 Annika C. Betzler
Annika C. Betzler Alexey Ushmorov2
Alexey Ushmorov2 Cornelia Brunner
Cornelia Brunner
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 February 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1125503
This article is part of the Research Topic Molecular and Cellular Control of B Cell responses: Germinal Center and Extrafollicular Responses for Cellular Outputs View all 6 articles
The germinal center (GC) reaction is a key process during an adaptive immune response to T cell specific antigens. GCs are specialized structures within secondary lymphoid organs, in which B cell proliferation, somatic hypermutation and antibody affinity maturation occur. As a result, high affinity antibody secreting plasma cells and memory B cells are generated. An effective GC response needs interaction between multiple cell types. Besides reticular cells and follicular dendritic cells, particularly B cells, T follicular helper (Tfh) cells as well as T follicular regulatory (Tfr) cells are a key player during the GC reaction. Whereas Tfh cells provide help to GC B cells in selection processes, Tfr cells, a specialized subset of regulatory T cells (Tregs), are able to suppress the GC reaction maintaining the balance between immune activation and tolerance. The formation and function of GCs is regulated by a complex network of signals and molecules at multiple levels. In this review, we highlight recent developments in GC biology by focusing on the transcriptional program regulating the GC reaction. This review focuses on the transcriptional co-activator BOB.1/OBF.1, whose important role for GC B, Tfh and Tfr cell differentiation became increasingly clear in recent years. Moreover, we outline how deregulation of the GC transcriptional program can drive lymphomagenesis.
Germinal centers (GCs) are transient structures that form within B cell follicles of secondary lymphoid organs (SLOs), including lymph nodes (LNs), spleen and Peyer’s patches (PPs), in response to T cell dependent antigens (Ags) (1). Within GCs, B cells undergo intensive proliferation, immunoglobulin (Ig) gene somatic hypermutation (SHM), class-switch recombination (CSR) and affinity maturation. As a result, high affinity antibody (Ab) secreting plasma cells and memory B cells are generated (2). An effective GC response needs interaction between multiple cell types. These are in addition to follicular dendritic cells (FDCs), which are stromal cells residing in B cell follicles of SLOs, specifically B cells, T follicular helper (Tfh) cells and T follicular regulatory (Tfr) cells (2).
In LNs, B cells are organized in follicles within the outer cortex, while T cells are located in the surrounding paracortical area (3). Follicles are proximate to the subcapsular sinus (SCS), where Ags are delivered from the upstream lymphatic vessels (4). Ag presentation to B cells can take place in the SCS, the follicle or the paracortex (5). The spleen is composed of two compartments, the blood-filtering red pulp and the lymphoid white pulp (6). Similar to LNs, the white pulp is composed of B cell follicles and surrounding T cell areas, but follicles are additionally surrounded by the marginal zone. B cell follicles are separated by interfollicular regions in LNs and by arterial vessels in the spleen (4). GCs form within the center of the B cell follicles. Organization of the distinct areas and GC formation relies on the coordinated expression of chemokines and interaction of various cell types, as outlined in more detail in the following.
Naïve B cells located in the follicles of SLOs can be activated by binding Ags via their BCRs (4). Several mechanisms for B cell Ag encounter depending on factors like Ag size or their route of entry have been described (4, 5). Small Ags can diffuse through the SCS or follicular conduits to the follicle and activate B cells (4, 7). Larger Ags can be presented to B cells on the surface of DCs, FDCs or macrophages (7). The different mechanisms of Ag presentation to B cells are thoroughly reviewed elsewhere (4, 5, 7).
Upon BCR signaling, the BCR/Ag-complex is internalized and the Ag degraded to present its peptides on major histocompatibility complex (MHC)-class II to T cells (8). BCR engagement also increases expression of CCR7, which initiates B cell migration towards the B/T cell border, as its ligands CCL19 and CCL21 are expressed in the T cell zone by dendritic cells (DCs) and stromal cells (9). At the same time, naïve T cells located in the T cell zone recognize Ag presented by DCs. This initiates the differentiation towards a Tfh cell phenotype, which is characterized by the upregulation of CXCR5 and downregulation of CCR7 (10). This induces migration to the B/T cell border, where CXCL13, the ligand for CXCR5, is abundantly secreted by FDCs (11). Additionally, both B and T cells increase expression of Epstein-Barr virus-induced G protein coupled receptor 2 (EBI2), which directs their migration to the B/T cell border (12, 13). At this site, B cells present peptides of the Ag on MHC-II to CD4+ T helper cells thereby providing survival and co-stimulatory signals. Ag primed B and T cells form long-lived interactions, which results in full B cell activation (14). Subsequently, activated B and Tfh cells migrate into the center of the follicle, where B cells begin to proliferate and seed the GC. Therefore, B and T cells downregulate CCR7 and EBI2 while constantly expressing CXCR5, which enables them to move into the center of the follicle (15, 16). Moreover, B cells upregulate S1P receptor 2 (S1PR2), which promotes B cell migration to the follicle by binding S1P present in the follicle (17). S1PR2 also seems to be required for retention of Tfh cells in the GC (18). However, a subset of activated B cells does not enter the follicle, but migrate to extrafollicular regions in the spleen or in the medullary cords of LNs and differentiate into short-lived plasmablasts (19). Plasmablasts provide immediate help in recognition and elimination of the Ag by IgM secretion and subsequently, by IgM-mediated pathways (19, 20). Consequently, extrafollicular responses generate Abs faster, but with lower affinity compared to GC responses (19).
After migration into the follicle, the GC expands fast as a consequence of rapid proliferation and GC B cells push aside the resident follicular B cells (11). By day 7, the GC is fully established and polarized into dark and light zones. The dark zone is named for its densely packed appearance as rapidly proliferating B cells, termed centroblasts, reside in this area and undergo SHM (2). Centroblasts express CXCR5 and CXCR4 and are retained in the dark zone by the expression of CXCL12 released by local stromal cells (21). Over time, centroblasts reduce their cell division rates as well as their CXCR4 expression. These B cells are then termed centrocytes. Loss of CXCR4 expression allows their entry into the less densely packed light zone, attracted by the FDC released CXCL13, the ligand of CXCR5 (11).. In the light zone, affinity-based selection and CSR takes place. B cells compete for available Ag and T cell help. Those B cell clones which developed high affinity towards the Ag have a survival advantage over lower affinity B cell clones, which undergo apoptosis (2). GC B cells experience multiple rounds of re-entry into the dark zone before they finally differentiate into either plasma cells or memory B cells (11).
The formation and function of GCs is regulated by a complex network of signals and molecules at multiple levels. In this review, we highlight recent developments in GC biology focusing on the transcriptional program regulating the GC reaction. Besides B cells, we will also highlight the transcriptional program essential for Tfh and Tfr cell differentiation and function.
Several excellent reviews exist describing the transcriptional regulation of B cells during GC initiation, expansion and terminal differentiation into plasma or memory B cells in detail (1, 11, 22, 23). Therefore, we will just briefly highlight the main TFs regulating GC B cell differentiation and function, and focus on the transcriptional co-activator BOB.1/OBF.1, which was recently described to be essential for the GC transcriptional program.
Throughout the GC reaction, multiple regulatory and transcriptional networks characterized by the expression of ‘master TFs’ take place (Figure 1). BCL6 (B cell lymphoma 6 protein) is regarded as the master regulator of the GC reaction and essential for its initiation. The expression of BCL6 is induced by IRF4 (Interferon-regulatory factor 4), IRF8 (Interferon-regulatory factor 8) and MEF2B (Myocyte-specific enhancer factor 2B) (24–26). BCL6 acts as a transcriptional repressor on multiple levels during the GC reaction allowing the establishment of the GC B cell program. Firstly, BCL6 represses cell cycle regulators (CDKN1A/p21) (27) and genes of the DNA damage response pathway (TP53, ATR, CHEK1) (28–30), which permits rapid proliferation and SHM (31). Secondly, BCL6 regulates a transcriptional program facilitating B cell migration into the GC (32, 33). Thirdly, BCL6 represses genes necessary for B cell differentiation into premature plasma cells, thereby facilitating effective affinity maturation of GC B cells (34). In the dark zone, AID (Activation-induced cytidine deaminase), encoded by Aicda, is the key enzyme for SHM (35). Its expression is positively regulated by PAX5 (Paired box protein 5), E2A and IRF8 (25, 36, 37). Moreover, FOXO1 (Forkhead-Box-Protein O1) is highly expressed in the nucleus of GC dark zone B cells (38, 39). The activity of FOXO proteins is regulated by posttranslational modifications thereby regulating e.g. its shuttling between the cytoplasm and the nucleus, its stability or DNA binding (40). Phosphorylation of FOXO1 by the PI3K/AKT pathway induces its translocation from the nucleus to the cytoplasm resulting in its transcriptional inactivity (40, 41). On the other hand, phosphorylation at different sites by other protein kinases like RAS/mitogen-activated protein kinase promote nuclear localization and therefore the transcriptional activity of FOXO (41, 42). Several studies revealed that FOXO1 is essential for the GC dark zone program, as Foxo1-deficient GCs lacked a dark zone (38, 39, 43). Moreover, FOXO1 and BCL6 both repress BLIMP1 (B lymphocyte maturation protein 1), which is essential for plasma cell differentiation, thereby maintaining the GC dark zone program (38). In the light zone, affinity maturation and CSR take place. IRF4 is upregulated in light zone B cells, which in turn represses BCL6, thereby ending the dark zone transcriptome and establishing the light zone program (23). FOXO1 also seems to be involved in regulating light zone programs, as Foxo1-deficient GC B cells were described with impaired affinity maturation and CSR (38). CSR also relies on the expression of AID. During CSR, IRF4, FOXO1, PAX5, E2A and BATF (B Cell Activating Transcription Factor) are involved in AID regulation (38, 44–46). Upregulated IRF4 expression also induces BLIMP1 (encoded by Prdm1) expression (44, 47), which is essential for plasma cell development (48, 49). BLIMP1 further represses the expression of Aicda, Bcl6 and Pax5, finally terminating the GC program (50, 51). BLIMP1 and IRF4 both activate XBP1 (X-box binding protein 1), which is required for Ab production and secretion (52–54). In contrast, ABF1 (Autonomously replicating sequence (ARS) binding factor 1) induces memory B cell differentiation and prevents plasma cell development (55). Another essential TF of the GC reaction is MYC. However, its role is incompletely understood. MYC is required during the very early phase of GC initiation, as its expression is induced immediately after immunization (56, 57). However, in dark zone B cells its expression is repressed by BCL-6. Later, MYC becomes re-expressed in a subset of light zone B cells and probably regulates their re-entry into the dark zone (56, 57).
