
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 January 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1118236
This article is part of the Research Topic Community Series in Antiviral Innate Immune Sensing, Regulation, and Viral Immune Evasion, volume II View all 16 articles
 Linhai Zhang1,2
Linhai Zhang1,2 Lijia Zhang1Fangjing Li1Wanyu Liu1Zhenzhen Tai1
Lijia Zhang1Fangjing Li1Wanyu Liu1Zhenzhen Tai1 Juan Yang1
Juan Yang1 Haiqing Zhang1
Haiqing Zhang1 Jinmei Tuo1,2*Changyin Yu1,2*
Jinmei Tuo1,2*Changyin Yu1,2* Zucai Xu1,2*
Zucai Xu1,2*Herpes simplex virus (HSV) is the most common pathogen of infectious encephalitis, accounting for nearly half of the confirmed cases of encephalitis. Its clinical symptoms are often atypical. HSV PCR in cerebrospinal fluid is helpful for diagnosis, and the prognosis is usually satisfactory after regular antiviral treatment. Interestingly, some patients with recurrent encephalitis have little antiviral effect. HSV PCR in cerebrospinal fluid is negative, but glucocorticoid has a significant effect after treatment. Specific antibodies, such as the NMDA receptor antibody, the GABA receptor antibody, and even some unknown antibodies, can be isolated from cerebrospinal fluid, proving that the immune system contributes to recurrent encephalitis, but the specific mechanism is still unclear. Based on recent studies, we attempt to summarize the relationship between herpes simplex encephalitis and innate immunity, providing more clues for researchers to explore this field further.
Viral infection causes encephalitis, an inflammation of the brain parenchyma accompanied by neurological dysfunction (1). Symptoms include headache, altered consciousness, seizures, focal dysfunction, papilledema, fever, myalgia, and respiratory or digestive symptoms (2). In general, the prognosis of viral encephalitis is determined by the pathogen and host immune status, but in a small number of cases, viral infection can lead to antibody-mediated autoimmune encephalitis (AE) (3, 4). Several neurological autoimmune diseases can be induced by HSV infection in individuals with selective innate immunodeficiency (5). Unlike adaptive immunity, innate immunity cannot establish and maintain immune memory against reinfection. To restrict viral infections, antiviral innate immunity acts in a non-specific manner when the body is exposed to pathogens (6), which has recently been challenged (7). There is extensive literature claiming that the innate immune system can create memory after infection and therefore be able to respond rapidly in the event of a second infection (8, 9), while pattern recognition receptors (PRRs) are a prerequisite for this ability, PRRs may also be key signals in the induction of autoimmune encephalitis (10). The purpose of this review was to identify the possible relationship between innate immunity and herpes simplex viral encephalitis (HSVE) and to offer new insight for clinical investigation.
An HSV-1 virus consists of a capsid, tegument, and envelope in a spherical shape (11). In addition to gD, gH, gL, and gB, the envelope contains 11 viral glycoproteins, among which the gB function as a fusogen to allow HSV to enter cells. It combined with heparan sulfate, herpesvirus entry mediator, and nectin on the surface of the host cell when cells are infected with HSV using the fusion mechanism involving gB, gD, gH/gL as the core (12, 13). The tegument and capsid enter the host cell after fusion, and the tegument recruits tubulin and dynein to transport the capsid to the nucleus (14, 15). Researchers have shown that the tegument proteins UL36 and UL37 trigger movement to the nucleus (16), releasing the genome into the nucleus (17). In addition, vp16 separates from the capsid and enters the nucleus to form a complex with host cell factor 1 (HCF-1) and octamer binding protein-1 (Oct-1), and finally, it binds to the promoter of the Immediate early (IE) gene, which drives gene expression (IE, E and L genes) (18, 19). HSV codes ICP0, ICP4, ICP22, ICP27, and ICP47 (20) genes can be activated or inhibited by them, thereby promoting or delaying a process. Ultimately, the IE gene produces proteins that regulate viral replication and cellular antigen presentation, and the E gene synthesizes viral DNA and packages proteins, the L gene produces proteins for virion assembly, and mature viruses exit the cell by exocytosis (21) (Figure 1).
