 Ibrahim Batal
Ibrahim Batal Pascale Khairallah
Pascale Khairallah Astrid Weins3
Astrid Weins3 Nicole K. Andeen
Nicole K. Andeen Michael B. Stokes
Michael B. Stokes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 23 February 2023
Sec. Alloimmunity and Transplantation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1124249
This article is part of the Research TopicRole of the Immune System in Renal Transplantation: Importance, Mechanism, and TherapyView all 12 articles
Primary focal segmental glomerulosclerosis (FSGS), typically characterized by diffuse podocyte foot process effacement and nephrotic syndrome (diffuse podocytopathy), is generally attributed to a circulating permeability factor. Primary FSGS can recur after transplantation where it manifests as diffuse foot process effacement in the early stages, with subsequent evolution of segmental sclerotic lesions. Previous published literature has been limited by the lack of stringent selection criteria to define primary FSGS. Although immunogenetic factors play an important role in many glomerular diseases, their role in recurrent primary FSGS post-transplantation has not been systematically investigated. To address this, we retrospectively studied a multicenter cohort of 74 kidney allograft recipients with end stage kidney disease due to primary FSGS, confirmed by clinical and histologic parameters. After adjusting for race/ethnicity, there was a numeric higher frequency of HLA-A30 antigen in primary FSGS (19%) compared to each of 22,490 healthy controls (7%, adjusted OR=2.0, P=0.04) and 296 deceased kidney donors (10%, OR=2.1, P=0.03). Within the group of transplant patients with end stage kidney disease due to primary FSGS, donor HLA-A30 was associated with recurrent disease (OR=9.1, P=0.02). Multivariable time-to-event analyses revealed that recipients who self-identified as Black people had lower risk of recurrent disease, probably reflecting enrichment of these recipients with APOL1 high-risk genotypes. These findings suggest a role for recipient and donor immunogenetic makeup in recurrent primary FSGS post-transplantation. Further larger studies in well-defined cohorts of primary FSGS that include high-resolution HLA typing and genome-wide association are necessary to refine these hereditary signals.
Focal segmental glomerulosclerosis (FSGS) is a histologic pattern of glomerular scarring associated with diverse forms of kidney damage (1). Primary FSGS is characterized by diffuse foot process effacement with nephrotic syndrome (diffuse podocytopathy; DP). The pathogenesis of primary FSGS remain unknown but clinical and experimental evidence support the existence of circulating permeability factor(s) that causes primary diffuse podocyte injury (1), which, lead to podocyte loss, segmental adhesion of the injured tuft to Bowman’s capsule (segmental glomerulosclerosis) and eventually to global glomerulosclerosis (2). Since these putative permeability factors remain poorly characterized, the diagnosis of primary FSGS currently relies on clinical and pathologic findings, and exclusion of known secondary causes.
Primary FSGS can recur following kidney transplantation and has a negative impact on graft survival. Recurrent primary FSGS manifests initially as nephrotic syndrome and diffuse foot process effacement, identical to minimal change disease (MCD), while segmental sclerotic lesions develop later (3, 4), probably reflecting cumulative podocyte depletion in susceptible individuals (5).
In addition to primary FSGS, the 2021 Kidney Disease Improving Global Outcomes (KDIGO) classification of FSGS includes three other categories (1) Secondary FSGS, caused by viruses, drugs, or, more commonly, adaptive responses to reduction of functioning nephrons and compensatory glomerular hyperfiltration, usually associated with focal foot process effacement and subnephrotic proteinuria (2) Genetic FSGS, which is most commonly monogenic in nature, and include familial, syndromic, and sporadic cases, and (3) Undetermined FSGS, characterized by focal foot process effacement, proteinuria without the nephrotic syndrome, and no identified secondary or genetic causes (6).
However, the current classification of FSGS is not ideal. The diagnosis of secondary FSGS can be difficult as, depending on the time of presentation, viral and drug-induced causes may go unnoticed. Diagnosing genetic FSGS is also challenging. While the most common monogenic causes of FSGS can now be efficiently identified using targeted gene panels, the list of genetic causes is continually expanding (7). Furthermore, it is increasingly apparent that common inherited variants, such as HLA polymorphisms, may also contribute to the risk of primary FSGS (8), which complicates the current definition of genetic FSGS. Equally important, classifying FSGS in patients with Apolipoprotein L1 (APOL1) kidney risk variants is particularly problematic (9). Nephropathic APOL1 variants occur with high frequency but low penetrance in populations with recent African ancestry, and may contribute to a higher incidence of kidney diseases from diverse etiologies (e.g., HIV-associated nephropathy, COVID-19 associated nephropathy, lupus nephritis, and hypertensive nephrosclerosis) (10, 11). That said, investigators from Mayo Clinic have shown that defining primary FSGS based on the presence of nephrotic syndrome and >80% foot process effacement is effective in excluding adaptive and monogenic forms of FSGS (12).
Despite the negative impact of recurrent DP on allograft survival (13), our knowledge of the immunologic and inherited factors associated with disease recurrence is extremely limited. This reflects the small sample size of published studies, the difficulty of completely excluding non-primary forms of FSGS, and the fact that FSGS in the allograft may arise from etiologies other than recurrent disease (e.g., adaptive FSGS from reduced nephron number and drug toxicity). In this report, we use the more expansive term DP to include cases of early recurrent primary FSGS that are characterized by nephrotic syndrome with complete or near-complete foot process effacement (≥ 80% of glomerular capillary surface area) but lack segmental sclerosis on light microscopy (14, 15).
