 Yinuo Gu1
Yinuo Gu1 Alan Chen-Yu Hsu2,3,4
Alan Chen-Yu Hsu2,3,4 Xu Zuo1
Xu Zuo1 Xiaoping Guo1
Xiaoping Guo1 Zhengjie Zhou1Shengyu Jiang1Zhuoer Ouyang1
Zhengjie Zhou1Shengyu Jiang1Zhuoer Ouyang1 Fang Wang1*
Fang Wang1*- 1Department of Pathogeny Biology, College of Basic Medical Sciences, Jilin University, Changchun, China
- 2Signature Research Program in Emerging Infectious Diseases, Duke - National University of Singapore (NUS) Graduate Medical School, Singapore, Singapore
- 3School of Medicine and Public Health, The University of Newcastle, Newcastle, NSW, Australia
- 4Viruses, Infections/Immunity, Vaccines and Asthma, Hunter Medical Research Institute, Newcastle, NSW, Australia
Influenza A virus (IAV) infection leads to severe inflammation, and while epithelial-driven inflammatory responses occur via activation of NF-κB, the factors that modulate inflammation, particularly the negative regulators are less well-defined. In this study we show that A20 is a crucial molecular switch that dampens IAV-induced inflammatory responses. Chronic exposure to low-dose LPS environment can restrict this excessive inflammation. The mechanisms that this environment provides to suppress inflammation remain elusive. Here, our evidences show that chronic exposure to low-dose LPS suppressed IAV infection or LPS stimulation-induced inflammation in vitro and in vivo. Chronic low-dose LPS environment increases A20 expression, which in turn positively regulates PPAR-α and -γ, thus dampens the NF-κB signaling pathway and NLRP3 inflammasome activation. Knockout of A20 abolished the inhibitory effect on inflammation. Thus, A20 and its induced PPAR-α and -γ play a key role in suppressing excessive inflammatory responses in the chronic low-dose LPS environment.
Introduction
IAV frequently causes severe infection with heightened inflammation that drives disease pathogenesis. While inflammation is essential in virus clearance, excessive inflammatory responses cause epithelial tissue destruction, acute lung injury and pneumonia (1, 2). Upon infection in airway epithelial cells, the primary site of IAV infection, IAV viral RNAs are recognized by the host pattern recognition receptors including retinoic acid inducible gene (RIG)-I and toll-like receptor (TLR)3. RIG-I binding to viral RNAs leads to activation of interferon regulatory factor (IRF)3 that facilitate the production of antiviral cytokines type I interferon (IFN). TLR3 promotes the activation of nuclear factor kappa-lightchain-enhancer of activated B cells (NF-κB), leading to the induction of pro-inflammatory cytokines such as interleukin (IL)-6 and tumour-necrosis-factor (TNF)α. Upon infection or stimulation, NLRP3, ASC, and pro-caspase-1 assembles to form an inflammasome complex, which activates pro-caspase-1 by cleavage. IL-1β is a potent inflammatory cytokine that is produced as a pro-protein that is then cleaved by caspase-1 (3).
It has been suggested that the host inflammatory response to IAV is more likely to have serious consequences than viral load (1).
The mechanisms that drive NF-κB pathway are well characterized, however the negative control factors that dampen NF-κB activation is less well studied. A recent study showed that chronic exposure of low-dose lipopolysaccharide (LPS) protected against the development of allergy and asthma (4, 5).
The molecular mechanisms underpinning this observation is unclear. We recently showed that A20 (also known as TNFα-induced protein 3) was crucial in limiting IAV-mediated NF-κB activation and IAV diseases in vitro and in vivo. The mechanisms by which low-grade LPS and A20 protect the host from heightened inflammation is unknown. In this study we determined that human airway epithelial cells and mice that were chronically exposed to low-dose LPS reduced subsequent IAV- and high-dose LPS-mediated NF-κB and NLRP3 activation, and this low-grade LPS tolerance was mediated by increased A20 and PPAR-α and PPAR-γ expression, the latter of which were also negative regulators of NF-κB.
Materials and methods
Cell culture and IAV
Human lung adenocarcinoma epithelial (A549) cells and Madin-Darby canine kidney (MDCK) cells were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). A549 cells were cultured in Ham’s F-12 K medium supplemented with 10% fetal bovine serum (FBS). MDCK cells were cultured in DMEM medium supplemented with 10% FBS. All cells were cultured with 5% CO2 at 37°C.
H1N1 influenza virus, A/Fort Monmouth/1/1947 virus (FM1), was propagated in the allantoic cavities of 9-11 day-old embryonated specific pathogen-free chicken eggs cultured at 37°C. After 48 h allantoic fluids of infected eggs were collected. Various dilutions of allantoic fluids were incubated with MDCK cells to determine the 50% tissue culture infectious dose (TCID50) calculated by the Reed-Muench method. TCID50 is 10-4.1. To determine the 50% lethal dose (LD50), the allantoic fluids were serially 10-fold diluted. The number of deaths and survivals in each dilution was used to calculate the LD50 by the Reed–Muench method. Based on the titrations, 1 LD50 of the allantoic fluids was determined to be 10-5.5.
Animals and animal experiments
Female BALB/c mice (6–8 weeks old) were purchased from the Yisi Laboratory Animal Technology Co., Ltd. (Changchun, China), and maintained in micro-isolator cages under specific pathogen-free conditions. The experimental manipulation of the mice was undertaken in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of Science & Technology of Jilin Province, China. All mice received human care in compliance with the 2011 Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The mouse experiments were approved by the ethics committee of The College of Basic Medical Sciences of Jilin University with the number 2022-467.
We intranasally administered with 100 ng LPS for chronic-LPSlo group or PBS for control group every other day. After 2 weeks, both groups of mice were treated with non-infectious allantoic fluid, H1N1 (10 LD50), and high-dose (10 µg/mL) LPS. Both H1N1 and high-dose LPS were inoculated intranasally after mice were anesthetized with 10% chloral hydrate intraperitoneally. Mice were sacrificed 24 h post infection points for sampling.
Histological analysis and haematoxykin & eosin staining
The lungs of the infected mice were fixed in 4% (weight/vol.) paraformaldehyde, embedded into paraffin and cut into 4-μm-thick sections. The sections were stained with haematoxylin & eosin, and observed under the microscope. Pathological scores of the lungs were determined based on the criteria: 0, normal lung tissue structure with no inflammatory cell infiltration; 1, normal alveolar structure, mild lung injury (< 25% of the lung) with little inflammatory cell infiltration; 2, alveolar collapse, moderate injury (25– 50% of the lung) with some inflammatory cell infiltration; 3, no alveolar structure, severe injury (> 50% of the lung) with massive inflammatory cell infiltration.
Chronic low-dose LPS stimulation, H1N1 infection and high-dose LPS stimulation in A549 cells
In the chronic-LPSlo group cells were exposed to low-dose LPS (1 ng/mL) every other day for 4 weeks by adding low-dose LPS to the culture the day after each passage, the control group cells exposed with PBS. After four weeks, cells in both groups were treated with media, H1N1, and high-dose (1µg/mL) LPS. A549 was inoculated with 102 TCID50/mL H1N1 for 2h, after which the inoculum was removed and fresh serum-free media was added and incubated at 37°C/5% CO2 for 24h.
Sample preparation and analysis of tandem mass tags labeled quantitative proteomics
A549 cells in control+H1N1 group, chronic-LPSlo+H1N1 group, control+acute-LPShi and chronic-LPSlo+acute-LPShi group were harvested for LC-MS analyses. Proteins were precipitated with 25mM DL-dithiothreitol, iodoacetamide, precooled acetone. After pellets we collected by centrifugation, the protein pellets were redissolved with enzymolysis diluent, followed by lyophilization and TMT labeling. Proteomic analysis was performed using a Q-Exactive mass spectrometer (Thermo, USA) after reversed-phase (RP) separation. Proteome Discover 2.4 (Thermo, USA) was used to process LC-MS/MS data, and credible proteins were screened using Score Sequest HT > 0 and unique peptide ≥ 1. Significant differences in proteins between the two groups were determined by performing Student’s t-test. Fold changes less than 0.67 or more than 1.5 and p-values less than 0.05 were considered significant. Annotation information of each identified protein was extracted using Uniprot database. Pathway enrichment analysis of differential proteins was performed based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Generation of A20 KO cells by CRISPR-Cas9
CRISPR−Cas9 system was used to knock out A20 expression in A549 cells. Briefly, two synthetic guide RNA (sgRNA) sequence targeting exon 3 of the A20 gene were designed online (https://www.benchling.com/crispr/) and then cloned into the CRISPR/Cas9 48138-puro vector. The sgRNA sequences were: sgRNA#1: AGGGGTACCCTATGCCCACC; sgRNA#2: CAGCCCTACTGCTATTCTAG. A20 cell clones were selected using green fluorescent protein (GFP) expression and restriction fragment length polymorphism.
