 Devi Gunasekera1,2†
Devi Gunasekera1,2† Pooja Vir
Pooja Vir Ahmad Faisal Karim
Ahmad Faisal Karim Margaret V. Ragni
Margaret V. Ragni Kathleen P. Pratt
Kathleen P. Pratt- 1Department of Medicine, Uniformed Services University of the Health Sciences, Bethesda, MD, United States
- 2Henry Jackson Foundation for the Advancement of Military Medicine, Bethesda, MD, United States
- 3Department of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
Background: Almost half of severe hemophilia A (HA) is caused by an intron 22 inversion mutation (Int22Inv), which disrupts the 26-exon F8 gene. Inverted F8 mRNA exons 1-22 are transcribed, while F8B mRNA, containing F8 exons 23-26, is transcribed from a promoter within intron 22. Neither FVIII activity nor FVIII antigen (cross-reacting material, CRM) are detectable in plasma of patients with an intron-22 inversion.
Objectives: To test the hypothesis that (putative) intracellular synthesis of FVIII proteins encoded by inverted F8 and F8B mRNAs confers T-cell tolerance to almost the entire FVIII sequence, and to evaluate the immunogenicity of the region encoded by the F8 exon 22-23 junction sequence.
Patients/Methods: Peripheral blood mononuclear cells (PBMCs) from 30 severe or moderate HA subjects (17 with an Int22Inv mutation) were tested by ELISPOT assays to detect cytokine secretion in response to FVIII proteins and peptides and to map immunodominant T-cell epitopes. Potential immunogenicity of FVIII sequences encoded by the F8 exon 22-23 junction region was also tested using peptide-MHCII binding assays.
Results: Eight of the Int22Inv subjects showed robust cytokine secretion from PBMCs stimulated with FVIII proteins and/or peptides, consistent with earlier publications from the Conti-Fine group. Peptide ELISPOT assays identified immunogenic regions of FVIII. Specificity for sequences encoded within F8 mRNA exons 1-22 and F8B mRNA was confirmed by staining Int22Inv CD4+ T cells with peptide-loaded HLA-Class II tetramers. FVIII peptides spanning the F8 exon 22-23 junction (encoding M2124-V2125) showed limited binding to MHCII proteins and low immunogenicity, with cytokine secretion from only one Int22Inv subject.
Conclusions: PBMCs from multiple subjects with an Int22Inv mutation, with and without a current FVIII inhibitor, responded to FVIII epitopes. Furthermore, the FVIII region encoded by the exon 22-23 junction sequence was not remarkably immunoreactive and is therefore unlikely to contain an immunodominant, promiscuous CD4+ T-cell epitope. Our results indicate that putative intracellular expression of partial FVIII proteins does not confer T-cell tolerance to FVIII regions encoded by inverted F8 mRNA or F8B mRNA.
Introduction
Almost half of severe hemophilia A (HA) patients have a F8 intron-22 inversion mutation (Int22Inv), which precludes expression of an intact, functional FVIII protein (1, 2). The F8 gene consists of 26 exons encoding FVIII domains A1-A2-B-A3-C1-C2 (3). An additional transcription start site within intron 22 produces F8B mRNA, which consists of a short exon encoding eight non-FVIII amino acid residues followed by F8 exons 23-26 encoding V2125-Y2332 (4). F8B mRNA is expressed in multiple tissues of individuals with and without HA, with the exception of HA patients having a deletion mutation within exons 23-26. In addition to F8B mRNA, individuals with an intron-22 inversion mutation express a truncated F8 mRNA consisting of F8 exons 1-22 (1, 2).
Approximately 30% of severe HA patients develop neutralizing anti-FVIII antibodies following replacement therapy with exogenous FVIII, which are clinically referred to as “inhibitors” (5–7) due to their interference with normal blood coagulation via blocking of FVIII cofactor activity. The development of anti-FVIII antibodies in HA patients follows a classic prime-boost response, with inhibitors generally developing within the initial 20 exposures to therapeutic FVIII (8). Initial inhibitor development after 150+ FVIII infusions is far less common. The role of CD4+ T cells in providing ongoing help necessary for anti-FVIII antibody generation was clearly shown in a 1993 study of HA patients infected with HIV. For those with an inhibitor, their titers declined as their CD4+ T-cell counts decreased (9). Proliferation and cytokine secretion of CD4+ T cells from HA subjects in response to FVIII protein and synthetic peptides was demonstrated in subsequent studies (10–15). More recently, specific HLA-restricted T-cell epitopes have been identified, with several confirmed by isolation of T-cell clones responding to specific FVIII peptides (16–21). Thus, the essential role of CD4+ T cells in development and persistence of inhibitor responses is well established.
A 2009 case-control study indicated that large F8 gene deletions were associated with increased risk of developing an inhibitor (22), and a 2012 meta-analysis further indicated that inhibitor risk in HA-Int22Inv patients was somewhat lower than in severe HA patients with a large structural alteration of the F8 gene, e.g., large deletion or early nonsense mutations (23). This provided some support for an earlier hypothesis that endogenous intracellular expression of FVIII partial proteins from these two mRNAs occurs, and that it may confer self-tolerance to the corresponding FVIII protein sequences, thereby decreasing the immunogenicity of FVIII replacement therapy in Int22Inv patients (24, 25). Since a substantial fraction of Int22Inv patients still develop inhibitors, a corollary of this hypothesis is that the FVIII C1 domain region encoded by mRNA spanning the exon 22-23 splice site (corresponding to FVIII residues ~I2103-A2146), which would be the only “non-self” region of therapeutic FVIII (Figure 1), is a promiscuous, highly immunogenic neo-epitope.
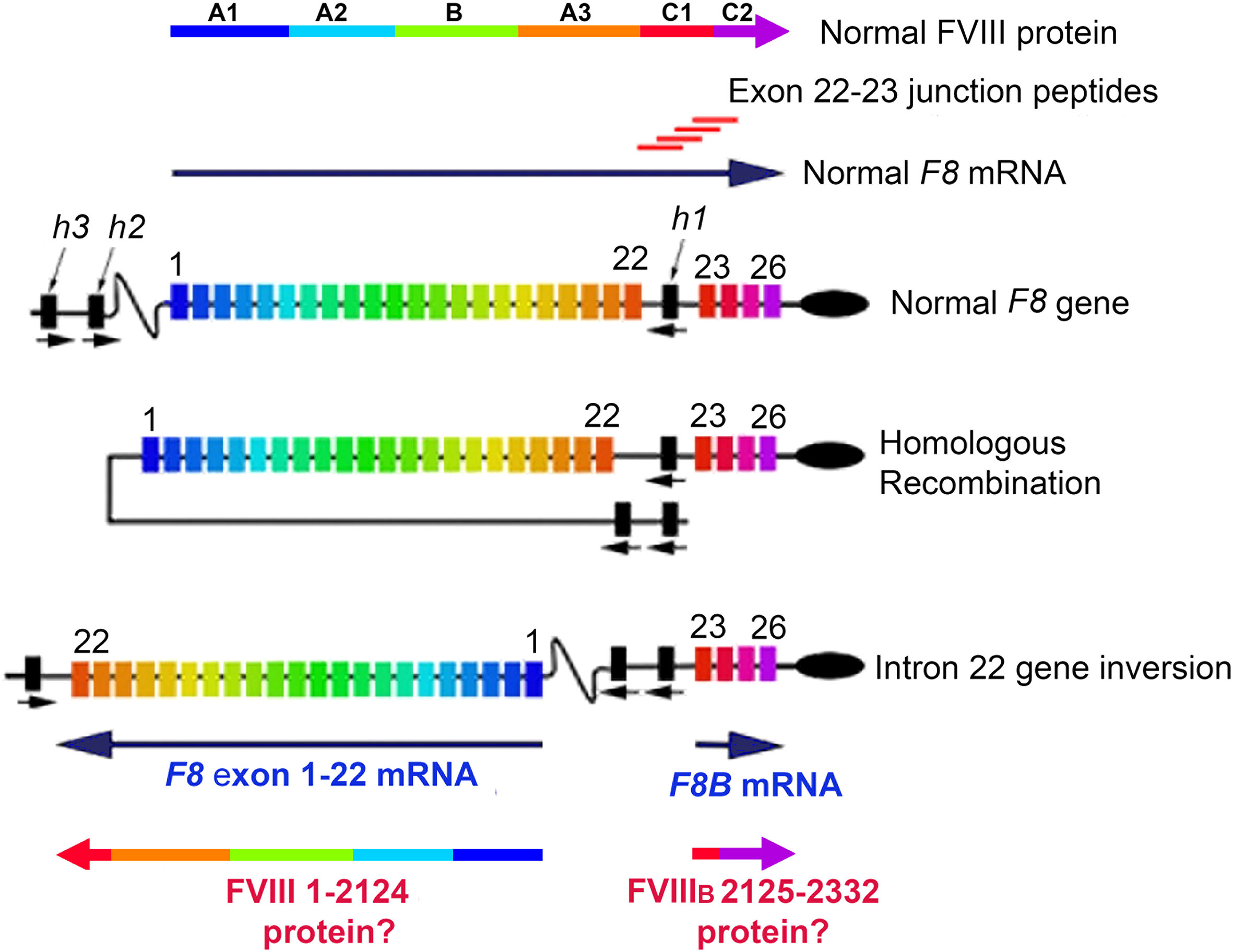
Figure 1 FVIII Structure, Int22Inv DNA, Int22Inv mRNA, and hypothesized partial FVIII proteins. Full-length FVIII consists of domains A1, A2, A3, B, C1 and C2, which are encoded by the 26-exon F8 gene. Following a type 1 or type 2 intron 22 inversion mutation, the primary F8 mRNA product consists of F8 exons 1-22, which encode the FVIII A1, A2 and B domains, plus the C1 domain sequence through M2124 and then terminating at a stop codon within intron 22. The shorter transcript F8B is comprised of 24 nucleotides followed by F8 exons 23-26, which encode FVIII residues 2125-2332. If these two partial FVIII proteins were expressed intracellularly, this could in principle promote tolerance to FVIII as a “self”-protein. The 20-mer exon 22-23 junction peptides, with F8 sequences overlapping by 12 residues, were designed to allow peptides to bind to the HLA-DRB1 binding grooves in multiple possible registers, in order to evaluate their potential ability to be presented to CD4+ T cells.
The present study tests this hypothesis and corollary by directly querying CD4+ T-effector responses of HA subjects with and without an Int22Inv mutation to recombinant (r)FVIII proteins and synthetic peptides. A primary goal was to map immunodominant epitopes that elicit CD4+ T-cell responses in the Int22Inv subjects and determine if the results indicate tolerance to FVIII regions encoded by inverted F8 exons 1-22 and/or by F8B. In addition, peptide-HLA Class II binding assays were employed to test the ability of peptides encoded by the F8 exon 22-23 junction region to be presented on a representative series of recombinant HLA-DRB1 proteins. The latter experiments would indicate if peptides encoded by this junction region, which cannot be expressed in patients with an Int22Inv mutation, could represent a promiscuous, immunodominant epitope responsible for inhibitor development in Int22Inv patients.
Materials and methods
Human subjects and blood samples
Blood samples from 11 severe and moderately severe HA subjects were donated as part of the “INHIBIT” feasibility study (5U34HL 114674, MVR, PI). Three of these were known Int22Inv subjects, while the HA-causing mutation was unknown for six of these subjects. PBMCs from severe HA and normal control subjects that were banked from earlier studies were also utilized (USU IRB#1, protocols MED-83-3442 and MED-83-3426). All subjects provided informed consent consistent with the Principles of Helsinki. Additional de-identified normal control samples were obtained from the NIH Blood Bank (Exempt E4). Both freshly prepared and frozen (7% DMSO in FBS) PBMC samples were analyzed.