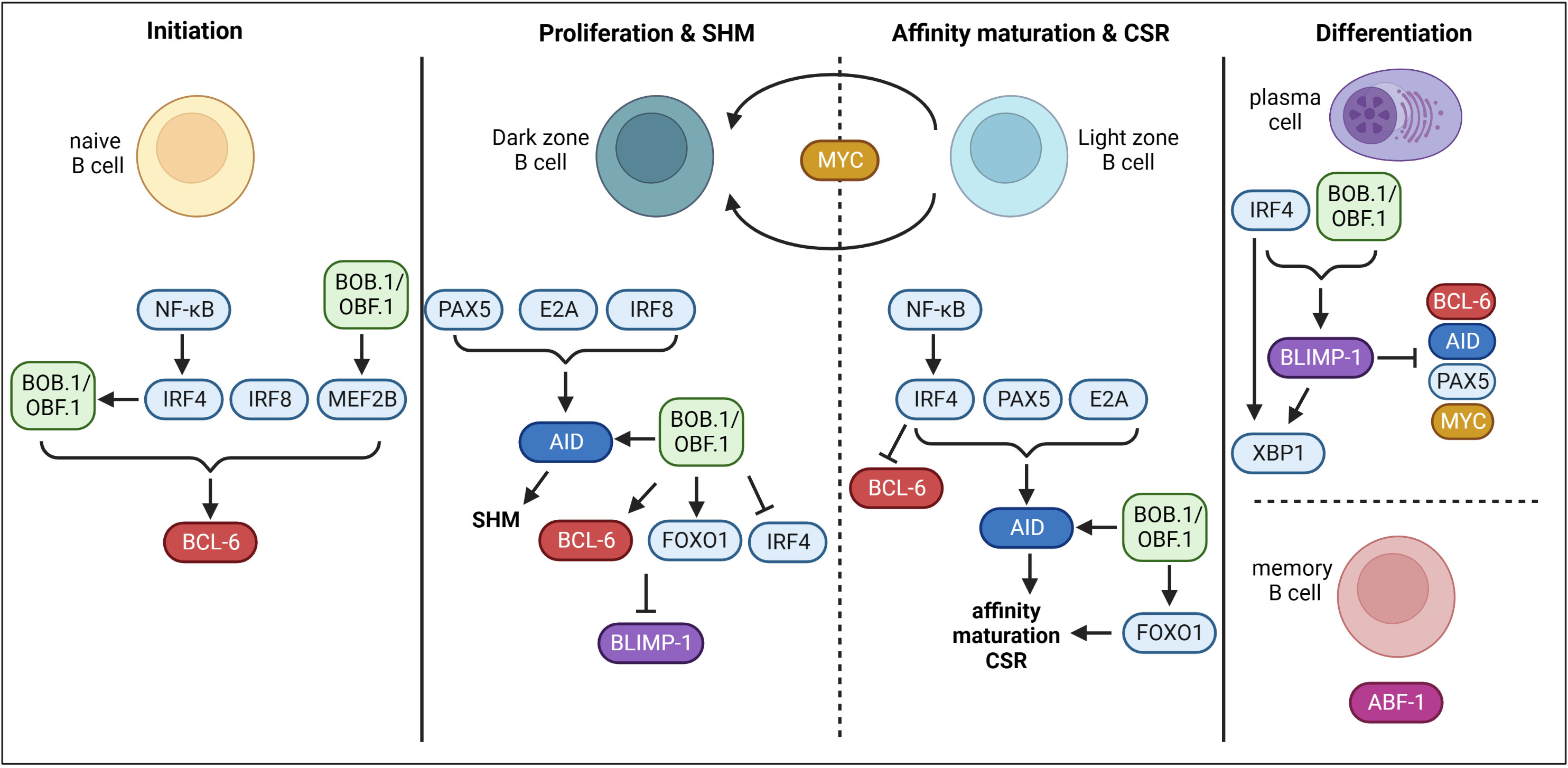
Figure 1 Transcriptional control of GC B cells. During GC initiation, TFs IRF4, IRF8 and MEF2B induce the expression of BCL-6. BOB.1/OBF.1 is also involved in the initiation of BCL-6 expression. Moreover, the transcriptional co-activator drives MEF2B expression. On the other hand, IRF4 seems to modulate the expression of BOB.1/OBF.1. In the dark zone, PAX5, E2A and IRF8 induce AID and therefore SHM. BOB.1/OBF.1 was also shown to be involved in AID, BCL-6 and FOXO1 induction. BCL-6 and FOXO1 inhibit BLIMP-1 and BOB.1/OBF.1 blocks IRF4 expression to prevent initiation of plasma cell differentiation at this stage. In the light zone, IRF4 is upregulated and prevents BCL-6 expression to terminate the dark zone program. IRF4, PAX5 and E2A promote AID expression and thereby affinity maturation and CSR. BOB.1/OBF.1 (POU2AF1) is again involved in regulation of AID and FOXO1 expression. The TF MYC probably regulates re-entry into the dark zone. IRF4 and BOB.1/OBF.1 are involved in driving BLIMP-1 expression. BLIMP-1 in turn represses BCL-6, AID, PAX5 and MYC and on the other hand facilitates expression of XBP1 together with IRF4 resulting in plasma cell differentiation. In contrast, ABF-1 induces memory B cell differentiation. This figure was created with BioRender.com.
Another factor essential for the GC reaction is BOB.1/OBF.1 (B cell Oct binding factor 1/Oct-binding factor 1), also known as OCA-B (octamer coactivator from B cells). BOB.1/OBF.1 is encoded by the Pou2af1 (POU domain class 2-associating factor 1) gene. BOB.1/OBF.1 is expressed in both B and T lymphocytes and acts as transcriptional co-activator of octamer-dependent transcription (58). The octamer motif is found in regulatory elements of B and T cell specific genes (59–61). BOB.1/OBF.1 interacts with TF of the Oct-family (Oct-1 and Oct-2) and enhances their binding selectivity to the octamer motif (5’-ATGCAAAT-3’) and their transcriptional activity (62–65). Thus, the ternary complex built by BOB.1/OBF.1 together with Oct-1 or Oct-2 on the DNA octamer motif regulates expression of genes essential for lymphocyte physiology (62, 65–70). Analysis of BOB.1/OBF.1-deficient mice revealed several defects in early and late B cell antigen-independent maturation. BOB.1/OBF.1 is critical for early B cell development in the bone marrow, as BOB.1/OBF.1-deficient mice feature a developmental block at the pro/pre B cell stage (71, 72). Consequently, the number of peripheral B cells is severely reduced in these mice (73). Additionally, BOB.1/OBF.1 is essential for the maturation of follicular B cells, since BOB.1/OBF.1-deficient mice show increased levels of immature but decreased numbers of mature B cells (71, 73). Besides, the number and function of marginal zone B cells is affected in the absence of BOB.1/OBF.1 (74). However, the most striking characteristics of BOB.1/OBF.1-deficient mice are the complete absence of GCs and consequently dramatically reduced levels of class-switched Igs (71–73, 75). However, it was completely unsolved at which stage of B cell development BOB.1/OBF.1 expression is essential for GC formation. By establishing a mouse system allowing the conditional deletion of BOB.1/OBF.1 at different time points of B-cell maturation, we have recently shown that an efficient GC reaction needs BOB.1/OBF.1 expression during the complete B-cell ontogeny (76). Notably, in Pou2af1fl/fl x Cγ1-Cre mice, in which BOB.1/OBF.1 is specifically deleted in GC B cells four days after immunization also lack GCs (76). This finding proves that this deficiency is a GC B-cell autonomous defect and not exclusively a consequence of defective early B-cell maturation. The TF IRF4 seems to regulate the induction of BOB.1/OBF.1 expression during the early GC reaction initiation (24). For a long time, GC-specific BOB.1/OBF.1 target genes were largely unknown. In 1998, NF-κB, Oct-2 and BOB.1/OBF.1 were shown to cooperatively regulate the promoter activity of the chemokine receptor Cxcr5 in B cells (77). Thus, reduced Cxcr5 expression observed in conventional BOB.1/OBF.1-deficient mice might contribute to the absence of GCs in these animals as a consequence of impaired B lymphocyte migration (69, 77). In addition, BOB.1/OBF.1-deficient B cells of postnatal day 9 showed a dramatic reduction in the frequency of migration towards Cxcl13 indicating an impaired Cxcr5 function in these cells (78). These findings in combination with observed diminished FDC numbers and reduced Cxcl13 expression in the absence of BOB.1/OBF.1 suggest that reduced lymphocyte numbers in SLOs of these mice could be a consequence of defective Cxcl13-Cxcr5 signaling (78). In 2006 Spi-B, which is essential for GC formation and maintenance (79), was revealed as BOB.1/OBF.1 target (80). Ten years later, a ternary complex on the Bcl6 promoter formed by BOB.1/OBF.1 and Oct-TFs was described, activating Bcl6 transcription and thereby promoting Tfh cell development (69). In 2020, it was demonstrated that BOB.1/OBF.1 also regulates BCL-6 expression in GC B cells (81). The authors found that BOB.1/OBF.1 forms a ternary complex with Oct-2 and the TF MEF2B, which occupies and regulates a locus control region that regulates BCL-6 expression (81). Most recently, combining multiple experimental approaches a map of direct BOB.1/OBF.1 target genes was defined. Precisely, transgenic mice expressing tagged versions of Oct-1, Oct-2 and BOB.1/OBF.1 were generated, which allowed for ChIP-sequencing and the identification of their DNA-binding sites (68). This strategy in combination with the analysis of specific histone marks to examine chromatin activity status as well as RNA-sequencing experiments allowed the identification of several BOB.1/OBF.1 target genes (68). These data show that BOB.1/OBF.1 and Oct-TFs regulate the GC transcriptional program by binding to multiple genes encoding TFs essential for GCs including Bcl6, Myc, Foxo1, Mef2b, Aicda and Spi1in primary B cells (68) (Figure 1). Moreover, the authors provide evidence that BOB.1/OBF.1 is also required for the maintenance of the GC transcriptional program. In the absence of BOB.1/OBF.1 the GC transcriptional program is disrupted as genes essential for GC maintenance (Bcl6) are downregulated and genes initiating the plasma cell program are upregulated (Irf4) (68). Together, these results highlight BOB.1/OBF.1 as a key player in maintaining the GC transcriptional program by activating BCL-6 and repressing IRF4. When the expression of BOB.1/OBF.1 is abolished, the BCL-6 pathway is diminished and IRF4 expression gets enhanced, which results in the termination of the GC program and the initiation of plasma cell differentiation (68). In addition, BOB.1/OBF.1 seems to be essential for the induction of Prdm1, thereby contributing to plasma cell differentiation (70).
The differentiation of Tfh cells is a multistage process, whereby multiple TFs that either promote or repress Tfh cell fate are involved (Figure 2). The first step of Tfh cell differentiation is the interaction of naïve CD4+ T cells with DCs in the T cell zone. CD4+ T cells undergo cell fate decision either towards T helper (TH) cell subsets or Tfh cells. There is evidence that Tfh cell development is initiated already during DC priming after viral infection (82). Thereby, ICOS (Inducible T-cell COStimulator) seems to be required to induce Bcl6 expression (82). Another study also suggested a direct link between TCR signal strength, IL-2 production and Tfh cell fate decision (83). Indeed, CD4+ T cell commitment towards Tfh cell fate was reported within first rounds of cell division only two days after viral infection (82). Cells either start to express Bcl6, the master regulator of Tfh cell differentiation, or its antagonist Blimp-1, which favors TH cell development. The expression of BCL-6 is induced on multiple levels. Several cytokines including IL-21, IL-6 and type I IFNs (IFN-α/β) induce Bcl6 expression via the activation of STAT1/3 (84–87). The TFs LEF-1 (Lymphoid enhancer binding factor 1) and TCF-1 (T cell factor-1) promote Bcl6 expression by targeting signaling molecules upstream of BCL-6 like IL-6R and ICOS (88). TCF-1 was also shown to directly bind to the Bcl6 promoter facilitating its expression (89). BCL-6 in turn represses Blimp-1 and TFs like T-bet, Gata3 and RORγt, which favor the development into TH subpopulations (90). BCL-6 and also the TF ASCL2 (Achaete-Scute Family BHLH Transcription Factor 2) induce the expression of CXCR5 and suppresses CCR7 expression inducing migration of pre-Tfh cells to the B/T cell border (91, 92). The second step of Tfh cell differentiation occurs upon interaction of pre-Tfh cells with Ag-specific B cells in the B/T cell border. B/T cell interaction functions as feedback loop as it further drives both Tfh as well as GC-B cell development. The B/T crosstalk happens in a peptide/MHC II and ICOS/ICOSL dependent manner. ICOS/ICOSL signaling represses the TF KLF2 (Krüppel-like Factor 2), which is required to maintain the Tfh cell phenotype as KLF2 promotes the expression of genes that mediate the migration to the T cell zones and away from the follicles (93). ICOS signaling also induces the TF c-MAF, which directly binds to the Il-21 promoter inducing the expression of this key-Tfh cytokine (94). Within GCs, SLAM/SAP binding is required to stabilize B/T cell interactions (95). Here, pre-Tfh cells differentiate into Tfh cells, a further polarized state characterized by highest expression levels of BCL-6, CXCR5, PD-1, ICOS and BTLA (96). What happens to Tfh cells after they have fully differentiated and provided help for GC B cells is very well understood. It is supposed that Tfh cells can exit a GC and (i) migrate to another GC, (ii) transitionally reside in an adjacent follicle and re-enter the same follicle again or (iii) completely exit the GC and acquire a memory-like phenotype (97, 98).
