 Linlin Zhang
Linlin Zhang Sangsu Bang
Sangsu Bang Qianru He
Qianru He Megumi Matsuda
Megumi Matsuda Xin Luo
Xin Luo Yong-Hui Jiang2
Yong-Hui Jiang2 Ru-Rong Ji
Ru-Rong Ji
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 08 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1124356
This article is part of the Research TopicPain, Immunity, and Neurological and Autoimmune DisordersView all 7 articles
Excessive inflammation has been implicated in autism spectrum disorder (ASD), but the underlying mechanisms have not been fully studied. SHANK3 is a synaptic scaffolding protein and mutations of SHANK3 are involved in ASD. Shank3 expression in dorsal root ganglion sensory neurons also regulates heat pain and touch. However, the role of Shank3 in the vagus system remains unknown. We induced systemic inflammation by lipopolysaccharide (LPS) and measured body temperature and serum IL-6 levels in mice. We found that homozygous and heterozygous Shank3 deficiency, but not Shank2 and Trpv1 deficiency, aggravates hypothermia, systemic inflammation (serum IL-6 levels), and sepsis mortality in mice, induced by lipopolysaccharide (LPS). Furthermore, these deficits can be recapitulated by specific deletion of Shank3 in Nav1.8-expressing sensory neurons in conditional knockout (CKO) mice or by selective knockdown of Shank3 or Trpm2 in vagal sensory neurons in nodose ganglion (NG). Mice with Shank3 deficiency have normal basal core temperature but fail to adjust body temperature after perturbations with lower or higher body temperatures or auricular vagus nerve stimulation. In situ hybridization with RNAscope revealed that Shank3 is broadly expressed by vagal sensory neurons and this expression was largely lost in Shank3 cKO mice. Mechanistically, Shank3 regulates the expression of Trpm2 in NG, as Trpm2 but not Trpv1 mRNA levels in NG were significantly reduced in Shank3 KO mice. Our findings demonstrated a novel molecular mechanism by which Shank3 in vagal sensory neurons regulates body temperature, inflammation, and sepsis. We also provided new insights into inflammation dysregulation in ASD.
Autism spectrum disorder (ASD) is defined by persistent deficits in the ability to initiate and to sustain reciprocal social interaction and social communication and characterized by a range of restricted, repetitive, and inflexible patterns of behavior, as well as various sensory abnormalities (1–5). SHANK3 is a scaffolding protein located at excitatory synapses, and mutations of SHANK3 (haploinsufficiency) may account for 1-2% of all ASD cases including Phelan-McDermid syndrome (PMS) (2, 6, 7). The disrupted striatum centered brain structures and synaptic transmission in this brain region are associated with ASD children with SHANK3 mutations and animal models of Shank3 deficiency (2, 4, 8, 9).
Accumulating evidence has indicated the link between ASD and generalized immune dysfunction (hyper-inflammation). For example, proinflammatory cytokines (e.g., TNF-α, IL-6, IL-8) are increased in the brains of ASD individuals (10). Peripheral blood mononuclear cells from ASD patients produced more TNF-α, IL-1β, and IL-6 than control cells (11). The plasma and serum concentrations of IL-1β, IL-6, and IL-8 are also elevated in ASD samples compared with the healthy control samples (12). Upregulations of IL‐6 and IL‐17 levels were reported in pregnant rodent mothers upon maternal immune activation and IL‐6 may induce the secretion of IL‐17 (3). IL-6 and IL-17 were shown to promote autistic behaviors in animal models (3, 13–15). By contrast, anti-IL-6 or anti-IL-17 treatments can alleviate the autistic behaviors induced by maternal immune activation (3, 14). Frequent respiratory infections were found in 57% of ASD patients and regarded as an ASD comorbidity (4). Anti-biotic treatment may improve ASD-related behaviors (16–18). Subacute neuropsychiatric syndrome in girls with SHANK3 mutations responds to immunomodulation (19). Preclinical study shows that acute inflammatory challenge induced by lipopolysaccharide (LPS) can unmask behavioral deficits of Shank3+/− mice (7). However, the mechanisms of excessive immune reaction in ASD are not fully understood.
Emerging roles for the gut microbiome and gut-brain axis have been implicated in ASD (18, 20, 21). Notably, the vagus system plays a critical role in regulating the gut-brain axis (18, 22, 23). Precision microbial-based therapy was shown to rescue social deficits in mouse models of ASD and, interestingly this rescue in Shank3B knockout mice depends upon the function of the vagus nerve (20). The molecular mechanisms underlying the vagus nerve modulation of ASD remain largely unknown. Recent studies have shown SHANK3 expression in the peripheral nervous system, such as primary sensory neurons of dorsal root ganglion (DRG); and furthermore, SHANK3 interacts with transient receptor potential ion channel V1 (TRPV1) and GABAA receptor in DRG neurons to regulate pain (24) and touch (25). Here we show that Shank3 is also expressed by vagal sensory neurons and Shank3 in nodose ganglion (NG) sensory neurons plays a crucial role in maintaining body temperature and protecting against systemic inflammation in responses to LPS challenge and temperature perturbations. Mechanistically, SHANK3 regulates the expression of TRPM2 (transient receptor potential melastatin 2), a nonselective Ca2+ permeable cation channel in NG vagal sensory neurons.
AAV.CMV.HI. eGFP-Cre.WPRE.SV40 was from Addgene (Plasmid #105545). Shank3 siRNA (Cat# AM16708, ID:194441), Trpv1 siRNA (# AM16708, ID: 223137), and Trpm2 siRNA (# 1320001, ID: mss219854) were purchased from Thermo scientific. Control siRNA was purchased from Santa cruz biotech (#sc-37007), and RVG peptide (# AS-62566) was from Anaspec (Fremont, CA). Menthol (# W266507), capsaicin (# M2028), and LPS (# L2630) were purchased from Sigma.
Shank3 knockout mice (exons 4 to 22 deletion, Δ4–22, C57BL/6 background RRID: MGI:5800311) (8), Shank3 flox/flox mice (RRID: MGI: 5800310) (26), and Shank2 flox/flox mice were generated at Yong-Hui Jiang’s laboratory at Duke University Medical Center. Both Shank2 and Shank3 floxed mice were backcrossed to C57BL/6J for more than 10 generations. To generate sensory neuron-specific deletion of Shank3 and Shank2, the flox mice were crossed with Nav1.8-cre mice, provided by Dr. Rohini Kuner of University of Heidelberg (27). Adult mice (males and females, 8–10 weeks) were used for behavioral and biochemical studies. CD1 mice (males and females, 8–10 weeks, Charles River Laboratories) were used for some siRNA knockdown experiments. All the mouse procedures were approved by the Institutional Animal Care & Use Committee of Duke University. Two to five mice were housed in each cage under a 12-hour alternating light-dark cycle with ad libitum access to water and food. Sample sizes were estimated based on our previous studies for similar types of behavioral and biochemical analyses (24, 28).
The siRNA preparation and injection were conducted as we previously described (29). For siRNA delivery to the vagus nerve, we mixed 100 nM siRNA with 1 µM RVG peptide that can facilitate siRNA uptake by neurons (30). For AAV9 virus delivery to vagus nerve, we mixed 1 x 109 virus in 10 µl of PBS. The vagal nerve was exposed as previously described (31). Mice were anesthetized with isoflurane and a midline cervical muscle and salivary glands were exposed by blunt dissection until the trachea was visible. The vagus nerve was gently separated from the artery, and bilateral peri-neural injections were conducted to deliver siRNA or virus (5 μl) using a glass electrode with tip size of 1 µm. The knockdown effects of siRNA and AAV9 virus were validated by examining the expression of Shank3, Trpm2, and Trpv1 in vagal sensory neurons in nodose ganglion (NG) using in situ hybridization.
The 5% menthol and 100 µM capsaicin were painted into left ear auricular acupoint or skin (50 µl) to measure the body temperature change. Mice were lightly anesthetized with 2.5% isoflurane and then painted by fine brush.
The systemic inflammation was generated by intraperitoneal injection of 1 mg/kg LPS (from Escherichia coli O111:B4) prepared in PBS (100 mg/ml stock). Body temperature measured before LPS injection, then at 0.5, 1, 3, 6, 24, 48, 72, and 96 hours after the LPS injection. The environmental temperature was maintained at 23 ± 0.5°C. To obtain a rectal temperature, the mouse is hand-restrained (briefly anesthetized) and placed on a horizontal surface (cage lid) with the tail lifted, and a probe (covered with Vaseline) was gently inserted into the rectum to a fixed depth of 2 cm. The rectal temperature was measured by the TA-29 flexible implantable probe and TC-344C temperature measuring unit (Warner Instruments., Hamden, CT, USA).
For ECG recording, the silver wires were implanted in the left abdomen below the heart and the right upper shoulder. Two electrodes were fixed to the incised tissues in mice using a 26 G needle under isoflurane anesthesia (32). The silver wires were exposed outside the neck region and put on the small jacket for the prevention of biting. The silver wires were exposed outside the neck region for the prevention of biting. After exposure to the menthol into ear acupoint region (concha area in outer ear), the ECG activity was measured in awake mice. The shaking induced electrical noise was excluded during data analysis. The analog electrical signal was amplified by a microelectrode ac amplifier (AM systems, model 1800) and converted by Digidata 1440A (Molecular device). Signals were filtered at 2 kHz and digitized at 5 kHz. Data were stored and analyzed with a personal computer using pCLAMP 10 (Molecular Devices) software.