Cytokine analysis
TNF-α, IL-6, IL-1β in the supernatant and mouse serum and IFN-α, IFN-β, IFN-γ and IL-18 in mouse alveolar lavage fluid were measured using ELISA kits (Lianke, Hangzhou, China), according to the manufacturer’s protocol.
Western blotting
Cells and lung tissues were lysed in RIPA buffer, and protein concentration determined by BCA protein assay kit (Beyotime, Shanghai, China). Denatured protein samples (20μg) were separated by 8% or 15% SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked in 5% nonfat milk for 3 h at room temperature, and probed with specific primary antibodies (1:1000) against p65 (ab16502), p-p65 (ab76302), A20 (ab92324), NLRP3 (ab214185), pro-caspase-1 (ab179515), caspase-1 (ab179515), PPAR-α (ab61182), PPAR-γ (ab209350) and β-actin (ab8227) (Abcam, Cambridge, UK) overnight at 4°C. Goat-derived anti-rabbit HRP conjugated antibody (1:2000) were used as secondary antibody for 1 h. Membranes were developed and analyzed using the Super Signal Chemiluminescent Substrate kit (Thermo Scientific, Rockford, USA) and ImageJ software.
mRNA analysis
Total RNAs from A549 cells and lung tissues were extracted using Trizol (Thermo Scientific, Rockford, USA). RNA was reverse-transcribed to cDNA using first strand cDNA synthesis kit (Thermo Scientific, Rockford, USA). qPCR assays were performed with SYBR Green PCR Master Mix (TaKaRa, Japan) and a Fast qPCR System (Applied Biosystems, USA). GAPDH was used as the internal reference. The relative mRNA level was calculated by 2−ΔΔCt method. The primers used were GAPDH (human), F; TCTTCTTTTGCGTCGCCAG, R; AGCCCCAGCCTTCTCCA, IL-1β (human), F; CTGTCCTGCGTGTTGAAAGA, R; TTGGGTAATTTTTGGGATCTACA, IL-6 (human), F; CACTGGTCTTTTGGAGTTTGAG, R; GGACTTTTGTACTCATCTGCAC, TNF-α (human), F; AGCCCTGGTATGAGCCCATCTATC, R; TCCCAAAGTAGACCTGCCCAGAC, TNFAIP3 (human), F; TGCACACTGTGTTTCATCGAG, R; ACGCTGTGGGACTGACTTTC, GAPDH (mouse), F; AGGTCGGTGTGAACGGATTTG, R; TGTAGACCATGTAGTTGAGGTCA, IL-1β (mouse), F; CACTACAGGCTCCGAGATGAACAAC, R; TGTCGTTGCTTGGTTCTCCTTGTAC, IL-6 (mouse), F; CTTCTTGGGACTGATGCTGGTGAC, R; TCTGTTGGGAGTGGTATCCTCTGTG, TNF-α (mouse), F; CGCTCTTCTGTCTACTGAACTTCGG, R; GTGGTTTGTGAGTGTGAGGGTCTG, TNFAIP3, F; TCCTCAGGCTTTGTATTTGAGC, R; TGTGTATCGGTGCATGGTTTTA.
Immunofluorescence
A549 cells were cultured in 24 well plate. The cells were fixed in 4% paraformaldehyde for 10 min and then permeabilized with 0.02% Triton X for 5 min. The cells were blocked with goat serum for 60 min. The cells were incubated with primary antibodies against p65 overnight at 4°C. The cells were incubated with the secondary antibodies for 60 min and then stained by DAPI. Fluorescent images were captured with an immunofluorescence microscope (IX71, Olympus, Japan).
Statistical analysis
Statistical analysis was performed using Prism software (GraphPad). Student’s t test or One-way analysis of variance (ANOVA), followed by Tukey’s post hoc analysis was used for calculation of statistical differences. p values <0.05 were considered statistically significant.
Results
Chronic low-dose LPS stimulation ameliorates symptoms and prolongs survival in H1N1-infected mice
Chronic-LPSlo stimulation has been well studied in childhood asthma and farm environments protect children from asthma through exposure to house dust mites and LPS (4, 5). To explore whether chronic-LPSlo stimulation is protective against H1N1 or high-dose LPS stimulation in vivo, we exposed mice to low-dose LPS or control PBS every other day for two weeks prior to H1N1 infection and high-dose LPS stimulation. We examined the lung histological changes of mice in each group, both H1N1 and acute-LPShi resulted in severe lung injury, diffuse swelling, alveolar cavity collapse, alveolar thickening, and severe infiltration of inflammatory cells (Figure 1A). Chronic-LPSlo stimulation significantly improved lung injury as assessed by pathology scores compared with control+H1N1 and control+acute-LPShi groups (Figure 1B).
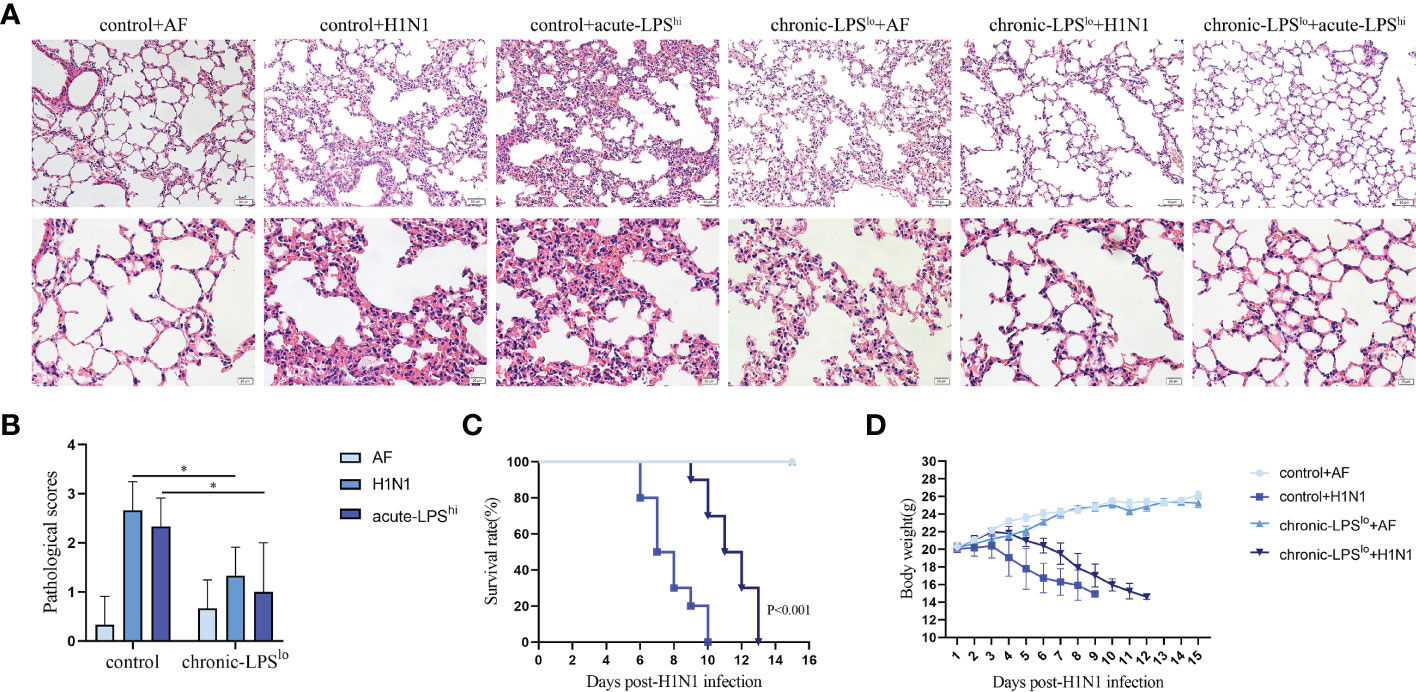
Figure 1 The effect of chronic low-dose LPS stimulation on lethal IAV infected mice. (A) Female BALB/c mice (n = 3 mice/group) were intranasally infected with 10 LD50 H1N1 influenza virus or an equivalent dilution of non-infectious allantoicfluid (AF). The lungs were isolated and sectioned for hematoxylin & eosin staining. (B) Pathological scores of the stained sections. (C) Survival data of female BALB/c mice (n = 10 mice/group) after intranasal infection with 10 LD50 H1N1. (D) Body weights of female BALB/c mice (n = 10 mice/group) after intranasal infection with 10 LD50 H1N1. *p< 0.05.
