and their lineage contributions are distinct. For example, Forte et al. discovered a small endocardial-derived fibroblast population expressing Wif1 and Dkk3 WNT signalling factors, relating to valve leaflets and endochondral specification toward the bone lineage, and a larger epicardial-derived fibroblast population presenting the expression of genes associated with metabolism, and cell migration (31).While other ScRNAseq studies on murine cardiac fibroblasts suggests two major sub populations based on the differential expression of Sca1. The Sca1high and Sca1low subpopulation both expressed canonical fibroblasts markers such as PDGFRα or Col1 and seems to have distinct adhesive and secretory phenotype. Moreover, the authors also distinguish a fibroblast population highly regulated by WNT signalling (32).
2.2.5 Lungs
Lung fibroblasts, like many other fibroblasts are marked by PDGFRα expression. Additional heterogeneity is observed within PDGFRα+ lung fibroblasts including a WNT subset which is also characterized by Axin2 expression (18). PDGFRα expression is increased upon Gli1+ (part of the Hedgehog signaling pathway) maturation of the Gli1+ progenitors, which label the mesenchymal lung cells, but not lipofibroblasts, and depend on Hedgehog signaling (16, 18, 33). In healthy lung fibroblasts, AXIN2+ PDGFRα+ are located towards the alveolar niche supporting the maintenance of the stem cell niche, whereas AXIN2+ PDGFRα- fibroblasts, a population associated with pathogenic remodeling in disease, are positioned towards the airways (3). In addition, αSMA+ fibroblasts express Tbx4 in response to lung injury (34).
The sub-epithelial regions of large lung airways are characterized by the expression of ACAT2+ in myofibroblasts secreting ECM and which show a dysregulated expansion in pulmonary fibrosis. The interstitial regions of the lungs express PLIN2+, responsible for phospholipid storage, a subset which are also elevated in pulmonary fibrosis (35).
2.2.6 Joints
The synovial membrane is histologically separate in two compartments: the lining layer and the sub-lining. Highly specific PRG4+CD55+CLIC5+ fibroblasts, are found in the synovial lining layer and are responsible for the secretion of synovial fluid (3, 36). By contrast, in resting condition the sub-lining fibroblasts are not well defined. However, significant increase in fibroblast heterogeneity is observed in the joints of patients with active synovial inflammation, contribution to both Inflammation and bone/cartilage erosion.
In resting conditions, fibroblasts modulate specific tissue niches by adapting to the needs of the surrounding microenvironment. Increasing scRNA-seq data on human fibroblasts in healthy individuals are unmasking new functions of fibroblasts beside the archetypal ECM formation.
2.3 Functional characterization of fibroblasts
Fibroblasts are critical for organizing functional tissue networks, due to ECM secretion, defining the tissue architecture and allowing cells to migrate and communicate. Thus, they have been implicated in the formation of specialized niches which support various processes including stem cell proliferation, haematopoiesis, and even joint lubrication (11, 37–40). Their multifaced properties also allow them to establish the functioning and positioning of other cell types. Following tissue damage fibroblasts are responsible for tissue healing, inflammation, and scarring (40).
Among those specialized functions, fibroblasts play a dominant role in immune system establishment from addressing alert signals during inflammation and participating in the T and B cell maturation and trafficking (38, 41).
Immune cell recruitment to peripheral tissues requires specific cues from endothelial cells. This includes cell interactions through adhesion molecules, including selectins and integrins, and activation molecules called chemokines (42). Due to their immunological properties, fibroblasts play a critical role in supporting the recruitment and retention of leukocytes within the tissue (43, 44), a process, that is essential for establishing an immune response (41). Stromal niches coordinate lymphocyte trafficking and survival in lymphatic tissues (45). In lymphoid tissue, fibroblastic reticular cells (FRCs) shape the stromal architecture of the secondary lymphoid organs (SLO) secreting chemokines, survival cues and the ECM network necessary for establishing the adaptative immunity (46). FRCs originate from the mesenchymal lymphoid tissue organizer cells (mLTo), expressing PDGFRα/β, which then differentiates into specialized FRC subsets in a “2 signals” model (47). The first signal, comprising lymphotoxin-β receptor (LTβR) and nuclear factor nuclear factor-kappa B (NF-κ B), commits to the FRC lineage differentiation, whereas the second signal provides FRC-specialized identities. Those FRC-specialized subsets form distinct niches of the SLO and are characterized by their localization and immune interactors. For instance, in the lymph node, FRC landscape comprise marginal reticular cells (MRC), follicular dendritic cells (FDCs) in the dark zone and in the light zone, T‐B border reticular cells (TBRC), interfollicular reticular cells (IRC), medullary reticular cells (MedRC), T cell reticular cells (TRC), and perivascular reticular cells (PRC) around blood vessels (47). While the second signal is not yet fully understood, some molecules such as tumor necrosis factor (TNF) or receptor activator of nuclear factor kappa-B ligand (RANKL) are involved in the establishment of FDC or MRC respectively (47, 48). FRCs also support immune cell communication by establishing chemokine gradients (CXCL13, CXCL12, CXCL21, CXCL19) and survival cues including IL-7, or RANKL (46). Thus, the stromal compartment comprising fibroblasts, is essential for many immune processes by elaborating a precise set of cytokines and chemokines to drive immune cell communication.
3.0 Fibroblast dysregulation in disease
3.1 Role in the immune response: The loss of homeostatic balance leading to inflammation
As already mentioned, fibroblasts support the recruitment and activation of immune cells, by secreting and responding to cytokines, chemokines, and other inflammatory stimuli. A multi-omics, cross-tissue study has demonstrated that fibroblasts, along with endothelial and epithelial cells, rapidly respond to immune activation based on epigenetic changes (49). Inflammation relies on an orchestrated series of events comprising the recruitment of immune cells, their activation, and a final resolution phase. It now appears that fibroblasts are critical in all these phases. Therefore, dysregulation of the immunomodulatory properties of fibroblasts can lead to persistent chronic inflammation and failed resolution. The resolution of the inflammation is vital for tissue homeostasis. Eliminating pro-inflammatory and survival signals initiates inflammation resolution, which is followed by an increase in apoptotic signalling and re-entry of the remaining inflammatory effectors in the circulatory and lymphatic system (50). This process can be perturbed by fibroblasts that remain primed to inflammatory signals leading to abnormal retention of lymphocytes within the peripheral tissue. This phenomenon is observed in rheumatoid arthritis (RA), where synovial fibroblasts (SFs) are primed by inflammatory signals, increasing their inflammatory potential. Indeed, upon tumor necrosis factor alpha (TNF-α) stimulation, SFs exhibit prolonged activation of NF-κ B leading to excessive interleukin 6 (IL-6) levels (51). This effect is increased upon re-stimulation confirming the priming potential of SFs in inflammation (52). Furthermore, a recent study has demonstrated that intracellular activation of the complement C3 component in rodent SFs induces glycolysis and activates the inflammasome (53). Therefore, primed SFs produce pro-inflammatory RANKL, IL-6, TNF and IL-1β, mediating inflammatory bone destruction. Evidence has also demonstrated the role of stromal cells in activating CXCR4, a chemokine receptor supporting the infiltration of synovial T cells, subsequently increasing their retention in the tissue via the endothelial expression of SDF-1 (CXCR4 ligand) (54, 55).
Other chemokines are also involved in CD4+ and CD8+ T cell accumulation and migration to the synovial microenvironment, including CCL5 or CXCL11 (56). Besides chemoattractant signals, apoptotic resistance is also increased in RA through T cell interaction with SFs via integrins (57). Excessive lymphocyte accumulation in RA is also mediated by the expression of pro-survival SF molecules, impairing accurate resolution. For instance, synovial fibroblasts secrete interleukin 7 (IL-7) and IL-15 inducing T cell proliferation (58). Altogether, this evidence suggests that synovial fibroblasts adopt FRC properties during chronic inflammation and permit the formation of ectopic-lymphoid structures, known as tertiary lymphoid structures (TLS) (17, 23, 59). By communicating with immune cells and shaping the microenvironment toward inflammation or resolution, fibroblasts appear as active contributors to inflammatory diseases, fibrosis, or cancer. Therefore, understanding the factors that drive fibroblast heterogeneity might help therapeutic targeting of inflammatory-mediated diseases.
3.2 Heterogeneity in the progression of disease across tissues
3.2.1 Rheumatoid arthritis
Fibroblasts cell states are dramatically altered in diseased tissues. For instance, fibroblasts in the joints of RA patients consist of discrete fibroblast populations which become pathogenic upon repetitive inflammatory signals. The synovium is a thin connective tissue at the interphase of the bones and is essential to maintain a low friction environment by secreting the synovial fluid into the synovial cavity. It comprises two distinct mesenchymal compartments: an epithelial-like lining layer formed by tissue resident macrophages and lubricin (PRG4) expressing lining layer fibroblasts and a sub-lining structure which contains fibroblasts the vascular system and the immune cells (39). During inflammation, the architecture of the sub-lining drastically changes, and the SL fibroblast population expands. Using data from the Accelerating Medicines Partnership Rheumatoid Arthritis/Systemic Lupus Erythematosus (AMP RA/SLE) Consortium, Zhang et al. identified 4 synovial fibroblast subsets: a perivascular and interstitial THY1+/HLA-DRhi, perivascular sub-lining CD34+ fibroblasts, a DKK3+/CADM1+ sub-lining population and CD55+PRG4+ lining layer fibroblasts (36). The CD55+ lining layer fibroblasts are the most transcriptionally distinct subset compared to the THY1+ sub-lining fibroblasts. Those results have been further confirming by Croft et al. and validated in the mouse synovium (60). Croft et al. also demonstrated functional heterogeneity of the fibroblast populations during arthritis. Indeed, intra-articular injection of FAP+THY+ fibroblasts resulted in severe and sustained inflammation, whereas injecting FAP+THY- increased osteoclast activity and bone damage via the activation of nuclear factor-κB ligand (RANKL) (60).