Mice were briefly anesthetized with 4% isoflurane and transcardially with PBS and 4% paraformaldehyde. Following perfusion, nodose ganglia (NG) were collected and postfixed in the same fixative overnight at 4°C. NG tissues were embedded in OCT medium (Tissue-Tek), sectioned (14 μm) in a cryostat, and thaw-mounted onto Superfrost Plus slides (VWR). In situ hybridization was performed using the RNAscope system (Advanced Cell Diagnostics) following the manufacturer’s instructions. The RNA scope probes for mouse Shank3 (# 417371), Trpm2 (# 316831-C3), and Trpv1 (# 313331-C3) were designed by Advanced Cell Diagnostics and the RNAscope multiplex fluorescent assays were performed according to the manufacturer’s instructions. Pre-hybridization and hybridization, and then washing were performed according to standard methods. All images were acquired with the same settings using a confocal microscope under 20x magnification. For quantification, four non-adjacent NG sections from each animal and four animals per group were included. The intensity score (0: no signal, 1: weak signal; 2: medium signal, and 3: strong signal) was used to semi-quantify the mRNA signals blindly. We also quantified the number of mRNA-labeled punctate dots (puncta) in each neuron using Image J.
Total RNAs from nodose ganglia were extracted using the Direct-zol™ RNA MiniPrep Kit (Zymo Research), and 0.5-1 μg of RNA was reverse-transcribed using the iScript cDNA Synthesis® (Bio-Rad). Specific primers, including Gapdh control, were designed using IDT SciTools Real-Time PCR software. We performed gene-specific mRNA analyses using the CFX96 Real-Time PCR system (BioRad). Quantitative PCR amplification reactions contained the same amount of Reverse transcription (RT) product, including 10 μl of 2× iQSYBR-green mix (BioRad) and 100-300 nM of forward and reverse primers in a final volume of 15 μl. Primer efficiency was obtained from the standard curve and integrated for calculation of the relative gene expression, which was based on real-time PCR threshold values of different transcripts and groups.
Trpm2 forward: TGCCTCACCTGCTCTTTGCC
Trpm2 reverse: TCTGTGTGTTCCTGCACCTA
TRPV1 forward: ATGTTCGTCTACCTCGTGTTCTTG
TRPV1 reverse: AGGCAGTGAGTTATTCTTCCCATCC
TRPM8 forward GTTGGACCTTGCCAGTGATGAG
TRPM8 reverse CCATTCTCCAGAAAGAGGCGGA
SCN10A forward ATGGAGGTCAGCCAGGACTACA
SCN10A reverse CTGTGAGGTTGTCCGCACTGAA
Gapdh forward: AGGTCGGTGTGAACGGATTTG
Gapdh reverse: GGGGTCGTTGATGGCAACA
To detect serum cytokine levels, we purchased mouse ELISA kits from R&D system (Minneapolis, MN, Cat# DY406 for IL-6). We collected whole blood from a facial vein (30 μl~60 μl). The collected blood was allowed to clot for 30 minutes at room temperature. The clot was then removed in a refrigerated centrifuge at 2,000 x g for 10 min to collect the supernatant (serum). To perform each ELISA assay, we used 5 μl of serum. We measured the absorbance of OD450 using a microplate reader (Bio-Rad) and then quantified the data by MPM6 software (BioRad).
All the data in the figures were expressed as mean ± SEM. We analyzed the biochemical or survival data using Prism GraphPad 6.0. One-Way ANOVA with Bonferroni post hoc or Two-Way ANOVA with Tukey’s posthoc test or unpaired t-test were used to assess biochemical, temperature, and ECG data. We analyzed the survival ratio using a Kaplan–Meier method and log-rank (Mantel-Cox) test. If the overall log-rank test was statistically significant (p < 0.05), we further performed pairwise comparisons of two groups. p < 0.05 was considered statistically significant.
To investigate an overall role of Shank3 in LPS-induced hypothermia, inflammation, and mortality, we employed Shank3 △e4-22 mutant mice, in which exons 4 to 22 were deleted and SHANK3 protein is completely lost (8). In the current study, we referred to △e4-22 mutant mice as Shank3 global knockout mice (GKO). First, we compared the baseline body temperature in wild-type (WT) and heterozygous (Shank3+/-) and homozygous (Shank3-/-) GKO mice. The rectal temperature analysis revealed that Shank3-/- mice exhibited no change in body core temperature as compared to WT and Shank3+/- mice (Figure 1A), suggesting that SHANK3 is not involved in the regulation of basal body temperature. Then, we challenged these mice with intraperitoneal (i.p.) injection of LPS (1 mg/kg) to induce sepsis (28). Herein, we found a rapid and transient hyperthermia in WT mice, as indicated by an elevation in rectal temperature at 30 minutes after LPS injection as compared to baseline (p = 0.0158, Figure 1A). Interestingly, this hyperthermia was completely compromised in both Shank3+/- and Shank3-/- mice (p = 0.0106 and p = 0.0004, respectively) when compared with WT mice (Figure 1A). We also revealed that LPS-induced hypothermia started at 3 hours (p = 0.0008), peaked at 6 hours (p < 0.0001), and continued for 24 hours (p=0.0488) after LPS intervention in WT mice. Furthermore, as compared to WT mice, this hypothermia was observed within 1 hour (p = 0.0411) and lasted for at least 48 hours (p < 0.0001) after LPS exposure in Shank3-/- mice. More strikingly, sepsis-induced hypothermia was significantly aggravated in both Shank3+/- and Shank3-/- mice (F (2, 408) = 48.24, p < 0.0001, two-way ANOVA, Figure 1A). There was also significant difference between heterozygous and homozygous mice (F (1, 245) = 11.35, p = 0.0011, two-way ANOVA, Figure 1A). This result suggested that loss of one copy of Shank3 is sufficient to cause hyperthermia defects and hypothermia exacerbation, which is reminiscent of autism patients with haploinsufficiency of SHANK3.
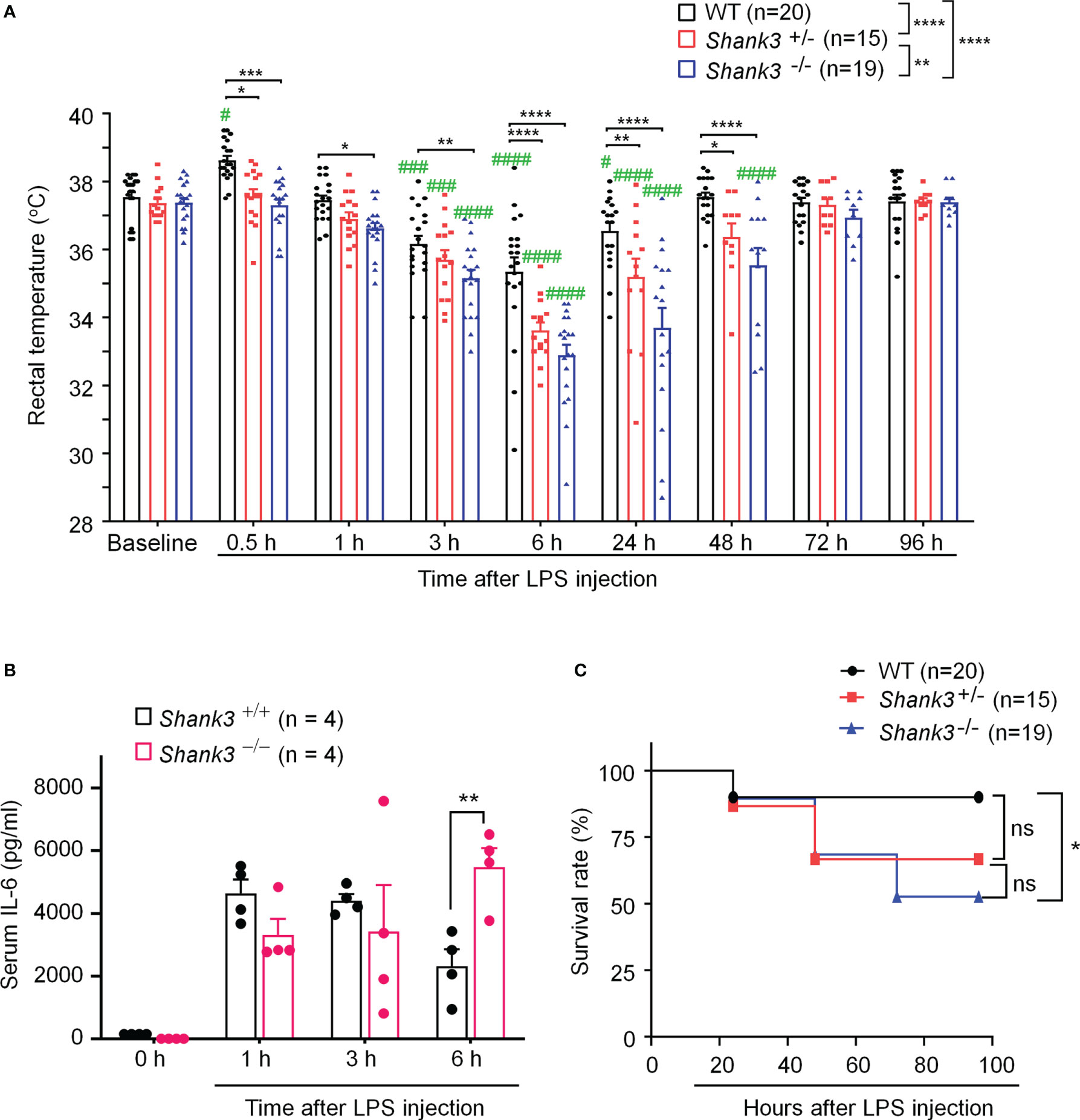
Figure 1 Shank3 global mutants exhibit exacerbation in LPS-induced hypothermia, systemic inflammation and mortality in C57BL/6 mice. (A) Time course of rectal temperature after LPS injection in Wild-type (WT, Shank3+/+) mice, heterozygous (Shank3+/−) mice and homozygous (Shank3−/−) mice. (B) Serum IL-6 levels, revealed by ELISA, in WT and Shank3−/− mice at different times with LPS. (C) Survival curves of WT, Shank3+/− and Shank3−/− mice treated with LPS. LPS (1mg/kg) was injected via intraperitoneal (i.p.) route. Data are expressed as mean ± SEM and analyzed by two-way ANOVA with Bonferroni post hoc test (A, B) and Mantel-Cox test (C). #p <0.05, ###p <0.001, ####p <0.0001 versus baseline. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001 versus WT mice. Sample sizes are indicated in brackets. n.s., not significant.