We recorded symptoms and body weight changes in H1N1-infected mice daily. The results showed that on day 2 after intranasal administration of H1N1, the mice began to show signs of decreased activity, loss of appetite, hunched back and ruffled fur. By day 5, mice may experience dyspnea and respiratory distress. Mice in the control group started dying on day 6 post-infection and all died within 10 days post-infection (Figure 1C). The mice gradually lost weight starting on the third day after infection until death (Figure 1D).
Chronic-LPSlo stimulation did not induce significant pathological changes in the lungs of mice, nor cause weight loss in mice, and ameliorated the aforementioned disease symptoms and delayed weight loss and prolonged survival.
Chronic low-dose LPS stimulation inhibits the activation of NF-κB signaling pathway and NLRP3 inflammasome
The activation of NF-κВ pathway and NLRP3 inflammasome during infection leads to upregulation of pro-inflammatory cytokines expression. To assess whether severe cytokine storms were generated during H1N1 infection or high-dose LPS stimulation and further clarify the mechanism by which chronic-LPSlo stimulation inhibits inflammation, we analyzed the effects of chronic-LPSlo stimulation on the regulation of NF-κВ pathway and NLRP3 inflammasome. In H1N1-infected or high-dose LPS-stimulated mice, chronic-LPSlo stimulation significantly decreased p65 phosphorylation (Figures 2A, B). We examined the production of TNF-α and IL-6 downstream of the NF-κВ pathway in mice lung tissues and sera. Compared with the control+AF group, H1N1 infection significantly increased the mRNA and protein levels of TNF-α and IL-6 (Figures 2C, S1 A, B).
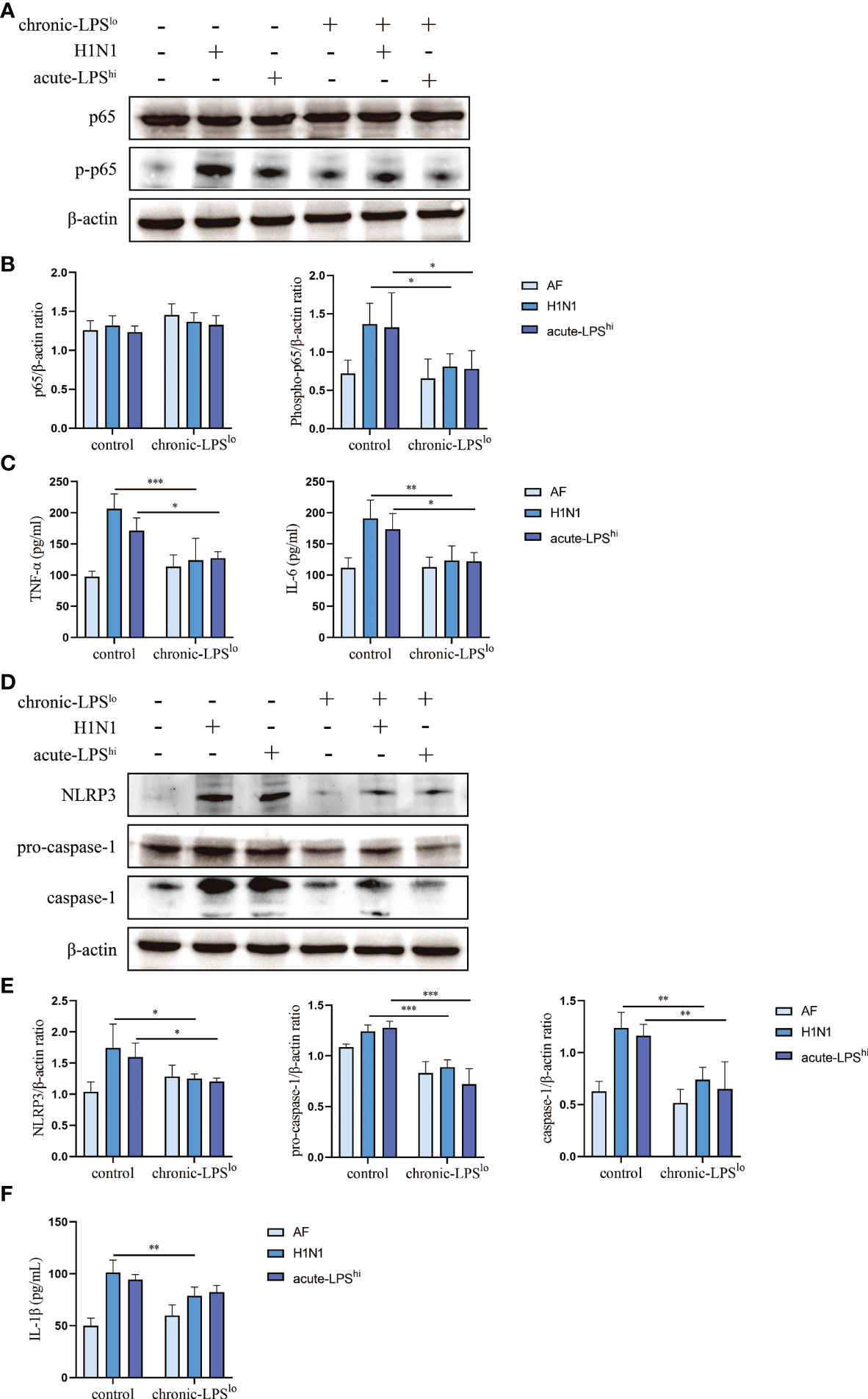
Figure 2 Effect of chronic low-dose LPS stimulation on NF-κB signaling pathway and NLRP3 inflammasome in high dose LPS-stimulated or H1N1-infected mice. (A, B) Relative protein levels of p65 and p-p65 in lung tissues were detected by western blot. (C) TNF-α and IL-6 in mice sera were measured by ELISA. (D, E) Relative protein levels of NLRP3, pro-caspase-1 and caspase-1 in lung tissues were detected by western blot. (F) IL-1β in mice sera were detected by ELISA. Data are mean ± SD, n = 3. *p< 0.05, **p< 0.01, ***p< 0.001.
Both H1N1 infection and acute-LPShi stimulation independently induced NLRP3 inflammasome activation, including increased NLRP3 and pro-caspase-1 protein expression and activated caspase-1 production. Chronic-LPSlo stimulation decreased NLRP3, pro-caspase-1 and caspase-1 at protein levels (Figures 2D, E). We examined the production of IL-1β downstream of the NLRP3 inflammasome in mice lung tissues and sera. Chronic-LPSlo stimulation significantly reduced the increase in the expression of IL-1β (Figures 2F, S1 C).
We also examined IFN-α, IFN-β, IFN-γ and IL-18 production in mouse alveolar lavage fluid. Chronic-LPSlo stimulation significantly reduced the increase in IFN-α and IFN-β expression, but had no significant effect on IFN-γ and IL-18 (Figure S2).
Our results showed that chronic-LPSlo stimulation inhibited H1N1 or acute LPShi induced activation of NLRP3 inflammasome and NF-κB and production of pro-inflammatory cytokines.
Chronic low-dose LPS stimulation upregulates A20 expression
A20 as a key protein in the protection mediated by chronic exposure to low-dose LPS environment is also a major determinant of progression and prognosis in several other inflammatory diseases (6–9). To observe the effects of chronic-LPSlo stimulation on human airway epithelial cells, we exposed A549 cells for 4 weeks to a low-dose of LPS (1ng/mL) or to control PBS before acute H1N1 infection or acute high-dose LPS stimulation. We next assessed the expression of A20 and showed that the mRNA and protein levels of A20 were significantly increased in the chronic-LPSlo group than in the control group in vivo (Figures 3A, B) and in vitro (Figures 3C, D).
