 Lu Pan1
Lu Pan1 Sirui Yang
Sirui Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 10 August 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1128754
This article is part of the Research TopicAutoimmune Diseases in ChildhoodView all 24 articles
Childhood-onset systemic lupus erythematosus (cSLE) is an autoimmune disease that results in significant damage and often needs more aggressive treatment. Compared to adult-onset SLE, cSLE has a stronger genetic background and more prevalent elevated type I Interferon expression. The management of cSLE is more challenging because the disease itself and treatment can affect physical, psychological and emotional growth and development. High dose oral glucocorticoid (GC) has become the rule for treating moderate to severe cSLE activity. However, GC-related side effects and potential toxicities are problems that cannot be ignored. Recent studies have suggested that GC pulse therapy can achieve disease remission rapidly and reduce GC-related side effects with a reduction in oral prednisone doses. This article reviews characteristics, including pathogenesis and manifestations of cSLE, and summarized the existing evidence on GC therapy, especially on GC pulse therapy in cSLE, followed by our proposal for GC therapy according to the clinical effects and pathogenesis.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that affects every organ system and causes heterogeneous clinical manifestations (1). This disease mostly occurs in adults, especially females (2). Approximately 15% to 20% of all SLE patients are diagnosed during childhood (3). Although childhood-onset SLE (cSLE) has similar pathogenesis, clinical manifestations, and immunologic disorders to adult-onset SLE (aSLE), there are some differences between them (4). There is evidence that cSLE has stronger genetic background and Interferon (IFN) signature (5). Previous studies have shown that compared to aSLE, cSLE has a more aggressive course and frequent damage accrual, with high morbidity and mortality (6). Moreover, patients with cSLE generally require more aggressive treatment to achieve lupus low disease activity state (LLDAS) than aSLE patients (7). Indeed, the management of cSLE is more challenging because the disease itself and treatment can affect physical, psychological and emotional growth and development.
Glucocorticoid (GC) remain the cornerstone of treatment for SLE patients and are commonly used for prolonged periods, especially in children. Daily high-dose oral GC is the most common therapeutic management for moderate to severe cSLE patients during the induction period (8). However, the side effects caused by long-term use of medium to high doses of GC are unfortunate for children who are growing, and this profile has led to a search for an optimized GC therapeutic management that has the least side effects. Recently, the intravenous pulse form of GC seems to have greater advantages in SLE. GC pulse is the essential element for effectively treating active SLE using lower doses of oral GC. In addition, GC pulse is not associated with most side effects, which were only related with the occurrence of cognitive impairment or psychosis (9, 10). Combined with the characteristics of cSLE, the possibility of GC pulse to reduce GC-related side effects while maintaining the disease remission offers significant promise. Therefore, this review summarized the characteristics of cSLE and described important developments in GC, especially in GC pulse, to develop new ideas for GC therapeutic management of cSLE from the perspective of pathogenesis and clinical practice.
Similar to aSLE, cSLE pathogenesis is complex and not fully understood. However, increasing severity with younger age and varying gender distribution in different age groups suggest variable pathogenic mechanisms between aSLE and cSLE.
Familial clusters, the impact of ethnicity on disease prognosis, age-specific differences in clinical and immunological phenotypes, and the high degree of concordance among monozygotic twins suggest a stronger involvement of genetic factors in cSLE. A recent study that included multi-ethnic SLE patients suggested that there is a negative association between non-HLA genetic risk and age of SLE diagnosis (11). Compared to aSLE, cSLE patients are more likely to carry novel/rare high-penetrance variants associated with monogenic lupus or may have a burden of low-penetrance common SLE susceptibility alleles (12).
Recently, cSLE has been found to be associated with single gene mutations, defining the concept of monogenic lupus. Genes linked to monogenic lupus belong to type I interferonopathies, the complement deficiencies, T and B cell tolerance breakdown, or other uncharacterized pathways (13).First, type I interferonopathies refer to a group of complex genetic disorders associated with imbalance of IFN mediated immune responses. The mutations identified so far cause IFN overexpression in three different pathways: defect in nucleases (TREX1, SAMHD1, ADAR1, RNASEH2), enhanced sensitivity of an innate immune sensor (IFIH, DDX58) or adaptor molecules downstream the innate sensors (TMEM173), and defective negative feedback of the IFN pathway (ISG15) (14). Furthermore, mutations in the ACP5 gene result in a deficiency of tartrate-resistant acid phosphatase (TRAP) enzyme, which eventually leads to excessive IFN production and the development of SLE (15). Mutations in genes involved in IFN signaling pathway are shown in Figure 1. Second, complement deficiency is another described subcategories of monogenic lupus and highlight the importance of apoptotic body clearance in lupus pathogenesis (16). Genetic deficiencies of C1q/r/s strongly predispose to SLE, with a penetrance of nearly 90% (17). Deficiencies in other complement component also promote SLE, but the risk is lower. Notably, complement deficiencies are associated with early-onset lupus, and has a less biased of sex ratio compared to aSLE. Third, tolerance breakdown caused by genetic mutations in B and/or T cell promotes SLE. Protein kinase C-δ (PKC-δ) is a serine/threonine kinase important in multiple apoptotic signaling cascades. Mutation in PKC-δ is associated with loss of B cell tolerance and has been identified in cSLE patients (18). Deficiency in RAG1/RAG2 lead to defects in T cell tolerance, and a patient with SLE was reported to have heterozygous mutation in RAG2 recently (19). Other genes involved in monogenic lupus including DNASE1/DNASE1L3 encoding proteins involved in the nucleic acid degradation pathway and CYBB gene causing chronic granulomatous disease.
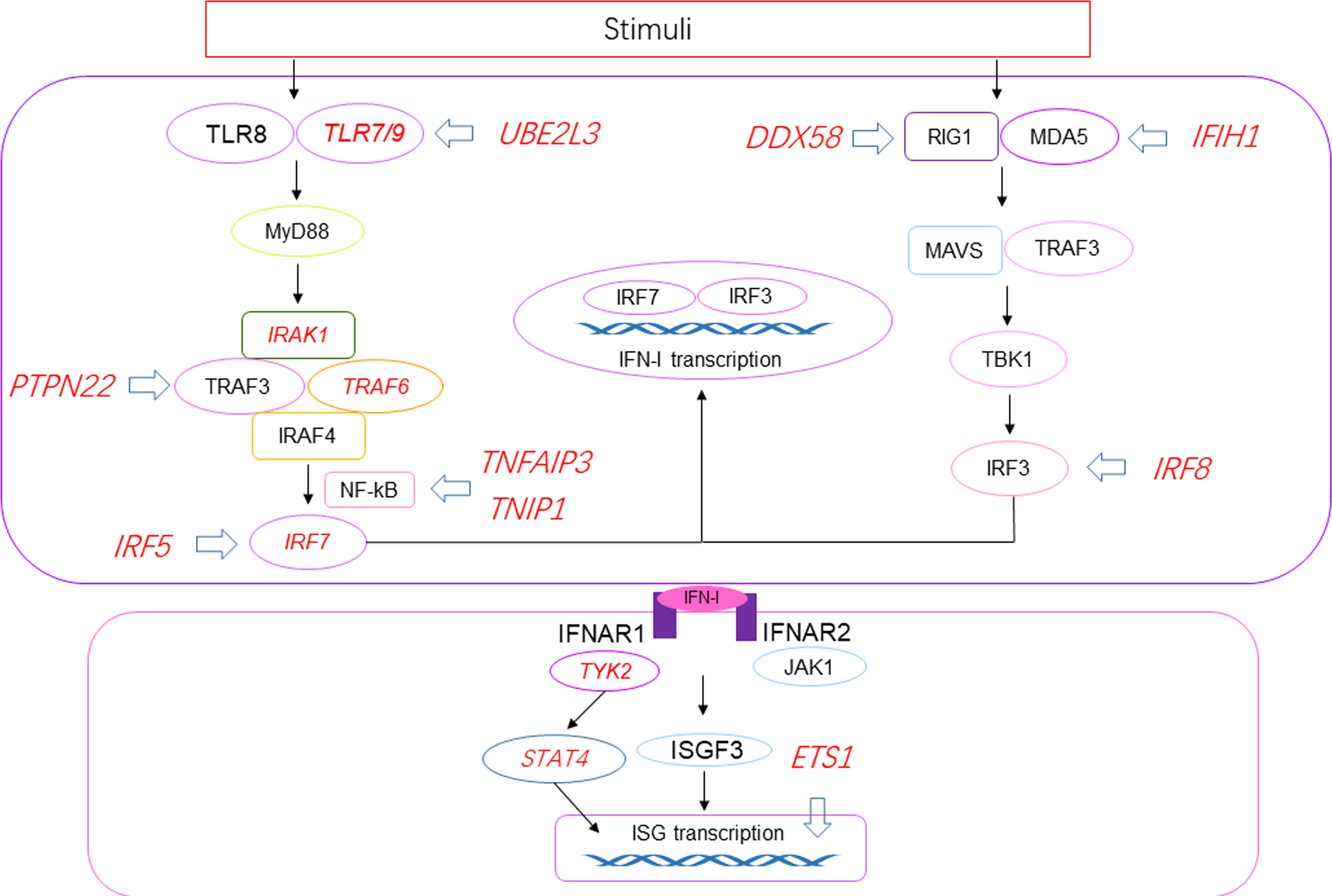
Figure 1 Susceptibility genes related to the production of IFN-I and IFN-I signaling pathway in SLE. Red italics represent susceptibility genes, and blue arrows represent proteins in the pathway affected by the susceptibility genes. UBE2L3 regulate the degradation of TLR4/9. High IFN-α in SLE patients is related to the risk allele of PTPN22 though TRAF3. TNFAIP3 and TNIP1 encode regulatory factors in IFN-I pathway, NF-κB activation. DDX58 is related to RIG1 hyperactivation. IFIH1 encodes MDA5 and is involved in elevated expression of IFN-induced genes. ETS1 is a negative regulator in SLE. IRF5/7/8 can directly induce transcription of proteins in IFN-I signaling pathway. IRAK1, TRAF6, STAT4 and TYK2 are related to the production of IFN-I. UBE2L3, ubiquitin-conjugating enzyme E2 L3; PTPN22, protein tyrosine phosphatase non-receptor type 22.