Previous studies by us and others showed an association between donor inherited factors and recurrence of both membranous nephropathy (16, 17) and IgA nephropathy (18). We hypothesized that immunogenetic background in the donors and/or the recipients contributes to the development of recurrent DP/primary FSGS. In this report, we examined the frequency of HLA antigens in a large multicenter cohort of kidney transplant patients with end stage kidney disease (ESKD) attributed to DP/primary FSGS. Here for the first time we show that patients with ESKD due to primary FSGS tend to be enriched with HLA-A30 antigen while donor HLA-A30 is associated with increased risk of recurrent disease after transplantation.
This study was a retrospective multicenter study that included the pathology archives of three North American medical centers: Columbia University Irving Medical Center (CUIMC, New York, NY), Brigham and Women’s Hospital (BWH, Boston, MA), and Oregon Health & Science University (OHSU, Portland, OR). Each center collected data under approval by their Institutional Review Boards.
In this report, strict inclusion criteria were used to select our cohort:
(1) Recurrent DP (n=47) was defined as recurrence of nephrotic syndrome (proteinuria ≥ 3.5 g and serum albumin ≤ 3.5 g/dL) in kidney allograft recipients with ESKD attributed to DP/FSGS, accompanied by allograft biopsies (performed in all but one recipient) showing foot process effacement involving ≥ 80% of glomerular capillary surface area. The remaining patient had a biopsy-proven MCD in the native kidney followed by FSGS, and presented 8 days after transplantation with nephrotic syndrome (urine protein/creatinine: 10 g/g and serum albumin 1.6 g/dL) and serum creatinine of 1.1 mg/dL. Native kidney biopsies and/or pathology reports were available for 29 of these subjects and showed FSGS (n=21) and MCD (n=8, including 7 patients with subsequent native kidney biopsies that showed FSGS). The remaining 18 cases had a clinical diagnosis of FSGS but pathologic materials were not available for re-review.
(2) Non-recurrent DP (n=27) was defined as absence of nephrotic syndrome post-transplantation in kidney transplant recipients who developed nephrotic syndrome in their native kidney (≥ 3.5 g of proteinuria and ≤ 3.5 g/dL of serum albumin) with a concurrent native kidney biopsy showing near complete foot process effacement (involving ≥ 80% of the glomerular capillary surface area). Of these, 24 subjects had native kidney biopsy showing FSGS and 3 demonstrated MCD (including 2 patients with subsequent native biopsies showing development of FSGS).
Serologic and/or molecular typing for HLA-A, -B, -DR, and -DQ was performed for both donors and recipients. The outcome was recurrence of DP in the kidney allograft and the subjects were censored at loss of follow-up or allograft failure. Studied variables including recipient demographics (age, sex, and race), donor demographics, allograft source, HLA-mismatch (A, B, and DR: scale 0-6), prior kidney transplant, and induction immunotherapy, were extracted from the medical record.
To test which HLA antigens that are potentially associated with DP, the most frequent HLA antigens in our transplant cohort were compared to two group of controls:
(1) External controls of healthy US residents (n=22,490) from National Bone Marrow Donor Program (19), providing HLA allelic frequencies for reference comparisons.
(2) Internal controls of deceased kidney donors whose kidneys were transplanted at CUIMC in 2010 onwards that were matched with our cohort of DP patients with regard to self-reported ancestry (n=296; for each patient with DP, four deceased donors were matched by ancestry).
Statistical analyses were performed using Prism 9 (Graphpad Inc, San Diego, CA) and SPSS Statistics, 26.0 (IBM, Armonk, NY). Categorical data were compared using Fisher Exact test while continuous data were compared using the Mann-Whitney test. With the except of discovery study, P less than 0.05 with two-sided hypothesis testing were considered statistically significant.
We identified 74 kidney allograft recipients who had ESKD due to DP/primary FSGS [CUIMC (n=60), OHSU (n=9), and BWH (n=5)]. Subjects were followed-up for a median of 68 (IQRs: 17, 107) months after transplantation. During follow-up, 47 (64%) patients developed recurrent disease [median 34 (IQRs: 12, 210) days post-transplantation] and 40 (54%) developed allograft failure [median 26 (IQRs: 14, 70) months after transplantation].
The median age at transplantation was 35 years (Supplemental Table 1). The cohort included 51% women, 27% Black recipients, 9% with prior kidney transplant, 40% recipients of living-related allografts, and a median donor-recipient HLA mismatch of 4. Donors had a median age of 35 years and included 57% women and 27% Black subjects. Most of the patients received induction therapy with a depleting agent (83%; including 76% with Thymoglobulin and 7% with Alemtuzumab) (Supplemental Table 1).
We initially identified the most frequent HLA antigens in all 74 recipients in this cohort [A2: 45%, A1: 20%, and A30: 19% in the A region; B44: 19%, B51: 18%, and B35: 16% in the B region; DR15: 28%, DR4: 27%, and DR7: 24% in the DR region; and DQ6: 47%, DQ7: 41%, and DQ2: 29% in the DQ region] (Supplemental Table 2).