In RA THY1+ fibroblasts expand from the perivascular niche in the direction of the lining layer and endothelial cells provide positional cues for fibroblasts via NOTCH3 signalling (61). NOTCH3 activation in fibroblasts also modulates the pathogenic sub-lining phenotype and increases their expansion. Genetic depletion and antibody blockade of NOTCH3 decreases synovial inflammation and damage, suggesting the important role of endothelial-fibroblast crosstalk in RA. By contrast, the lining layer fibroblast identity is maintained in the absence of endothelial positional cues (62). However, a recent study have demonstrated a novel transcriptional regulation which direct the fate of SFs toward a ECM degrading phenotype. Indeed, Yan et al. identified the transcription factor ETS1 as a positive regulator of Tnfsf11 (coding for RANKL), MMP13 and MMP3 in arthritic SFs (63). This discovery gives new lines of thought in fate decision process of pathological fibroblasts besides the identified environmental cues.
3.2.2 Ulcerative colitis
In the gut, most fibroblast subsets exist in both healthy individuals and in patients with ulcerative colitis (UC), with inflammatory fibroblast populations in UC expanding (21). These inflammatory associated fibroblasts are characterised by the expression of the colitis and fibrosis related gene, IL-11, and share markers with cancers-associated fibroblasts (CAFs), such as the fibroblast activated protein (FAP), or WNT2 (63), suggesting a putative shared state/origin between them. Furthermore, OSMR (Oncostatin M) is highly expressed in the inflammatory-associated fibroblasts, highlighting their role in anti-TNF therapy resistance (64).
3.2.3 Periodontitis
Periodontitis is an inflammatory disease affecting the gingiva due to a dysbiosis on the tooth surface. Transcriptional analysis on periodontitis tissue indicates a decrease of the stromal cells (endothelial/fibroblast) proportion, albeit presenting transcriptional similarities compared to healthy tissue (22). Nevertheless, a transcriptional shift toward inflammation is observed in the epithelial and stromal population including the fibroblasts clusters previously described in health. Indeed, genes associated with antimicrobial response are enriched in the fibroblast populations. Moreover, the increased expression of CXCL1, CXCL2 and CXCL3 by the fibroblasts reflect an active recruitment of neutrophil. A parallel study also demonstrated the increased of inflammatory fibroblasts proportions in mild and severe periodontitis gingiva (65). In addition, the authors report a disruption of fibroblast population known to be involved in tissue repair. Notably characterized by a diminution of collagen VI in severe periodontitis tissue (65)
3.2.4 Pulmonary fibrosis
Habermann et al. have identified different fibroblast states in the lungs of pulmonary fibrosis patients compared to non-fibrotic donors, including ACTA2+ myofibroblasts, PLIN2+ lipofibroblast-like, HAS1 high populations (35). Overall, fibroblast populations are expanded in fibrotic lung tissue compared to controls and are located in specific niches. Indeed, the aSMA encoding Acta2-expressing myofibroblasts are expanded in the sub-epithelial regions of the large airways, while PLIN2+ populations are diffusely distributed in the interstitial regions around the alveoli and HAS1+ populations in the sub pleural regions. Dysregulated gene expression in these fibroblast niches promote pathologic ECM expansion.
The presence of various fibroblast populations in disease supports the concept that fibroblasts are key players in many pathologies by sustaining inflammation or increasing fibrosis. Recognising that fibroblast heterogeneity depends on their environmental cues, it is critical to determine if the activation of pathogenic fibroblast subsets is tissue-dependent or shared across diseases.
3.3 Cross tissue pathological implications
Novel statistical models for integrative clustering of scRNA- seq datasets allow an assessment of the cellular state across species, tissues, and diseases. In line with the evidence of a universal dpt+ fibroblasts in the mouse (12), Korsunsky et al. elucidated the presence of shared fibroblasts activation states across human diseases and tissues (66). The authors compared the transcriptomic profiles of fibroblasts from multiple organs using individual adult scRNA-seq datasets including the Adult Human Cell Atlas (AHCA) (67) and Tabula Sapiens (TS) (68). They found over 256 genes from AHCA and 357 from TS as universal fibroblast markers across tissues, generating de novo scRNA-seq data to characterize inflammatory fibroblasts from the gut, the lungs, the synovium, and the salivary glands. It is important to note that this study is pioneering in its use of scRNA-seq on salivary glands from patients with Sjögren’s syndrome and confirms the presence of PDPN+CD34+ and CD34-CCL19+ populations as identified through multi-channel flow cytometry (23). Between those 4 organs the authors identified 5 clusters, with shared markers, including SPARC+COL3A1+, FBLN1+, PTGS2+SEMA4A+, CD34+MFAP5+, and CXCL10+CCL19+. Among those clusters SPARC+COL3A1+ and CXCL10+CCL19+ were significantly expand in all the inflamed but not normal tissues (44) (66). Gene ontology (GO) and transcription factor analysis of the SPARC+COL3A1+ indicated enriched expression of the ECM and Notch signalling pathways, whereas CXCL10+CCL19+ was enriched in genes regulating lymphocytes chemotaxis, T cell proliferation, NF-κ B and interferon (IFN) signature. The interaction between SPARC+COL3A1+ and endothelial cell via NOTCH has been demonstrated in vitro, consolidating the role of endothelial/fibroblasts crosstalk in priming synovial tissue to increase lymphocyte infiltration. This SPARC+COL3A1+ population closely corresponds with NOTCH-activated THY1+ SFs during RA (6, 8, 36, 69). SPARC+COL3A1+ fibroblasts might expand before CXCL10+CCL19+, suggesting a later differentiation in favour of lymphocyte interaction after endothelial activation. Additionally, HLA-DRhi synovial fibroblasts might be related to the CXCL10+CCL19+ due to their strong IFN response. Recent evidence supports the concept that SFs-induced CXCL10 expression is achieved following stimulation by TNF-α and IFN, which subsequently activated T cell via CXCR3 (70). Overlapping activation states of fibroblasts is found across tissues and diseases but may not be exclusive to inflammation. Indeed, a recent study identified the crosstalk between epithelial cells and fibroblasts through SPARC promoting a chronic wound healing phenotype in idiopathic pulmonary fibrosis (71). This highlights that the state of fibroblast activation is potentially responsible for driving disease by communicating with different cell types through similar signals. In summary, integrative clustering studies on stromal cells has revealed new approaches to understand common cellular activation mechanisms leading to potential cross-disease therapies (Figure 2).
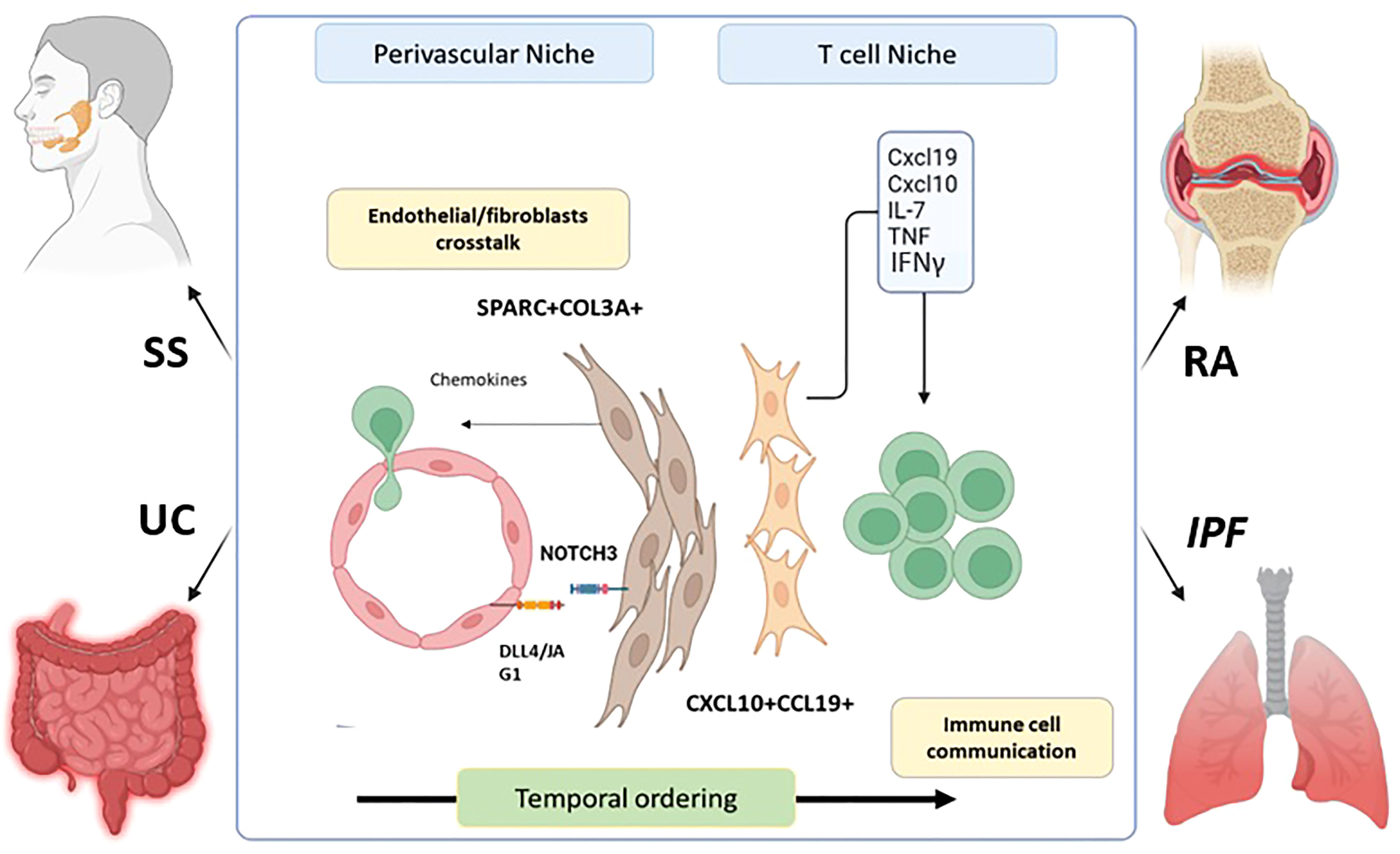
Figure 2 Fibroblasts shared an inflammatory phenotype across diseases and tissues. SPARC + COL3A + fibroblasts expend in the perivascular niche during inflammation after crosstalk with endothelial cells via NOTCH signaling pathway. This leads to their accumulation and the expression of chemokine’s promoting T and B cell infiltration in the tissue. Later, the CXCL10+CCL19 + fibroblasts population emerged and promote the retention and survival of T cell with in the tissue by the expression if IFNy, IL-7, TNF, and various chemokine’s. This establishment is temporal as evidence in mouse colitis models shows the transitory apparition of SPARC+COL3A+ followed by CXCL10+CCL19 + population. This mechanism was characterized in RA. However, SPARC + COL3A _ CXCL10+CCL19 + and CXCL10 +CCL19 _ fibroblasts are expended during RA (Rheumatoid Arthritis), SS (Sjogren’s syndrome), IPF(idiopathic pulmonary Fibrosis) and UC (Ulcerative Colitis).