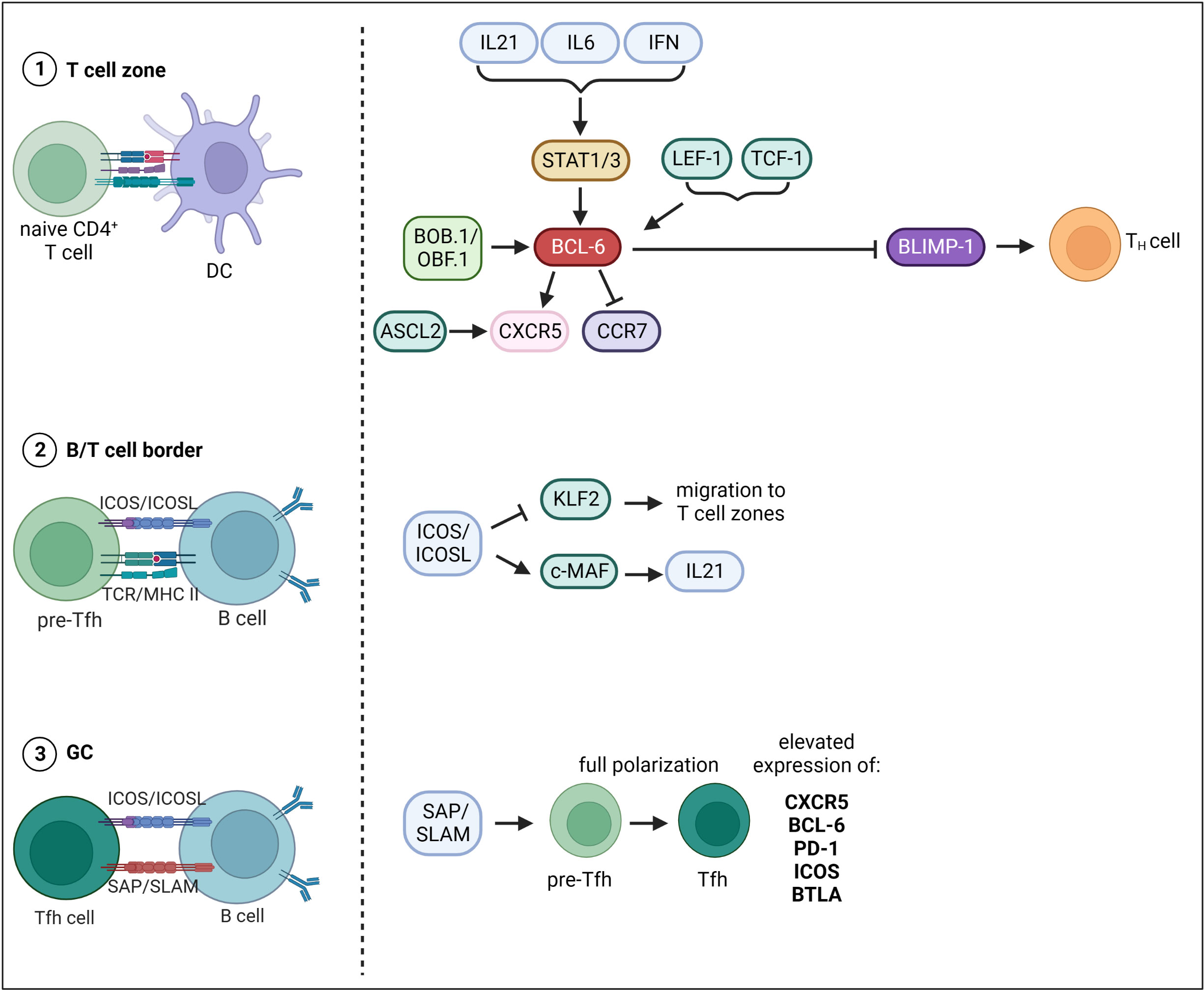
Figure 2 Transcriptional control of Tfh cell differentiation. Tfh cell differentiation occurs in three main steps. The first step of Tfh cell differentiation is the interaction of naïve CD4+ T cells with DCs in the T cell zone. The expression of BCL-6, master TF of Tfh cells, is induced on multiple levels. Cytokines including IL-21, IL-6 and type I IFNs (IFN-a/b) drive Bcl-6 expression via the activation of STAT1/3. Besides, TFs LEF-1 and TCF-1 as well as the transcriptional co-activator BOB.1/OBF.1 together with TFs Oct-1/Oct-2 induce Bcl- 6 expression. BCL-6 in turn represses Blimp-1 and TFs favoring development into TH subpopulations. BCL-6 and the TF ASCL2 induce the expression of CXCR5 and suppresses CCR7 expression inducing migration of pre-Tfh cells to the B/T cell border. The second step of Tfh cell differentiation then occurs upon interaction of pre-Tfh cells with Ag-specific B cells in the B/T cell border. ICOS/ICOSL signaling represses the TF KLF2 thereby inhibiting migration back to the T cell zones. ICOS signaling also induces the TF c-MAF, which facilitates IL21 expression. The third step of differentiation occurs within GCs, where SLAM/SAP binding is required to stabilize B/T cell interactions. Here, pre-Tfh cells differentiate into Tfh cells, a further polarized state characterized by highest expression levels of BCL-6, CXCR5, PD-1, ICOS and BTLA. This figure was created with BioRender.com.
The transcriptional co-activator BOB.1/OBF.1 plays a central role in controlling the transcriptional program of GC B cells as outlined above. BOB.1/OBF.1 has long been considered a B cell specific factor, but in recent years a contribution of BOB.1/OBF.1 expression in T cells to the GC reaction became evident. Several recent studies revealed a high BOB.1/OBF.1 expression in murine and human Tfh cells (69, 99–101) and BOB.1/OBF.1 ablation results in reduced Tfh cell numbers in mice (69, 102–105). Most recently, a patient with mutation in the Pou2af1 gene resulting in the absence of BOB.1/OBF.1 protein was reported who also featured reduced Tfh cell numbers (103). Moreover, BOB.1/OBF.1 deficiency prevented the differentiation of CD4+ T cells into Tfh cells in an autoimmune mouse model (106). In line, BOB.1/OBF.1-deficient mice revealed a significantly reduced Tfh cell compartment in response to viral infection (102). However, when mice were reconstituted with BOB.1/OBF.1-deficient T and WT B cells prior to influenza infection Tfh cells developed normally (102). In contrast, mice reconstituted with BOB.1/OBF.1-deficient B and WT T cells revealed impaired Tfh cell differentiation, which initially led to the conclusion that this defect might be B-cell mediated (102). Nonetheless, Stauss et al. reported a prominent reduction of Tfh cells in BOB.1/OBF.1-deficient mice upon SRBC immunization and revealed a CD4+ T cell autonomous defect by performing reconstitution experiments (69). In addition, loss of BOB.1/OBF.1 was found to be associated with reduced Bcl-6 expression in Tfh cells from mixed bone marrow chimeras. ChIP experiments of BOB-deficient CD4+ T cells maintained under Tfh inducing conditions revealed binding of BOB.1/OBF.1 together with Oct-1 and Oct-2 to an octamer motif within the Bcl6 promoter (69). In the same year, the study by Yamashita et al. contradicted these findings (99). They reported increased Tfh percentages in BOB.1/OBF.1-deficient animals suggesting a BOB.1/OBF.1 related mechanism that limits TCR-mediated Tfh cell expansion (99). Different experimental settings regarding immunization and mouse strains might account for this discrepancy. Yamashita et al. only analyzed PD1+ CXCR5+ Tfh cells of spleens by flow cytometry and their further experimental approach was predominantly in vitro (99). Stauss et al., reported a reduction of multiple Tfh cell subsets (PD1+, ICOS+ BTLA+) in spleen, LNs and PPs of BOB.1/OBF.1-deficient mice by flow cytometry and immunofluorescence (69). Moreover, they showed that BOB.1/OBF.1 transactivates Bcl6 and Btla promoters (69). In 2022, Lombard-Vadnais et al., could show that Tfh differentiation is blocked at the early Tfh maturation stage when BOB.1/OBF.1 is absent in hematopoietic cells (104). However, in line with the observation of Karnowski et al. (102), the authors here also suggested that BOB.1/OBF.1 plays a T cell extrinsic role in Tfh cell differentiation and that instead its expression in B cells promotes Tfh cell maturation (104). In the same year, a study by us revealed reduced Tfh cell numbers when BOB.1/OBF.1 was deleted tissue-specific in a CD4- or IL21-Cre dependent manner (105). In particular, the reduction in Pou2af1fl/fl x IL21-Cre mice, which deletes BOB.1/OBF.1 predominantly in Tfh cells, provides evidence that Tfh cell development and function requires T-cell specific BOB.1/OBF.1 expression. Since IL21 is also required for Tfh cell differentiation (84), crossing Pou2af1fl/fl mice to IL21-Cre mice results in BOB.1/OBF.1 deficiency already in pre-Tfh cells. Thus, BOB.1/OBF.1 might play a role already in pre-Tfh cells and their subsequent differentiation. Moreover, T and Tfh cell-specific BOB.1/OBF.1 deletion also resulted in impaired GC formation (105). Thus, BOB.1/OBF.1 expression in T cells is also required for efficient GC formation. Consequently, the contribution of BOB.1/OBF.1 to the GC reaction cannot be exclusively attributed to its expression in B cells. The fact that the reduction of GCs was more prominent in Pou2af1fl/fl x CD4-Cre mice suggests that BOB.1/OBF.1 expression in T cells is required early during the process of GC initiation. This is in line with findings of Pou2af1 being highly expressed in early stage GC-Tfh cells (88). Altogether, the role of BOB.1/OBF.1 for Tfh cell differentiation was controversially discussed in the past years due to different experimental approaches. However, tissue-specific deletion of BOB.1/OBF.1 emphasizes the Tfh cell intrinsic role of BOB.1/OBF.1 for Tfh cell maturation and function (105). Yet, since signals from GC B cells are required for Tfh maintenance, a combination of both the B cell and Tfh cell intrinsic role of BOB.1/OBF.1 might contribute to both efficient GC B and Tfh cell development.
Tfr cells are a subpopulation of regulatory T cells (Tregs) and are found within GCs in mice and humans (107–112). Their assumed function is the suppression of the GC reaction by repressing excessive Tfh and GC B cell proliferation thereby promoting selection of high affinity B cell clones (107–109, 113). Tfr cells share characteristics of both Tregs and Tfh cells and it is supposed that Tfr cells have different phenotypes at different stages of maturation or in various locations (111, 114, 115). Tfr cells express FoxP3 (Forkhead box P3), CTLA-4 (Cytotoxic T-lymphocyte-associated Protein 4), GITR (Glucocorticoid-induced TNFR-related gene) and BLIMP-1 similar to Tregs (108, 111, 116, 117). On the other hand, they express BCL-6, CXCR5, ICOS, PD1 and SAP typical for Tfh cells (108, 111, 117). It is assumed that the majority of Tfr cells arises from natural Tregs (107, 108). However, there is evidence that they can also derive from naïve FoxP3-negative CD4+ T cells (118).