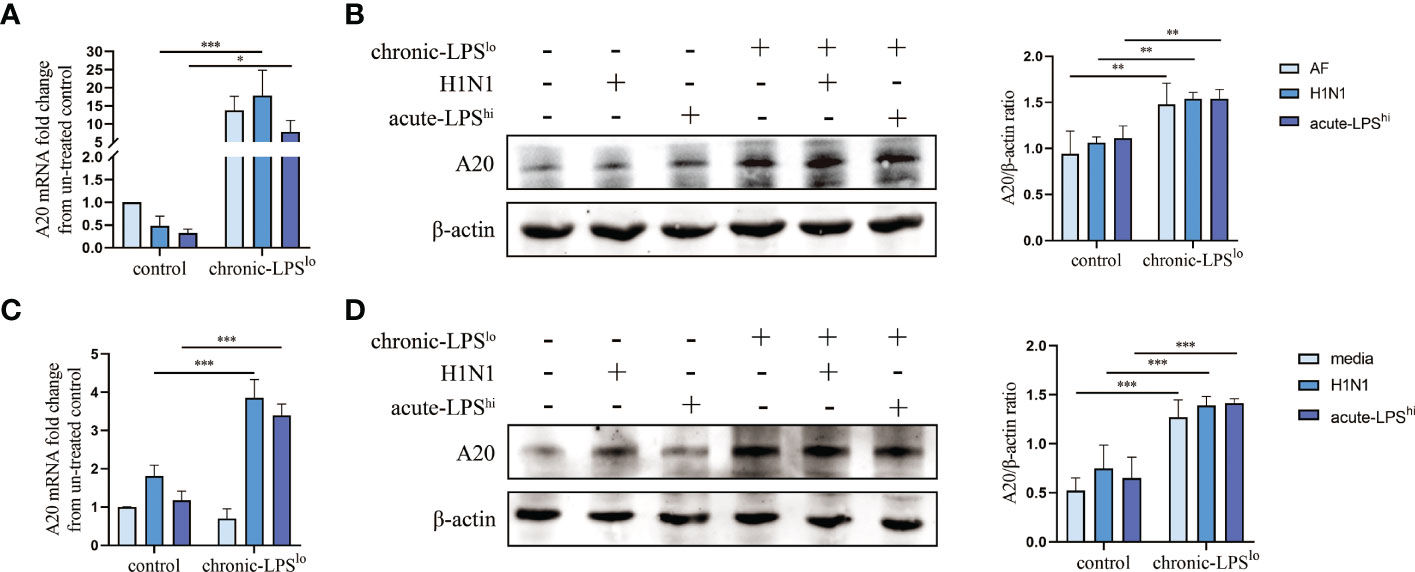
Figure 3 Effect of chronic low-dose LPS stimulation on A20. (A) A20 mRNA in lung tissues was detected by qPCR. (B) Relative protein levels of A20 in lung tissues were detected by western blot. (C) A20 mRNA in A549 cells was detected by qPCR. (D) Relative protein levels of A20 in A549 cells were detected by western blot. Data are mean ± SD, n = 3. *p< 0.05, **p< 0.01, ***p< 0.001.
A20 knockout abolishes the anti-inflammatory effects by chronic low-dose LPS stimulation
To further assess the roles of A20 in dampening H1N1 and acute-LPShi-mediated inflammatory responses, we generated A20-deficient A549 cells (A549A20-KO) using CRISPR-Cas9 gene editing, and showed a complete lack of A20 protein expression (Figure 4A). A549A20-KO cells proliferated at similar rate as wild-type cells (A549WT) and did not show significant morphological differences.
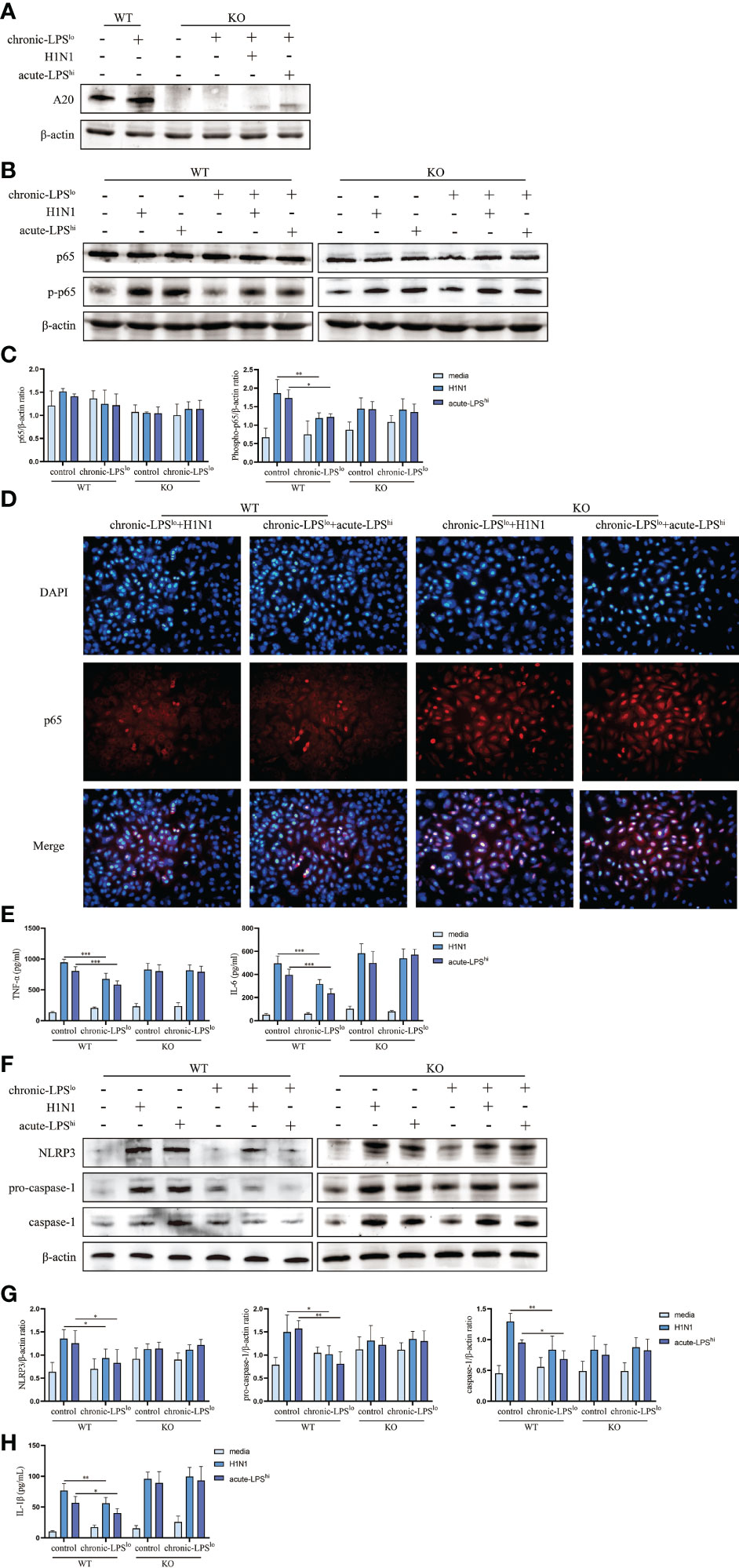
Figure 4 Effect of chronic low-dose LPS stimulation on NF-κB signaling pathway and NLRP3 inflammasome in high dose LPS-stimulated or H1N1-infected A549WT and A549A20-KO cells. (A) Relative protein levels of A20 in A549A20-KO cells were detected by western blot. (B, C) Relative protein levels of p65 and p-p65 in A549WT cells and A549A20-KO cells were detected by western blot. (D) Immunofluorescent images of the localization of p65 in A549WT and A549A20-KO cells. (E) TNF-α and IL-6 in A549WT and A549A20-KO cells culture supernatants were measured by ELISA. (F, G) Relative protein levels of NLRP3, pro-caspase-1 and caspase-1 in A549WT and A549A20-KO cells were detected by western blot. (H) IL-1β in A549WT and A549A20-KO cells culture supernatants were measured by ELISA. Data are mean ± SD, n = 3. *p< 0.05, **p< 0.01.
We measured the levels of activity NF-κB by assessing the levels of phosphorylated p65 (p-p65). H1N1 infection or LPS stimulation significantly increased the activation of p-p65 in both control H1N1 and control acute-LPShi groups. The inhibition of chronic LPSlo stimulation to nuclear translocation of p65 in H1N1-infected and high-dose LPS-stimulated A549A20-KO cells was lost (Figures 4B–D). There was also no significant difference in the production of TNF-α and IL-6 between the control group and the chronic-LPSlo group (Figures 4E, S3 A, B). These results indicated that the inhibitions of chronic-LPSlo stimulation on NF-κB signaling pathway in H1N1-infected and high-dose LPS-stimulated A549 cells were A20 dependent.