Inhibitor titers
Inhibitor titers, expressed as Bethesda units (BU)/mL, were from clinical records for the subjects, which reported results of CLIA-certified clotting or chromogenic substrate assays (26). Both clotting and chromogenic inhibitor assays are functional assays that measure the inhibition of FVIII cofactor activity in the presence of anti-FVIII antibodies. The clotting assay measures plasma clotting time [using a 1-stage assay (27)], while the chromogenic assay measures cleavage of a chromogenic substrate for factor X; normal factor X kinetics require a normal FVIII level. In either assay, serial dilutions of the patient plasma are mixed with a normal pooled plasma sample, and the residual FVIII activity, compared to a parallel assay using the normal pooled plasma plus buffer, is measured and reported in BU/mL. One BU/mL is the reciprocal of the patient plasma dilution that results in 50% normal FVIII activity in the assay. The lower limit of quantification is generally considered to be 0.6 BU/mL, and titers > 5.0 BU/mL are considered high-titer inhibitors.
Reagents, buffers and instruments
Recombinant (r)FVIII (Kogenate 27NZPO) was from Bayer (Leverkusen, Germany) or Baxter (Westlake Village, CA, USA; Recombinate TRH07504AA). Recombinant FVIII-C2 protein was purified in-house as described (28). Tetanus and Diptheria (TD) toxoid adsorbed was from Mass Biologics, (Boston, MA, USA) and Remel PHA (R30852801) (100μg/ml) were from Thermo Scientific (Rockford, IL, USA). Benzonase nuclease, 250 U/μl (021M0852) and 0.4% Trypan blue (T8154) were from Sigma Aldrich (St Louis, MO, USA). RPMI 1640 with 25 mM HEPES, DMEM/F-12(1(1:1), MEM nonessential amino acids (100X), 1X D-PBS (Ca/Mg-free) and 10X D-PBS were from Life Technologies (Grand Island, NY, USA). Ficoll-Paque PLUS was from GE Healthcare Life Sciences (Piscataway, NJ, USA). HCl was from JT Baker Tyco Products (Phillipsburg, NJ, USA). Human IFN-gamma ELISPOT pair (551849), Human IL-5 ELISPOT pair (551886), Human IL-10 ELISPOT pair (551883), AEC substrate set (551951) and TMB substrate were from BD Biosciences (San Jose, CA, USA). IL-7 (0.5ng/ml) was from Peprotech (Rocky Hill, NJ, USA). Fetal bovine serum was from Hyclone (Logan, UT, USA). ImmunO human serum type AB was from MP Biomedicals (Solon, OH, USA). Recombinant, extracellular domains of 10 HLA-DRB1 protein monomers, HLA Class II tetramers loaded with the relevant peptides and anti-HLA-DR antibody L243 were provided by the Tetramer Core Facility at the Benaroya Research Institute (Seattle, WA, USA). AIM-V medium and goat anti human IgG-HRP were from Invitrogen (Carlsbad, CA, USA). Sheep anti-human FVIII was from Cedarlane labs (Burlington, NC, USA). ELISA 12 well strips were from E & K Scientific (Santa Clara, CA, USA). Wallac enhancement solution, fluorescence assay buffer and europium-labeled straptavidin were from Wallac-Perkin Elmer (Turku, Finland). 96-well polypropylene plates and Corning Costar high-binding ELISA plates were from Sigma Aldrich (St. Louis, MO, USA), cat # CLS3799 and CLS2592, respectively). Recombinant human IL-2 (Chiron Il-2, Proleukin, 70 IU/ml) was from R & D systems, cat #202-IL-010)
FVIII peptides and peptide pools
Individual 20-mer peptides spanning the FVIII A2, C1 and C2 sequences, as well as equimolar pools containing 2-5 of these peptides, were from Anaspec, Inc. (Fremont, CA, USA, Table S1). Additional 15-mer peptides spanning the FVIII A1, A2, A3, C1 and C2 domain sequences, with 12-residue overlaps, were from Mimotopes, Inc. (Mulgrave, Victoria, Australia), which also provided large, medium and small equimolar pools of these peptides (Tables S2–S5). An intron 22-23 ‘junction peptide pool’ was created by combining four 20-mer peptides (Mimotopes, Inc.) with overlapping sequences spanning the F8 exon 22-23 – encoded region (FVIII 2103-2146) in DMSO (Table 1). All peptides were ordered at >70% advertised purity and their quality was verified by mass spectrometry.
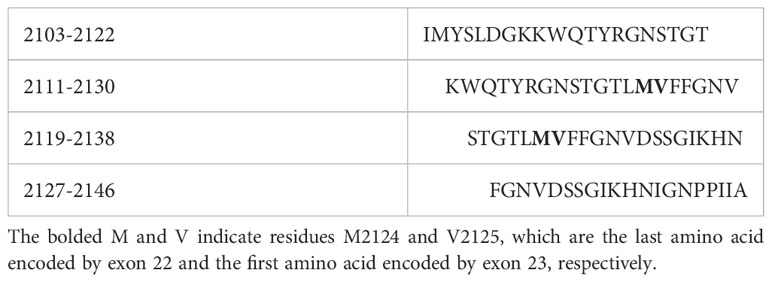
Table 1 FVIII peptides spanning the F8 exon 22-23 junction region.
Peptide-MHCII binding assays and predictions
Quantitative peptide-MHC competition binding assays were carried out to determine binding avidities of four 20-mer peptides spanning the F8 exon 22-23 junction region to 10 HLA-DRB1 proteins using methodology described previously (29). Briefly, 1-µl aliquots of serially diluted (50nM – 50μM in DMSO) non-biotinylated FVIII peptides, were added to triplicate wells of 96-well polypropylene plates. Serially diluted non-FVIII reference peptides known to bind to specific HLA-DRB1 (Table S6) were added to separate wells as positive controls. The HLA-DRB1 proteins were diluted to 50 nM in 150 mM citrate-phosphate, pH 5.4, 0.75% n-octyl-beta-D-glucopyranoside, 1 mM PMSF. 50 μl of the HLA-DRB1 solution was added per well, and the plates were sealed and incubated at 37°C for 30 min to allow peptide binding to the HLA-DRB1 protein. Next, 1 μl of the appropriate biotinylated reference peptide, at a concentration in the known binding range for its respective HLA-DRB1 (Table S6), was added to each well, the solutions were mixed, and the plates were incubated at 37°C for 18-24 hrs. 100 μl aliquots of the anti-HLA-DR antibody L243 (10 μg/mL in 12.5 mM borate buffer, pH 8.2) were added to 96-well ELISA plates, and the plates were incubated at 4°C for 18-24 hrs, washed, and blocked with 1xDPBS containing 5% FBS for 3 hrs. The plates were washed again and 50 μl neutralization solution (50 mM Tris, pH 8.0, 0.75% n-octyl-beta-D-glucopyranoside) was added to each well. The contents of wells containing the peptide-HLA-DRB1 binding reactions were then transferred to wells of the ELISA plates containing the neutralization solution and the ELISA plates were incubated for 18-24 hrs at 4°C. After washing the plates 5X with 300 ul/well of 1XPBS, 0.05% Tween-20, 100 ul of Europium-labeled streptavidin (diluted 1:1,000 into Wallac assay buffer) was added to each well. The plates were covered with a black polystyrene lid, incubated at room temperature for 4 hrs and washed 5X with 300 ul/well of 1XPBS, 0.05% Tween-20. Wallac enhancement solution (100 μl/well) was then added, and the plates were incubated at room temperature for 30 min and then read on a Victor 2D time-resolved fluorometer. Sigmoidal binding curves were simulated and IC50 values calculated for the FVIII peptides, based on their competition with the reference peptides for each HLA-DRB1.
Predicted affinities of the four FVIII junction peptides for the 10 HLA-DRB1 alleles were obtained using the Immune Epitope Database (IEDB) server, using the NetMHCIIPan 4.0 prediction algorithm (30).
In vitro expansion of FVIII-specific T cells
FVIII-specific CD4+ T cells were expanded by culturing PBMCs with or without rFVIII, rFVIII-C2 protein, or pooled or individual FVIII peptides, as follows. One vial of frozen PBMCs (~10 million cells) was thawed by slowly adding 9 ml of AIM-V medium containing 10% human serum + 1.8 μl of 25K benzonase nuclease. The cells were washed with AIM-V medium containing 10% human serum and re-suspended in AIM-V medium containing 15% human serum, and then plated at 1x106 cells per well in 48-well cell culture plates (1 mL/well) with one of the following: rFVIII protein (1 μg/mL ~8nM), rFVIII-C2 protein (2 μg/mL = 50 nM), pooled FVIII peptides (50 nM and 70 nM), FVIII pooled junction peptides (200 nM), or individual FVIII peptides (50 or 100 nM). Positive control wells were stimulated with 0.02Lf (5 μl/mL) TD toxoid, PHA (10 μg/mL) and negative controls with 5 μl DMSO. The plates were incubated for five days at 37°C in a 5% CO2 incubator. At day 6, cells were harvested and re-suspended in AIM-V medium (no serum) containing 0.5ng/mL IL-7 and then directly plated on 96-well flat-bottom ELISPOT plates. For some individual peptide ELISPOT assays, expansions were continued for an additional 3 days, adding fresh AIM-V medium and IL-2 (3.5 IU/mL final concentration).
Epitope mapping by ELISPOT assays
ELISPOT plates were coated with capture antibody solutions (anti-IFN-γ, anti-IL-5 or anti-IL-10) diluted to 5 μg/ml in DPBS and incubated at 4C overnight. Wells were then washed once with 200 μl/well Blocking Solution (AIM-V medium containing 10% human serum). Another 200 μl Blocking Solution was then added to each well and the plates were incubated for 2 hrs at room temp and the Blocking solution discarded. The expanded PBMCs were then added in serial dilutions (carried out in duplicate when sufficient cells were available) with final concentrations of 2X105, 1x105 and 5x104 cells/well, and incubated with the same antigenic stimulants as before. The plates were incubated for 24 hrs for IFN-γ ELISPOTS or 48 hrs for IL-5 and IL-10 ELISPOTS at 37C, 5% CO2. The suspended cells were then discarded and the plates washed with 200 μl/well dH2O (2X) and 3x with PBS containing 0.05% Tween 20 (wash buffer I). The biotinylated anti-human antibody provided in the kits (0.25 μg/ml, diluted in PBS + 10% human serum) was then added and the plates were incubated for 2 hrs at room temperature. The plates were washed 3X with 200 μl/well wash buffer I, washed 2X with 200 μl/well PBS, and developed with streptavidin-HRP (BD Biosciences, #557630) diluted 1:100 in PBS containing 10% human serum for 1 hr at room temperature. Plates were then washed 4X with 200 μl/well wash buffer I, and washed 2X with 200 μl/well PBS. Spots were then developed by adding 100 μl/well of BD ELISPOT AEC substrate and incubated for 5-30 minutes in the dark. The wells were then washed 2X with 200 μl/well dH20 and air-dried at room temp for 2 hrs. IFN-γ, IL-5 and IL-10-specific spot-forming cells (SFC) were then counted using software on a CTL Immunospot S6 Ultimate Image Analyzer. Stimulants were: rFVIII 5 μl/mL = 5 nM; rFVIII-C2 1μg/mL = 50 nM; FVIII peptides (pooled or individual): 50-100 nM; TT; 5 μl/mL; PHA: 5 μl/mL. The criteria for antigen-specific (positive) responses were: a minimum of 25 spots per million cells (based on spots per 200,000 cells/well) and wells with the stimulant had ≥3X the average number of spots counted for the unstimulated wells. In addition, antigen-specific responses to large FVIII peptide pools were “decoded”, when sufficient cells were available, by subsequent ELISPOT assays using smaller peptide pools and then with individual peptides, to map immunogenic FVIII regions and specific epitopes contributing to the cytokine response.
