
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 February 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1131588
This article is part of the Research Topic Community Series in Hepatic Immune Response underlying Liver Cirrhosis and Portal Hypertension, volume II View all 14 articles
 Nana Zhang1,2,3†
Nana Zhang1,2,3† Huimin Yao1,2,3†
Huimin Yao1,2,3† Zhixuan Zhang1,2,3†
Zhixuan Zhang1,2,3† Zhuoqun Li2,3,4
Zhuoqun Li2,3,4 Xue Chen2,3
Xue Chen2,3 Yan Zhao1,2,3
Yan Zhao1,2,3 Ran Ju1,2,3
Ran Ju1,2,3 Jiayi He1,2,3
Jiayi He1,2,3 Heli Pan1,2,3
Heli Pan1,2,3 Xiaoli Liu1,2,3*
Xiaoli Liu1,2,3* Yi Lv1,2,3,4*
Yi Lv1,2,3,4*Hepatic fibrosis is often secondary to chronic inflammatory liver injury. During the development of hepatic fibrosis, the damaged hepatocytes and activated hepatic stellate cells (HSCs) caused by the pathogenic injury could secrete a variety of cytokines and chemokines, which will chemotactic innate and adaptive immune cells of liver tissue and peripheral circulation infiltrating into the injury site, mediating the immune response against injury and promoting tissue reparation. However, the continuous release of persistent injurious stimulus-induced inflammatory cytokines will promote HSCs-mediated fibrous tissue hyperproliferation and excessive repair, which will cause hepatic fibrosis development and progression to cirrhosis even liver cancer. And the activated HSCs can secrete various cytokines and chemokines, which directly interact with immune cells and actively participate in liver disease progression. Therefore, analyzing the changes in local immune homeostasis caused by immune response under different pathological states will greatly enrich our understanding of liver diseases’ reversal, chronicity, progression, and even deterioration of liver cancer. In this review, we summarized the critical components of the hepatic immune microenvironment (HIME), different sub-type immune cells, and their released cytokines, according to their effect on the development of progression of hepatic fibrosis. And we also reviewed and analyzed the specific changes and the related mechanisms of the immune microenvironment in different chronic liver diseases.Moreover, we retrospectively analyzed whether the progression of hepatic fibrosis could be alleviated by modulating the HIME.We aimed to elucidate the pathogenesis of hepatic fibrosis and provide the possibility for exploring the therapeutic targets for hepatic fibrosis.
Hepatic fibrosis is a reversible wound-healing response to liver injury, which is a major feature of any form of the chronic liver disease progressing to cirrhosis and liver failure (1). The main manifestations of hepatic fibrosis are chronic liver injury and accumulation of extracellular matrix (ECM) proteins (2). The continuous accumulation of ECM proteins could disrupt the normal function of the liver, and persistent hepatic fibrosis will progress to liver cirrhosis and even hepatocellular carcinoma (3). However, no effective drugs have been approved to target hepatic fibrosis. The therapeutic approaches to alleviate hepatic fibrosis mainly include: controlling or removing the underlying causes of hepatic fibrosis, preventing the activation and proliferation of HSCs, inhibiting the overexpression of pro-fibrotic cytokine TGF-β, and promoting fibrous tissue degradation. An emerging stem cell therapy is that the stem cells can transform into functionally active hepatocyte-like cells and participate in liver function repair and reconstruction (4).
It has been confirmed that hepatic fibrosis is driven by the activation of HSCs (5), which is the main source of ECM (6). HSCs are located in the perisinusoidal space and maintain a non-proliferative and quiescent phenotype in the normal liver tissue. The HSCs will transdifferentiate into proliferative fibrotic myofibroblasts, acquiring pro-inflammatory and pro-fibrotic properties under the chronic injury caused by many various pathogenic factors such as pathogenic microorganisms, alcohol, chemical drugs, and lipid deposition (7). The myofibroblasts transformed from HSCs account for about 82%-96% of myofibroblasts in various chronic liver diseases (8). Therefore, activated HSCs are often regarded as the main therapeutic target.
HIME is a dynamic network system composed of a variety of immune cells, immune cytokines, and related ECM (9), and it closely affects the onset and progression of hepatic fibrosis. Various immune cells are enriched in liver tissue and involved in the physiological and pathological process of hepatic metabolism and disease (10). Immune cells in the liver are diverse at a stable state and evolve further during the development of chronic liver disease, directly affecting the severity of the disease (11, 12). The recruitment and activation of immune cells in the diseased liver tissue are often initiated by the massive release and activation of cytokines and chemokines. Hepatic kuffer cells (KCs) are activated by various damage-associated molecular patterns (DAMPs, such as free DNA, ATP, high mobility group box 1) and pathogen-associated molecular patterns (such as LPS or viral DNA) that interact with Toll-like receptors (TLRs) or recombinant purinergic receptor P2X, ligand-gated ion channel 7 when hepatocytes are damaged (13, 14). This leads to the activation of the inflammasome, and the release of interleukin-1β (IL-1β), IL-18, and various other pro-inflammatory cytokines as well as chemokines. It will promote the recruitment of circulating leukocytes (monocytes and neutrophils), and activate adaptive immune T cells function. Once the infection or injury is controlled, the KCs and macrophages transform into anti-inflammatory and tissue repair phenotypes to control excessive tissue-damaging inflammatory responses (15). However, the persistent injurious stimulus in the liver will result in inflammatory cytokines continuously releasing that promote HSCs-mediated fibrous tissue hyperproliferation and excessive repair, which causes hepatic fibrosis development and progression to cirrhosis (16).
Hepatic fibrosis is an intermediate link and reversible stage in the process of chronic liver disease developing into liver cirrhosis. Intervention in this link will determine the prognostic direction of the various liver diseases. However, there is no efficient treatment strategy for hepatic fibrosis, which is mainly because profound awareness of the changes in the microenvironment of hepatic fibrosis still lacks, and no targeting and efficient intervention on the hepatic fibrosis microenvironment. Exploring the changes in local immune homeostasis and microenvironment caused by immune response under this pathological state could greatly enrich our understanding of liver disease reversal, chronicity, and even deterioration to liver sclerosis. Therefore, it is of great significance to elucidate the HIME for the study of hepatic fibrosis and the development of therapeutic strategies. In this review, we first summarized the critical components of HIME, the alteration and function of different sub-type immune cells, and their related mechanisms according to their effect on the development of progression of hepatic fibrosis. The specific changes and the related mechanisms of HIME in different chronic liver diseases were also reviewed and analyzed. Finally, we also summarized the therapeutic approaches and targets for HIME to reverse the progression of hepatic fibrosis, and we aimed to provide new research ideas and the possibility of exploring the therapy for hepatic fibrosis.
The immune cells are central members of the immune microenvironment in the progression of hepatic fibrosis, and they can mediate the immune response by synthesizing and secreting different chemical inflammatory mediators. The immune cells affect the function and number of HSCs mainly through direct or indirect effects, thus influencing the progression of hepatic fibrosis. Simultaneously, they constitute a dynamically evolving microenvironment and interact as well as restrict each other, forming a complex network system, which jointly affects the process of hepatic fibrosis.
Hepatic macrophages include liver-resident KCs and monocyte-derived macrophages (MDMs) (17). The MDMs, with high heterogeneity and plasticity, can differentiate into functionally distinct macrophage subsets. In mice, the circulating macrophages can be divided into the Ly6Chi CCR2hi CX3CR1lo monocytes which are considered to be potent pro-inflammatory cells, and Ly6Clo CCR2lo CX3CR1hi monocytes, which with an anti-inflammatory patrolling function (18). Ly6Chi monocytes in response to liver chronic injury are recruited to the liver and differentiate into MDM. These Ly6Chi MDMs have been shown to promote liver fibrosis, and they could promote HSCs activation as well as proliferation and ECM production by expressing a large number of pro-fibrotic mediators. The MDMs are also capable of differentiating into Ly6Clo restorative macrophage phenotypes, which promote ECM degradation and fibrosis resolution (19). KCs, the settled macrophages in the liver, have been identified as key regulators of hepatic inflammation. The DAMPs released from damaged hepatocytes can activate KCs and promote circulating macrophage infiltration in the liver (12). The activated macrophages have been reported to possess a bidirectional effect on the onset, progression, and reversal of hepatic fibrosis (17). Activated KCs could activate the HSCs by secreting large amounts of pro-inflammatory factors such as TGF-β1, IL-1β, and tumor necrosis factor-α (TNF-α) as well as the chemokines (20, 21). The TGF-β1 and platelet-derived growth factor (PDGF) are the most important inflammatory mediators in hepatic fibrosis. TGF-β1 regulates the activation of HSCs through the Smad pathway and inhibits the degradation of the ECM (22). PDGF activates HSCs and promotes their proliferation through the subsequent phosphatidylinositol 3 kinase (PI3K) and extracellular signal-regulated kinase pathways (23). Additionally, macrophages also mediate the reversal of fibrosis. Firstly, macrophages secrete a series of matrix metallic-proteinases (MMPs), which specifically degrade collagen and non-collagen extracellular matrix (24). Secondly, macrophages mediate HSCs apoptosis by expressing TNF-related apoptosis-inducing ligand (25), leading to the reduction of ECM and alleviation of hepatic fibrosis (26). Relaxin is an anti-fibrotic peptide hormone that can directly reverse the activation of HSCs to promote the resolution of hepatic fibrosis (27). A study showed that hepatic macrophages express primary relaxin receptors and they switch from a profibrogenic to the pro-resolution phenotype upon binding with relaxin. The latter releases exosomes that promote relaxin-mediated quiescence of activated hepatic astrocytes via miR-30a-5p (28). This indicates that macrophages have great potential in anti-fibrosis research and can be used as a target for anti-fibrosis therapy or reversal of fibrosis.
Dendritic cells (DCs) are the specialized antigen-presenting cells, with the function of transforming immune tolerance to immune activation, regulating the direction of the immune response in HIME of hepatic fibrosis (29). During hepatic fibrosis, DCs proliferate and undergo phenotypic changes. They stimulate adjacent T cells and natural killer(NK) cells through TNF-α, and up-regulate intercellular adhesion molecule-1 and CD40 expression of HSCs, thus, promoting the HSCs proliferation and activation (30, 31). DCs also aggravate liver inflammation by inhibiting infiltration of Treg cells and activating TLRs to produce a large number of cytokines (32). And the DCs depletion completely abrogates the elevated levels of many inflammatory mediators that are produced in the fibrotic liver (33). A similar study found knockout of DCs reduced the clearance of activated HSCs and delayed the fibrotic reversal process during the reversal phase of CCL4-induced hepatic fibrosis in mice (34).However, it also reported that although DCs can promote the activation of HSCs, the depletion of DCs does not affect the evolution of liver fibrosis in CCL4-induced hepatic fibrosis (35). Moreover, the application of FMS-liketyrosinekinase3 ligand induced the proliferation of DCs, accompanied by increased MMP-9 secretion from DCs. And the MMP-9 not only directly degrades collagen but also recruits innate immune cells such as macrophages and neutrophils, secreting MMP-8 and MMP-13 to degrade collagen, facilitating the reversal of hepatic fibrosis (34). Besides, long-term (12 weeks) alcohol intake can specifically recruit plasmacytoid dendritic cells to the liver in female mice. These plasmacytoid dendritic cells are characterized by secreting IFN-α and with anti-fibrosis function (36). These results suggest the effect of DCs in hepatic fibrosis is still controversial and need to be revealed and defined in the future research.