4.0 Fibroblasts and ageing
4.1 Cellular senescence of fibroblasts
Cellular senescence, irreversible cell growth arrest, was initially documented in 1961 by Hayflick and Moorhead with their experiments on primary human fibroblasts, which demonstrated a finite proliferation lifespan in vitro and a gradual exhaustion of their replicative potential (72). The subsequent identification of senescent cells in evolutionarily conserved embryological organs through major animal lineages revealed their phenotypic ancestry and gave rise to several theories regarding their contribution to ageing (73–78). Since then, senescence has been shown to be a complex phenomenon with beneficial functions in embryonic development (73, 75, 79), wound healing (80–82), tissue repair (83) and cancer prevention (84, 85), but also critical deleterious roles in organismal ageing (86, 87), age-related diseases (87), abnormal immune responses, inflammation, and even tumorigenesis primarily detected later in life (88, 89). It has been proposed that ageing is caused by the depletion of stem and progenitor cells alongside the effects of senescent cells’ inflammatory phenotype (84, 90). Comprehensive research has been conducted to reveal the molecular triggers, mechanisms, and functionality underpinning senescent cells in tissues and how these contribute towards ageing and age-related diseases.
Cellular senescence refers to the genetically programmed and stable proliferation arrest state that cells undergo in response to detrimental stimuli (89, 91, 92). These stimuli, arising from intrinsic and extrinsic stresses, limit the propagation of damaged and stressed cells, causing them to enter a permanent cell cycle pause state (93). In quiescent skin fibroblasts, permanent senescence is induced by cell-autonomous (cell cycle exit), non-cell autonomous (ability to modulate surrounding tissues) as well as exogenous (e.g. tobacco smoking, UV irradiation) stressors, leading to various responses (94–96). Telomere attrition, or so-called telomere shortening, one of the first molecular senescence triggers reported, is caused by the progressive shortening at the chromosomal ends with repeated cell divisions (97, 98). This is characteristic of fibroblast senescence and is referred to as replicative senescence (RS), induced by replicative exhaustion (72, 93). Additionally, senescence in dermal fibroblasts is marked by mitochondrial dysfunction, also known as mitophagy (99–102); DNA damage and subsequent activation of the DNA damage response (DDR) signaling pathway (103, 104); loss of proteostasis caused by the dysregulated homeostasis of the cellular proteome driven by aberrant protein synthesis, folding and degradation (105–107); excessive oxidative DNA damage and oxidative stress (102); chromosomal and epigenetic aberrations, including post-translational modifications like cross-linking and oxidation (107); metabolic reprogramming and impairment in the DNA repair mechanisms (107, 108).
These triggers provoke the upregulation or downregulation of corresponding proteins, driving fibroblast senescence, which enhance tissue ageing. Mitochondrial dysfunction, for example, associated with the formation of reactive oxygen species (ROS) in ageing fibroblasts, amplifies the expression of activation-protein 1 (AP-1) and NF-κB dependent signaling pathways leading to excessive DNA damage in the skin and elevated accumulation of superoxide ions, which have been indicated as inducers of fibroblast senescence and accelerated skin ageing (109–111).
It is also well-established that elevated cyclin-dependent kinase (CDK) levels are another common feature of cellular senescence in dermal fibroblasts (107). Among those, p16Ink4a/Arf (CDKN2A) and p21 (CDKN1A), two of best well-known senescence markers, alongside the tumor suppressor protein p53 and the senescence-associated β-galactosidase (SA-β-gal), associated with lysosomal activity, present a significantly increased expression during fibroblast senescence, playing a causal role in skin ageing and age-related diseases (85, 87, 112–114). Importantly, clearance of p16Ink4a/Arf positive senescent cells using senolytic treatment in both BuRB1 progeroid ATTAC transgenic mouse models reverses the effects of senescence in tissues and prevents the accumulation of age-related diseases (87, 112). Besides that, fibroblast senescent cells also exhibit enhanced phosphorylated γ-H2AX, a DNA Damage Response (DDR) marker, the inactivation of which causes senescent cells to resume DNA replication (113, 115, 116).
4.2 Tissue ageing: The role of fibroblasts
The ability of the human body to resolve inflammation decreases with advancing age, causing an imbalance between pro-inflammatory and anti-inflammatory cellular profiles (117). This gives rise to “inflammaging” - the chronic low-grade pro-inflammatory state that accelerates with older age and is predominantly observed in age-related diseases, such as osteoarthritis, cardiac disease and even several tumorigenesis types as well as other non-age related disease like diabetes (117, 118).
Tissue ageing is characterized by the systemic increase in multiple pro-inflammatory cytokines caused by the accumulation of senescent cells (117). Although these are stable in cellular division and growth, they remain viable and metabolically active, implementing a complex response known as the senescence-associated secretory phenotype (SASP) (119, 120), characterized by the secretion of a range of pro-inflammatory molecules including chemokines (macrophage inflammatory proteins (MIPs), monocyte chemoattractant proteins (MCPs)), interleukins (IL-1, IL-6, IL-8, IL-18), receptor ligands, matrix metalloproteases (MMPs), ECM components, growth factors (transforming growth factor β (TGF-β)), proteases, and cytokines, that can induce inflammation, alter the microenvironment and modulate neighboring cells (121–123).
Previous studies have documented that SASP is the primary mechanism by which senescent cells detrimentally impact on the skin and other tissues (124). SASP is required for the clearance of senescent cells, which they accomplish by recruiting and secreting immune factors that initiate phagocytosis (91). Therefore, it plays a critical role in directing cells of the innate and adaptive immune system towards fibroblast senescent cells, promoting their clearance, and terminating inflammation (124). However, aged tissues accumulate excessive senescent cell numbers contributing to age-related tissue deterioration (125). This feature, along with impaired immune activities, stem and progenitor cells exhaustion observed with advanced age, results in the accumulation of persistent senescent cells that aggravate damage and contribute to tissue ageing (91). For instance, inefficient clearance leads to loss of homeostasis and imbalance between senescence and clearance, resulting in deficiencies and age-related diseases (90, 126, 127). Previous research has shown that Nrf2, a redox regulator used to induce senescence in vitro and in vivo, creates an aberrant ECM when overexpressed in fibroblasts (128). This aberrant ECM, rather than promoting wound healing, acts in a pro-tumorigenic manner, suggesting that the beneficial effects of senescent cells are context-dependent (94). Therefore, if not cleared from the tissue, senescent cells can dominate and cause detrimental effects (94).
Senescent fibroblasts with an abnormal SASP can also disrupt the interactions between the dermis and the epidermis (129). Fibroblasts derived from the dermis release an insulin-like growth factor (IGF-1) which is essential for the mesenchymal stem cell niches (MSC) and regulates the balance of epidermal cell proliferation and differentiation (129). During fibroblast senescence, IGF-1 signaling is downregulated because of enhanced superoxide ion production accumulating from the dysfunctional mitochondria of the aged human and murine dermal fibroblasts (130). Later evidence indicates that deprived IGF-1 and inhibition of the relaying IGF-1 signal causes suppression of collagen synthesis in the dermis and subsequent epidermal atrophy due to enhanced DNA damage induction, γH2AX and p16Ink4a/Arf production in the epidermal cells, consolidating the data underlying the vitality of IGF-1 in the human skin (129, 131, 132).
It is also vital to note that, the pathophysiology and inflammaging of the skin is a multifactorial rather than a single event, created by the complex network of cellular cross-link communication among fibroblasts, keratinocytes and melanocytes, as well as their interactions with the external environmental stressors (117).
Interestingly, besides the production of pro-inflammatory cytokines and interleukins derived by SASP, recent evidence highlights the role of inflammation in fibroblast senescent cells’ generation from an alternative, intriguing ankle. For instance, data from metabolically downregulated C3-/- mice, injected with monosodium urate signals (MSU), present fibroblast senescence, which is elevated in aged-cultured cells, marked by enhanced expression of senescence-associated β-galactosidase, as well as p15, p16 and p21 senescent markers (53). Therefore, when the complement 3 component of the innate immune system is abolished, fibroblasts cannot be primed and become senescent (53). Similar effects are also observed when mTOR and HIFα are pharmacologically inhibited using rapamycin and the translational inhibitor KC7F2, respectively (53). The authors, therefore, propose that senescence induction via pharmaceutical inhibition of mTOR or HIFα, or C3 depletion, create an immune-regulatory phenotype, prohibiting uncontrolled complement activation, ameliorating the detrimental inflammatory effects recurring at specific previously affected sites (53). However, it would also be beneficial to determine whether this C3 abolishment creates any adverse effects in the long term or measure the exact levels of senescence that create this anti-inflammatory effect.