Indeed, single locus confers only a small disease risk. Most individuals carry risk alleles and develop SLE with the influence of environment and time. Genome-wide association studies (GWAS) have identified more than 100 risk loci, accelerating the discovery of common single nucleotide polymorphisms (SNPs) (20). Compared with aSLE, cSLE are less affected by environment and time, but have more severe clinical manifestations, which may be related to increased genetic risk. The calculation of genetic risk score (GRS) by counting the number of SLE-associated risk alleles weighted odds ratios (ORs) has more power to predict disease susceptibility (21). Studies in Gullah and African-Americans showed an increased number of SLE-associated polymorphisms in cSLE compared with aSLE (22). Similarly, a Korean study showed that cSLE had a higher genetic risk scores (GRS) than aSLE (23). A recent study showed that there was a trend for stronger associations between both GRS and LN risk in Europeans with cSLE compared with aSLE (24). The evidence for common variants being directly pathogenic is limited. Next generation sequencing (NGS) technologies, including whole genome sequencing (WGS) and whole exome sequencing (WES), have led to increasing recognition that rare/novel variants are more deleterious than common variants (25). A previous study explored rare variants by undertaking WES of SLE patients showed that 14 missense de-novo variants were identified in SLE probands (26). In recent years, more and more rare variants that cause SLE have been identified, such as novel peripheral gene MEF2D, germline rare P2RY8 missense variants and SAT1 LOF variants (27–29). A study in Mexicans showed that catalytically impaired TYK2 variants were protective against SLE, but there is no difference between aSLE and cSLE (30). The genetic variants in cSLE differed from those in aSLE are showed in Table 1.
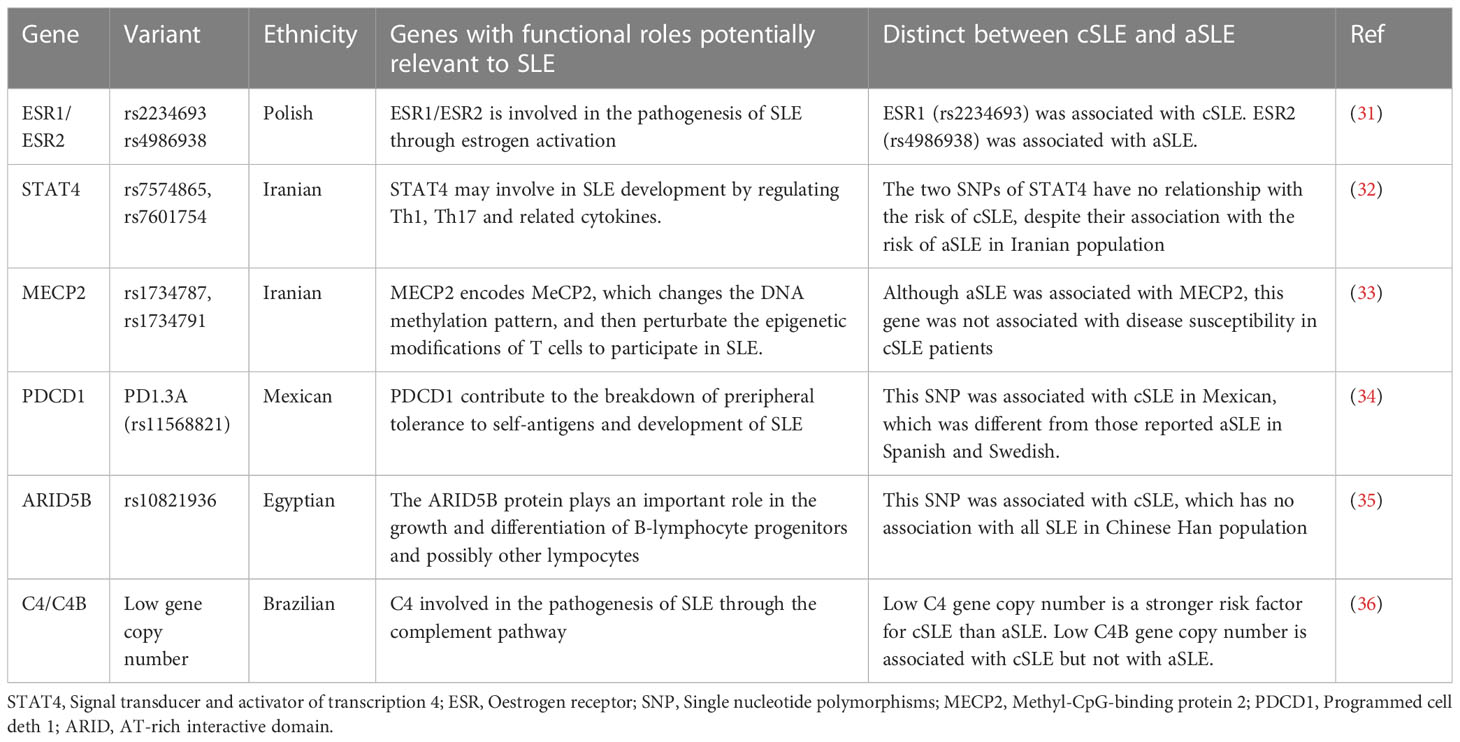
Table 1 Different genetic variants between cSLE and aSLE.
The clinical variability of SLE is further influenced by genetic modifiers, epigenetics and environmental factors. We will only describe the epigenetic mechanisms briefly. DNA methylation, posttranslational histone modifications, and non-coding RNAs are the main epigenetic mechanisms studied. DNA hypomethylation plays an important role in T cell activation and contributes to SLE pathogenesis (37). It is worth noting that some DNA methylation, such as long interspersed nuclear element-1 (LINE-1), differs between aSLE and cSLE (38). Abnormal histone acetylation and altered microRNA (miRNA) expression also play crucial roles in the pathological processes of SLE. Increased histone acetylation in monocytes and CD4+ T cells has been detected in SLE patients and inhibition of histone acetylation can alleviate disease activity (39). In recent years, there have been many studies on the progress of miRNA in SLE, such as miR-146a, miR-31 and miR-98 (40, 41). In addition, E2F transcription factor 2 (E2F2)-miR-17-5p was found to increase autoantibody production by upregulating IL-10 (42). Furthermore, miR-448 was involved in SLE by targeting the suppressor of cytokine signaling 5 (SOCS5) to promote helper T cell (Th)17 activation (43). Taken together, genetic factors play an important role in SLE, and the available data suggested that cSLE is more strongly influenced by genetic factors.
Type I IFN (IFN-I) is a multifunctional immune factor that bridges innate and adaptive immunity. It has been reported that IFN-I can promote the expansion of autoantibody-secreting cells (44) and more than half of the SLE-associated genetic loci are connected to the IFN-I pathway (45). Of note, the overexpression of IFN-regulated genes in the peripheral blood monocytes of almost all active cSLE patients was observed only in approximately 50% of aSLE patients (46, 47), revealing that cSLE is more closely related to IFN-I.
Plasmacytoid dendritic cells (pDCs) are the primary source of IFN-I. Under normal conditions, IFN-I production is strictly regulated for it ceases after pathogens have been cleared. However, many SLE patients demonstrate chronic overactivity in IFN-I pathways. Endogenous stimuli act upon a susceptible genetic background to result in IFN-I production.
In SLE, pDCs are activated by specific IFN immune complexes (ICs), derived from autoantibodies and endogenous or exogenous nucleic acid-binding proteins, and produce IFN-I (48). Interferogenics ICs are endocytosed through Fc gamma receptor IIa (FcγRIIa) on pDC and interact with TLR7 or TLR8/9 and initiate an activation chain that includes myeloid differentiation factor 88 (MyD88). MyD88 activates interleukin-1 receptor-associated kinase (IRAK) 4, then triggers IRAK1, tumor necrosis factor receptor associated factor (TRAF) 3 and TRAF6. Next, the interferon regulatory family (IRF) 7 is activated, resulting the initiate transcription of IFN-I (49). RIG-I and melanoma differentiation-associated gene 5 (MDA5) recognize ds-RNA and bind to mitochondrial antiviral signaling protein (MAVS), which activates TRAF3 and TANK-binding kinase 1 (TBK1) resulting the activation of IRF3 (50). Neutrophil extracellular traps (NETs) are another mechanism that triggers IFN-I generation. NETs accumulate in SLE patients, resulting in long-term exposure to the body and externalization of their antigens, thus producing high levels of IFN-I in a TLR9-dependent manner (51). In addition, mitochondrial reactive oxygen species (ROS) can initiate NETs formation (NETosis), activate the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, and induce IFN-I production. IFN-I production further exerts its effects by ligating the IFN-α/β receptor (IFNAR). The Janus activating kinase (JAK)-STAT pathway is the most common signaling pathway. IFN-I combines with IFNAR to activate JAK1 and tyrosine kinase 2 (TYK2), and phosphorylated STAT1 and STAT2 form a transcription factor complex with IFN regulatory factor 9 (IRF9), which translocate to the nucleus, binds to IFN regulator elements in the promoters of IFN-regulated genes and initiates transcription genes (52). In SLE, IFN signaling-induced IFN-regulated genes participate in a positive feedback loop of autoimmunity, causing permanent autoimmune inflammation. Gene mutations related to IFN-I production or IFN-I signaling pathway (Figure 1) can lead to the production of self-derived IFN inducers and suppress the negative feedback signals that downregulate the IFN response.