NK cells in liver were subdivided into CD49a+DX5 – liver-resident natural killer (lrNK) cells and CD49a- DX5+ conventional natural killer (cNK) cells. Both of them could kill activated HSCs in a tumor necrosis factor-related apoptosis-inducing ligand dependent manner (37). Compared to cNK cells, lrNK cells are less mature at a rest statue, but it have enhanced activity upon pathogenic stimuli and exhibit higher cytotoxicity (38). It suggested that the enrichment of lrNK cells in the liver may be to prepare for a special function against liver injury. It also was reported that lrNK cells can highly express CD107a and perforin, work together with a variety of activated receptors, release interferon-γ (INF-γ) while splitting infected hepatocytes, and inhibit the activation of HSCs in the human viral hepatitis (39). In the early stage of hepatic fibrosis, cNK cells infiltrate into the liver parenchyma and alleviate or reverse hepatic fibrosis by directly killing or inducing apoptosis of HSCs through degranulation and TNF-related apoptosis-inducing ligand expression (40). Moreover, the activated NK cells release IFN-γ through the JAK-STAT pathway to antagonize hepatic fibrosis by exerting a killing effect on the activated HSCs (41, 42). The activated NK cells can also secrete IL-10 and IL-22,which promote the senescence of activated HSCs and alleviate liver fibrosis through signal transducer-activator 3 (STAT3)/p53/p21 pathway (43, 44). And the activation of metatropic gluamate receptor 5 in NK cells can also reduce liver fibrosis by increasing their cytotoxicity and IFN-γ production (45).The NKp46(+)NK· cells were reported to attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice (46). In patients with chronic HVB infection, the terminally differentiated NK cells often highly express CD57 and DNAM-1, while NKp46 and NKG2A expression at low levels. These NK cells can kill activated HCSs through tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) dependent and CD44-osteopontin dependent manners (47). In contrast, during the progressive phase of hepatic fibrosis, persistent activation of HSCs will inhibit NK cell activity and weaken its anti-fibrotic effect. It is attributed to the increased metabolism of vitamin A. Its metabolites, retinoic acid, and retinol can inhibit the activation of IFN-γ on the transcriptional STAT-1 pathway by secreting the suppressor of cytokine signaling 1 (SOCS1) (41, 48).
NKT cells, a subset of T cells, possess NK and T cell receptors and display both NK cell and T cell properties. Similar to the NK cells, NKT cells also inhibit hepatic fibrosis by directly killing the activated HSC cells or producing IFN-γ to exert the inhibiting effect on HSCs. In the CCL4-induced acute liver injury mouse model, the hepatic NKT can slow down the onset of acute liver injury and liver inflammation by inhibiting activated HSCs (49). However, the anti-fibrotic effect of NKT cells is limited to the acute liver injury period, and in persistent chronic liver injury, the anti-fibrotic effect of NKT is diminished due to functional failure of NKT and immune tolerance of the liver (50). Additionally, Park et al. found that α-galactosyl neurosphingosamine overactivated NKT cells will accelerate CCL4-induced acute liver injury, inflammation, and fibrosis (51). In the progression of the alcoholic liver, NKT cell activation can exacerbate liver injury and promote an inflammatory response (52). Another study found that NKT cells from viral-infected patients could secrete more pro-inflammatory factors IL-4 and IL-13 compared with those from non-infected patients. It suggests that NKT cells may promote fibrosis by producing pro-inflammatory factors IL-4 and IL-13 to promote the development of fibrosis in patients with chronic viral infection (53).
Different subtypes of T cells have different effects on hepatic fibrosis. Th1 and Th2 cells play the core role in this process, and they tend to exhibit mutually antagonistic effects on hepatic fibrosis. IFN-γ secreted by Th1 cells regulates the balance of MMP and tissue inhibitor of metalloproteinase (TIMP), which can inhibit HSCs activation and proliferation, induce HSCs apoptosis, and inhibit TGF-β expression in a variety of cells (e.g., hepatocytes, KCs, HSCs) to exert anti-fibrotic effects (54, 55). IL-13 secreted by Th2 induces the production of collagen I, collagen III, α-smooth muscle actin, and TIMP-1 to induce TGF-β secretion, activate HSCs, and inhibit HSCs apoptosis thus promoting the progression of hepatic fibrosis (56). Moreover, IL-4, IL-5, and IL-13 secreted by Th2 cells reduce the liver inflammatory response, but also inhibit the Th1-mediated cellular immune response and prevent the clearance of viruses and parasites, leading to the persistence of viruses and inflammation (57). In addition, Th1 and Th2 cells regulate cellular collagen synthesis by antagonistically regulating nitric oxide synthase 2/arginase activity (58). In a non-alcoholic steatohepatitis (NASH) mouse model, mice lacking the IFN-γ (typical Th1 cytokine) showed significant protection against liver damage and fibrosis (59).
CD8+ T cells mainly produce cytotoxic molecules such as IFN-γ, TNF, and perforin (60). During NASH in mice, the number of CD8+ T cells in the liver was increased, and lipid-conditioned CXCR6+ CD8+ T cells induce hepatocyte killing in a perforin-independent, FasL (CD95L)-dependent manner (61). Consequently, in a diet-induced mouse model of NASH, CD8+ T cell depletion blunted liver injury (62). Thus, CD8+ T cells are likely to promote hepatic injury during NASH. But the mechanism by which T cell subsets are activated and interact to promote liver inflammation is not well understood. And more research is needed to investigate the specific mechanism and how to target these pathways without compromising immune defenses (63). Additionally, hepatic tissue-resident memory T cells (TRM cells) stably occupy tissues and participate in hepatic fibrosis progression differing from T cells in the circulation. CD8+T cells characterized by CD69 and CD103 are defined as CD8+ TRM cells (64, 65). Koda Y et al. found that CD8+ TRM cells were able to attract HSCs in a CCR5-dependent manner and mediated apoptosis of activated HSCs through the Fas- FasL pathway (66). Although little latest work had been done to verify the exist of CD4+TRM fraction in human liver, its relationship with liver fibrosis has not been systematically studied and documented (67).
Gamma-delta T cells(γδ T cells), as liver tissue-resident T cells, are characterized by the gamma and delta chains of T cell receptors (68, 69). Hepatic γδ T cells account approximately for 3% - 5% of the hepatic lymphocytes and 15% - 25% of the hepatic T cells, which are identified as liver-resident cells with predominant production of IL-17as well as the IFN-γ (70). And the IFN-γ will activate macrophages to release IL-15, prompting γδ T cells to accumulate at the infectious site and participate in local anti-inflammatory and anti-fibrotic effects, whereas IL-17, a major pro-inflammatory factor, often plays a pro-fibrotic effect on the fibrosis progress (70, 71). In the CCl4-induced hepatic fibrosis murine model, γδ T cells could activate the mTOC2 signaling pathway in response to macrophages, promoted CXCR3 transcription, and drove γδ T cell accumulation in the liver (72). It also documented that hepatic γδ T cells could kill the activated HSCs in a TRAIL-or FasL-dependent manner and secrete IFN-γ to inhibit differentiation of pro-fibrotic Th 17 cells to relieve hepatic fibrosis. And it also promoted HSC lysis by enhancing the cytotoxicity of conventional NK cells and liver-resident NK cells on activated HSCs (37). However, hepatocyte-derived exosomes mediated TLR3 activation in HSCs and increased IL-17 production from γδ T cells, and the IL-17 strongly stimulated the expression of a-SMA, TGF-β, IL-6, and collagen type I-α1 in HSCs and aggravated liver fibrosis (73).
B cells are classified as B-1 cells, B-2 cells, effector B cells, and regulatory B cells (Bregs) based on their surface molecular, localization, and functional characteristics. Relatively few studies indicated that B cells can influence the development and progression of hepatic fibrosis in an antibody-independent manner (74). It has been demonstrated that B cells knockout mice given CCL4 injections had significantly lower hepatic fibrosis than normal control mice, suggesting that B cells have a pro-fibrotic effect and this effect is independent of antibodies and T cells (75). Conversely, B cells can also exacerbate hepatic fibrosis by indirectly affecting T lymphocyte function and contact with fibroblasts, phagocytes, and NK cells through the secretion of IL-1, IL-6, and IL-4 (76). Secondly, it has been found by retrospective analysis that the number of B lymphocytes was significantly lower in the cirrhotic group than in the hepatitis group and healthy individuals, which also implies that B lymphocytes are affected in the progression of cirrhosis and are involved in the development of cirrhosis (5). In addition, similarly to T cells, regulatory B cells (Bregs) also secrete cytokines inducing immune tolerance and maintaining environmental homeostasis. Bregs from patients with chronic hepatitis B or hepatitis B virus(HBV)-associated hepatic fibrosis can inhibit the function of Th1 and Th17 through intercellular contact and secretion of IL-10. Bregs can also induce the conversion of CD4+CD25-T cells to Tregs, resulting in inflammatory response suppression and inflammatory repair (77).
In summary, the immune cells directly act on HSCs through the interaction of ligands and receptors or by secreting cytokines to regulate the activity of HSCs. Additionally, immune cells play an immunomodulatory role or indirect effect on HSCs regulating the inflammatory response through the synthesis and secretion of different cytokines and chemokines, thus promoting or inhibiting the formation and progression of hepatic fibrosis. We depicted how the main sub-types of immune cells in the HIME influence the HSCs in Figure 1.