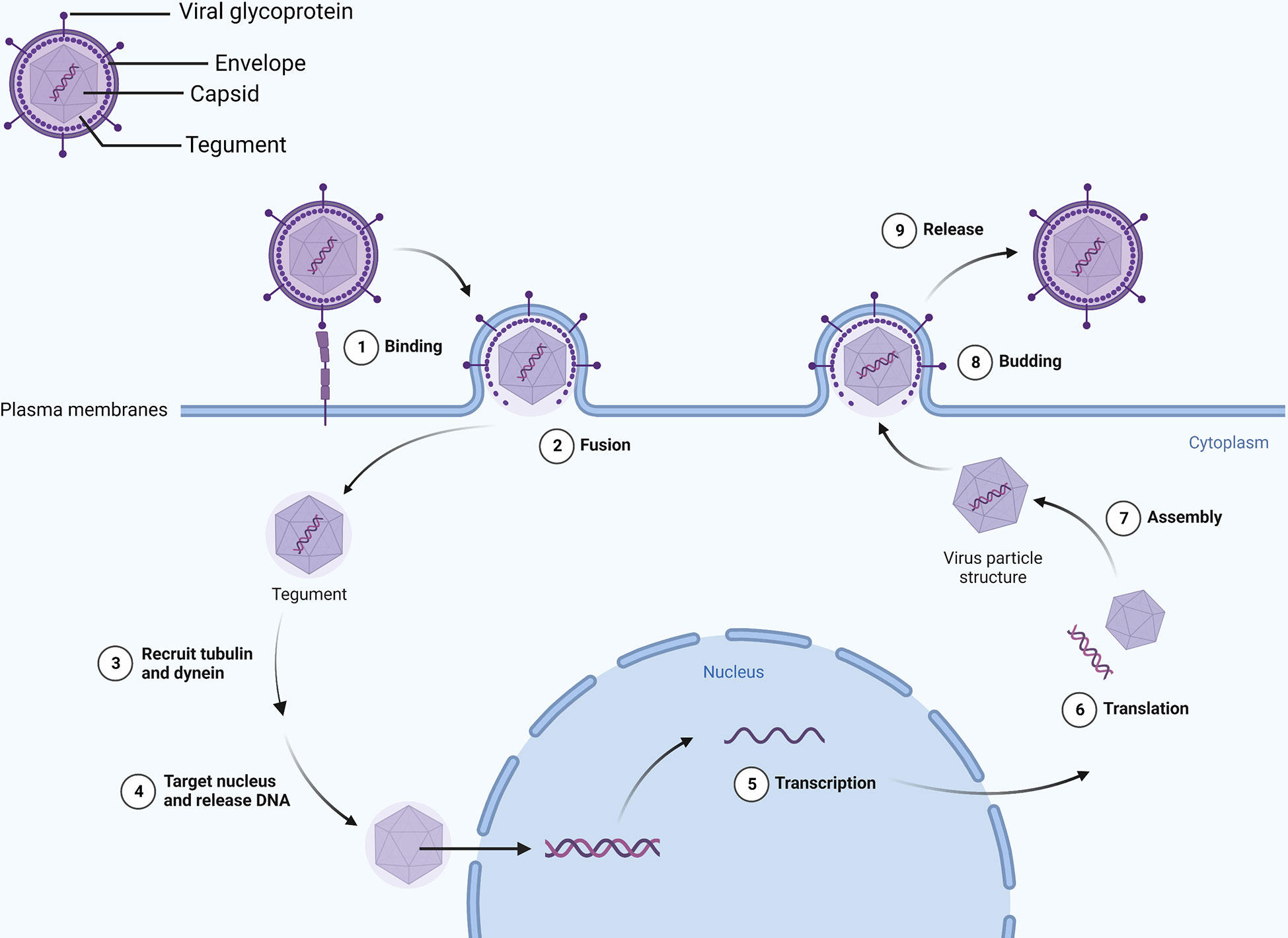
Figure 1 Viral envelope glycoproteins mediate binding with the cell membrane. Viral DNA enters the nucleus and transcribes and translates viral particle accessories as the envelope merges with the cell membrane. Eventually, intact virions leave the cell by exocytosis.
Through reverse axoplasmic transport, the virus in the exposed area enters nerve endings and reaches the neuronal cell body after lytic infection (22). Although it has been reported in the vagus nerve and superior cervical ganglion, it is also usually latent in the trigeminal ganglion (23). IE, E, and L genes start to express approximately 24-72 h after infection. IE and E transcription decreases while latency-associated transcript (LAT) gradually increases, thus forming latent infection (21). The mechanism may be related to promoting viral genome silencing by LAT (24). Local latent viruses can be reactivated and replicated by fever, emotional or hormonal imbalance, trauma, or immunosuppression and locally produce blisters, sores, or ulcers (25, 26).
For a virus to enter the brain, it must cross the blood-brain barrier, which is different from a peripheral infection. Transcellular transport is severely limited by the tight junctions between cells within the blood-brain barrier, which separates the central nervous system from peripheral blood circulation (27). However, HSV can be transported reversely along the nerve, bypassing the blood-brain barrier and entering the central nervous system, activating innate immune cells and generating an innate immune response (28), Its viral genomic DNA and some RNA intermediates become the true pathogen-related molecular patterns (PAMPs) of pattern recognition receptors (PRRs).