IFN-I has a significant impact on the immune system. IFN-α promotes the expression of MHC-II and costimulatory molecules and stimulates monocytes to differentiate into mature dendritic cells (DCs) (53). Mature DCs and IFN-α activate B cells and increase autoantibody production. B cells interact with pDC through the platelet endothelial cell adhesion molecule (PECAM)-1 receptor to promote IFN-α secretion. IFN-α can also promote the differentiation of CD4+ T cells into Th1 and Th17, inhibit the development of Th2 and regulatory T cells (Treg), and enhance the cytotoxicity of CD8+ T cells. However, persistent IFN-α stimulation inhibits Th1 differentiation and promotes the development of T follicular helper (Tfh) cells, which supports B cell activation (54). Regarding the innate immune system, IFN-α enhances macrophage phagocytosis and natural killer (NK) cell cytotoxicity, and NK cells have a particularly strong effect on pDC through lymphocyte-associated antigen-1 (LFA-1)-dependent cell-cell interaction (55). Therefore, activation of the IFN-I system in SLE will widely affect the immune system (Figure 2), drive autoimmune response and chronic inflammation, and eventually cause tissue and organ damage. What’s more, children with SLE had elevated TLRs expression, and TLRs can induce neutrophil apoptosis, which lends more support to the role of IFN-I in cSLE (56).
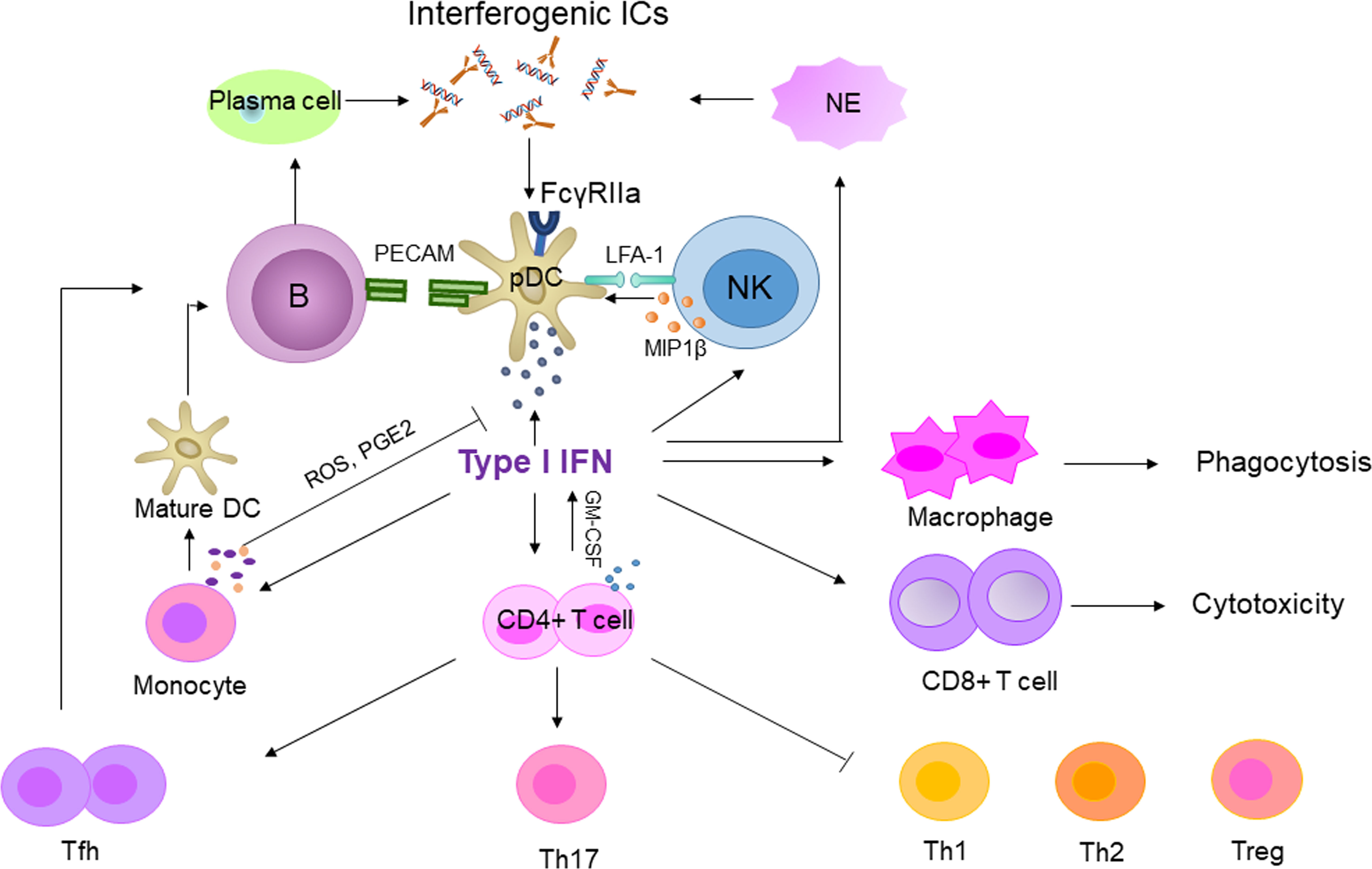
Figure 2 The central role of type I IFN in SLE. Interferogenic ICs stimulate pDCs and produce type I IFN. The secreted type I IFN acted on innate and adaptive immune cells to amplify the autoimmune response. B, T and NK enhanced type I IFN production by activated pDCs via PECAM, GM-CSF and LFA-1, respectively. In contrast, activated monocytes suppressed the secreted type I IFN by pDC via ROS and PGE2. GM-CSF, Granulocyte-macrophage colony stimulating factor; PGE2, Prostaglandin E2; NE, neutrophil.
aSLE and cSLE differ in some disease phenotypes. Children have less gender bias in favor of females than adults. Moreover, children have higher SLE disease activity index (SLEDAI) scores than adults, and the earlier the age of onset, the greater the disease activity (57). This heterogeneity is broader in terms of clinical manifestations.
Systemic presentations, such as neurological, renal, and haematological involvement, are more significant with cSLE, whereas Raynaud’s phenomenon, pulmonary involvement, and photosensitivity are more common with aSLE (58) (Table 2). Among these, nephritis is the most important variation. A previous study showed that crescents on biopsy were more common in the cSLE (69). Another study reported the proportion of adults and children with lupus nephritis receiving kidney transplantation to be 1.9% and 3%, respectively (70). Neurological involvement is another important variation. A recent meta-analysis of neuropsychiatric symptoms in cSLE revealed that neuropsychiatric events were common in cSLE, including headaches, cognitive dysfunction, mood disorders and psychosis (71). Childhood-onset lupus remains a strong predictor of mortality. Serologically, cSLE and aSLE have similar positivity rates for most circulating antinuclear antibodies. However, a recent study showed that children had more anti-double-stranded DNA (dsDNA) and anticardiolipin immunoglobulin M (IgM) but less anti–Sjögren’s-syndrome-related antigen A (anti-Ro) and anti–Sjögren’s-syndrome-related antigen B (anti-La) antibodies (62). Antiphospholipid antibody-related thrombosis is uncommon in children but common in adults (72). In addition, rheumatoid factor positivity is more frequently encountered in aSLE, whereas arthritis is more common in cSLE (4).