Next, we detected the effect of chronic-LPSlo stimulation on NLRP3 inflammasome after knockout of A20. Knockout of A20 made the inhibition of NLRP3 signaling pathway by chronic low-dose LPS stimulation lost (Figures 4F, G). The inhibitory effect of chronic-LPSlo stimulation on the production of IL-1β also disappeared (Figures 4H, S3 C). These results indicated that the inhibitions of chronic-LPSlo stimulation on NLRP3 inflammasome in H1N1-infected and high-dose LPS-stimulated A549 cells were A20 dependent.
Chronic low-dose LPS stimulation inhibits H1N1-infected or high-dose LPS-stimulated inflammation via PPAR
To assess the global changes induced by chronic-LPSlo stimulation at the protein level, we subjected the cells from control+H1N1 group, chronic-LPSlo+H1N1 group, control+acute-LPShi and chronic-LPSlo+acute-LPShi group to TMT-labeled quantitative proteomics. We identified 6,178 quantitative proteins in the control+H1N1 group and chronic-LPSlo+H1N1 group, of which 136 proteins had significantly increased or decreased expression. We identified 6,228 quantified proteins in control+acute-LPShi and chronic-LPSlo+acute-LPShi group, of which 86 proteins had significantly increased or decreased expression. Cluster analysis and heat map showed differentially expressed proteins control+H1N1 group compared with chronic-LPSlo+H1N1 group (Figure 5A) and control+acute-LPShi group compared with chronic-LPSlo+acute-LPShi group (Figure 5B).
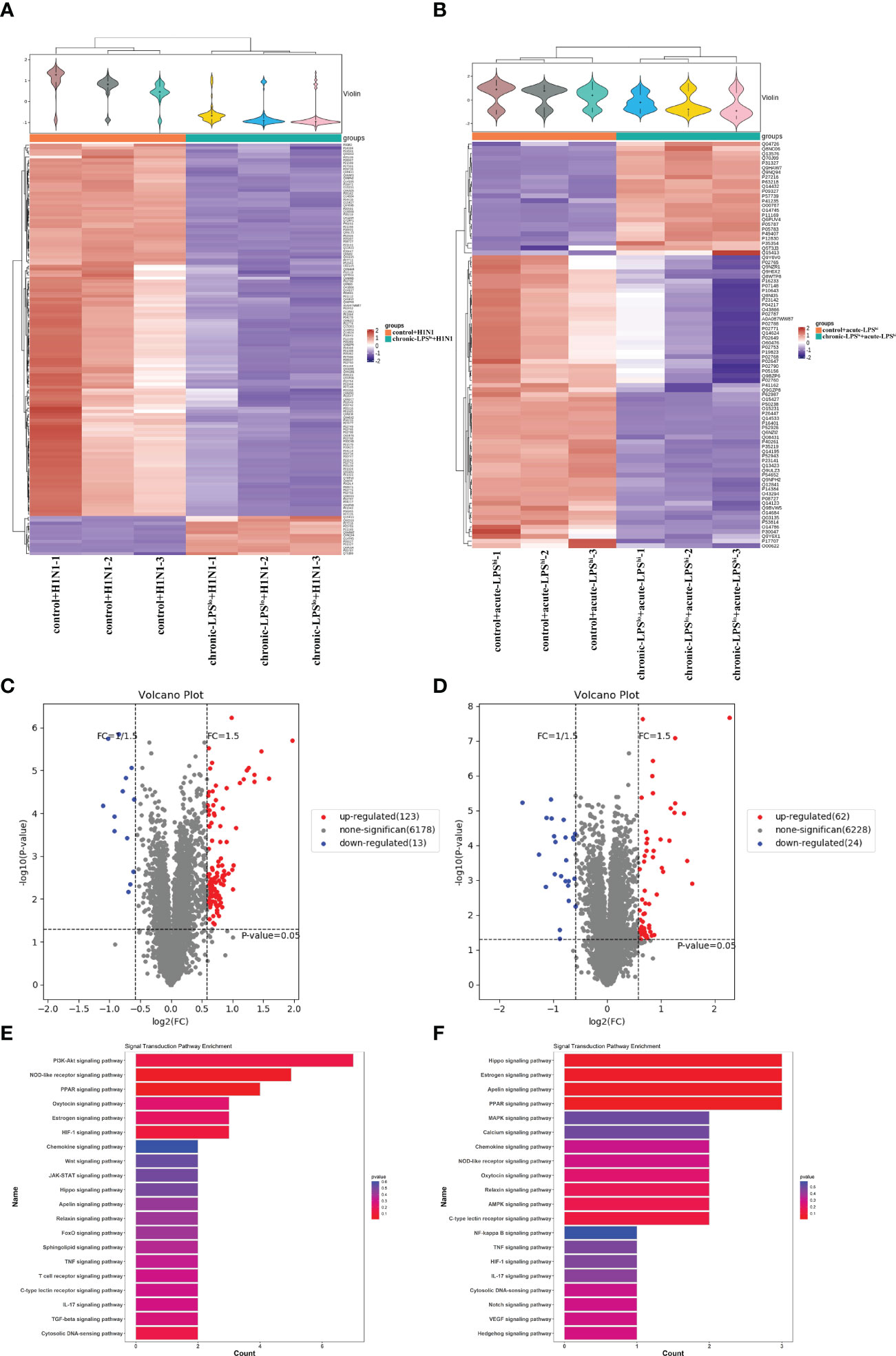
Figure 5 Inhibition of inflammation by chronic low-dose LPS stimulation is related to PPAR. (A) Hierarchical clustering analysis heat map in control+H1N1 group compared with chronic-LPSlo+H1N1 group. (B) Hierarchical clustering analysis heat map in control+acute-LPShi group compared with chronic-LPSlo+acute-LPShi group. (C) The differentially expressed proteins analyzed by volcano plots in control+H1N1 group compared with chronic-LPSlo+H1N1 group (vertical dotted lines, fold change > 1.5-fold; horizontal dotted line, p < 0.05). (D) The differentially expressed proteins analyzed by volcano plots in control+acute-LPShi group compared with chronic-LPSlo+acute-LPShi group (vertical dotted lines, fold change > 1.5-fold; horizontal dotted line, p < 0.05). (E) Top 20 of signaling transduction of KEGG pathway enrichment analysis in control+H1N1 group compared with chronic-LPSlo+H1N1 group. (F) Top 20 of signaling transduction of KEGG pathway enrichment analysis in control+acute-LPShi group compared with chronic-LPSlo+acute-LPShi group.
The volcano plot showed that 123 proteins that were increased and 13 proteins that were decreased in the 136 differential proteins in chronic-LPSlo+H1N1 group compared with control+H1N1 group (Figure 5C). 62 proteins were increased and 24 proteins were decreased in the 86 differential proteins in chronic-LPSlo+acute-LPShi group compared with control+acute-LPShi group (Figure 5D).
KEGG pathway analysis showed the top 20 pathways that are well characterized in both H1N1 infection and LPS stimulation, including the PPAR, NOD-like receptor, NF-κB, TNF and chemokine signaling pathways. Interestingly we found that PPAR was highly enriched in chronic-LPSlo+H1N1 group (Figure 5E) and chronic-LPSlo+acute-LPShi group (Figure 5F).
The increase of PPAR expression induced by chronic low-dose LPS stimulation is regulated by A20
Both PPAR-α and PPAR-γ inhibit NF-κB activation like A20 and PPAR-α has been reported to be positively regulated by A20 (10–12). In addition, PPAR-α/γ also exerts anti-inflammatory effects during influenza infection (13, 14). Despite the inflammatory signaling pathway is well characterized, whether PPAR-α/γ is involved in the mechanism by which A20 suppresses the excessive inflammatory response of IAV remains unclear.
To explore whether PPAR plays a key role in the inhibitory effect of chronic-LPSlo stimulation on inflammation, we measured the protein levels of PPAR-α and -γ in A549WT cells and A549A20-KO cells. Consistent with the proteomics, western blot analysis confirmed the increased expression of PPAR-α and -γ in chronic-LPSlo groups. Furthermore, this elevated PPAR-α and -γ expression induced in chronic low-dose LPS stimulation was abolished in A549A20-KO cells (Figures 6A, B), indicating that PPAR-α and -γ expression was regulated by A20, and loss of which resulted in exaggerated inflammatory responses by H1N1 or high-dose LPS.