Epitope mapping by HLA Class II tetramer staining
PBMCs were added to 48-well plates in 1ml of 10% human serum RPMI medium (1 million PBMCs per well) and then stimulated with individual or pooled 20-mer or 15-mer FVIII peptides (10 μM) and incubated for up to 19 days at 37C, 5% CO2. At day 7, 50 μl of IL-2 was added (35 IU/mL final concentration) without removing any medium. At day 9, 400-500 μL supernatant was removed and replaced with fresh 15% human serum RPMI medium containing 50 μl IL-2 after which cells were fed with 50 μl IL-2 every 48-72 hours, splitting as needed. After 17-19 days, the cells were re-suspended, 100 μL aliquots were transferred to FACS tubes, and 1.5 μl of the appropriate peptide-loaded, PE-labeled HLA Class II tetramer (~10 μg/mL final concentration) was added. The tubes were incubated in the dark at 37C for 2 hrs and then put on ice. A mixture of anti-CD3-PerCP + anti-CD4 APC (3.75 μL each from stock solutions) was added to each tube, and tubes with no antibody added served as a negative staining control. The tubes were then incubated in the dark at 4C for 30 min. Additional tubes containing beads for compensation (ThermoFisher (ebiosciences), Rockville, MD, USA), were stained by adding 5 μL of anti-CD3-PerCP, 1.5 μl of anti-CD4-APC and 1.5 μl of anti-CD4-PE and tubes were incubated in the dark for 30 min. Sample and control cells were then washed and re-suspended in ice-cold FACS buffer, stored on ice in a covered container, and analyzed by flow cytometry and/or FACS acquisition. The gating strategy was: Singlets (FSC-H/FSC-A), Lymphocytes (SSC-A/FSC-A), followed by gating on anti-CD3-PerCP-high, followed by tetramer-PE/CD4-APC to detect antigen-specific CD4+ T cells.
Isolation of FVIII-specific T-cell clones
To isolate FVIII-specific T-cell clones, CD4hiTetramerhi cells were single-cell sorted into 96-well round-bottom plates containing 100 μl RPMI medium per well. To each well, (200,000) irradiated HLA-mismatched PBMC were added as feeder cells in a volume of 100 μL RPMI Medium containing 15% human serum AB plus PHA (5 μg/mL). The cells were incubated for 24 hrs at 37C, 5% CO2, and then IL-2 (10 IU/mL) in 25 μl RPMI medium was added to each well. The cells were then incubated at 37C in 5% CO2 for another 14-21 days. For wells containing expanding clones, tetramer staining was performed to confirm their specificity. If clones were not expanding, the cells were re-stimulated with irradiated, HLA-mismatched PBMCs and PHA as before, adding fresh medium and IL-2 (3.5 IU/mL final concentration) as needed to continue the expansion or else frozen in 10% DMSO in FBS.
Results
Peptide-MHCII binding assays and predictions
The experimental FVIII peptide-HLA-DRB1 affinities are in Table 2A and their predicted affinities are in Table 2B. Only HLA-DR0404, DR0701, DR0901 and DR1501 showed moderate or strong binding affinity to one or more peptides spanning the F8 exon 22-23-encoded region of FVIII, while weak binding was observed to HLA-DR0101 and DR0401. The MHCIIpan algorithm (30) predicted moderate-to-strong binding affinity to additional HLA-DR proteins. Based on the experimental results, ELISPOT assays were then carried out using PBMCs from nine HA subjects with an intron 22 inversion mutation who were also known to have HLA-DR1, DR4, DR7, DR9 or DR15 alleles.
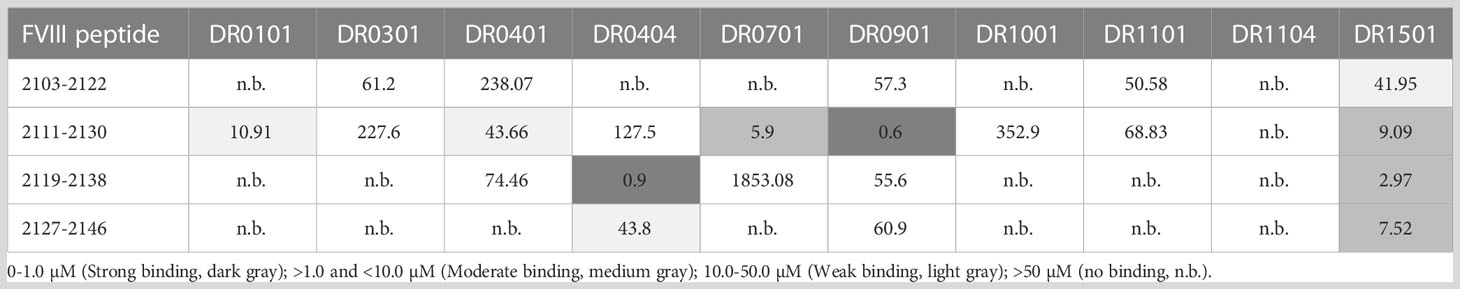
Table 2A Experimental binding affinities (IC50 values) of FVIII exon 22-23 junction peptides.
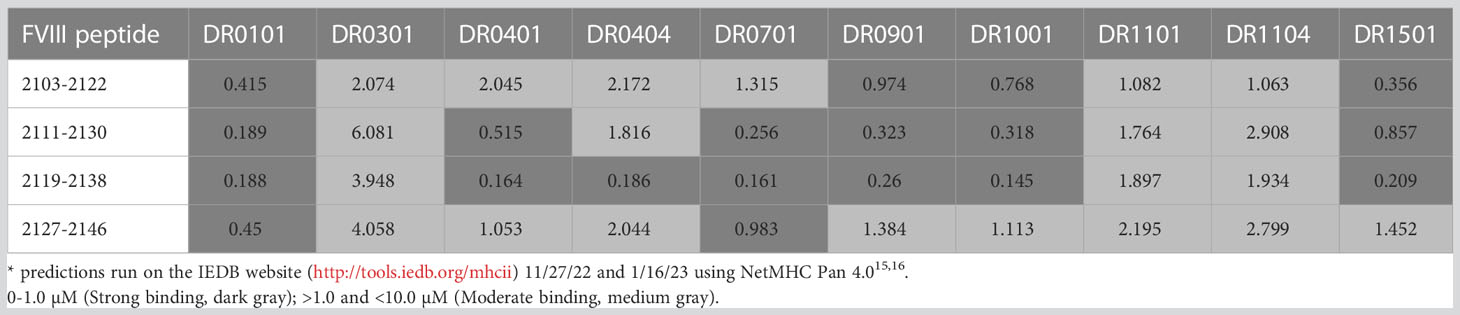
Table 2B Predicted binding affinities (IC50 values)* of FVIII exon 22-23 junction peptides.
Epitope mapping by ELISPOT assays
Several strategies were used to map T-cell epitopes, in order to test specific hypotheses and/or to rationally choose which peptides to utilize when there were not sufficient PBMCs to systematically test responses to larger and then smaller peptide pools followed by individual peptides (due to competing studies and resulting blood volume limitations in subjects with hemophilia A). The number of PBMCs per subject was highly variable, as samples were obtained from both adults and children, and the amounts of blood drawn also reflected the stated preferences of the subjects. Therefore, the number of experiments and replicate wells per experiment varied somewhat according to the number of cells available for the assays.
rFVIII and/or rFVIII-C2 domain protein ELISPOTs were included as a specific control in almost all ELISPOT experiments. For positive responders, IFN-γ secretion was generally more robust when cells were stimulated with 50 nM rFVIII-C2 than with 8 nM rFVIII. Unfortunately, FVIII protein becomes toxic to CD4+ T cells at higher concentrations. The stronger responses to rFVIII-C2, and to pooled or individual FVIII peptides in some assays, may simply be due to stimulation of lower-avidity CD4+ T cells by the higher concentrations of rFVIII-C2 and FVIII peptides. Of note, the rFVIII-C2 preps were routinely tested to confirm low endotoxin levels (31).
Exon 22-23 junction peptides
Potential CD4+ T-cell responses to exon 22-23 junction sequences were tested using samples from nine HA subjects with an intron-22 inversion mutation who also had one of the HLA-DRB1 alleles shown experimentally to bind peptides with these sequences: HLA-DR1, DR4, DR7, DR9 or DR15 (Table 2A). Eight of these subjects failed to show a response to the pooled FVIII exon 22-23 junction peptides when tested using IFN-γ ELISPOT assays (Table 3). Five of them had a current inhibitor and four had no inhibitor history. The one subject who showed a response to the junction peptides, G4, had a low-titer inhibitor. He had an intron 22 inversion mutation as well as a missense mutation, H1499Y, and his HLA-DRB1 alleles were HLA-DRB1*0301, 1501. Therefore, additional MHCII binding predictions were made using the IEDB analysis resource Consensus tool (32, 33) with the input sequence FVIII 1485-1513, lpktsgkvellpkvhiyqkdlfptetsng, which contains all possible 15-mer sequences containing residue H1499. Interestingly, the IEDB prediction algorithm identified two overlapping 9-mer core sequences containing H1499 as potential HLA-DRB1*1501-restrictred CD4+ T-cell epitopes, ranking them in the top 15-20% based on predicted binding affinity (Table S7).
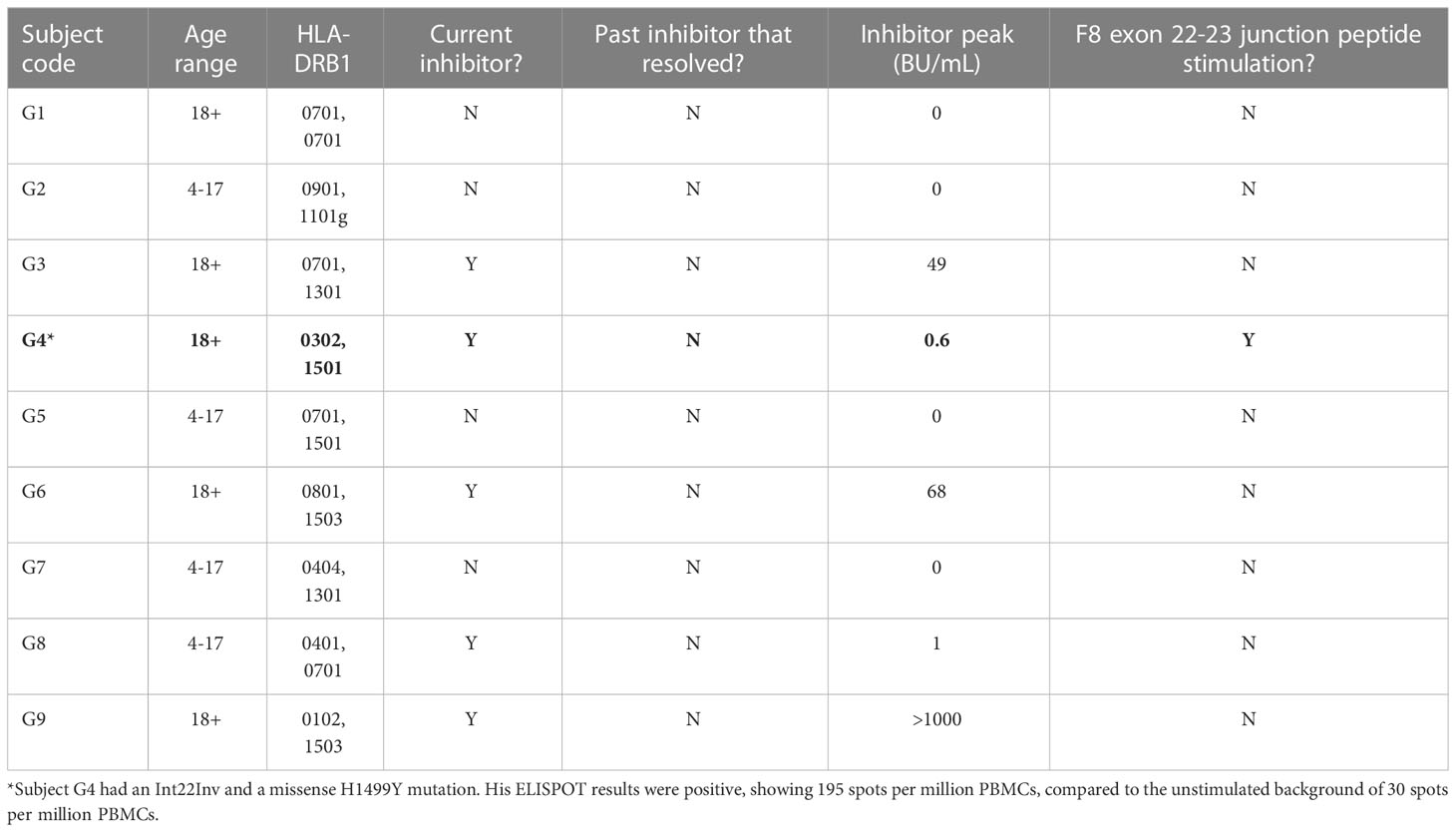
Table 3 IFN-γ ELiSpot responses of Int22Inv PBMCs to pooled junction peptides (FVIII 2103-2146).