Next, we investigated whether sepsis-induced systemic inflammation was altered in Shank3 mutant mice. Pro-inflammatory cytokine IL-6 is well recognized as a biomarker for sepsis and sepsis-associated death (28). We detected robust increases of serum IL-6 levels at 6 hours after LPS injection in Shank3-/- mice as compared to WT mice (F (1, 24) = 0.1450, p = 0.008226, two-way ANOVA, Figure 1B).
Also, we evaluated survival rate following LPS injection among three genotypes. Low-dose LPS at 1 mg/kg resulted in a very low mortality (10%, Figure 1C) in WT mice. As compared to WT mice, we found a slight increase (23.33%) in mortality in LPS-treated Shank3+/- mice (p = 0.1062, Figure 1C) but a significant increase (37.37%) in mortality in LPS-treated Shank3-/- mice (p = 0.0152, Figure 1C). Collectively, these data indicate that SHANK3 confers protection against hypothermia, systemic inflammation and septic death following LPS injection.
Vagus nerve stimulation has been shown to protect against sepsis and systemic inflammation (22). Recent studies reported Shank3 expression in peripheral sensory neurons of DRG (24, 25). However, the expression and role of SHANK3 in the vagus system is unclear. Using a sensitive RNA scope assay, we found extensive Shank3 mRNA expression in majority of vagal sensory neurons in nodose ganglia (NG, Figure 2A). To evaluate an underlying contribution of peripheral SHANK3 to hypothermia regulation, we generated Shank3 conditional knockout mice by crossing Shank3-floxed mice with Nav1.8-Cre mice, leading to specific deletion of Shank3 in Nav1.8-expressing peripheral sensory neurons including NG neurons (33). In this study, we referred to Shank3f/f mice as littermate control wildtype (WT) mice, and referred to Nav1.8-cre; Shank3f/+ and Nav1.8-cre; Shank3f/f mice as Shank3 conditional knockout (CKO) heterozygous and homozygous mice. Notably, Shank3 expression was substantially reduced in CKO mice (Nav1.8-cre; Shank3f/f mice) as compared to Shank3f/f mice (p = 0.0017, Figures 2A, B). This result indicates a selective expression of Shank3 in NG sensory neurons and also validated Shank3 CKO mice.
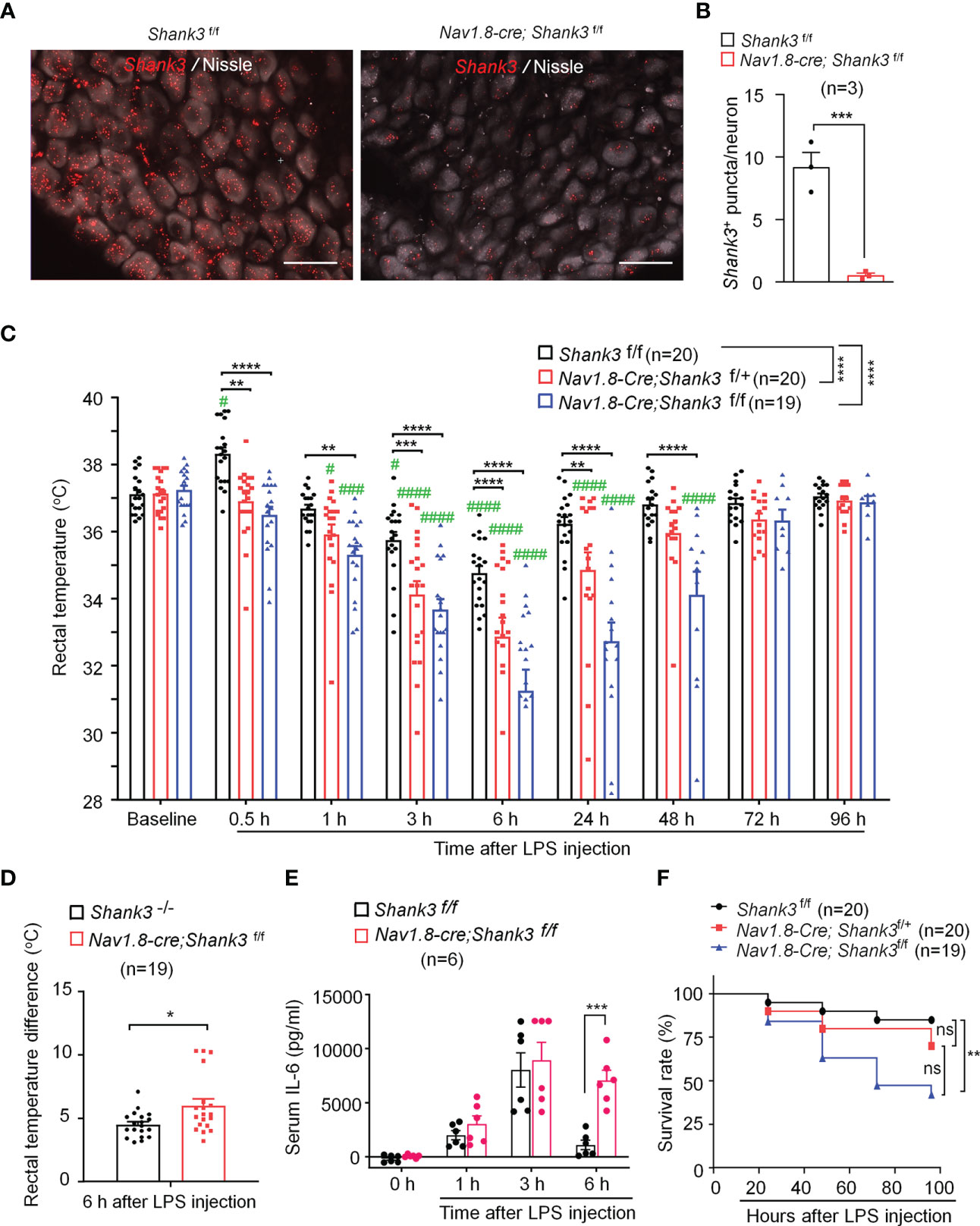
Figure 2 LPS-induced hypothermia, systemic inflammation and mortality are aggravated in Shank3 CKO C57BL/6 mice. Shank3 CKO mice were generated by crossing Shank3-floxed mice with Nav1.8-Cre mice, leading to specific loss of Shank3 in Nav1.8-expressing sensory neurons. (A) In situ hybridization (ISH) images showing Shank3 mRNA (red) expression in vagal sensory neurons in nodose ganglia (NG) of Shank3f/f mice and Nav1.8-cre; Shank3f/f mice. Scale bar, 50 μm. (B) Quantification of the number of Shank3 puncta in NG neurons. (C) Time course of rectal temperature after LPS (1mg/kg, i.p.) injection in Shank3f/f mice, Nav1.8-cre; Shank3f/+ mice and Nav1.8-cre; Shank3f/f mice. (D) Changes (reduction) of rectal temperature at 6 hours following LPS injection in GKO (Shank3−/−) mice and CKO (Nav1.8-cre; Shank3f/f) mice. (E) Serum IL-6 levels in Shank3f/f mice and Nav1.8-cre; Shank3f/f mice treated with LPS. (F) Survival curves of three genotypes mice treated with LPS. Data are expressed as mean ± SEM and analyzed by unpaired two-tailed t test (B, D), two-way ANOVA with Bonferroni post hoc test (C, E), and Mantel-Cox test (F). #p < 0.05, ###p < 0.001, ####p < 0.0001 versus baseline. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus Shank3f/f mice. Sample sizes are indicated in brackets. n.s., not significant.
As in GKO mice, body temperature analysis revealed no difference of basal body temperature between WT and CKO mice (Figure 2C), suggesting that Shank3 in sensory neurons does not contribute to basal body temperature. We next compared hypothermia phenotypes after LPS injection in three genotypes of WT and CKO mice. Herein, we reported a rapid and transient hyperthermia in Shank3f/f mice, as indicated by the elevation in rectal temperature at 30 minutes after LPS injection as compared to baseline (p = 0.0473, Figure 2C). Interestingly, this hyperthermia was completely compromised in both heterozygous and homozygous CKO mice (p = 0.0025 and p < 0.0001, respectively) when compared with Shank3f/f mice. Furthermore, homozygous CKO mice exhibited more rapid (within 1 hour, p=0.0038) and more lasting (>48 hours, p < 0.0001) hypothermia, as compared to Shank3f/f mice (Figure 2C), suggesting that Shank3 deficiency in sensory neurons accelerates the induction of hypothermia and delays the resolution of hypothermia after sepsis. Intriguingly, sepsis-induced hypothermia was significantly aggravated in both heterozygous and homozygous CKO mice (F (2, 448) = 58.38, p < 0.0001, two-way ANOVA, Figure 2C). There was also significant difference between heterozygous and homozygous CKO mice (F (1, 286) = 16.47, p < 0.0001, two-way ANOVA, Figure 2C). More strikingly, Shank3 CKO mice exhibited more remarkable changes (reductions) in rectal temperature when compared with Shank3 GKO (p = 0.0136, Figure 2D), suggesting a dominant role of peripheral SHANK3 in temperature regulation.
Next, we investigated whether LPS-induced systemic inflammation would also be affected in Shank3 CKO mice. Serum IL-6 levels did not differ at 3 hours and 6 hours after LPS stimulation in WT and CKO mice. At 24 hours, IL-6 levels returned to baseline in WT mice but remained at the peak in CKO (F (3, 40) = 29.21, p = 0.0002, two-way ANOVA, Figure 2E). Survival rate analysis showed that LPS significantly increased mortality (42.90%) in homozygous CKO mice (p=0.0083, vs. WT, Figure 2F). Collectively, these data demonstrate that SHANK3 in Nav1.8-expressing peripheral sensory neurons confers protection against hypothermia, systemic inflammation and septic death after sepsis.