Senescence is also induced via activation of the melanocortin type 1 receptor (MC1), which alternates the genetic expression, increasing MMP expression, deteriorating collagen production and altering the remodeling phase of wound healing. Nevertheless, recent evidence shows that selective agonism of MC1 through activation of G-protein coupled receptor (GPCR) and dependence on the ERK1/2 downstream phosphorylation ameliorates inflammation in the joints and protects the cartilage via inducing senescence in the synovial tissue (133). Therefore, selective MRC1 expression inducing senescence is proposed as a promising therapeutic target against inflammatory arthritis (133).
This evidence thus indicates the vitality of harmonized senescence fibroblasts, strengthening the hypothesis that the absolute depletion of senescent cells can also be detrimental to the tissues.
4.3 Fibroblast heterogeneity during ageing
Biological ageing can be considered as a disease on its own (134). Ageing leads to the accumulation of degenerative biological processes involving a progressive decline in organismal homeostasis (135). By driving modifications in the tissue, including increased inflammation, loss of tissue regeneration, fibrosis, and metabolic imbalance, ageing is a major comorbid factor in many diseases. While cellular senescence participates in many processes, other ageing factors that drive age-related disease are not yet fully understood. To address this issue, several scRNA-seq analysis have tried to elucidate the transcriptional heterogeneity of stromal cells in ageing. A study on mouse dermis revealed that old fibroblasts exhibit transcriptional noise and partially lose their identity while adopting adipocyte characteristics (136). A GO analysis comparing young and aged dermal fibroblasts indicated an upregulation of genes related with the immune response, a characteristic of the ageing cells (24). These mechanisms are not skin exclusive, as old murine cardiac fibroblasts also upregulate pathways associated with inflammation or ECM regulation. Interestingly, old cardiac fibroblasts seem to upregulate genes related to osteogenesis, indicating their potential involvement in epicardial layer calcification observed in elderly individuals (137). These data suggest that ageing fibroblasts may lose their specialized identity leading to abnormal functions. However, fibroblasts may not converge toward a common ageing phenotype, suggesting that tissue specificity remains. Future single-cell transcriptomic studies on other tissues and diseases will help to elucidate a potentially shared aging phenotype.
5.0 Therapeutic perspectives of targeting fibroblasts in inflammation and ageing
Many efforts have been made to control the pathological phenotype of fibroblasts in different tissues in order to develop new approaches for the treatment of several diseases. In this section, we discuss the strategies that have been employed to develop a fibroblast targeted therapy.
Targeting cytokines, that are involved in the impaired functions of fibroblast, is considered of value for managing arthritis and other fibroblast-related conditions. Current therapeutic strategies that block fibroblast-activating signals such as anti- TNF therapy and anti- IL-6 receptor blocking antibodies, have shown clinical relevance for the treatment of chronic inflammatory conditions (138). Moreover, a range of strategies (such as monoclonal neutralising antibodies and small-molecule inhibitors) to treat cancer and fibrosis have been developed against TGF-β1; highly expressed and produced by fibroblasts (139, 140). However, targeting a single stimulus is not considered an efficient approach in blocking fibroblast activation with the presence of other stimulating factors. Moreover, this approach can prevent the activation of other cell types and induce side effects under certain conditions (3, 139). For this reason, intracellular proteins and signalling pathways, such as MAP and JAK (Janus kinase), have also attracted attention as targets to control fibroblast activation in RA regardless of stimulator type (141). The inhibition of transcriptional activators YAP/TAZ in fibroblasts is also considered as a promising strategy to explore for RA therapy. Indeed, the suppression of YAP/TAZ in SFs led to a reduction in their resistance to apoptosis, their inflammatory phenotype, proliferation, and ability to invade (142, 143). Further research highlights the use of YAP/TAZ inhibitors as a treatment for Crohn’s disease and for controlling intestinal fibrosis (144). In addition, recent findings have demonstrated the advantages of suppressing YAP/TAZ in fibroblasts by activating the dopamine receptor D1 (DRD1) in mouse models of lung and liver fibrosis (145).
These inhibitory approaches are now making progress with several preclinical studies aiming to design small inhibitor molecules with a high degree of selectivity. However, the risk of nonspecific and off-target effects requires deep consideration when developing these molecules. Targeting cytokine signal transduction may not be sufficient, especially in chronic conditions where multiple factors can promote the abnormal behaviour of fibroblasts (146). In this context, epigenetic dysregulation such as histone modification and miRNA expression are also implicated in the pathogenic phenotype of RA FLS (147, 148). Thus, manipulating the epigenetic mechanisms of fibroblasts could be beneficial to develop new drugs for RA and other pathologies. In fact, induction of protective miRNAs expression and inhibition of HDACs (histone deacetylase) as well as BET proteins have shown promising results in RA SFs and animal models of arthritis (149, 150). Beside epigenetic changes, miRNAs can target specific pathways involved in disease progression. For instance, miR-17 supress the IL-6 family autocrine loop by targeting JAK1/STAT3 pathway leading to anti-inflammatory and anti-erosion responses (151). Furthermore, inducing MiR-613 reduce the expression of the targeted transcript encoding for DKK1 in RA SFs which altered their proliferation, apoptotic resistance, and aggressiveness (152). On the other hand, targeting dysregulated miRNAs in CAFs might be promising for the development of novel anti- cancer drugs (153, 154). Several studies have also established the beneficial effect of inhibiting DNA methylation and HDACs in the suppression of the pro-fibrotic phenotype by inhibiting the myo-fibroblasts trans differentiation of hepatic stem cells (HSCs) (155), or by directly targeted HSC derived myofibroblasts and their apoptotic resistance (156, 157).
In addition to the epigenetic machinery, strategies that target the imbalanced metabolic pathways in both RA SFs and CAFs have been employed to reduce RA severity and inhibit tumour growth respectively (158, 159)). Interestingly, a recent study proposes a new strategy to control fibrosis and hypertrophic scars formation using an anti-glycolytic agent that regulates fibroblasts activity (160). Glycolysis inhibition has also been shown to reduce cardiac fibroblasts activation in-vitro and to control cardiac fibrosis in mice with myocardial infarction (69). Targeted inhibition of glycolysis in fibroblasts is therefore considered as a promising approach for the treatment of several fibroblasts -related diseases.
Cellular senescence is another feature of RA fibroblasts that contribute to the chronicity of arthritis by triggering a pro-inflammatory phenotype (161). However, a recent study introduced fibroblast senescence induction as a new way to control joint inflammation through the selective activation of G-protein coupled receptor, MC1R, that inhibits FLS proliferation and promotes the acquisition of a pro-reparative phenotype with anti-arthritic effects in K/BxN arthritis model (133). Furthermore, senescence inhibition has been shown to have anti-fibrotic effects on IPF fibroblasts. Additionally, some anti-fibrosis and anti-cancer drugs are known to alleviate the senescence of lung fibroblasts (162).
The development of single-cell RNA-sequencing technologies highlighted the presence of distinct subsets of fibroblasts associated with different pathological conditions. Therefore, specific elimination of aberrant fibroblasts based on a specific cell marker may inform new opportunities to investigate novel drug targets in multiple diseases such as fibrosis, cancer, and chronic inflammatory diseases (3) (163). As described earlier, CXCL10+CCL19+ immune-interacting and SPARC+COL3A1+ vascular-interacting fibroblasts were identified as two fibroblast subtypes shared between inflamed tissues of four inflammatory diseases involving lung, intestine, salivary gland, and synovium. These inflammatory clusters are therefore suggested as novel therapeutic targets which may provide new approaches to develop common therapies for multiple chronic inflammatory diseases (66).
Deletion of systemic and local FAPα+ fibroblasts lead to protection associated with reduced leukocyte infiltration, inflammatory mediators, and joint damage in mouse models of arthritis (60). FAP expression on myofibroblasts of mice with IPF permits to selectively target fibroblasts which are promoting tissue fibrosis. Indeed, treatment with FAP-targeted PI3K inhibitor decreased collagen production and deposition and increased mouse survival (164). In this same context, COL1+ fibroblasts have been recently identified as a good tool to test the effect of inhibiting genetically STAT 3 in mouse model of colitis-associated colorectal cancer, where it led to a decreased proliferation of tumor cells (165).
Similarly, targeting NOTCH3 receptor expressed on fibroblasts genetically or therapeutically promotes anti-arthritic effects in mice by significantly reducing joint swelling and bone erosion (61).
Cadherin 11 also represents a potential candidate to control the altered behaviour of fibroblasts in several pathologies such as arthritis and fibrosis. Different strategies such as antibody-mediated blockade and genetic deletion of this surface marker have been tested and shown their effectiveness (166, 167).
All together, these findings suggest that targeting fibroblast specific molecules could be a therapeutic avenue for RA and other chronic diseases that involve fibroblasts. However, specific depletion of pathogenic subset of fibroblasts based on their surface markers without side effects on tissue homeostasis and other organs is challenging (141, 168).