We compared the frequency of these antigens to external healthy controls (n=22,490) (19). Since 12 HLA antigens have been compared, a Bonferroni-corrected significance cutoff of 0.05/12 = 0.004 was utilized to define statistical significance. HLA-A30 was the only antigen that was more prevalent in recipients with DP (OR=3.0, P=0.0008). However, this particular antigen, which is typically more prevalent in Black people (Supplemental Table 2), did not reach Bonferroni-threshold for statistical significance after adjusting for race/ethnicity using Cochran–Mantel–Haenszel test (adjusted OR=2.0, P=0.04) (Figure 1A). Using conditional logistic regression conditioned on self-reported race/ethnicity matching, HLA-A30 was more common in DP compared to a second set of controls composed of 296 deceased kidney donors matched by race/ethnicity [14/74 (19%) vs. 29/296 (10%), OR=2.1, P=0.03] (Figure 1A).
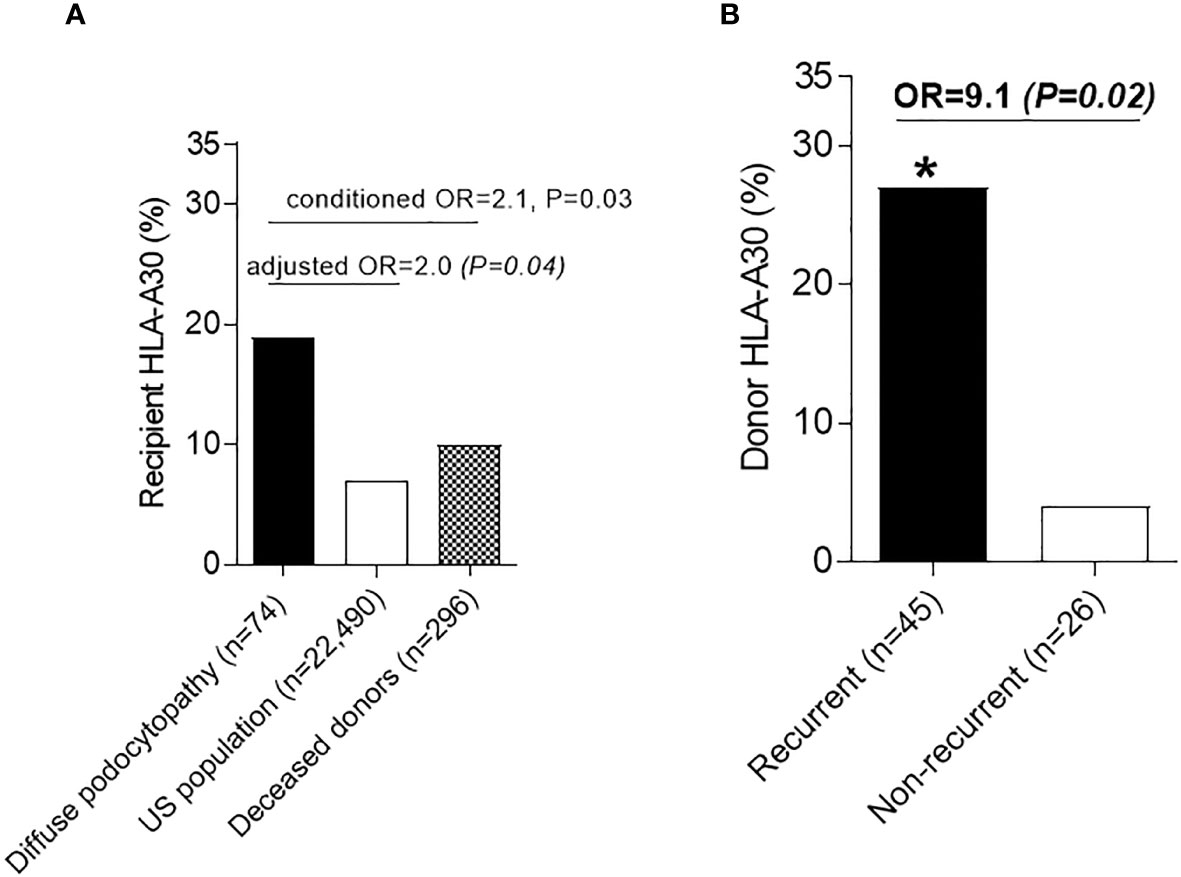
Figure 1 Human Leukocyte antigens in diffuse podocytopathy (A) Prevalence of HLA-A30 antigen in cases with diffuse podocytopathy (patients with end stage kidney disease due to diffuse podocytopathy, n=74) compared to US population (n=22,490) and deceased kidney donors matched to cases with regard to race/ethnicity (n=296). (B) Prevalence of HLA-A30 in the kidney donors of patients with recurrent (n=42) vs. non-recurrent (n=22) diffuse podocytopathy. Donor typing for class-I was not available for 2 donors with recurrent diseases and 1 without recurrent disease. * (significant P value).