To investigate a specific role of peripheral SHANK3 in regulating body temperature, we challenged WT and Shank3 CKO mice in different conditions of hypothermia and hyperthermia, including cold room habituation at 4°C (Figure 3A), 5% isoflurane anesthesia (Figure 3B), intrathecal injection of PGE2 (200 ng, Figure S3), 5% menthol application to the back and auricular region (Figures 3C, D), and auricular application of 100 μM capsaicin (Figure 3E). Cold room testing showed deficits in homozygous CKO mice (F (1, 60) = 19.49, p < 0.0001, two-way ANOVA, vs. WT) and heterozygous CKO mice (F (1, 56) = 5.568, p = 0.0168 vs. WT) in maintaining core temperature (Figure 3A). Interestingly, CKO mice exhibited rapid hypothermia compared to WT mice (F (2, 92) = 10.54, p < 0.05 vs. WT, Figure 3A). Brief anesthesia to 5% isoflurane produced a rapid drop in body temperature in 10 min, but no difference was found between three genotypes (F (2, 42) = 3.094, p = 0.0558, two-way ANOVA, Figure 3B). Intrathecal PGE2 induced hyperthermia and fever in WT mice, which was impaired in Shank3 CKO mice (F (2, 48) = 3.835, p = 0.0285, two-way ANOVA, Figure S1). Next, we observed a rapid and transient hyperthermia after 5% menthol smearing in body trunk in Shank3f/f mice (Figure 3C). This hyperthermia at 30 min was partially attenuated in homozygous CKO mice (p = 0.0218, vs. WT), although we did not observe any significant difference among three genotypes (F (2, 80) = 1.776, p = 0.176, two-way ANOVA, Figure 3C). Next, we smeared 5% menthol to auricular vagal region to mimic vagus nerve stimulation (aVNS, Figure 3D). This cold/cool stimulation induced hyperthermia in WT mice but not in homozygous CKO mice (F (1, 56) = 12.85, p = 0.0007, two-way ANOVA, Figure 3D), which was consistent with the results in Shank3 GKO mice (Figure S2A). Similarly, aVNS with 100 μM capsaicin smearing produced hyperthermia in Shank3f/f mice, which was compromised in both heterozygous and homozygous CKO mice (F (2, 92) = 22.51, p < 0.0001, two-way ANOVA, Figure 3E). The same result was also observed in Shank3 GKO mice (Figure S2B).
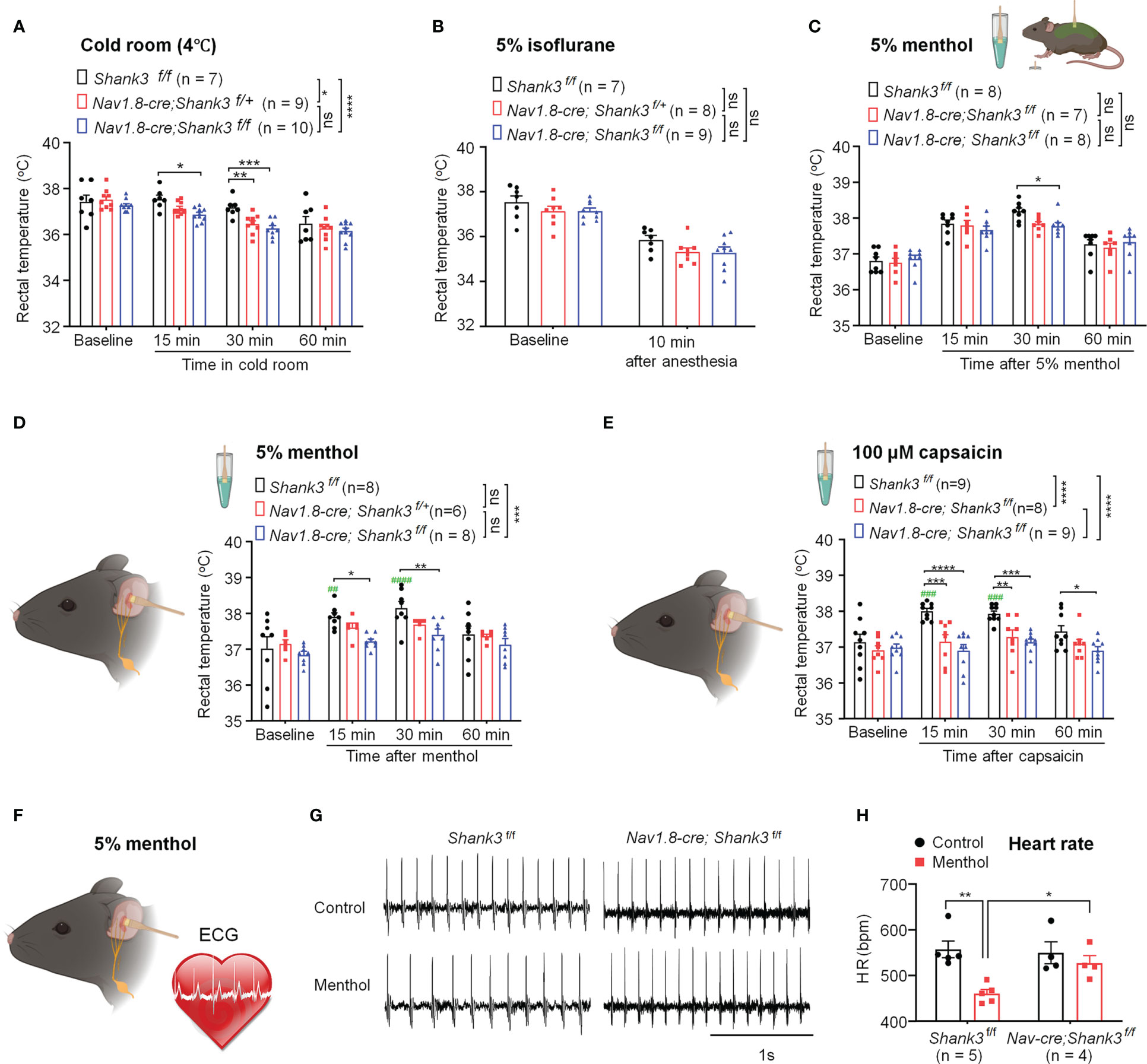
Figure 3 Shank3 in sensory neurons is required for the maintenance of body temperature after perturbations in C57BL/6 mice. (A) Time course of rectal temperature after acute exposure to a cold room (4°C) in three genotypes. (B) Time course of rectal temperature after brief anesthesia to 5% isoflurane in three genotypes. (C) Time course of rectal temperature after 5% menthol smearing on body trunk in Shank3f/f mice, Nav1.8-cre; Shank3f/+ mice and Nav1.8-cre; Shank3f/f mice. (D) Time course of rectal temperature after auricular painting of 5% menthol in three genotypes. (E) Time course of rectal temperature after auricular painting of 100 μM capsaicin in three genotypes. (F) Experimental design for ECG recording after auricular painting of 5% menthol in littermate control WT and CKO mice. (G) Representative traces of ECG in Shank3f/f mice and Nav1.8-cre; Shank3f/f mice following vehicle or menthol treatment. (H) Heart rate (HR) in Shank3f/f mice and Nav1.8-cre; Shank3f/f mice following vehicle or menthol treatment. Data are expressed as mean ± SEM and analyzed by two-way or one-way ANOVA with Bonferroni post hoc comparisons. ##p < 0.01, ###p < 0.001, ####p < 0.0001, ####p < 0.0001, versus baseline. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus Shank3f/f mice. Sample sizes are indicated in brackets. n.s., not significant.
Next, we recorded electrocardiogram (ECG) to assess VNS-induced heart rate (HR) change following auricular menthol smearing (Figure 3F). As expected, menthol-induced VNS resulted in a significant decrease of HR in WT Shank3f/f mice, which was abolished in homozygous CKO mice (F (1, 14) = 11.73, p = 0.0041, two-way ANOVA, Figures 3G, H). Collectively, these detailed results suggest that peripheral Shank3 in vagal sensory nerve plays an important role in maintaining core temperature following perturbations.
SHANK3 is expressed by different types of peripheral sensory neurons in mouse DRG, NG, and trigeminal ganglia (34). To confirm a specific role of peripheral SHANK3 in vagal nerve for regulating the LPS-induced effects, we used Shank3 siRNA to knockdown endogenous SHANK3 expression in vagal sensory neurons in NG. A short peptide derived from rabies virus glycoprotein (RVG) has been utilized to facilitate siRNA uptake by neurons including sensory neurons and enhance knockdown efficiency of siRNA (29, 30). We performed bilateral peri-vagal nerve delivery of Shank3 siRNA (mixed with the RVG peptide, 1:10) in CD1 mice (Figure 4A). In situ hybridization (ISH) revealed a profound reduction in Shank3 mRNA in NG neurons following Shank3 siRNA (100 nM) treatment (F (2, 9) = 48.70, p = 0.0002, one-way ANOVA, Figure 4B), suggesting the successful establishment of SHANK3 knockdown by siRNA. We next challenged these mice with LPS on 3 days following siRNA injection. Intriguingly, we found that LPS-induced transient hyperthermia in control siRNA treated mice was completely lost after Shank3 siRNA treatment (p < 0.0001, Figure 4C). Moreover, LPS-induced hypothermia was significantly aggravated by vagal SHANK3 knockdown (F (1, 75) = 90.52, p < 0.0001, two-way ANOVA, vs. control siRNA, Figure 4C). Peri-vagal Shank3 siRNA (50 nM, 100 nM and 200 nM) enhanced LPS-induced hypothermia in a dose-dependent manner (F (5, 297) = 53.45, p < 0.0001, two-way ANOVA, Figure S3A). Furthermore, we detected a robust increase of serum IL-6 levels at 3 hours after LPS injection (p = 0.0027, Shank3 siRNA vs. control siRNA, Figure 4D). However, we did not detect any difference of mortality between control siRNA and Shank3 siRNA treatment, as CD1 mice are more resistant to sepsis than C57BL/6 mice (Figures S3B–D).