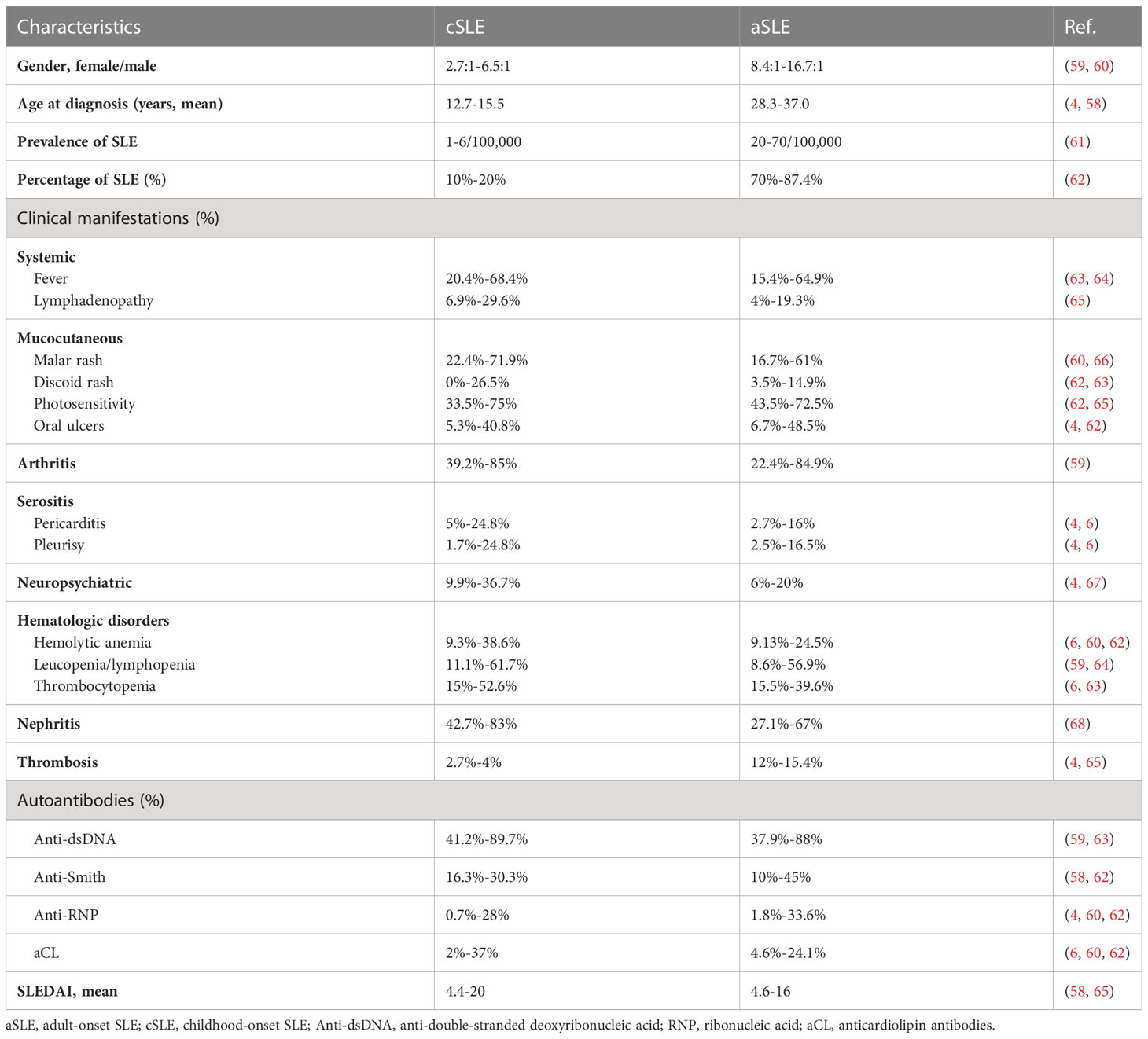
Table 2 Clinical and immunological characteristics in cSLE and aSLE.
SLE cannot be cured, disease activity fluctuates with periods of flare and remission. Long-term maintenance of LLDAS is the goal of current treatment. Previous research has shown that cSLE is less likely to achieve LLDAS than aSLE is (7). Additionally, the specific physical and psychological characteristics of children and adolescents render cSLE patients more vulnerable to the long-term effects of the disease. Hydroxychloroquine and immunosuppressive drugs have improved the prognosis of SLE. The efficacy of “new” biologic agents, such as belimumab and telitacicept, is exciting. However, GC remains the mainstay of SLE. GCs are required by 97% and 70% of cSLE and aSLE patients, respectively, and their average doses are higher in children than in adults (73). cSLE patients not only have a higher Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) Damage Index (SDI), but also accumulate damage faster (74), which is mainly related to GC toxicity, although time is considered. Given that GC play an irreplaceable role in the treatment of cSLE, their proper use is particularly important.
The intensity of GC is as follows: low dose ≤7.5mg/d prednisone equivalent; medium dose >7.5mg/d, but ≤30mg/d prednisone equivalent; high dose >30mg/d, but ≤ 100mg/d prednisone equivalent; pulse therapy ≥250mg/d for one or a few days (75). The use of high dose oral GC have become the rule for treating moderate to severe SLE activity (76). Recent data suggested that GC pulse therapy may have a biologic rational in the treatment of SLE, especially of cSLE.
The anti-inflammatory and immunosuppressive actions of GC are exerted by two different mechanisms. Classic genomic effects are mediated by cytosolic GC receptors (GR). GC diffuse through the plasma membrane into the cytoplasm, forming a multiprotein complex with GR (GC-GR). The activated GC-GR complex moves to the nucleus and binds to specific GC-responsive elements (GRE), activating the transcription of specific genes and the synthesis of specific regulator proteins; this process is known as transactivation (Figure 3). Alternately, the GC-GR complex inhibits the activity of transcription factors such as activator protein 1 (AP-1) or NF-κB through direct or indirect action, thereby reducing the production of proinflammatory factors (77); this process is known as transrepression (Figure 3). In most cases, the anti-inflammatory and immunomodulatory effects of GC are achieved through transrepression, and transactivation mediates its side effects. A prednisone dose of >100 mg/day led to complete saturation of cGR, and non-genomic effects were activated. Three main modes of action exist for non-genomic effects. The GC-GR complex directly blocks the activation of phospholipase A2, thereby inhibiting the release of arachidonic acid. In contrast, GC rapidly activate GR on cell membranes, mediating rapid signal transduction via p38 MAPK. In addition, GC can alter cellular function by influencing cation transport, which contributes to rapid immunosuppression (78). The clinical impacts of non-genomic effects are rapid and play an important role in mediating GC pulse therapy. In contrast, genomic effects are often not immediate and are the primary activation mechanism of daily oral GC therapy. The toxicity and anti-inflammatory effects of the genomic effect increase in parallel with the dose. However, activating non-genomic effects provides additional benefits to patients with no increased toxicity (79). Ruiz-Arruza et al. found that the mean daily prednisone dose was higher in patients accruing GC-attributable damage (11 vs 7 mg/day, p=0.04) and MP pulses are not associated with damage accrual, suggesting that the dose of MP pulse is not included in the cumulative dese, which may be attributed to its rapid metabolism, as it is administered for a short time rather than continuously every day. Therefore, high-dose GC pulse can fully exert non-genomic effects and exert anti-inflammatory effect more quickly and fully. This is critical for children with SLE who require long-term management, as reducing organ damage in the early stage of disease helps to improve prognosis.
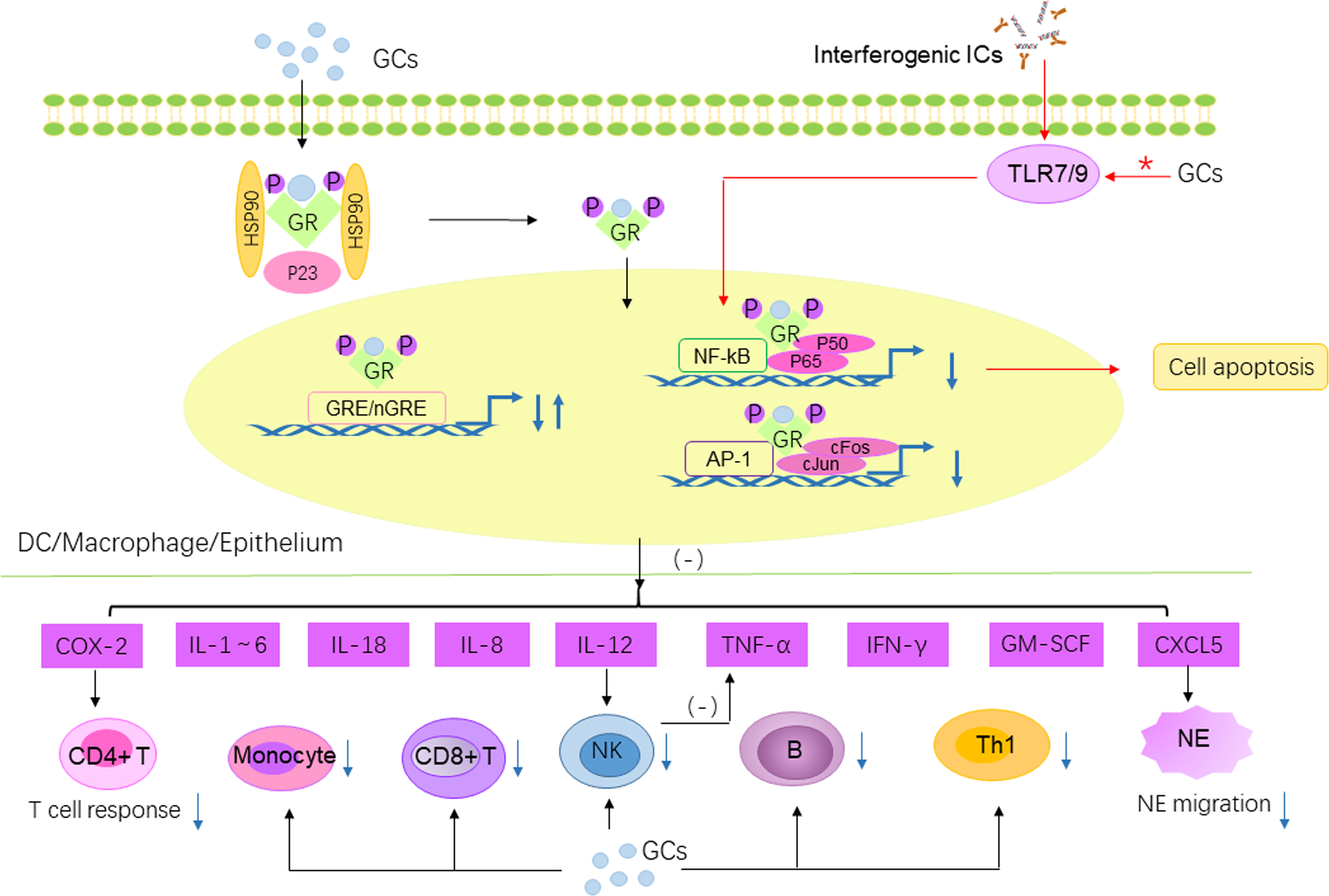
Figure 3 The anti-inflammatory/immunosuppressive effects of GC and difference between oral and pulsed GC. The effects of GC are transmitted intracellularly through the binding of GR to GRE/nGRE to suppress the expression of pro-inflammatory genes. Furthermore, GR can interact with p65 subunit of NF-κB to repress NF-κB regulated gene expression. GR can also interact with c-Jun to repress AP-1 regulated gene transcription. Then the synthesis of IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-12, IL-18, GM-CSF, TNF-α, IFN-γ are decreased. The decreased IL-12 further reduced the secretion of TNF-α by NK cells. GR negatively regulates CXCL5, which in turn decreases NE migration. On the other hand, GR suppress the excessive inflammatory response mediated by T cell response by inhibiting COX-2. Finally, GC directly decreased the number of multiple immune cells including NK, CD8+ T cells, B cells, Th1 cells and monocytes. Pathway marked by red asterisks and red arrows is the different pathway affected by oral and pulse GC. The GC pulse regimen affects TLR7/9 in the IFN pathway, thereby affecting the activation of NF-κB, which in turn mediates pDC apoptosis and reduces IFN-α production. IFN pathway is not significantly affected by oral GCs. GRE, glucocorticoid response elements; nGRE, negative GRE; GM-CSF, granulocyte macrophage-colony stimulating factor; NE, neutrophil; TNF-α, tumor necrosis factor-α.
