 Yang Du
Yang Du Zhiqiang Hu
Zhiqiang Hu Yien Luo4
Yien Luo4 Helen Y. Wang
Helen Y. Wang Xiao Yu
Xiao Yu Rong-Fu Wang
Rong-Fu Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 February 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1130423
This article is part of the Research Topic cGAS-STING Pathway in Infectious and Non-Infectious Diseases View all 5 articles
The efficacious detection of pathogens and prompt induction of innate immune signaling serve as a crucial component of immune defense against infectious pathogens. Over the past decade, DNA-sensing receptor cyclic GMP-AMP synthase (cGAS) and its downstream signaling adaptor stimulator of interferon genes (STING) have emerged as key mediators of type I interferon (IFN) and nuclear factor-κB (NF-κB) responses in health and infection diseases. Moreover, both cGAS-STING pathway and pathogens have developed delicate strategies to resist each other for their survival. The mechanistic and functional comprehension of the interplay between cGAS-STING pathway and pathogens is opening the way for the development and application of pharmacological agonists and antagonists in the treatment of infectious diseases. Here, we briefly review the current knowledge of DNA sensing through the cGAS-STING pathway, and emphatically highlight the potent undertaking of cGAS-STING signaling pathway in the host against infectious pathogenic organisms.
The innate immune system serves as the first line of host defense against infectious diseases by detecting the pathogen-associated molecular patterns (PAMPs) of the pathogens. This task relies on the germline-encoded pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), RIG-I-like receptors, Nod-like receptors (NLRs), C-type lectin receptors and intracellular DNA and RNA sensors (1–3). Upon detection of PAMPs from pathogens, signaling cascades through these receptors lead to the production of inflammatory cytokines and chemokines, as well as the initiation of cell death to eliminate infected cells (3–5).
Sensing the microbial DNA, best known as the blueprint of life, acts as a key element in triggering host defense, and spans across a wide variety of species. In mammalian cells, three major innate immune receptors have been identified as responsible for DNA sensing, including TLR9, absent in melanoma 2 (AIM2), and cyclic GMP–AMP synthase (cGAS). TLR9 localizes on the endosomal membrane and recognizes single-stranded DNA (ssDNA) specifically containing unmethylated cytidine-phosphate-guanosine (CpG) motifs from bacteria or viruses (6, 7). AIM2 contributes to the recognition of double-stranded DNA (dsDNA) in the cytosolic compartment through the formation of inflammasomes, inducing the activation of cysteine-aspartic proteases 1 (Caspase 1), resulting in the proteolytic maturation of the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18, as well as cell pyroptosis (8–13). Recently, dsDNA sensing, which is mediated by cyclic-GMP-AMP (cGAMP) synthase (cGAS)-stimulator of interferon genes (STING) pathway, has been considered as a crucial element for linking the recognition of pathogens DNA to the establishment of host innate immune defense state (1, 14, 15). Upon binding to dsDNA, the catalytic activity of cGAS is triggered, leading to the production of 2’3’-cGAMP, which acts as the secondary messenger that subsequently binds and activates the adaptor protein STING on the endoplasmic reticulum (ER) membrane, and mediates downstream pathway to produce an array of type I and type III interferons (IFNs) (16, 17). The existence of homologs of cGAS and/or STING in various organisms and the sequence-independent manner of sensing dsDNA from pathogens confer cGAS-STING signaling pathway a powerful role in the innate immune system (18). In this review, we briefly summarize the current understanding of DNA sensing through the cGAS-STING pathway and mainly focus on the potent capacity of cGAS-STING signaling pathway in the host immunity against infectious diseases.
DNA is mostly squeezed in the nucleus and mitochondria and expeditiously degraded by cytosolic and lysosomal DNA-degrading nucleases in normal circumstances (19). Upon infections, increased amounts of foreign intracellular DNA are sensed by cGAS (20, 21), which belongs to the nucleotidyltransferase (NTase) enzyme family (22). cGAS is composed of a C-terminal NTase domain and an N-terminal highly positively charged domain (21, 23–25). cGAS binds to dsDNA in a sequence-independent manner. Upon interacting with DNA, the catalytic domain of cGAS undergoes a structure conformational change, which induces the synthesis of 2’3’-cGAMP from adenosine triphosphate (ATP) and guanosine triphosphate (GTP) (24, 26, 27). Although cGAS could bind with short dsDNA (~20 bp), it prefers longer length of DNA (>45 bp), which could facilitate cGAS to form more stable dimers and trigger a stronger enzymatic activity (28, 29). Notably, ssDNA could also interact with cGAS (30–32). However, such binding cannot lead to the activation of cGAS, suggesting precise enzymatic activation of cGAS by dsDNA. Besides the C-terminal NTase domain, the N-terminal domain is reported to facilitate the synthesis of 2’3’-cGAMP by increasing the enzymic activation of cGAS via induction of cGAS-DNA liquid–liquid phase separation (29). The occurrence of such phase separation is dependent on a high concentration of cGAS and DNA (each exceeded 30 nM) (29), illustrating that only when the amount of cytosolic DNA reaches a sufficient level, cGAS is activated. This critical threshold of activation of cytosolic cGAS by foreign intracellular DNA renders a proper safeguard mechanism for the host. However, in the case of exposure to self-DNA, cGAS can cause auto-inflammation and pathology (31, 33–36). Besides cytoplasm, cGAS is also present in the nucleus which is rich in genomic DNA (37, 38). The nuclear localization of cGAS relies on a structurally more accessible cGAS catalytic domain which is also necessary for cGAS tethering to chromatin (38). Based on this, emerging studies have focused on why nuclear cGAS is unable to be activated by self-genomic DNA. In 2020, several groups employed Cryo-EM and revealed 11 structures of cGAS bound to the nucleosome in total, and these interactions are mainly dependent on histones H2A/H2B, which suggested how cGAS is tethered to and suppressed by chromatin inside nuclear and thus prevent autoreactivity (39–44). Consistently, Li et al. reported that cGAS activity could be suppressed via hyperphosphorylation at the N terminus and inhibition of oligomerization due to chromatin tethering during mitosis, which could prevent cGAS phase separation into liquid droplets to synthesize cGAMP (45). Generally, these findings identify cGAS as a vital guard in maintaining cell homeostasis and defending against danger from outside.
2’3’-cGAMP, then binds to STING, on the ER membrane (16, 26, 46). STING contains an N-terminal transmembrane segment, and a cytoplasmic ligand-binding domain (LBD), followed by a C-terminal tail (CTT) (47–51). In the absence of a ligand, STING forms a homo-dimer through two CTTs, exhibiting as a V-shaped binding pocket facing the cytosol. In the presence of cGAMP, cGAMP binding leads to the closure of the ligand-binding pocket and subsequently triggers a 180° rotation of LBD relative to the transmembrane domain. This rotation is accompanied by a conformational rearrangement on the side of the LBD dimer, inducing the formation of the STING tetramer and higher-order oligomers (51). This ligand-induced oligomerization then triggers the translocation of STING from ER to ER-Golgi intermediate compartment (ERGIC) and the Golgi (46, 52). The ER-to-Golgi traffic is dependent on the GTPase SAR1A, coat protein complex-II (COP-II), and ADP-ribosylation factor (ARF) GTPases (53, 54). So far, what signal within STING is sensed to regulate this trafficking process remains poorly understood. It is reported that STING is retained on the ER membrane by Ca2+ sensor stromal interaction molecule 1 (STIM1), restricting the activation of STING via its translocation from the ER to the ER-Golgi intermediate compartment (55). On the contrary, STEEP facilitates STING ER exit through elevating phosphatidylinositol-3-phosphate (PtdIns(3)P) production and ER membrane curvature formation (56). Recently, abnormal Golgi-to-ER STING retrieval is reported in COPA syndrome, which is characterized by chronic up-regulation of type I IFN signaling. Further mechanism study shows that mutations in COPA result in the accumulation of ER-resident STING at the Golgi, leading to enhanced type I IFN signaling (57).
Once trafficking to the Golgi compartments, STING recruits TANK-binding kinase 1 (TBK1) to initiate downstream signaling (16). STING is known to undergo diverse post-translational modifications, including phosphorylation, palmitoylation, ubiquitination, SUMOylation, nitro-alkylation, glycosylation, carbonylation, reversible oxidation, and et al. (58–60). Palmitoylation of STING at the trans-Golgi network (TGN) is essential for the activation of STING. Mutation at cysteine residues 88/91 (Cys88/91) attenuates the palmitoylation and abolishes the induction of type I IFN response (61). In addition, covalent small-molecule inhibitors that target the Cys91 block the activation-induced palmitoylation of STING, which is essential for the clustering of STING at the Golgi apparatus and, in turn, for the recruitment of TBK1 (62). Following STING trafficking to the Golgi compartments, STING oligomers recruit TBK1 through the conserved PLPLRT/SD amino acid binding motif within CTT, which triggers the activation of TBK1 via dimerization-mediated TBK1 autophosphorylation at Ser172, a residue that is important for TBK1 activation (63–65). In turn, STING is phosphorylated by TBK1 at Ser366 within CTT, which is part of pLxIS (p, hydrophilic residue; x, any residue; S, phosphorylation site). This phosphorylated STING then recruits IRF3, facilitating the phosphorylation of IRF3 by TBK1 (66). Notably, either S366A STING or L374A STING abrogates the interaction between STING and IRF3, and subsequent phosphorylation of IRF3 by TBK1 (65). Activated IRF3 dimers then translocate to the nucleus to activate the transcription of type I IFNs, which in turn leads to the production of diverse IFN-stimulated genes (ISGs), establishing a robust anti-infection state (67). Besides type I IFN signaling cascades, STING also activates nuclear factor-κB (NF-κB) through a poorly discovered mechanism. Emerging evidence suggests STING induces NF-κB activity via a TBK1-independent mechanism, which is distinct from IRF3 activation. NF-κB activation through the miniCTT (subdomain in the C-terminal domain), distinct from the TBK1 binding domain, suggests the activation is not fully dependent on the CTT, which rules out a role for TBK1 (68). Blocking K224 and K288 ubiquitination of STING strongly impairs IRF3 but not NF-κB activation. Mechanism studies show that K224 and K288 on STING are critical for trafficking from ER to Golgi, illustrating that STING may activate the NF-κB pathway prior to its trafficking to the Golgi compartment for TBK1 interaction (69, 70). Besides, TBK1 is dispensable for NF-κB activation downstream of STING in myeloid cells. TBK1 acts redundantly with IκB kinase ε (IKKε, also known as IKKi) to induce NF-κB upon STING activation. Notably, TAK1 and IKKβ are essential for STING-induced-NF-κB signaling. A study in zebrafish shows that the conserved PxExxD motif at the CTT of zebrafish STING could directly associate with tumor necrosis factor receptor associated factor 6 (TRAF6) to trigger NF-κB signaling (71). In addition, in an etoposide-induced DNA damage context, STING-mediated NF-κB activation relies on TRAF6-dependent K63-linked ubiquitylation of STING, in which process involves interferon-γ-inducible factor 16 (IFI16) and tumor suppressor p53, and is independent on TBK1 (72), suggesting in certain contexts, STING may dictate cytokine production through alternative routes. Taken together, the cGAS-STING pathway utilizes diverse downstream factors to eliminate intracellular pathogens (Figure 1).
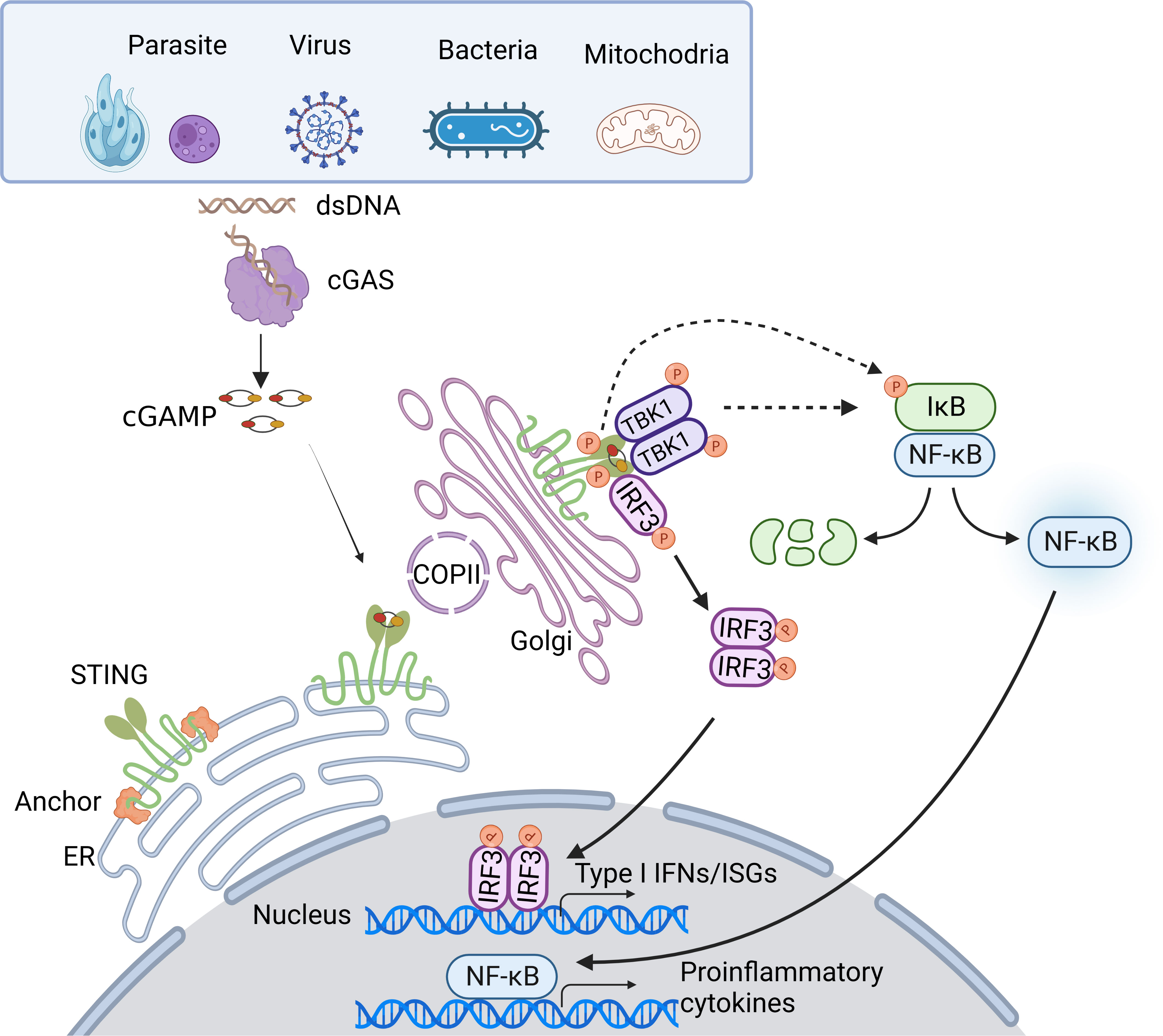
Figure 1 Overview of the cGAS-STING DNA-sensing pathway. Cytosolic DNA sensing by cGAS-STING pathway is implicated in pathogen infections. DNA from pathogens or infections-induced mitochondria release can enter the cytosol and initiate the cGAS-STING pathway. Upon binding dsDNA, cGAS is activated and produces 2’3’-cGAMP. cGAMP, the secondary messenger, subsequently binds STING, leading to conformational rearrangement, which induces the formation of the STING tetramer and higher-order oligomers release from anchor proteins, and translocation from the ER to Golgi via COPII. At Golgi, STING oligomers recruit TBK1, triggering TBK1 autophosphorylation. In turn, STING is phosphorylated by TBK1. Phosphorylated STING then recruits IRF3, facilitating the phosphorylation of IRF3 by TBK1. Activated IRF3 dimers then translocate to the nucleus to activate the transcription of type I IFNs, which in turn leads to the production of diverse IFN-stimulated genes (ISGs). Besides, STING induces NF-κB activity via TBK1-dependent and independent manners, resulting in the IκBα phosphorylation and subsequently translocation of NF-κB to the nucleus, leading to the production of inflammatory cytokines.