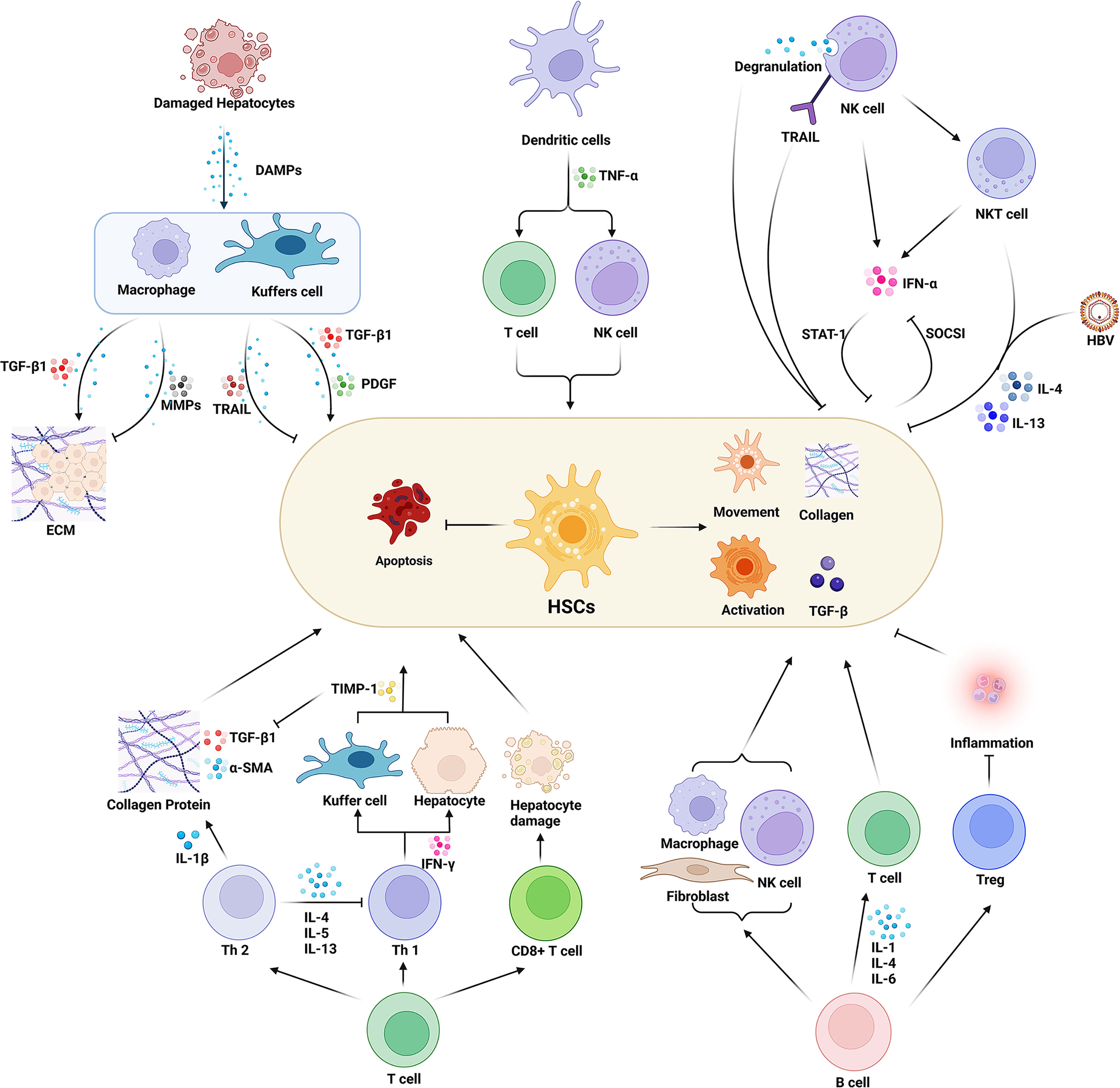
Figure 1 Effect of various immune cells in the hepatic immune microenvironment (HIME) on hepatic fibrosis. Various immune cells affect the process of hepatic fibrosis by promoting or inhibiting the function of HSCs. Macrophages play a dual role in this process. The peripheral macrophages and the resident liver macrophages (Kuffer cells) are activated under the damage-associated molecular pattern released from the damaged hepatocytes during injury or inflammatory conditions. Activated macrophages secrete proinflammatory factors such as TGF-β to promote ECM formation and activate HSCs. On the other hand, macrophages can also inhibit hepatic fibrosis by expressing MMPs and TRAIL. Dendritic cells secrete TNF-α to activate HSCs by enhancing the immune function of T cells and NK cells. And the activated NK cells inhibit the function of HSCs and prevent hepatic fibrosis progression. While NK cells also can directly damage HSCs through degranulation and up-regulation of TRAIL expression. Moreover, it can secrete INF-γ, and synergize NKT cells to inhibit HSCs activation through the STAT signaling pathway. However, HSCs can inhibit INF-γ secretion of NK cells via the SOCSI pathway. The balance of Th1/Th2 cells plays an important role in the process of hepatic fibrosis. Th1 cells secrete INF-γ to increase the expression of TIMP in macrophages and inhibit the activity of HSCs. Th2 cells can secrete a variety of cytokines to activate HSCs and inhibit the HSCs’ apoptosis as well as the activity of Th1 cells. CD8+ T cells secrete cytotoxic factors to mediate hepatocyte injury, thereby activating HSCs. B cells regulate macrophages, fibroblasts, and T cells and promote HSCs activation by secreting interleukins. And B cells are also able to regulate Treg cells to inhibit inflammatory responses and HSCs’ activation. Created with BioRender.com.
Viral hepatitis is a type of infectious disease caused by the hepatitis virus and characterized by hepatocyte degeneration, necrosis, and apoptosis. Chronic hepatitis, especially chronic hepatitis B, undergoes fibrosis with the virus-induced long-term inflammation and leading to the evolutionary pathway of “hepatitis–cirrhosis–liver cancer” (78, 79).
The major mechanisms of viral hepatitis progression to cirrhosis are the direct and indirect activation of HSCs (6). The Hepatitis virus can promote HSCs activation by disturbing immune cell functions, thereby exacerbating hepatic fibrosis (80). Xie et al. (81) found that the core antigen HBeAg of HBV is the most important component of HBV-induced macrophage activation. HBeAg binds with macrophages through the TLR-2 receptor, activates the NF-κB signaling pathway in macrophages, and releases a large number of cytokines and chemokines to inhibit viral proliferation and regulate HSCs’ function, producing different pro-fibrotic effects. On the one hand, TNF-α and IL-1β can inhibit HSCs apoptosis by up-regulating the expression of TIMP-1 and down-regulating the expression of BMP and activin membrane-bound inhibitor of HSCs (82–85). Macrophages are also able to up-regulate PI3K-AKT-mTOR and p38 mitogen-activated protein kinase (p38 MAPK) pathway in HSCs to promote the HSCs movement, and also activate TGF-β/smad pathway to promote their proliferation and contraction, and promote the production of type I and type III collagen (86). Moreover, the activated HSCs further affect the function of macrophages, and they interactively promote the progression of hepatic fibrosis. Akt/PKB (protein kinase B) in HSCs promotes fibrogenic M2 polarization in macrophages (87). HBeAg directly induces up-regulate the secretion of TGF-β of HSCs, and the secreted TGF-β positively feeds back to HSCs themselves and mediates their activation and proliferation (88). The HBV and its related proteins reduce the expression of TLR in macrophages, leading to HBV immune tolerance and persistent infection. The described mechanisms of macrophage and HSCs interacted in the liver for a long time, allowing hepatic fibrosis occurrence and progress to cirrhosis or even hepatocellular carcinoma (78, 89).
Viral infection could inhibit NK cells to produce the anti-fibrotic cytokine IFN-γ, promoting the development of hepatic fibrosis. A study found that HBV infection down-regulates the expression of NK cell activating receptors NKG2D and 2B4 on NK cells via TGF-β1 secreted by hepatocytes, resulting in a decreased expression of their intracellular adaptor proteins DAP10 and SAP, which impair NK cell-mediated cytotoxicity ability and IFN-γ production (90). Another study found that ability to the production of IFN-γ of CD56 (dim) NK cells in patients with chronic HBV infection is impaired, leading to an increased NKG2A expression and decreased CD16 expression, thereby preventing NK cells activation and promoting hepatic fibrosis (91). Activation of the PDGF/PDGF-β receptor also plays a critical role in hepatic fibrosis. HBV core protein and HBV regulatory X protein are two proteins expressed by HBV, which can promote PDGF production by increasing the transcription of PDGF in hepatocytes. PDGF will bind to the PDGF-β receptor, a receptor located on HSCs, induce receptor phosphorylation, and initiate various intracellular signaling pathways activation such as the JAK/STAT, PI3K, PLC-γ, or MAPK pathways, leading to HSC migration, proliferation, and ECM secretion (92–94).
In the virus-infected HIEM, exosomes secreted by hepatocytes, containing miRNAs, also can regulate the function of HSCs. MiR-19, an exosome secreted from HCV-infected hepatocytes, could reduce SOCS3 production of HSC. And the SOCS3 could block JAK kinase, thus the reduction of SOCS3 will activate the JAK-STAT3 pathway, increase cyclin D1 transcription, and stimulate HSC proliferation (95). HCV-infected hepatocytes also secrete miR-192-containing exosomes and exert an effect on HSCs, increase TGF-β1 expression, and stimulate HSCs activation and differentiation into myofibroblasts phenotype (96). Figure 2 summarized the effect of viral hepatitis-induced HIME changes on the development and progression of hepatic fibrosis.
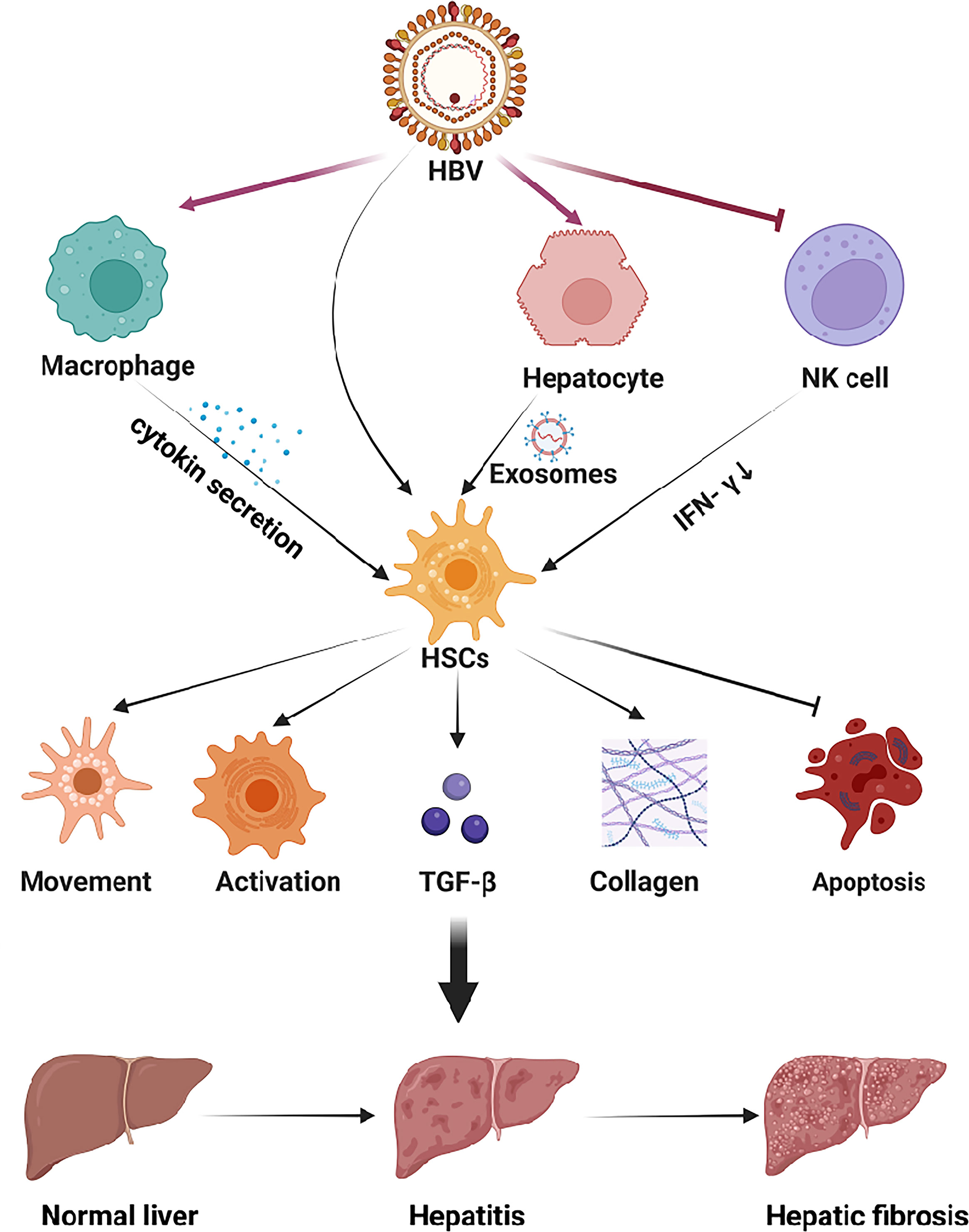
Figure 2 The HIME of HBV-induced viral hepatitis. The hepatitis virus replicates within hepatocytes and is secreted into the extracellular space via e exosomes. Viral antigens can exert multiple effects in HSCs to promote hepatic fibrosis. In addition, the virus can be recognized by macrophages and NK cells, which will up-regulate or down-regulate the secretion of cytokines to activate HSCs. Subsequently, HSCs activation aggravates hepatitis and promotes the progression of hepatic fibrosis. Created with BioRender.com.
Autoimmune liver diseases (AILD) are mainly characterized by liver injury along with elevated serum immunoglobulins and the presence of multiple auto-antibodies in the blood, which is associated with autoimmunity. It can be divided into autoimmune hepatitis, primary sclerosing cholangitis, and primary biliary cholangitis (PBC), according to the cell type involved (97).