Almost all primary and secondary immune cells are target for GC effects. As mentioned above, GC act by binding to GR that stimulates or inhibits gene expression. The anti-inflammatory and immunosuppressive effects of GC include inhibiting the synthesis of inflammatory cytokines and inducing the apoptosis of immune cells (See Figure 3 for details). Clinical effects of GC are strongly dependent on the solubility, the rate of absorption, the metabolic rate, the affinity of GC to its receptor and the dose administered. Prednisone is commonly used as oral therapy, whereas methylprednisolone (MP) is often used as pulse therapy for its high bioavailability and wide distribution. In general, more than 90% of circulating GC is bound to transcortin (80). When transcortin is saturated, GC bind to albumin or remain unbound. Prednisone shows nonlinear protein binding, whereas MP is strictly linear, possibly because it binds albumin and not transcortin (81). In terms of GC absorption, although GC have a high oral absorption rate and bioavailability of > 60%, intravenous administration promotes a more rapid onset of action and higher bioavailability (82). And what’s more, compared with adults, the total body clearance (CL) of MP is often higher in children administered high-dose MP pulse therapy, indicating that individual children are more tolerant to MP pulse therapy (83).
Although GC pulse therapy is considered to have lesser systemic side effects, there are few studies in literature comparing the effects of GC pulse therapy versus daily oral GC on the pathogenesis of SLE. Guiducci et al. (84) found that the daily oral prednisone returned to normal multiple transcriptional modules, except for the IFN pathway. In contrast, intravenous MP (IVMP) pulse therapy normalized the IFN signature. Consistent with this, the number of pDCs in the IVMP pulse group was significantly reduced, accompanied by decreased IFN-α levels (Figure 3). In this study, further experiments confirmed that stimulation of pDCs though TLR7/9 can account for the reduced activity of GC to inhibit the IFN pathway in SLE. And activation of NF-κB is crucial for the survival of pDCs. Interestingly, recognition of self-nucleic acid by TLR7/9 is an important step in the pathogenesis of cSLE, promoting the production of IFN-I. Combined with the overexpression of IFN-regulated genes and TLRs are more closely related to cSLE (46, 56), suggesting that IVMP pulse therapy may be more suitable for cSLE.
Children are more susceptible to the side effects of GC than adults, especially regarding growth, development, mental and psychological aspects. GC-related toxicity is highly dependent on the time and dose of exposure. A safe dose has not been established, and GC-related damage occurs even at doses of 4.5-7.5mg/day (85). The effect of GC on bone is worth noting, as it is closely related to osteoporosis and osteonecrosis. The persistent use of GC in children can lead to delayed puberty and short stature in adulthood (86). This may be related to the GC-induced inhibition of the hypothalamic-pituitary-adrenal (HPA) axis and the decrease in bone mineral density. Some studies have demonstrated that GC pulse therapy does not lead to a decrease in bone mineral density and is not independently correlated with osteonecrosis (87). Additionally, a study on multiple sclerosis (MS) using GC pulse therapy found no prolonged suppressive effect on the HPA axis (88). This might explain the reduced severity of Cushing’s syndrome in children treated with GC pulse therapy compared with those treated with daily oral therapy (89). Neuropsychiatric symptoms are another common adverse effect of GC in children. Epileptic seizures, behavioral abnormalities, and cognitive impairment are the most common neuropsychiatric manifestations in cSLE patients treated with GC. However, there is evidence that neuropsychiatric symptoms are not associated with GC pulse therapy (90). In addition to organ damage caused by persistent disease activity, GC is an independent risk factor for cSLE damage. And studies on aSLE have shown that GC pulse therapy is not associated with damage accrual. Infection and hyperglycemia are other common adverse reactions. Although fasting blood glucose (FBG) levels are elevated during GC pulse therapy, FBG levels in non-diabetic patients slowly return to normal after the pulse ends (90). Major infections can occurre at a median oral prednisone dose of only 7.5mg, and the risk of infection increased 11-fold for every 10mg daily increase in prednisone dose (91). Infection appears to be the side effect associated with GC pulse therapy that requires special attention. However, some researchers suggest that GC pulses have no independent effect on infections (92).
GC pulses were first used for the treatment of SLE in the 1970s and have been used ever since. An observational cohort study showed that IVMP pulse therapy reduced the subsequent oral doses of prednisone (93). The use of reduced oral prednisone doses decreased GC-related damage and improved cardiovascular prognosis without increasing SLE-induced damage (94). These findings suggest that IVMP pulse therapy is superior to high-dose daily oral prednisone therapy. The Lupus-Cruces protocol, based on the above evidence, has attracted much attention in recent years. This protocol can be summarized as intermittent IVMP pulses combined with low-dose oral prednisone. Compared to traditional GC treatment regimens, intermittent IVMP pulses reduce the dose of oral prednisone and enhance the clinical response (95). A recent observational cohort study showed a higher rate of prolonged remission in the Lupus-Cruces group (96). All of the above suggested that intermittent IVMP pulse combined with low-dose oral prednisone have good application prospects for the treatment of SLE. Details of this GC administration pattern are provided in Figure 4. It should be noted that there is significant variation in individual patient responsiveness to GC therapy, especially in patients with associated immunodeficiency. Agents targeting genes and genetic pathways are currently under more intensive investigation for patients with immunodeficiency in cSLE (97).
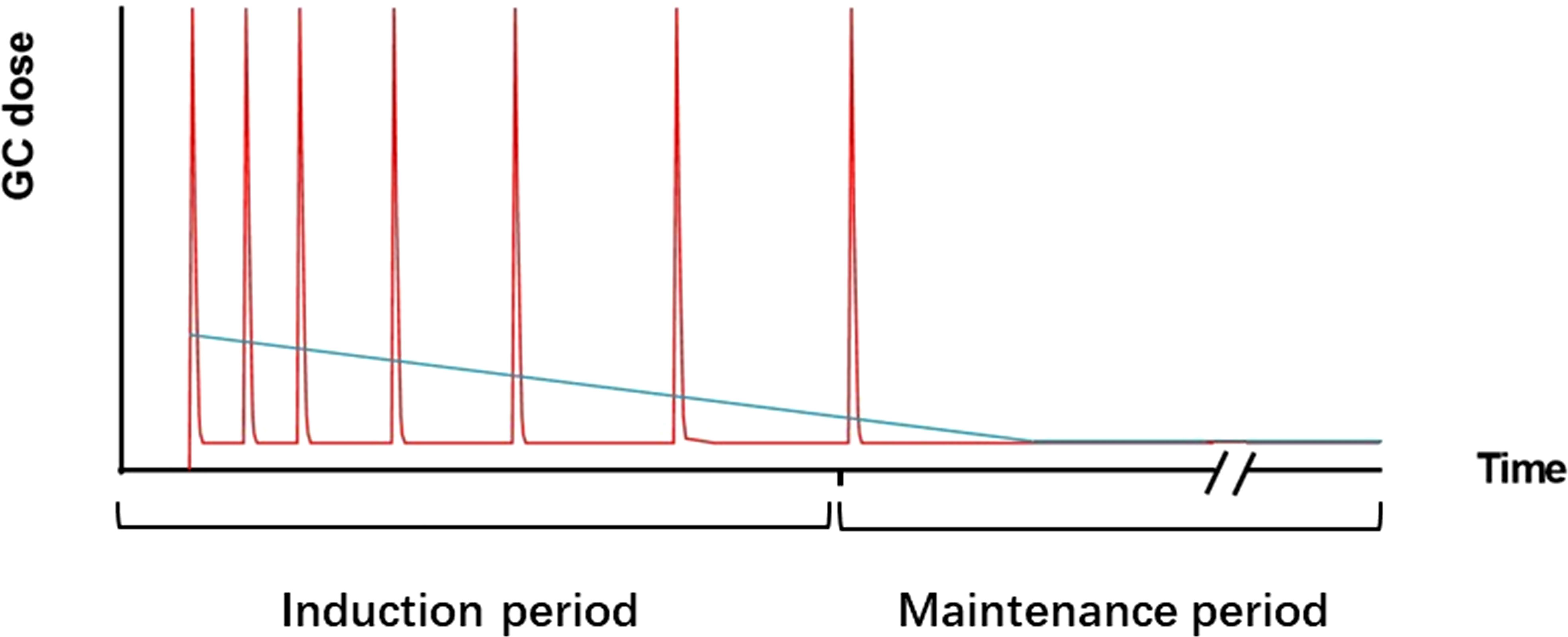
Figure 4 Pattern-trend plot of GC therapy. The red line represents the repeated IVMP pulses combined with low-dose oral prednisone therapy. The blue line represents the daily oral prednisone therapy. In daily oral therapy, prednisone doses decreased gradually but mostly remained at higher levels. In intermittent IVMP pulses combined with low-dose oral prednisone therapy, MP doses were high during the pulses, and intermittent and maintenance periods were given a low dose of GCs.