Parasitic diseases, caused by worms and protozoa, are one of the common infectious zoonoses that severely threaten host health. Anti-parasite innate immunity is the key weapon for the host defense against parasitic invasion, which also decides the end of parasitosis’s clinical outcome. In the process of parasite invasion, DNA released from pathogens is sensed by multiple nucleic acid sensors. Among them, cGAS-STING signaling is the most widely studied immune pathway participating in innate immunity against Plasmodium, Toxoplasma gondii (T. gondii), Trypanosoma cruzi (T. cruzi), and Schistosoma mansoni (S. mansoni) infection (Figure 2).
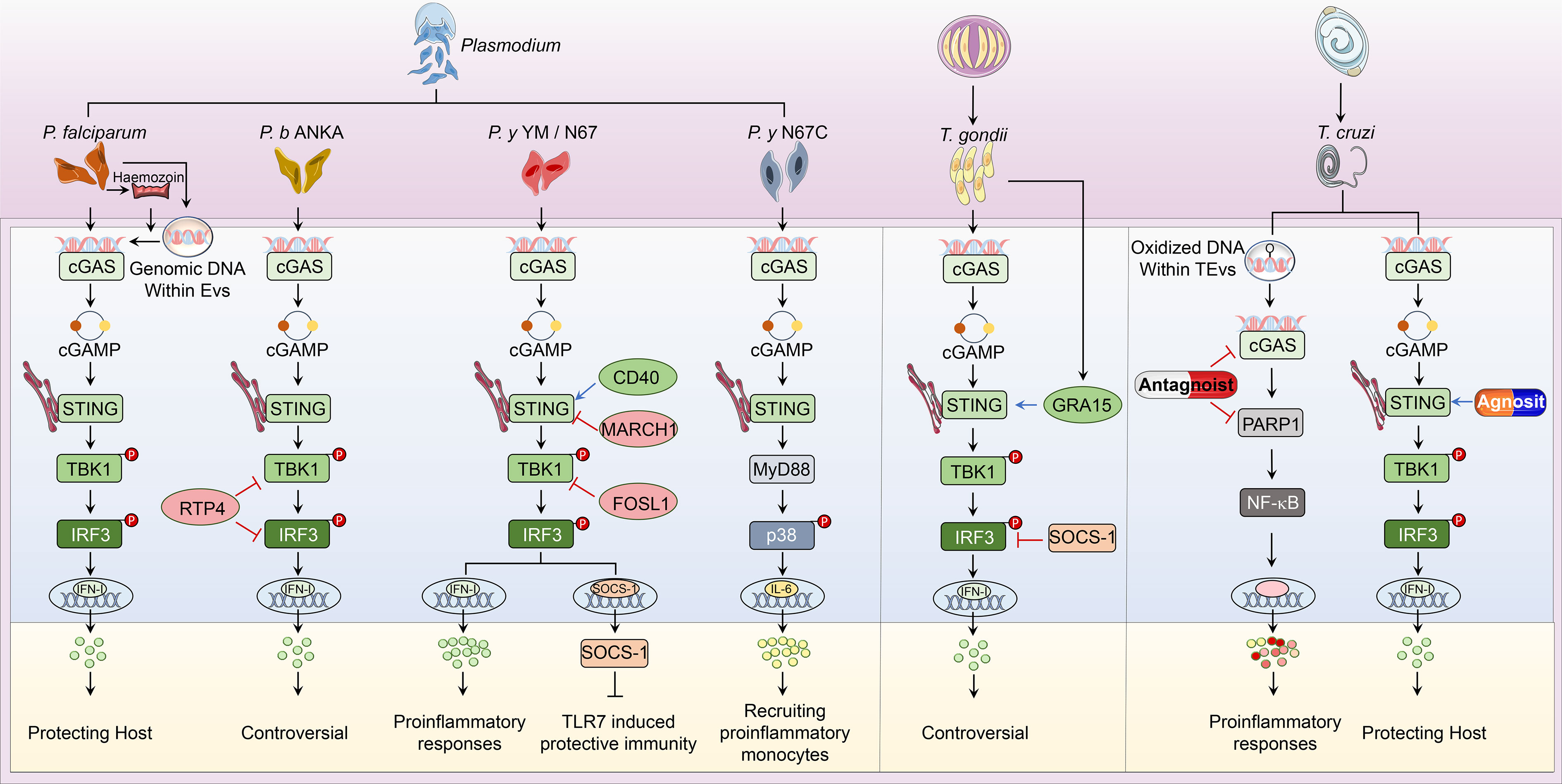
Figure 2 Overview of cGAS-STING pathway in host defense against parasitic infection. i. The classical cGAS-STING-type I IFN pathway. cGAS is an innate immune sensor that recognizes a diverse array of parasitic genomic DNA of Plasmodium, T. gondii, and T. cruzi, as well as EVs containing P. falciparum. Upon binding parasitic gDNA, cGAS oligomerizes with these gDNAs and performs its catalytic role to synthesize 2’3’ cGAMP. cGAMP then binds to STING at the ER membrane and stimulates the oligomerization of STING. Following, STING translocates from the ER to Golgi and recruits TBK1, which induces TBK1 autophosphorylation. Activated TBK1 phosphorylates STING, which in turn recruits IRF3 for TBK1-mediated phosphorylation. Phosphorylated IRF3 dimers translocate to the nucleus and trigger type I IFN production, which determines the established or disputed disease outcomes. ii. The nonclassical cGAS-STING pathway. During P. y YM infection, cGAS-STING signaling induces a negative regulator SOCS1 expression to inhibit TLR7-MyD88-mediated type I IFN responses in pDCs. For P. y N67C infection, cGAS-STING-MyD88-p38 signaling specifically induces late IL-6 production, which expands CD11b+Ly6Chi proinflammatory monocytes to inhibit immunity. Upon T. cruzi infection, EVs containing T. cruzi gDNA activate cGAS and trigger the PARP1-dependent NF-κB signaling, which induces proinflammatory responses. iii. Regulators of cGAS-STING pathway. RTP4, acting as a negative regulator, targets both TBK1 and IRF3 during P. b ANKA infection. Upon P. y YM/N67 infection, MARCH1 and FOSL1 negatively adjust STING and TBK1 respectively. Besides, CD40 is a positive regulator of STING-promoting type I IFN responses. For T. gondii infection, SOCS1 interacts with IRF3 to suppress cGAS-STING-type I IFN signaling, but GRA15 secreted from this parasite enhances type I IFN responses by activating STING. At the phase of T. cruzi infection, cGAS or PARP1-specific antagonists have been used to suppress cGAS-PARP1-NF-κB signaling, which limits harmful proinflammatory responses. Besides, vaccines containing STING agonists could also enhance anti-T. cruzi immunity by promoting type I IFN signaling.
Malaria caused by Plasmodium infection is a deadly infectious disease that affects 247 million people worldwide; it resulted in about 619,000 global deaths in 2021 [WHO, 2022]. During malaria infection, multiple Plasmodium biological products, such as GPI-anchors (73–77), heamozoin (78–82), genomic DNA (gDNA) (78, 83–88) and RNA (87, 89, 90), are acting as PAMPs recognized by host PRRs existing in innate immune cells including monocytes, macrophages, conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs) (84, 91–96). Among these PAMPs-PPRs interactions, cGAS is a non-negligible sensor sensing Plasmodium gDNA in addition to TLR9. Early studies revealed Plasmodium gDNA, which is rich in AT-motif (97), could induce type I IFN responses via an uncharted DNA receptor but not these well-known sensors like TLR9, DAI, RNA polymerase-III or IFI16/p204 (86). To explore this hidden character recognizing AT-rich DNA, we built a Plasmodium yoelii (P. y) YM infection mouse model using Mb21d1–/– (coding for cGAS), and Tmem173gt mice. Tmem173gt is an I199N missense mutant allele of the STING gene (formerly, Tmem173). Tmem173gt mice cannot produce IFN-β in response to cyclic dinucleotides (98). In line with our in vitro study, we found that cGAS functions as a DNA sensor in pDCs for detecting YM gDNA at the early infection stage and primarily induces STING-mediated type I IFN signaling (99). Consistently, by using human monocytes, another research group also identified cGAS as the cytosolic sensor of P. falciparum (P. f.) gDNA, and access of parasitic gDNA to the cytosolic compartment was mediated by Plasmodium hemozoin (100). In addition, extracellular vesicles (EVs) secreted by P. f. infected RBCs are confirmed as a vehicle transferring parasitic genomic DNA to immune cells, which then stimulate STING-TBK1-IRF3-dependent gene induction and type I IFN production (101). Overall, these researches indicate that cGAS is essential for Plasmodium gDNA detection and STING-triggered type I IFN responses during malaria.
Although cGAS-STING mediated activation of type I IFN signaling is solid, the effects of cGAS-STING signaling on the pathological outcome of Plasmodium-infected hosts are still controversial (102). With regards to the role of type I IFN in P. falciparum infection, several clinical studies have indicated that children with mild malaria show a higher IFN-α level than those with severe malaria (103). Lower circulating IFN-α is also observed in children from Kenya with severe malaria anemia (SMA). Two polymorphisms [IFN-α2 (A173T) and IFN-α8 (T884A)] in IFN-α promoter regions, which reduce IFN-α generation, is related to increased susceptibility to SMA (104). As for the mouse malaria model, we firstly reported activation of cGAS-STING, and MDA5-MAVS by P. y YM infection, triggered IRF3-mediated limited production of IFN-α/β, which then induced negative regulator SOCS1 inhibit RNA-TLR7-MyD88-dependent more puissant type I IFN responses (99). Furthermore, we found SOCS1 expression is markedly reduced in Ifih1-/-, Mavs-/-, and Tmem173gt pDCs, allowing the activation of robust MyD88-dependent type I IFN production, which reveals a detrimental role of cGAS-STING signaling during YM infection (99). Consistently, Spaulding et al. also reported robust type I IFN production during YM infection requires priming of pDCs by activated CD169+ macrophages upon STING-mediated sensing of parasites in the bone marrow, and this type I IFN response causes severe disease outcomes (105). Although both groups show that cGAS-STING mediated signaling at the early YM infection stage is harmful to the host, the roles of STING in triggering type I IFN expression are quite different, which need to be further investigated. Recently, by using a lethal P. y N67C model, we further demonstrated the detrimental role of cGAS-STING signaling. The cGAS/STING activation recruits myeloid differentiation factor 88 (MyD88) and specifically activates the p38-dependent signaling pathway to produce late IL-6, which expands CD11b+Ly6Chi proinflammatory monocytes to inhibit anti-malaria immunity (88). However, when it comes to P. b ANKA infection, STING plays a unique disputable role due to the controversial function of type I IFN. Sharma et al. reported that STING is harmful to the host as it induced TBK1-IRF-type I IFN signaling, causing the short survival of the host during P. b ANKA infection (86), but we recently found rescuing the STING-induced type I IFN could lower parasitemia and alleviate neurologic symptoms (106). Generally, these findings establish that cGAS-STING-induced type I IFN accelerates inflammation pathology and promotes host mortality.
However, in some cases, cGAS-STING signaling enhances immunity in some non-lethal malaria models. Hahn et al. reported P. y 17XNL infection leads to a protective type I IFN response in WT control, while Mb21d1–/– and Tmem173gt mice hold higher parasitemia and worsened outcomes (107). They further compared the survival of Mb21d1–/– mice between lethal and nonlethal P. y infections and reached a coincident conclusion that cGAS-STING is harmful to YM-infected mice (107). Generally, cGAS-STING is a double edge sword for the host to fight against malaria, which strongly relies on parasite strains and host models.