As the autoimmune tolerance of patients with AILD is impaired, a number of auto-antibodies are produced and accumulated in the HIME, including antinuclear antibodies in autoimmune hepatitis, antimyosmooth muscle antibodies, and antimitochondrial antibodies in PBC. Driven by humoral immunity, the activated CD4+ T cells (including Th1 and Th2) stimulate B cells to produce antibodies against auto-antigens by directly T-B cell membranes contacting and releasing cytokines, and with chronic stimulation on hepatocytes. The damaged hepatocytes could recruit the NK cells and macrophage infiltration that exacerbate the inflammatory response within the liver (63, 98). Wu et al. (99) showed that the transcription of POU6F1 in biliary epithelial cells activates monocyte chemotactic protein-1 (MCP-1) and promotes peripheral M1 and M2 macrophage recruitment and infiltration into peribiliary gland (PBG) niche, producing chronic inflammation, resulting in dilated PBG compartments and the formation of “onionskin” fibrosis characterized by multifocal fibrosis around intrahepatic and extrahepatic bile ducts in a mouse model of primary sclerosing cholangitis.
Regulatory T cells (Tregs)/Th17 cell imbalance is critical for immune disorders in patients with AILD. Tregs are circulating auto-reactive T cells that limit autoimmune damage. A significant decrease in Tregs was found in the peripheral blood and liver of PBC patients (100, 101). In addition, Zhu et al. (102) found that knockout of AMP-activated protein kinase α1 and Tregs-specific deletion could weaken the suppressive activity of Tregs and lead to the development of alcoholic liver disease(ALD) in mice. Th17 cells were significantly increased in the bile ducts of patients with primary biliary cholangitis (103). Th17 cells are helper T cells differentiated from Th0 cells stimulated by IL-6 and IL-23, which can secrete a large number of pro-inflammatory factors such as IL-17 and IL-22, activate TNF or Fas/FasL pathway to mediate hepatocyte apoptosis, and activate HSCs to promote the development of hepatic fibrosis (104, 105). Figure 3 presented the main alterations of the HIME in the AILD.
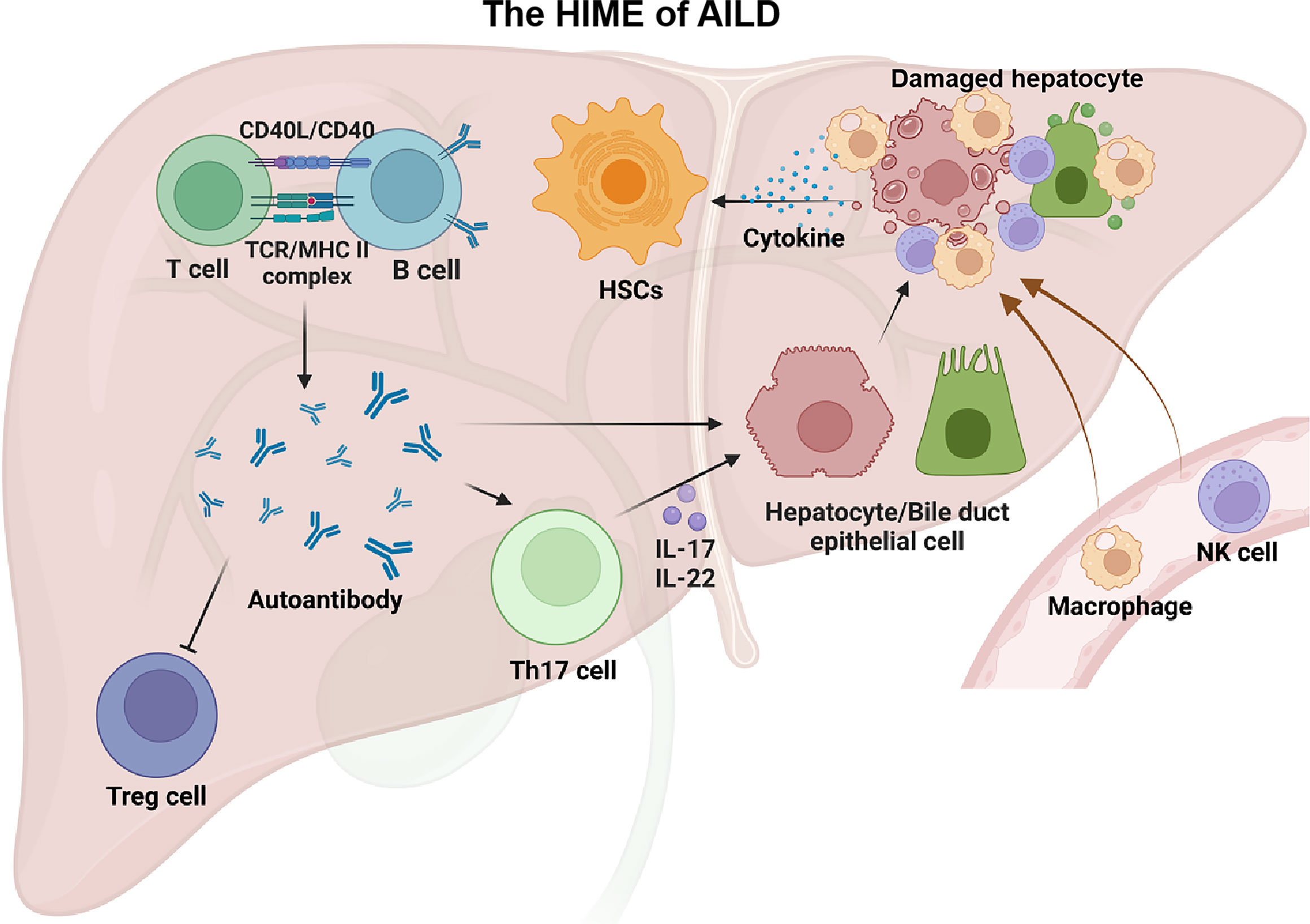
Figure 3 The HIME of AILD. Autoimmune liver disease happens when a large number of autoantigens are present in the liver. With the assistance of T cells, B cells can generate a large number of autoantibodies that recognize these autoantigens. These autoantibodies can impair the function of Treg cells and lead to autoimmune damage. In addition, autoantibodies can directly, or indirectly through Th17 cells, exert an effect on targeted cells (e.g., hepatocytes, biliary epithelial cells) and recruit peripheral macrophages and NK cells, causing liver injury. These injured cells release large amounts of cytokines thereby driving the HSCs activation. Created with BioRender.com.
Fatty liver disease is mainly caused by triglyceride accumulation in the liver leading to continuous hepatocyte stimulation and arousing long-term inflammatory responses. According to whether exposure to long-term excessive alcohol consumption fatty liver disease can be divided into ALD and non-alcoholic fatty liver disease (NAFLD).
ALD As ALD patients often consume amounts of alcohol for a long time, the majority of ethanol is metabolized in the liver. Acetaldehyde, the intermediate product of ethanol, can directly damage hepatocytes and bind with the intracellular proteins as antigens to cause humoral immune responses (106, 107). This tissue stress injury leads to an aseptic inflammation (108), which further activates the KCs, initiating the immune response and recruiting peripheral monocytes into the liver. And the KCs are capable of releasing the number of inflammatory factors and chemokines such as TGF-β, TNF-α, and IL-1β to stimulate HSCs activation. Furthermore, macrophages of the M2 type possess a powerful activating effect on HSCs (109, 110). Ethanol-induced telomerase reverses transcriptase expression in macrophages and promotes macrophage polarized into M1 type through the NF-κB signaling pathway (111). And ethanol can activate HSCs to induce collagen deposition by up-regulating P2X7 receptor (P2X7R) expression-an ATP-gated, non-selective cation channel receptor belongs to the P2X family of P2 purine receptor in human macrophages leading to NLRP3 inflammasome activation and releasing the IL-1β. Inhibition of NLRP3 inflammasome activation effectively suppresses further deterioration of alcoholic steatohepatitis and attenuates alcohol-induced liver injury (112–114). Another study found P2X7R can also mediate acetaldehyde-induced HSC activation via the PKC-dependent GSK3β pathway (115). Additionally, ethanol can also directly affect the expression of epigenetic regulators during HSC transdifferentiation. The histone modifying enzymes such as H3K4 and methyltransferase MLL1 are upregulated in HSCs, when exposed to ethanol and recruited to the elastin gene promoter, resulting in the up-regulation of pro-fibrotic elastin genes expression and increased ECM protein expression.
ROS also plays an important role in hepatic fibrosis resulting from alcoholic liver disease. Macrophages exposed to chronic ethanol can induce ROS release, which acts as an inducer of the TGF-β signaling pathway, thereby promoting the activation of HSCs. The dual effects of ethanol and ROS promote hepatocyte lysis and death, releasing the DAMPs to the extracellular matrix, stimulating macrophage activation, and initiating liver regeneration as well as fibrosis processes (116, 117). ATP, as DAMPs released during cell lysis, acts as an endogenous danger signal to activate intracellular inflammatory response through P2X7R and exacerbate cell damage (118).
A recent study found that alcohol intake increases intestinal permeability, promotes the transfer of endotoxins such as lipopolysaccharide (LPS) from Gram-negative bacteria to the portal vein, and reaches the liver with the bloodstream. LPS activation of various TLR ligands was shown to induce miR-155 in macrophages. miR-155 plays a critical role in promoting macrophage M2 polarization. miR-155 also targets and activates SMAD2/5, Snail1, STAT3 genes that are involved in fibrosis. And the miR-155 knockout protects mice from alcohol-induced steatosis and inflammation (119–121).
NAFLD NAFLD is characterized by excessive lipids accumulation in the liver, which can affect bile acid metabolism, hepatocyte metabolism, and the function of macrophages and HSCs. And it can promote the progression of NAFLD to NASH or even cirrhosis by activating the metabolism of intrahepatic immune cells (122)(Figure 4). Innate immune activation is a key factor in the exacerbation of hepatic inflammation in NAFLD/NASH. The TLR was reported to mediate B-cell activation through activation of myeloid differentiation primary response protein 88 (MyD88) in a high-fat high-carbohydrate diet-fed mouse model of NASHA. The activated B cells secreting pro-inflammatory cytokines, modulating neighboring immune cells, and differentiating into antibody-secreting cells will result in the progression of NAFLD. A study reported that transplantation of gut microbiota from NAFLD patients into recipient mice can also increase B cell accumulation and activation in the liver (123). Studies have shown that insulin resistance is often accompanied by NAFLD-associated hepatic fibrosis, and NK cells contribute to the development of obesity-associated insulin resistance. Lipid accumulation promotes IL-6R expression in mouse NK cells, and IL-6/STAT3-dependent myeloid NK cell subsets are a critical determinant of NAFLD-associated insulin resistance in vivo (124). Almost all CD4+ T cells are involved in the sterile inflammation associated with NASH. IL-17 secreted by Th17 cells increases c-Jun N-terminal kinase (JNK) activation in steatotic hepatocytes and exacerbates hepatocyte injury, while IL-22 secreted by Th22 cells protects hepatocytes by inhibiting PI3K/Akt-mediated JNK activation. And the balance breaking between IL-17 and IL-22 will lead to hepatocyte injury and accelerate NAFLD progression (125). Ghazarian et al. (126) reported that high-fat diet (HFD) feeding increased the expression of IFN-αR on CD8+ T cells and upregulated the key transcription factors of IFN-I in the liver of the NAFLD mouse model. While the activation of IFN-I responses drives the expansion of intrahepatic pathogenic CD8+ T cells, leading to the NAFLD progression. In mouse models of NAFLD, saturated free fatty acid (FFA) levels are increased in the blood, and they can bind to TLR2 and TLR4 of liver-resident Kupffer cells and peripherally infiltrating macrophages, triggering multiple mechanisms to produce the ROS, such as mitochondrial damage, endoplasmic reticulum stress, and nicotinamide adenine dinucleotide phosphate oxidase. Nicotinamide adenine dinucleotide phosphate oxidase2-derived ROS could stimulate macrophages to produce pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, which promote the development of hepatic fibrosis by up-regulating NF-κB pathway and activate JNK activator protein-1 in HSCs (127). Another study reported a high-fat diet-induced increased bile acid secretion, resulting in gut microbiota disturbances and intestinal barrier dysfunction in a mouse model of NAFLD (128). It will lead to an increase of LPS and other bacteria-derived compounds in portal blood, promoting the activation of TLR and other pattern recognition receptors in the liver, and triggering local inflammatory and fibrotic responses. In addition, in a mouse model of induced NASH-fed diets rich in palmitate, cholesterol, and sucrose, the palmitate of hepatocyte specifically promotes the overexpression of the Notch pathway ligand Jag1 by activating TLR4 to upregulate NF-κB signaling. And the activation of the Notch signaling pathway in hepatocytes drives the expression of genes encoding osteopontin and secreted phosphoprotein 1 (Spp1), which promotes HSCs activation and hepatic fibrosis (129).