cSLE has more severe clinical manifestations and injuries earlier than aSLE does. Compared to adult-onset SLE, cSLE has a stronger genetic background and more prevalent elevated IFN-I expression. Guidelines and consensus recommendations for moderate to severe cSLE are high-dose daily oral GC. In view of the side effects and irreplaceability of GC, the key approach for a reasonable and successful systematic GC treatment is to minimize the dose of the administered GC in order to reduce the occurrence of side effects. Of course, immunosuppressive agents are necessary regardless of GC application regimen. GC pulse therapy can rapidly exert anti-inflammatory and immunosuppressive effects with reduced GC-related toxicity and allowing a reduction in oral prednisone doses. Combined with the pharmacology, activation mechanism and the specific effect on IFN of the GC pulse, intermittent IVMP pulse therapy may be the preferred treatment for cSLE rather than the current daily oral therapy. Further clinical and experimental studies are required to support this idea.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This work was supported by the grant from Department of Science and Technology of Jilin Province, China, under Grant YDZJ202101ZYTS082; and Department of Finance of Jilin Province, China, under Grant JLSWSRCZX2021-025.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pan L, Lu MP, Wang JH, Xu M, Yang SR. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J Pediatr (2020) 16(1):19–30. doi: 10.1007/s12519-019-00229-3
2. Ahamada MM, Jia Y, Wu X. Macrophage polarization and plasticity in systemic lupus erythematosus. Front Immunol (2021) 12:734008. doi: 10.3389/fimmu.2021.734008
3. Silva CA, Avcin T, Brunner HI. Taxonomy for systemic lupus erythematosus with onset before adulthood. Arthritis Care Res (Hoboken) (2012) 64(12):1787–93. doi: 10.1002/acr.21757
4. El-Garf K, El-Garf A, Gheith R, Badran S, Salah S, Marzouk H, et al. A comparative study between the disease characteristics in adult-onset and childhood-onset systemic lupus erythematosus in Egyptian patients attending a large university hospital. Lupus (2021) 30(2):211–8. doi: 10.1177/0961203320972778
5. Wright TB, Punaro M. Paediatric systemic lupus erythematosus: insights from translational research. Rheumatol (Oxf) (2017) 56(suppl_1):i24–31. doi: 10.1093/rheumatology/kew447
6. Fatemi A, Matinfar M, Smiley A. Childhood versus adult-onset systemic lupus erythematosus: long-term outcome and predictors of mortality. Clin Rheumatol (2017) 36(2):343–50. doi: 10.1007/s10067-016-3509-1
7. Fiorot FJ, Islabão AG, Pereira RM, Terreri MT, Saad-Magalhães C, Novak GV, et al. Disease presentation of 1312 childhood-onset systemic lupus erythematosus: influence of ethnicity. Clin Rheumatol (2019) 38(10):2857–63. doi: 10.1007/s10067-019-04631-0
8. Ruiz-Irastorza G, Bertsias G. Treating systemic lupus erythematosus in the 21st century: new drugs and new perspectives on old drugs. Rheumatol (Oxf) (2020) 59(Suppl5):v69–81. doi: 10.1093/rheumatology/keaa403
9. Ruiz-Irastorza G, Ugarte A, Ruiz-Arruza I, Khamashta M. Seventy years after Hench’s Nobel prize: revisiting the use of glucocorticoids in systemic lupus erythematosus. Lupus (2020) 29(10):1155–67. doi: 10.1177/0961203320930099
10. Ruiz-Arruza I, Ugarte A, Cabezas-Rodriguez I, Medina JA, Moran MA, Ruiz-Irastorza G. Glucocorticoids and irreversible damage in patients with systemic lupus erythematosus. Rheumatol (Oxf) (2014) 53(8):1470–6. doi: 10.1093/rheumatology/keu148
11. Dominguez D, Kamphuis S, Beyene J, Wither J, Harley JB, Blanco I, et al. Relationship between genetic risk and age of diagnosis in systemic lupus erythematosus. J Rheumatol (2021) 48(6):852–8. doi: 10.3899/jrheum.200002
12. Demirkaya E, Zhou Q, Smith CK, Ombrello MJ, Deuitch N, Tsai WL, et al. Brief report: deficiency of complement 1r subcomponent in early-onset systemic lupus erythematosus: the role of disease-modifying alleles in a monogenic disease. Arthritis Rheumatol (2017) 69(9):1832–9. doi: 10.1002/art.40158
13. Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New horizons in the genetic etiology of systemic lupus erythematosus and lupus-like disease: monogenic lupus and beyond. J Clin Med (2020) 9(3):712. doi: 10.3390/jcm9030712
14. Omarjee O, Picard C, Frachette C, Moreews M, Rieux-Laucat F, Soulas-Sprauel P, et al. Monogenic lupus: Dissecting heterogeneity. Autoimmun Rev (2019) 18(10):102361. doi: 10.1016/j.autrev.2019.102361
15. An J, Briggs TA, Dumax-Vorzet A, Alarcón-Riquelme ME, Belot A, Beresford M, et al. Tartrate-resistant acid phosphatase deficiency in the predisposition to systemic lupus erythematosus. Arthritis Rheumatol (2017) 69(1):131–42. doi: 10.1002/art.39810
16. Alperin JM, Ortiz-Fernández L, Sawalha AH. Monogenic lupus: A developing paradigm of disease. Front Immunol (2018) 9:2496. doi: 10.3389/fimmu.2018.02496
17. Sturfelt G, Truedsson L. Complement in the immunopathogenesis of rheumatic disease. Nat Rev Rheumatol (2012) 8(8):458–68. doi: 10.1038/nrrheum.2012.75
18. Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, et al. Protein kinase cδ deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis Rheum (2013) 65(8):2161–71. doi: 10.1002/art.38008
19. Walter JE, Lo MS, Kis-Toth K, Tirosh I, Frugoni F, Lee YN, et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J Allergy Clin Immunol (2015) 135(1):272–3. doi: 10.1016/j.jaci.2014.07.063
20. Charras A, Smith E, Hedrich CM. Systemic lupus erythematosus in children and young people. Curr Rheumatol Rep (2021) 23(3):20. doi: 10.1007/s11926-021-00985-0
21. Deng Y, Tsao BP. Updates in lupus genetics. Curr Rheumatol Rep (2017) 19(11):68. doi: 10.1007/s11926-017-0695-z
22. Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis (2011) 70(1):151–6. doi: 10.1136/ard.2010.141697
23. Joo YB, Lim J, Tsao BP, Nath SK, Kim K, Bae SC. Genetic variants in systemic lupus erythematosus susceptibility loci, XKR6 and GLT1D1 are associated with childhood-onset SLE in a Korean cohort. Sci Rep (2018) 8(1):9962. doi: 10.1038/s41598-018-28128-z
24. Webber D, Cao J, Dominguez D, Gladman DD, Levy DM, Ng L, et al. Association of systemic lupus erythematosus (SLE) genetic susceptibility loci with lupus nephritis in childhood-onset and adult-onset SLE. Rheumatol (Oxf) (2020) 59(1):90–8. doi: 10.1093/rheumatology/kez220
25. Jiang SH, Stanley M, Vinuesa CG. Rare genetic variants in systemic autoimmunity. Immunol Cell Biol (2020) 98(6):490–9. doi: 10.1111/imcb.12339
26. Pullabhatla V, Roberts AL, Lewis MJ, Mauro D, Morris DL, Odhams CA, et al. De nov mutations implicate novel genes in systemic lupus erythematosus. Hum Mol Genet (2018) 27(3):421–9. doi: 10.1093/hmg/ddx407
27. He Y, Gallman AE, Xie C, Shen Q, Ma J, Wolfreys FD, et al. P2RY8 variants in lupus patients uncover a role for the receptor in immunological tolerance. J Exp Med (2022) 219(1):e20211004. doi: 10.1084/jem.20211004
28. Farias FHG, Dahlqvist J, Kozyrev SV, Leonard D, Wilbe M, Abramov SN, et al. A rare regulatory variant in the MEF2D gene affects gene regulation and splicing and is associated with a SLE sub-phenotype in Swedish cohorts. Eur J Hum Genet (2019) 27(3):432–41. doi: 10.1038/s41431-018-0297-x