Given the importance of cGAS-STING and downstream signaling in anti-malaria immunity, emerging studies have focused on the regulation of this pathway (108). We have indicated that upon P. y N67 or P. y YM infection, mice deficient in MARCH1 had significantly better survival rates than WT mice, which indicated the negative function of MARCH1 in generating protective immunity (87). We further showed that MARCH1 could interact with STING and lower its protein level (109). In addition to MARCH1, CD40 is another vital positive regulator we reported helping to defend against malaria as the increase of CD40 expression caused by P. y N67 infection could enhance the protein level of STING, which in turn enhances the type I IFN production at the early stage of infection and prolongs host survival (110). Mechanistically, CD40 induced by iRBCs, parasite DNA/RNA, and various TLR ligands could compete with STING to bind TRAF2/3 and/or TRAF6 to weaken the ubiquitination of STING, which promotes the stability of STING (110). Displaying as a key downstream molecule of cGAS-STING signaling, TBK1 is the central kinase to activate type I IFN (111, 112). Activated TBK1, in turn, phosphorylates STING, which allows the STING-TBK1 complex to recruit and phosphorylate IRF3, thus inducing type I IFN and ISGs expression (5, 66). We recently suggested that the type I IFN could promote RTP4 expression post P. b ANKA infection (106). Moreover, RTP4 specially binds to TBK1 and inhibits type I IFN response by inhibiting TBK1 and IRF3 expression and activation thereby weakening type I IFN production (106). Inhibition of RTP4 expression may help lower parasitemia and be beneficial to alleviate symptoms of cerebral malaria (CM) and other neuropathology (106). Besides RTP4, we also discovered FOSL1 as another suppressive regulator function on TBK1 through resisting the formation of TBK1/TRAF3/TRIF (TRIF, Toll/4ll-1 Receptor-domain-containing adapter-inducing interferon-beta) complexes by limiting K63 ubiquitination of TRAF3 and TRIF and finally leads to suppression of type I IFN production at early infection and liver stages, which eventually results in P. y N67 parasite fast growth and host death (113). Obviously, the function of these regulators targeting STING depends on the particular role of STING in different malaria models. Although many efforts have been paid for studies about the regulation of cGAS-STING, there are still lots of research gaps to be explored for the potential value of this signaling pathway in the prophylaxis and treatment of malaria.
Toxoplasma gondii (T. gondii) is an obligate intracellular eukaryotic parasite from the phylum Apicomplexa that infects up to one-third of the global population (114). Although it normally only causes mild illness in healthy hosts, toxoplasmosis is a common opportunistic infectious disease with high mortality in individuals who are immunocompromised like AIDS patients (115). It has been reported that specific Toxoplasma strain–host combinations may lead to detrimental outcomes during pregnancy (116).
Innate immunity acts as the first line of host defense and observes pathogen infection, which is essential for resisting T. gondii infection. Particularly, cGAS-STING is the considerable immune signaling devoted to anti-T. gondii innate immunity. Wang et al. initially employed mice deficient in cGAS or STING to explore their role in a mouse toxoplasmosis model and found that cGAS is necessary for the activation of anti-T. gondii immune signaling. Consistently, STING knockout mice are much more susceptible to T. gondii infection than WT mice due to the impaired protective type I IFN production (117). Interestingly, they also found that mice deficient in STING exhibited more severe toxoplasmosis symptoms than cGAS-deficient mice, which suggests that there might be some other sensors getting involved in the activation of STING (117). Besides, STING could protect the host from toxoplasmosis in another IDO1-dependent way. During toxoplasmosis, AKT phosphorylated STING forms a heterodimer with TICAM2 to promote IRF3-dependent transcription of indoleamine-pyrrole-2,3-dioxygenase-1 (IDO1), which is a critical kinase limiting parasite replication (118). However, some studies exhibited an opposing role of IRF3 that promotes the replication of T. gondii by using Irf3-/- mice and IRF3 knockout cells. They found that ISGs rather than type I IFN induced by parasite-activated IRF3 were indeed essential. Moreover, they defined parasite-IRF3 signaling activation (PISA) as a novel pro-parasitic signaling pathway, which is dependent on the cGAS-STING-TBK1 signaling and requires the adaptor TRIF (119, 120). Recently, we found that type I IFN induced during toxoplasmosis was harmful to the host by promoting PD-1 expression in T cells and destroying its function on secreting IFN-γ, but the underlying mechanism of how type I IFN generated needs to be further investigated (121).
The regulation of cGAS-STING during T. gondii infection can be normally divided into two types: the first regulation is induced by parasitic effectors, while another one is mediated by intrinsic regulators. Wang et al. found the T. gondii dense granule protein GRA15 enhanced STING polyubiquitination at Lys337 and promoted STING oligomerization in a TRAF-dependent manner, and they also verified GRA15-/- T. gondii was more virulent resulting in higher mortality of WT mice (117). A recent report expounded that T. gondii virulence factor TgROP18I can not only interact with IRF3 and inhibit IFN-β production, but also suppress the recruitment of ubiquitin, p62, and LC3 to the parasitophorous vacuole membrane (PVM) thus deactivating the autophagic inhibition of T. gondii proliferation (122). Furthermore, some intrinsic regulators can be induced during toxoplasmosis and act on cGAS-STING signaling. Gao et al. found that FAF1 expression level was downregulated by T. gondii infection via a PI3K/AKT dependent manner, which is correlated with enhanced IRF3 transcription activity. They further found that inhibiting PI3K/AKT pathway was essential for IRF3 nuclear translocation to activate the transcription of ISGs, thereby facilitating T. gondii proliferation (123). Caspase-1-induced IL-1β is beneficial to protecting the host against T. gondii infection, we recently found that IL-1/IL-1R signaling could improve the expression of SOCS1 which could interact with IRF3 to restrain type I IFN production and maintain T cell function (121). Hence, any effort to control and eradicate toxoplasmosis needs a better understanding of the regulation of innate immune responses to T. gondii infection, which is also required for the development of new effective vaccines and drugs.
Trypanosoma cruzi (T. cruzi) is another zoonotic protozoan parasite, which is the etiological agent of American trypanosomiasis, or Chagas disease, and is transmitted when the infected feces of the triatomine vector are inoculated through a bite site or an intact mucous membrane of the mammalian host (124, 125). T. cruzi infection is lifelong in the absence of effective treatment. The most important consequence of T. cruzi infection is cardiomyopathy, which occurs in 20% to 30% of infected persons (126). Similar to other protozoons, the surface of T. cruzi is decorated with a variety of PAMPs, which are mainly composed of glycosylated molecules that are attached to the cell membrane via a glycosylphosphatidylinositol (GPI) anchor, acting as potent inducers of immune response (127). Except for the GPI-induced innate immunity, Choudhuri et al. recently revealed oxidized DNA encapsulated by T. cruzi-induced extracellular vesicles (TEvs) is an important PAMP sensed by cGAS rather than TLR9, which was necessary for PARP1-dependent NF-κB activation and proinflammatory macrophages response leading to severe chronic inflammatory pathology in Chagas disease (128). Furthermore, these researches showed short-term treatment with cGAS antagonists or PARP1 inhibitors was sufficient to potently suppress TEv-induced cytokines’ expression in macrophages, which offered a potential therapy for controlling Chagas disease (128). However, a more recent study identified STING signaling as a pivotal role in splenic IFN-β and IL-6 expression at early infection (129). STING deficiency led to a weakened production of splenic parasite-specific IFN-γ as well as a reduction of IFN-γ/perforin-producing CD8+ T cells (129). Though this protective role of STING signaling was recently proposed, studies concentrating on STING agonists applied for the treatment of Chagas disease were first reported by Malchiodi’s group in 2017. Initially, this group found STING agonist c-di-AMP is an efficient adjuvant, which couples with Tc52, the parasitic antigen, could induce stronger Th17 plus Th1 specific cellular and humoral immune responses against T. cruzi infection (130). Subsequently, they employed c-di-AMP combined with another engineered chimeric parasitic antigen Traspain as a novel vaccine, which showed a strong protection during the whole course of the infection (131). In a recent study, they demonstrated that CpG/c-di-AMP as adjuvants could contribute to T cell priming and polyfunctional CD4+ and CD8+ T cell-mediated anti-T. cruzi immunity (132). Overall, these studied profoundly demonstrate that cGAS-STING acts as a protagonist of innate immunity against T. cruzi infection, and the vaccines formulated based on this signaling may be effective in the Chagas disease treatment.
Although some efforts have been made to explore the vital role of cGAS-STING signaling and the related regulation, strategies for parasitic pathogens counteracting cGAS-STING signaling are still less known. Regulators like host MARCH1, RTP4, SOCS1, and FOSL1, as well as pathogen TgROP18I that we discussed above are capable of suppressing cGAS-STING signaling during parasitic infections (99, 106, 109, 113, 121, 122). However, whether there are some other host or parasitic proteins responsible for pathogens to control cGAS-STING signaling in the manner of camouflaging cytosolic foreign DNA ligands, post-translational modification of cGAS-STING signaling molecules, degradation of the 2’3’-cGAMP, or some other means still needs further studied.
Innate immunity is essential to control viral infection at the early stage of host antiviral immunity. Viral infection triggers cGAS-STING signaling pathway that induces activation of type I IFN and NF-κB signaling to restrict viral infection and to sustain homeostasis. The feature that cGAS senses DNA in a sequence-independent manner enables cGAS to detect a broad range of intracellular viral nucleic acids, thus rendering a powerful role of cGAS in the anti-viral immune response. On the other hand, viruses have developed diverse strategies to counteract cGAS-STING axis for their survival (Figure 3).
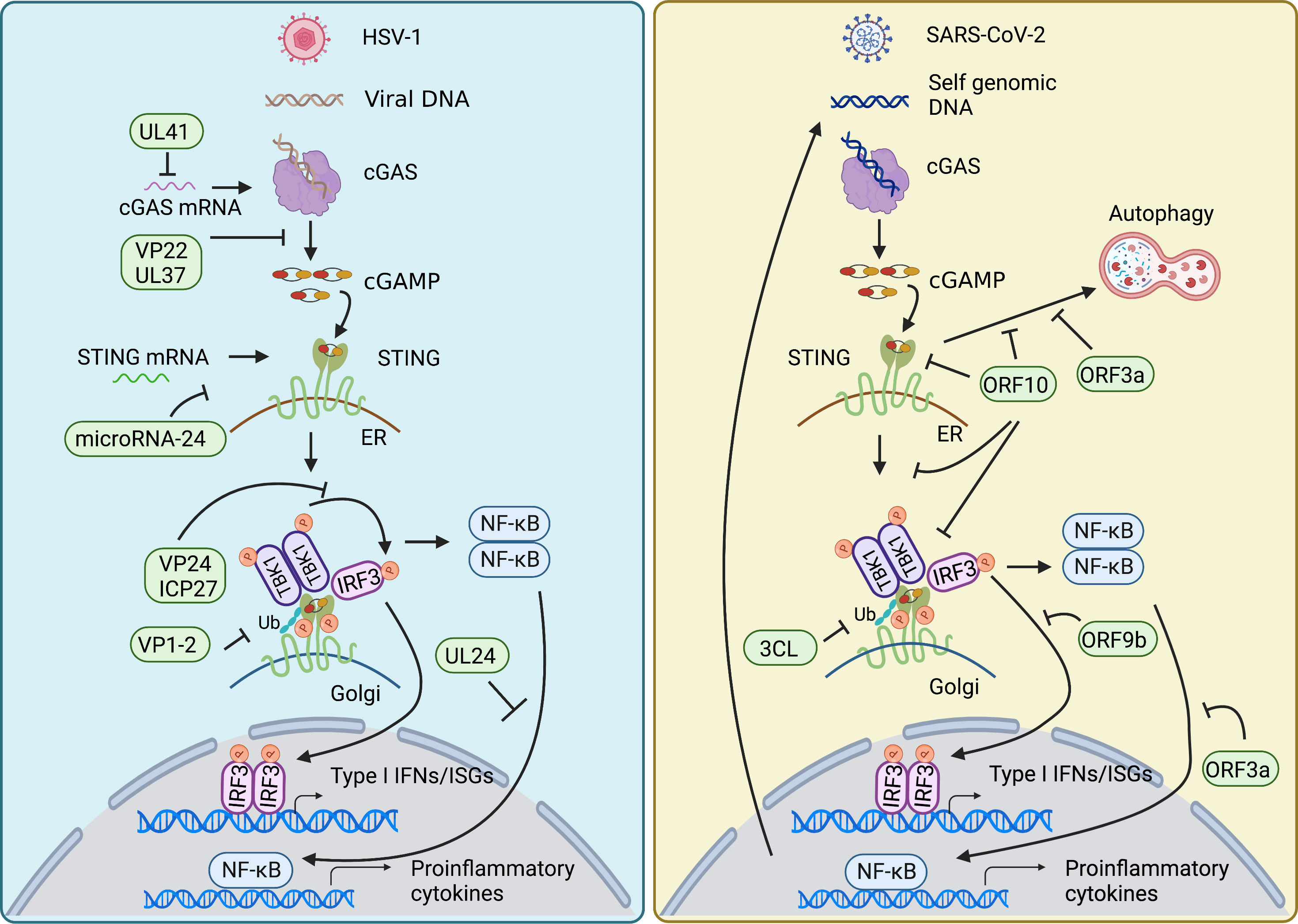
Figure 3 Viral evasion strategies in cGAS-STING pathway. HSV-1 and SARS-CoV-2 have developed diverse strategies to counteract cGAS-STING axis for their survival. Both mainly target signal transduction processes of the cGAS-STING pathway, such as decreasing the mRNA level of cGAS, damping the enzymatic activity of cGAS, impairing the translation of STING, restricting the translocation of STING from ER to Golgi, affecting the modification of STING, inhibiting the assembly of TBK1-activated STING signalosome, as well as nuclear translocation of NF-κB factor p65 and IRF3. In addition, SARS-CoV-2 also employs its encoding protein to intervene in STING-mediated autophagy to facilitate its replication.