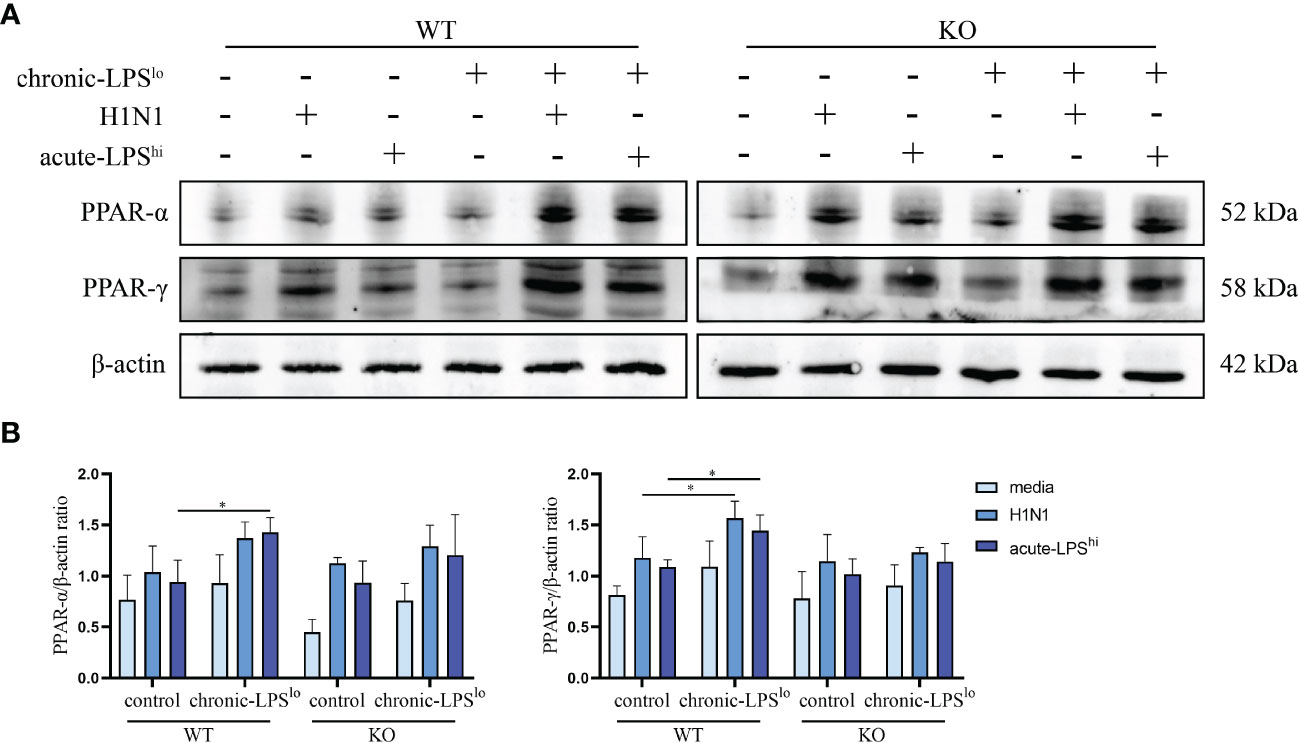
Figure 6 Effect of chronic low-dose LPS stimulation on PPAR-α and -γ. (A, B) Relative protein levels of PPAR-α and -γ in A549WT and A549A20-KO cells were detected by western blot. Data are mean ± SD, n = 3. *p< 0.05.
Discussion
Here, we demonstrate that H1N1 infection and high-dose LPS stimulation induce inflammation; however, the inflammatory response, NF-κB and NLRP3 activation were inhibited in chronic low-dose LPS environment. We show that A20 is a negative regulator of NF-κB and NLRP3-mediated inflammation and that A20 gene and protein levels are upregulated in a chronic low-dose LPS environment. Impaired inflammatory response in the chronic low-dose LPS environment was attributed to increased A20 and PPAR-α and -γ expression. Elevated A20 levels increased PPAR expression, leading to dampen of NF-κB and NLRP3 activity and inflammation. A20 knockout resulted in loss of chronic low-dose LPS-mediated suppression of inflammation. Thus, chronic low-dose LPS environment increases A20 and PPAR-α and -γ expression, in turn inhibits NF-κB and NLRP3 induced inflammation following IAV infection or high-dose LPS stimulation (Figure 7).
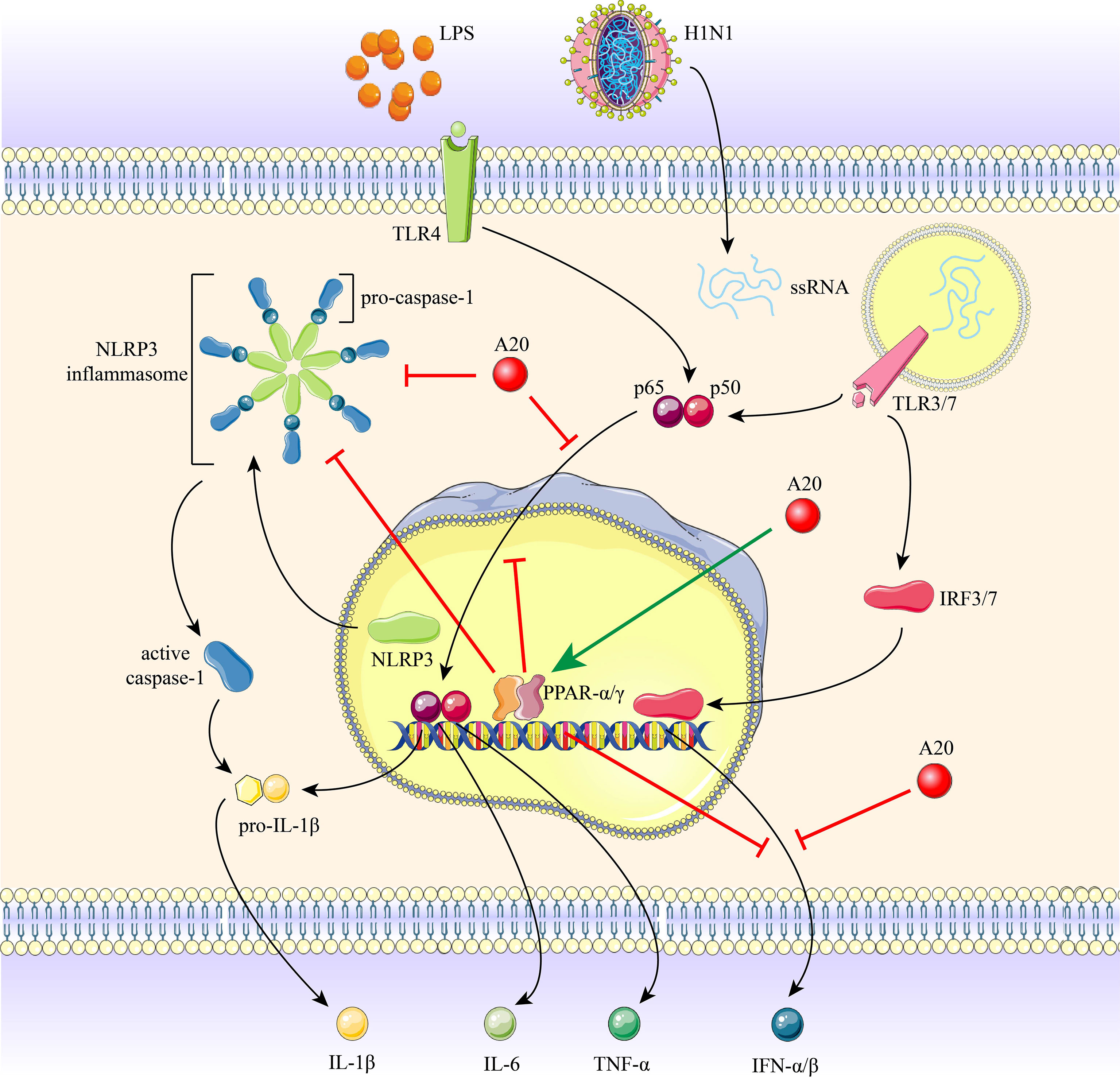
Figure 7 The role of A20 and PPAR-α and -γ in H1N1 infection and LPS stimulation induced inflammation. Recognition of H1N1 ssRNA or LPS by TLRs initiates the activation of NF-κB and NLRP3 inflammasome, promoting the production of downstream pro-inflammatory cytokines. Chronic low-dose LPS environment induced a marked increase in A20 expression, resulting in the restriction of NLRP3 inflammasome, caspase-1 cleavage, nuclear translocation of p65 subunit and IFNs expression. A20 increases PPAR-α and -γ expression, and PPAR-α and -γ also inhibit the above-mentioned inflammatory pathway.