Conclusions and perspectives
From their embryonic origins in primary mesenchymal tissue to their specialized sub population observed in specific niches, fibroblasts have colonized and shaped the entire organism by connecting organs and cells together. Their contribution to ECM secretion is essential to maintain tissue function and create precise networks for cell communication. However, fibroblasts are not just structural cells. Single cell RNA sequencing reveals the transcriptomic heterogeneity of fibroblasts underpinning essential regulation in health, aging and diseases (Figure 3). The characterization of fibroblasts subsets and their positional identities in many organs highlight their role in supporting very specific functions within the tissue. However, those processes are perturbed during diseases such as rheumatoid arthritis, pulmonary fibrosis, or ulcerative colitis where specific fibroblast subsets can lead to disease progression. The identification of overlapping sub populations across inflammatory diseases reveals shared mechanism influencing fibroblasts’ behaviours and leading to aberrant inflammatory responses. Communication between endothelial and fibroblasts presents a critical role in the establishment of pathogenic fibroblasts subset which then participate to the recruitment and retention of immune cells, impairing resolution of inflammation. The discovery of common activation mechanisms driving phenotypic changes in fibroblasts across diseases and organs might suggest a plasticity of this cell type. Despite the proven existence of a universal “reservoir” fibroblast population (12), its biological relevance might be difficult to study due to plasticity. However, the potential or limitation of this plasticity needs to be further described particularly when developing induced pluripotent stem cells from fibroblasts (169, 170). Interestingly, recent studies on ageing fibroblasts report that they adopt other cell type traits such as adipocytes in the dermis, or osteoblasts in cardiac tissue. In contrast, fibroblast-driven inflammation is common during disease or ageing. While pathogenic sub populations emerge after repeated inflammatory stimuli, the ageing processes also induce changing in the aged fibroblasts leading towards inflammation. However, those changes do not increase heterogeneity in distinct populations but rather in stochastic transcriptional noise. One can therefore suggest that tissue ageing acts as a potent environmental cue driving fibroblasts to an increasingly common inflammatory phenotype and loss of their tissue distinct expression patterns. To address this, the transcriptomic profiles and functional characteristics of fibroblasts need to be further investigated across a range of tissues in both health and disease. Finally, the identification of transcription factors regulating the fate of fibroblasts toward a destructive or pro fibrotic phenotype remains unclear. The recent evidence of ETS1 as a transcription factor inducing ECM degradation programme in SFs leads to a better understanding the opposing role of fibroblasts in destructive/inflammatory diseases compared to pro fibrotic pathologies (171). Interestingly, the transcription factor PU.1 has been identified as a regulator of pro fibrotic phenotype in fibroblasts (172). Moreover, ETS1 and PU.1 are mutually exclusive, and their expression does not overlap in fibroblasts in both IPF and arthritic synovium (171). This highlights an important fate decision mechanism in fibroblasts by driving a pro destructive or pro fibrotic phenotype. Nonetheless, the mechanisms that control those fate decision pathway remains unexplored.
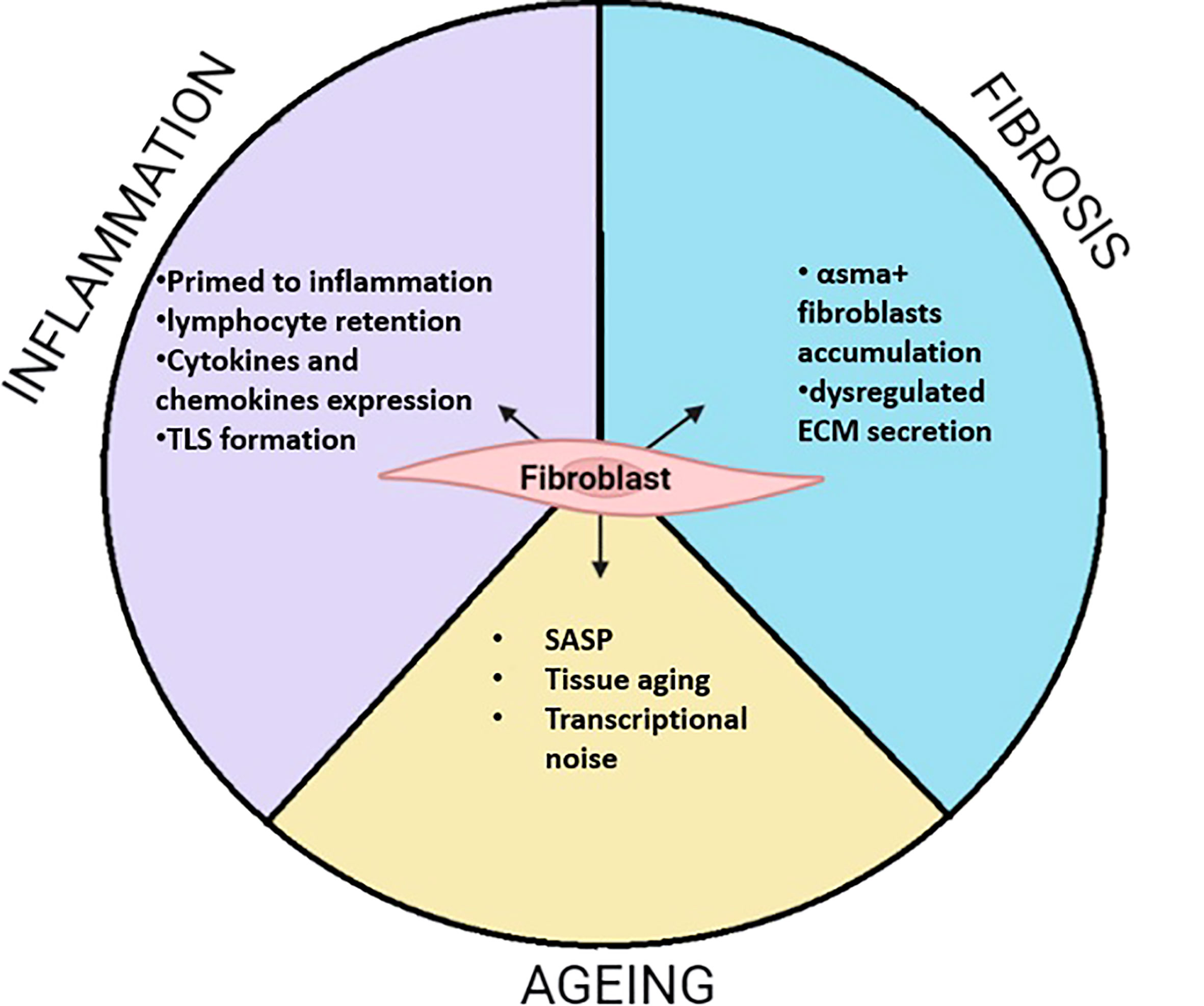
Figure 3 Summarized function of fibroblasts during inflammation, fibrosis and Aging. During inflammation fibroblasts adopt activate phenotypes. This often leads to the priming of fibroblasts which remains potent to inflammatory stimuli. This is characterized by the secretion of pro-inflammatory cytokines and chemokines. Those properties can lead to local accumulation of lymphocytes where fibroblast support their survival by tertiary lymphoid structure (TLS). In fibrosis, my fibroblasts are accumulating and secreting abnormal ECM modules leading to matrix stiffness enabling proper network communication. During aging, fibroblasts highly participate to low grade inflammation call inflammaging through the release of SASP containing pro inflammatory cytokines. Evidence shows that fibroblasts transcriptome changes toward a less define transcriptomic identity called transcriptional noise. While other old fibroblasts, adopt other cell type characteristics such as osteoclasts in cardiac fibroblast or adipocyte in the skin.
Author contributions
VG and MK contributed equally. All authors contributed to the article and approved the submitted version.
Funding
This work was Versus Arthritis grants 22617 to GA, Medical Research Council Versus Arthritis Centre for Musculoskeletal Ageing Research (CMAR) to VG.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Virchow RLK. Die cellularpathologie in ihrer begründung auf physiologische und pathologische gewebelehre: A. hirshcwald. (Berlin: Verlag von August Hirschwald) (1859).
2. Ziegler E. General pathology: Or, the science of the causes, nature and course of the pathological disturbances which occur in the living subject: William wood and company. (1900).
3. Davidson S, Coles M, Thomas T, Kollias G, Ludewig B, Turley S, et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat Rev Immunol (New York: William Wood and Company) (2021) 21(11):704–17. doi: 10.1038/s41577-021-00540-z
4. Denu RA, Nemcek S, Bloom DD, Goodrich AD, Kim J, Mosher DF, et al. Fibroblasts and mesenchymal Stromal/Stem cells are phenotypically indistinguishable. Acta Haematol (2016) 136(2):85–97. doi: 10.1159/000445096
5. LeBleu VS, Neilson EG. Origin and functional heterogeneity of fibroblasts. FASEB J (2020) 34(3):3519–36. doi: 10.1096/fj.201903188R
6. Frank-Bertoncelj M, Trenkmann M, Klein K, Karouzakis E, Rehrauer H, Bratus A, et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Commun (2017) 8:14852. doi: 10.1038/ncomms14852
7. Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. (2002) 99(20):12877–82. doi: 10.1073/pnas.162488599
8. Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature (2020) 588(7838):466–72. doi: 10.1038/s41586-020-2797-4
9. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature (2020) 587(7835):619–25. doi: 10.1038/s41586-020-2922-4
10. Ascensión AM, Fuertes-Álvarez S, Ibañez-Solé O, Izeta A, Araúzo-Bravo MJ. Human dermal fibroblast subpopulations are conserved across single-cell RNA sequencing studies. J Invest Dermatol (2021) 141(7):1735–44.e35. doi: 10.1016/j.jid.2020.11.028
11. Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. (2018) 175(2):372–86.e17. doi: 10.1016/j.cell.2018.08.067
12. Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-tissue organization of the fibroblast lineage. Nature (2021) 593(7860):575–9. doi: 10.1038/s41586-021-03549-5
13. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol (2010) 225(3):631–7. doi: 10.1002/jcp.22322
14. Koliaraki V, Prados A, Armaka M, Kollias G. The mesenchymal context in inflammation, immunity and cancer. Nat Immunol (2020) 21(9):974–82. doi: 10.1038/s41590-020-0741-2
15. Lynch MD, Watt FM. Fibroblast heterogeneity: Implications for human disease. J Clin Invest. (2018) 128(1):26–35. doi: 10.1172/JCI93555
16. Kugler MC, Loomis CA, Zhao Z, Cushman JC, Liu L, Munger JS. Sonic hedgehog signaling regulates myofibroblast function during alveolar septum formation in murine postnatal lung. Am J Respir Cell Mol Biol (2017) 57(3):280–93. doi: 10.1165/rcmb.2016-0268OC
17. Yoshitomi H. Regulation of immune responses and chronic inflammation by fibroblast-like synoviocytes. Front Immunol (2019) 10:1395. doi: 10.3389/fimmu.2019.01395
18. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. (2011) 108(52):E1475–83. doi: 10.1073/pnas.1117988108
19. Janson DG, Saintigny G, van Adrichem A, Mahe C, El Ghalbzouri A. Different gene expression patterns in human papillary and reticular fibroblasts. J Invest Dermatol (2012) 132(11):2565–72. doi: 10.1038/jid.2012.192
20. He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol (2020) 145(6):1615–28. doi: 10.1016/j.jaci.2020.01.042
21. Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell (2019) 178(3):714–30 e22. doi: 10.1016/j.cell.2019.06.029
22. Williams DW, Greenwell-Wild T, Brenchley L, Dutzan N, Overmiller A, Sawaya AP, et al. Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell. (2021) 184(15):4090–104 e15. doi: 10.1016/j.cell.2021.05.013
23. Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, et al. Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc Natl Acad Sci U S A. (2019) 116(27):13490–7. doi: 10.1073/pnas.1905301116