Herpes simplex virus 1 (HSV-1), a member of the alphaherpesvirus family, is an enveloped virus with a linear dsDNA genome encoding at least 84 proteins. HSV-1 infection causes a variety of pathologies, ranging from benign cold sores to fatal encephalitis (133). Several reports demonstrated that ablation of cGAS or STING rendered mice sensitive to HSV-1 infection due to the deficiency of type I IFN production (46, 134, 135), suggesting the pivotal role of cGAS-STING-IFN axis during HSV-1 infection. Furthermore, autophagy induction via cGAMP-mediated STING’s trafficking from ER to Golgi is shown to be crucial for clearing HSV-1 in the cytosol of infected cells in vitro (54), implying that STING may contribute to antiviral responses in a type I IFN-independent manner. STING-S365A mice are shown to be resistant to HSV-1 infection (136–138). Since phosphorylation of serine 365 (S365) at CTT of STING by TBK1 is required for the recruitment of IRF3, which is essential for the phosphorylation of IRF3 and induction of type I IFNs (63, 65), the antiviral response in STING-S365A mice suggests the existence of type I IFN-independent mechanism. Among these findings, one study showed that the type I IFN-independent antiviral response is contributed by STING-induced, TBK1-dependent autophagy. CTT deficiency renders the mice incapable of autophagy induction, and susceptible to HSV-1 infection (137). Whereas another group observed that L373A STING mice, which lacked TBK1/IKKε activation but preserved autophagy induction were still susceptible to HSV-1 infection, implying that autophagy alone is not enough to power the antiviral response by STING. Moreover, they suggested that the elevated transcription of NF-κB-driven genes (CXCL1 [C-X-C Motif Chemokine Ligand 1], CXCL2, and 4-1BBL [4-1BB ligand]) in the cells from STING-S365A mice may contribute to viral resistance, indicating that NF-κB is a strong candidate for the IFN-independent STING-induced antiviral responses (138).
Although the host has evolved the delicate antiviral responses, such as inductions of type I IFN, NF-κB, and autophagy, it still could lead to a lifelong latent infection in the trigeminal ganglia, due to its ability to counteract the host’s innate antiviral response (139–141). HSV-1 tegument protein UL41, an endoribonuclease with the activity of mRNA-specific RNase, has been shown to reduce the mRNA level of cGAS, thus lowing the protein level of cGAS, and subsequently decreasing cGAS/STING-mediated IFN-β production (142). Besides the mRNA level, the enzymatic activity of cGAS is inhibited by tegument protein VP22 (143). Meanwhile, HSV-1 tegument protein UL37 deamidates human and mouse cGAS, restricting the ability of cGAS to catalyze cGAMP synthesis, which is important for downstream immune response. Notably, the deamidation site is not conserved in non-human primates, implying that HSV-1 utilizes species sequence variation to counteract host defenses (144). To antagonize the STING-mediated immune response, HSV-1 VP1-2 directly deubiquitinates STING and impairs its downstream signaling (145). In addition, HSV-1 VP24 and ICP27 intervene in the association between TBK1 and IRF3 and the TBK1-activated STING signalosome, respectively, impeding IRF3 activation and type I IFN induction (146, 147). Moreover, besides affecting type I IFN induction, HSV-1 UL24 selectively restricts NF-κB promoter activation through binding NF-κB subunits p65 and p50 (148). Of note, besides HSV-1 encoded proteins, microRNA-24, which is induced by HSV-1 infection, targets the 3’ untranslated region of STING mRNA and impairs its translation (149).
Human papillomavirus (HPV) is a circular double-stranded DNA virus, which can cause benign and malignant lesions in humans (150–153). The study of HPV has mainly been driven by the severity of HPV-associated pathologies. HPVs are the infectious agents of benign lesions, as well as anogenital and oropharyngeal cancers. About 99% of cervical cancers are caused by the infection of high-risk HPVs (HR-HPVs) (154). There are 15 identified HR-HPV types, including HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, and 82 (155). HPV-driven malignant transformation in cervical cancer is mainly correlated with biological processes mediated by the HPV oncogenes. HPV18 E7 has been shown to antagonize DNA sensing by using the LXCXE motif to interact with STING in cervical squamous cell carcinoma (156). Besides, HPV16 E7 is reported to interfere with cGAS-STING response in head and neck squamous cell carcinomas (HNSCCs) and oropharyngeal squamous cell carcinomas (OPSCC) (157, 158). However, the underlying mechanism through HPV16 E7 remains unknown. To figure out the mechanism, Xiaobo Luo et al. reported that HPV16 E7 in HNSCC shares low homology with HPV18 E7, and promotes autophagy-dependent degradation of STING via interacting with NLRX1 (159). Besides autophagy, HPV oncogenes also employ epigenetic factors to manipulate cGAS-STING response. H3K9-specific DNA methyltransferase SUV39H1 has been shown to be upregulated by HPV E7 to silence the expression of both cGAS and STING (160). In addition, HPV evolves a unique vesicular trafficking pathway to protect itself from cGAS/STING surveillance (161). Although some progress has been achieved in understanding the cGAS-STING pathway manipulated by HPV, novel mechanisms need to be illuminated. Acquiring an in-depth understanding of the cellular proteins and pathways overthrown by HPV during infection, and especially during carcinogenesis, will assist in the development of novel therapeutic agents.
Coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has spread around the world and possesses significant threats to public health worldwide. SARS-CoV-2 is a positive-sense single-stranded RNA (ssRNA). It has been demonstrated that cGAS-STING could not only recognize dsDNA from DNA viruses but also engage in RNA virus infection. The contribution in RNA virus infection involves the recognition of viral signatures or cellular DNA released from mitochondria or nuclei induced by cellular stress (162, 163). Emerging evidence suggests that cGAS-STING pathway is involved during SARS-CoV-2 infection (164–167). Intranasal administration of diABZI-4, a STING agonist, before or even after SARS-CoV-2 infection provides complete protection from severe respiratory disease in K18-ACE2 transgenic mice. Mechanistically, intranasal treatment with diABZI-4 induced the oligomerization of STING, resulting in subsequent expression of a variety of IFN-stimulated genes (ISGs), including Cxcl10, interferon-induced protein with tetratricopeptide repeats 1 (Ifit1), Isg15, Mx dynamin like GTPase 1(Mx1), and signal transducer and activator of transcription 1 (Stat1), as well as myeloid cells and lymphocytes activation in the lung (164). Consistently, Minghua Li et al. found that treatment utilizing diABZI-4 could restrict SARS-CoV-2 infection via transiently stimulating IFN signaling in primary human bronchial epithelial cells and mice (165). In addition, SARS-CoV-2 infection could induce cell fusion in a spike protein- and ACE2-dependent manner, and subsequently triggers the generation of cytoplasmic genomic DNA, which then activates cGAS-STING axis to contribute interferon and pro-inflammatory gene expression (166, 168). Targeting cGAS-STING pathway via diABZI-4 inhibits viral replication in vitro (166). Interestingly, another team showed SARS-CoV-2 activates cGAS-STING signaling in endothelial cells by provoking mitochondrial DNA release, leading to cell death and type I IFN production. Furthermore, the administration of H-151, a small-molecule STING inhibitor, significantly decreases severe lung inflammation induced by SARS-CoV-2 and improves disease outcomes. This finding uncovers an unexpected immunopathology role of type I IFNs-induced by the cGAS-STING-axis in COVID-19 (169). Most recently, Christopher J et al. have observed a significant upregulation of inflammatory cytokines, in severe COVID-19 patients and SARS-CoV-2 infected lung epithelial cells. Further study shows that such inflammatory cytokines are induced downstream of NF-κB activation, which is mediated by cGAS-STING but not RNA sensors. Pharmacological inhibition of cGAS-STING axis by utilizing STING inhibitors dampens SARS-CoV-2-mediated inflammatory gene activation in vitro (170). To date, some reports show that induction of type I IFNs is inadequate in autopsy samples from COVID-19 patients, and SARS-CoV-2–infected cells in culture (171). However, some reports show that type I IFNs are elevated in the lungs of COVID-19 patients (169, 172). These differences between these studies may reflect differential timing of sampling and severity of disease, which also suggests cytokine induction timing may dictate the disease outcome. Thus, it is crucial to reconsider the therapeutic potential of STING agonists or inhibitors at different SARS-CoV-2 infection stages.
As a highly transmissible viral pathogen, SARS-CoV-2 has adopted multiple strategies to restrict and evade the antiviral IFN response, which has been demonstrated by several studies. 3CL of SARS-CoV-2 interrupts K63-ubiquitin modification of STING to impair the assembly of functional STING complex, which is crucial for the induction of type I IFNs (167). SARS-CoV-2 accessory protein ORF3a has been shown to interact with STING and impair the nuclear translocation of NF-κB factor p65, leading to the downregulation of NF-κB activation, thus impeding IFN promoter activation (167). In addition, SARS-CoV-2 ORF3a has also been shown to disrupt the STING-LC3 interaction, thus impeding cGAS-STING-induced autophagy to facilitate its replication (173). Most recently, SARS-CoV-2 ORF10 is found to antagonize STING-dependent antiviral response in two ways. On one hand, ORF10 restricts STING-mediated autophagy. On the other hand, ORF10 dampens STING-dependent type I IFN activation by impairing STING oligomerization and aggregation, ER to Golgi trafficking, and functional STING complex formation with TBK1 (174). Meanwhile, SARS-CoV-2 ORF9b is reported to suppress the phosphorylation and nuclear translocation of IRF3, thus subsequently reducing the induction of type I IFNs (175).
To date, HIV-1 infection is still considered a global public health issue. HIV is an RNA retrovirus that has (+) ssRNA and is dependent on reverse transcription for replication (176). It has been shown that HIV could be sensed by cGAS-STING axis, which is crucial for mediating type I IFN induction (177). Besides, activated CD4+ T cells utilize cGAS to sense HIV for establishing a bioactive IFN-I response, which could be potentiated by HIV accessory protein Vpr (178). Whereas IFN-I induction is thought to be lacking in HIV-1 target cells due to effective evasion mechanisms (179–181). Recently, HIV-1 nonstructural protein viral infectivity factor (Vif) is reported to inhibit type I IFN production to facilitate immune evasion. Mechanism study shows that HIV infection induces the association of Vif with SHP-1, which further promotes the recruitment of SHP-1 to STING, resulting in the inhibition of K63-linked ubiquitination of STING at Lys337 by dephosphorylating STING at Tyr162 (182). In addition, HIV Vpu impairs the cGAS-dependent type I IFN response to HIV-1 in primary CD4+ T cells (178). It has been shown that HIV, with intact capsids, is transported across the cytoplasm and through nuclear pores before integration, implying a pivotal role of HIV capsids in shielding viral DNA from cytosolic sensors (183–189). Further study shows that disrupting HIV-1 capsid formation, by utilizing capsid destabilizing small molecule PF-74, leads to a cGAS-dependent IFN response (190). In line with these, Lorena et al. found that the genetic reversal of two specific amino acid adaptations in the capsid of pandemic HIV-1(M) enables activation of cGAS and innate immune responses (181). These findings demonstrate an antagonistic role for HIV accessory proteins and capsids against the host’s innate immune response activation.
Viral hepatitis has long been a hot spot for its high morbidity and mortality. The main cause of death in patients with viral hepatitis is the infection of the hepatitis B virus (HBV). HBV belongs to the DNA retrovirus, carrying a dsDNA genome, the relaxed-circular DNA (rcDNA), which is repaired into a covalently closed circular DNA (cccDNA) in the nuclei of infected cells. The cccDNA then synthesizes a (+)ssRNA strand called pregenomic RNA (pgRNA) in the nucleus, which is then reverse-transcribed in the cytosol for genomic DNA replication (191, 192). STING is shown highly expressed in immortalized human hepatocyte NKNT-3/NTCP cell-derived cell clones, which are resistant to HBV, via induction of type III IFN (193). Most recently, STING activation is found to efficiently inhibit HBV replication through epigenetic suppression of cccDNA (194). In addition, activation of cGAS-STING pathway in both HepG2 cells and mice by dsDNA or cGAMP results in significant inhibition of HBV replication (195). Of note, in this study, the researcher artificially transfected the pHBV1.3 plasmids into the cells and mice to mimic the infection of HBV instead of using the alive HBV. In line with this finding, recently, it has been shown that hepatocytes can respond to HBV rcDNA in a cGAS-dependent manner, but not to live HBV infection (192, 196), suggesting HBV escapes the sensing of its DNAs by cGAS/STING. Verrier et al. imply the underlying escaping mechanism is that HBV infection impairs the expression of cGAS and its effector gene expression (196). Although emerging evidence suggests cGAS-STING signaling pathway in hepatocytes may inhibit HBV infection, most of them use artificial transfection of HBV DNA into cells or mice, which may not represent the live HBV infection context, thus raising the concern for data authenticity. Therefore, more in vivo studies using live HBV infection need to be conducted to uncover the role of cGAS-STING pathway in HBV infection.
Influenza is an infectious respiratory disease, which is mainly caused by influenza A virus (IAV) and influenza B virus (IBV) in humans. Influenza virus impacts the respiratory tract by direct viral infection (197). Christian et al. reported that the production of interferon is impaired in STING-deficient THP-1 cells, but not in cGAS-deficient THP-1 cells after IAV infection, suggesting that IAV initiates a STING-dependent, cGAS-independent pathway, which is critical for full interferon production. To counteract the host’s antiviral defense, IAV utilizes FP (the fusion peptide), which is highly conserved among IAV and IBV strains, to impair STING dimerization and TBK1 phosphorylation in response to membrane fusion through binding STING in the region of the dimerization interphase (198). In addition, it has been shown that cytosolic mitochondrial DNA (mtDNA) plays important roles in cGAS-mediated antiviral immune responses after infection with RNA viruses, such as dengue virus and lymphocytic choriomeningitis virus (LCMV) (199–201). In the influenza viral infection context, Miyu et al. found that influenza virus M2 protein, a proton-selective ion channel, which is critical for the viral uncoating during viral entry and budding, triggers cytosolic mtDNA release in a MAVS-dependent manner, and activates cGAS- and DDX41-mediated IFN-β production. To evade host immunological surveillance, the influenza virus employs nonstructural protein 1 to interact with mtDNA to evade the STING-dependent antiviral immunity. Of note, cGAS deficiency did not markedly alter the viral titer in the lung. By contrast, STING deficiency significantly elevated the viral titer in the lung compared to that from WT mice. These findings suggest the presence of other DNA sensors that activate redundant signaling pathways to limit viral replication (202).