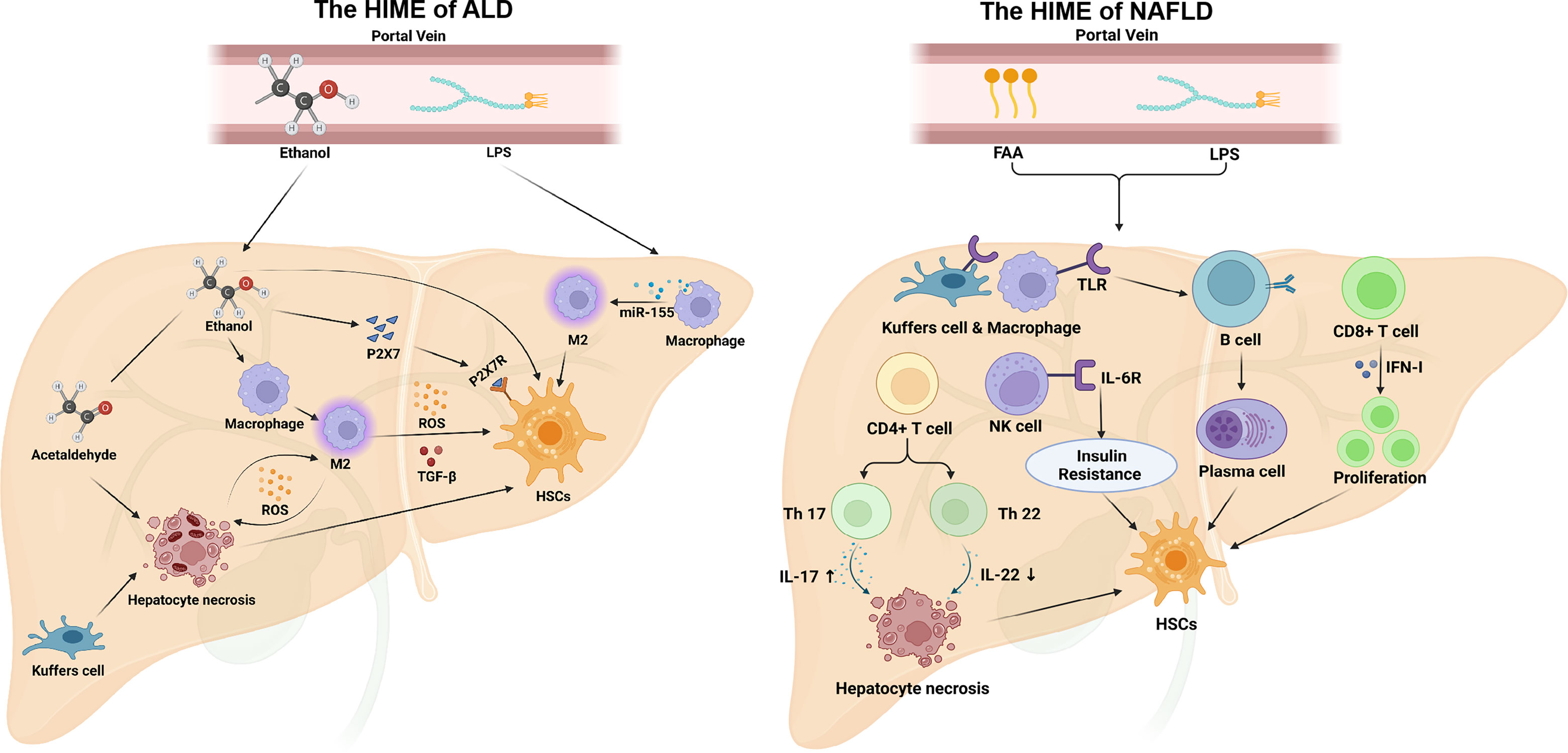
Figure 4 The HIME of ALD and NAFLD. In ALD, ethanol is absorbed through the intestine and entry the portal vein to the liver. Ethanol can stimulate M2 macrophage polarization that secretes reactive oxygen species and TGF-β to promote hepatic fibrosis. Ethanol also increases P2X7R(P2X7 receptor) expression on HSCs, and P2X7 can promote HSCs activation. Acetaldehyde, an intermediate product of ethanol, induces hepatocyte necrosis. And necrotic hepatocytes directly activate HSCs and release ROS to activate M1 macrophages. Additionally, ethanol can disrupt the intestinal barrier, causing LPS to enter the liver via the portal vein and induce M2 macrophage polarization. In NAFLD, free fatty acid and LPS from the peripheral circulation induce TLR expression in macrophages, transform B cells into plasma cells, and secrete antibodies, which activate HSCs activation. In addition, free fatty acid and LPS are also able to stimulate CD4+ T cells, regulate the balance of Th17/Th22 cells, and induce hepatocyte necrosis by increasing IL-17 and reducing IL-22. Moreover, CD8+ T cells and NK cells can also promote HSCs activation under this HIME. Created with BioRender.com.
The importance of immune cell populations in the development of chronic liver disease is self-evident. However, most traditional treatments for liver fibrosis are still focused on inhibiting the activation of HSCs. The role of HIME in liver fibrosis should not be underestimated. Therefore, blocking the infiltration of immune cells in the blood or altering the function of infiltrating immune cells or cytokines may be a potential target for the treatment of liver fibrosis. Table 1 summarized the details of the effects of traditional Chinese medicine(TCM) in modulating the HIME for preventing hepatic fibrosis of chronic liver disease. In four preclinical studies, researchers regulated the functions of HSCs (130, 132), macrophages or KCs (25, 131, 134–136, 138), regulatory T cells (133), DCs (30), and NK cells (25, 137) by different treatment approach in the CCl4 or bile duct ligation -induced liver fibrosis murine model. And these interventions can effectively alleviate hepatic fibrosis and tissue inflammation. Another study found inhibiting the chemokines CCR2/5 could significantly reduce circulating Ly6Chi monocytes and hepatic monocyte-derived macrophage infiltration in murine models of NASH, and attenuate hepatic inflammation and fibrosis (140). A chemical agent, dopamine receptor D2 antagonism, was screened to target the Yes-associated protein signaling pathway in macrophages. It antagonizes Yes-associated protein-dependent fibrotic crosstalk between selectively targeted macrophages and CTGF + VCAM1 + vascular niche, blocks fibrosis, and restores liver architecture in rodent and large animal models of NASH (139).
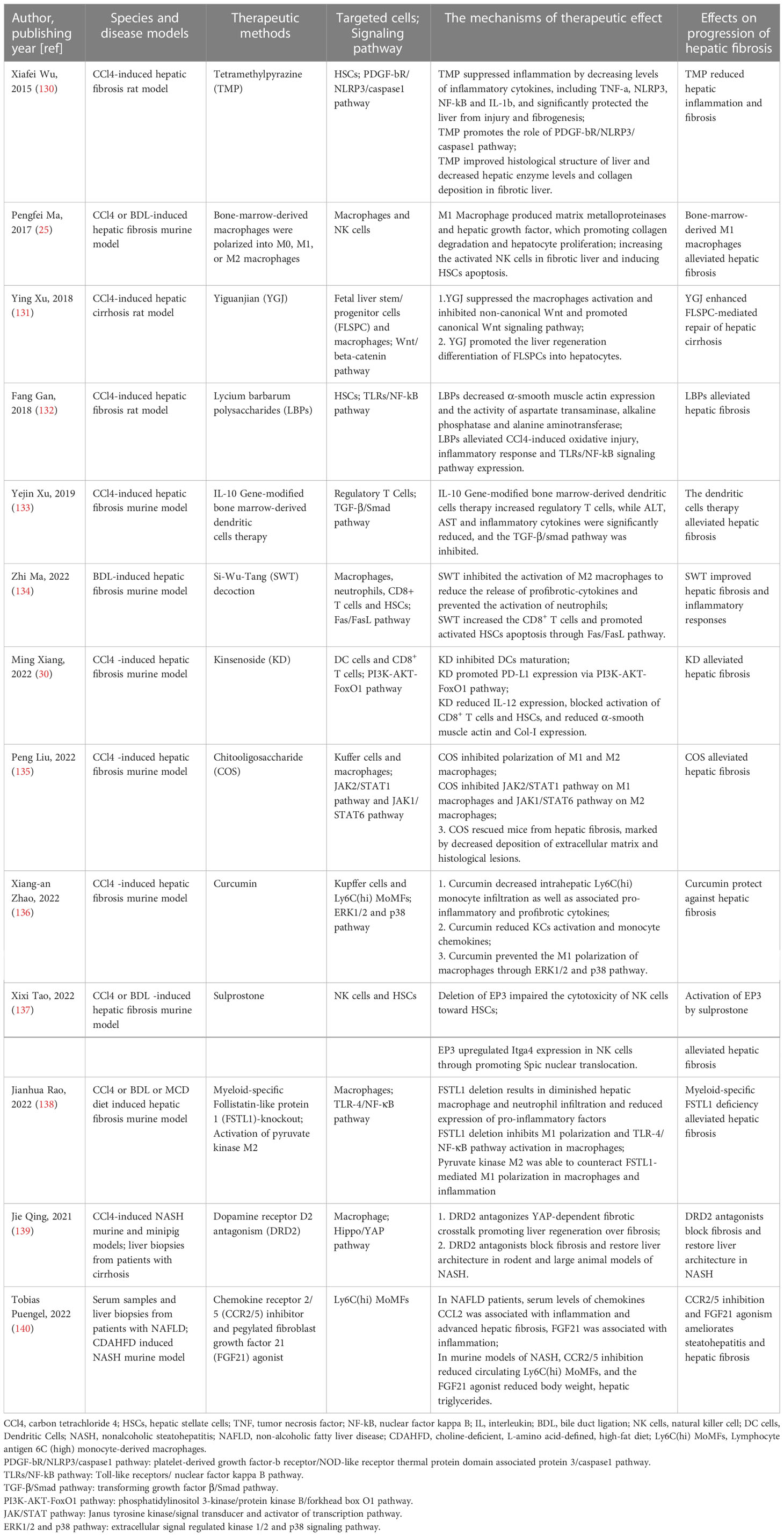
Table 1 The effects of regulating the HIME on the progression of hepatic fibrosis.