Systematic epitope mapping using pooled and individual FVIII peptides
Subjects P1 and P11 (both with an intron-22 inversion) generously donated sufficient blood to carry out fairly comprehensive epitope mapping by ELISPOT assays. P1 had a past inhibitor that had resolved (peak titer 15.2 BU/mL approximately 10 years prior to donating this sample). IFN-γ ELISPOT assays showed robust responses to both rFVIII and rFVIII-C2 protein (Figure 2A). His responses to A2 and C2 domain peptides were tested next, in order to test the hypotheses that tolerance to A2 epitopes was conferred by translation from the inverted F8 mRNA (exons 1-22, containing the A2 domain sequence) and/or from F8B mRNA (exons 23-26, containing the C2 domain sequence). No responses to pooled A2 domain peptides were revealed under these assay conditions (not shown). However, his PBMCs responded to one of the pools of 20-mer peptides spanning the C2 domain sequence (Figure 2B). Decoding of this response by testing the individual peptides comprising this pool identified robust responses to peptides corresponding to C2-2210-2229, C2-2226-2245 and C2-2242-2261 (Figure 2C).
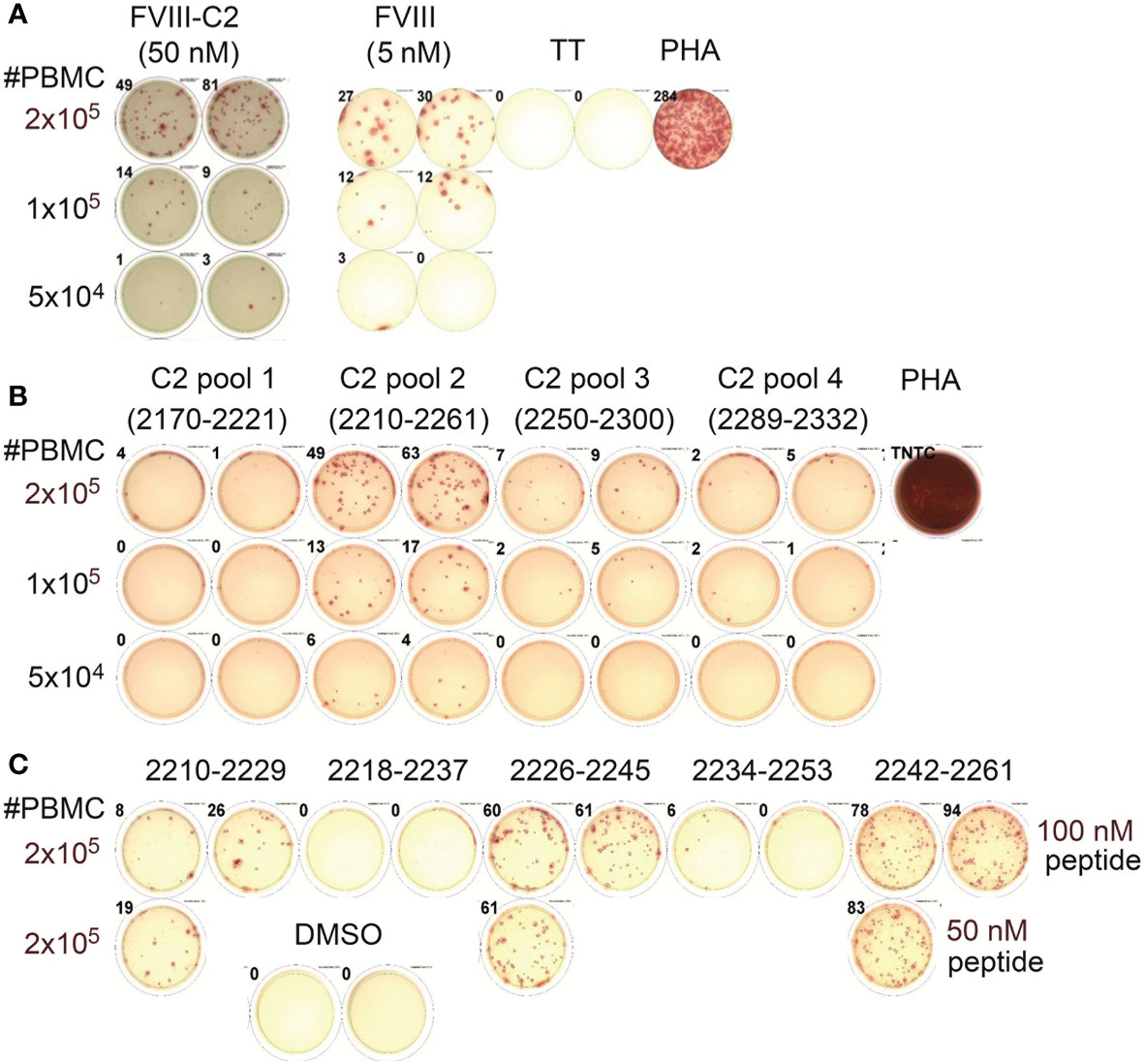
Figure 2 Mapping of CD4+ T-cell epitopes in the FVIII C2 domain by peptide ELISPOT assays (Int22Inv subject P1). (A) This subject’s PBMCs showed robust IFN-γ secretion when stimulated with rFVIII and rFVIII-C2 proteins. The FVIII-C2 ELISPOTs were carried out on different plates and read separately, hence the different background color in this image. (B) Decoding of his response to the FVIII C2 domain using pools of 20-mer peptides spanning the C2 domain sequence indicated that C2 pool #2 contained one or more immunodominant epitopes. (C) Further decoding by stimulations with the individual 20-mer peptides comprising C2 pool 2 indicated that he had specific responses to three epitopes in the FVIII C2 domain. His PBMCs did not respond to pooled peptides spanning the FVIII A2 domain (not shown).
Subject P11 also had a previous inhibitor that resolved four years before donating this blood sample. His PBMCs showed robust IFN-γ secretion when stimulated with rFVIII, rFVIII-C2 protein, peptide pools spanning the FVIII A2 domain sequence, and peptide pools spanning the FVIII C2 domain sequence (Figures 3A–C). His PBMCs also responded to rFVIII stimulation with secretion of IL-5 and IL-10 (Figure 3D).
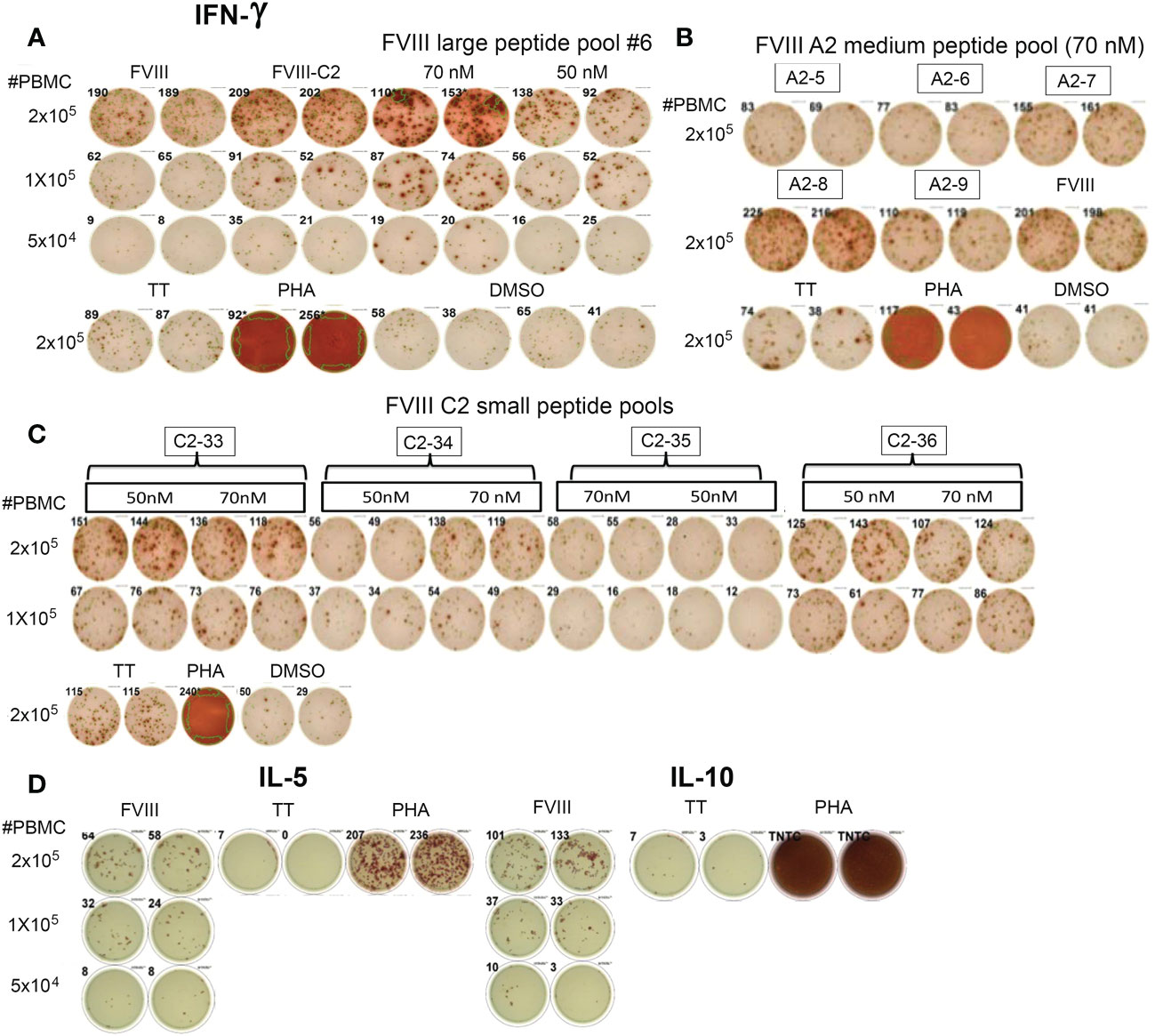
Figure 3 CD4+ T cells from Int22Inv subject P11 respond to epitopes in the FVIII A2 and C2 domains. (A) He showed a strong IFN-γ response to rFVIII, rFVIII-C2 protein, and FVIII large peptide pool #6 (which contained peptides spanning the C2 domain sequence). (B) His PBMCs also responded to FVIII A2 domain medium peptide pools A2-7, A2-8 and A2-9. (C) Decoding of his response to rFVIII-C2 protein and FVIII large peptide pool #6 by stimulating his PBMCs with small peptide pools spanning the FVIII C2 domain sequence. His PBMCs responded to epitopes in pools C2-33, C2-34 and C2-36. (D) This subject also showed Th2 (IL-5) and Tr1 (IL-10) responses to rFVIII stimulation. His responses to multiple epitopes in the A2 and C2 domains indicate that the inverted F8 mRNA containing exons 1-22 and F8B mRNA (encoding the putative FVIIIB protein containing the FVIII C2 domain sequence) did not confer tolerance to the FVIII A2 or C2 domains. TT = tetanus toxoid. PHA = phytohemagglutinin. Negative control: DMSO (5 μl/well). rFVIII: 5 nM, rFVIII-C2: 50 nM.
Querying Int22Inv T-cell responses to a known HLA-DRB1*1101-restricted epitope in FVIII
IFN-γ ELISPOT assays were carried out to determine if PBMCs from six severe HA subjects with an HLA-DR11 allele would respond to a known HLA-DRB1*1101-restricted T-cell epitope in the FVIII A2 domain, FVIII-589-608, which was identified in a HA subject with missense substitution R593C (20). The overlapping 20-mer peptides FVIII-A2-28 and/or A2-29 both contained the epitope of interest. Two of the subjects had an Int22Inv mutation, two had a frameshift mutation, and the HA-causing mutation was unknown for the other two. One of the Int22Inv subjects responded to peptide A2-29, and both of the Int22Inv subjects also showed a robust response to the FVIII-C2 protein (Figure S1). Together, these results indicated the Int22Inv mutation did not confer tolerance to either the FVIII A2 or C2 domains.