Besides viral DNA, cGAS is reported to recognize bacterial DNA and synthesize cGAMP to activate downstream STING (203–209). Furthermore, stimuli other than cGAS induced-cGAMP, including cyclic dinucleotides (CDNs) [cyclic diGMP, cyclic diAMP], and ER stress, which could be induced by bacterial infection, can also activate STING (210–212). All these suggest the intricate interplay between bacterial infection and cGAS-STING responses. Of note, the activation of cGAS-STING signaling pathway has pleiotropic roles in the bacterial infection process.
Chlamydia trachomatis (C. trachomatis) is a gram-negative bacterium. C. trachomatis infection causes the most common sexually transmitted disease, with serious complications such as pelvic inflammatory disease, infertility, and ectopic pregnancy in women, and epididymitis and orchitis in men (213). It is well known that type I IFN is pivotal in antiviral response. However, the interplay between type I IFNs and bacterial infections seems somewhat pleiotropic and contexts dependent (214). Type I IFN signaling has been shown to exacerbate host pathology during C. trachomatis infection. It is reported that cGAS is recruited to the chlamydial inclusion membrane and mediates the generation of cGAMPs, which subsequently initiate STING-induced IFN-β expression during C. trachomatis infection. Moreover, the colocalization of cGAS and STING on the cytosolic side of the chlamydial inclusion membrane suggests that chlamydial DNA seems to be recognized outside the inclusion. This study also provides evidence that cGAMPs could transfer from infected cells to adjacent cells and thus upregulate IFN-β expression in adjacent uninfected cells during in vivo infection, escalating the pathogenesis (215). On the contrary, a recent in vivo study shows that mice deficient in either cGAS or STING significantly increased the yields of live C. trachomatis in the lower genital tract, demonstrating a beneficial role of cGAS-STING response during C. trachomatis infection (216). Moreover, STING also engages in the induction of type I IFN response via the detection of bacterial metabolite cyclic di-AMP during C. trachomatis infection (217). Further study shows that such type I IFN is essential for inflammasome activation (218). These studies suggest STING/interferon pathway may be a potent therapeutic target to treat C. trachomatis infection and its associated inflammatory pathology. Staphylococcus aureus (S. aureus) is a gram-positive bacterium that is the leading cause of infective endocarditis, as well as skin, soft tissue, and pleuropulmonary infections (219). S. aureus DNA is demonstrated to be sensed by cGAS. Recently, cGAS is identified as a DNA sensor for recognizing the extracellular pathogen S. aureus and Pseudomonas aeruginosa (P. aeruginosa) (a gram-negative bacterium), inducing the production of type I IFNs. Though the cGAS-STING-IFN axis is shown to be critical for restricting the P. aeruginosa infection, the biological function of this type I IFN in S. aureus replication remains unclear in this study (209). Intriguingly, Casey et al. report cyclic di-AMP, which is released extracellularly from S. aureus biofilm, induces a STING-dependent type I IFN response in macrophages, facilitating S. aureus intracellular survival in macrophages (220). Since type I IFNs could induce the activation of STAT3, which is a key mediator of anti-inflammatory signaling (221), the cGAMPs-STING-type I IFNs axis may be the underlying mechanism, through which biofilms render the host an anti-inflammatory state, thereby resulting in the persistent biofilm-mediated infections in an immunocompetent host (222). Moreover, STING and TLR pathways are activated at the early stage of infection with live S. aureus. However, they display opposite roles in host defense to S. aureus. On one hand, IL-1β induction via TLR signaling protects the host against S. aureus infection. On the other hand, type I IFNs induced by STING signaling could inhibit the transcription of IL-1β as well as the processing of pro-IL-1β protein into mature IL-1β through the inflammasome. The orchestrated immune response operated by TLR and STING signaling contributes to establish an effective anti-microbe response, meanwhile, protects the host from overwhelming inflammatory response (223). In addition, STING was shown to enhance host defense against S. aureus infection via blocking the necroptosis of macrophages (224), although previous studies show that cGAS-STING contributes to the necroptosis in macrophages in a type I IFN-dependent manner (225, 226), suggesting the specific role of cGAS-STING in distinct contexts. Listeria monocytogenes (L. monocytogenes) is a gram-positive intracellular foodborne pathogen and the causative agent of listeriosis (227, 228). The innate immune system is a double edge sword, that is it not only may contribute to defending against infection, but also could lead to pathology. DNA from L. monocytogenes is sorted into EVs in infected cells through a STING-TBK1-MVB12b (multivesicular body protein) pathway and delivered to bystander cells to stimulate cGAS-STING pathway, which facilitates the spread of infection signs across tissues prior to the actual infection process. Moreover, the EVs from infected macrophages could promote FasL-stimulated apoptosis of splenic T lymphocytes, which is also dependent on the presence of cGAS and STING in the T lymphocytes, suggesting the exquisite immune evasion strategy employed by L. monocytogenes (229). It is reported that type I IFN, which is induced by L. monocytogenes infection is deleterious to the host’s anti-bacterial response via L. monocytogenes-mediated apoptosis of leukocytes (230–232). The complement anaphylatoxins, C5a and C3a that are generated during activation of the complement cascade in response to infection are shown to inhibit the production of detrimental IFN-β by damping the expression of DEAD-box helicase 41 (DDX41), STING, phosphorylated TBK1, and phosphorylated p38 mitogen-activated protein kinase (MAPK), which play critical roles in the type I IFN induction upon L. monocytogenes infection (233). Interestingly, Alexander et al. reported that L. monocytogenes-derived c-di-AMP activated STING and induced type I IFN signaling. Deficiency of STING decreased the influx of inflammatory monocytes and increased systemic bacterial burden during enterocolitis (234). In addition, STING deficiency impaired the Ly6Chi monocytes, increasing bacterial burden in the liver during L. monocytogenes infection (235). Streptococcus pneumoniae (S. pneumoniae) is a leading cause of pneumonia, bacteremia, meningitis, and otitis media (236). S. pneumoniae produces c-di-AMP via CdaA, a diadenylate cyclase (237). And phosphodiesterase 1 (Pde1) and Pde2 are responsible for the degrading of c-di-AMP (237). Pneumococci deficient in Pde2 led to increased concentrations of c-di-AMP in the mutant pneumococci and resulted in the hyperactivation of STING and excessive IFN-β expression, as well as rapid cytotoxicity (238). However, another study showed that cGAS-STING pathway had no contribution to the immune response against S. pneumoniae in mice and humans, although pneumococcal DNA could be detected by cGAS and then initiated type I IFN production through STING (207). Moreover, it is reported that STING promoted coagulation in a type I IFN response-independent manner, increasing the risk of severe sepsis caused by S. pneumoniae (239). In addition to type I IFN, IFN-γ could also be induced upon S. pneumoniae infection. Elevated plasma IFN-γ was correlated with increased mortality in patients with S. pneumoniae sepsis (240). Deficiency of IFN-γ rendered mice more resistant to developing pneumococcal meningitis (241). It has been shown that cGAS-STING pathway cooperates with MyD88 pathway in Ly6Chi monocytes to promote late-stage lung IFN-γ production via the production of IL-12p70 during pulmonary pneumococcal infection (242). Mycobacterium tuberculosis (M. tuberculosis) is the etiological agent of tuberculosis (TB) and the leading cause of death due to its transmissible and drug-resistant features (243). M. tuberculosis has been shown to initiate the cytosolic DNA surveillance pathway via phagosome disruption mediated by the mycobacterial protein secretion system ESX-1 (244). Multiple reports showed that the exposed mycobacterial DNA in the cytosol via ESX-1 system could be sensed by cGAS-STING pathway and activate type I IFN production and autophagy process, suggesting an unanticipated cross-talk between DNA surveillance and autophagy, meanwhile indicating a major role for selective autophagy process in resistance to M. tuberculosis infection (204, 206). On the contrary, M. tuberculosis has adopted delicate strategies to evade host DNA surveillance to protect its survival (245–247). For instance, M. tuberculosis coding protein Rv0753c (MmsA) has been shown to interact with STING and promote STING for p62-mediated autophagic degradation, thus damping STING-mediated type I IFN production (245). Besides, M. tuberculosis phosphodiesterase (PDE) CdnP was identified to restrict STING activation and the type I IFN response through hydrolysis of both bacterial-derived c-di-AMP and host-derived cGAMP (247). Due to the severity and infectivity of TB, although CDNs-adjuvanted protein subunit vaccine (248) and the inhibitor, that targeting M. tuberculosis PDE (246) can protect the host from M. tuberculosis infection, there is a lot of room remaining to uncover to pave the way for TB therapy.
Fungi and mammals share a co−evolutionary history and are involved in a complex web of interactions (249). Superficial and invasive fungal infections lead to diseases that range from irritating to life-threatening, developing invasive infections during their lives, and mortality for these infections often exceeds 50% (250). Antifungal immunity is mainly composed of cellular innate-, adaptive-, humoral-, and mucosal-immune mechanisms. And considerable PRRs, like TLRs, NOD2, Dectin families, and CLR families have been proven momentous for the detection of fungal PAMPs (249, 251–253). However, the role of cGAS-STING signaling acting on antifungal immunity is less known. Although Majer et al. reported that type I IFN induced by Candida albicans infection promotes sepsis as type I IFN recruits and activates inflammatory monocytes/DCs in a CCL2-dependent way, which causes high host-destructing potency, the mechanism of type I IFN production and whether it depends on cGAS-STING are still ill-defined (254). Besides, β-Glucans, the major cell wall structural components in fungi, have been applied to anti-tumor research. Kalafati et al. found β-glucan-induced trained immunity was related to transcriptomic and epigenetic rewiring of granulopoiesis and neutrophil reprogramming toward an anti-tumor phenotype, which is type I IFN dependent (252, 255). Given the unequivocal function of cGAS-STING as a bridge to link pathogens’ PAMPs and type I IFN responses, we have reasons to believe cGAS-STING possesses a potentially important role in antifungal immunity, which would be interesting to comprehend the physiological function and underlying mechanism of the cGAS-STING pathway in the fungi infection.
In the past few years, we have obtained a mounting knowledge of the structures and biomedical functions in cGAS-STING pathway, which provides a new framework for understanding the immune stimulatory capacity of dsDNA and c-GAMPs. The exquisite sensing of the pathway to foreign nucleic acids and CDNs enables the development of robust anti-microbe responses to protect the host from invading pathogens. Whereas, due to the pleiotropic biological functions of activation of cGAS-STING pathway and type I IFNs as well as inflammatory cytokines in distinct infection contexts, continuing efforts will need to obtain more in-depth knowledge on the effects of the cGAS-STING pathway in infectious disease pathogenesis in specific scenarios. These efforts will advance side by side with in vivo studies by using conditional knockout mice or in ex vivo studies by using human samples. Furthermore, emerging evidence from functional and mechanistic studies suggests that multiple intracellular and extracellular information, such as pathogen DNA contained-EVs, mitochondrial DNA release, ER stress, and activation of other signaling pathways, cooperates with cGAS-STING to initiate an efficacious detection of invading pathogens and subsequent prompt activation of innate immune signaling. However, the underlying mechanism of manipulating cGAS-STING pathway to avoid aberrant and excessive immune response is still not fully understood. In this regard, more efforts need to be put into the regulation mechanism of cGAS-STING pathway on molecule and cell scales. Accordingly, to avoid the deleterious consequences of aberrant activation of cGAS-STING pathway, usage of agonists or antagonists of cGAS and STING in certain infection contexts may be instructive. As we discussed above, cytokine induction timing may dictate the disease outcome. Thus, it is crucial to revalue the therapeutic potential of agonists or antagonists at different infection stages. Moreover, the hallmark of the interplay between innate immune response and infectious microbes is the pathogen evasion process. Various counteraction mechanisms involved in cGAS-STING pathway have already been elucidated. However, how pathogen factors (pathogen proteins, nucleic acids, and metabolites) interact with cGAS and STING, and how pathogen factors manipulate the structure and signal transduction of this innate immune pathway are still not fully understood. New insight into evasion strategies employed by pathogens would deepen our knowledge of infectious disease pathogenesis, thereby providing new opportunities for developing therapeutic interventions.