29. Xu L, Zhao J, Sun Q, Xu X, Wang L, Liu T, et al. Loss-of-function variants in SAT1 cause X-linked childhood-onset systemic lupus erythematosus. Ann Rheum Dis (2022) 81(12):1712–21. doi: 10.1136/ard-2022-222795
30. Contreras-Cubas C, García-Ortiz H, Velázquez-Cruz R, Barajas-Olmos F, Baca P, Martínez-Hernández A, et al. Catalytically impaired TYK2 variants are protective against childhood- and adult-onset systemic lupus erythematosus in Mexicans. Sci Rep (2019) 9(1):12165. doi: 10.1038/s41598-019-48451-3
31. Kisiel BM, Kosinska J, Wierzbowska M, Rutkowska-Sak L, Musiej-Nowakowska E, Wudarski M, et al. Differential association of juvenile and adult systemic lupus erythematosus with genetic variants of oestrogen receptors alpha and beta. Lupus (2011) 20(1):85–9. doi: 10.1177/0961203310381514
32. Salmaninejad A, Mahmoudi M, Aslani S, Poursani S, Ziaee V, Rezaei N. Association of STAT4 gene single nucleotide polymorphisms with Iranian juvenile-onset systemic lupus erythematosus patients. Turk J Pediatr (2017) 59(2):144–9. doi: 10.24953/turkjped.2017.02.005
33. Mahmoudi M, Aslani S, Hamzeh E, Ziaee V, Poursani S, Nicknam MH, et al. Association study of MECP2 gene single nucleotide polymorphisms in juvenile-onset systemic lupus erythematosus patients from Iran. Fetal Pediatr Pathol (2017) 36(6):423–31. doi: 10.1080/15513815.2017.1367871
34. Velázquez-Cruz R, Orozco L, Espinosa-Rosales F, Carreño-Manjarrez R, Solís-Vallejo E, López-Lara ND, et al. Association of PDCD1 polymorphisms with childhood-onset systemic lupus erythematosus. Eur J Hum Genet (2007) 15(3):336–41. doi: 10.1038/sj.ejhg.5201767
35. Mosaad YM, Hammad A, AlHarrass MF, Sallam R, Shouma A, Hammad E, et al. ARID5B rs10821936 and rs10994982 gene polymorphism and susceptibility to juvenile systemic lupus erythematosus and lupus nephritis. Lupus (2021) 30(8):1226–32. doi: 10.1177/09612033211010338
36. Pereira KM, Faria AG, Liphaus BL, Jesus AA, Silva CA, Carneiro-Sampaio M, et al. Low C4, C4A and C4B gene copy numbers are stronger risk factors for juvenile-onset than for adult-onset systemic lupus erythematosus. Rheumatol (Oxf) (2016) 55(5):869–73. doi: 10.1093/rheumatology/kev436
37. Coit P, Roopnarinesingh X, Ortiz-Fernández L, McKinnon-Maksimowicz K, Lewis EE, Merrill JT, et al. Hypomethylation of miR-17-92 cluster in lupus T cells and no significant role for genetic factors in the lupus-associated DNA methylation signature. Ann Rheum Dis (2022) 81(10):1428–37. doi: 10.1136/annrheumdis-2022-222656
38. Huang X, Su G, Wang Z, Shangguan S, Cui X, Zhu J, et al. Hypomethylation of long interspersed nucleotide element-1 in peripheral mononuclear cells of juvenile systemic lupus erythematosus patients in China. Int J Rheum Dis (2014) 17(3):280–90. doi: 10.1111/1756-185X.12239
39. von Knethen A, Heinicke U, Weigert A, Zacharowski K, Brüne B. Histone deacetylation inhibitors as modulators of regulatory T cells. Int J Mol Sci (2020) 21(7):2356. doi: 10.3390/ijms21072356
40. Perez-Hernandez J, Martinez-Arroyo O, Ortega A, Galera M, Solis-Salguero MA, Chaves FJ, et al. Urinary exosomal miR-146a as a marker of albuminuria, activity changes and disease flares in lupus nephritis. J Nephrol (2021) 34(4):1157–67. doi: 10.1007/s40620-020-00832-y
41. Yuan S, Tang C, Chen D, Li F, Huang M, Ye J, et al. miR-98 modulates cytokine production from human PBMCs in systemic lupus erythematosus by targeting IL-6 mRNA. J Immunol Res (2019) 2019:9827574. doi: 10.1155/2019/9827574
42. Xu L, Wang L, Shi Y, Deng Y, Oates JC, Kamen DL, et al. Up-regulated interleukin-10 induced by E2F transcription factor 2-microRNA-17-5p circuitry in extrafollicular effector B cells contributes to autoantibody production in systemic lupus erythematosus. Arthritis Rheumatol (2022) 74(3):496–507. doi: 10.1002/art.41987
43. Zhang J, Guo Y, Sun Y, Chang L, Wang X. Inhibition of microRNA-448 suppresses CD4(+) T cell inflammatory activation via up-regulating suppressor of cytokine signaling 5 in systemic lupus erythematosus. Biochem Biophys Res Commun (2022) 596:88–96. doi: 10.1016/j.bbrc.2022.01.097
44. Liu M, Guo Q, Wu C, Sterlin D, Goswami S, Zhang Y, et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol Immunol (2019) 16(4):367–79. doi: 10.1038/s41423-018-0010-6
45. Gallucci S, Meka S, Gamero AM. AbnorMalities of the type I interferon signaling pathway in lupus autoimmunity. Cytokine (2021) 146:155633. doi: 10.1016/j.cyto.2021.155633
46. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197(6):711–23. doi: 10.1084/jem.20021553
47. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA (2003) 100(5):2610–5. doi: 10.1073/pnas.0337679100
48. Luo S, Wang Y, Zhao M, Lu Q. The important roles of type I interferon and interferon-inducible genes in systemic lupus erythematosus. Int Immunopharmacol (2016) 40:542–9. doi: 10.1016/j.intimp.2016.10.012
49. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis (2018) 77(10):1507–15. doi: 10.1136/annrheumdis-2018-212988
50. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol (2007) 7(5):353–64. doi: 10.1038/nri2079
51. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi: 10.1126/scitranslmed.3001201
52. Chyuan IT, Tzeng HT, Chen JY. Signaling pathways of type I and type III interferons and targeted therapies in systemic lupus erythematosus. Cells (2019) 8(9):963. doi: 10.3390/cells8090963
53. Eloranta ML, Rönnblom L. Cause and consequences of the activated type I interferon system in SLE. J Mol Med (Berl) (2016) 94(10):1103–10. doi: 10.1007/s00109-016-1421-4
54. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci USA (2014) 111(20):7409–14. doi: 10.1073/pnas.1401662111
55. Hagberg N, Berggren O, Leonard D, Weber G, Bryceson YT, Alm GV, et al. IFN-α production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1β and LFA-1. J Immunol (2011) 186(9):5085–94. doi: 10.4049/jimmunol.1003349
56. Midgley A, Thorbinson C, Beresford MW. Expression of Toll-like receptors and their detection of nuclear self-antigen leading to immune activation in JSLE. Rheumatol (Oxf) (2012) 51(5):824–32. doi: 10.1093/rheumatology/ker400
57. Aggarwal A, Srivastava P. Childhood onset systemic lupus erythematosus: how is it different from adult SLE? Int J Rheum Dis (2015) 18(2):182–91. doi: 10.1111/1756-185X.12419
58. Bundhun PK, Kumari A, Huang F. Differences in clinical features observed between childhood-onset versus adult-onset systemic lupus erythematosus: A systematic review and meta-analysis. Med (Baltimore) (2017) 96(37):e8086. doi: 10.1097/MD.0000000000008086
59. das Chagas Medeiros MM, Bezerra MC, Braga FN, da Justa Feijão MR, Gois AC, Rebouças VC, et al. Clinical and immunological aspects and outcome of a Brazilian cohort of 414 patients with systemic lupus erythematosus (SLE): comparison between childhood-onset, adult-onset, and late-onset SLE. Lupus (2016) 25(4):355–63. doi: 10.1177/0961203315606983
60. Sassi RH, Hendler JV, Piccoli GF, Gasparin AA, da Silva Chakr RM, Brenol JC, et al. Age of onset influences on clinical and laboratory profile of patients with systemic lupus erythematosus. Clin Rheumatol (2017) 36(1):89–95. doi: 10.1007/s10067-016-3478-4
61. Kliegman RM, Shah SS, Tasker RC, Wilson KM, Behrman RE. Nelson Textbook of Pediatrics. (Beijing: World Book Inc.) (2017).