Mapping epitopes restricted to HLA-DR3
ELISPOT assays were first carried out for subject G21, who had an Int22Inv mutation and was monogenic for HLA-DRB1*0301. His PBMCs showed positive IFN-γ responses to rFVIII and to large peptide pools LP1, LP2 and LP3, which together span FVIII residues 1-737 (Figure 4A). The FVIII heavy chain consists of the A1 and A2 domains, residues 1-740. There were not enough PBMCs to allow systematic decoding via ELISPOT assays using smaller peptide pools and then individual peptides covering 740 residues. Therefore, the Immune Epitope Database (IEDB) server (33) was used to predict peptides within the FVIII A1 and A2 domains that would bind with high affinity to HLA-DR0301. Twenty-five 15-mer FVIII peptides from our library contained a total of 18 unique 9-mer motifs that were predicted to be in the top 2% for binding to HLA-DR0301 (Table S8). In several cases where overlapping 15-mer peptides contained the same motif the 2-3 peptides were pooled, generating a total of 18 individual or pooled FVIII peptides for further testing (Table S9). Stimulation of his PBMCs with four of these individual peptides, A1-41, A1-58, A2-47 and A2-59, produced IFN-γ secretion slightly above background levels (Figure 4B).
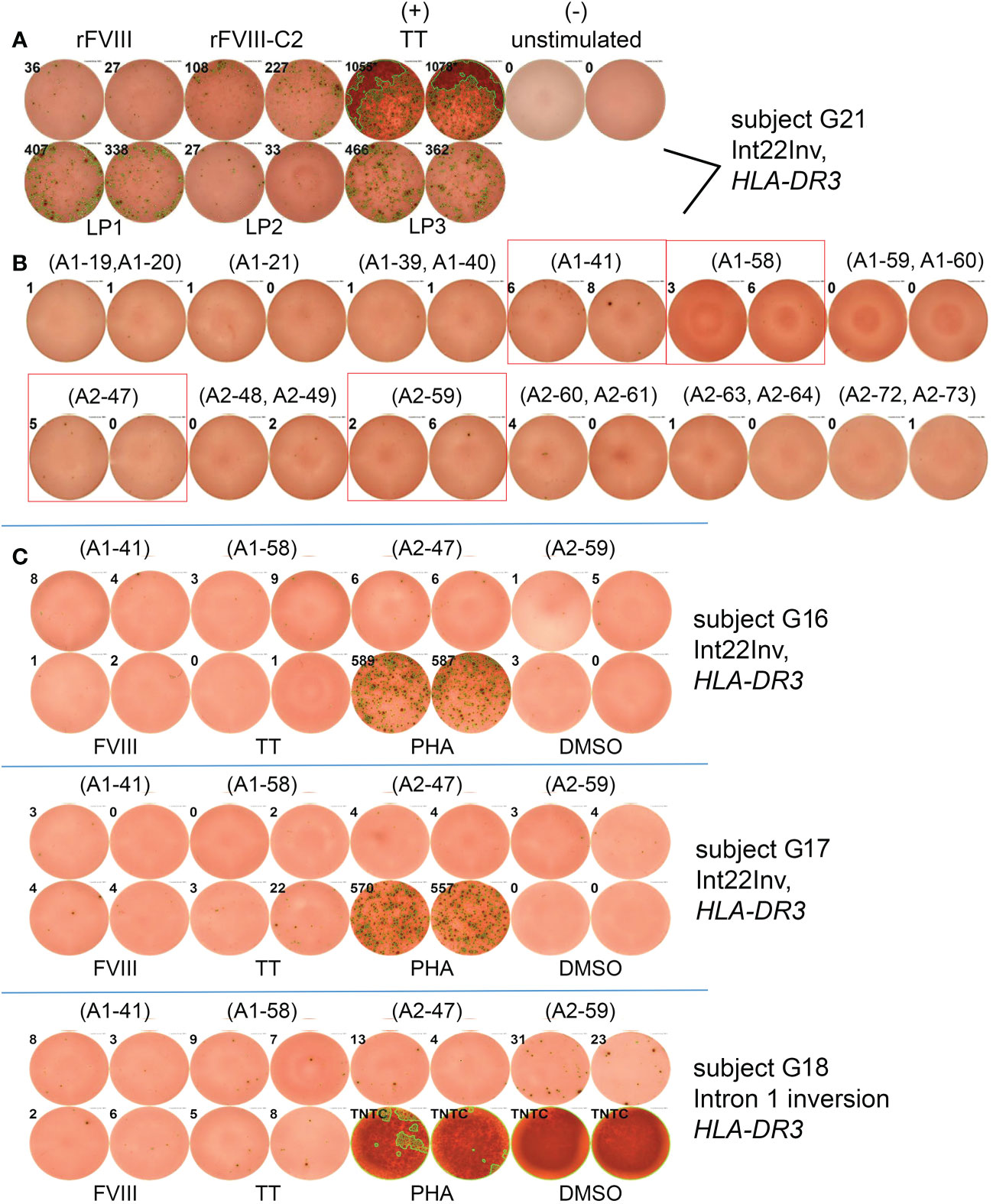
Figure 4 Epitope mapping of HLA-DRB1*0301-restricted responses to FVIII A1 and A2 domains by ELISPOT assays. (A) PBMCs from Int22Inv subject G21 responded to rFVIII and rFVIII-C2 domain protein, tetanus/diptheria toxin, and FVIII large peptide pools LP1, LP2 and LP3. Peptide pools LP1-LP3 consist of overlapping 15-mer peptides spanning the FVIII A1 and the A2 domains (Table S3). and LP2 (B) PBMCs from the same subject also responded to individual 15-mer FVIII peptides A1-41, A1-58, A2-47 and A2-59. Results for the remaining 6 peptides or peptide pools were negative and are not pictured. (C) PBMCs from an additional three Int22Inv subjects with an HLA-DR3 allele (G16, G17 and G18) showed responses above background for one or more of the 4 FVIII A1 or A2 domain peptides. The response of the final subject to peptide FVIII-A2-59 (FVIII 605-619) was the most promising.
Next, PBMCs from three additional Int22Inv HA subjects who all had a current inhibitor and an HLA-DR3 allele, G16, G17 and G18, were tested for IFN-γ responses to these 4 peptides by ELISPOT assays (Figure 4C). The strongest response was from G18, responding to peptide A2-59. Therefore, HLA-DR0301 tetramers loaded with the 4 pooled peptides, and with individual peptide A2-59, were ordered.
Other HA ELISPOT assays
PBMCs from 11 severe and moderate HA subjects, three of whom had a confirmed Int22Inv mutation, were tested for responses to rFVIII and rFVIII-C2 protein (Figure S2). Eight of them showed robust cytokine secretion in response to rFVIII and 9 responded to rFVIII-C2. In general, comparing results of ELISPOT experiments testing reactivity to rFVIII and rFVIII-C2, among the positive responders IFN-γ secretion was usually more robust when cells were stimulated with 50 nM rFVIII-C2 than with 8 nM rFVIII. Unfortunately, FVIII protein becomes toxic to CD4 T cells at higher concentrations. The stronger responses to rFVIII-C2, and to pooled or individual FVIII peptides in some assays, may simply be due to stimulation of lower-avidity CD4 T cells by the higher concentrations of rFVIII-C2 and FVIII peptides. Of note, the rFVIII-C2 preps were routinely tested to confirm low endotoxin levels.
Non-HA control ELISPOT assays
Control ELISPOT experiments indicated that IFN-γ secretion from non-HA PBMCs in response rFVIII stimulation under these assay conditions (Figure S3) was rare, while responses of non-HA PBMCs to rFVIII-C2 protein (included as a control in several experiments) were seen more frequently but still were not common.
Background proliferation
Two severe HA and two non-HA control subjects showed high background IFN-γ secretion (seen in ELISPOT wells with DMSO alone added); they were therefore excluded from this study and are not included in any figures or tables.
Epitope mapping by HLA Class II tetramer staining and isolation of FVIII-specific T-cell clones
HLA-Class II tetramer staining was carried out using CD4+ T cells isolated from four subjects with an Int22Inv mutation. Subject G16 was an adult inhibitor subject who had an HLA-DRB1*0301, 1201g haplotype. Subject G19 was an adult inhibitor subject with HLA-DRB1*0302, 0901 haplotype. Subject G20 (age range 4-17 years) had the HLA-DRB1*0301, 1101 haplotype and a current inhibitor. Subject G21 (age range 4-17 years) had the HLA-DRB1*0301, 0301 haplotype and no inhibitor history. Tetramer staining was also carried out for subject G18, an adult with an intron 1 inversion mutation, a current inhibitor, and HLA-DRB1*0301, 1401g haplotype, in order to further test for HLA-DRB1*0301-restricted epitopes in FVIII. Tetramer staining of CD4+ T cells isolated from these subjects’ PBMCs was followed by sorting of tetramer-hi cells and expansion of clones in culture.
Tetramer staining to test for T-cell responses to epitopes in the FVIII A2 domain
PBMCs from Int22Inv subject G20 were queried using HLA-DR1101 tetramers loaded with peptide pool A2-6, which consisted of four 20-mer peptides spanning FVIII residues 565-616. This region contains a known HLA-DRB1*1101-restricted T-cell epitope that was recognized by HA subjects with an R593C missense mutation (20). Figure 5A shows staining of representative T-cell clones with HLA-DR1101 tetramers loaded with peptide pool A2-6, obtained following expansion of CD4+ T cells using this same peptide pool. Results clearly indicate recognition of one or more epitopes in the region FVIII 565-616.