24. Sole-Boldo L, Raddatz G, Schutz S, Mallm JP, Rippe K, Lonsdorf AS, et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun Biol (2020) 3(1):188. doi: 10.1038/s42003-020-0922-4
25. Tabib T, Morse C, Wang T, Chen W, Lafyatis R. SFRP2/DPP4 and FMO1/LSP1 define major fibroblast populations in human skin. J Invest Dermatol (2018) 138(4):802–10. doi: 10.1016/j.jid.2017.09.045
26. Philippeos C, Telerman SB, Oules B, Pisco AO, Shaw TJ, Elgueta R, et al. Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J Invest Dermatol (2018) 138(4):811–25. doi: 10.1016/j.jid.2018.01.016
27. Vorstandlechner V, Laggner M, Kalinina P, Haslik W, Radtke C, Shaw L, et al. Deciphering the functional heterogeneity of skin fibroblasts using single-cell RNA sequencing. FASEB J (2020) 34(3):3677–92. doi: 10.1096/fj.201902001RR
28. Gaffen SL, Moutsopoulos NM. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci Immunol (2020) 5(43). doi: 10.1126/sciimmunol.aau4594
29. Moutsopoulos NM, Konkel JE. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol (2018) 39(4):276–87. doi: 10.1016/j.it.2017.08.005
30. Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. (2021) 184(15):3852–72. doi: 10.1016/j.cell.2021.06.024
31. Forte E, Skelly DA, Chen M, Daigle S, Morelli KA, Hon O, et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep (2020) 30(9):3149–63 e6. doi: 10.1016/j.celrep.2020.02.008
32. Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife (2019) 8. doi: 10.7554/eLife.43882
33. Li C, Li M, Li S, Xing Y, Yang CY, Li A, et al. Progenitors of secondary crest myofibroblasts are developmentally committed in early lung mesoderm. Stem Cells (2015) 33(3):999–1012. doi: 10.1002/stem.1911
34. Arora R, Metzger RJ, Papaioannou VE. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PloS Genet (2012) 8(8):e1002866. doi: 10.1371/journal.pgen.1002866
35. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv (2020) 6(28). doi: 10.1126/sciadv.aba1972
36. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol (2019) 20(7):928–42. doi: 10.1038/s41590-019-0378-1
37. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell wnt signaling niches maintain stemness of alveolar type 2 cells. Science (2018) 359(6380):1118–23. doi: 10.1126/science.aam6603
38. Rougier F, Dupuis F, Denizot Y. Human bone marrow fibroblasts–an overview of their characterization, proliferation and inflammatory mediator production. Hematol Cell Ther (1996) 38(3):241–6. doi: 10.1007/s00282-996-0241-3
39. Marsh LJ, Kemble S, Reis Nisa P, Singh R, Croft AP. Fibroblast pathology in inflammatory joint disease. Immunol Rev (2021) 302(1):163–83. doi: 10.1111/imr.12986
40. Buckley CD. Fibroblast cells reveal their ancestry. Nature (2021) 593(7860):511–2. doi: 10.1038/d41586-021-01204-7
41. Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, et al. A stromal address code defined by fibroblasts. Trends Immunol (2005) 26(3):150–6. doi: 10.1016/j.it.2004.11.014
42. Ebert LM, Schaerli P, Moser B. Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol (2005) 42(7):799–809. doi: 10.1016/j.molimm.2004.06.040
43. McGettrick HM, Ward LS, Rainger GE, Nash GB. Mesenchymal stromal cells as active regulators of lymphocyte recruitment to blood vascular endothelial cells. Methods Mol Biol (2017) 1591:121–42. doi: 10.1007/978-1-4939-6931-9_9
44. McGettrick HM, Buckley CD, Filer A, Rainger GE, Nash GB. Stromal cells differentially regulate neutrophil and lymphocyte recruitment through the endothelium. Immunology (2010) 131(3):357–70. doi: 10.1111/j.1365-2567.2010.03307.x
45. Chang JE, Turley SJ. Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol (2015) 36(1):30–9. doi: 10.1016/j.it.2014.11.003
46. Acton SE, Onder L, Novkovic M, Martinez VG, Ludewig B. Communication, construction, and fluid control: Lymphoid organ fibroblastic reticular cell and conduit networks. Trends Immunol (2021) 42(9):782–94. doi: 10.1016/j.it.2021.07.003
47. Lutge M, Pikor NB, Ludewig B. Differentiation and activation of fibroblastic reticular cells. Immunol Rev (2021) 302(1):32–46. doi: 10.1111/imr.12981
48. Cheng HW, Onder L, Novkovic M, Soneson C, Lutge M, Pikor N, et al. Origin and differentiation trajectories of fibroblastic reticular cells in the splenic white pulp. Nat Commun (2019) 10(1):1739. doi: 10.1038/s41467-019-09728-3
49. Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, Lercher A, et al. Structural cells are key regulators of organ-specific immune responses. Nature. (2020) 583(7815):296–302. doi: 10.1038/s41586-020-2424-4
50. Fullerton JN, Gilroy DW. Resolution of inflammation: A new therapeutic frontier. Nat Rev Drug Discovery (2016) 15(8):551–67. doi: 10.1038/nrd.2016.39
51. Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, et al. Tumor necrosis factor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol (2013) 65(4):928–38. doi: 10.1002/art.37853
52. Crowley T, O’Neil JD, Adams H, Thomas AM, Filer A, Buckley CD, et al. Priming in response to pro-inflammatory cytokines is a feature of adult synovial but not dermal fibroblasts. Arthritis Res Ther (2017) 19(1):35. doi: 10.1186/s13075-017-1248-6
53. Friscic J, Bottcher M, Reinwald C, Bruns H, Wirth B, Popp SJ, et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity (2021) 54(5):1002–21 e10. doi: 10.1016/j.immuni.2021.03.003
54. Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol (2000) 165(6):3423–9. doi: 10.4049/jimmunol.165.6.3423
55. Bradfield PF, Amft N, Vernon-Wilson E, Exley AE, Parsonage G, Rainger GE, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis Rheumatol (2003) 48(9):2472–82. doi: 10.1002/art.11219
56. Burman A, Haworth O, Hardie DL, Amft EN, Siewert C, Jackson DG, et al. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J Immunol (2005) 174(3):1693–700. doi: 10.4049/jimmunol.174.3.1693
57. Salmon M, Scheel-Toellner D, Huissoon AP, Pilling D, Shamsadeen N, Hyde H, et al. Inhibition of T cell apoptosis in the rheumatoid synovium. J Clin Invest (1997) 99(3):439–46. doi: 10.1172/JCI119178
58. Harada S, Yamamura M, Okamoto H, Morita Y, Kawashima M, Aita T, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheumatol (1999) 42(7):1508–16. doi: 10.1002/1529-0131(199907)42:7<1508::AID-ANR26>3.0.CO;2-L
59. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: A novel target in chronic inflammation. Front Immunol (2016) 7:477. doi: 10.3389/fimmu.2016.00477
60. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature (2019) 570(7760):246–51. doi: 10.1038/s41586-019-1263-7
61. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature (2020) 582(7811):259–64. doi: 10.1038/s41586-020-2222-z
62. Kiener HP, Watts GF, Cui Y, Wright J, Thornhill TS, Skold M, et al. Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ culture. Arthritis Rheumatol (2010) 62(3):742–52. doi: 10.1002/art.27285
63. Kramer N, Schmollerl J, Unger C, Nivarthi H, Rudisch A, Unterleuthner D, et al. Autocrine WNT2 signaling in fibroblasts promotes colorectal cancer progression. Oncogene (2017) 36(39):5460–72. doi: 10.1038/onc.2017.144
64. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin m drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med (2017) 23(5):579–89. doi: 10.1038/nm.4307
65. Caetano AJ, Yianni V, Volponi A, Booth V, D’Agostino EM, Sharpe P. Defining human mesenchymal and epithelial heterogeneity in response to oral inflammatory disease. Elife. (2021) 10. doi: 10.7554/eLife.62810
66. Korsunsky I, Wei K, Pohin M, Kim EY, Barone F, Major T, et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med (N Y) (2022) 3(7):481–518 e14. doi: 10.1016/j.medj.2022.05.002
67. He S, Wang LH, Liu Y, Li YQ, Chen HT, Xu JH, et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol (2020) 21(1):294. doi: 10.1186/s13059-020-02210-0
68. Tabula Sapiens C, Jones RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR, et al. The tabula sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science (2022) 376(6594):eabl4896. doi: 10.1126/science.abl4896
69. Chen ZT, Gao QY, Wu MX, Wang M, Sun RL, Jiang Y, et al. Glycolysis inhibition alleviates cardiac fibrosis after myocardial infarction by suppressing cardiac fibroblast activation. Front Cardiovasc Med (2021) 8:701745. doi: 10.3389/fcvm.2021.701745
70. Ueno A, Yamamura M, Iwahashi M, Okamoto A, Aita T, Ogawa N, et al. The production of CXCR3-agonistic chemokines by synovial fibroblasts from patients with rheumatoid arthritis. Rheumatol Int (2005) 25(5):361–7. doi: 10.1007/s00296-004-0449-x
71. Yao L, Zhou Y, Li J, Wickens L, Conforti F, Rattu A, et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J Biol Chem (2021) 297(3):101096. doi: 10.1016/j.jbc.2021.101096
72. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res (1961) 25(3):585–621. doi: 10.1016/0014-4827(61)90192-6
73. Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell (2013) 155(5):1104–18. doi: 10.1016/j.cell.2013.10.019