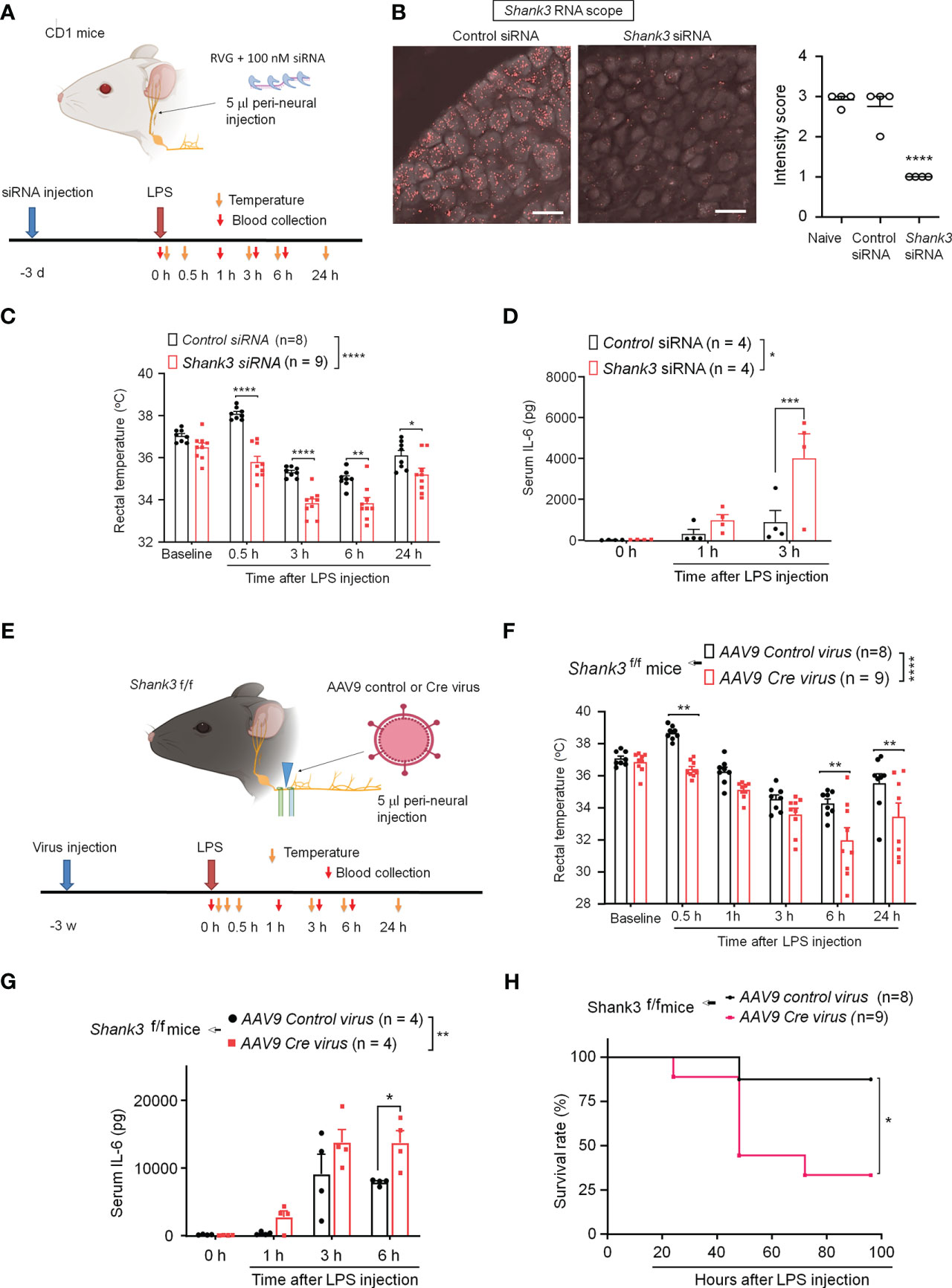
Figure 4 SHANK3 knockdown in vagal sensory neurons aggravates LPS-induced hypothermia, systemic inflammation and mortality in CD1 mice (A–D) and C57BL/6 mice (E–G). (A) Experimental design for SHANK3 knockdown, LPS injection, body temperature measurement and blood collection in CD1 mice. Peri-neural injection of 100 nM Shank3 siRNA (5 μl, mixed with RVG peptide, 1:10) was made to knockdown SHANK3 expression in vagal sensory neurons in NG. (B) ISH images showing Shank3 mRNA (red) expression in NG following control siRNA and Shank3 siRNA treatment. Scale bar, 50 μm. (C) Time course of rectal temperature after LPS injection (1mg/kg, i.p.) in control siRNA and Shank3 siRNA treated mice. (D) Serum IL-6 levels after LPS exposure in control siRNA and Shank3 siRNA treated mice. (E) Experimental design for SHANK3 knockdown, LPS injection, body temperature measurement and blood collection in Shank3f/f mice. Peri-neural AAV9-Cre virus injection was made to knockdown SHANK3 expression in vagal sensory neurons in NG in Shank3f/f mice. (F) Time course of rectal temperature after LPS (1mg/kg) injection in AAV9 control virus and AAV9 Cre virus treated mice. (G) Serum IL-6 levels and (H) Survival curves after LPS exposure in AAV9 control virus and AAV9 Cre virus treated mice. Data are expressed as mean ± SEM and analyzed by one-way ANOVA with Bonferroni post hoc test (B), two-way ANOVA with Bonferroni post hoc test (C, D, F, G), and Mantel-Cox test (H). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus mice treated control siRNA or AAV9 control virus. Sample sizes are indicated in brackets. n.s., not significant.
Next, we employed GFP fluorescence-labeled AAV9-Cre virus to produce a more persistent knockdown of endogenous SHANK3 expression in vagal sensory neurons, by using bilateral peri-vagal nerve delivery of AAV9 Cre virus in Shank3f/f mice (Figure 4E). Peri-vagal nerve injection of GFP-labeled AAV9 Cre virus resulted in uptake of AAV9 Cre virus in NG neurons as a result of axonal transport from the vagal nerve (Figure S4A). Simultaneously, we detected a profound reduction in Shank3 mRNA in NG neurons following the same treatment (p = 0.0002, Figures S4A–C), manifesting the successful establishment of SHANK3 knockdown by Cre virus in Shank3f/f mice. We next challenged these mice with LPS at 2 weeks following the virus injection. We found that the Cre virus treatment abolished the LPS-induced transient hyperthermia (p = 0.002 vs. control virus) and exacerbated the LPS-induced hypothermia (F (1, 89) = 36.12, p < 0.0001, two-way ANOVA, vs. control virus, Figure 4F). Moreover, the Shank3 virus treated mice exhibited a significant increase of serum IL-6 levels (F (1, 24) = 10.18, p = 0.0039, Two-Way ANOVA, vs. control virus, Figure 4G) following LPS challenge. Vagal SHANK3 knockdown by Cre virus treatment also resulted in a significant reduction in survival rate after LPS intervention in Shank3f/f mice (p = 0.0274, vs. control virus, Figure 4H). Collectively, these data indicate that SHANK3 in vagal sensory neurons confers protection against LPS-induced hypothermia, systemic inflammation and septic death.
TRP channels are thermosensors in peripheral nervous system and detect ambient temperature changes (35, 36). First, we employed quantitative RT-PCR to measure mRNA expressions of Trpm2, Trpv1, Trpv4, Trpm8 and Scn10A (encoding Nav1.8) in NG of WT and GKO mice. Interestingly, we detected a robust reduction in Trpm2 (p = 0.00754) but not Trpv1, Trpv4, Trpm8 and Scn10A mRNA levels in NG of Shank3−/− mice (Figure 5A). ISH revealed a broad expression of Trpm2 mRNA in majority of NG neurons in WT mice, but this expression was significantly lower in Shank3−/− mice (p < 0.05, vs. WT, Figure 5B).