The occurrence and progression of liver fibrosis often involve a variety of cells and multiple signaling pathways. Clinical TCM treatments are advanced in action on multi-cellular and multi-target, which might be a new choice for alleviating liver fibrosis. Ginsenoside (KD), the active ingredient in the TCM ginseng, was reported could reduce hepatic histopathological damage, proinflammatory cytokine release, and extracellular matrix deposition in CCl4-induced hepatic fibrosis. The KD restrained the hepatic fibrosis-driven rise in CD86, MHC-II, and CCR7 levels, while upregulated PD-L1 expression on DCs, which blocked CD8+ T cell activation. Additionally, KD reduced DC glycolysis, maintained DCs immature, and was accompanied by an IL-12 decrease in DCs. These effects disturbed the communication of DCs and HSCs with the expression or secretion of α-smooth muscle actin and Col-I declined in the liver (30). Lycium barbarum polysaccharides (LBPs) supplementation was reported to reduce CCl4-induced oxidative injury, inflammatory response, and TLRs/NF-kB signaling pathway expression, which alleviated CCl4-induced liver fibrosis (132). Moreover, two Chinese herbal formulas Si-Wu-Tang (134)and Yi Guan Jian (131) were reported to regulate the activation and polarization of macrophage, which significantly improved liver fibrosis and inflammatory responses. Additionally, Si-Wu-Tang also can inhibit the M1 from inhibiting the infiltration of neutrophils and promote the CD8+ organization to reside memory T cells cytotoxic effect inducing the HSCs apoptosis (134). The Yi guan Jian can enhance the therapeutic effects of fetal liver stem/progenitor cells that promoted the liver regeneration differentiation of fetal liver stem/progenitor cells into hepatocytes (131). Collectively the data supports the notion that multi-target therapeutics may be an effective option for t liver fibrosis treatment (Table 1).
The immune cells and HSCs could synthesize and secrete several chemical mediators under chronic liver injury, and they constitute a dynamic HIME of an immune cells-cytokines-chemokines network system that affects the processes of hepatic fibrosis. It can be seen the HIME of hepatic fibrosis is complex not only because there are a variety of cells involved, but also because there are different sub-types of immune cells, which play different roles at different stages of liver disease progression. Moreover, in addition to local immune cells in the liver, a variety of immune cells in the circulation also can be chemotactic into the liver tissue and differentiate into different cell subtypes, exerting an effect on the initiation, progression, and regression of hepatic fibrosis. According to the summary and analysis in this review, most sub-types of immune cells have bidirectional effects on the development of hepatic fibrosis, because the immune cells interact with each other and also with other stromal cells in the microenvironment through diverse and complex cytokines networks, and eventually achieve a balance. While this balance will be broken with the persistence or elimination of the disease’s causes, then these cells will form a new balance through the other signaling pathways and exert a promoting or inhibiting role in hepatic fibrosis to a different degree. Therefore, it is necessary not only to explore the function of a certain group of immune cells, but also to explore cells’ diversity, and accurately analyze the cells’ specific phenotype and role in different liver diseases, different stages of the same disease, and different HIME caused by different pathogenic factors stimulus. Recently, single-cell sequencing can clarify the specific role of single cells. If this detection method is applied to the investigation of the immune microenvironment of hepatic fibrosis, it may greatly enrich the existing immune network and provide comprehensive and accurate ideas for the study of hepatic fibrosis. Additionally, the specific mechanisms for the immune cells of a specific phenotype to play different roles at different stages of chronic liver disease need to be elucidated, so that a precise intervention can be performed. However, the study on specific mechanisms is currently a major vacancy for the effect of HIME on hepatic fibrosis, thus, future studies need to focus on a deeper and more specific regulatory pathway to achieve an accurate intervention target, thereby delaying or avoiding the occurrence and progression of hepatic fibrosis.
NZ, HY, and ZZ conceived of the subject matter and wrote the logic of this review, searched, categorized, and summarized all relevant literature, and wrote the initial draft. ZL, XC, YZ, and RJ helped to check relevant information check the content of the initial draft. JH and HP edited the second draft and composed the manuscript. XL and YL reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 82202297, Grant No. 81900573), and the Natural Science Basic Research Program of Shaanxi Province (Grant No. 2022JQ-914).
We would like to thank the website of https://app.biorender.com/for providing cell morphological elements that were used to complete our Figures. And the four figures of this review are created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
HSCs, hepatic stellate cells; HIME, hepatic immune microenvironment; ECM, extracellular matrix;KCs, kuffer cells; DAMP, damage-associated molecular pattern; TLR, Toll-like receptor; IL, interleukin; MDM, monocyte-derived macrophage; TNF, tumor necrosis factor; PDGF, platelet-derived growth factor; MMP, matrix metallic-proteinases; DCs, dendritic cells; NK, natural killer; lrNK, liver-resident natural killer;cNK, conventional natural killer;STAT, signal transducer-activator; SOCS, suppressor of cytokine signaling; TIMP, tissue inhibitor of metalloproteinase; NASH, non-alcoholic steatohepatitis; HBV, hepatitis B virus; MAPK, mitogen-activated protein kinase; AILD, autoimmune liver diseases; PBC, primary biliary cholangitis; ALD, alcoholic liver disease; NAFLD, non-alcoholic fatty liver disease; P2X7R, P2X7 receptor; LPS, lipopolysaccharide; JNK, Jun N-terminal kinase; TCM, traditional Chinese medicine; KD, kinsenoside.
1. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
2. Dhar D, Baglieri J, Kisseleva T, Brenner DA. Mechanisms of liver fibrosis and its role in liver cancer. Exp Biol Med (Maywood N.J.) (2020) 245:96–108. doi: 10.1177/1535370219898141
3. Aydın MM, Akçalı KC. Liver fibrosis. Turkish J Gastroenterol (2018) 29:14–21. doi: 10.5152/tjg.2018.17330
4. Deng Y, Xia B, Chen Z, Wang F, Lv Y, Chen G. Stem cell-based therapy strategy for hepatic fibrosis by targeting intrahepatic cells. Stem Cell Rev Rep (2021) 18:77–93. doi: 10.1007/s12015-021-10286-9
5. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
6. Cai X, Wang J, Wang J, Zhou Q, Yang B, He Q, et al. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol Res (2020) 155:104720. doi: 10.1016/j.phrs.2020.104720
7. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U.S.A. (2012) 109:9448–53. doi: 10.1073/pnas.1201840109
8. Yavuz BG, Pestana RC, Abugabal YI, Krishnan S, Chen J, Hassan MM, et al. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget (2020) 11:1186–201. doi: 10.18632/oncotarget.27532
9. Jiajia S, Shuangshuang L, Yanning L, Min Z. Extracellular vesicles participate in macrophage-involved immune responses under liver diseases. Life Sci (2020) 240:117094. doi: 10.1016/j.lfs.2019.117094
10. Wang R, Tang R, Li B, Ma X, Schnabl B, Tilg H. Gut microbiome, liver immunology, and liver diseases. Cell Mol Immunol (2020) 18.4–17. doi: 10.1038/s41423-020-00592-6
11. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol (2020) 18:151–66. doi: 10.1038/s41575-020-00372-7
12. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest (2017) 127:55–64. doi: 10.1172/JCI88881
13. Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Trans Res (2017) 3:170–88. doi: 10.18053/jctres.03.2017S1.003
14. Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando) (2012) 26:103–14. doi: 10.1016/j.trre.2011.10.006
15. Ju C, Mandrekar P. Macrophages and alcohol-related liver inflammation. Alcohol Res (2015) 37:251–62.
16. Khanam A, Saleeb PG, Kottilil S. Pathophysiology and treatment options for hepatic fibrosis: Can it be completely cured? Cells (2021) 10:1097. doi: 10.3390/cells10051097
17. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol (2014) 60:1090–96. doi: 10.1016/j.jhep.2013.12.025
18. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17:306–21. doi: 10.1038/nri.2017.11
19. Papachristoforou E, Ramachandran P. Macrophages as key regulators of liver health and disease. Int Rev Cell Mol Biol (2022) 368:143–212. doi: 10.1016/bs.ircmb.2022.04.006
20. Bartneck M, Koppe C, Fech V, Warzecha KT, Kohlhepp M, Huss S, et al. Roles of CCR2 and CCR5 for hepatic macrophage polarization in mice with liver parenchymal cell-specific NEMO deletion. Cell Mol Gastroenterol Hepatol (2021) 11:327–47. doi: 10.1016/j.jcmgh.2020.08.012
21. Cheng D, Chai J, Wang H, Fu L, Peng S, Ni X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int (2021) 41:2279–94. doi: 10.1111/liv.14940
22. Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, et al. TGF-beta and the tissue microenvironment: Relevance in fibrosis and cancer. Int J Mol Sci (2018) 19:1294. doi: 10.3390/ijms19051294
23. Lan T, Zhuang L, Li S, Yang G, Xuan Y, Guo J. Polydatin attenuates hepatic stellate cell proliferation and liver fibrosis by suppressing sphingosine kinase 1. BioMed Pharmacother (2020) 130:110586. doi: 10.1016/j.biopha.2020.110586