62. Bortolini MFF, Pereira VP, Gomes Dos Santos TA, Nisihara R, Skare TL. Systemic lupus erythematosus in children and adults: A retrospective study in Brazilian patients. Lupus (2021) 30(7):1197–202. doi: 10.1177/09612033211010330
63. Feng JB, Ni JD, Yao X, Pan HF, Li XP, Xu JH, et al. Gender and age influence on clinical and laboratory features in Chinese patients with systemic lupus erythematosus: 1,790 cases. Rheumatol Int (2010) 30(8):1017–23. doi: 10.1007/s00296-009-1087-0
64. Carreño L, López-Longo FJ, Monteagudo I, Rodríguez-Mahou M, Bascones M, González CM, et al. Immunological and clinical differences between juvenile and adult onset of systemic lupus erythematosus. Lupus (1999) 8(4):287–92. doi: 10.1191/096120399678847786
65. Kim H, Levy DM, Silverman ED, Hitchon C, Bernatsky S, Pineau C, et al. A comparison between childhood and adult onset systemic lupus erythematosus adjusted for ethnicity from the 1000 Canadian Faces of Lupus Cohort. Rheumatol (Oxf) (2019). doi: 10.1093/rheumatology/kez006
66. Gormezano NW, Kern D, Pereira OL, Esteves GC, Sallum AM, Aikawa NE, et al. Autoimmune hemolytic anemia in systemic lupus erythematosus at diagnosis: differences between pediatric and adult patients. Lupus (2017) 26(4):426–30. doi: 10.1177/0961203316676379
67. Sousa S, Gonçalves MJ, Inês LS, Eugénio G, Jesus D, Fernandes S, et al. Clinical features and long-term outcomes of systemic lupus erythematosus: comparative data of childhood, adult and late-onset disease in a national register. Rheumatol Int (2016) 36(7):955–60. doi: 10.1007/s00296-016-3450-2
68. Amaral B, Murphy G, Ioannou Y, Isenberg DA. A comparison of the outcome of adolescent and adult-onset systemic lupus erythematosus. Rheumatol (Oxf) (2014) 53(6):1130–5. doi: 10.1093/rheumatology/ket488
69. Sato VA, Marques ID, Goldenstein PT, Carmo LP, Jorge LB, Titan SM, et al. Lupus nephritis is more severe in children and adolescents than in older adults. Lupus (2012) 21(9):978–83. doi: 10.1177/0961203312443421
70. Mina R, Brunner HI. Update on differences between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Res Ther (2013) 15(4):218. doi: 10.1186/ar4256
71. Santos FPST, Nascimento BR, Calderaro DC, Ferreira GA, Correa H. Neuropsychiatric syndromes in childhood-onset systemic lupus erythematosus: A systematic review and meta-analysis. J Clin Rheumatol (2021) 27(5):206–14. doi: 10.1097/RHU.0000000000001029
72. Islabão AG, Mota LMH, Ribeiro MCM, Arabi TM, Cividatti GN, Queiroz LB, et al. Childhood-onset systemic lupus erythematosus-related antiphospholipid syndrome: A multicenter study with 1519 patients. Autoimmun Rev (2020) 19(12):102693. doi: 10.1016/j.autrev.2020.102693
73. Deng J, Chalhoub NE, Sherwin CM, Li C, Brunner HI. Glucocorticoids pharmacology and their application in the treatment of childhood-onset systemic lupus erythematosus. Semin Arthritis Rheum (2019) 49(2):251–9. doi: 10.1016/j.semarthrit.2019.03.010
74. Kifer N, Sestan M, Frkovic M, Kifer D, Kozmar A, Padjen I, et al. 2019 ACR/EULAR classification criteria and therapy in predicting organ damage accrual in patients with childhood-onset systemic lupus erythematosus: A retrospective study over the last 29 years. Lupus (2022) 31(7):828–36. doi: 10.1177/09612033221094707
75. Buttgereit F, da Silva JA, Boers M, Burmester GR, Cutolo M, Jacobs J, et al. Standardised nomenclature for glucocorticoid dosages and glucocorticoid treatment regimens: current questions and tentative answers in rheumatology. Ann Rheum Dis (2002) 61(8):718–22. doi: 10.1136/ard.61.8.718
76. Piga M, Floris A, Sebastiani GD, Prevete I, Iannone F, Coladonato L, et al. Risk factors of damage in early diagnosed systemic lupus erythematosus: results of the Italian multicentre Early Lupus Project inception cohort. Rheumatol (Oxf) (2020) 59(9):2272–81. doi: 10.1093/rheumatology/kez584
77. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med (2005) 353(16):1711–23. doi: 10.1056/NEJMra050541
78. Panettieri RA, Schaafsma D, Amrani Y, Koziol-White C, Ostrom R, Tliba O. Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol Sci (2019) 40(1):38–49. doi: 10.1016/j.tips.2018.11.002
79. Strehl C, Buttgereit F. Unraveling the functions of the membrane-bound glucocorticoid receptors: first clues on origin and functional activity. Ann N Y Acad Sci (2014) 1318:1–6. doi: 10.1111/nyas.12364
80. Brien TG. Human corticosteroid binding globulin. Clin Endocrinol (Oxf) (1981) 14(2):193–212. doi: 10.1111/j.1365-2265.1981.tb00616.x
81. Czock D, Keller F, Rasche FM, Häussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet (2005) 44(1):61–98. doi: 10.2165/00003088-200544010-00003
82. Tóth GG, Westerlaken BO, Eilders M, Laseur M, Jonkman MF, Uges DR. Dexamethasone pharmacokinetics after high-dose oral therapy for pemphigus. Ann Pharmacother (2002) 36(6):1108–9. doi: 10.1345/aph.1A047
83. Ito S, Kusunoki Y, Oka T, Ito Y, Okuno A, Yoshioka H. Pharmacokinetics of high-dose methylprednisolone in children. Dev Pharmacol Ther (1992) 19(2-3):99–105. doi: 10.1159/000457470
84. Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature (2010) 465(7300):937–41. doi: 10.1038/nature09102
85. Apostolopoulos D, Kandane-Rathnayake R, Raghunath S, Hoi A, Nikpour M, Morand EF. Independent association of glucocorticoids with damage accrual in SLE. Lupus Sci Med (2016) 3(1):e000157. doi: 10.1136/lupus-2016-000157
86. Jongvilaikasem P, Rianthavorn P. Longitudinal growth patterns and final height in childhood-onset systemic lupus erythematosus. Eur J Pediatr (2021) 180(5):1431–41. doi: 10.1007/s00431-020-03910-2
87. Sinha A, Bagga A. Pulse steroid therapy. Indian J Pediatr (2008) 75(10):1057–66. doi: 10.1007/s12098-008-0210-7
88. Lević Z, Micić D, Nikolić J, Stojisavljević N, Sokić D, Janković S, et al. Short-term high dose steroid therapy does not affect the hypothalamic-pituitary-adrenal axis in relapsing multiple sclerosis patients. Clinical assessment by the insulin tolerance test. J Endocrinol Invest (1996) 19(1):30–4. doi: 10.1007/BF03347855
89. Klein-Gitelman MS, Pachman LM. Intravenous corticosteroids: adverse reactions are more variable than expected in children. J Rheumatol (1998) 25(10):1995–2002.
90. Koncak G, Tolunay O, Unal A, Celiloglu C, Celik U. Short-term side effects of pulse steroid treatment in children. J Coll Phys Surg Pak (2022) 32(2):262–4. doi: 10.29271/jcpsp.2022.02.262
91. Ruiz-Irastorza G, Olivares N, Ruiz-Arruza I, Martinez-Berriotxoa A, Egurbide MV, Aguirre C. Predictors of major infections in systemic lupus erythematosus. Arthritis Res Ther (2009) 11(4):R109. doi: 10.1186/ar2764
92. Golder V, Hoi A. Systemic lupus erythematosus: an update. Med J Aust (2017) 206(5):215–20. doi: 10.5694/mja16.01229
93. Ruiz-Irastorza G, Garcia M, Espinosa G, Caminal L, Mitjavila F, González-León R, et al. First month prednisone dose predicts prednisone burden during the following 11 months: an observational study from the RELES cohort. Lupus Sci Med (2016) 3(1):e000153. doi: 10.1136/lupus-2016-000153
94. Ruiz-Arruza I, Lozano J, Cabezas-Rodriguez I, Medina JA, Ugarte A, Erdozain JG, et al. Restrictive use of oral glucocorticoids in systemic lupus erythematosus and prevention of damage without worsening long-term disease control: an observational study. Arthritis Care Res (Hoboken) (2018) 70(4):582–91. doi: 10.1002/acr.23322
95. Ruiz-Irastorza G, Ugarte A, Saint-Pastou Terrier C, Lazaro E, Iza A, Couzi L, et al. Repeated pulses of methyl-prednisolone with reduced doses of prednisone improve the outcome of class III, IV and V lupus nephritis: An observational comparative study of the Lupus-Cruces and lupus-Bordeaux cohorts. Autoimmun Rev (2017) 16(8):826–32. doi: 10.1016/j.autrev.2017.05.017
96. Ruiz-Irastorza G, Ruiz-Estevez B, Lazaro E, Ruiz-Arruza I, Duffau P, Martin-Cascon M, et al. Prolonged remission in SLE is possible by using reduced doses of prednisone: An observational study from the Lupus-Cruces and Lupus-Bordeaux inception cohorts. Autoimmun Rev (2019) 18(9):102359. doi: 10.1016/j.autrev.2019.102359
Keywords: childhood-onset systemic lupus erythematosus, genetic factors, type I interferon, glucocorticoids, intravenous methylprednisolone pulse therapy
Citation: Pan L, Liu J, Liu C, Guo L, Punaro M and Yang S (2023) Childhood-onset systemic lupus erythematosus: characteristics and the prospect of glucocorticoid pulse therapy. Front. Immunol. 14:1128754. doi: 10.3389/fimmu.2023.1128754
Received: 21 December 2022; Accepted: 21 July 2023;
Published: 10 August 2023.
Edited by:
Magda Carneiro-Sampaio, University of São Paulo, BrazilReviewed by:
Sezgin Sahin, Istanbul University-Cerrahpasa, TürkiyeCopyright © 2023 Pan, Liu, Liu, Guo, Punaro and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sirui Yang, c3J5YW5nQGpsdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.