To test whether HLA-A30 is associated with recurrent DP, we compared the frequencies of this antigen in both recipients and donors with recurrent and non-recurrent disease. While the proportion of recipient HLA-A30 was not significantly different [6/47 (13%) recurrent vs. 8/27 (30%) non-recurrent, P=0.12], the frequency of donor HLA-A30 was higher in recurrent disease [12/45 (27%) recurrent DP vs. 1/26 (4%) non-recurrent DP, OR=9.1, P=0.02)] (Figure 1B). Despite the small sample size, the results for the association between HLA-A30 and recurrent disease remained significant even after adjustment for donor age and donor Black race (binary logistic regression: OR=11.0, P=0.03). The frequency of other HLA antigens did not differ between recurrent and non-recurrent diseases (Supplemental Table 3). Overall, donor HLA-A30 had 96% specificity, 27% sensitivity, 92% positive predictive value, and 43% negative predictive value for predicting recurrence of DP (Supplemental Table 4). The breakdown for the association of HLA-A30 in the recipients and donors with recurrent disease is also presented in Supplemental Table 5.
To identify independent predictors for recurrent DP, all transplant patients with ESKD due to DP (n=74) were studied in a time-to event model, where DP recurrence was considered as the outcome of interest (Table 1). Univariable and multivariable analyses showed that Black recipient race was the only independent variable associated with the outcome, where it was protective against recurrent disease [(aHR)=0.31, P=0.008] (Table 1). When the relation between recipient Black recipients and recurrent DP was explored further, we found that 4/5 (80%) of self-identified Black recipients with non-recurrent disease that were genotyped for APOL1 had high-risk genotypes (3 G1/G1 and 1 G1/G2).
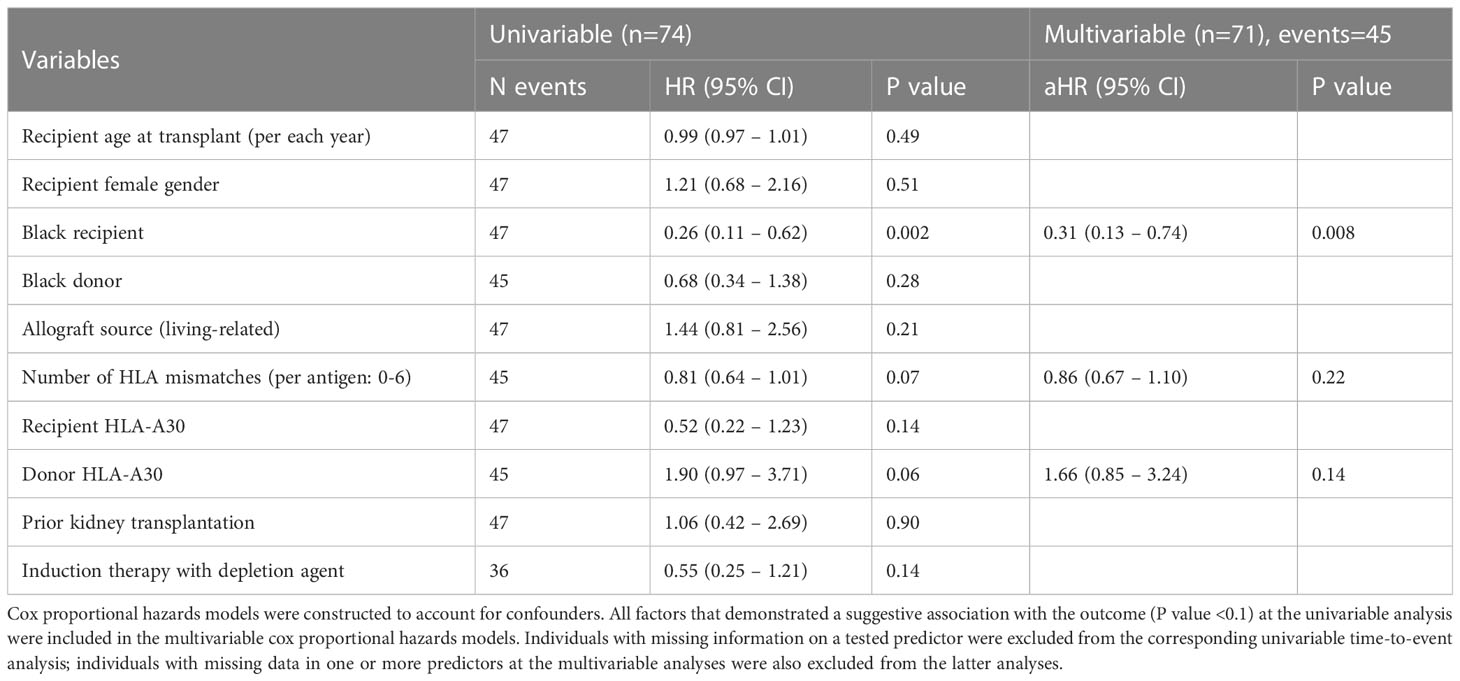
Table 1 Univariable and multivariable analyses of the associations with recurrent disease in patients with end stage kidney disease due to diffuse podocytopathy.
Primary FSGS and MCD are DP, characterized by diffuse foot process effacement and nephrotic syndrome (6). In the native kidney, there is growing recognition that abnormalities of T and B cell immunity are common in DP and may play a pathogenic role (20). This includes the association with allergy, immunization, and B cell neoplasms, as well as responsiveness to immunomodulatory therapies, including corticosteroid therapy and B cell depletion (21, 22). With regard to inherited factors, genome-wide association studies (GWAS) have shown that steroid-sensitive nephrotic syndrome, including MCD and primary FSGS, is associated with genomic susceptibility loci in HLA regions (23–25) and non-HLA regions (23, 26), most of which are linked to the immune system. Lastly, in addition to other associations, nephropathic APOL1 genotypes have also been linked to FSGS (27), sometimes with diffuse foot process effacement. Together, these findings suggest that DP/primary FSGS is a polygenic disease associated with immune dysregulation and immunogenetic susceptibility.