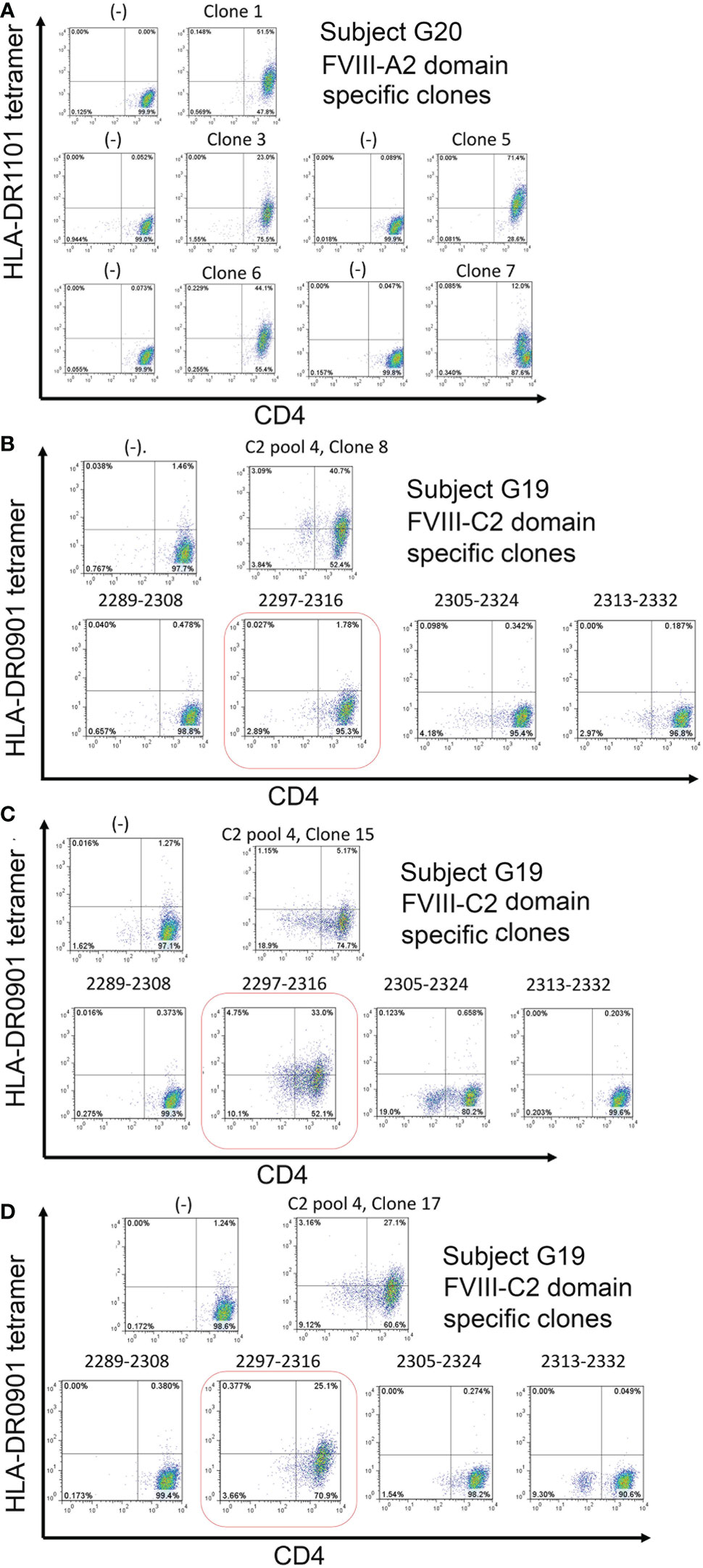
Figure 5 Tetramer staining identifies CD4+ T-cell clones restricted to FVIII A2 and C2 domain peptides. A. CD4+ T-cell clones restricted to one or more epitopes in the FVIII A2 domain were isolated from inhibitor subject G20 (Int22Inv, HLA-DRB1*0301, 1101) PBMCs by staining his CD4+ T cells with DR1101 tetramers loaded with A2 peptide pool #6 (four 20-mer peptides spanning FVIII residues 565-616). The tetramer-hi cells were then single-cell sorted, expanded in culture, and the expanded clones were stained with the same tetramer, following standard protocols in our laboratory (29). All five clones showed high-avidity tetramer binding. B, C, D. CD4+ T-cell clones recognizing pooled and individual peptides corresponding to the FVIII C2 domain were isolated from inhibitor subject G19 (Int22Inv, HLA-DRB1*0302, 0901) following a similar protocol. Clones recognizing C2 pool 4 peptides (spanning FVIII residues 2238-2332) were isolated. Decoding of this response using DR0901 peptides loaded with the individual peptides comprising this pool identified FVIII 2297-2316 as the immunodominant epitope. Negative controls: staining using the same HLA-DR tetramers loaded with irrelevant (tetanus) peptides did not produce tetramer-hi signals, indicating that these tetramers did not bind nonspecifically to CD4+ T cells.
Tetramer staining to test for T-cell responses to epitopes in the FVIII C2 domain
CD4+ T-cell clones were isolated from Int22Inv subject G19 following expansion of CD4+ T cells using a 20-mer peptide pool spanning the FVIII C2 domain region 2289-2332. Figures 5B–D shows tetramer staining of representative HLA-DRB1*0901-restricted T-cell clones recognizing these pooled peptides. A second staining of expanded clones using tetramers loaded with the individual FVIII-C2 peptides comprising this pool identified FVIII-2297-2316 as an HLA-DRB1*0901-restricted T-cell epitope recognized by CD4+ T cells from this subject.
HLA-DR3-restricted T-cell clones recognizing another FVIII A2 domain epitope
PBMCs from three Int22Inv subjects and one intron 1 subject were stimulated with a pool of four immunogenic peptides identified by ELISPOT assays: A1-41, A1-58, A2-47 and A2-59 (Figure 4B). The expanded cells were then stained and single-cell sorted using HLA-DR0301 tetramers loaded with these pooled peptides (not shown). Clones were obtained from three of these subjects. Specificity of the staining was then confirmed using an HLA-DR0301 tetramer loaded with peptide A2-59. This tetramer produced strong staining for two clones expanded from Int22Inv subject G16, two clones from Int22Inv subject G21, and three clones from intron 1 inversion subject G18, confirming that this peptide contained a FVIII epitope. Representative tetramer staining results are in Figure 6. The gating strategy and staining of the remaining clones are in Figure S4.
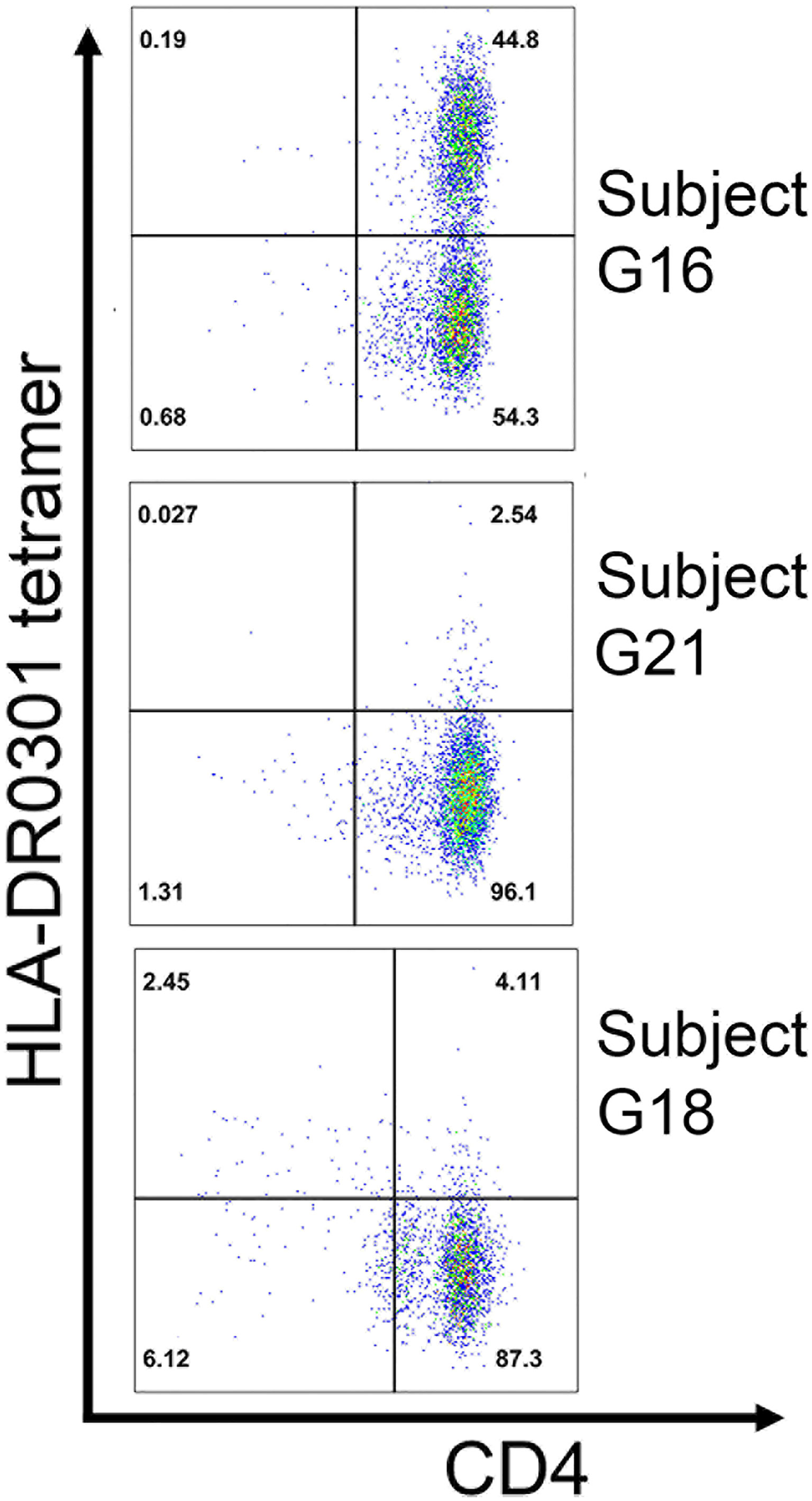
Figure 6 Confirmation of an HLA-DRB1*0301-restricted FVIII-A2 domain epitope by tetramer staining. T-cell clones expanded by stimulation with 15-mer peptide FVIII-A2-59 (FVIII 605-619) from three unrelated HA subjects: G16 (Int22Inv, current inhibitor), G18 (intron 1 inversion, current inhibitor), and G21 (Int22Inv, no inhibitor history). All three had an HLA-DRB1*0301 allele, and their clones were stained with an HLA-DR0301 tetramer loaded with peptide FVIII-A2-59. The results indicate that this peptide contains an immunodominant, HLA-DRB1*0301-resticted T-cell epitope recognized by CD4+ T cells from these subjects.
Positive and negative controls for tetramer-based assays
Positive controls stimulating PBMCs from subjects with tetanus-diptheria toxoid peptides demonstrated the validity of tetramer-based epitope mapping via isolation of multiple TT-specific T-cell clones (Figure S5). Negative controls for all tetramer experiments consisted of tetramers loaded with an irrelevant peptide to rule out nonspecific binding of the tetramer to CD4+ T cells.
Summary of FVIII immunogenicity assays
Results of all assays, grouped by assay type and presented as per-subject results, as well as clinical and demographic data, are summarized in Table 4.
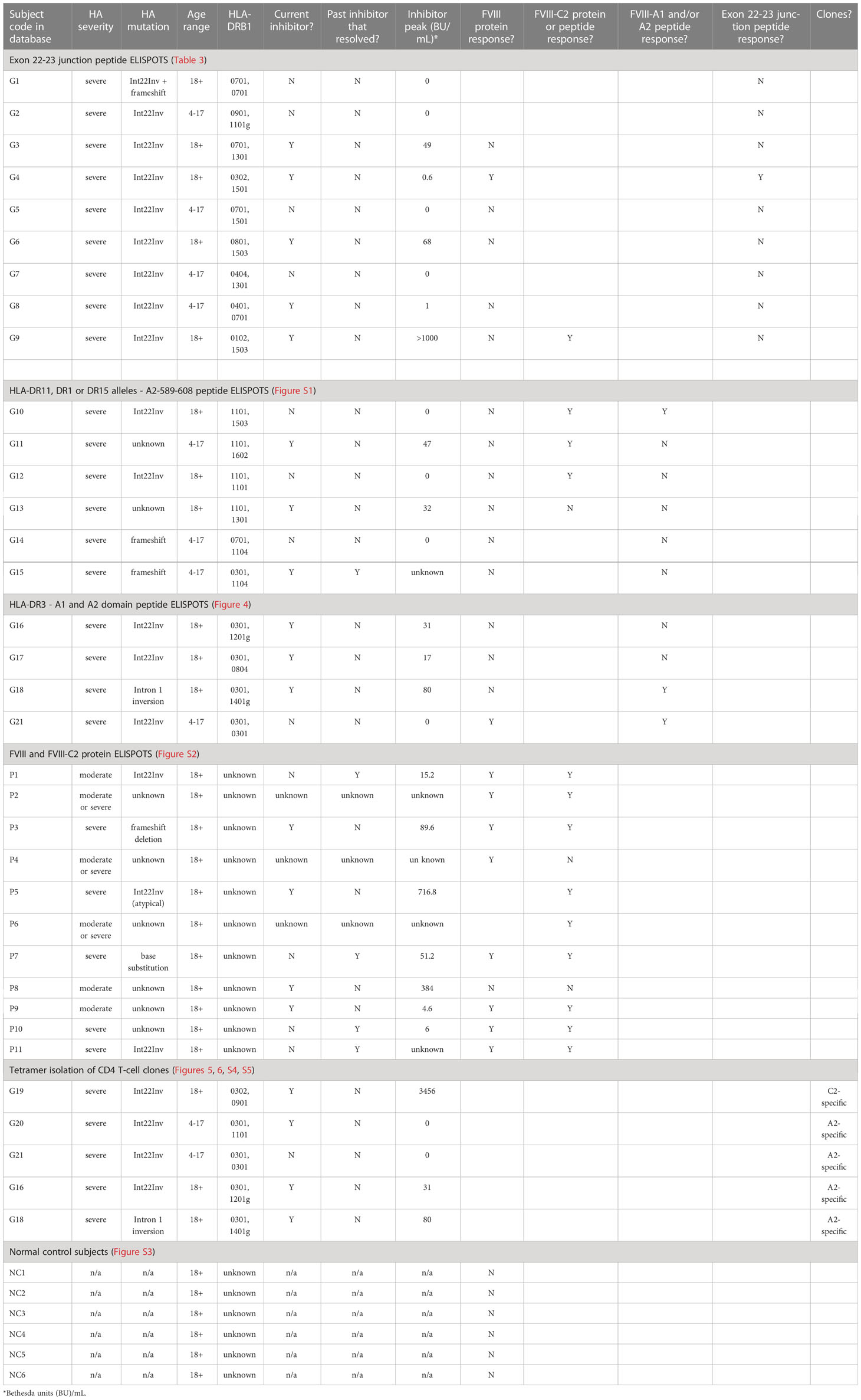
Table 4 Per-subject summary of clinical, demographic and experimental data.