R-FW, XY, YD, ZH and YL designed and wrote the manuscript. YD, ZH, YL, HW, XY, and R-FW discussed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was in part supported by grants from the NCI, NIH (R01CA101795, R01CA246547 and U54CA210181), Department of Defense (DoD) CDMRP BCRP (BC151081) and LCRP (LC200368) to R-FW. This work was also partially supported by grants from National Natural Science Foundation of China (82171741), Guangdong Basic and Applied Basic Research Foundation (2019B1515120033 and 2021A1515012140) to XY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54:289–96. doi: 10.1016/j.molcel.2014.03.040
2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
3. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
4. Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
5. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
6. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
7. Lind NA, Rael VE, Pestal K, Liu B, Barton GM. Regulation of the nucleic acid-sensing toll-like receptors. Nat Rev Immunol (2022) 22:224–35. doi: 10.1038/s41577-021-00577-0
8. Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
9. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
10. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
11. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323:1057–60. doi: 10.1126/science.1169841
12. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
13. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
14. Liu N, Pang X, Zhang H, Ji P. The cGAS-STING pathway in bacterial infection and bacterial immunity. Front Immunol (2021) 12:814709. doi: 10.3389/fimmu.2021.814709
15. Fan YM, Zhang YL, Luo H, Mohamud Y. Crosstalk between RNA viruses and DNA sensors: Role of the cGAS-STING signalling pathway. Rev Med Virol (2022) 32:e2343. doi: 10.1002/rmv.2343
16. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
17. Deb P, Dai J, Singh S, Kalyoussef E, Fitzgerald-Bocarsly P. Triggering of the cGAS-STING pathway in human plasmacytoid dendritic cells inhibits TLR9-mediated IFN production. J Immunol (2020) 205:223–36. doi: 10.4049/jimmunol.1800933
18. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science (2019) 363. doi: 10.1126/science.aat8657
19. Kawane K, Motani K, Nagata S. DNA Degradation and its defects. Cold Spring Harb Perspect Biol (2014) 6. doi: 10.1101/cshperspect.a016394
20. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi: 10.1126/science.1229963
21. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
22. Kuchta K, Knizewski L, Wyrwicz LS, Rychlewski L, Ginalski K. Comprehensive classification of nucleotidyltransferase fold proteins: Identification of novel families and their representatives in human. Nucleic Acids Res (2009) 37:7701–14. doi: 10.1093/nar/gkp854
23. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature (2013) 498:332–7. doi: 10.1038/nature12305
24. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
25. Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep (2013) 3:1362–8. doi: 10.1016/j.celrep.2013.05.008
26. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498:380–4. doi: 10.1038/nature12306
27. Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep (2013) 3:1355–61. doi: 10.1016/j.celrep.2013.05.009
28. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
29. Du M, Chen ZJ. DNA-Induced liquid phase condensation of cGAS activates innate immune signaling. Science (2018) 361:704–9. doi: 10.1126/science.aat1022
30. Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kübler K, Wittmann S, et al. Sequence-specific activation of the DNA sensor cGAS by y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol (2015) 16:1025–33. doi: 10.1038/ni.3267
31. Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature (2018) 557:57–61. doi: 10.1038/s41586-018-0050-1
32. Emam A, Wu X, Xu S, Wang L, Liu S, Wang B. Stalled replication fork protection limits cGAS-STING and p-body-dependent innate immune signalling. Nat Cell Biol (2022) 24:1154–64. doi: 10.1038/s41556-022-00950-8
33. Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. Acetylation blocks cGAS activity and inhibits self-DNA-Induced autoimmunity. Cell (2019) 176:1447–1460.e14. doi: 10.1016/j.cell.2019.01.016
34. Crow YJ. Type I interferonopathies: Mendelian type I interferon up-regulation. Curr Opin Immunol (2015) 32:7–12. doi: 10.1016/j.coi.2014.10.005
35. Xiao N, Wei J, Xu S, Du H, Huang M, Zhang S, et al. cGAS activation causes lupus-like autoimmune disorders in a TREX1 mutant mouse model. J Autoimmun (2019) 100:84–94. doi: 10.1016/j.jaut.2019.03.001
36. Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, et al. RNase H2 catalytic core aicardi-goutières syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J Exp Med (2016) 213:329–36. doi: 10.1084/jem.20151464
37. Bai J, Liu F. Nuclear cGAS: Sequestration and beyond. Protein Cell (2022) 13:90–101. doi: 10.1007/s13238-021-00869-0
38. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife (2019) 8. doi: 10.7554/eLife.47491
39. Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature (2020) 587:673–7. doi: 10.1038/s41586-020-2749-z
40. Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature (2020) 587:678–82. doi: 10.1038/s41586-020-2748-0
41. Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, et al. Structural basis for the inhibition of cGAS by nucleosomes. Science (2020) 370:455–8. doi: 10.1126/science.abd0237
42. Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, et al. Structural basis of nucleosome-dependent cGAS inhibition. Science (2020) 370:450–4. doi: 10.1126/science.abd0609
43. Cao D, Han X, Fan X, Xu R-M, Zhang X. Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res (2020) 30:1088–97. doi: 10.1038/s41422-020-00422-4
44. Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature (2020) 587:668–72. doi: 10.1038/s41586-020-2750-6
45. Li T, Huang T, Du M, Chen X, Du F, Ren J, et al. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science (2021) 371. doi: 10.1126/science.abc5386
46. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
47. Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol (2012) 19:722–4. doi: 10.1038/nsmb.2331
48. Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity (2012) 36:1073–86. doi: 10.1016/j.immuni.2012.03.019
49. Huang YH, Liu XY, Du XX, Jiang ZF, Su XD. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat Struct Mol Biol (2012) 19:728–30. doi: 10.1038/nsmb.2333
50. Shang G, Zhu D, Li N, Zhang J, Zhu C, Lu D, et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol (2012) 19:725–7. doi: 10.1038/nsmb.2332
51. Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
52. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci United States America (2009) 106:20842–6. doi: 10.1073/pnas.0911267106
53. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe (2015) 18:157–68. doi: 10.1016/j.chom.2015.07.001
54. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
55. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol (2019) 20:152–62. doi: 10.1038/s41590-018-0287-8
56. Zhang BC, Nandakumar R, Reinert LS, Huang J, Laustsen A, Gao ZL, et al. STEEP mediates STING ER exit and activation of signaling. Nat Immunol (2020) 21:868–79. doi: 10.1038/s41590-020-0730-5
57. Lepelley A, Martin-Niclos MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA lead to abnormal trafficking of STING to the golgi and interferon signaling. J Exp Med (2020) 217. doi: 10.1084/jem.20200600
58. Kang J, Wu J, Liu Q, Wu X, Zhao Y, Ren J. Post-translational modifications of STING: A potential therapeutic target. Front Immunol (2022) 13:888147. doi: 10.3389/fimmu.2022.888147
59. Hong Z, Ma T, Liu X, Wang C. cGAS-STING pathway: Post-translational modifications and functions in sterile inflammatory diseases. FEBS J (2022) 289:6187–208. doi: 10.1111/febs.16137
60. Liu J, Rui K, Peng N, Luo H, Zhu B, Zuo X, et al. The cGAS-STING pathway: Post-translational modifications and functional implications in diseases. Cytokine Growth factor Rev (2022) 68:69–80. doi: 10.1016/j.cytogfr.2022.09.003
61. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
62. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
63. Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
64. Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature (2019) 569:718–22. doi: 10.1038/s41586-019-1228-x
65. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signaling (2012) 5:ra20. doi: 10.1126/scisignal.2002521
66. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
67. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
68. Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg MC, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med (2017) 214:1769–85. doi: 10.1084/jem.20161674
69. Ni G, Konno H, Barber GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol (2017) 2. doi: 10.1126/sciimmunol.aah7119
70. Stempel M, Chan B, Juranic Lisnic V, Krmpotic A, Hartung J, Paludan SR, et al. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-kappaB signaling and STING's dual role during MCMV infection. EMBO J (2019) 38. doi: 10.15252/embj.2018100983
71. de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, Kranzusch PJ. Modular architecture of the STING c-terminal tail allows interferon and NF-kappaB signaling adaptation. Cell Rep (2019) 27:1165–1175 e5. doi: 10.1016/j.celrep.2019.03.098
72. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Mol Cell (2018) 71:745–760 e5. doi: 10.1016/j.molcel.2018.07.034
73. Schofield L, Hewitt MC, Evans K, Siomos MA, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature (2002) 418:785–9. doi: 10.1038/nature00937
74. Kamena F, Tamborrini M, Liu X, Kwon YU, Thompson F, Pluschke G, et al. Synthetic GPI array to study antitoxic malaria response. Nat Chem Biol (2008) 4:238–40. doi: 10.1038/nchembio.75
75. Gowda DC. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol (2007) 23:596–604. doi: 10.1016/j.pt.2007.09.003
76. Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem (2005) 280:8606–16. doi: 10.1074/jbc.M413541200
77. Schofield L, Hackett F. Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J Exp Med (1993) 177:145–53. doi: 10.1084/jem.177.1.145
78. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to toll-like receptor 9. Proc Natl Acad Sci U.S.A. (2007) 104:1919–24. doi: 10.1073/pnas.0608745104
79. Jaramillo M, Bellemare MJ, Martel C, Shio MT, Contreras AP, Godbout M, et al. Synthetic plasmodium-like hemozoin activates the immune response: A morphology - function study. PloS One (2009) 4:e6957. doi: 10.1371/journal.pone.0006957
80. Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and syk kinases. PloS Pathog (2009) 5:e1000559. doi: 10.1371/annotation/abca067d-b82b-4de6-93c5-0fcc38e3df05
81. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PloS One (2009) 4:e6510. doi: 10.1371/journal.pone.0006510
82. Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med (2005) 201:19–25. doi: 10.1084/jem.20041836
83. Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, et al. Combined action of nucleic acid-sensing toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to toxoplasma gondii in mice. Cell Host Microbe (2013) 13:42–53. doi: 10.1016/j.chom.2012.12.003
84. Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, et al. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a toll-like receptor 9-dependent pathway. J Immunol (2004) 172:4926–33. doi: 10.4049/jimmunol.172.8.4926
85. Wu X, Gowda NM, Kumar S, Gowda DC. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol (2010) 184:4338–48. doi: 10.4049/jimmunol.0903824
86. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the plasmodium falciparum genome. Immunity (2011) 35:194–207. doi: 10.1016/j.immuni.2011.05.016
87. Wu J, Tian L, Yu X, Pattaradilokrat S, Li J, Wang M, et al. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc Natl Acad Sci U.S.A. (2014) 111:E511–20. doi: 10.1073/pnas.1316467111
88. Du Y, Luo Y, Hu Z, Lu J, Liu X, Xing C, et al. Activation of cGAS-STING by lethal malaria N67C dictates immunity and mortality through induction of CD11b(+) Ly6C(hi) proinflammatory monocytes. Adv Sci (Weinh) (2022) 9:e2103701. doi: 10.1002/advs.202103701
89. Liehl P, Zuzarte-Luís V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for plasmodium activate innate immunity against liver-stage infection. Nat Med (2014) 20:47–53. doi: 10.1038/nm.3424
90. Baccarella A, Fontana MF, Chen EC, Kim CC. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun (2013) 81:4431–42. doi: 10.1128/IAI.00923-13
91. Dobbs KR, Crabtree JN, Dent AE. Innate immunity to malaria-the role of monocytes. Immunol Rev (2020) 293:8–24. doi: 10.1111/imr.12830
92. Chua CL, Brown G, Hamilton JA, Rogerson S, Boeuf P. Monocytes and macrophages in malaria: protection or pathology? Trends Parasitol (2013) 29:26–34. doi: 10.1016/j.pt.2012.10.002
93. Hirako IC, Assis PA, Galvão-Filho B, Luster AD, Antonelli LR, Gazzinelli RT. Monocyte-derived dendritic cells in malaria. Curr Opin Microbiol (2019) 52:139–50. doi: 10.1016/j.mib.2019.08.002
94. Osii RS, Otto TD, Garside P, Ndungu FM, Brewer JM. The impact of malaria parasites on dendritic cell-T cell interaction. Front Immunol (2020) 11:1597. doi: 10.3389/fimmu.2020.01597
95. Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O'Garra A, et al. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J Exp Med (2006) 203:1427–33. doi: 10.1084/jem.20052450
96. Liu Y-J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633
97. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite plasmodium falciparum. Nature (2002) 419:498–511. doi: 10.1038/nature01097
98. Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, et al. The n-ethyl-N-nitrosourea-induced goldenticket mouse mutant reveals an essential function of sting in the in vivo interferon response to listeria monocytogenes and cyclic dinucleotides. Infect Immun (2011) 79:688–94. doi: 10.1128/IAI.00999-10
99. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
100. Gallego-Marin C, Schrum JE, Andrade WA, Shaffer SA, Giraldo LF, Lasso AM, et al. Cyclic GMP-AMP synthase is the cytosolic sensor of plasmodium falciparum genomic DNA and activates type I IFN in malaria. J Immunol (Baltimore Md 1950) (2018) 200:768–74. doi: 10.4049/jimmunol.1701048