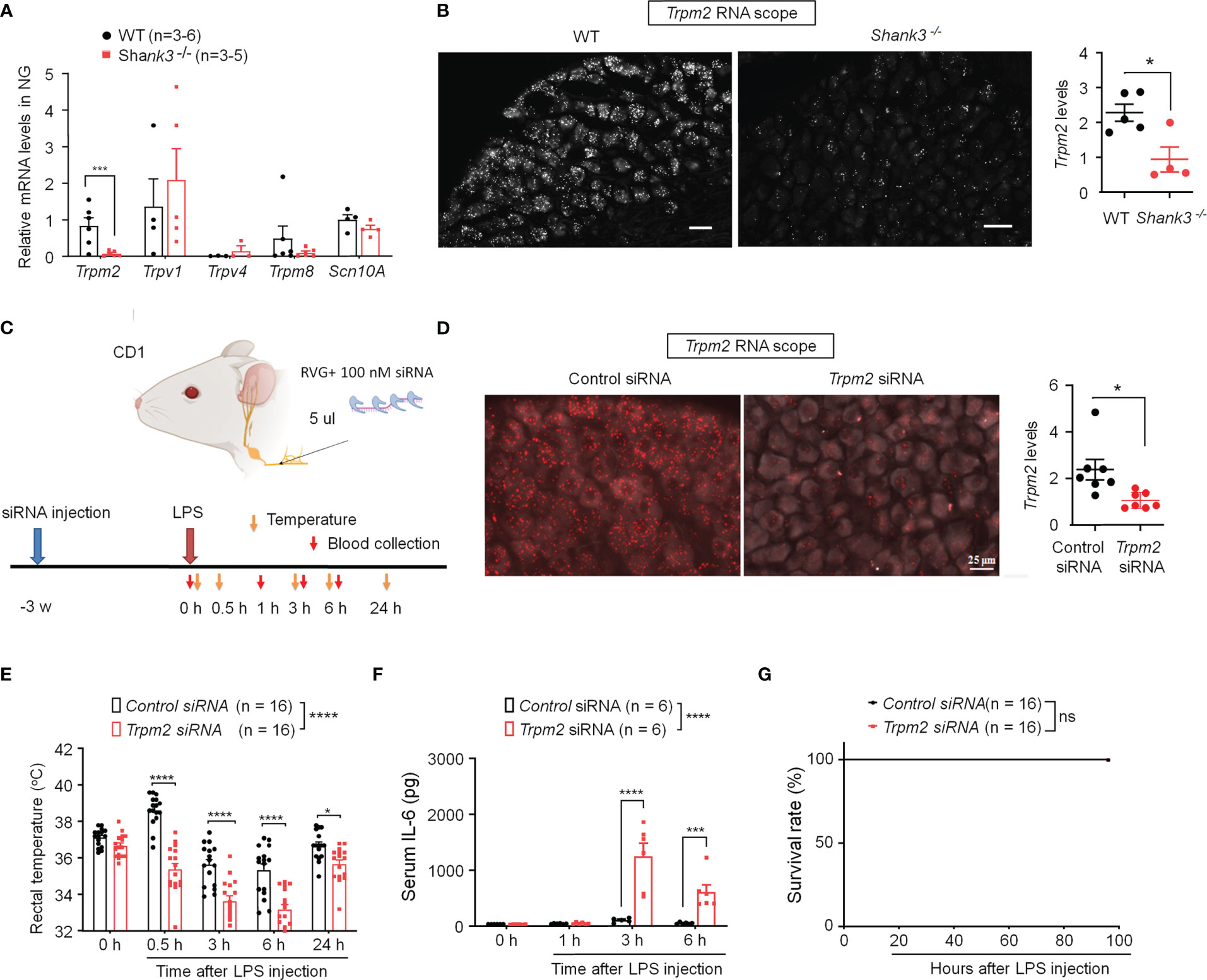
Figure 5 SHANK3 regulates body temperature and inflammation via TRPM2 in C57BL/6 mice (A, B) and CD1 mice (C–G). (A) RT-PCR analysis showing the expression of Trpm2, Trpv1, Trpv4, Trpm8 and Scn10A in NG of WT mice and Shank3−/− mice. (B) ISH images showing Trpm2 mRNA expression in NG neurons of WT mice and Shank3−/− mice. Scale bar, 50 μm. Right, quantification of Shank3 mRNA levels. *p < 0.05. (C) Experimental design for TRPM2 knockdown, LPS injection, body temperature measurement and blood collection in CD1 mice. Peri-neural injection of 100 nM Trpm2 siRNA was made to knockdown TRPM2 expression in NG neurons. (D) ISH images showing Trpm2 mRNA expression in NG neurons following control siRNA and Trpm2 siRNA injection. Scale bar, 50 μm. Right, quantification of Trpm2 mRNA levels. *p < 0.05. (E) Time course of rectal temperature after LPS injection (1mg/kg, i.p.) in control siRNA and Trpm2 siRNA treated mice. (F) Serum IL-6 levels and (G) survival curves after LPS exposure in control siRNA and Trpm2 siRNA treated mice. Data are expressed as mean ± SEM and analyzed by unpaired two-tailed t test (B, D), two-way ANOVA with Bonferroni post hoc test (E, F), and Mantel-Cox test (G). *p < 0.05, ***p < 0.001, ****p < 0.0001 versus WT mice or mice treated control siRNA. Sample sizes are indicated in brackets. ns, not significant.
To further verify the potential role of peripheral TRPM2 in vagal nerve in sepsis-induced hypothermia, Trpm2 siRNA (100 nM) was employed to knockdown endogenous TRPM2 expression in vagal sensory neurons of CD1 mice through bilateral peri-vagal nerve delivery (Figure 5C). ISH detected a significant reduction in Trpm2 mRNA in NG neurons following the siRNA treatment (p = 0.01331, Figure 5D). Following LPS challenge, Trpm2 siRNA treatment exacerbated hypothermia (F (1, 150) = 120.4, P < 0.0001, vs. control siRNA, Figure 5E) and increased serum IL-6 levels (F (1, 39) = 39.31, p < 0.0001, vs. control siRNA, Figure 5F), without affecting mortality in CD1 mice (Figure 5G). Together, these data indicate that 1) TRPM2 is a downstream event of SHANK3 signaling in NG neurons and 2) loss of TRPM2 in vagal sensory neurons can recapitulate some of the key features of SHANK3 knockdown, including hypothermia and systemic IL-6 increase.
Dysfunction of SHANK2 protein is also a critical step for the development of autistic-like behaviors (2, 37). We therefore investigated whether SHANK2 is involved in LPS and menthol induced thermal regulation using Shank2 CKO mice with Shank2 deletion in Nav1.8+ sensory neurons. We did not find significant differences in LPS-induced hypothermia (F (2, 124) = 0.1497, p = 0.8611, two-way ANOVA, Figure 6A) and survival rate (Figure S5A) between littermate control mice (Shank2f/f) and CKO mice (Nav1.8-cre; Shank2f/+ and Nav1.8-cre; Shank2f/f). In parallel, we did not observe significant differences in menthol-induced hyperthermia (F (2, 64) = 0.3973, p = 0.6738, two-way ANOVA, Figure 6B), capsaicin-induced hyperthermia (Figure S5B), and anesthesia-induced hypothermia (Figure S5C). Thus, Shank2 in peripheral sensory neurons is not involved in the regulation of body core temperature.
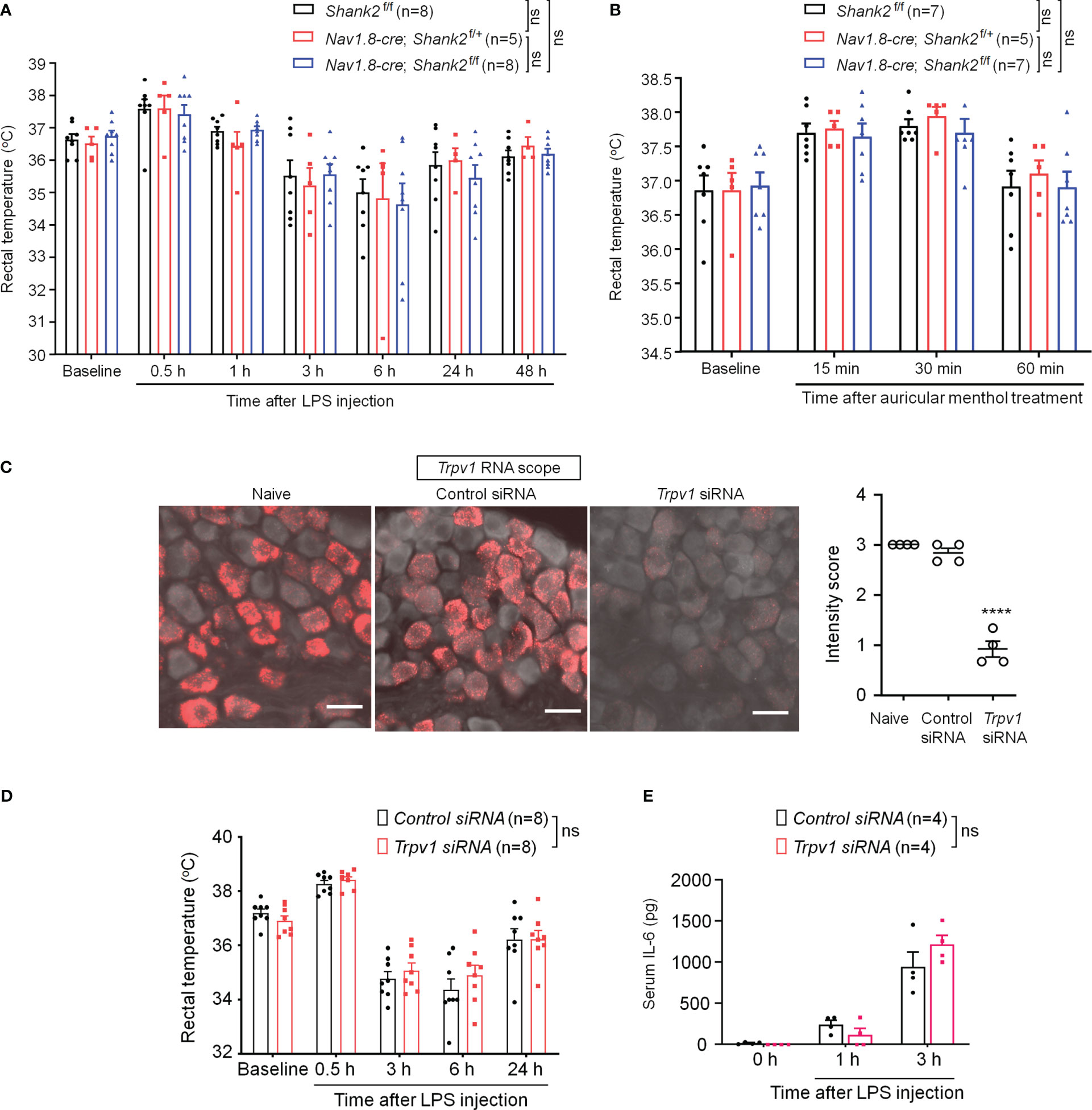
Figure 6 SHANK2 and TRPV1 are dispensable in LPS-induced hypothermia and systemic inflammation in C57BL/6 mice. Shank2 conditional knockout mice were generated by crossing Shank2-floxed mice with Nav1.8-Cre mice, leading to specific loss of Shank2 in Nav1.8-expressing sensory neurons. (A) Time course of rectal temperature after LPS injection (1mg/kg, i.p.) in Shank2f/f mice, Nav1.8-cre; Shank2f/+ mice and Nav1.8-cre; Shank2f/f mice. (B) Time course of rectal temperature after auricular painting of 5% menthol in Shank2f/f mice, Nav1.8-cre; Shank2f/+ mice and Nav1.8-cre; Shank2f/f mice. (C) Peri-neural injection of 100 nM Trpv1 siRNA was made to knockdown TRPV1 expression in vagal sensory neurons in NG. ISH images showing Trpv1 mRNA (red) expression in NG neurons following control siRNA and Trpv1 siRNA injection. Scale bar, 50 μm. ****p < 0.0001, unpaired t-test. (D) Time course of rectal temperature after LPS injection (1mg/kg, (i) p.) in control siRNA and Trpv1 siRNA treated mice. (E) Serum IL-6 levels after LPS exposure in control siRNA and Trpv1 siRNA treated mice. Data are expressed as mean ± SEM and analyzed by two-way or one-way ANOVA with Bonferroni post hoc test. Sample sizes are indicated in brackets. Not significant, n.s. ****p < 0.0001 versus naive mice.