Similar to Tfh cells, the Tfr cell differentiation process is initiated by Ag presentation by DCs (Figure 3). However, it is not completely solved which Ag signals initiate Tfr cell differentiation since Tfr cells respond to both foreign and self-Ags, but more strongly to self-Ags (118, 119). Besides Ag-receptor stimulation, Tfr cell differentiation depends on co-stimulation through CD28 and ICOS (108, 120). However, Tfr and Tfh cells were shown to have different TCR repertoires and that of Tfr cells resembles more that of Tregs (121). This allows the conclusion that Tfr cells develop in a polyclonal and Ag-independent manner from Tregs (113). Initial Tfr cell differentiation relies on CD28 and ICOS signaling, whereas interaction with B cells via SLAM/SAP is required for their full differentiation (90, 108, 120). Besides Tfh cells, BCL-6 is also the key TF regulating Tfr cell differentiation. In contrast to Tfh cells, Tfr cells express both BCL-6 and BLIMP-1 (108). It is assumed that the balanced regulation by these two opposing TFs is essential for efficient Tfr differentiation and function, since BCL-6 ablation results in the absence of Tfr cells (108), while lack of Blimp-1 increases Tfr cell numbers, however, with reduced suppressive capacity (122). Therefore, it is hypothesized that BCL-6 is essential for Tfr generation, whereas BLIMP-1 is required to limit and control Tfr cell numbers (122). Another key TF of Tfr cells is FoxP3, which is the known master regulator of Treg cells. FoxP3 modifies the Tfh transcriptional program to induce a Tfr-like functional state (123) as well as to maintain the Tfr cell transcriptional program (123). Thus, FoxP3 is required for Tfr cell identity and suppressive function (123). GC homing in Tfr cells is initiated by the TF NFAT2 (Nuclear factor of activated T cells 2), which upregulates CXCR5 expression, thus playing a comparable role to ASCL2 in Tfh cells (124). In this context, store-operated Ca2+ entry (SOCE) through Ca2+ release-activated Ca2+ (CRAC) channels mediated by stromal interacting molecules (STIM) and ORAI proteins were shown to promote Tfr cell differentiation through NFAT-mediated IRF4, BATF, and BCL-6 TFs (125). Subsequently, CXCR5 expression is maintained by BCL-6 (107, 108). BCL-6 in turn is regulated by ICOS and mTORC1 in Tfr cells. ICOS induces PI3 kinase and its subunit p85α interacts with osteopontin (OPN-i) (126). OPN-i translocates to the nucleus preventing BCL-6 from ubiquitin-dependent proteasome degradation (126). Additionally, mTORC1 was shown to promote Tfr cell differentiation via STAT3/TCF-1/BCL-6 pathway (127). TFs Id2/Id3 (Inhibitor of DNA Binding 2/3) are also involved in Tfr cell differentiation. Id2 and Id3 expression decreases upon TCR stimulation allowing the induction of the Tfr cell specific transcriptional program including CXCR5 and IL10 expression (128). The sclerostin domain-containing protein 1 (SOSTDC1) was shown to block the WNT-β-catenin axis thereby facilitating Tfr cell differentiation, possibly by upregulating FoxP3 and CXCR5 (129). In addition, the ablation of c-Maf, which is also involved in Tfh cell differentiation, resulted in reduced Tfr cell numbers (130). However, the exact mechanisms by which c-Maf regulates Tfr cell differentiation remain to be elucidated.
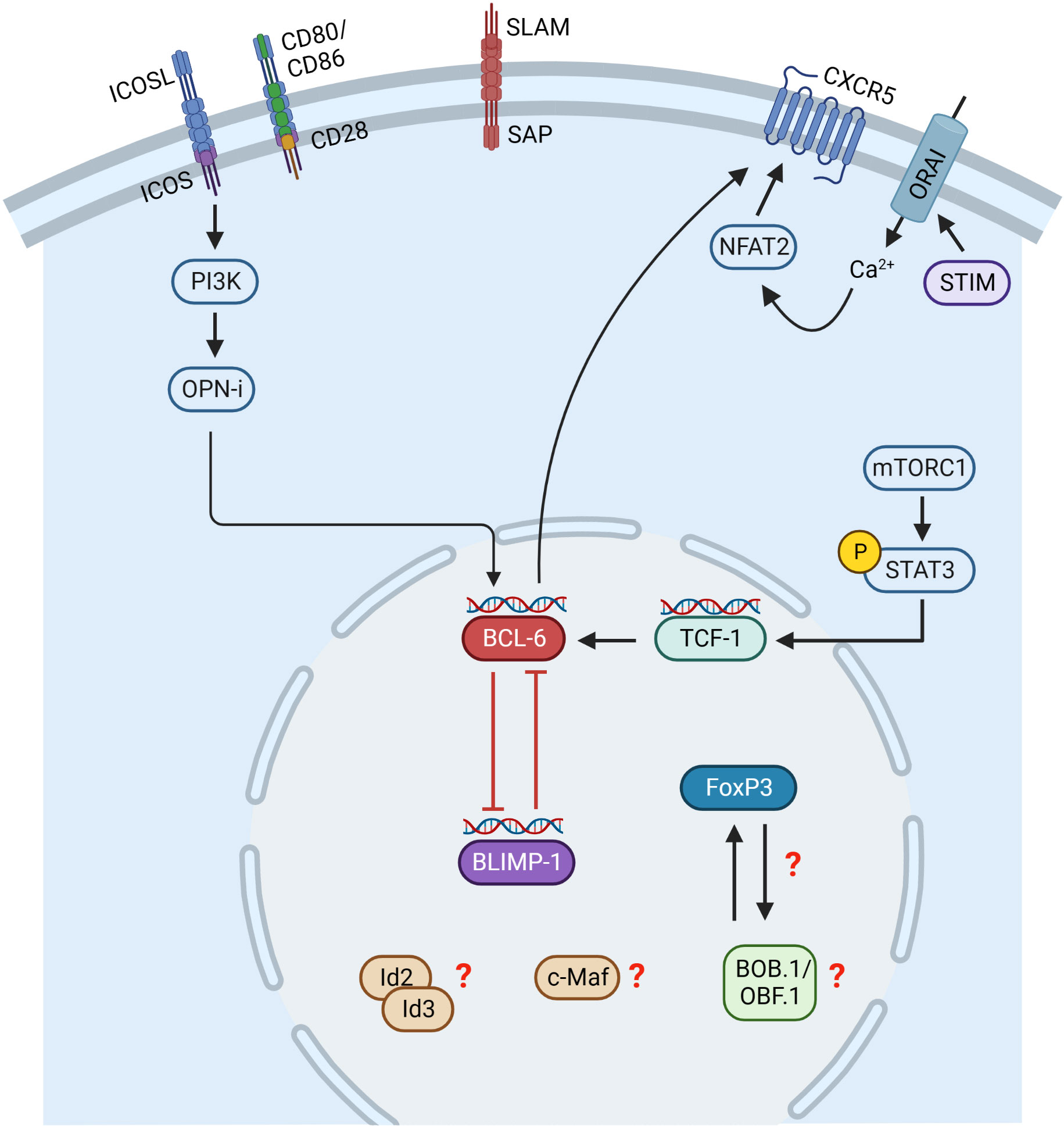
Figure 3 Transcriptional control of Tfr cell differentiation. Initial Tfr cell differentiation depends on CD28 and ICOS signaling, whereas interaction with B cells via SLAM/SAP is required for their full differentiation. Tfr cells express both BCL-6 and BLIMP-1 and possibly their balanced regulation is essential for Tfr cell differentiation. BCL-6 is regulated by ICOS and mTORC1. ICOS induces PI3K and its subunit p85α interacts with osteopontin (OPN-i). OPN-i translocates to the nucleus preventing BCL-6 from ubiquitin-dependent proteasome degradation. mTORC1 induces phosphorylation of STAT3, activating TCF-1 expression, which upregulates BCL-6. Stromal interacting molecules (STIM) and ORAI proteins mediate Ca2+ entry inducing NFAT2, which upregulates CXCR5. Id2/Id3, c-Maf and BOB.1/OBF.1 are also involved in Tfr cell differentiation, but the exact mechanisms are incompletely understood so far. This figure was created with BioRender.com.
As BOB.1/OBF.1 was previously shown to be required for Tfh cell differentiation by regulating BCL-6 expression, as outlined in the previous chapter, it is likely that BOB.1/OBF.1 might also be involved in Tfr cell differentiation. BOB.1/OBF.1 was already linked to FoxP3 and Tregs, since an altered expression of Pou2af1 in Tregs was revealed (111, 131, 132). In detail, POU2AF1 was shown to be up-regulated in circulating human blood Tfr cells compared to Tfh and Treg cells (111). Additionally, Pou2af1 expression is suppressed in stimulated FoxP3+ cells in comparison to FoxP3- cells and FoxP3 occupies the Pou2af1 promoter (131). On the other hand, co-culture experiments of T cells with Tregs resulted in an upregulation of Pou2af1 expression in suppressed compared to non-suppressed T cells (133). Metenou et al. reported a downregulation of Pou2af1 in Tregs from patients with chronic infection compared to healthy individuals (132). B cells co-cultured with Tfr cells downregulate Pou2af1 expression (134), which further suggests a relation between Tfr cells and Pou2af1 expression in a broader context. Ablation of BOB.1/OBF.1 in T cells using Pou2af1fl/fl x CD4-Cre mice resulted in an increase of Treg cells in our study (105). Additionally, these mice revealed an impaired Ag-specific immune response (105). These results imply that BOB.1/OBF.1 expression in both Tfh and Tfr cells might balance affinity maturation. Indeed, the exact role of Tfr cells for affinity maturation is not clear. Originally, Tfr cells were thought to control the production of Ag-specific Abs, while more recent studies suggest Tfr cells to restrain affinity maturation (15). However, the exact role of BOB.1/OBF.1 in Tregs and Tfr cells especially in terms of affinity maturation needs to be further elucidated. So far, the role of BOB.1/OBF.1 for Treg/Tfr cell development and function and whether its expression in these cell subsets promotes or restrains affinity maturation is not clear. RNA-seq analysis of BOB.1/OBF.1-sufficient compared to BOB.1/OBF.1-deficient CD4+ T cells of immunized mice revealed the histone methyltransferase Kmt2d (Lysine Methyltransferase 2d) among the top differentially expressed genes. We found increased Kmt2d expression in BOB.1/OBF.1-deficient CD4+ T cells compared to WT CD4+ T cells (105). Kmt2d deficiency was reported to compromise Treg cell development (135), possibly due to its requirement for the induction of FoxP3 expression (135). Mechanistically, Kmt2d catalyzed H3K4 methylation at distant enhancers via chromatin looping thereby contributing to the induction of FoxP3 expression (135).
Altogether, only little is known about the function of BOB.1/OBF.1 in Tregs so far. There is evidence that BOB.1/OBF.1 has a role in Tregs and/or Tfr cells, as several studies described altered Pou2af1 expression in these cells in different experimental settings, as outlined above. However, there is no definite knowledge about the role of BOB.1/OBF.1 in these cells at the current time. Some studies report an induction of Pou2af1 expression in Tregs or Tfr cells, while others describe a suppression. Further studies are required to get deeper insights into how BOB.1/OBF.1 expression is regulated, particularly in Tfr cells, as well as how Treg-specific BOB1.OBF.1 contributes to the balanced regulation of induction and suppression of an ongoing GC reaction.
The GC reaction relies on the interplay of multiple TFs regulating distinct phases of GC formation, as described in the preceding chapters. These TFs can be hijacked to drive tumorigenesis of GC-derived lymphomas. The majority of non-Hodgkin lymphomas (NHLs) derive from GC B cells including diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL) and Burkitt lymphoma (BL) (136, 137). The exact signaling pathways and mechanisms driving B cell lymphomagenesis are thoroughly reviewed elsewhere (136–140). In this chapter, we highlight the contribution of key TFs of the GC reaction as well as of the transcriptional co-activator BOB.1/OBF.1 to malignant transformation.