74. Romer A, Parsons TS. The vertebrate body. Philadelphia. PA: Holt-Saunders International (1977) p. 555–56.
75. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell (2013) 155(5):1119–30. doi: 10.1016/j.cell.2013.10.041
76. Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution (1957) 11(4):398–411. doi: 10.2307/2406060
77. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol (2011) 192(4):547–56. doi: 10.1083/jcb.201009094
78. Giaimo S, d’Adda di Fagagna F. Is cellular senescence an example of antagonistic pleiotropy? Aging Cell (2012) 11(3):378–83. doi: 10.1111/j.1474-9726.2012.00807.x
79. Muñoz-Espín D, Serrano M. Cellular senescence: From physiology to pathology. Nat Rev Mol Cell Biol (2014) 15(7):482–96. doi: 10.1038/nrm3823
80. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell (2014) 31(6):722–33. doi: 10.1016/j.devcel.2014.11.012
81. Darby IA, Laverdet B, Bonté F, Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol (2014) 7:301–11. doi: 10.2147/CCID.S50046
82. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol (2010) 12(7):676–85. doi: 10.1038/ncb2070
83. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell (2008) 134(4):657–67. doi: 10.1016/j.cell.2008.06.049
84. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: Aging, cancer, and injury. Physiol Rev (2019) 99(2):1047–78. doi: 10.1152/physrev.00020.2018
85. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88(5):593–602. doi: 10.1016/S0092-8674(00)81902-9
86. Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol (2008) 10(7):825–36. doi: 10.1038/ncb1744
87. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature (2011) 479(7372):232–6. doi: 10.1038/nature10600
88. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: A key player in cancer development. J Hematol Oncol (2020) 13(1):151. doi: 10.1186/s13045-020-00986-z
89. Campisi J, d’Adda di Fagagna F. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
90. van Deursen JM. The role of senescent cells in ageing. Nature (2014) 509(7501):439–46. doi: 10.1038/nature13193
91. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
92. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell (2007) 130(2):223–33. doi: 10.1016/j.cell.2007.07.003
93. Pawlikowski JS, Adams PD, Nelson DM. Senescence at a glance. J Cell Sci (2013) 126(Pt 18):4061–7. doi: 10.1242/jcs.109728
94. Demaria M, Desprez PY, Campisi J, Velarde MC. Cell autonomous and non-autonomous effects of senescent cells in the skin. J Invest Dermatol (2015) 135(7):1722–6. doi: 10.1038/jid.2015.108
95. Toutfaire M, Bauwens E, Debacq-Chainiaux F. The impact of cellular senescence in skin ageing: A notion of mosaic and therapeutic strategies. Biochem Pharmacol (2017) 142:1–12. doi: 10.1016/j.bcp.2017.04.011
96. Wang AS, Dreesen O. Biomarkers of cellular senescence and skin aging. Front Genet (2018) 9:247. doi: 10.3389/fgene.2018.00247
97. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature (1990) 345(6274):458–60. doi: 10.1038/345458a0
98. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science (1998) 279(5349):349–52. doi: 10.1126/science.279.5349.349
99. Berneburg M, Gattermann N, Stege H, Grewe M, Vogelsang K, Ruzicka T, et al. Chronically ultraviolet-exposed human skin shows a higher mutation frequency of mitochondrial DNA as compared to unexposed skin and the hematopoietic system. Photochem Photobiology (1997) 66(2):271–5. doi: 10.1111/j.1751-1097.1997.tb08654.x
100. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol Ther (2018) 183:34–49. doi: 10.1016/j.pharmthera.2017.10.005
101. Birch-machin MA, Tindall M, Turner R, Haldane F, Rees JL. Mitochondrial DNA deletions in human skin reflect photo- rather than chronologic aging. J Invest Dermatol (1998) 110(2):149–52. doi: 10.1046/j.1523-1747.1998.00099.x
102. Passos JF, Saretzki G, von Zglinicki T. DNA Damage in telomeres and mitochondria during cellular senescence: Is there a connection? Nucleic Acids Res (2007) 35(22):7505–13. doi: 10.1093/nar/gkm893
103. d’Adda di Fagagna F. Living on a break: Cellular senescence as a DNA-damage response. Nat Rev Cancer (2008) 8(7):512–22. doi: 10.1038/nrc2440
104. Hoeijmakers JH. DNA Damage, aging, and cancer. N Engl J Med (2009) 361(15):1475–85. doi: 10.1056/NEJMra0804615
105. Chondrogianni N, Gonos ES. Proteasome function determines cellular homeostasis and the rate of aging. In: Tavernarakis N, editor. Protein metabolism and homeostasis in aging. Boston, MA: Springer US (2010). p. 38–46.
106. Norsgaard H, Clark BFC, Rattan SIS. Distinction between differentiation and senescence and the absence of increased apoptosis in human keratinocytes undergoing cellular aging in vitro. Exp Gerontology (1996) 31(5):563–70. doi: 10.1016/0531-5565(96)00011-3
107. Tigges J, Krutmann J, Fritsche E, Haendeler J, Schaal H, Fischer JW, et al. The hallmarks of fibroblast ageing. Mech Ageing Dev (2014) 138:26–44. doi: 10.1016/j.mad.2014.03.004
108. da Silva PFL, Schumacher B. DNA Damage responses in ageing. Open Biol (2019) 9(11):190168. doi: 10.1098/rsob.190168
109. Krutmann J, Schroeder P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J Investig Dermatol Symp Proc (2009) 14(1):44–9. doi: 10.1038/jidsymp.2009.1
110. Meyer P, Maity P, Burkovski A, Schwab J, Müssel C, Singh K, et al. A model of the onset of the senescence associated secretory phenotype after DNA damage induced senescence. PloS Comput Biol (2017) 13(12):e1005741. doi: 10.1371/journal.pcbi.1005741
111. Treiber N, Maity P, Singh K, Kohn M, Keist AF, Ferchiu F, et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell (2011) 10(2):239–54. doi: 10.1111/j.1474-9726.2010.00658.x
112. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature (2016) 530(7589):184–9. doi: 10.1038/nature16932
113. d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature (2003) 426(6963):194–8. doi: 10.1038/nature02118
114. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci (1995) 92(20):9363–7. doi: 10.1073/pnas.92.20.9363
115. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol (2012) 14(4):355–65. doi: 10.1038/ncb2466
116. Takai H, Smogorzewska A, de Lange T. DNA Damage foci at dysfunctional telomeres. Curr Biol (2003) 13(17):1549–56. doi: 10.1016/S0960-9822(03)00542-6
117. Lee YI, Choi S, Roh WS, Lee JH, Kim TG. Cellular senescence and inflammaging in the skin microenvironment. Int J Mol Sci (2021) 22(8). doi: 10.3390/ijms22083849
118. Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, et al. An update on inflamm-aging: Mechanisms, prevention, and treatment. J Immunol Res (2016) 2016:8426874. doi: 10.1155/2016/8426874
119. Calhoun C, Shivshankar P, Saker M, Sloane LB, Livi CB, Sharp ZD, et al. Senescent cells contribute to the physiological remodeling of aged lungs. J Gerontol A Biol Sci Med Sci (2016) 71(2):153–60. doi: 10.1093/gerona/glu241
120. Gey C, Seeger K. Metabolic changes during cellular senescence investigated by proton NMR-spectroscopy. Mech Ageing Dev (2013) 134(3-4):130–8. doi: 10.1016/j.mad.2013.02.002
121. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
122. Pérez-Mancera PA, Young AR, Narita M. Inside and out: The activities of senescence in cancer. Nat Rev Cancer (2014) 14(8):547–58. doi: 10.1038/nrc3773
123. Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J Cell Sci (2005) 118 3):485–96. doi: 10.1242/jcs.01635
124. Ghosh K, Capell BC. The senescence-associated secretory phenotype: Critical effector in skin cancer and aging. J Invest Dermatol (2016) 136(11):2133–9. doi: 10.1016/j.jid.2016.06.621
125. Gruber F, Kremslehner C, Eckhart L, Tschachler E. Cell aging and cellular senescence in skin aging - recent advances in fibroblast and keratinocyte biology. Exp Gerontol (2020) 130:110780. doi: 10.1016/j.exger.2019.110780
126. Davan-Wetton CSA, Pessolano E, Perretti M, Montero-Melendez T. Senescence under appraisal: Hopes and challenges revisited. Cell Mol Life Sci (2021) 78(7):3333–54. doi: 10.1007/s00018-020-03746-x
127. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J Clin Invest. (2013) 123(3):966–72. doi: 10.1172/JCI64098
128. Hiebert P, Wietecha MS, Cangkrama M, Haertel E, Mavrogonatou E, Stumpe M, et al. Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev Cell (2018) 46(2):145–61.e10. doi: 10.1016/j.devcel.2018.06.012
129. Wlaschek M, Maity P, Makrantonaki E, Scharffetter-Kochanek K. Connective tissue and fibroblast senescence in skin aging. J Invest Dermatol (2021) 141(4s):985–92. doi: 10.1016/j.jid.2020.11.010
130. Singh K, Maity P, Krug L, Meyer P, Treiber N, Lucas T, et al. Superoxide anion radicals induce IGF-1 resistance through concomitant activation of PTP1B and PTEN. EMBO Mol Med (2015) 7(1):59–77. doi: 10.15252/emmm.201404082
131. Anisimov VN, Bartke A. The key role of growth hormone–insulin–IGF-1 signaling in aging and cancer. Crit Rev Oncology/Hematology (2013) 87(3):201–23. doi: 10.1016/j.critrevonc.2013.01.005