24. Geervliet E, Moreno S, Baiamonte L, Booijink R, Boye S, Wang P, et al. Matrix metalloproteinase-1 decorated polymersomes, a surface-active extracellular matrix therapeutic, potentiates collagen degradation and attenuates early liver fibrosis. J Control Release (2021) 332:594–607. doi: 10.1016/j.jconrel.2021.03.016
25. Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ, Zhao Y, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol (2017) 67:770–79. doi: 10.1016/j.jhep.2017.05.022
26. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol (2021) 18:151–66. doi: 10.1038/s41575-020-00372-7
27. Fallowfield JA, Ramachandran P. A relaxin-based nanotherapy for liver fibrosis. Nat Nanotechnol (2021) 16:365–66. doi: 10.1038/s41565-020-00832-w
28. Hu M, Wang Y, Liu Z, Yu Z, Guan K, Liu M, et al. Hepatic macrophages act as a central hub for relaxin-mediated alleviation of liver fibrosis. Nat Nanotechnol (2021) 16:466–77. doi: 10.1038/s41565-020-00836-6
29. Rahman AH, Aloman C. Dendritic cells and liver fibrosis. Biochim Biophys Acta (2013) 1832:998–1004. doi: 10.1016/j.bbadis.2013.01.005
30. Xiang M, Liu T, Tian C, Ma K, Gou J, Huang R, et al. Kinsenoside attenuates liver fibro-inflammation by suppressing dendritic cells via the PI3K-AKT-FoxO1 pathway. Pharmacol Res (2022) 177:106092. doi: 10.1016/j.phrs.2022.106092
31. Xu W, Wu M, Chen B, Wang H. Myeloid cells in alcoholic liver diseases: Mechanism and prospect. Front Immunol (2022) 13:971346. doi: 10.3389/fimmu.2022.971346
32. Vonghia L, Van Herck MA, Weyler J, Francque S. Targeting myeloid-derived cells: New frontiers in the treatment of non-alcoholic and alcoholic liver disease. Front Immunol (2019) 10:563. doi: 10.3389/fimmu.2019.00563
33. Xu R, Zhang Z, Wang FS. Liver fibrosis: mechanisms of immune-mediated liver injury. Cell Mol Immunol (2012) 9:296–301. doi: 10.1038/cmi.2011.53
34. Jiao J, Sastre D, Fiel MI, Lee UE, Ghiassi-Nejad Z, Ginhoux F, et al. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology (2012) 55:244–55. doi: 10.1002/hep.24621
35. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol (2014) 96:657–65. doi: 10.1189/jlb.6A0114-004RR
36. Alharshawi K, Fey H, Vogle A, Klenk T, Kim M, Aloman C. Sex specific effect of alcohol on hepatic plasmacytoid dendritic cells. Int Immunopharmacol (2021) 90:107166. doi: 10.1016/j.intimp.2020.107166
37. Liu M, Hu Y, Yuan Y, Tian Z, Zhang C. gammadeltaT cells suppress liver fibrosis via strong cytolysis and enhanced NK cell-mediated cytotoxicity against hepatic stellate cells. Front Immunol (2019) 10:477. doi: 10.3389/fimmu.2019.00477
38. Zhang Y, Wu Y, Shen W, Wang B, Yuan X. Crosstalk between NK cells and hepatic stellate cells in liver fibrosis (Review). Mol Med Rep (2022) 25:181–94. doi: 10.3892/mmr.2022.12724
39. Wang Y, Zhang C. The roles of liver-resident lymphocytes in liver diseases. Front Immunol (2019) 10:1582. doi: 10.3389/fimmu.2019.01582
40. Huang YH, Chen YX, Zhang LJ, Chen ZX, Wang XZ. Hydrodynamics-based transfection of rat interleukin-10 gene attenuates porcine serum-induced liver fibrosis in rats by inhibiting the activation of hepatic stellate cells. Int J Mol Med (2014) 34:677–86. doi: 10.3892/ijmm.2014.1831
41. Shah R, Reyes-Gordillo K, Arellanes-Robledo J, Lechuga CG, Hernandez-Nazara Z, Cotty A, et al. TGF-beta1 up-regulates the expression of PDGF-beta receptor mRNA and induces a delayed PI3K-, AKT-, and p70(S6K) -dependent proliferative response in activated hepatic stellate cells. Alcohol Clin Exp Res (2013) 37:1838–48. doi: 10.1111/acer.12167
42. Glassner A, Eisenhardt M, Kramer B, Korner C, Coenen M, Sauerbruch T, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest (2012) 92:967–77. doi: 10.1038/labinvest.2012.54
43. Fan Y, Zhang W, Wei H, Sun R, Tian Z, Chen Y. Hepatic NK cells attenuate fibrosis progression of non-alcoholic steatohepatitis in dependent of CXCL10-mediated recruitment. Liver Int (2020) 40:598–608. doi: 10.1111/liv.14307
44. Huang YH, Chen MH, Guo QL, Chen ZX, Chen QD, Wang XZ. Interleukin-10 induces senescence of activated hepatic stellate cells via STAT3-p53 pathway to attenuate liver fibrosis. Cell Signal (2020) 66:109445. doi: 10.1016/j.cellsig.2019.109445
45. Choi WM, Ryu T, Lee JH, Shim YR, Kim MH, Kim HH, et al. Metabotropic glutamate receptor 5 in natural killer cells attenuates liver fibrosis by exerting cytotoxicity to activated stellate cells. Hepatology (2021) 74:2170–85. doi: 10.1002/hep.31875
46. Tosello-Trampont AC, Krueger P, Narayanan S, Landes SG, Leitinger N, Hahn YS. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology (2016) 63:799–812. doi: 10.1002/hep.28389
47. Wijaya RS, Read SA, Schibeci S, Eslam M, Azardaryany MK, El-Khobar K, et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis b. J Hepatol (2019) 71:252–64. doi: 10.1016/j.jhep.2019.03.012
48. Yoneda A, Sakai-Sawada K, Niitsu Y, Tamura Y. Vitamin a and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp Cell Res (2016) 341:8–17. doi: 10.1016/j.yexcr.2016.01.012
49. Zheng S, Yang W, Yao D, Tang S, Hou J, Chang X. A comparative study on roles of natural killer T cells in two diet-induced non-alcoholic steatohepatitis-related fibrosis in mice. Ann Med (2022) 54:2233–45. doi: 10.1080/07853890.2022.2108894
50. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol (2014) 14:181–94. doi: 10.1038/nri3623
51. Khan HA, Ahmad MZ, Khan JA, Arshad MI. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat Dis Int (2017) 16:245–56. doi: 10.1016/s1499-3872(17)60014-6
52. Wang H, Yin S. Natural killer T cells in liver injury, inflammation and cancer. Expert Rev Gastroenterol Hepatol (2015) 9:1077–85. doi: 10.1586/17474124.2015.1056738
53. Marrero I, Maricic I, Feldstein AE, Loomba R, Schnabl B, Rivera-Nieves J, et al. Complex network of NKT cell subsets controls immune homeostasis in liver and gut. Front Immunol (2018) 9:2082. doi: 10.3389/fimmu.2018.02082
54. Fujishima S, Shiomi T, Yamashita S, Yogo Y, Nakano Y, Inoue T, et al. Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch Pathol Lab Med (2010) 134:1136–42. doi: 10.5858/2009-0144-OA.1
55. Xu Y, Liang P, Bian M, Chen W, Wang X, Lin J, et al. Interleukin-13 is involved in the formation of liver fibrosis in clonorchis sinensis-infected mice. Parasitol Res (2016) 115:2653–60. doi: 10.1007/s00436-016-5012-7
56. Lee HL, Jang JW, Lee SW, Yoo SH, Kwon JH, Nam SW, et al. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci Rep (2019) 9:3260. doi: 10.1038/s41598-019-40078-8
57. Luo XY, Takahara T, Kawai K, Fujino M, Sugiyama T, Tsuneyama K, et al. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol (2013) 305:G891–99. doi: 10.1152/ajpgi.00193.2013
58. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell (2014) 26:549–64. doi: 10.1016/j.ccell.2014.09.003
59. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature (2021) 592:444–49. doi: 10.1038/s41586-021-03233-8
60. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol (2022) 22:429–43. doi: 10.1038/s41577-021-00639-3
61. Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology (2018) 67:1970–85. doi: 10.1002/hep.29636
62. Wang XM, Holz LE, Chowdhury S, Cordoba SP, Evans KA, Gall MG, et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol Cell Biol (2017) 95:443–53. doi: 10.1038/icb.2016.116
63. Cargill T, Culver EL. The role of b cells and b cell therapies in immune-mediated liver diseases. Front Immunol (2021) 12:661196. doi: 10.3389/fimmu.2021.661196
64. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 194:2059–63. doi: 10.4049/jimmunol.1402256
65. Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol Rev (2013) 255:165–81. doi: 10.1111/imr.12087
66. Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y, et al. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun (2021) 12:4474. doi: 10.1038/s41467-021-24734-0
67. Pallett LJ, Maini MK. Liver-resident memory T cells: life in lockdown. Semin Immunopathol (2022) 44:813–25. doi: 10.1007/s00281-022-00932-w
68. Wang X, Tian Z. Gammadelta T cells in liver diseases. Front Med (2018) 12:262–68. doi: 10.1007/s11684-017-0584-x
69. Qu G, Wang S, Zhou Z, Jiang D, Liao A, Luo J. Comparing mouse and human tissue-resident gammadelta T cells. Front Immunol (2022) 13:891687. doi: 10.3389/fimmu.2022.891687
70. Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology (2008) 47:729–36. doi: 10.1002/hep.22034
71. Liu Q, Yang Q, Wu Z, Chen Y, Xu M, Zhang H, et al. IL-1beta-activated mTORC2 promotes accumulation of IFN-gamma(+) gammadelta T cells by upregulating CXCR3 to restrict hepatic fibrosis. Cell Death Dis (2022) 13:289. doi: 10.1038/s41419-022-04739-3
72. Hammerich L, Bangen JM, Govaere O, Zimmermann HW, Gassler N, Huss S, et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology (2014) 59:630–42. doi: 10.1002/hep.26697
73. Seo W, Eun HS, Kim SY, Yi HS, Lee YS, Park SH, et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology (2016) 64:616–31. doi: 10.1002/hep.28644
74. Floreani A, Liberal R, Vergani D, Mieli-Vergani G. Autoimmune hepatitis: Contrasts and comparisons in children and adults - a comprehensive review. J Autoimmun (2013) 46:7–16. doi: 10.1016/j.jaut.2013.08.004
75. Rosser EC, Mauri C. Regulatory b cells: origin, phenotype, and function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
76. Liu Y, Cheng LS, Wu SD, Wang SQ, Li L, She WM, et al. IL-10-producing regulatory b-cells suppressed effector T-cells but enhanced regulatory T-cells in chronic HBV infection. Clin Sci (Lond) (2016) 130:907–19. doi: 10.1042/CS20160069
77. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, et al. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol (2020) 11:1169. doi: 10.3389/fimmu.2020.01169