Given a functional link between SHANK3 and TRPV1 in DRG primary sensory neurons and heat sensation (24), we evaluated whether peripheral TRPV1 is important for sepsis-induced hypothermia. Trpv1 siRNA was employed to knockdown endogenous TRPV1 expression in NG neurons via peri-vagal nerve delivery in CD1 mice. ISH detected a substantial reduction in Trpv1 mRNA in NG neurons following Trpv1 siRNA (100 nM) injection [F (2, 9) = 115.8, p < 0.0001, Figure 6C]. Compared to the control siRNA, Trpv1 siRNA had no significant effects on LPS-induced hypothermia (F (1, 70) = 0.648, p = 0.4235, two-way ANOVA, Figure 6D) and serum expressions of IL-6 (F (1, 18) = 0.3131, p = 0.5827, Figure 6E). These findings suggest that TRPV1 in vagal sensory neurons is not required for sepsis-induced hypothermia, systemic inflammation, and mortality.
In this study, using Shank3 complete knockout mice with deletion of exons 4 to 22 (Δe4-22 Shank3─/─) (8, 24), we have made several interesting findings. First, SHANK3 is broadly expressed in vagal sensory neurons in NG. Second, Shank3 regulates Trpm2 expression in vagal sensory neurons. Third, Shank3 deficiency, but not Shank2 and Trpv1 deficiency, exacerbates hypothermia, systemic inflammation (serum IL-6 levels), and mortality in mice, induced by a low dose of lipopolysaccharide (1 mg/kg). Fourth, Shank3 CKO mice with Shank3 deficiency in Nav1.8-expressing sensory neurons can recapitulate all these detrimental effects of LPS in GKO mice. Strikingly, selective knockdown of Shank3 or Trpm2 in vagal sensory neurons is sufficient to mediate these actions of SHANK3. Our study highlights a novel role of vagal sensory neurons in mediating SHANK3-dependent disorders.
Mutations in the SHANK3 gene have been recognized as a genetic risk factor for ASD (2). Notably, heterozygous SHANK3 mutations are typically associated with idiopathic ASD in patients, but heterozygous deletion of Shank3 gene in mice does not commonly induce ASD-related behavioral deficit (7, 8). Using △e4-22 Shank3 full knockout strategy, Shank3 haploinsufficiency was sufficient to cause marked deficits in TRPV1-medicated heat pain. These pain deficits were fully recapitulated in Shank3+/- CKO mice with Shank3 deficiency in sensory neurons (24). In the present study, Shank3+/- GKO and CKO mice also exhibited significant deficits in LPS-induced hypothermia and systemic inflammation, albeit Shank3-/- GKO and CKO mice had more robust phenotypes. Furthermore, genetically vulnerable Shank3+/- mice, when challenged with LPS to induce an acute inflammatory response, showed robust circuit and behavioral alterations related to ASD (7).
Apart from genetic risk factors, studies also support the hypothesis of immune dysregulation in ASD (3). Especially, IL-6 and IL-17 were upregulated in ASD individuals or animal models of ASD and contribute to autistic behaviors in animal models (3, 13–15). Furthermore, blocking these cytokines with neutralizing antibodies were shown to alleviate autistic behaviors (3, 14). Frequent respiratory infections were found in 57% of ASD patients and regarded as an ASD comorbidity (4). Devastating symptoms were reported in some PMS patients after acute infections or stressful environmental challenges (38). Several clinical studies reported improvements in ASD behaviors following antibiotic treatments (16–18) and immunomodulation (19). Low dose LPS challenge (1-2 mg/kg) induces systemic inflammation (“cytokine storm”) and neuroinflammation and exacerbates ASD symptoms in Shank3+/- mice (7). We found that IL-6 is significantly dysregulated (upregulated) after Shank3 deficiency, in response to LPS challenge. It will be of great interest to examine the changes of these cytokines in the brain and CSF, which could drive some ASD symptoms.
Accumulating evidence suggests that the vagus system plays a critical role in regulating the gut-brain axis (18, 22, 23), and dysregulation of this axis contributes to ASD (18, 20, 21). Microbial-based therapy has been used to treat social deficits in mouse models of ASD and the vagus nerve is required for this therapeutic effect in Shank3B knockout mice (20). VNS is a safe and effective therapy for pediatric patients with drug-resistant epilepsy. There is some evidence that VNS, when performed for epilepsy, may improve behavior in ASD individuals (39). An observational study revealed that VNS not only controlled epilepsy but also improved the autistic behaviors, especially in language, social and self-help (40). It appears that this behavioral improvement occurs independently of VNS’ modulation of seizure (39). Our findings suggest that SHANK3 in vagal sensory neurons may regulate vagal activity. One important vagal regulation is to regulate heart rate. However, this regulatory function of the vagus nerve is impaired after Shank3 deficiency. This notion is supported by our ECG data demonstrating that auricular VNS reduced heart rate in WT mice but not in Shank3 KO mice. Vagus nerve regulates the hemostasis of various physiological functions in our body, including body temperature and inflammation. VNS and peripheral nerve stimulation (e.g., acupuncture) confer protection against LPS-induced hypothermia, systemic inflammation, and septic death (22, 41, 42).
Using sensitive RNAscope ISH, we revealed broad expression of Shank3 mRNA in majority of NG neurons. Importantly, selective knockdown of Shank3 by siRNA and AAV Cre virus, via a perineural delivery approach (29, 30), was sufficient to produce the phenotypes (hypothermia and excessive inflammation) in Shank3 GKO and CKO mice. To our knowledge this is the first report to demonstrate the function of Shank3 in vagal sensory neurons. Single-cell RNAseq showed low level expression of Shank3 in some vagal sensory neurons (43). Earlier study showed that SHANK3 expression in primary sensory neurons regulates thermal pain and heat hyperalgesia (24). As a scaffold protein, SHANK3 interacts with TRPV1 and regulates the surface expression of this heat sensor. Thus, capsaicin-induced cellular responses and pain were largely abolished in mice with Shank3 deficiency (24). NG sensory neurons also express multiple TRP channels, including TRPV1, TRPA1, TRPM8, and TRPM2 (43). Interestingly, Shank3 deficiency resulted in reduced expression of Trpm2, without changing the expression of Trpv1, Trpv4, Trpm8, and SCN10A (encoding Nav1.8). This raises a possibility of transcriptional regulation of Trpm2 by SHANK3. Interestingly, SHANK3 was found in the nuclei of primary sensory neurons (24). Future studies are needed to investigate how SHANK3 regulates the transcription of Trpm2. TRPM2 is a non-selective cation channel. In hypothalamic neurons, TRPM2 channel is a heat sensor. TRPM2 in hypothalamus can limit fever and drive hypothermia (44). TRPM2 is also expressed abundantly in immune cells and is important in inflammatory processes (45). Especially, TRPM2 acts as an oxidant sensor and oxidant sensing by TRPM2 inhibits neutrophil migration and mitigates inflammation (46). We speculate that TRPM2 in vagal sensory neurons could sense the LPS-induced inflammatory oxidants (e.g., ROS). Thus, activation of TRPM2 by stress and oxidants in NG neurons will result in calcium influx and activation of vagal sensory neurons to mediate the anti-inflammatory actions of the VNS. It is noteworthy that basal body temperature under the non-stressful normal conditions does not require Shank3 and Trmp2 in vagal sensory neurons. However, SHANK3 or/and TRPM2 play an important role in maintaining body temperature, under stressful conditions with temperature perturbations, such as LPS challenge, VNS by auricular application of menthol and capsaicin, cold environment, and PGE2-induced fever.
Our results also showed that Shank3 deficiency in GKO and CKO mice or Shank3 knockdown in vagal sensory neurons lead to increased mortality in response to systemic LPS challenge (1 mg/kg, i.p.). This low-dose LPS challenge produced very low mortality (10%) in WT mice but resulted in 50% mortality in global and conditional Shank -/- mice. This septic death after Shank3 deficiency may result from a substantial loss of body temperature (hypothermia, <32°C body temperature was closely associated with septic death) and excessive inflammation by cytokine storm. LPS-induced IL-6 upregulation is highly correlated to septic death (28). Among the inflammatory cytokines we measured in this study, only IL-6 is significantly dysregulated after Shank3 and Trpm2 deficiency, in response to LPS challenge. A recent study in 2021 was conducted in 9754 autism pupils in Scotland, which is 1.2% out of 787,666 pupils examined. The authors cautiously concluded that the death rate in the current generation of children and young adults with autism is no higher than for other children, in part due to improved diagnosis and care (47). It is noteworthy that ASD children with SHANK3 mutations exhibit frequent respiratory infections in 57% cases (4). It will be of great interest to study if individuals with SHANK3 mutations have a higher risk of mortality than ASD individuals with other genetic mutations.
In summary, our findings demonstrate a novel role of SHANK3 in vagal sensory neurons in maintaining body temperature after stress-related perturbations. Furthermore, vagal SHANK3 can limit excessive inflammation after LPS challenge, offering new molecular insights into inflammation dysregulation in some ASD individuals. Finally, we provided a novel link between SHANK3 and TRPM2, an oxidant/stress sensor, in vagal sensory neurons. Recent study connected SHANK to TRPV4 in the Nucleus Accumbens, as TRPV4 inhibition could alleviate autistic behaviors in Shank3+/- mice (7). It remains to be investigated whether impairment of SHANK3-medated vagal modulation is sufficient to drive behavioral changes in ASD, moreover, whether TRPM2 agonist can improve the ASD symptoms.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Duke University IACUC.