Although the pathogenesis of recurrent DP post-transplantation is unknown, the existence of a circulating glomerular permeability factor is supported by several observations, including rapid recurrence of the nephrotic syndrome following transplantation, induction of proteinuria in the rat using serum from a patient with recurrent FSGS (28), and resolution of recurrent nephrotic syndrome following re-transplantation of the kidney into a different recipient (29).
Uffing et al. published the largest study to date of recurrent FSGS in the kidney allograft, which included 176 kidney transplant recipients who developed ESKD from FSGS (13). In addition to the association with White recipient race, older recipient age, and history of bilateral nephrectomy, this study showed a significant association between recurrent FSGS and lower recipient body mass index (BMI) (13). The latter finding may reflect the association between higher BMI and adaptive FSGS secondary to obesity in the native kidney, which is unlikely to recur early after transplantation in the form of nephrotic syndrome. Interestingly, a few prior studies have shown that young recipient age, rather than old recipient age as shown by Uffing et al., was a risk factor for FSGS recurrence (30, 31). Furthermore, depending on the study, recurrence rate for primary FSGS in the kidney allograft varies from 20% to 52% of cases (32, 33). These contradicting results suggest contamination of the previously studied cohorts by secondary forms of FSGS and support the crucial need for using rigorous criteria to exclude non-primary cases of FSGS when studying disease recurrence after transplantation.
Although HLA antigens have emerged as important immunogenetic risk factors in many immune-mediated kidney diseases (34–37), their role in recurrent disease has not been systematically examined in a pure cohort of DP/primary FSGS. One study attempted to assess this issue in a pediatric transplant population with ESKD attributed to FSGS and found an association between recurrent FSGS and a few HLA class-II antigens, including DQ2, DR7, and DR53 (38). However, that study was registry-based and lacked data on clinical and histologic parameters at time of diagnosis of FSGS in the native kidney, which further increases the possibility of selection bias and contamination by non-primary forms of FSGS, which in our experience comprise the bulk of cases labeled as FSGS-induced ESKD.
In this report, we used stringent inclusion criteria requiring the presence of both nephrotic syndrome (≥ 3.5 g of proteinuria, ≤ 3.5 g/dL serum albumin) and ≥ 80% of foot process effacement to assemble a relatively pure cohort of DP/primary FSGS (15). Despite the contribution of three major transplant centers, we only could identify 74 transplant patients fulfilling our rigorous selection criteria. Notably, our study showed that several patients with ESKD due to primary FSGS had prior native kidney biopsies that demonstrated MCD. These findings further support the intimate association between these two morphologic manifestations of DP.
Although not reaching statistical significance by Bonferroni criteria in this relatively small sample, our data suggested an association between primary FSGS in the native kidney and HLA-A30 antigen. Prior studies have shown an association between HLA-A30 and several autoimmune diseases, including type 1 diabetes mellitus, vitiligo, and systemic sclerosis (39–41), which suggest that HLA-A30 may contribute to immune dysregulation. Our study also demonstrated an association between HLA-A30 in the donor and recurrent primary FSGS. It is conceivable that donor HLA antigens may influence antigen presentation by donor immune cells or podocytes, which can function as antigen-presenting cells (42), cross reactivity/molecular mimicry, or acceleration of podocyte death in immunologically susceptible patients.
Despite the high positive predictive value (92%) of HLA-A30 in predicting recurrent disease, it was not surprising that this relatively low frequency HLA antigen, which has a prevalence of 7% in the general population and 19% in patients with ESKD secondary to primary FSGS, did not reach statistical significance as an independent predictor for recurrent disease in this small cohort.
With regard to recipient inherited factors, a few prior studies have shown that White recipient race is a risk factor for developing recurrent FSGS (13, 43, 44). We propose that White recipient race may not be a real risk factor but rather may be confounded by the genetic make-up in Black recipients, in whom FSGS may be less likely to recur after receiving a kidney from a donor with a different genomic constitution. Indeed, multivariable analyses in the current study showed lower risk of recurrence in Black subjects with FSGS/DP. This might reflect the increased prevalence of high-risk APOL1 genotypes (80%) in Black recipients with non-recurrent DP. It is now well accepted that donor APOL1 kidney risk variants may be a risk factor for podocyte damage in the kidney allograft (45, 46) while APOL1 status in the recipient is less likely to influence recurrent disease. Importantly, while it is still unclear how to classify FSGS in patients with APOL1 high-risk genotypes, it would be interesting in the future to systematically assess APOL1 genotypes in recipients with vs. without recurrent DP/primary FSGS.
Our findings should be interpreted in light of several limitations, including small sample size, low resolution of HLA typing, lack of HLA-DQ typing in several recipients and donors, lack of APOL1 genotyping in most Black recipients, and the lack of detailed clinical history (e.g., recipient BMI and whether native kidney nephrectomies were performed or not).