132. Gallagher Emily J, LeRoith D. Is growth hormone Resistance/IGF-1 reduction good for you? Cell Metab (2011) 13(4):355–6. doi: 10.1016/j.cmet.2011.03.003
133. Montero-Melendez T, Nagano A, Chelala C, Filer A, Buckley CD, Perretti M. Therapeutic senescence via GPCR activation in synovial fibroblasts facilitates resolution of arthritis. Nat Commun (2020) 11(1):745. doi: 10.1038/s41467-020-14421-x
134. Khaltourina D, Matveyev Y, Alekseev A, Cortese F, Iovita A. Aging fits the disease criteria of the international classification of diseases. Mech Ageing Dev (2020) 189:111230. doi: 10.1016/j.mad.2020.111230
135. Wagner KH, Cameron-Smith D, Wessner B, Franzke B. Biomarkers of aging: From function to molecular biology. Nutrients (2016) 8(6). doi: 10.3390/nu8060338
136. Salzer MC, Lafzi A, Berenguer-Llergo A, Youssif C, Castellanos A, Solanas G, et al. Identity noise and adipogenic traits characterize dermal fibroblast aging. Cell (2018) 175(6):1575–90 e22. doi: 10.1016/j.cell.2018.10.012
137. Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight (2019) 4(22). doi: 10.1172/jci.insight.131092
138. Wei K, Nguyen HN, Brenner MB. Fibroblast pathology in inflammatory diseases. J Clin Invest (2021) 131(20). doi: 10.1172/JCI149538
139. Jenkins BH, Buckingham JF, Hanley CJ, Thomas GJ. Targeting cancer-associated fibroblasts: Challenges, opportunities and future directions. Pharmacol Ther (2022) 240:108231. doi: 10.1016/j.pharmthera.2022.108231
140. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol Cancer (2022) 21(1):104. doi: 10.1186/s12943-022-01569-x
141. Jose Alcaraz M. New potential therapeutic approaches targeting synovial fibroblasts in rheumatoid arthritis. Biochem Pharmacol (2021) 194:114815. doi: 10.1016/j.bcp.2021.114815
142. Zhou W, Shen Q, Wang H, Yang J, Zhang C, Deng Z, et al. Knockdown of YAP/TAZ inhibits the migration and invasion of fibroblast synovial cells in rheumatoid arthritis by regulating autophagy. J Immunol Res (2020) 2020:9510594. doi: 10.1155/2020/9510594
143. Caire R, Audoux E, Courbon G, Michaud E, Petit C, Dalix E, et al. YAP/TAZ: Key players for rheumatoid arthritis severity by driving fibroblast like synoviocytes phenotype and fibro-inflammatory response. Front Immunol (2021) 12:791907. doi: 10.3389/fimmu.2021.791907
144. Ou W, Xu W, Liu F, Guo Y, Huang Z, Feng T, et al. Increased expression of yes-associated protein/YAP and transcriptional coactivator with PDZ-binding motif/TAZ activates intestinal fibroblasts to promote intestinal obstruction in crohn’s disease. EBioMedicine (2021) 69:103452. doi: 10.1016/j.ebiom.2021.103452
145. Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N, et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med (2019) 11(516). doi: 10.1126/scitranslmed.aau6296
146. Sandhu G, Thelma BK. New druggable targets for rheumatoid arthritis based on insights from synovial biology. Front Immunol (2022) 13:834247. doi: 10.3389/fimmu.2022.834247
147. Klein K, Gay S. Epigenetics in rheumatoid arthritis. Curr Opin Rheumatol (2015) 27(1):76–82. doi: 10.1097/BOR.0000000000000128
148. Hammaker D, Firestein GS. Epigenetics of inflammatory arthritis. Curr Opin Rheumatol (2018) 30(2):188–96. doi: 10.1097/BOR.0000000000000471
149. Park JK, Shon S, Yoo HJ, Suh DH, Bae D, Shin J, et al. Inhibition of histone deacetylase 6 suppresses inflammatory responses and invasiveness of fibroblast-like-synoviocytes in inflammatory arthritis. Arthritis Res Ther (2021) 23(1):177. doi: 10.1186/s13075-021-02561-4
150. Krishna V, Yin X, Song Q, Walsh A, Pocalyko D, Bachman K, et al. Integration of the transcriptome and genome-wide landscape of BRD2 and BRD4 binding motifs identifies key superenhancer genes and reveals the mechanism of bet inhibitor action in rheumatoid arthritis synovial fibroblasts. J Immunol (2021) 206(2):422–31. doi: 10.4049/jimmunol.2000286
151. Najm A, Masson FM, Preuss P, Georges S, Ory B, Quillard T, et al. MicroRNA-17-5p reduces inflammation and bone erosions in mice with collagen-induced arthritis and directly targets the JAK/STAT pathway in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol (2020) 72(12):2030–9. doi: 10.1002/art.41441
152. Liu L, Zuo Y, Xu Y, Zhang Z, Li Y, Pang J. MiR-613 inhibits proliferation and invasion and induces apoptosis of rheumatoid arthritis synovial fibroblasts by direct down-regulation of DKK1. Cell Mol Biol Lett (2019) 24:8. doi: 10.1186/s11658-018-0130-0
153. Chatterjee A, Jana S, Chatterjee S, Wastall LM, Mandal G, Nargis N, et al. MicroRNA-222 reprogrammed cancer-associated fibroblasts enhance growth and metastasis of breast cancer. Br J Cancer (2019) 121(8):679–89. doi: 10.1038/s41416-019-0566-7
154. Wang R, Sun Y, Yu W, Yan Y, Qiao M, Jiang R, et al. Downregulation of miRNA-214 in cancer-associated fibroblasts contributes to migration and invasion of gastric cancer cells through targeting FGF9 and inducing EMT. J Exp Clin Cancer Res (2019) 38(1):20. doi: 10.1186/s13046-018-0995-9
155. Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ (2007) 14(2):275–85. doi: 10.1038/sj.cdd.4401979
156. Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J (2014) 43(5):1448–58. doi: 10.1183/09031936.00095113
157. Zeybel M, Luli S, Sabater L, Hardy T, Oakley F, Leslie J, et al. A proof-of-Concept for epigenetic therapy of tissue fibrosis: Inhibition of liver fibrosis progression by 3-deazaneplanocin a. Mol Ther (2017) 25(1):218–31. doi: 10.1016/j.ymthe.2016.10.004
158. Aghakhani S, Zerrouk N, Niarakis A. Metabolic reprogramming of fibroblasts as therapeutic target in rheumatoid arthritis and cancer: Deciphering key mechanisms using computational systems biology approaches. Cancers (Basel) (2020) 13(1). doi: 10.3390/cancers13010035
159. Chu CQ. Highlights of strategies targeting fibroblasts for novel therapies for rheumatoid arthritis. Front Med (Lausanne) (2022) 9:846300. doi: 10.3389/fmed.2022.846300
160. Meng X, Yu Z, Xu W, Chai J, Fang S, Min P, et al. Control of fibrosis and hypertrophic scar formation via glycolysis regulation with IR780. Burns Trauma (2022) 10:tkac015. doi: 10.1093/burnst/tkac015
161. Del Rey MJ, Valin A, Usategui A, Ergueta S, Martin E, Municio C, et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun Ageing (2019) 16:29. doi: 10.1186/s12979-019-0169-4
162. Lin Y, Xu Z. Fibroblast senescence in idiopathic pulmonary fibrosis. Front Cell Dev Biol (2020) 8:593283. doi: 10.3389/fcell.2020.593283
163. Deng CC, Hu YF, Zhu DH, Cheng Q, Gu JJ, Feng QL, et al. Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases. Nat Commun (2021) 12(1):3709. doi: 10.1038/s41467-021-24110-y
164. Hettiarachchi SU, Li YH, Roy J, Zhang F, Puchulu-Campanella E, Lindeman SD, et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci Transl Med (2020) 12(567). doi: 10.1126/scitranslmed.aay3724
165. Heichler C, Schmied A, Enderle K, Scheibe K, Murawska M, Schmid B, et al. Targeting STAT3 signaling in COL1+ fibroblasts controls colitis-associated cancer in mice. Cancers (Basel) (2022) 14(6). doi: 10.3390/cancers14061472
166. Sfikakis PP, Vlachogiannis NI, Christopoulos PF. Cadherin-11 as a therapeutic target in chronic, inflammatory rheumatic diseases. Clin Immunol (2017) 176:107–13. doi: 10.1016/j.clim.2017.01.008
167. Pedroza M, Welschhans RL, Agarwal SK. Targeting of cadherin-11 decreases skin fibrosis in the tight skin-1 mouse model. PloS One (2017) 12(11):e0187109. doi: 10.1371/journal.pone.0187109
168. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol (2020) 16(6):316–33. doi: 10.1038/s41584-020-0413-5
169. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
170. Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature (2019) 574(7779):553–8. doi: 10.1038/s41586-019-1658-5
171. Yan M, Komatsu N, Muro R, Huynh NC, Tomofuji Y, Okada Y, et al. ETS1 governs pathological tissue-remodeling programs in disease-associated fibroblasts. Nat Immunol (2022) 23(9):1330–41. doi: 10.1038/s41590-022-01285-0
Keywords: fibroblast, health, diseases, ageing, inflammation
Citation: Gauthier V, Kyriazi M, Nefla M, Pucino V, Raza K, Buckley CD and Alsaleh G (2023) Fibroblast heterogeneity: Keystone of tissue homeostasis and pathology in inflammation and ageing. Front. Immunol. 14:1137659. doi: 10.3389/fimmu.2023.1137659
Received: 04 January 2023; Accepted: 14 February 2023;
Published: 28 February 2023.
Edited by:
Mojca Frank Bertoncelj, BioMed X Institute, GermanyReviewed by:
Aleksander M. Grabiec, Jagiellonian University, PolandMarkus H. Hoffmann, University of Lübeck, Germany
Andreas Ramming, University of Erlangen Nuremberg, Germany
Copyright © 2023 Gauthier, Kyriazi, Nefla, Pucino, Raza, Buckley and Alsaleh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher D. Buckley, Y2hyaXN0b3BoZXIuYnVja2xleUBrZW5uZWR5Lm94LmFjLnVr; Ghada Alsaleh, Z2hhZGEuYWxzYWxlaEBuZG9ybXMub3guYWMudWs=
†These authors have contributed equally to this work