Discussion
The hypothesis that HA patients with an intron-22 inversion (Int22Inv) mutation have a lower risk of developing a neutralizing anti-FVIII antibody (“inhibitor”) response (24, 25) has gained fairly wide traction in the hemophilia A community. Well-conducted earlier studies have indeed indicated that severe HA patients with an intron-22 inversion mutation had a lower inhibitor incidence compared to patients with other large structural changes in the F8 gene such as large frameshifts or deletions, early stop codons, etc. (22, 23). However, an important point to note is that the sizes of these respective HA cohorts differ substantially: almost half of severe HA patients have an intron-22 inversion mutation, while the other large structural changes are a heterogeneous group of mutations that together comprise only ~6% of all mutations resulting in severe HA (34) (Ahmed and Pratt, J Thromb Haemost, in press). Furthermore, a significant fraction of patients with specific, rare large structural F8 changes are related, compared to the overall low relatedness among the intron-22 inversion population. A family history of inhibitor development has been noted as a risk factor in multiple studies, indicating roles for other genetic factors influencing immune and/or inflammatory responses (35–37).
Individuals with an Int22Inv mutation express an mRNA from the inverted locus that contains F8 exons 1-22 spliced to an additional 16 in-frame codons, followed by a stop codon (1, 2). An alternative F8 isoform expressed in multiple human tissues, termed F8B mRNA, is a 2.6-kb transcript initiated from a start site within intron 22 and containing F8 exons 23-26 (4). HA-Int22Inv patients do not circulate measurable FVIII antigenic material (often referred to as cross-reactive material, or FVIII-CRM+). Almost all of them have FVIII clotting activity (FVIII:C) levels <1% normal, i.e., by definition they have severe HA. (The rare exceptions, which generally report FVIII levels of 1-2% normal, are likely due to experimental variations or unusually high activity of other non-FVIII clotting factors). It has been hypothesized that partial FVIII proteins are expressed intracellularly from these two partial F8 transcripts, and that they contain FVIII residues 1-2124 (encoded by F8 exons 1-22) and FVIII residues 2125-2332 (encoded by F8B mRNA containing F8 exons 23-26) (24, 38). It has been further hypothesized that this intracellular expression of the entire FVIII sequence, contained in 2 partial FVIII proteins, confers immune tolerance to these proteins, thereby explaining the apparently lower inhibitor risk associated with Int22Inv mutations (25). If such tolerance is conferred to Int22Inv patients, then their CD4+ T-cell responses (providing help for anti-FVIII antibody production) to infused, therapeutic wild-type FVIII would be restricted to a neoepitope containing FVIII residues M2124 and V2125, as the mRNA encoding this short region is interrupted by the inversion mutation, precluding translation of this region (24, 25). In support of this hypothesis, Pandey et al. reported detection of intracellular FVIII-CRM+ in both liver tissues and circulating cells from Int22Inv and nonhemophilic subjects, using antibody staining and LC-MS/MS analysis of cellular immunoprecipitates (24, 38). Our laboratory carried out similar experiments, using carefully validated antibodies to evaluate human and canine Int22-Inv liver tissues and cellular samples via immunofluorescence, immunohistochemistry, western blots and LC-MS/MS of immunprecipitates. We have been unable to detect intracellular expression of FVIII-CRM+ proteins using these sensitive assays, leading us to suggest that antibodies used in the earlier studies were in fact binding nonspecifically to other antigens besides FVIII (39).
The most relevant data addressing the question of possible tolerance to FVIII is the actual patient outcomes. Our lab recently carried out a systematic regression analyses of data from >6,000 HA subjects enrolled in the “My Life Our Future” study in the U.S., of whom 1,075 had an Int22Inv mutation (Ahmed and Pratt, J Thromb Haemost, in press). A major conclusion of this study was that inhibitor risk associated with Int22Inv mutations was indistinguishable from that associated with other large structural changes in the F8 gene. We attribute the apparent discrepancy of this result with reports from earlier case-control and meta-analysis studies (22, 23) to heterogeneity in the respective, much smaller cohorts (compared to the Int22Inv cohort) in each study that had mutations entailing large structural F8 changes. Thus, the recently obtained statistical data from a large cohort in the U.S. indicate that individuals with an Int22Inv mutation are as likely to develop an inhibitor response as those with HA due to other major F8 gene disruptions.
The present study directly addresses the question of whether Int22Inv patients are tolerized to FVIII, with the exception of a hypothesized neoepitope encoded by the F8 exon 22-23 junction region. First, the binding affinities of 20-mer peptides spanning the exon 22-23 junction regions to ten recombinant HLA-DR proteins were determined using an established peptide-HLA competition binding assay (17, 29, 31). Predicted peptide-HLA-DR affinities were also obtained using a recent update of the same algorithm (MHCIIPan) used by Sauna et al. to predict the immunogenicity of this region (30). As in their study, medium-to-strong affinity binding of these peptides to multiple HLA-DR was predicted. However, the peptide binding assays revealed far fewer high- or medium-affinity interactions (Figure 2). Our experimental results showed high- or medium-affinity MHCII binding of these peptides by HLA-DR7, DR9 and DR15, but not by the other 7 HLA alleles that were tested; these 10 HLA-DR alleles were broadly representative of the U.S. population.
Although the IEDB and other prediction algorithms are extremely useful for applications such as determining prior exposure to a given pathogen or antigen, or peptide-based vaccine design, or for generating candidates for experimental tests of peptide immunogenicity, experimental validation is important before investing too many resources on the basis of predictions alone. MHC Class II and T-cell epitope prediction algorithms are continually being improved, and data such as those in the present study, characterizing both binding and non-binding of peptides to specific MHC alleles, can be utilized to train prediction algorithms and further improve their accuracy (40). Based on the present study’s experimental results, we conclude that the exon 22-23 junction-encoded region, encompassing the FVIII C1 domain sequence extending 9-12 residues on either side of M2124-V2125, is unlikely to comprise an immunodominant, promiscuous T-cell epitope driving anti-FVIII immune responses in most HA-Int22Inv patients. The one subject who showed positive ELISPOT results (IFN-γ secretion in response to stimulation with exon 22-23 junction peptides), subject G4, had a current low-titer inhibitor, and this result indicated that an epitope in the exon 22-23 junction region contributed to his anti-FVIII immune response. He also had a missense F8 mutation, H1499Y, although the relevance of this second mutation in an individual with an Int22Inv mutation is not clear, given that FVIII intact would not be expressed. The mutation H1499Y was not found in the CHAMPS hemophilia A mutation database (34) (accessed 01/24/2023, https://www.cdc.gov/ncbddd/hemophilia/champs.html), so we found no independent data regarding a potential association with inhibitor development. The remaining eight Int22Inv subjects (one also had a frameshift mutation) did not secrete IFN-γ in response to stimulation with the exon 22-23 junction peptides (Tables 3, 4).
Do individuals with an Int22Inv mutation have immune tolerance to FVIII proteins encoded by inverted F8 exons 1-22 and/or F8B exons 23-36? This question was addressed by experiments to test the null hypothesis using independent, complementary methods: ELISPOT assays and HLA-DR tetramer staining. CD4+ T-cell responses to rFVIII proteins and peptides were queried using PBMCs obtained from HA-Int22Inv subjects, as well as HA subjects with other F8 mutations and healthy non-HA controls. Positive controls for these assays included stimulation with tetanus/diptheria toxin (TT) and phytohaemagluttinin (PHA), while negative controls included stimulation with the dimethylsulfoxide (DMSO) carrier solution for peptides, or incubating cells with an irrelevant peptide-loaded tetramer, or comparisons of Int22Inv cellular responses with those of healthy non-HA normal controls. The ELISPOT assays showed interferon-γ secretion in response to rFVIII and/or rFVIII-C2 proteins in almost all of the HA-Int22Inv experiments, whereas anti-FVIII responses were rare in the non-HA control samples. Further epitope mapping using pooled and individual FVIII peptides as stimulants clearly identified HA-Int22Inv immune responses to the FVIII A2 and C2 domains.
HLA Class II tetramer staining was carried out as a stringent, independent test to identify HLA-restricted CD4+ T-cell responses to specific epitopes in FVIII, using PBMCS from Int22Inv subjects. An HLA-DRB1*1101-restricted epitope in FVIII was characterized earlier, identifying the wild-type FVIII A2 domain sequence 498-503 as a neoepitope recognized by CD4+ T cells from two unrelated HA subjects with missense mutation FVIII-R593C (20). Thus, using tetramers loaded with FVIII-A2 peptides, we were able to test the hypothesis that a subject with an Int22Inv mutation and HLA-DRB1*1101 allele would be tolerized to a confirmed HLA-DRB1*1101-restricted epitope that contributed to the anti-FVIII immune responses of HA subjects with a missense mutation at this site. Staining using HLA-DR1101 tetramers loaded with pooled A2 domain peptides, followed by isolation of multiple CD4+ T-cell clones, indeed confirmed that Int22Inv subject G21 responded to one or more HLA-DRB1*1101-restricted epitopes within the FVIII A2 domain region 565-616 (Figure 5A).
Staining of CD4+ T cells from a second Int22Inv subject, G19, using HLA-DR0901 tetramers loaded with pooled C2 domain peptides, produced positive staining using C2 peptide pool #4 (containing peptides spanning FVIII 2289-2332). Multiple clones were again isolated. Three of these pooled peptide responses were decoded by a second staining using tetramers loaded with the individual FVIII-C2 peptides comprising the pool. All three decoding experiments identified FVIII-2297-2316 as an HLA-DRB1*0901-restricted T-cell epitope contributing to the anti-FVIII T-cell response of this subject (Figures 5B–D). Finally, FVIII-specific T-cell clones recognizing an HLA-DRB1*0301-restricted epitope in the FVIII A2 domain were isolated from two Int22Inv subjects and one intron 1 inversion subject with the same HLA-DRB1 allele (Figure 6). One of the Int22Inv subjects had a current inhibitor, one had no inhibitor history, and the intron 1 inversion subject had a current inhibitor. Two of these subjects also responded to FVIII A2 domain epitopes in ELISPOT assays (Figure 4). To summarize, CD4+ T-cell clones recognizing epitopes in the FVIII A2 or C2 domain were isolated from four unrelated Int22Inv subjects.
Approximately 20 years ago, the Conti-Fine group characterized CD4+ T-cell responses to FVIII in HA and non HA subjects, primarily through T-cell proliferation and ELISPOT assays employing FVIII and pools of synthetic FVIII peptides spanning several FVIII domains (10, 12–15, 41). The present study builds on their earlier work, focusing on specific epitopes recognized by HA subjects with an Int22Inv mutation. We also tested the hypothesis that HA-Int22Inv patients have been tolerized to FVIII sequences encoded by mRNAs containing F8 exons 1-22 and/or 23-26. Results of this study provide evidence in support of the null hypothesis: rather than being tolerized, CD4+ T-effector cells from multiple Int22Inv subjects readily responded to multiple epitopes in FVIII. Together with recent statisitical/epidemiological evidence (Ahmed and Pratt, J Thromb Haemostas, in press) and our failure to detect FVIII-CRM+ proteins in liver tissues or circulating cells from Int22Inv subjects (39), the present study indicates that Int22Inv patients should be monitored just as closely as other severe HA patients for development of an inhibitor, especially during initial FVIII infusions.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Uniformed Services University IRB #1. Written informed consent to participate in this study was provided by the participants or by their legal guardian/next of kin.