The gene encoding the TF MYC was the first to be linked to GC B cell lymphomagenesis even before its role in GC physiology was resolved (141, 142). MYC is expressed during the early initiation phase of the GC and subsequently repressed by BCL-6. However, it becomes re-expressed in a subset of light zone B cells later on (56, 57). The ectopic and constitutive expression of MYC is a frequent feature of GC-derived lymphomas. Chromosomal translocations of MYC are found in all BLs and in a proportion of DLBCLs (143, 144). In most cases, the MYC gene translocates into the Ig heavy chain locus and is brought under the transcriptional control of the Ig enhancer elements resulting in its transcriptional deregulation (141–143). Moreover, it is thought that translocations remove BCL-6 binding sites on the MYC promoter (56). Consequently, repression of MYC by BCL-6 is prevented, leading to ectopic MYC expression. A further link between MYC and cancer is that functions of the TF include controlling proliferation, cell growth, apoptosis and DNA replication (136, 145). Aberrant MYC expression thereby drives tumorigenesis.
As BCL-6 is a key TF for GC reaction, its dysregulation is also involved in the pathogenesis of GC-derived lymphomas. Aberrant BCL-6 expression is triggered by different mechanisms. Translocations or mutations of BCL-6 are found in DLBCL and FL (146). The most frequent cause of altered BCL-6 expression is translocation. These translocations replace the regulatory region of BCL-6 with promoters from Ig and non-Ig genes, preventing downregulation of BCL-6 (147–149). In addition, GC-derived lymphomas display various somatic mutations in the BCL-6 regulatory region that occur separately from translocations, avoiding the auto-regulatory mechanisms of BCL-6 to repress its own expression or block binding of suppressive IRF4 (150–152). BCL-6 dysregulation is also caused by indirect mechanisms including mutations of MEF2B. These mutations are commonly found in DLBCL and FL. MEF2B transactivates BCL-6. Thus, MEF2B mutations contribute to lymphomagenesis by deregulating BCL-6 expression (26, 153).
FOXO1 is a well-known tumor suppressor, but recent studies also reported tumor-promoting functions of the TF for different tumor entities (154–157). In GCs, FOXO1 instructs the dark zone program (38, 39). Recurrent somatic mutations of FOXO1 are common in B-NHL of GC origin including GCB-DLBCL, FL and BL (157–161), but their pathogenetic significance might differ in these entities. In GCB-DLBCL, the FOXO1 mutations are associated with poor prognosis and they are more frequent in relapsed or primary refractory cases (159, 160, 162), whereas in BL, there is no correlation with outcome or progression of the disease (161). The mechanisms regulated by the FOXO1 mutations are also obscure. Importantly, both activating (repressing FOXO1 inactivating phosphorylation) and inactivating, (targeting DNA-binding domains) are common in GCB-DLBCL (163). Physiologically, PI3K/AKT phosphorylates FOXO1 leading to its nuclear export and transcriptional inactivation (162, 164). Consequently, it has been hypothesized that in BL and GCB-DLBCL, mutations in the N-terminal hotspot help FOXO1 to escape the AKT-mediated inactivation (157). Nevertheless, following studies in BL cell lines and original tumors demonstrated strong nucleal FOXO1 expression virtually in all cases independently on the mutational status (161, 165). Moreover, the postulation on high AKT activity in BL has been questioned (166). Overall, there is a lack of correlation between PI3K/AKT signaling status, FOXO1 mutations, and subcellular localization, suggesting that selection of FOXO1 mutations during lymphomagenesis cannot be explained solely by the effects of PI3K/AKT on FOXO1.
Although the mechanisms underlying the oncogenic effects of activating and inactivating FOXO1 mutations are still obscure, the essential role of FOXO1 in maintenance of BL is clearly documented (165). A recent study provides evidence that FOXO1 mutations encode for proteins that mimic signaling and transcriptional features of positively selected B cells (167). Consequently, expansion of FOXO1 mutant B cells is favoured (167).
Disrupted PRDM1 gene expression is frequently found in Activated B-cell-like (ABC) DLBCLs, which is a post-GC malignancy. However, it is supposed that the disease is caused by a blockade of the terminal differentiation at the plasmablast stage (136, 138). This blockade seems to be due to BLIMP-1 inactivation, caused by truncations, mutations or frameshift deletions of the PRDM1 gene (168–170). As PRDM1 is directly repressed by BCL-6, dysregulated BCL-6 expression can constitutively repress PRDM1 (34). The TF Spi-B also represses PRDM1 expression (171). Spi-B is highly upregulated in ABC-DLBCLs promoting PRDM1 inactivation (172, 173). Experiments using mouse models further suggest that development of ABC DLBCL is a consequence of both BLIMP-1 inactivation and constitutive NF-κB activation (174).
Dysregulated BOB.1/OBF.1 expression in GC-derived lymphomas including FL, BL, DLBCL and HL has been reported by multiple studies. High expression levels of BOB.1/OBF.1 have been found in GC-derived lymphomas (FL, BL, DLBCL), whereas its expression could not be detected in lymphomas that represent other stages of B cell development (175–177). A study by Wang et al. identified BOB.1/OBF.1 as essential for BL cell lines (178). Moreover, others described a requirement of BOB.1/OBF.1 for proliferation and survival of DLBCL cells (179, 180). Chapuy et al. identified a DLBCL-specific super-enhancer near the BOB.1/OBF.1 promoter controlling its expression (179). Inhibition of this super-enhancer impaired DLBCL proliferation (179). Later it was shown that BOB.1/OBF.1 seems to mediate its effects in DLBCL by interaction with Oct-2 (180). Thus, targeting the BOB.1/OBF.1-Oct-2 interaction could be an effective therapeutic strategy in DLBCL (180). Most recently, Song et al. revealed that the fast proliferation of GC-derived lymphoma cells is dependent on BOB.1/OBF.1 (68). Mechanistically, BOB.1/OBF.1 is repressing IRF4 thereby driving GC-derived lymphoma cell proliferation (68). Interestingly, lymphoma cells with reduced BOB.1/OBF.1 expression adopt features associated with favorable prognosis suggesting the transcriptional co-activator a valuable prognostic marker (68).
In contrast, BOB.1/OBF.1 expression is aberrantly silenced in classical Hodgkin lymphoma (cHL). Hodgkin and Reed-Sternberg cells (HRS) are a malignant component of cHL. These cells are characterized by the extinction of B cell program including Ig production (181). The HRS cells lack expression of B cell specific TFs BOB.1/OBF.1 and Oct-2 (182–185). Mechanistically, there is evidence that downregulation of B cell specific genes including BOB.1/OBF.1 in HRS cells is a result of promoter hypermethylation (186). Possibly, the silencing of BOB.1/OBF.1 combined with additional mutations might have a causative role in classical HL.
Besides aberrant expression, BOB.1/OBF.1 chromosomal rearrangements or mutations involved in lymphomagenesis have also been reported. Fusion of LAZ3/BCL-6 and BOB.1/OBF.1 genes by chromosomal translocation have been revealed (187–189). Additionally, a germline variation in the 3′-untranslated region of the POU2AF1 gene was shown to be associated with susceptibility to lymphoma (190).
In total, all of these studies underline the involvement of BOB.1/OBF.1 in lymphomagenesis, highlighting its diagnostic and therapeutic potential.
Besides lymphomagenesis, deregulation of the transcriptional program regulating the GC reaction can also lead to imbalanced immune responses driving autoimmunity. In particular, deregulated expression of BOB.1/OBF.1 was shown to contribute to the pathogenesis of several autoimmune diseases (191–194). As its contribution to autoimmunity is thoroughly reviewed elsewhere (195), we rather focused on its role in driving lymphomagenesis in this review.
Understanding of the GC biology has made considerable progress in recent years. An effective GC reaction needs interaction of several cell types including reticular cells, FDCs, DCs, GC B, Tfh and Tfr cells. This complex network is coordinated by multiple distinct TFs, which became increasingly clear over the past years. However, it is still not completely resolved how exactly these TFs perform their function. Also, further regulatory factors including co-activators, co-repressors, miRNAs as well as post-transcriptional or epigenetic regulations need to be considered. The requirement of the transcriptional co-activator BOB.1/OBF.1 for the GC reaction not only in B, but also in T cells became more and more evident in recent years. Nevertheless, further studies are required to understand its role especially in Tfr cells. Uncovering mechanisms that have not yet been fully understood, will also help to improve our understanding of dysregulations driving GC-derived lymphomagenesis and therefore help to define new therapeutic approaches to treat GC-derived B malignancies.