H1N1 causes serious health problems worldwide as a major infectious pathogen (15, 16). IAV infections cause severe airway inflammation and cytokine storms that lead to high morbidity and mortality (17, 18). Despite recent progress in the fight against IAV, control of inflammation remains a major challenge for severely ill patients (19–21). There are no effective treatments for the excessive inflammation and severe consequences in IAV, and the mechanisms of these events are poorly understood. Significant differences in influenza prevalence between metropolitan and rural areas suggest that environmental exposures may impact influenza severity (22). Possibly metropolitan cities have high hygiene standards, whereas people from small towns are protected because of continuous exposure to LPS or other microbial components (23, 24). Modulators targeting autoimmunity as preventive strategies such as chronic exposure to low-dose LPS environment is emerging as a promising strategy for a variety of inflammatory diseases, including IAV (4, 5).
A20 as a key protein in the protection mediated by chronic exposure to low-dose LPS environment is also a major determinant of progression and prognosis in several other inflammatory diseases (6–9). Environmental protection and A20 are well studied in childhood asthma, and farm environments protect children from asthma by increasing A20 expression (4, 5). Interestingly, A20 has been shown to inhibit NF-κB signaling pathway and NLRP3 inflammasome activation as well as autophagy, thereby preventing pulmonary fibrosis and arthritis (25–29). Here, we show that environment-mediated protection of A20 suppresses IAV or LPS induced inflammation and report new evidence for the molecular mechanisms that may be involved.
Here, we found that chronic-LPSlo environment-mediated increases in A20 expression in cells significantly suppressed NF-κB signaling pathway activity and pro-inflammatory cytokine production to suppress IAV-induced hyper inflammation. Similar findings were reported in myeloid cells and lung epithelial cells (30, 31). Worth mentioning as the first line of defense against IAV infection, cytokines are a double-edged sword. Hyper induction of pro-inflammatory cytokine production also known as ‘cytokine storm’, it correlated directly with magnified inflammation and an unfavorable prognosis of IAV (18, 32, 33). Although cytokines play an important role in the antiviral response, if cytokines form a cytokine storm will hinder anti-virus immunity (34–36). Therefore, the exact role of A20 during viral infection requires further study. We could not rule out that other factors may also influence the relationship between A20 expression and IAV prognosis.
In addition to its key role in regulating NF-κB, several lines of evidence suggest that A20 regulates NLRP3 activation (37–39). For example, excessive Nlrp3 inflammasome activation drives arthritis pathogenesis in A20 knockout mice due to A20 putting a brake on Nlrp3 inflammasome activation by reducing LPS-induced Nlrp3 expression levels (29). Here, we consistently demonstrate that environment-mediated increases in A20 expression exhibit NLRP3 inflammasome suppression and IL-1beta expression downregulation upon infection with IAV or LPS stimulation, which is consistent with previous reports. Yet, we also explicitly show that A20 knockout cells no longer have the protection mediated by the chronic low-dose LPS environment. Our results combined with previous studies highlight the importance of increased A20 expression through the environment as a potential new therapeutic option for reducing IAV-mediated inflammation and cytokine storm.
We also found that PPAR-α and -γ plays an essential role in the protection of environment-mediated A20 elevation against inflammation, and PPAR-α and -γ are positively regulated by A20 and exert a similar inhibitory effect on inflammation as A20. Studies have shown that A20 protects against restenosis after carotid artery injury in rats through PPAR-α inhibition of NF-κB pathway activation (10). In addition, A20 can protect mice from lethal hepatic ischemia-reperfusion injury by increasing PPAR-α expression and inhibiting NF-κB activation (40). Unlike the tight link between PPAR-alpha and NF-κB, PPAR-gamma mainly inhibits NLRP3 inflammasome (41–44). Considering excessive inflammation is tied to IAV related mortality, PPAR-α/γ has been considered as a therapeutic target to limit such harmful inflammation. In a recent study, IAV infection was shown to reduce PPAR-γ mRNA levels in mouse alveolar macrophages (45). In addition, significant activation of PPAR-α and -γ by drugs can protect mice from IAV infection and pneumonia (14). It is crucial that cytokine production is finely tuned during anti-viral responses, in which it provides optimal protection while avoiding unwanted inflammation and tissue damage (46, 47). In this study the role of PPAR-α and -γ in environmental-mediated protection of chronic low-dose LPS the positive regulation of PPAR-gamma by A20 were explored for the first time. Whether the balance between A20 and inflammation is dependent on PPAR-α/γ, or whether there is crosstalk between A20 and PPAR-α/γ remains unclear; additional studies are required to resolve these points.
Collectively, our results suggest that inflammatory responses stimulated by H1N1 infection or high-dose LPS in vitro and in vivo were ameliorated in a chronic low-dose LPS environment. Increased levels of PPAR-α and -γ by A20 inhibits NF-κB and NLRP3 inflammasome activation and subsequently downregulate inflammatory cytokine production. A20 is increasingly recognized as a therapeutic target for a variety of diseases, therefore, long-term exposure to low-dose LPS environmental may help promote the resolution of IAV or LPS induced inflammation, thereby improving adverse clinical outcomes both in lung tissues.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors. The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: http://proteomecentral.proteomexchange.org; PXD035611.
Ethics statement
The animal study was reviewed and approved by the ethics committee of The College of Basic Medical Sciences of Jilin University.
Author contributions
A-YH and FW designed and supervised the study. YG reviewed the literature and wrote the manuscript. YG, XZ, XG and ZZ performed experiments and analyzed data. SJ and ZO performed statistical analysis. A-YH revised the manuscript. FW obtained funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Key Laboratory of Precision Infectious Diseases of Jilin Province (Grant no.20200601011JC), Jilin Provincial Key Laboratory of Pathogen Biology (Grant no.3D5200117426), Jilin Provincial Development and Reform Commission Common Disease Precision Prevention and Control Engineering Laboratory, Jilin Provincial Science and Technology Innovation Center of Secondary Development of Chinese Traditional Patent Medicine (Grant no.20210502005ZP).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1119473/full#supplementary-material
References
1. Oshansky CM, Gartland AJ, Wong SS, Jeevan T, Wang D, Roddam PL, et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am J Respir Crit Care Med (2014) 189(4):449–62. doi: 10.1164/rccm.201309-1616OC
2. Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: The role of macrophage-expressed tnf-related apoptosis-inducing ligand. J Exp Med (2008) 205(13):3065–77. doi: 10.1084/jem.20080201
3. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Sci (New York NY) (2015) 349(6245):316–20. doi: 10.1126/science.aaa8064
4. Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Sci (New York NY) (2015) 349(6252):1106–10. doi: 10.1126/science.aac6623
5. Krusche J, Twardziok M, Rehbach K, Böck A, Tsang MS, Schröder PC, et al. Tnf-α-Induced protein 3 is a key player in childhood asthma development and environment-mediated protection. J Allergy Clin Immunol (2019) 144(6):1684–96.e12. doi: 10.1016/j.jaci.2019.07.029
6. Ma A, Malynn BA. A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol (2012) 12(11):774–85. doi: 10.1038/nri3313
7. Priem D, van Loo G, Bertrand MJM. A20 and cell death-driven inflammation. Trends Immunol (2020) 41(5):421–35. doi: 10.1016/j.it.2020.03.001