101. Sisquella X, Ofir-Birin Y, Pimentel MA, Cheng L, Abou Karam P, Sampaio NG, et al. Malaria parasite DNA-harbouring vesicles activate cytosolic immune sensors. Nat Commun (2017) 8:1985. doi: 10.1038/s41467-017-02083-1
102. He X, Xia L, Tumas KC, Wu J, Su XZ. Type I interferons and malaria: A double-edge sword against a complex parasitic disease. Front Cell Infect Microbiol (2020) 10:594621. doi: 10.3389/fcimb.2020.594621
103. Luty AJ, Perkins DJ, Lell B, Schmidt-Ott R, Lehman LG, Luckner D, et al. Low interleukin-12 activity in severe plasmodium falciparum malaria. Infect Immun (2000) 68:3909–15. doi: 10.1128/IAI.68.7.3909-3915.2000
104. Kempaiah P, Anyona SB, Raballah E, Davenport GC, Were T, Hittner JB, et al. Reduced interferon (IFN)-α conditioned by IFNA2 (-173) and IFNA8 (-884) haplotypes is associated with enhanced susceptibility to severe malarial anemia and longitudinal all-cause mortality. Hum Genet (2012) 131:1375–91. doi: 10.1007/s00439-012-1175-1
105. Spaulding E, Fooksman D, Moore JM, Saidi A, Feintuch CM, Reizis B, et al. STING-licensed macrophages prime type I IFN production by plasmacytoid dendritic cells in the bone marrow during severe plasmodium yoelii malaria. PloS Pathog (2016) 12:e1005975. doi: 10.1371/journal.ppat.1005975
106. He X, Ashbrook AW, Du Y, Wu J, Hoffmann HH, Zhang C, et al. RTP4 inhibits IFN-I response and enhances experimental cerebral malaria and neuropathology. Proc Natl Acad Sci U.S.A. (2020) 117:19465–74. doi: 10.1073/pnas.2006492117
107. Hahn WO, Butler NS, Lindner SE, Akilesh HM, Sather DN, Kappe SH, et al. cGAS-mediated control of blood-stage malaria promotes plasmodium-specific germinal center responses. JCI Insight (2018) 3. doi: 10.1172/jci.insight.94142
108. Cai C, Hu Z, Yu X. Accelerator or brake: Immune regulators in malaria. Front Cell Infect Microbiol (2020) 10:610121. doi: 10.3389/fcimb.2020.610121
109. Wu J, Xia L, Yao X, Yu X, Tumas KC, Sun W, et al. The E3 ubiquitin ligase MARCH1 regulates antimalaria immunity through interferon signaling and T cell activation. Proc Natl Acad Sci U.S.A. (2020) 117:16567–78. doi: 10.1073/pnas.2004332117
110. Yao X, Wu J, Lin M, Sun W, He X, Gowda C, et al. Increased CD40 expression enhances early STING-mediated type I interferon response and host survival in a rodent malaria model. PloS Pathog (2016) 12:e1005930. doi: 10.1371/journal.ppat.1005930
111. Herhaus L. TBK1 (TANK-binding kinase 1)-mediated regulation of autophagy in health and disease. Matrix Biol (2021) 100-101:84–98. doi: 10.1016/j.matbio.2021.01.004
112. Galluzzi L, Vanpouille-Box C, Bakhoum SF, Demaria S. SnapShot: CGAS-STING signaling. Cell (2018) 173:276–276.e1. doi: 10.1016/j.cell.2018.03.015
113. Cai B, Wu J, Yu X, Su XZ, Wang RF. FOSL1 inhibits type I interferon responses to malaria and viral infections by blocking TBK1 and TRAF3/TRIF interactions. mBio (2017) 8. doi: 10.1128/mBio.02161-16
115. Matta SK, Rinkenberger N, Dunay IR, Sibley LD. Toxoplasma gondii infection and its implications within the central nervous system. Nat Rev Microbiol (2021) 19:467–80. doi: 10.1038/s41579-021-00518-7
116. Arranz-Solís D, Mukhopadhyay D, Saeij JJP. Toxoplasma effectors that affect pregnancy outcome. Trends Parasitol (2021) 37:283–95. doi: 10.1016/j.pt.2020.10.013
117. Wang P, Li S, Zhao Y, Zhang B, Li Y, Liu S, et al. The GRA15 protein from toxoplasma gondii enhances host defense responses by activating the interferon stimulator STING. J Biol Chem (2019) 294:16494–508. doi: 10.1074/jbc.RA119.009172
118. Majumdar T, Sharma S, Kumar M, Hussain MA, Chauhan N, Kalia I, et al. Tryptophan-kynurenine pathway attenuates β-catenin-dependent pro-parasitic role of STING-TICAM2-IRF3-IDO1 signalosome in toxoplasma gondii infection. Cell Death Dis (2019) 10:161. doi: 10.1038/s41419-019-1420-9
119. Majumdar T, Chattopadhyay S, Ozhegov E, Dhar J, Goswami R, Sen GC, et al. Induction of interferon-stimulated genes by IRF3 promotes replication of toxoplasma gondii. PloS Pathog (2015) 11:e1004779. doi: 10.1371/journal.ppat.1004779
120. Wang X, Majumdar T, Kessler P, Ozhegov E, Zhang Y, Chattopadhyay S, et al. STING requires the adaptor TRIF to trigger innate immune responses to microbial infection. Cell Host Microbe (2016) 20:329–41. doi: 10.1016/j.chom.2016.08.002
121. Hu Z, Wu D, Lu J, Zhang Y, Yu SM, Xie Y, et al. Inflammasome activation dampens type I IFN signaling to strengthen anti-toxoplasma immunity. mBio (2022) 13:e0236122. doi: 10.1128/mbio.02361-22
122. Chen M, Yao L, Zhou L, Yang P, Zou W, Xu L, et al. Toxoplasma gondii ROP18(I) inhibits host innate immunity through cGAS-STING signaling. FASEB J (2022) 36:e22171. doi: 10.1096/fj.202101347R
123. Gao FF, Quan JH, Choi IW, Lee YJ, Jang SG, Yuk JM, et al. FAF1 downregulation by toxoplasma gondii enables host IRF3 mobilization and promotes parasite growth. J Cell Mol Med (2021) 25:9460–72. doi: 10.1111/jcmm.16889
124. Rassi A Jr., Rassi A, Marin-Neto JA. Chagas disease. Lancet (2010) 375:1388–402. doi: 10.1016/S0140-6736(10)60061-X
126. Rassi A Jr., Rassi A, Marcondes de Rezende J. American Trypanosomiasis (Chagas disease). Infect Dis Clin North Am (2012) 26:275–91. doi: 10.1016/j.idc.2012.03.002
127. Magalhães LMD, Gollob KJ, Zingales B, Dutra WO, diversity P. Immunity, and the fate of infections: Lessons learned from trypanosoma cruzi human-host interactions. Lancet Microbe (2022) 3:e711–22. doi: 10.1016/S2666-5247(21)00265-2
128. Choudhuri S, Garg NJ. PARP1-cGAS-NF-κB pathway of proinflammatory macrophage activation by extracellular vesicles released during trypanosoma cruzi infection and chagas disease. PloS Pathog (2020) 16:e1008474. doi: 10.1371/journal.ppat.1008474
129. Vieira RS, Nascimento MS, Noronha IH, Vasconcelos JRC, Benvenuti LA, Barber GN, et al. STING signaling drives production of innate cytokines, generation of CD8(+) T cells and enhanced protection against trypanosoma cruzi infection. Front Immunol (2021) 12:775346. doi: 10.3389/fimmu.2021.775346
130. Matos MN, Cazorla SI, Schulze K, Ebensen T, Guzmán CA, Malchiodi EL. Immunization with Tc52 or its amino terminal domain adjuvanted with c-di-AMP induces Th17+Th1 specific immune responses and confers protection against trypanosoma cruzi. PloS Negl Trop Dis (2017) 11:e0005300. doi: 10.1371/journal.pntd.0005300
131. Sanchez Alberti A, Bivona AE, Cerny N, Schulze K, Weißmann S, Ebensen T, et al. Engineered trivalent immunogen adjuvanted with a STING agonist confers protection against trypanosoma cruzi infection. NPJ Vaccines (2017) 2:9. doi: 10.1038/s41541-017-0010-z
132. Sanchez Alberti A, Bivona AE, Matos MN, Cerny N, Schulze K, Weißmann S, et al. Mucosal heterologous Prime/Boost vaccination induces polyfunctional systemic immunity, improving protection against trypanosoma cruzi. Front Immunol (2020) 11:128. doi: 10.3389/fimmu.2020.00128
133. Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet (2001) 357:1513–8. doi: 10.1016/S0140-6736(00)04638-9
134. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science (2013) 341:1390–4. doi: 10.1126/science.1244040
135. Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun (2016) 7:13348. doi: 10.1038/ncomms13348
136. Wu J, Dobbs N, Yang K, Yan N. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity (2020) 53:115–126 e5. doi: 10.1016/j.immuni.2020.06.009
137. Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun (2020) 11:3382. doi: 10.1038/s41467-020-17156-x
138. Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2100225118
139. Su C, Zhan G, Zheng C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J (2016) 13:38. doi: 10.1186/s12985-016-0495-5
140. Zheng C. Evasion of cytosolic DNA-stimulated innate immune responses by herpes simplex virus 1. J Virol (2018) 92. doi: 10.1128/JVI.00099-17
141. Bhowmik D, Zhu F. Evasion of intracellular DNA sensing by human herpesviruses. Front Cell Infect Microbiol (2021) 11:647992. doi: 10.3389/fcimb.2021.647992
142. Su C, Zheng C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J Virol (2017) 91. doi: 10.1128/JVI.02414-16
143. Huang J, You H, Su C, Li Y, Chen S, Zheng C. Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J Virol (2018) 92. doi: 10.1128/JVI.00841-18
144. Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe (2018) 24:234–248 e5. doi: 10.1016/j.chom.2018.07.004
145. Bodda C, Reinert LS, Fruhwurth S, Richardo T, Sun C, Zhang BC, et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J Exp Med (2020) 217. doi: 10.1084/jem.20191422
146. Zhang D, Su C, Zheng C. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J Virol (2016) 90:5824–9. doi: 10.1128/JVI.00186-16
147. Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J (2016) 35:1385–99. doi: 10.15252/embj.201593458
148. Xu H, Su C, Pearson A, Mody CH, Zheng C. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-kappaB activation. J Virol (2017) 91. doi: 10.1128/JVI.00025-17
149. Sharma N, Wang C, Kessler P, Sen GC. Herpes simplex virus 1 evades cellular antiviral response by inducing microRNA-24, which attenuates STING synthesis. PloS Pathog (2021) 17:e1009950. doi: 10.1371/journal.ppat.1009950
150. Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, et al. The biology and life-cycle of human papillomaviruses. Vaccine (2012) 30(Suppl 5):F55–70. doi: 10.1016/j.vaccine.2012.06.083
151. Galloway DA, Laimins LA. Human papillomaviruses: shared and distinct pathways for pathogenesis. Curr Opin Virol (2015) 14:87–92. doi: 10.1016/j.coviro.2015.09.001
152. Scarth JA, Patterson MR, Morgan EL, Macdonald A. The human papillomavirus oncoproteins: A review of the host pathways targeted on the road to transformation. J Gen Virol (2021) 102. doi: 10.1099/jgv.0.001540
153. McBride AA. Mechanisms and strategies of papillomavirus replication. Biol Chem (2017) 398:919–27. doi: 10.1515/hsz-2017-0113
154. Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol (1999) 189:12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F
155. Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, et al. International agency for research on cancer multicenter cervical cancer study, epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med (2003) 348:518–27. doi: 10.1056/NEJMoa021641
156. Lau L, Gray EE, Brunette RL, Stetson DB. DNA Tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science (2015) 350:568–71. doi: 10.1126/science.aab3291
157. Shaikh MH, Bortnik V, McMillan NA, Idris A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb Pathog (2019) 132:162–5. doi: 10.1016/j.micpath.2019.05.004
158. Bortnik V, Wu M, Julcher B, Salinas A, Nikolic I, Simpson KJ, et al. Loss of HPV type 16 E7 restores cGAS-STING responses in human papilloma virus-positive oropharyngeal squamous cell carcinomas cells. J Microbiol Immunol Infect (2021) 54:733–9. doi: 10.1016/j.jmii.2020.07.010
159. Luo X, Donnelly CR, Gong W, Heath BR, Hao Y, Donnelly LA, et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J Clin Invest (2020) 130:1635–52. doi: 10.1172/JCI129497
160. Lo Cigno I, Calati F, Borgogna C, Zevini A, Albertini S, Martuscelli L, et al. Human papillomavirus E7 oncoprotein subverts host innate immunity via SUV39H1-mediated epigenetic silencing of immune sensor genes. J Virol (2020) 94. doi: 10.1128/JVI.01812-19
161. Uhlorn BL, Jackson R, Li S, Bratton SM, Van Doorslaer K, Campos SK. Vesicular trafficking permits evasion of cGAS/STING surveillance during initial human papillomavirus infection. PloS Pathog (2020) 16:e1009028. doi: 10.1371/journal.ppat.1009028
162. Ma Z, Damania B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe (2016) 19:150–8. doi: 10.1016/j.chom.2016.01.010
163. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
164. Humphries F, Shmuel-Galia L, Jiang Z, Wilson R, Landis P, Ng SL, et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Sci Immunol (2021) 6. doi: 10.1126/sciimmunol.abi9002
165. Li M, Ferretti M, Ying B, Descamps H, Lee E, Dittmar M, et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci Immunol (2021) 6. doi: 10.1126/sciimmunol.abi9007
166. Zhou Z, Zhang X, Lei X, Xiao X, Jiao T, Ma R, et al. Sensing of cytoplasmic chromatin by cGAS activates innate immune response in SARS-CoV-2 infection. Signal Transduct Target Ther (2021) 6:382. doi: 10.1038/s41392-021-00800-3
167. Rui Y, Su J, Shen S, Hu Y, Huang D, Zheng W, et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct Target Ther (2021) 6:123. doi: 10.1038/s41392-021-00515-5
168. Liu X, Wei L, Xu F, Zhao F, Huang Y, Fan Z, et al. SARS-CoV-2 spike protein-induced cell fusion activates the cGAS-STING pathway and the interferon response. Sci Signaling (2022) 15:eabg8744. doi: 10.1126/scisignal.abg8744
169. Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature (2022) 603:145–51. doi: 10.1038/s41586-022-04421-w
170. Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee JY, Plociennikowska A, et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kappaB. Commun Biol (2022) 5:45. doi: 10.1038/s42003-021-02983-5
171. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell (2020) 181:1036–1045 e9. doi: 10.1016/j.cell.2020.04.026
172. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe (2020) 27:883–890 e2. doi: 10.1016/j.chom.2020.04.017
173. Su J, Shen S, Hu Y, Chen S, Cheng L, Cai Y, et al. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J Med Virol (2022) 95:e28175. doi: 10.1002/jmv.28175
174. Han L, Zheng Y, Deng J, Nan ML, Xiao Y, Zhuang MW, et al. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J Med Virol (2022) 94:5174–88. doi: 10.1002/jmv.27965
175. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol (2021) 93:5376–89. doi: 10.1002/jmv.27050
176. Tan X, Sun L, Chen J, Chen ZJ. Detection of microbial infections through innate immune sensing of nucleic acids. Annu Rev Microbiol (2018) 72:447–78. doi: 10.1146/annurev-micro-102215-095605
177. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science (2013) 341:903–6. doi: 10.1126/science.1240933
178. Vermeire J, Roesch F, Sauter D, Rua R, Hotter D, Van Nuffel A, et al. HIV Triggers a cGAS-dependent, vpu- and vpr-regulated type I interferon response in CD4(+) T cells. Cell Rep (2016) 17:413–24. doi: 10.1016/j.celrep.2016.09.023
179. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature (2013) 503:402–5. doi: 10.1038/nature12769
180. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol (2010) 11:1005–13. doi: 10.1038/ni.1941
181. Zuliani-Alvarez L, Govasli ML, Rasaiyaah J, Monit C, Perry SO, Sumner RP, et al. Evasion of cGAS and TRIM5 defines pandemic HIV. Nat Microbiol (2022) 7:1762–76. doi: 10.1038/s41564-022-01247-0
182. Wang Y, Qian G, Zhu L, Zhao Z, Liu Y, Han W, et al. HIV-1 vif suppresses antiviral immunity by targeting STING. Cell Mol Immunol (2022) 19:108–21. doi: 10.1038/s41423-021-00802-9
183. Burdick RC, Li C, Munshi M, Rawson JMO, Nagashima K, Hu WS, et al. HIV-1 uncoats in the nucleus near sites of integration. Proc Natl Acad Sci U.S.A. (2020) 117:5486–93. doi: 10.1073/pnas.1920631117
184. Bejarano DA, Peng K, Laketa V, Borner K, Jost KL, Lucic B, et al. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. Elife (2019) 8. doi: 10.7554/eLife.41800
185. Zila V, Margiotta E, Turonova B, Muller TG, Zimmerli CE, Mattei S, et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell (2021) 184:1032–1046 e18. doi: 10.1016/j.cell.2021.01.025
186. Li C, Burdick RC, Nagashima K, Hu WS, Pathak VK. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2019467118
187. Muller TG, Zila V, Peters K, Schifferdecker S, Stanic M, Lucic B, et al. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. Elife (2021) 10. doi: 10.7554/eLife.64776
188. Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity (2013) 39:1132–42. doi: 10.1016/j.immuni.2013.11.002
189. Elsner C, Ponnurangam A, Kazmierski J, Zillinger T, Jansen J, Todt D, et al. Absence of cGAS-mediated type I IFN responses in HIV-1-infected T cells. Proc Natl Acad Sci U.S.A. (2020) 117:19475–86. doi: 10.1073/pnas.2002481117
190. Sumner RP, Harrison L, Touizer E, Peacock TP, Spencer M, Zuliani-Alvarez L, et al. Disrupting HIV-1 capsid formation causes cGAS sensing of viral DNA. EMBO J (2020) 39:e103958. doi: 10.15252/embj.2019103958
191. Rehermann B, Nascimbeni M. Immunology of hepatitis b virus and hepatitis c virus infection. Nat Rev Immunol (2005) 5:215–29. doi: 10.1038/nri1573
192. Lauterbach-Riviere L, Bergez M, Monch S, Qu B, Riess M, Vondran FWR, et al. Hepatitis b virus DNA is a substrate for the cGAS/STING pathway but is not sensed in infected hepatocytes. Viruses (2020) 12. doi: 10.3390/v12060592
193. Dansako H, Imai H, Ueda Y, Satoh S, Shimotohno K, Kato N. High-level expression of STING restricts susceptibility to HBV by mediating type III IFN induction. FASEB Bioadv (2019) 1:67–80. doi: 10.1096/fba.1022
194. Li Y, He M, Wang Z, Duan Z, Guo Z, Wang Z, et al. STING signaling activation inhibits HBV replication and attenuates the severity of liver injury and HBV-induced fibrosis. Cell Mol Immunol (2022) 19:92–107. doi: 10.1038/s41423-021-00801-w
195. He J, Hao R, Liu D, Liu X, Wu S, Guo S, et al. Inhibition of hepatitis b virus replication by activation of the cGAS-STING pathway. J Gen Virol (2016) 97:3368–78. doi: 10.1099/jgv.0.000647
196. Verrier ER, Yim SA, Heydmann L, El Saghire H, Bach C, Turon-Lagot V, et al. Hepatitis b virus evasion from cyclic guanosine monophosphate-adenosine monophosphate synthase sensing in human hepatocytes. Hepatology (2018) 68:1695–709. doi: 10.1002/hep.30054
197. Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, et al. Influenza. Nat Rev Dis Primers (2018) 4:3. doi: 10.1038/s41572-018-0002-y
198. Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, et al. Influenza a virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun (2016) 7:10680. doi: 10.1038/ncomms10680
199. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
200. Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol (2017) 2:17037. doi: 10.1038/nmicrobiol.2017.37
201. Sun B, Sundstrom KB, Chew JJ, Bist P, Gan ES, Tan HC, et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep (2017) 7:3594. doi: 10.1038/s41598-017-03932-1
202. Moriyama M, Koshiba T, Ichinohe T. Influenza a virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat Commun (2019) 10:4624. doi: 10.1038/s41467-019-12632-5
203. Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to francisella infection. J Immunol (2015) 194:3236–45. doi: 10.4049/jimmunol.1402764
204. Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for mycobacterium tuberculosis. Cell Host Microbe (2015) 17:820–8. doi: 10.1016/j.chom.2015.05.005
205. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe (2015) 17:799–810. doi: 10.1016/j.chom.2015.05.003
206. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The cytosolic sensor cGAS detects mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe (2015) 17:811–9. doi: 10.1016/j.chom.2015.05.004
207. Ruiz-Moreno JS, Hamann L, Jin L, Sander LE, Puzianowska-Kuznicka M, Cambier J, et al. The cGAS/STING pathway detects streptococcus pneumoniae but appears dispensable for antipneumococcal defense in mice and humans. Infect Immun (2018) 86. doi: 10.1128/IAI.00849-17
208. Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, et al. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J (2014) 33:1654–66. doi: 10.15252/embj.201488029
209. Zhou CM, Wang B, Wu Q, Lin P, Qin SG, Pu QQ, et al. Identification of cGAS as an innate immune sensor of extracellular bacterium pseudomonas aeruginosa. iScience (2021) 24:101928. doi: 10.1016/j.isci.2020.101928
210. Yin Q, Tian Y, Kabaleeswaran V, Jiang X, Tu D, Eck MJ, et al. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol Cell (2012) 46:735–45. doi: 10.1016/j.molcel.2012.05.029
211. Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: The first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev (2013) 77:1–52. doi: 10.1128/MMBR.00043-12
212. Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell (2017) 171:809–823 e13. doi: 10.1016/j.cell.2017.09.034
213. Mishori R, McClaskey EL, WinklerPrins VJ. Chlamydia trachomatis infections: screening, diagnosis, and management. Am Fam Physician (2012) 86:1127–32.
214. Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe (2016) 19:760–9. doi: 10.1016/j.chom.2016.05.016
215. Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, et al. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during chlamydia trachomatis infection. J Immunol (2014) 193:2394–404. doi: 10.4049/jimmunol.1302718
216. Su X, Xu H, French M, Zhao Y, Tang L, Li XD, et al. Evidence for cGAS-STING signaling in the female genital tract resistance to chlamydia trachomatis infection. Infection Immun (2022) 90:e0067021. doi: 10.1128/iai.00670-21
217. Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, et al. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during chlamydia trachomatis infection. mBio (2013) 4:e00018–13. doi: 10.1128/mBio.00018-13
218. Webster SJ, Brode S, Ellis L, Fitzmaurice TJ, Elder MJ, Gekara NO, et al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to chlamydia trachomatis infection. PloS Pathog (2017) 13:e1006383. doi: 10.1371/journal.ppat.1006383
219. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev (2015) 28:603–61. doi: 10.1128/CMR.00134-14
220. Gries CM, Bruger EL, Moormeier DE, Scherr TD, Waters CM, Kielian T. Cyclic di-AMP released from staphylococcus aureus biofilm induces a macrophage type I interferon response. Infect Immun (2016) 84:3564–74. doi: 10.1128/IAI.00447-16
221. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12:125–35. doi: 10.1038/nri3133
222. Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol (2011) 186:6585–96. doi: 10.4049/jimmunol.1002794
223. Scumpia PO, Botten GA, Norman JS, Kelly-Scumpia KM, Spreafico R, Ruccia AR, et al. Opposing roles of toll-like receptor and cytosolic DNA-STING signaling pathways for staphylococcus aureus cutaneous host defense. PloS Pathog (2017) 13:e1006496. doi: 10.1371/journal.ppat.1006496
224. Liu ZZ, Yang YJ, Zhou CK, Yan SQ, Ma K, Gao Y, et al. STING contributes to host defense against staphylococcus aureus pneumonia through suppressing necroptosis. Front Immunol (2021) 12:636861. doi: 10.3389/fimmu.2021.636861
225. Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ (2019) 26:332–47. doi: 10.1038/s41418-018-0122-7
226. Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J Immunol (2018) 200:2748–56. doi: 10.4049/jimmunol.1701492
227. Ramaswamy V, Cresence VM, Rejitha JS, Lekshmi MU, Dharsana KS, Prasad SP, et al. Listeria–review of epidemiology and pathogenesis. J Microbiol Immunol Infect (2007) 40:4–13.
228. Barbuddhe SB, Chakraborty T. Listeria as an enteroinvasive gastrointestinal pathogen. Curr Top Microbiol Immunol (2009) 337:173–95. doi: 10.1007/978-3-642-01846-6_6
229. Nandakumar R, Tschismarov R, Meissner F, Prabakaran T, Krissanaprasit A, Farahani E, et al. Intracellular bacteria engage a STING-TBK1-MVB12b pathway to enable paracrine cGAS-STING signalling. Nat Microbiol (2019) 4:701–13. doi: 10.1038/s41564-019-0367-z
230. Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to listeria monocytogenes. J Exp Med (2004) 200:527–33. doi: 10.1084/jem.20040976
231. O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to listeria monocytogenes infection. J Exp Med (2004) 200:437–45. doi: 10.1084/jem.20040712
232. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to listeria infection. J Exp Med (2004) 200:535–40. doi: 10.1084/jem.20040769
233. Mueller-Ortiz SL, Calame DG, Shenoi N, Li YD, Wetsel RA. The complement anaphylatoxins C5a and C3a suppress IFN-beta production in response to listeria monocytogenes by inhibition of the cyclic dinucleotide-activated cytosolic surveillance pathway. J Immunol (2017) 198:3237–44. doi: 10.4049/jimmunol.1601420
234. Louie A, Bhandula V, Portnoy DA. Secretion of c-di-AMP by listeria monocytogenes leads to a STING-dependent antibacterial response during enterocolitis. Infect Immun (2020) 88. doi: 10.1128/IAI.00407-20
235. Jin L, Getahun A, Knowles HM, Mogan J, Akerlund LJ, Packard TA, et al. STING/MPYS mediates host defense against listeria monocytogenes infection by regulating Ly6C(hi) monocyte migration. J Immunol (2013) 190:2835–43. doi: 10.4049/jimmunol.1201788
236. Loughran AJ, Orihuela CJ, Tuomanen EI. Streptococcus pneumoniae: Invasion and inflammation. Microbiol Spectr (2019) 7. doi: 10.1128/microbiolspec.GPP3-0004-2018
237. Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, et al. Two DHH subfamily 1 proteins in streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J Bacteriol (2013) 195:5123–32. doi: 10.1128/JB.00769-13
238. Wooten AK, Shenoy AT, Arafa EI, Akiyama H, Martin IMC, Jones MR, et al. Unique roles for streptococcus pneumoniae phosphodiesterase 2 in cyclic di-AMP catabolism and macrophage responses. Front Immunol (2020) 11:554. doi: 10.3389/fimmu.2020.00554
239. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe (2020) 27:556–570 e6. doi: 10.1016/j.chom.2020.02.004
240. Bjerre A, Brusletto B, Hoiby EA, Kierulf P, Brandtzaeg P. Plasma interferon-gamma and interleukin-10 concentrations in systemic meningococcal disease compared with severe systemic gram-positive septic shock. Crit Care Med (2004) 32:433–8. doi: 10.1097/01.CCM.0000104950.52577.97
241. Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, et al. Inflammasome-dependent IFN-gamma drives pathogenesis in streptococcus pneumoniae meningitis. J Immunol (2012) 189:4970–80. doi: 10.4049/jimmunol.1201687
242. Patel S, Tucker HR, Gogoi H, Mansouri S, Jin L. cGAS-STING and MyD88 pathways synergize in Ly6C(hi) monocyte to promote streptococcus pneumoniae-induced late-stage lung IFNgamma production. Front Immunol (2021) 12:699702. doi: 10.3389/fimmu.2021.699702
243. Ehrt S, Schnappinger D, Rhee KY. Metabolic principles of persistence and pathogenicity in mycobacterium tuberculosis. Nat Rev Microbiol (2018) 16:496–507. doi: 10.1038/s41579-018-0013-4
244. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe (2012) 11:469–80. doi: 10.1016/j.chom.2012.03.007
245. Sun Y, Zhang W, Dong C, Xiong S. Mycobacterium tuberculosis MmsA (Rv0753c) interacts with STING and blunts the type I interferon response. mBio (2020) 11. doi: 10.1128/mBio.03254-19
246. Karanja CW, Yeboah KS, Sintim HO. Identification of a mycobacterium tuberculosis cyclic dinucleotide phosphodiesterase inhibitor. ACS Infect Dis (2021) 7:309–17. doi: 10.1021/acsinfecdis.0c00444
247. Dey RJ, Dey B, Zheng Y, Cheung LS, Zhou J, Sayre D, et al. Inhibition of innate immune cytosolic surveillance by an m. tuberculosis phosphodiesterase. Nat Chem Biol (2017) 13:210–7. doi: 10.1038/nchembio.2254
248. Van Dis E, Sogi KM, Rae CS, Sivick KE, Surh NH, Leong ML, et al. STING-activating adjuvants elicit a Th17 immune response and protect against mycobacterium tuberculosis infection. Cell Rep (2018) 23:1435–47. doi: 10.1016/j.celrep.2018.04.003
249. Iliev ID, Leonardi I. Fungal dysbiosis: Immunity and interactions at mucosal barriers. Nat Rev Immunol (2017) 17:635–46. doi: 10.1038/nri.2017.55
250. Wheeler ML, Limon JJ, Underhill DM. Immunity to commensal fungi: Detente and disease. Annu Rev Pathol (2017) 12:359–85. doi: 10.1146/annurev-pathol-052016-100342
251. Netea MG, Joosten LA, van der Meer JW, Kullberg BJ, van de Veerdonk FL. Immune defence against candida fungal infections. Nat Rev Immunol (2015) 15:630–42. doi: 10.1038/nri3897
252. Brown GD, Gordon S. Fungal beta-glucans and mammalian immunity. Immunity (2003) 19:311–5. doi: 10.1016/S1074-7613(03)00233-4
253. Underhill DM, Iliev ID. The mycobiota: Interactions between commensal fungi and the host immune system. Nat Rev Immunol (2014) 14:405–16. doi: 10.1038/nri3684
254. Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during candida infections. PloS Pathog (2012) 8:e1002811. doi: 10.1371/journal.ppat.1002811
Keywords: cGAS, STING, infectious diseases, immune regulation, innate immunity
Citation: Du Y, Hu Z, Luo Y, Wang HY, Yu X and Wang R-F (2023) Function and regulation of cGAS-STING signaling in infectious diseases. Front. Immunol. 14:1130423. doi: 10.3389/fimmu.2023.1130423
Received: 23 December 2022; Accepted: 24 January 2023;
Published: 06 February 2023.
Edited by:
Tuo Li, University of Texas Southwestern Medical Center, United StatesReviewed by:
Sheng Sean Su, University of Texas Southwestern Medical Center, United StatesCopyright © 2023 Du, Hu, Luo, Wang, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong-Fu Wang, cm9uZ2Z1d2FAdXNjLmVkdQ==; Xiao Yu, eGlhb3l1NTIzQHNtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.