AB wrote the manuscript and prepared visualizations. AU and CB reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15(3):137–48. doi: 10.1038/nri3804
2. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2022) 40(1):413–42. doi: 10.1146/annurev-immunol-120419-022408
3. Chang JE, Turley SJ. Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol (2015) 36(1):30–9. doi: 10.1016/j.it.2014.11.003
4. Turner JS, Benet ZL, Grigorova IL. Signals 1, 2 and b cell fate or: Where, when and for how long? Immunol Rev (2020) 296(1):9–23. doi: 10.1111/imr.12865
5. Phan TG, Gray EE, Cyster JG. The microanatomy of B cell activation. Curr Opin Immunol (2009) 21(3):258–65. doi: 10.1016/j.coi.2009.05.006
6. Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol (2005) 5(8):606–16. doi: 10.1038/nri1669
7. Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol (2009) 9(1):15–27. doi: 10.1038/nri2454
8. McShane AN, Malinova D. The ins and outs of antigen uptake in B cells. Front Immunol (2022) 13:892169. doi: 10.3389/fimmu.2022.892169
9. Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Förster R, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. (2002) 416(6876):94–9. doi: 10.1038/416094a
10. Hardtke S, Ohl L, Förster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood. (2005) 106(6):1924–31. doi: 10.1182/blood-2004-11-4494
11. Recaldin T, Fear DJ. Transcription factors regulating B cell fate in the germinal centre. Clin Exp Immunol (2016) 183(1):65–75. doi: 10.1111/cei.12702
12. Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and nonlymphoid tissues. J Immunol (Baltimore Md 1950). (2011) 187(6):3026–32. doi: 10.4049/jimmunol.1101262
13. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T Follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity. (2015) 42(4):704–18. doi: 10.1016/j.immuni.2015.03.002
14. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. (2011) 34(6):947–60. doi: 10.1016/j.immuni.2011.03.024
15. Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the germinal center response. Front Immunol (2018) 9:2469. doi: 10.3389/fimmu.2018.02469
16. Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int Immunol (2010) 22(6):413–9. doi: 10.1093/intimm/dxq047
17. Green JA, Cyster JG. S1PR2 links germinal center confinement and growth regulation. Immunol Rev (2012) 247(1):36–51. doi: 10.1111/j.1600-065X.2012.01114.x
18. Moriyama S, Takahashi N, Green JA, Hori S, Kubo M, Cyster JG, et al. Sphingosine-1-phosphate receptor 2 is critical for follicular helper T cell retention in germinal centers. J Exp Med (2014) 211(7):1297–305. doi: 10.1084/jem.20131666
19. Elsner RA, Shlomchik MJ. Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity. (2020) 53(6):1136–50. doi: 10.1016/j.immuni.2020.11.006
20. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zúñiga E, et al. Extrafollicular antibody responses. Immunol Rev (2003) 194:8–18. doi: 10.1034/j.1600-065X.2003.00058.x
21. Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol (2009) 9(9):618–29. doi: 10.1038/nri2588
22. Laidlaw BJ, Cyster JG. Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol (2021) 21(4):209–20. doi: 10.1038/s41577-020-00446-2
23. Song S, Matthias PD. The transcriptional regulation of germinal center formation. Front Immunol (2018) 9:2026–. doi: 10.3389/fimmu.2018.02026
24. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. (2013) 38(5):918–29. doi: 10.1016/j.immuni.2013.04.009
25. Lee CH, Melchers M, Wang H, Torrey TA, Slota R, Qi CF, et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med (2006) 203(1):63–72. doi: 10.1084/jem.20051450
26. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol (2013) 14(10):1084–92. doi: 10.1038/ni.2688
27. Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center b cells. Nat Immunol (2005) 6(10):1054–60. doi: 10.1038/ni1245
28. Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre b cells. Nature. (2004) 432(7017):635–9. doi: 10.1038/nature03147
29. Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol (2007) 8(7):705–14. doi: 10.1038/ni1478
30. Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood cells molecules diseases. (2008) 41(1):95–9. doi: 10.1016/j.bcmd.2008.02.003
31. Basso K, Saito M, Sumazin P, Margolin AA, Wang K, Lim W-K, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. (2010) 115(5):975–84. doi: 10.1182/blood-2009-06-227017
32. Huang C, Gonzalez David G, Cote Christine M, Jiang Y, Hatzi K, Teater M, et al. The BCL6 RD2 domain governs commitment of activated B cells to form germinal centers. Cell Rep (2014) 8(5):1497–508. doi: 10.1016/j.celrep.2014.07.059
33. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. (2011) 34(6):961–72. doi: 10.1016/j.immuni.2011.03.025
34. Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by bcl-6 inhibits plasmacytic differentiation. J Immunol (2004) 173(2):1158–65. doi: 10.4049/jimmunol.173.2.1158
35. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. (2000) 102(5):553–63. doi: 10.1016/S0092-8674(00)00078-7
36. Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol (2003) 4(6):586–93. doi: 10.1038/ni923
37. Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med (2003) 198(9):1427–37. doi: 10.1084/jem.20030802
38. Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, et al. The FOXO1 transcription factor instructs the germinal center dark zone program. Immunity. (2015) 43(6):1064–74. doi: 10.1016/j.immuni.2015.10.015
39. Sander S, Chu Van T, Yasuda T, Franklin A, Graf R, Calado Dinis P, et al. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity. (2015) 43(6):1075–86. doi: 10.1016/j.immuni.2015.10.021
40. Brown AK, Webb AE. Regulation of FOXO factors in mammalian cells. Curr topics Dev Biol (2018) 127:165–92. doi: 10.1016/bs.ctdb.2017.10.006
41. Graves DT, Milovanova TN. Mucosal immunity and the FOXO1 transcription factors. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02530
42. Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, et al. Mitogen-activated protein kinases, erk and p38, phosphorylate and regulate Foxo1. Cell signalling. (2007) 19(3):519–27. doi: 10.1016/j.cellsig.2006.08.015
43. Inoue T, Shinnakasu R, Ise W, Kawai C, Egawa T, Kurosaki T. The transcription factor Foxo1 controls germinal center b cell proliferation in response to T cell help. J Exp Med (2017) 214(4):1181–98. doi: 10.1084/jem.20161263
44. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. (2006) 25(2):225–36. doi: 10.1016/j.immuni.2006.07.009
45. Hauser J, Grundström C, Kumar R, Grundström T. Regulated localization of an AID complex with E2A, PAX5 and IRF4 at the igh locus. Mol Immunol (2016) 80:78–90. doi: 10.1016/j.molimm.2016.10.014
46. Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both b cells and T cells. Nat Immunol (2011) 12(6):536–43. doi: 10.1038/ni.2037
47. Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim H-P, Oh J, Tunyaplin C, et al. Analysis of interleukin-21-Induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. (2009) 31(6):941–52. doi: 10.1016/j.immuni.2009.10.008
48. Turner CA Jr., Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell. (1994) 77(2):297–306. doi: 10.1016/0092-8674(94)90321-2
49. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. (2003) 19(4):607–20. doi: 10.1016/S1074-7613(03)00267-X
50. Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, et al. Multifunctional role of the transcription factor blimp-1 in coordinating plasma cell differentiation. Nat Immunol (2016) 17(3):331–43. doi: 10.1038/ni.3349
51. Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of pax-5 is required for differentiation of b cells to immunoglobulin m-secreting plasma cells. Mol Cell Biol (2002) 22(13):4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002
52. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. (2001) 412(6844):300–7. doi: 10.1038/35085509
53. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol (2006) 7(7):773–82. doi: 10.1038/ni1357
54. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. (2004) 21(1):81–93. doi: 10.1016/j.immuni.2004.06.010
55. Chiu YK, Lin IY, Su ST, Wang KH, Yang SY, Tsai DY, et al. Transcription factor ABF-1 suppresses plasma cell differentiation but facilitates memory B cell formation. J Immunol (Baltimore Md 1950). (2014) 193(5):2207–17. doi: 10.4049/jimmunol.1400411
56. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol (2012) 13(11):1083–91. doi: 10.1038/ni.2428
57. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, et al. The cell-cycle regulator c-myc is essential for the formation and maintenance of germinal centers. Nat Immunol (2012) 13(11):1092–100. doi: 10.1038/ni.2418
58. Brunner C, Wirth T. BOB.1/OBF.1 - A critical regulator of B cell function. Curr Immunol Rev (2006) 2(1):3–12. doi: 10.2174/157339506775471901
59. Falkner FG, Zachau HG. Correct transcription of an immunoglobulin kappa gene requires an upstream fragment containing conserved sequence elements. Nature. (1984) 310(5972):71–4. doi: 10.1038/310071a0
60. Parslow TG, Blair DL, Murphy WJ, Granner DK. Structure of the 5' ends of immunoglobulin genes: A novel conserved sequence. Proc Natl Acad Sci U S A. (1984) 81(9):2650–4. doi: 10.1073/pnas.81.9.2650
61. Mueller K, Quandt J, Marienfeld RB, Weihrich P, Fiedler K, Claussnitzer M, et al. Octamer-dependent transcription in T cells is mediated by NFAT and NF-κB. Nucleic Acids Res (2013) 41(4):2138–54. doi: 10.1093/nar/gks1349
62. Gstaiger M, Georgiev O, van Leeuwen H, van der Vliet P, Schaffner W. The b cell coactivator Bob1 shows DNA sequence-dependent complex formation with Oct-1/Oct-2 factors, leading to differential promoter activation. EMBO J (1996) 15(11):2781–90. doi: 10.1002/j.1460-2075.1996.tb00638.x
63. Luo Y, Roeder RG. Cloning, functional characterization, and mechanism of action of the b-cell-specific transcriptional coactivator OCA-b. Mol Cell Biol (1995) 15(8):4115–24. doi: 10.1128/MCB.15.8.4115
64. Cepek KL, Chasman DI, Sharp PA. Sequence-specific DNA binding of the B-cell-specific coactivator OCA-b. Genes Dev (1996) 10(16):2079–88. doi: 10.1101/gad.10.16.2079
65. Chasman D, Cepek K, Sharp PA, Pabo CO. Crystal structure of an OCA-B peptide bound to an Oct-1 POU domain/octamer DNA complex: Specific recognition of a protein-DNA interface. Genes Dev (1999) 13(20):2650–7. doi: 10.1101/gad.13.20.2650
66. Brunner C, Laumen H, Nielsen PJ, Kraut N, Wirth T. Expression of the aldehyde dehydrogenase 2-like gene is controlled by BOB.1/OBF.1 B lymphocytes. J Biol Chem (2003) 278(46):45231–9. doi: 10.1074/jbc.M302539200
67. Brunner C, Sindrilaru A, Girkontaite I, Fischer KD, Sunderkötter C, Wirth T. BOB.1/OBF.1 controls the balance of TH1 and TH2 immune responses. EMBO J (2007) 26(13):3191–202. doi: 10.1038/sj.emboj.7601742
68. Song S, Cao C, Choukrallah MA, Tang F, Christofori G, Kohler H, et al. OBF1 and Oct factors control the germinal center transcriptional program. Blood. (2021) 137(21):2920–34. doi: 10.1182/blood.2020010175
69. Stauss D, Brunner C, Berberich-Siebelt F, Hopken UE, Lipp M, Muller G. The transcriptional coactivator Bob1 promotes the development of follicular T helper cells via Bcl6. EMBO J (2016) 35(8):881–98. doi: 10.15252/embj.201591459
70. Corcoran LM, Hasbold J, Dietrich W, Hawkins E, Kallies A, Nutt SL, et al. Differential requirement for OBF-1 during antibody-secreting cell differentiation. J Exp Med (2005) 201(9):1385–96. doi: 10.1084/jem.20042325
71. Hess J, Nielsen PJ, Fischer KD, Bujard H, Wirth T. The B lymphocyte-specific coactivator BOB.1/OBF.1 is required at multiple stages of b-cell development. Mol Cell Biol (2001) 21(5):1531–9. doi: 10.1128/MCB.21.5.1531-1539.2001
72. Schubart DB, Rolink A, Schubart K, Matthias P. Cutting edge: Lack of peripheral B cells and severe agammaglobulinemia in mice simultaneously lacking bruton’s tyrosine kinase and the b cell-specific transcriptional coactivator OBF-1. J Immunol (2000) 164(1):18–22. doi: 10.4049/jimmunol.164.1.18
73. Nielsen PJ, Georgiev O, Lorenz B, Schaffner W. B lymphocytes are impaired in mice lacking the transcriptional co-activator Bob1/OCA-B/OBF1. Eur J Immunol (1996) 26(12):3214–8. doi: 10.1002/eji.1830261255