In conclusion, a strict definition of recurrent DP/primary FSGS is an initial step to advance our understanding of recurrent disease after transplantation. Our pilot study represents the first attempt to systematically assess immunogenetic predictors of recurrent disease in a well-defined cohort of transplant patients with ESKD attributed to DP/primary FSGS. Our results suggest that donor HLA-A30 is a potential risk factors for recurrent DP/primary FSGS while Black recipient race may be a potential protective factor. However, it is still possible that the observed association between HLA-A30 and primary FSGS is affected by the differential distribution of HLA-A30 across worldwide populations. Therefore, future larger GWAS studies that properly control for genetic ancestry are crucial to examine the effects of HLA-A30 in a more comprehensive manner, to refine the donor and recipient genomic signals contributing to recurrent primary FSGS, and to obtain better insights into the pathobiology of recurrent DP (47). The ultimate goals of such efforts would be to guide the clinician towards improving donor-recipient matching and develop targeted approaches to prevent recurrence and improve allograft survival.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Columbia University Institutional Review Board. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
IB participated in research design, performance of research, data analyses, and in the writing of the manuscript. PK participated in performance of research and writing of the manuscript. AW participated in research design, performance of research, and writing the manuscript. NA participated in research design, performance of research, and writing the manuscript. MBS participated in research design and writing of manuscript. PK is currently at the Baylor College of Medicine Houston, TX. All authors contributed to the article and approved the submitted version.
IB is supported by the 2022 Nelson Faculty Development Award. AW is supported by a NephCure Kidney International – NephCure-Neptune Ancillary Study Award.
We thank Carley Shaut (Medicine, Division of Nephrology, Oregon Health & Science University), Melissa Yeung (Medicine, Division of Nephrology, Brigham and Women’s Hospital and Harvard Medical School), Xiaoyi Ye (Medicine, Division of Nephrology, Hartford Hospital), Emily Daniel, Hilda Fernandez, and Francesca Zanoni (Medicine, Division of Nephrology, Columbia University Irving Medical Center), and Geo Serban and Elena-Rodica Vasilescu (Pathology and Cell Biology, Columbia University Irving Medical Center) for providing missing clinical and laboratory data. We thank Krzysztof Kiryluk for his advices regarding statistical analyses.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1124249/full#supplementary-material
BMI, body mass index; BWH, Brigham and Women’s Hospital; CUIMC, Columbia University Irving Medical Center; DP, diffuse podocytopathy; ESKD, end stage kidney disease; FSGS, focal segmental glomerulosclerosis; GWAS, genome-wide association studies; KDIGO, Kidney Disease Improving Global Outcomes; OHSU, Oregon Health & Science University.
1. D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med (2011) 365(25):2398–411. doi: 10.1056/NEJMra1106556
2. Lim BJ, Yang JW, Do WS, Fogo AB. Pathogenesis of focal segmental glomerulosclerosis. J Pathol Transl Med (2016) 50(6):405–10. doi: 10.4132/jptm.2016.09.21
3. Nadasdy T, Ivanyi B, Marofka F, Orvos H, Mohacsi G, Ormos J. Two cases of recurrent focal and segmental glomerulosclerosis in renal allografts. Nephrol Dial Transplant. (1991) 6(5):375–6. doi: 10.1093/ndt/6.5.375
4. Schachter ME, Monahan M, Radhakrishnan J, Crew J, Pollak M, Ratner L, et al. Recurrent focal segmental glomerulosclerosis in the renal allograft: Single center experience in the era of modern immunosuppression. Clin Nephrol. (2010) 74(3):173–81. doi: 10.5414/CNP74173
5. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. (2016) 12(12):768–76. doi: 10.1038/nrneph.2016.147
6. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM. Kidney disease: Improving global outcomes glomerular diseases work g. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int (2021) 100(4S):S1–S276. doi: 10.1016/j.kint.2021.05.021
7. Sambharia M, Rastogi P, Thomas CP. Monogenic focal segmental glomerulosclerosis: A conceptual framework for identification and management of a heterogeneous disease. Am J Med Genet C Semin Med Genet (2022) 190(3):377–98. doi: 10.1002/ajmg.c.31990