LZ, SB and R-RJ designed research; LZ, SB, QH, MM, and XL performed research and analyzed data; LZ, SB, Y-HJ and R-RJ wrote the manuscript and other authors edited the manuscript. All authors contributed to the article and approved the submitted version.
This study is supported by Duke University Anesthesiology Research Funds and NIH R01grant DE17794. Y-HJ is supported by R01 MH117289 and R01HD088007.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1124356/full#supplementary-material
1. Kaiser T, Feng G. Modeling psychiatric disorders for developing effective treatments. Nat Med (2015) 21:979–88. doi: 10.1038/nm.3935
2. Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron (2013) 78:8–27. doi: 10.1016/j.neuron.2013.03.016
3. Azhari A, Azizan F, Esposito G. A systematic review of gut-immune-brain mechanisms in autism spectrum disorder. Dev Psychobiol (2019) 61:752–71. doi: 10.1002/dev.21803
4. Liu C, Li D, Yang H, Li H, Xu Q, Zhou B, et al. Altered striatum centered brain structures in SHANK3 deficient Chinese children with genotype and phenotype profiling. Prog Neurobiol (2021) 200:101985. doi: 10.1016/j.pneurobio.2020.101985
5. Orefice LL. Outside-in: Rethinking the etiology of autism spectrum disorders. Science (2019) 366:45–6. doi: 10.1126/science.aaz3880
6. Sarasua SM, Boccuto L, Sharp JL, Dwivedi A, Chen CF, Rollins JD, et al. Clinical and genomic evaluation of 201 patients with phelan-McDermid syndrome. Hum.Genet (2014) 133:847–59. doi: 10.1007/s00439-014-1423-7
7. Tzanoulinou S, Musardo S, Contestabile A, Bariselli S, Casarotto G, Magrinelli E, et al. Inhibition of Trpv4 rescues circuit and social deficits unmasked by acute inflammatory response in a Shank3 mouse model of autism. Mol Psychiatry (2022) 27:2080–94. doi: 10.1038/s41380-021-01427-0
8. Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, et al. Altered mGluR5-homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun (2016) 7:11459. doi: 10.1038/ncomms11459
9. Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature (2011) 472:437–42. doi: 10.1038/nature09965
10. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol (2009) 207:111–6. doi: 10.1016/j.jneuroim.2008.12.002
11. Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol (2001) 120:170–9. doi: 10.1016/S0165-5728(01)00421-0
12. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol Psychiatry (2015) 20:440–6. doi: 10.1038/mp.2014.59
13. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
14. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (2016) 351:933–9. doi: 10.1126/science.aad0314
15. Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature (2017) 549:528–32. doi: 10.1038/nature23910
16. Sandler RH, Finegold SM, Bolte ER, Buchanan CP, Maxwell AP, Vaisanen ML, et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J Child Neurol (2000) 15:429–35. doi: 10.1177/088307380001500701
17. Urbano M, Okwara L, Manser P, Hartmann K, Herndon A, Deutsch SI. A trial of d-cycloserine to treat stereotypies in older adolescents and young adults with autism spectrum disorder. Clin Neuropharmacol (2014) 37:69–72. doi: 10.1097/WNF.0000000000000033
18. Vuong HE, Hsiao EY. Emerging roles for the gut microbiome in autism spectrum disorder. Biol Psychiatry (2017) 81:411–23. doi: 10.1016/j.biopsych.2016.08.024
19. Bey AL, Gorman MP, Gallentine W, Kohlenberg TM, Frankovich J, Jiang YH, et al. Subacute neuropsychiatric syndrome in girls with SHANK3 mutations responds to immunomodulation. Pediatrics (2020) 145:e20191490. doi: 10.1542/peds.2019-1490
20. Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA, et al. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron (2019) 101:246–59.e6. doi: 10.1016/j.neuron.2018.11.018
21. Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa-Mattioli M. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell (2016) 165:1762–75. doi: 10.1016/j.cell.2016.06.001
23. Yu CD, Xu QJ, Chang RB. Vagal sensory neurons and gut-brain signaling. Curr Opin Neurobiol (2020) 62:133–40. doi: 10.1016/j.conb.2020.03.006
24. Han Q, Kim YH, Wang X, Liu D, Zhang ZJ, Bey AL, et al. SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron (2016) 92(6):1279–93. doi: 10.1016/j.neuron.2016.11.007
25. Orefice LL, Mosko JR, Morency DT, Wells MF, Tasnim A, Mozeika SM, et al. Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cell (2019) 178:867–86.e24. doi: 10.1016/j.cell.2019.07.024
26. Bey AL, Wang X, Yan H, Kim N, Passman RL, Yang Y, et al. Brain region-specific disruption of Shank3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl Psychiatry (2018) 8(1):94. doi: 10.1038/s41398-018-0142-6
27. Agarwal N, Offermanns S, Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis (2004) 38:122–9. doi: 10.1002/gene.20010
28. Bang S, Donnelly CR, Luo X, Toro-Moreno M, Tao X, Wang Z, et al. Activation of GPR37 in macrophages confers protection against infection-induced sepsis and pain-like behaviour in mice. Nat Commun (2021) 12:1704. doi: 10.1038/s41467-021-21940-8
29. Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lu N, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest (2014) 124:1173–86. doi: 10.1172/JCI72230
30. Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature (2007) 448:39–43. doi: 10.1038/nature05901
31. Zanos TP, Silverman HA, Levy T, Tsaava T, Battinelli E, Lorraine PW, et al. Identification of cytokine-specific sensory neural signals by decoding murine vagus nerve activity. Proc Natl Acad Sci USA (2018) 115:E4843–52. doi: 10.1073/pnas.1719083115
32. Mishra V, Gautier NM, Glasscock E. Simultaneous video-EEG-ECG monitoring to identify neurocardiac dysfunction in mouse models of epilepsy. J Vis Exp (2018) (131):57300. doi: 10.3791/57300-v
33. Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH, et al. Nociceptor-specific gene deletion using heterozygous NaV1.8-cre recombinase mice. Pain (2005) 113:27–36. doi: 10.1016/j.pain.2004.08.015
34. Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, et al. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain (2012) 153:2017–30. doi: 10.1016/j.pain.2012.04.022
35. Vriens J, Nilius B, Voets T. Peripheral thermosensation in mammals. Nat Rev Neurosci (2014) 15:573–89. doi: 10.1038/nrn3784
36. Patapoutian A, Peier AM, Story GM, Viswanath V. ThermoTRP channels and beyond: Mechanisms of temperature sensation. Nat Rev Neurosci (2003) 4:529–39. doi: 10.1038/nrn1141
37. Pappas AL, Bey AL, Wang X, Rossi M, Kim YH, Yan H, et al. Deficiency of Shank2 causes mania-like behavior that responds to mood stabilizers. JCI Insight (2017) 2:e92052. doi: 10.1172/jci.insight.92052
38. Kolevzon A, Delaby E, Berry-Kravis E, Buxbaum JD, Betancur C. Neuropsychiatric decompensation in adolescents and adults with phelan-McDermid syndrome: a systematic review of the literature. Mol Autism (2019) 10:50. doi: 10.1186/s13229-019-0291-3
39. van Hoorn A, Carpenter T, Oak K, Laugharne R, Ring H, Shankar R. Neuromodulation of autism spectrum disorders using vagal nerve stimulation. J Clin Neurosci (2019) 63:8–12. doi: 10.1016/j.jocn.2019.01.042
40. Wang Z, Yuan X, Zhang Q, Wen J, Cheng T, Qin X, et al. Effects of stable vagus nerve stimulation efficacy on autistic behaviors in ten pediatric patients with drug resistant epilepsy: An observational study. Front Pediatr (2022) 10:846301. doi: 10.3389/fped.2022.846301
41. Torres-Rosas R, Yehia G, Pena G, Mishra P, del Rocio Thompson-Bonilla M, Moreno-Eutimio MA, et al. Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat Med (2014) 20:291–5. doi: 10.1038/nm.3479
42. Liu S, Wang Z, Su Y, Qi L, Yang W, Fu M, et al. A neuroanatomical basis for electroacupuncture to drive the vagal-adrenal axis. Nature (2021) 598:641–5. doi: 10.1038/s41586-021-04001-4
43. Kupari J, Haring M, Agirre E, Castelo-Branco G, Ernfors P. An atlas of vagal sensory neurons and their molecular specialization. Cell Rep (2019) 27:2508–23.e4. doi: 10.1016/j.celrep.2019.04.096
44. Song K, Wang H, Kamm GB, Pohle J, Reis FC, Heppenstall P, et al. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science (2016) 353:1393–8. doi: 10.1126/science.aaf7537
45. Haraguchi K, Kawamoto A, Isami K, Maeda S, Kusano A, Asakura K, et al. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci (2012) 32:3931–41. doi: 10.1523/JNEUROSCI.4703-11.2012
46. Wang G, Cao L, Liu X, Sieracki NA, Di A, Wen X, et al. Oxidant sensing by TRPM2 inhibits neutrophil migration and mitigates inflammation. Dev Cell (2016) 38:453–62. doi: 10.1016/j.devcel.2016.07.014
Keywords: autism, nodose ganglion, sepsis, TRPM2, TRPV1, vagus nerve, pain, inflammation
Citation: Zhang L, Bang S, He Q, Matsuda M, Luo X, Jiang Y-H and Ji R-R (2023) SHANK3 in vagal sensory neurons regulates body temperature, systemic inflammation, and sepsis. Front. Immunol. 14:1124356. doi: 10.3389/fimmu.2023.1124356
Received: 15 December 2022; Accepted: 25 January 2023;
Published: 08 February 2023.
Edited by:
Ping-Heng Tan, Chi Mei Medical Center, TaiwanReviewed by:
Haitham Amal, Hebrew University of Jerusalem, IsraelCopyright © 2023 Zhang, Bang, He, Matsuda, Luo, Jiang and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ru-Rong Ji, cnUtcm9uZy5qaUBkdWtlLmVkdQ==
†These authors have contributed equally to this work
†ORCID: Ru-Rong Ji, https://orcid.org/0000-0002-9355-3688
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.