74. Samardzic T, Marinkovic D, Nielsen PJ, Nitschke L, Wirth T. BOB.1/OBF.1 deficiency affects marginal-zone b-cell compartment. Mol Cell Biol (2002) 22(23):8320–31. doi: 10.1128/MCB.22.23.8320-8331.2002
75. Schubart DB, Rolink A, Kosco-Vilbois MH, Botteri F, Matthias P. B-cell-specific coactivator OBF-1/OCA-B/Bob1 required for immune response and germinal centre formation. Nature. (1996) 383:538. doi: 10.1038/383538a0
76. Betzler AC, Fiedler K, Hoffmann TK, Fehling HJ, Wirth T, Brunner C. BOB.1/OBF.1 is required during b-cell ontogeny for B-cell differentiation and germinal center function. Eur J Immunol (2022) 52(3):404–17. doi: 10.1002/eji.202149333
77. Wolf I, Pevzner V, Kaiser E, Bernhardt G, Claudio E, Siebenlist U, et al. Downstream activation of a TATA-less promoter by Oct-2, Bob1, and NF-kappaB directs expression of the homing receptor BLR1 to mature B cells. J Biol Chem (1998) 273(44):28831–6. doi: 10.1074/jbc.273.44.28831
78. Betzler AC, Fiedler K, Kokai E, Wirth T, Hoffmann TK, Brunner C. Impaired peyer's patch development in BOB.1/OBF.1-deficient mice. Eur J Immunol (2021) 51(7):1860–3. doi: 10.1002/eji.202048578
79. Su GH, Chen HM, Muthusamy N, Garrett-Sinha LA, Baunoch D, Tenen DG, et al. Defective B cell receptor-mediated responses in mice lacking the ets protein, spi-b. EMBO J (1997) 16(23):7118–29. doi: 10.1093/emboj/16.23.7118
80. Bartholdy B, Du Roure C, Bordon A, Emslie D, Corcoran LM, Matthias P. The ets factor spi-b is a direct critical target of the coactivator OBF-1. Proc Natl Acad Sci (2006) 103(31):11665–70. doi: 10.1073/pnas.0509430103
81. Chu CS, Hellmuth JC, Singh R, Ying HY, Skrabanek L, Teater MR, et al. Unique immune cell coactivators specify locus control region function and cell stage. Mol Cell (2020) 80(5):845–61.e10. doi: 10.1016/j.molcel.2020.10.036
82. Choi Youn S, Kageyama R, Eto D, Escobar Tania C, Johnston Robert J, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. (2011) 34(6):932–46. doi: 10.1016/j.immuni.2011.03.023
83. DiToro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science (2018) 361(6407):eaao2933. doi: 10.1126/science.aao2933
84. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. (2008) 29(1):138–49. doi: 10.1016/j.immuni.2008.05.009
85. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol (Baltimore Md 1950). (2013) 190(7):3049–53. doi: 10.4049/jimmunol.1203032
86. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
87. Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, et al. Type I IFN induces binding of STAT1 to Bcl6: Divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol (Baltimore Md 1950). (2014) 192(5):2156–66. doi: 10.4049/jimmunol.1300675
88. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate TFH differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol (2015) 16(9):980–90. doi: 10.1038/ni.3226
89. Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, et al. The transcription factor TCF-1 initiates the differentiation of TFH cells during acute viral infection. Nat Immunol (2015) 16(9):991–9. doi: 10.1038/ni.3229
90. Ribeiro F, Perucha E, Graca L. T Follicular cells: The regulators of germinal center homeostasis. Immunol Lett (2022) 244:1–11. doi: 10.1016/j.imlet.2022.02.008
91. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor bcl-6 directs T follicular helper cell lineage commitment. Immunity. (2009) 31(3):457–68. doi: 10.1016/j.immuni.2009.07.002
92. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature. (2014) 507(7493):513–8. doi: 10.1038/nature12910
93. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating krüppel-like factor 2. J Exp Med (2015) 212(2):217–33. doi: 10.1084/jem.20141432
94. Hiramatsu Y, Suto A, Kashiwakuma D, Kanari H, S-i K, Ikeda K, et al. C-maf activates the promoter and enhancer of the IL-21 gene, and TGF-β inhibits c-maf-induced IL-21 production in CD4+ T cells. J leukocyte Biol (2010) 87(4):703–12. doi: 10.1189/jlb.0909639
95. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal germinal center responses require a multistage T cell: B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity. (2010) 32(2):253–65. doi: 10.1016/j.immuni.2010.01.010
96. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
97. Crotty S. T Follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
98. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T Follicular helper cell dynamics in germinal centers. Science. (2013) 341(6146):673–7. doi: 10.1126/science.1241680
99. Yamashita K, Kawata K, Matsumiya H, Kamekura R, Jitsukawa S, Nagaya T, et al. Bob1 limits cellular frequency of T-follicular helper cells. Eur J Immunol (2016) 46(6):1361–70. doi: 10.1002/eji.201545499
100. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T Follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol (2004) 173(1):68–78. doi: 10.4049/jimmunol.173.1.68
101. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke Mark A, Arlehamn Cecilia L, et al. Human circulating PD-1+CXCR3-CXCR5+ memory tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. (2013) 39(4):758–69. doi: 10.1016/j.immuni.2013.08.031
102. Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D'Costa K, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med (2012) 209(11):2049–64. doi: 10.1084/jem.20111504
103. Kury P, Staniek J, Wegehaupt O, Janowska I, Eckenweiler M, Korinthenberg R, et al. Agammaglobulinemia with normal B-cell numbers in a patient lacking Bob1. J Allergy Clin Immunol (2021) 147(5):1977–80. doi: 10.1016/j.jaci.2021.01.027
104. Lombard-Vadnais F, Lacombe J, Chabot-Roy G, Ferron M, Lesage S. OCA-b does not act as a transcriptional coactivator in T cells. Immunol Cell Biol (2022) 100(5):338–51. doi: 10.1111/imcb.12543
105. Betzler AC, Ezić J, Abou Kors T, Hoffmann TK, Wirth T, Brunner C. T Cell specific BOB.1/OBF.1 expression promotes germinal center response and T helper cell differentiation. Front Immunol (2022) 13:889564. doi: 10.3389/fimmu.2022.889564
106. Chevrier S, Kratina T, Emslie D, Karnowski A, Corcoran LM. Germinal center-independent, IgM-mediated autoimmunity in sanroque mice lacking Obf1. Immunol Cell Biol (2014) 92(1):12–9. doi: 10.1038/icb.2013.71
107. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi: 10.1038/nm.2426
108. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
109. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (Baltimore Md 1950). (2011) 187(9):4553–60. doi: 10.4049/jimmunol.1101328
110. Amé-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: Role in the survival of malignant B cells. Leukemia. (2012) 26(5):1053–63. doi: 10.1038/leu.2011.301
111. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, et al. A distinct subpopulation of CD25(-) T-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci U S A (2017) 114(31):E6400–e9. doi: 10.1073/pnas.1705551114
112. Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romão VC, et al. Human blood t(fr) cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol (2017) 2(14). doi: 10.1126/sciimmunol.aan1487
113. Xie MM, Dent AL. Unexpected help: Follicular regulatory T cells in the germinal center. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01536
114. Jacobsen JT, Hu W, BRC T, Solem S, Galante A, Lin Z, et al. Expression of Foxp3 by T follicular helper cells in end-stage germinal centers. Science (2021) 373(6552). doi: 10.1126/science.abe5146
115. Hao H, Nakayamada S, Tanaka Y. Differentiation, functions, and roles of T follicular regulatory cells in autoimmune diseases. Inflammation Regeneration. (2021) 41(1):14. doi: 10.1186/s41232-021-00164-9
116. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. (2014) 41(6):1026–39. doi: 10.1016/j.immuni.2014.12.005
117. Fahlquist Hagert C, Degn SE. T Follicular regulatory cells: Guardians of the germinal centre? Scandinavian J Immunol (2020) 92(4):e12942. doi: 10.1111/sji.12942
118. Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, et al. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun (2016) 7:10579. doi: 10.1038/ncomms10579
119. Ritvo PG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, et al. T(fr) cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of t(fh) cells. Sci Immunol (2017) 2(15). doi: 10.1126/sciimmunol.aan0368
120. Zhang R, Sage PT, Finn K, Huynh A, Blazar BR, Marangoni F, et al. B cells drive autoimmunity in mice with CD28-deficient regulatory T cells. J Immunol (Baltimore Md 1950). (2017) 199(12):3972–80. doi: 10.4049/jimmunol.1700409
121. Maceiras AR, Almeida SCP, Mariotti-Ferrandiz E, Chaara W, Jebbawi F, Six A, et al. T Follicular helper and T follicular regulatory cells have different TCR specificity. Nat Commun (2017) 8:15067. doi: 10.1038/ncomms15067
122. Yang G, Yang X, Zhang J, Li G, Zheng D, Peng A, et al. Transcriptional repressor Blimp1 regulates follicular regulatory T-cell homeostasis and function. Immunology. (2018) 153(1):105–17. doi: 10.1111/imm.12815
123. Hou S, Clement RL, Diallo A, Blazar BR, Rudensky AY, Sharpe AH, et al. FoxP3 and Ezh2 regulate tfr cell suppressive function and transcriptional program. J Exp Med (2019) 216(3):605–20. doi: 10.1084/jem.20181134
124. Vaeth M, Müller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, et al. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J Exp Med (2014) 211(3):545–61. doi: 10.1084/jem.20130604
125. Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich-Siebelt F, et al. Store-operated Ca2+ entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity. (2016) 44(6):1350–64. doi: 10.1016/j.immuni.2016.04.013
126. Leavenworth JW, Verbinnen B, Yin J, Huang H, Cantor H. A p85α-osteopontin axis couples the receptor ICOS to sustained bcl-6 expression by follicular helper and regulatory T cells. Nat Immunol (2015) 16(1):96–106. doi: 10.1038/ni.3050
127. Xu L, Huang Q, Wang H, Hao Y, Bai Q, Hu J, et al. The kinase mTORC1 promotes the generation and suppressive function of follicular regulatory T cells. Immunity. (2017) 47(3):538–51.e5. doi: 10.1016/j.immuni.2017.08.011
128. Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu L-F, et al. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol (2014) 15(8):767–76. doi: 10.1038/ni.2928
129. Wu X, Wang Y, Huang R, Gai Q, Liu H, Shi M, et al. SOSTDC1-producing follicular helper T cells promote regulatory follicular T cell differentiation. Science. (2020) 369(6506):984–8. doi: 10.1126/science.aba6652
130. Wheaton JD, Yeh CH, Ciofani M. Cutting edge: c-maf is required for regulatory T cells to adopt RORγt(+) and follicular phenotypes. J Immunol (Baltimore Md 1950). (2017) 199(12):3931–6. doi: 10.4049/jimmunol.1701134
131. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. (2007) 445(7130):931–5. doi: 10.1038/nature05478
132. Metenou S, Coulibaly YI, Sturdevant D, Dolo H, Diallo AA, Soumaoro L, et al. Highly heterogeneous, activated, and short-lived regulatory T cells during chronic filarial infection. Eur J Immunol (2014) 44(7):2036–47. doi: 10.1002/eji.201444452
133. Sukiennicki TL, Fowell DJ. Distinct molecular program imposed on CD4+ T cell targets by CD4+CD25+ regulatory T cells. J Immunol (Baltimore Md 1950). (2006) 177(10):6952–61. doi: 10.4049/jimmunol.177.10.6952
134. Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W, et al. Suppression by T(FR) cells leads to durable and selective inhibition of B cell effector function. Nat Immunol (2016) 17(12):1436–46. doi: 10.1038/ni.3578
135. Placek K, Hu G, Cui K, Zhang D, Ding Y, Lee JE, et al. MLL4 prepares the enhancer landscape for Foxp3 induction via chromatin looping. Nat Immunol (2017) 18(9):1035–45. doi: 10.1038/ni.3812
136. Basso K, Dalla-Favera R. Germinal centres and b cell lymphomagenesis. Nat Rev Immunol (2015) 15(3):172–84. doi: 10.1038/nri3814
137. Mlynarczyk C, Fontán L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev (2019) 288(1):214–39. doi: 10.1111/imr.12755
138. Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol (2012) 30:565–610. doi: 10.1146/annurev-immunol-020711-075027