8. Gerbase-DeLima M, Pereira-Santos A, Sesso R, Temin J, Aragao ES, Ajzen H. Idiopathic focal segmental glomerulosclerosis and HLA antigens. Braz J Med Biol Res (1998) 31(3):387–9. doi: 10.1590/S0100-879X1998000300010
9. Dummer PD, Limou S, Rosenberg AZ, Heymann J, Nelson G, Winkler CA, et al. APOL1 kidney disease risk variants: An evolving landscape. Semin Nephrol. (2015) 35(3):222–36. doi: 10.1016/j.semnephrol.2015.04.008
10. Daneshpajouhnejad P, Kopp JB, Winkler CA, Rosenberg AZ. The evolving story of apolipoprotein L1 nephropathy: The end of the beginning. Nat Rev Nephrol. (2022) 18(5):307–20. doi: 10.1038/s41581-022-00538-3
11. Larsen CP, Beggs ML, Saeed M, Ambruzs JM, Cossey LN, Messias NC, et al. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod Pathol (2015) 28(1):95–102. doi: 10.1038/modpathol.2014.92
12. Miao J, Pinto EVF, Hogan MC, Erickson SB, El Ters M, Bentall AJ, et al. Identification of genetic causes of focal segmental glomerulosclerosis increases with proper patient selection. Mayo Clin Proc (2021) 96(9):2342–53. doi: 10.1016/j.mayocp.2021.01.037
13. Uffing A, Perez-Saez MJ, Mazzali M, Manfro RC, Bauer AC, de Sottomaior Drumond F, et al. Recurrence of FSGS after kidney transplantation in adults. Clin J Am Soc Nephrol. (2020) 15(2):247–56. doi: 10.2215/CJN.08970719
14. Jacobs-Cacha C, Vergara A, Garcia-Carro C, Agraz I, Toapanta-Gaibor N, Ariceta G, et al. Challenges in primary focal segmental glomerulosclerosis diagnosis: from the diagnostic algorithm to novel biomarkers. Clin Kidney J (2021) 14(2):482–91. doi: 10.1093/ckj/sfaa110
15. De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: A clinicopathologic approach. J Am Soc Nephrol. (2018) 29(3):759–74. doi: 10.1681/ASN.2017090958
16. Batal I, Vasilescu ER, Dadhania DM, Adel AA, Husain SA, Avasare R, et al. Association of HLA typing and alloimmunity with posttransplantation membranous nephropathy: A multicenter case series. Am J Kidney Dis (2020) 76(3):374–83. doi: 10.1053/j.ajkd.2020.01.009
17. Berchtold L, Letouze E, Alexander MP, Canaud G, Logt AV, Hamilton P, et al. HLA-d and PLA2R1 risk alleles associate with recurrent primary membranous nephropathy in kidney transplant recipients. Kidney Int (2021) 99(3):671–85. doi: 10.1016/j.kint.2020.08.007
18. Kavanagh CR, Zanoni F, Leal R, Jain NG, Stack MN, Vasilescu ER, et al. Clinical predictors and prognosis of recurrent IgA nephropathy in the kidney allograft. Glomerular Dis (2022) 2(1):42–53. doi: 10.1159/000519834
19. Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the united states population. Hum Immunol (2007) 68(9):779–88. doi: 10.1016/j.humimm.2007.04.005
20. Campbell RE, Thurman JM. The immune system and idiopathic nephrotic syndrome. Clin J Am Soc Nephrol (2022) 17(12):1823–34. doi: 10.2215/CJN.07180622
21. Caster DJ, Magalhaes B, Pennese N, Zaffalon A, Faiella M, Campbell KN, et al. Efficacy and safety of immunosuppressive therapy in primary focal segmental glomerulosclerosis: A systematic review and meta-analysis. Kidney Med (2022) 4(8):100501. doi: 10.1016/j.xkme.2022.100501
22. Tedesco M, Mescia F, Pisani I, Allinovi M, Casazza G, Del Vecchio L, et al. The role of rituximab in primary focal segmental glomerular sclerosis of the adult. Kidney Int Rep (2022) 7(8):1878–86. doi: 10.1016/j.ekir.2022.05.024
23. Dufek S, Cheshire C, Levine AP, Trompeter RS, Issler N, Stubbs M, et al. Genetic identification of two novel loci associated with steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. (2019) 30(8):1375–84. doi: 10.1681/ASN.2018101054
24. Debiec H, Dossier C, Letouze E, Gillies CE, Vivarelli M, Putler RK, et al. Transethnic, genome-wide analysis reveals immune-related risk alleles and phenotypic correlates in pediatric steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. (2018) 29(7):2000–13. doi: 10.1681/ASN.2017111185
25. Jia X, Horinouchi T, Hitomi Y, Shono A, Khor SS, Omae Y, et al. Strong association of the HLA-DR/DQ locus with childhood steroid-sensitive nephrotic syndrome in the Japanese population. J Am Soc Nephrol. (2018) 29(8):2189–99. doi: 10.1681/ASN.2017080859
26. Jia X, Yamamura T, Gbadegesin R, McNulty MT, Song K, Nagano C, et al. Common risk variants in NPHS1 and TNFSF15 are associated with childhood steroid-sensitive nephrotic syndrome. Kidney Int (2020) 98(5):1308–22. doi: 10.1016/j.kint.2020.05.029
27. Friedman DJ, Pollak MR. APOL1 nephropathy: From genetics to clinical applications. Clin J Am Soc Nephrol. (2021) 16(2):294–303. doi: 10.2215/CJN.15161219
28. Zimmerman SW. Increased urinary protein excretion in the rat produced by serum from a patient with recurrent focal glomerular sclerosis after renal transplantation. Clin Nephrol. (1984) 22(1):32–8.