Author contributions
KP: Conceived the project and designed experiments, analyzed data, supervised the project and wrote the paper. DG, PV and AK: Performed experiments, analyzed data and edited the paper. MR: Enrolled subjects, consulted, and edited the paper. All authors contributed to the article and approved the submitted version.
Funding
Funded by R01 HL 130448 (KP), a Grifols investigator-initiated grant “Mechanisms of Immune Tolerance to Factor VIII” (KP), NHLBI U34 HL11674 (MR), 1RC2 HL 101851 (KP), and intramural funds to KP (Uniformed Services University). Grifols had no role in the study design, sample or data collection, sample analysis, interpretation, or writing of the manuscript.
Acknowledgments
We thank Dr. Eddie James (Benaroya Research Institute, Seattle WA) for help with HLA-DRB1 reagents and tests and Dr. Maochang Liu (Benaroya Research Institute) for carrying out pilot experiments. We thank Dr. Kateryna Lund (Uniformed Services University) for analysis of tetramer staining data. We thank all subjects for their generous blood donations and all clinical staff who participated in this study.
Conflict of interest
Author KP is an inventor on patents related to FVIII immunogenicity.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The opinions or assertions contained herein are the private ones of the author and are not to be construed as official or reflecting the views of the Department of Defense or the Uniformed Services University of the Health Sciences.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1128641/full#supplementary-material
References
1. Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia a. Nat Genet (1993) 5:236–41. doi: 10.1038/ng1193-236
2. Naylor J, Brinke A, Hassock S, Green PM, Giannelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia a is due to large DNA inversions. Hum Mol Genet (1993) 2:1773–8. doi: 10.1093/hmg/2.11.1773
3. Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, et al. Characterization of the human factor VIII gene. Nature (1984) 312:326–30. doi: 10.1038/312326a0
4. Levinson B, Kenwrick S, Gamel P, Fisher K, Gitschier J. Evidence for a third transcript from the human factor VIII gene. Genomics (1992) 14:585–9. doi: 10.1016/s0888-7543(05)80155-7
5. Gouw SC, Ter Avest PC, Van Helden PM, Voorberg J, Van Den Berg HM. Discordant antibody response in monozygotic twins with severe haemophilia a caused by intensive treatment. Haemophilia (2009) 15:712–7. doi: 10.1111/j.1365-2516.2009.01998.x
6. Gouw SC, Van Den Berg HM, Fischer K, Auerswald G, Carcao M, Chalmers E, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia a: The RODIN study. Blood (2013) 121:4046–55. doi: 10.1182/blood-2012-09-457036
7. Johnsen JM, Fletcher SM, Dove A, Mccracken H, Martin BK, Kircher M, et al. Results of genetic analysis of 11 314 participants enrolled in the my life, our future hemophilia genotyping initiative in the united states. J Thromb Haemost (2022) 20:2022–34. doi: 10.1111/jth.15805
8. Reipert BM, Gangadharan B, Hofbauer CJ, Berg V, Schweiger H, Bowen J, et al. The prospective hemophilia inhibitor PUP study reveals distinct antibody signatures prior to FVIII inhibitor development. Blood Adv (2020) 4:5785–96. doi: 10.1182/bloodadvances.2020002731
9. Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, et al. Loss of high-responder inhibitors in patients with severe hemophilia a and human immunodeficiency virus type 1 infection: A report from the multi-center hemophilia cohort study. Am J Hematol (1993) 42:375–9. doi: 10.1002/ajh.2830420408
10. Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Key NS, et al. CD4+ T cell response to factor VIII in hemophilia a, acquired hemophilia, and healthy subjects. Thromb Haemost (1999) 82:509–15. doi: 10.1055/s-0037-1615873
11. Reding MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1- and Th2-induced anti-factor VIII IgG subclasses in congenital and acquired hemophilia patients. Thromb Haemost (2002) 88:568–75.
12. Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Human CD4+ T-cell epitope repertoire on the C2 domain of coagulation factor VIII. J Thromb Haemost (2003) 1:1777–84. doi: 10.1046/j.1538-7836.2003.00251.x
13. Hu GL, Okita DK, Conti-Fine BM. T Cell recognition of the A2 domain of coagulation factor VIII in hemophilia patients and healthy subjects. J Thromb Haemost (2004) 2:1908–17. doi: 10.1111/j.1538-7836.2004.00918.x
14. Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope repertoire of human CD4(+) T cells on the A3 domain of coagulation factor VIII. J Thromb Haemost (2004) 2:1385–94. doi: 10.1111/j.1538-7836.2004.00850.x
15. Hu G, Guo D, Key NS, Conti-Fine BM. Cytokine production by CD4+ T cells specific for coagulation factor VIII in healthy subjects and haemophilia a patients. Thromb Haemost (2007) 97:788–94.
16. Jacquemin M, Vantomme V, Buhot C, Lavend'homme R, Burny W, Demotte N, et al. CD4+ T-cell clones specific for wild-type factor VIII: A molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia a. Blood (2003) 101:1351–8. doi: 10.1182/blood-2002-05-1369
17. James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-Cell responses over time in a mild hemophilia a inhibitor subject: Epitope identification and transient immunogenicity of the corresponding self-peptide. J Thromb Haemost (2007) 5:2399–407. doi: 10.1111/j.1538-7836.2007.02762.x
18. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood (2009) 114:1423–8. doi: 10.1182/blood-2009-01-200725
19. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. HLA-DR-restricted T-cell responses to factor VIII epitopes in a mild haemophilia a family with missense substitution A2201P. Haemophilia (2010) 16:44–55. doi: 10.1111/j.1365-2516.2008.01905.x
20. James EA, Van Haren SD, Ettinger RA, Fijnvandraat K, Liberman JA, Kwok WW, et al. T-Cell responses in two unrelated hemophilia a inhibitor subjects include an epitope at the factor VIII R593C missense site. J Thromb Haemost (2011) 9:689–99. doi: 10.1111/j.1538-7836.2011.04202.x
21. Ettinger RA, Paz P, James EA, Gunasekera D, Aswad F, Thompson AR, et al. T Cells from hemophilia a subjects recognize the same HLA-restricted FVIII epitope with a narrow TCR repertoire. Blood (2016) 128:2043–54. doi: 10.1182/blood-2015-11-682468
22. Ragni MV, Ojeifo O, Feng J, Yan J, Hill KA, Sommer SS, et al. Risk factors for inhibitor formation in haemophilia: A prevalent case-control study. Haemophilia (2009) 15:1074–82. doi: 10.1111/j.1365-2516.2009.02058.x
23. Gouw SC, Van Den Berg HM, Oldenburg J, Astermark J, De Groot PG, Margaglione M, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia a: Systematic review and meta-analysis. Blood (2012) 119:2922–34. doi: 10.1182/blood-2011-09-379453
24. Pandey GS, Yanover C, Miller-Jenkins LM, Garfield S, Cole SA, Curran JE, et al. Endogenous factor VIII synthesis from the intron 22-inverted F8 locus may modulate the immunogenicity of replacement therapy for hemophilia a. Nat Med (2013) 19:1318–24. doi: 10.1038/nm.3270
25. Sauna ZE, Lozier JN, Kasper CK, Yanover C, Nichols T, Howard TE. The intron-22-inverted F8 locus permits factor VIII synthesis: Explanation for low inhibitor risk and a role for pharmacogenomics. Blood (2015) 125:223–8. doi: 10.1182/blood-2013-12-530113
26. Miller CH, Boylan B, Shapiro AD, Lentz SR, Wicklund BM, Hemophilia Inhibitor Research Study, I. Limit of detection and threshold for positivity of the centers for disease control and prevention assay for factor VIII inhibitors. J Thromb Haemost (2017) 15:1971–6. doi: 10.1111/jth.13795
27. Potgieter JJ, Damgaard M, Hillarp A. One-stage vs. chromogenic assays in haemophilia a. Eur J Haematol (2015) 94(Suppl 77):38–44. doi: 10.1111/ejh.12500
28. Lin JC, Ettinger RA, Schuman JT, Zhang AH, Wamiq-Adhami M, Nguyen PC, et al. Six amino acid residues in a 1200 A2 interface mediate binding of factor VIII to an IgG4kappa inhibitory antibody. PloS One (2015) 10:e0116577. doi: 10.1371/journal.pone.0116577
29. Gunasekera D, Ettinger RA, Nakaya Fletcher S, James EA, Liu M, Barrett JC, et al. Factor VIII gene variants and inhibitor risk in African American hemophilia a patients. Blood (2015) 126:895–904. doi: 10.1182/blood-2014-09-599365
30. Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res (2020) 48:W449–W54. doi: 10.1093/nar/gkaa379
31. Ettinger RA, Liberman JA, Gunasekera D, Puranik K, James EA, Thompson AR, et al. FVIII proteins with a modified immunodominant T-cell epitope exhibit reduced immunogenicity and normal FVIII activity. Blood Adv (2018) 2:309–22. doi: 10.1182/bloodadvances.2017013482
32. Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PloS Comput Biol (2008) 4:e1000048. doi: 10.1371/journal.pcbi.1000048
33. Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinf (2010) 11:568. doi: 10.1186/1471-2105-11-568
34. Payne AB, Miller CH, Kelly FM, Michael Soucie J, Craig Hooper W. The CDC hemophilia a mutation project (CHAMP) mutation list: A new online resource. Hum Mutat (2013) 34:E2382–91. doi: 10.1002/humu.22247
35. Astermark J, Berntorp E, White GC, Kroner BL, Group, MS. The malmo international brother study (MIBS): Further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia (2001) 7:267–72. doi: 10.1046/j.1365-2516.2001.00510.x
36. Hay CR. The epidemiology of factor VIII inhibitors. Haemophilia (2006) 12(Suppl 6):23–8; discussion 8-9. doi: 10.1111/j.1365-2516.2006.01362.x
37. Gouw SC, van den Berg HM. The multifactorial etiology of inhibitor development in hemophilia: Genetics and environment. Semin Thromb Hemost (2009) 35:723–34. doi: 10.1055/s-0029-1245105
38. Pandey GS, Tseng SC, Howard TE, Sauna ZE. Detection of intracellular factor VIII protein in peripheral blood mononuclear cells by flow cytometry. BioMed Res Int (2013) 2013:793502. doi: 10.1155/2013/793502
39. Gunasekera D, Vir P, Karim AF, Ragni MV, Merricks EP, Nichols TC, et al. FVIII protein is not detectable in human PBMCs or livers from dogs with an intron-22 inversion mutation: Implications for FVIII immunogenicity and tolerance. Blood (2019) 134. doi: 10.1182/blood-2019-124880
40. Lundegaard C, Lund O, Nielsen M. Predictions versus high-throughput experiments in T-cell epitope discovery: Competition or synergy? Expert Rev Vaccines (2012) 11:43–54. doi: 10.1586/erv.11.160
Keywords: hemophilia A, immune tolerance, intron-22 inversion mutation, epitope mapping, factor VIII
Citation: Gunasekera D, Vir P, Karim AF, Ragni MV and Pratt KP (2023) Hemophilia A subjects with an intron-22 gene inversion mutation show CD4+ T-effector responses to multiple epitopes in FVIII. Front. Immunol. 14:1128641. doi: 10.3389/fimmu.2023.1128641
Received: 21 December 2022; Accepted: 20 February 2023;
Published: 01 March 2023.
Edited by:
Stephen Robert Daley, Queensland University of Technology, AustraliaReviewed by:
Qizhen Shi, Medical College of Wisconsin, United StatesSeema Patel, Emory University, United States
Copyright © 2023 Gunasekera, Vir, Karim, Ragni and Pratt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen P. Pratt, a2F0aGxlZW4ucHJhdHRAdXN1aHMuZWR1
†These authors have contributed equally to this work