8. Guo W, Ma J, Guo S, Wang H, Wang S, Shi Q, et al. A20 regulates the therapeutic effect of anti-Pd-1 immunotherapy in melanoma. J immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-001866
9. Martens A, van Loo G. A20 at the crossroads of cell death, inflammation, and autoimmunity. Cold Spring Harbor Perspect Biol (2020) 12(1). doi: 10.1101/cshperspect.a036418
10. Meng Z, Gao P, Chen L, Peng J, Huang J, Wu M, et al. Artificial zinc-finger transcription factor of A20 suppresses restenosis in sprague dawley rats after carotid injury Via the pparα pathway. Mol Ther Nucleic Acids (2017) 8:123–31. doi: 10.1016/j.omtn.2017.06.010
11. Toobian D, Ghosh P, Katkar GD. Parsing the role of ppars in macrophage processes. Front Immunol (2021) 12:783780. doi: 10.3389/fimmu.2021.783780
12. Ju Z, Su M, Li D, Hong J, Im DS, Kim S, et al. An algal metabolite-based ppar-Γ agonist displayed anti-inflammatory effect Via inhibition of the nf-κb pathway. Mar Drugs (2019) 17(6). doi: 10.3390/md17060321
13. Gopal R, Mendy A, Marinelli MA, Richwalls LJ, Seger PJ, Patel S, et al. Peroxisome proliferator-activated receptor gamma (Pparγ) suppresses inflammation and bacterial clearance during influenza-bacterial super-infection. Viruses (2019) 11(6). doi: 10.3390/v11060505
14. Bei Y, Tia B, Li Y, Guo Y, Deng S, Huang R, et al. Anti-influenza a virus effects and mechanisms of emodin and its analogs Via regulating Pparα/Γ-Ampk-Sirt1 pathway and fatty acid metabolism. BioMed Res Int (2021) 2021:9066938. doi: 10.1155/2021/9066938
16. Enserink M, Cohen J. Virus of the year. The novel H1n1 influenza. Sci (New York NY) (2009) 326(5960):1607. doi: 10.1126/science.326.5960.1607
17. Mudd PA, Crawford JC, Turner JS, Souquette A, Reynolds D, Bender D, et al. Distinct inflammatory profiles distinguish covid-19 from influenza with limited contributions from cytokine storm. Sci Adv (2020) 6(50). doi: 10.1126/sciadv.abe3024
18. Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol (2016) 13(1):3–10. doi: 10.1038/cmi.2015.74
19. Ramiro S, Mostard RLM, Magro-Checa C, van Dongen CMP, Dormans T, Buijs J, et al. Historically controlled comparison of glucocorticoids with or without tocilizumab versus supportive care only in patients with covid-19-Associated cytokine storm syndrome: Results of the chic study. Ann rheumatic Dis (2020) 79(9):1143–51. doi: 10.1136/annrheumdis-2020-218479
20. Wang Q, Fang P, He R, Li M, Yu H, Zhou L, et al. O-Glcnac transferase promotes influenza a virus-induced cytokine storm by targeting interferon regulatory factor-5. Sci Adv (2020) 6(16):eaaz7086. doi: 10.1126/sciadv.aaz7086
21. Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin immunopathol (2017) 39(5):541–50. doi: 10.1007/s00281-017-0636-y
22. Dalziel BA-O, Kissler S, Gog JA-O, Viboud CA-O, Bjørnstad OA-O, Metcalf CA-O, et al. Urbanization and humidity shape the intensity of influenza epidemics in U.S. cities. Science (2018) 362(6410):1095–9203. doi: 10.1126/science.aat6030
23. Bach JF. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat Rev Immunol (2018) 18(2):105–20. doi: 10.1038/nri.2017.111
24. Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol (2017) 18(10):1076–83. doi: 10.1038/ni.3829
25. Ventura S, Cano F, Kannan Y, Breyer F, Pattison MJ, Wilson MS, et al. A20-binding inhibitor of nf-κb (Abin) 2 negatively regulates allergic airway inflammation. J Exp Med (2018) 215(11):2737–47. doi: 10.1084/jem.20170852
26. Liu SS, Lv XX, Liu C, Qi J, Li YX, Wei XP, et al. Targeting degradation of the transcription factor C/Ebpβ reduces lung fibrosis by restoring activity of the ubiquitin-editing enzyme A20 in macrophages. Immunity (2019) 51(3):522–34.e7. doi: 10.1016/j.immuni.2019.06.014
27. Municio C, Dominguez-Soto Á, Fuentelsaz-Romero S, Lamana A, Montes N, Cuevas VD, et al. Methotrexate limits inflammation through an A20-dependent cross-tolerance mechanism. Ann rheumatic Dis (2018) 77(5):752–9. doi: 10.1136/annrheumdis-2017-212537
28. Razani B, Whang MI, Kim FS, Nakamura MC, Sun X, Advincula R, et al. Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis-like disease and inflammation. Nat Immunol (2020) 21(4):422–33. doi: 10.1038/s41590-020-0634-4
29. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the Nlrp3 inflammasome by A20 protects against arthritis. Nature (2014) 512(7512):69–73. doi: 10.1038/nature13322
30. Maelfait J, Roose K, Bogaert P, Sze M, Saelens X, Pasparakis M, et al. A20 (Tnfaip3) deficiency in myeloid cells protects against influenza a virus infection. PloS Pathog (2012) 8(3):e1002570. doi: 10.1371/journal.ppat.1002570
31. Maelfait J, Roose K, Vereecke L, Mc Guire C, Sze M, Schuijs MJ, et al. A20 deficiency in lung epithelial cells protects against influenza a virus infection. PloS Pathog (2016) 12(1):e1005410. doi: 10.1371/journal.ppat.1005410
32. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
33. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol reviews: MMBR (2012) 76(1):16–32. doi: 10.1128/mmbr.05015-11
34. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
35. Biron CA. Interferons alpha and beta as immune regulators–a new look. Immunity (2001) 14(6):661–4. doi: 10.1016/s1074-7613(01)00154-6
36. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun (2014) 5:3864. doi: 10.1038/ncomms4864
37. Huang X, Feng Z, Jiang Y, Li J, Xiang Q, Guo S, et al. Vsig4 mediates transcriptional inhibition of Nlrp3 and il-1β in macrophages. Sci Adv (2019) 5(1):eaau7426. doi: 10.1126/sciadv.aau7426
38. Xue Z, Xi Q, Liu H, Guo X, Zhang J, Zhang Z, et al. Mir-21 promotes Nlrp3 inflammasome activation to mediate pyroptosis and endotoxic shock. Cell Death Dis (2019) 10(6):461. doi: 10.1038/s41419-019-1713-z
39. Mouton-Liger F, Rosazza T, Sepulveda-Diaz J, Ieang A, Hassoun SM, Claire E, et al. Parkin deficiency modulates Nlrp3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia (2018) 66(8):1736–51. doi: 10.1002/glia.23337
40. Ramsey HE, Da Silva CG, Longo CR, Csizmadia E, Studer P, Patel VI, et al. A20 protects mice from lethal liver Ischemia/Reperfusion injury by increasing peroxisome proliferator-activated receptor-alpha expression. Liver Transplant (2009) 15(11):1613–21. doi: 10.1002/lt.21879
41. Yang CC, Wu CH, Lin TC, Cheng YN, Chang CS, Lee KT, et al. Inhibitory effect of pparγ on Nlrp3 inflammasome activation. Theranostics (2021) 11(5):2424–41. doi: 10.7150/thno.46873
42. Liu Y, Bi X, Zhang Y, Wang Y, Ding W. Mitochondrial Dysfunction/Nlrp3 inflammasome axis contributes to angiotensin ii-induced skeletal muscle wasting Via ppar-Γ. Lab investigation; J Tech Methods Pathol (2020) 100(5):712–26. doi: 10.1038/s41374-019-0355-1
43. Fu J, Zhao B, Ni C, Ni H, Xu L, He Q, et al. Rosiglitazone alleviates mechanical allodynia of rats with bone cancer pain through the activation of ppar-Γ to inhibit the nf-κb/Nlrp3 inflammatory axis in spinal cord neurons. PPAR Res (2021) 2021:6086265. doi: 10.1155/2021/6086265
44. Meng QQ, Feng ZC, Zhang XL, Hu LQ, Wang M, Zhang HF, et al. Ppar-Γ activation exerts an anti-inflammatory effect by suppressing the Nlrp3 inflammasome in spinal cord-derived neurons. Mediators Inflammation (2019) 2019:6386729. doi: 10.1155/2019/6386729
45. Huang S, Zhu B, Cheon IS, Goplen NP, Jiang L, Zhang R, et al. Ppar-Γ in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J Virol (2019) 93(9). doi: 10.1128/jvi.00030-19
46. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for Trim29 in the control of innate immunity in the respiratory tract. Nat Immunol (2016) 17(12):1373–80. doi: 10.1038/ni.3580
Keywords: A20 (TNFAIP3), IAV, PPAR, NF-κB, NLRP3 inflammasome
Citation: Gu Y, Hsu AC-Y, Zuo X, Guo X, Zhou Z, Jiang S, Ouyang Z and Wang F (2023) Chronic exposure to low-level lipopolysaccharide dampens influenza-mediated inflammatory response via A20 and PPAR network. Front. Immunol. 14:1119473. doi: 10.3389/fimmu.2023.1119473
Received: 08 December 2022; Accepted: 02 January 2023;
Published: 16 January 2023.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Yaling Dou, Texas A&M Health Science Center, United StatesGuangchuan Wang, Jinzhou Medical University, China
Copyright © 2023 Gu, Hsu, Zuo, Guo, Zhou, Jiang, Ouyang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Wang, d2ZAamx1LmVkdS5jbg==