139. Küppers R. Mechanisms of b-cell lymphoma pathogenesis. Nat Rev Cancer. (2005) 5(4):251–62. doi: 10.1038/nrc1589
140. Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. (2010) 141(1):27–38. doi: 10.1016/j.cell.2010.03.016
141. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in burkitt lymphoma cells. Proc Natl Acad Sci U S A (1982) 79(24):7824–7. doi: 10.1073/pnas.79.24.7824
142. Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A (1982) 79(24):7837–41. doi: 10.1073/pnas.79.24.7837
143. Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in burkitt lymphoma. Cold Spring Harbor Perspect Med (2014) 4(2). doi: 10.1101/cshperspect.a014282
144. Li S, Lin P, Young KH, Kanagal-Shamanna R, Yin CC, Medeiros LJ. MYC/BCL2 double-hit high-grade B-cell lymphoma. Adv anatomic pathology. (2013) 20(5):315–26. doi: 10.1097/PAP.0b013e3182a289f2
145. Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-myc. Nature. (2007) 448(7152):445–51. doi: 10.1038/nature05953
146. Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol (2010) 105:193–210. doi: 10.1016/S0065-2776(10)05007-8
147. Ye BH, Chaganti S, Chang CC, Niu H, Corradini P, Chaganti RS, et al. Chromosomal translocations cause deregulated BCL6 expression by promoter substitution in B cell lymphoma. EMBO J (1995) 14(24):6209–17. doi: 10.1002/j.1460-2075.1995.tb00311.x
148. Chen W, Iida S, Louie DC, Dalla-Favera R, Chaganti RS. Heterologous promoters fused to BCL6 by chromosomal translocations affecting band 3q27 cause its deregulated expression during B-cell differentiation. Blood. (1998) 91(2):603–7. doi: 10.1182/blood.V91.2.603
149. Ichinohasama R, Miura I, Funato T, Sato I, Suzuki C, Saito Y, et al. A recurrent nonrandom translocation (3;7)(q27;p12) associated with BCL-6 gene rearrangement in B-cell diffuse large cell lymphoma. Cancer Genet cytogenetics. (1998) 104(1):19–27. doi: 10.1016/S0165-4608(97)00412-3
150. Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. (2003) 101(8):2914–23. doi: 10.1182/blood-2002-11-3387
151. Wang X, Li Z, Naganuma A, Ye BH. Negative autoregulation of BCL-6 is bypassed by genetic alterations in diffuse large B cell lymphomas. Proc Natl Acad Sci U S A (2002) 99(23):15018–23. doi: 10.1073/pnas.232581199
152. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell (2007) 12(3):280–92. doi: 10.1016/j.ccr.2007.08.011
153. Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell (2018) 34(3):453–65.e9. doi: 10.1016/j.ccell.2018.08.006
154. Coomans de Brachène A, Demoulin J-B. FOXO transcription factors in cancer development and therapy. Cell Mol Life Sci (2016) 73(6):1159–72. doi: 10.1007/s00018-015-2112-y
155. Hornsveld M, Smits LMM, Meerlo M, van Amersfoort M, Groot Koerkamp MJA, van Leenen D, et al. FOXO transcription factors both suppress and support breast cancer progression. Cancer Res (2018) 78(9):2356–69. doi: 10.1158/0008-5472.CAN-17-2511
156. Sykes Stephen M, Lane Steven W, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. (2011) 146(5):697–708. doi: 10.1016/j.cell.2011.07.032
157. Kabrani E, Chu VT, Tasouri E, Sommermann T, Baßler K, Ulas T, et al. Nuclear FOXO1 promotes lymphomagenesis in germinal center B cells. Blood. (2018) 132(25):2670–83. doi: 10.1182/blood-2018-06-856203
158. Grande BM, Gerhard DS, Jiang A, Griner NB, Abramson JS, Alexander TB, et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic burkitt lymphoma. Blood. (2019) 133(12):1313–24. doi: 10.1182/blood-2018-09-871418
159. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep (2014) 6(1):130–40. doi: 10.1016/j.celrep.2013.12.027
160. Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, et al. Genetic landscapes of relapsed and refractory diffuse Large B-cell lymphomas. Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(9):2290–300. doi: 10.1158/1078-0432.CCR-15-2123
161. Zhou P, Blain AE, Newman AM, Zaka M, Chagaluka G, Adlar FR, et al. Sporadic and endemic burkitt lymphoma have frequent FOXO1 mutations but distinct hotspots in the AKT recognition motif. Blood advances. (2019) 3(14):2118–27. doi: 10.1182/bloodadvances.2018029546
162. Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJ, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. (2013) 121(18):3666–74. doi: 10.1182/blood-2013-01-479865
163. Sablon A, Bollaert E, Pirson C, Velghe AI, Demoulin J-B. FOXO1 forkhead domain mutants in B-cell lymphoma lack transcriptional activity. Sci Rep (2022) 12(1):1309. doi: 10.1038/s41598-022-05334-4
164. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. (1999) 96(6):857–68. doi: 10.1016/S0092-8674(00)80595-4
165. Gehringer F, Weissinger SE, Swier LJ, Moller P, Wirth T, Ushmorov A. FOXO1 confers maintenance of the dark zone proliferation and survival program and can be pharmacologically targeted in burkitt lymphoma. Cancers (Basel). (2019) 11(10):1427. doi: 10.3390/cancers11101427
166. Gehringer F, Weissinger SE, Moller P, Wirth T, Ushmorov A. Physiological levels of the PTEN-PI3K-AKT axis activity are required for maintenance of burkitt lymphoma. Leukemia. (2020) 34(3):857–71. doi: 10.1038/s41375-019-0628-0
167. Roberto MP, Varano G, Vinas-Castells R, Holmes AB, Kumar R, Pasqualucci L, et al. Mutations in the transcription factor FOXO1 mimic positive selection signals to promote germinal center B cell expansion and lymphomagenesis. Immunity. (2021) 54(8):1807–24.e14. doi: 10.1016/j.immuni.2021.07.009
168. Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large b cell lymphoma. Cancer Cell (2010) 18(6):568–79. doi: 10.1016/j.ccr.2010.10.030
169. Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV, et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med (2006) 203(2):311–7. doi: 10.1084/jem.20052204
170. Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. (2006) 107(10):4090–100. doi: 10.1182/blood-2005-09-3778
171. Schmidlin H, Diehl SA, Nagasawa M, Scheeren FA, Schotte R, Uittenbogaart CH, et al. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood. (2008) 112(5):1804–12. doi: 10.1182/blood-2008-01-136440
172. Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W, Wright GW, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large b cell lymphoma. J Exp Med (2007) 204(3):633–43. doi: 10.1084/jem.20062041
173. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. (2008) 105(36):13520–5. doi: 10.1073/pnas.0804295105
174. Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, et al. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell (2010) 18(6):580–9. doi: 10.1016/j.ccr.2010.11.024
175. Greiner A, Muller KB, Hess J, Pfeffer K, Muller-Hermelink HK, Wirth T. Up-regulation of BOB.1/OBF.1 expression in normal germinal center B cells and germinal center-derived lymphomas. Am J Pathol (2000) 156(2):501–7. doi: 10.1016/S0002-9440(10)64754-2
176. Loddenkemper C, Anagnostopoulos I, Hummel M, Jöhrens-Leder K, Foss H-D, Jundt F, et al. Differential eµ enhancer activity and expression of BOB.1/OBF.1, Oct2, PU.1, and immunoglobulin in reactive B-cell populations, B-cell non-Hodgkin lymphomas, and Hodgkin lymphomas. J Pathol (2004) 202(1):60–9. doi: 10.1002/path.1485
177. Sáez A-I, Artiga M-J, Sánchez-Beato M, Sánchez-Verde L, García J-F, Camacho F-I, et al. Analysis of octamer-binding transcription factors Oct2 and Oct1 and their coactivator BOB.1/OBF.1 in lymphomas. Modern Pathol (2002) 15(3):211–20. doi: 10.1038/modpathol.3880518
178. Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, et al. Identification and characterization of essential genes in the human genome. Science. (2015) 350(6264):1096–101. doi: 10.1126/science.aac7041
179. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell (2013) 24(6):777–90. doi: 10.1016/j.ccr.2013.11.003
180. Hodson DJ, Shaffer AL, Xiao W, Wright GW, Schmitz R, Phelan JD, et al. Regulation of normal b-cell differentiation and malignant B-cell survival by OCT2. Proc Natl Acad Sci (2016) 113(14):E2039–E46. doi: 10.1073/pnas.1600557113
181. Küppers R, Klein U, Schwering I, Distler V, Bräuninger A, Cattoretti G, et al. Identification of Hodgkin and reed-sternberg cell-specific genes by gene expression profiling. J Clin Invest. (2003) 111(4):529–37. doi: 10.1172/JCI200316624
182. Re D, Müschen M, Ahmadi T, Wickenhauser C, Staratschek-Jox A, Holtick U, et al. Oct-2 and bob-1 deficiency in Hodgkin and reed sternberg cells. Cancer Res (2001) 61(5):2080–4.
183. Stein H, Marafioti T, Foss H-D, Laumen H, Hummel M, Anagnostopoulos I, et al. Down-regulation of BOB.1/OBF.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant Hodgkin disease correlates with immunoglobulin transcription. Blood (2001) 97(2):496–501. doi: 10.1182/blood.v97.2.496
184. Theil J, Laumen H, Marafioti T, Hummel M, Lenz G, Wirth T, et al. Defective octamer-dependent transcription is responsible for silenced immunoglobulin transcription in reed-sternberg cells. Blood. (2001) 97(10):3191–6. doi: 10.1182/blood.V97.10.3191
185. Hertel CB, Zhou XG, Hamilton-Dutoit SJ, Junker S. Loss of b cell identity correlates with loss of B cell-specific transcription factors in Hodgkin/Reed-sternberg cells of classical Hodgkin lymphoma. Oncogene. (2002) 21(32):4908–20. doi: 10.1038/sj.onc.1205629
186. Ushmorov A, Leithäuser F, Sakk O, Weinhäsel A, Popov SW, Möller P, et al. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood. (2006) 107(6):2493–500. doi: 10.1182/blood-2005-09-3765
187. Galiègue Zouitina S, Quief S, Hildebrand MP, Denis C, Lecocq G, Collyn-d'Hooghe M, et al. The b cell transcriptional coactivator BOB1/OBF1 gene fuses to the LAZ3/BCL6 gene by t(3;11)(q27;q23.1) chromosomal translocation in a B cell leukemia line (Karpas 231). Leukemia (1996) 10(4):579–87.
188. Galiègue-Zouitina S, Quief S, Hildebrand MP, Denis C, Lecocq G, Collyn-d'Hooghe M, et al. Fusion of the LAZ3/BCL6 and BOB1/OBF1 genes by t(3; 11) (q27; q23) chromosomal translocation. C R Acad Sci III (1995) 318(11):1125–31.
189. Yuille MA, Galiegue-Zouitina S, Hiorns LR, Jadayel D, De Schouwer PJ, Catovsky D, et al. Heterogeneity of breakpoints at the transcriptional co-activator gene, BOB-1, in lymphoproliferative disease. Leukemia. (1996) 10(9):1492–6.
190. Zhai K, Chang J, Hu J, Wu C, Lin D. Germline variation in the 3'-untranslated region of the POU2AF1 gene is associated with susceptibility to lymphoma. Mol carcinogenesis. (2017) 56(8):1945–52. doi: 10.1002/mc.22652
191. Ikegami I, Takaki H, Kamiya S, Kamekura R, Ichimiya S. Bob1 enhances RORγt-mediated IL-17A expression in Th17 cells through interaction with RORγt. Biochem Biophys Res Commun (2019) 514(4):1167–71. doi: 10.1016/j.bbrc.2019.05.057
192. Levels MJ, Van Tok MN, Cantaert T, Cañete JD, Kroese FG, Germar K, et al. The transcriptional coactivator Bob1 is associated with pathologic B cell responses in autoimmune tissue inflammation. Arthritis Rheumatol (Hoboken NJ). (2017) 69(4):750–62. doi: 10.1002/art.39993
193. Kim U, Qin XF, Gong S, Stevens S, Luo Y, Nussenzweig M, et al. The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. (1996) 383(6600):542–7. doi: 10.1038/383542a0
194. Zuo J, Ge H, Zhu G, Matthias P, Sun J. OBF-1 is essential for the generation of antibody-secreting cells and the development of autoimmunity in MRL-lpr mice. J Autoimmun (2007) 29(2-3):87–96. doi: 10.1016/j.jaut.2007.05.001
Keywords: germinal center (GC), GC B cells, Tfh cells, Tfr cells, transcriptional regulation, BOB.1/OBF.1, lymphoma
Citation: Betzler AC, Ushmorov A and Brunner C (2023) The transcriptional program during germinal center reaction - a close view at GC B cells, Tfh cells and Tfr cells. Front. Immunol. 14:1125503. doi: 10.3389/fimmu.2023.1125503
Received: 16 December 2022; Accepted: 24 January 2023;
Published: 03 February 2023.
Edited by:
Dessi Malinova, Queen’s University Belfast, Belfast, United KingdomReviewed by:
Irina Grigorova, Michigan Medicine, University of Michigan, United StatesCopyright © 2023 Betzler, Ushmorov and Brunner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cornelia Brunner, Y29ybmVsaWEuYnJ1bm5lckB1bmlrbGluaWstdWxtLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.