29. Gallon L, Leventhal J, Skaro A, Kanwar Y, Alvarado A. Resolution of recurrent focal segmental glomerulosclerosis after retransplantation. N Engl J Med (2012) 366(17):1648–9. doi: 10.1056/NEJMc1202500
30. Francis A, Trnka P, McTaggart SJ. Long-term outcome of kidney transplantation in recipients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2016) 11(11):2041–6. doi: 10.2215/CJN.03060316
31. Hickson LJ, Gera M, Amer H, Iqbal CW, Moore TB, Milliner DS, et al. Kidney transplantation for primary focal segmental glomerulosclerosis: Outcomes and response to therapy for recurrence. Transplantation (2009) 87(8):1232–9. doi: 10.1097/TP.0b013e31819f12be
32. Dall'Amico R, Ghiggeri G, Carraro M, Artero M, Ghio L, Zamorani E, et al. Prediction and treatment of recurrent focal segmental glomerulosclerosis after renal transplantation in children. Am J Kidney Dis (1999) 34(6):1048–55. doi: 10.1016/S0272-6386(99)70010-7
33. Tejani A, Stablein DH. Recurrence of focal segmental glomerulosclerosis posttransplantation: A special report of the north American pediatric renal transplant cooperative study. J Am Soc Nephrol. (1992) 2(12 Suppl):S258–63. doi: 10.1681/ASN.V212s258
34. Doxiadis II, De Lange P, De Vries E, Persijn GG, Claas FH. Protective and susceptible HLA polymorphisms in IgA nephropathy patients with end-stage renal failure. Tissue Antigens (2001) 57(4):344–7. doi: 10.1034/j.1399-0039.2001.057004344.x
35. Klouda PT, Manos J, Acheson EJ, Dyer PA, Goldby FS, Harris R, et al. Strong association between idiopathic membranous nephropathy and HLA-DRW3. Lancet (1979) 2(8146):770–1. doi: 10.1016/S0140-6736(79)92118-4
36. Phelps RG, Rees AJ. The HLA complex in goodpasture's disease: a model for analyzing susceptibility to autoimmunity. Kidney Int (1999) 56(5):1638–53. doi: 10.1046/j.1523-1755.1999.00720.x
37. Andeen NK, Smith KD, Vasilescu ER, Batal I. Fibrillary glomerulonephritis is associated with HLA-DR7 and HLA-B35 antigens. Kidney Int Rep (2020) 5(8):1325–7. doi: 10.1016/j.ekir.2020.05.010
38. Shaw BI, Ochoa A, Chan C, Nobuhara C, Gbadegesin R, Jackson AM, et al. HLA loci and recurrence of focal segmental glomerulosclerosis in pediatric kidney transplantation. Transplant Direct. (2021) 7(10):e748. doi: 10.1097/TXD.0000000000001201
39. Deng C, Tong N, Li X. [Association of HLA-A0205 and HLA-A30 with latent autoimmune diabetes in adults]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. (2010) 27(5):1089–94.
40. Finco O, Cuccia M, Martinetti M, Ruberto G, Orecchia G, Rabbiosi G. Age of onset in vitiligo: relationship with HLA supratypes. Clin Genet (1991) 39(1):48–54. doi: 10.1111/j.1399-0004.1991.tb02984.x
41. Del Rio AP, Sachetto Z, Sampaio-Barros PD, Marques-Neto JF, Londe AC, Bertolo MB. HLA markers for poor prognosis in systemic sclerosis Brazilian patients. Dis Markers. (2013) 35(2):73–8. doi: 10.1155/2013/301415
42. Goldwich A, Burkard M, Olke M, Daniel C, Amann K, Hugo C, et al. Podocytes are nonhematopoietic professional antigen-presenting cells. J Am Soc Nephrol. (2013) 24(6):906–16. doi: 10.1681/ASN.2012020133
43. Abbott KC, Sawyers ES, Oliver JD 3rd, Ko CW, Kirk AD, Welch PG, et al. Graft loss due to recurrent focal segmental glomerulosclerosis in renal transplant recipients in the united states. Am J Kidney Dis (2001) 37(2):366–73. doi: 10.1053/ajkd.2001.21311
44. Nehus EJ, Goebel JW, Succop PS, Abraham EC. Focal segmental glomerulosclerosis in children: multivariate analysis indicates that donor type does not alter recurrence risk. Transplantation (2013) 96(6):550–4. doi: 10.1097/TP.0b013e31829c2431
45. Santoriello D, Husain SA, De Serres SA, Bomback AS, Crew RJ, Vasilescu ER, et al. Donor APOL1 high-risk genotypes are associated with increased risk and inferior prognosis of de novo collapsing glomerulopathy in renal allografts. Kidney Int (2018) 94(6):1189–98. doi: 10.1016/j.kint.2018.06.024
46. Oniszczuk J, Moktefi A, Mausoleo A, Pallet N, Malard-Castagnet S, Fourati S, et al. De novo focal and segmental glomerulosclerosis after COVID-19 in a patient with a transplanted kidney from a donor with a high-risk APOL1 variant. Transplantation (2021) 105(1):206–11. doi: 10.1097/TP.0000000000003432
Keywords: recurrent focal segmental glomerular sclerosis, kidney allograft, HLA, diffuse podocytopathy, focal segment glomerulosclerosis
Citation: Batal I, Khairallah P, Weins A, Andeen NK and Stokes MB (2023) The role of HLA antigens in recurrent primary focal segmental glomerulosclerosis. Front. Immunol. 14:1124249. doi: 10.3389/fimmu.2023.1124249
Received: 14 December 2022; Accepted: 06 February 2023;
Published: 23 February 2023.
Edited by:
Long Zheng, The second Affiliated Hospital of Zhejiang University School of Medicine, ChinaCopyright © 2023 Batal, Khairallah, Weins, Andeen and Stokes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ibrahim Batal, aWIyMzQ5QGN1bWMuY29sdW1iaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.