78. Global hepatitis report (2017). Available at: https://apps.who.int/iris/handle/10665/255016.
79. The L. Viral hepatitis elimination: A challenge, but within reach. Lancet (2022) 400:251. doi: 10.1016/S0140-6736(22)01377-0
80. Nagatsuma K, Hano H, Murakami K, Shindo D, Matsumoto Y, Mitobe J, et al. Hepatic stellate cells that coexpress LRAT and CRBP-1 partially contribute to portal fibrogenesis in patients with human viral hepatitis. Liver Int (2014) 34:243–52. doi: 10.1111/liv.12255
81. Xie X, Lv H, Liu C, Su X, Yu Z, Song S, et al. HBeAg mediates inflammatory functions of macrophages by TLR2 contributing to hepatic fibrosis. BMC Med (2021) 19:247. doi: 10.1186/s12916-021-02085-3
82. Liu C, Chen X, Yang L, Kisseleva T, Brenner DA, Seki E. Transcriptional repression of the transforming growth factor beta (TGF-beta) pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by nuclear factor kappaB (NF-kappaB) p50 enhances TGF-beta signaling in hepatic stellate cells. J Biol Chem (2014) 289:7082–91. doi: 10.1074/jbc.M113.543769
83. Tao L, Xue D, Shen D, Ma W, Zhang J, Wang X, et al. MicroRNA-942 mediates hepatic stellate cell activation by regulating BAMBI expression in human liver fibrosis. Arch Toxicol (2018) 92:2935–46. doi: 10.1007/s00204-018-2278-9
84. Cong M, Liu T, Wang P, Fan X, Yang A, Bai Y, et al. Antifibrotic effects of a recombinant adeno-associated virus carrying small interfering RNA targeting TIMP-1 in rat liver fibrosis. Am J Pathol (2013) 182:1607–16. doi: 10.1016/j.ajpath.2013.01.036
85. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology (2010) 139:323–34. doi: 10.1053/j.gastro.2010.03.052
86. Wang S, He L, Xiao F, Gao M, Wei H, Yang J, et al. Upregulation of GLT25D1 in hepatic stellate cells promotes liver fibrosis via the TGF-beta1/SMAD3 pathway In vivo and In vitro. J Clin Transl Hepatol (2023) 11:1–14. doi: 10.14218/JCTH.2022.00005
87. Da SML, Marson RF, Solari M, Nardi NB. Are liver pericytes just precursors of myofibroblasts in hepatic diseases? insights from the crosstalk between perivascular and inflammatory cells in liver injury and repair. Cells (2020) 9:188. doi: 10.3390/cells9010188
88. Zan Y, Zhang Y, Tien P. Hepatitis b virus e antigen induces activation of rat hepatic stellate cells. Biochem Biophys Res Commun (2013) 435:391–6. doi: 10.1016/j.bbrc.2013.04.098
89. Wang P, Lin T, Yang P, Yeh C, Pan T. Hepatic stellate cell modulates the immune microenvironment in the progression of hepatocellular carcinoma. Int J Mol Sci (2022) 23:10777. doi: 10.3390/ijms231810777
90. Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, et al. TGF-beta1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PloS Pathog (2012) 8:e1002594. doi: 10.1371/journal.ppat.1002594
91. Tjwa ET, van Oord GW, Hegmans JP, Janssen HL, Woltman AM. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis b. J Hepatol (2011) 54:209–18. doi: 10.1016/j.jhep.2010.07.009
92. Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev (2016) 28:53–61. doi: 10.1016/j.cytogfr.2015.10.002
93. Kocabayoglu P, Lade A, Lee YA, Dragomir AC, Sun X, Fiel MI, et al. Beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J Hepatol (2015) 63:141–47. doi: 10.1016/j.jhep.2015.01.036
94. Bai Q, An J, Wu X, You H, Ma H, Liu T, et al. HBV promotes the proliferation of hepatic stellate cells via the PDGF-B/PDGFR-beta signaling pathway in vitro. Int J Mol Med (2012) 30:1443–50. doi: 10.3892/ijmm.2012.1148
95. Devhare PB, Sasaki R, Shrivastava S, Di Bisceglie AM, Ray R, Ray RB. Exosome-mediated intercellular communication between hepatitis c virus-infected hepatocytes and hepatic stellate cells. J Virol (2017) 91:e02225-16. doi: 10.1128/JVI.02225-16
96. Kim JH, Lee CH, Lee SW. Exosomal transmission of MicroRNA from HCV replicating cells stimulates transdifferentiation in hepatic stellate cells. Mol Ther Nucleic Acids (2019) 14:483–97. doi: 10.1016/j.omtn.2019.01.006
98. Langeneckert AE, Lunemann S, Martrus G, Salzberger W, Hess LU, Ziegler AE, et al. CCL21-expression and accumulation of CCR7(+) NK cells in livers of patients with primary sclerosing cholangitis. Eur J Immunol (2019) 49:758–69. doi: 10.1002/eji.201847965
99. Wu S, Cao Y, Lu H, Qi X, Sun J, Ye Y, et al. Aberrant peribiliary gland niche exacerbates fibrosis in primary sclerosing cholangitis and a potential therapeutic strategy. BioMed Pharmacother (2022) 153:113512. doi: 10.1016/j.biopha.2022.113512
100. Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC, et al. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology (2014) 59:1007–15. doi: 10.1002/hep.26583
101. Sebode M, Peiseler M, Franke B, Schwinge D, Schoknecht T, Wortmann F, et al. Reduced FOXP3(+) regulatory T cells in patients with primary sclerosing cholangitis are associated with IL2RA gene polymorphisms. J Hepatol (2014) 60:1010–16. doi: 10.1016/j.jhep.2013.12.027
102. Zhu H, Liu Z, An J, Zhang M, Qiu Y, Zou MH. Activation of AMPKalpha1 is essential for regulatory T cell function and autoimmune liver disease prevention. Cell Mol Immunol (2021) 18:2609–17. doi: 10.1038/s41423-021-00790-w
103. Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, et al. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol (2010) 184:2886–98. doi: 10.4049/jimmunol.0901216
104. Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E, et al. Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology (2013) 58:1084–93. doi: 10.1002/hep.26447
105. Feng TT, Zou T, Wang X, Zhao WF, Qin AL. Clinical significance of changes in the Th17/Treg ratio in autoimmune liver disease. World J Gastroenterol (2017) 23:3832–38. doi: 10.3748/wjg.v23.i21.3832
106. Chakrabarty RP, Chandel NS. Mitochondria as signaling organelles control mammalian stem cell fate. Cell Stem Cell (2021) 28:394–408. doi: 10.1016/j.stem.2021.02.011
107. Navas LE, Carnero A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct Target Ther (2021) 6:2. doi: 10.1038/s41392-020-00354-w
108. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143:1158–72. doi: 10.1053/j.gastro.2012.09.008
109. Park J, Shao M, Kim MY, Baik SK, Cho MY, Utsumi T, et al. An endoplasmic reticulum protein, nogo-b, facilitates alcoholic liver disease through regulation of kupffer cell polarization. Hepatol (Baltimore Md.) (2017) 65:1720–34. doi: 10.1002/hep.29051
110. Matsuda M, Seki E. Hepatic stellate cell-macrophage crosstalk in liver fibrosis and carcinogenesis. Semin Liver Dis (2020) 40:307–20. doi: 10.1055/s-0040-1708876
111. Wu X, Yang Y, Li W, Cheng Y, Li X, Huang C, et al. Telomerase reverse transcriptase acts in a feedback loop with NF-κB pathway to regulate macrophage polarization in alcoholic liver disease. Sci Rep (2016) 6:77–93. doi: 10.1038/srep18685
112. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatol (Baltimore Md.) (2014) 59:77–93. doi: 10.1002/hep.26592
113. Yujia Z, Sufan W, Ting W, Yuanling H, Nengzhi P, Xuye J, et al. Cyanidin-3-O-β-glucoside inactivates NLRP3 inflammasome and alleviates alcoholic steatohepatitis via SirT1/NF-κB signaling pathway. Free Radic Biol Med (2020) 160:77–93. doi: 10.1016/j.freeradbiomed.2020.08.006
114. Liu X, Wang Y, Wu D, Li S, Wang C, Han Z, et al. Magnolol prevents acute alcoholic liver damage by activating PI3K/Nrf2/PPARgamma and inhibiting NLRP3 signaling pathway. Front Pharmacol (2019) 10:1459. doi: 10.3389/fphar.2019.01459
115. Wu X, Wang Y, Wang S, Xu R, Lv X. Purinergic P2X7 receptor mediates acetaldehyde-induced hepatic stellate cells activation via PKC-dependent GSK3beta pathway. Int Immunopharmacol (2017) 43:164–71. doi: 10.1016/j.intimp.2016.12.017
116. Wu XQ, Yang Y, Li WX, Cheng YH, Li XF, Huang C, et al. Telomerase reverse transcriptase acts in a feedback loop with NF-kappaB pathway to regulate macrophage polarization in alcoholic liver disease. Sci Rep (2016) 6:18685. doi: 10.1038/srep18685
117. Tom L, Neil K, Robert FS. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology (2014) 147:765–83. doi: 10.1053/j.gastro.2014.07.018
118. Luiz EBS, Paola DAM, Vanessa RF, Thiago FDAA, Patrícia TS, Suellen DSO, et al. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J Hepatol (2017) 67:716–26. doi: 10.1016/j.jhep.2017.05.021
119. Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut (2009) 58:704–20. doi: 10.1136/gut.2008.156307
120. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med (2007) 13:1324–32. doi: 10.1038/nm1663
121. Shashi B, Timea C, Banishree S, James Z, Karen K, Donna C, et al. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J Hepatol (2016) 64:1378–87. doi: 10.1016/j.jhep.2016.01.035
122. Maurice J, Manousou P. Non-alcoholic fatty liver disease. Clin Med (London England) (2018) 18:245–50. doi: 10.7861/clinmedicine.18-3-245
123. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota-driven activation of intrahepatic b cells aggravates nonalcoholic steatohepatitis through innate and adaptive signaling. Hepatol (Baltimore Md.) (2021) 74:704–22. doi: 10.1002/hep.31755
124. Sebastian T, Eva T, Ruth H, Adelheid ML, Jan M, Katharina T, et al. IL-6/Stat3-Dependent induction of a distinct, obesity-associated NK cell subpopulation deteriorates energy and glucose homeostasis. Cell Metab (2017) 26:171–84. doi: 10.1016/j.cmet.2017.05.018
125. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (London Engl 1979) (2016) 130:193–203. doi: 10.1042/CS20150405
126. Magar G, Xavier SR, Mark KN, Helen L, Kejing Z, Helena L, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol (2017) 2:eaai7616. doi: 10.1126/sciimmunol.aai7616
127. Kim SY, Jeong J, Kim SJ, Seo W, Kim M, Choi W, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun (2017) 8:2247. doi: 10.1038/s41467-017-02325-2
128. Liu HX, Keane R, Sheng L, Wan YJ. Implications of microbiota and bile acid in liver injury and regeneration. J Hepatol (2015) 63:1502–10. doi: 10.1016/j.jhep.2015.08.001
129. Yu J, Zhu C, Wang X, Kim K, Bartolome A, Dongiovanni P, et al. Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Sci Transl Med (2021) 13:eabe1692. doi: 10.1126/scitranslmed.abe1692
130. Wu X, Zhang F, Xiong X, Lu C, Lian N, Lu Y, et al. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway. IUBMB Life (2015) 67:312–21. doi: 10.1002/iub.1348
131. Xu Y, Fan W, Xu W, Jiang S, Chen G, Liu C, et al. Yiguanjian decoction enhances fetal liver stem/progenitor cell-mediated repair of liver cirrhosis through regulation of macrophage activation state. World J Gastroenterol (2018) 24:4759–72. doi: 10.3748/wjg.v24.i42.4759
132. Gan F, Liu Q, Liu Y, Huang D, Pan C, Song S, et al. Lycium barbarum polysaccharides improve CCl(4)-induced liver fibrosis, inflammatory response and TLRs/NF-kB signaling pathway expression in wistar rats. Life Sci (2018) 192:205–12. doi: 10.1016/j.lfs.2017.11.047
133. Xu Y, Tang X, Yang M, Zhang S, Li S, Chen Y, et al. Interleukin 10 gene-modified bone marrow-derived dendritic cells attenuate liver fibrosis in mice by inducing regulatory T cells and inhibiting the TGF-beta/Smad signaling pathway. Mediators Inflammation (2019) 2019:4652596. doi: 10.1155/2019/4652596
134. Ma Z, Xue X, Bai J, Cai Y, Jin X, Jia K, et al. Si-Wu-Tang ameliorates bile duct ligation-induced liver fibrosis via modulating immune environment. BioMed Pharmacother (2022) 155:113834. doi: 10.1016/j.biopha.2022.113834
135. Liu P, Li H, Gong J, Geng Y, Jiang M, Xu H, et al. Chitooligosaccharides alleviate hepatic fibrosis by regulating the polarization of M1 and M2 macrophages. Food Funct (2021) 13:753–68. doi: 10.1039/D1FO03768D
136. Zhao XA, Chen G, Liu Y, Chen Y, Wu H, Xiong Y, et al. Curcumin reduces Ly6C(hi) monocyte infiltration to protect against liver fibrosis by inhibiting kupffer cells activation to reduce chemokines secretion. BioMed Pharmacother (2018) 106:868–78. doi: 10.1016/j.biopha.2018.07.028
137. Tao X, Zhang R, Du R, Yu T, Yang H, Li J, et al. EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J Exp Med (2022) 219:e20212414. doi: 10.1084/jem.20212414
138. Rao J, Wang H, Ni M, Wang Z, Wang Z, Wei S, et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut (2022) 71:2539–50. doi: 10.1136/gutjnl-2021-325150
139. Qing J, Ren Y, Zhang Y, Yan M, Zhang H, Wu D, et al. Dopamine receptor D2 antagonism normalizes profibrotic macrophage-endothelial crosstalk in non-alcoholic steatohepatitis. J Hepatol (2022) 76:394–406. doi: 10.1016/j.jhep.2021.09.032
Keywords: hepatic fibrosis, hepatic immune microenvironment, chronic liver diseases, hepatic stellate cells (HSCs), immune cells
Citation: Zhang N, Yao H, Zhang Z, Li Z, Chen X, Zhao Y, Ju R, He J, Pan H, Liu X and Lv Y (2023) Ongoing involvers and promising therapeutic targets of hepatic fibrosis: The hepatic immune microenvironment. Front. Immunol. 14:1131588. doi: 10.3389/fimmu.2023.1131588
Received: 25 December 2022; Accepted: 02 February 2023;
Published: 16 February 2023.
Edited by:
Jinhang Gao, Sichuan University, ChinaCopyright © 2023 Zhang, Yao, Zhang, Li, Chen, Zhao, Ju, He, Pan, Liu and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Liu, bGl1eGlhb2xpMDEwOEB4anR1LmVkdS5jbg==; Yi Lv, bHV5aTE2OUAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.