After the virus enters the brain, nucleic acid sensing is important to detect the virus. Microglia express cyclic-GMP-AMP synthase (cGAS), a nucleotidyl transferase and an important cytoplasmic sensor that recognizes DNA ligands in different cell types (29, 30).
Compared to wild-type mice, cGAS- and STING-deficient mice had significantly higher viral loads in brain tissue, according to Reinert et al. (29). cGAS is activated after binding to viral double-stranded DNA (dsDNA) and utilizes ATP and GTP to form cyclic GMP-AMP (cGAMP). CGAMP further activates the stimulator of interferon gene (STING), which is transported to the Golgi by COPII (31), activation of TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1) after palmitoylation, leading to the activation of interferon regulatory factor 3 (IRF3) and interferon production (32, 33). A zebrafish model of HSV-1 infection induces robust interferon production and depends on STING expression, but cGAS seems dispensable for the STING signaling, whereas DDX41 and DHX9 were found to be more closely related to interferon production in zebrafish (34).
An immune response is triggered by the DNA-dependent activator of IFN-regulatory factors (DAI), a recently discovered DNA sensor that detects nucleic acids exposed during cell damage or infection. Using artificially induced DAI and B-DNA stimulation of L929 cells, IFN was found to be expressed earlier and at higher levels than controls, which was associated with the synergy of IRF3 and TBK1 and independent of TLR9 (35). Thanh et al. demonstrated that in DAI knockdown HepG2 cells, HSV-1 viral gene and ICP0 expression were increased, but DAI knockdown did not affect cytoplasmic DNA stimulation-mediated interferon release, suggesting that there may be other pathways that can promote interferon expression (36). The receptor-interacting protein 1 (RIP1) and the receptor-interacting protein 3 (RIP3) can also be recruited by DAI through its receptor-interacting protein homotypic interaction motifs (RHIMs), then activating NF-κB (37). A new DNA sensor, IFI16, has been discovered in the cytoplasm, similar to DAI. It is a member of the PYHIN protein family with two DNA-binding domains that can directly bind to viral DNA and recruit STING (38). A nuclear localization signal allows IFI16 to recognize HSV DNA and acetylate itself, which recruits STING and induces the production of IFN (39, 40).
A cytoplasmic-localized RNA sensor, Retinoic acid-inducible gene (RIG)-I-like receptor (RLR) includes RIG-I, MDA5, and LGP2, whose enhanced expression is induced by viral infections and interferon stimulation, which leads to antiviral effects (41–43). HSV replication in mutated human hepatoma cells inactivated by RIG-I demonstrates their relationship (44). According to Emma et al., RIG-I expression is parallel with intracellular DNA load, and RIG-I cooperates with DAI to exert an antiviral effect on HSV through RNA polymerase 3 (45), it is unclear, however, whether RIG-I recognizes RNA transcribed by HSV.
Also, Toll-like receptors (TLRs) play a crucial role in recognition of viruses by the host. HSVE pathogenesis is linked to TLR3 deficiency (46). In the TLR3 molecule, an ectodomain (ECD) is present inside the endosome, and an extracellular Toll/interleukin-1 receptor domain (TIR) is present outside the endosome. The ligand-binding ECD domain promotes the phosphorylation of TLR3, and the TIR domain recruits adaptor proteins, which are important for downstream signaling (47). Multiple cells express TLR3, which recognizes double-stranded RNA (dsRNA), an intermediate in viral replication (48). When TLR3 binds to its ligand, it recruits its only adaptor-TRIF (or TICAM1)-triggering downstream signaling that activates TBK1, an inhibitor of κB (IκB) kinase-related kinase-ϵ (IKK-ϵ), and phosphorylates IRF- 3, while phosphorylated IRF-3 is translocated to the nucleus to induce interferon gene transcription (49–52). TLR2, 7, 9, and other subtypes also contribute to viral recognition (53, 54) (Figure 2). UNC93B1 is a multi-transmembrane protein that plays a crucial role in necleic-acid-sensing TLR signaling (55). Studies have shown that the UNC93B1 regulates the TLR7/9 signaling pathway by transferring TLR7 and 9 to endolysosomes (56). UNC93B1 prevents the STING from hyperactivation, thus inhibiting the cGAS-STING pathway and its subsequent interferon production, this was shown in UNC93B1-deficient mice that UNC93B1 deficiency strengthens the host immune responses to the cytosolic DNA stimulation and UNC93B1-deficient mice are more resistant to HSV-1 infection (57).
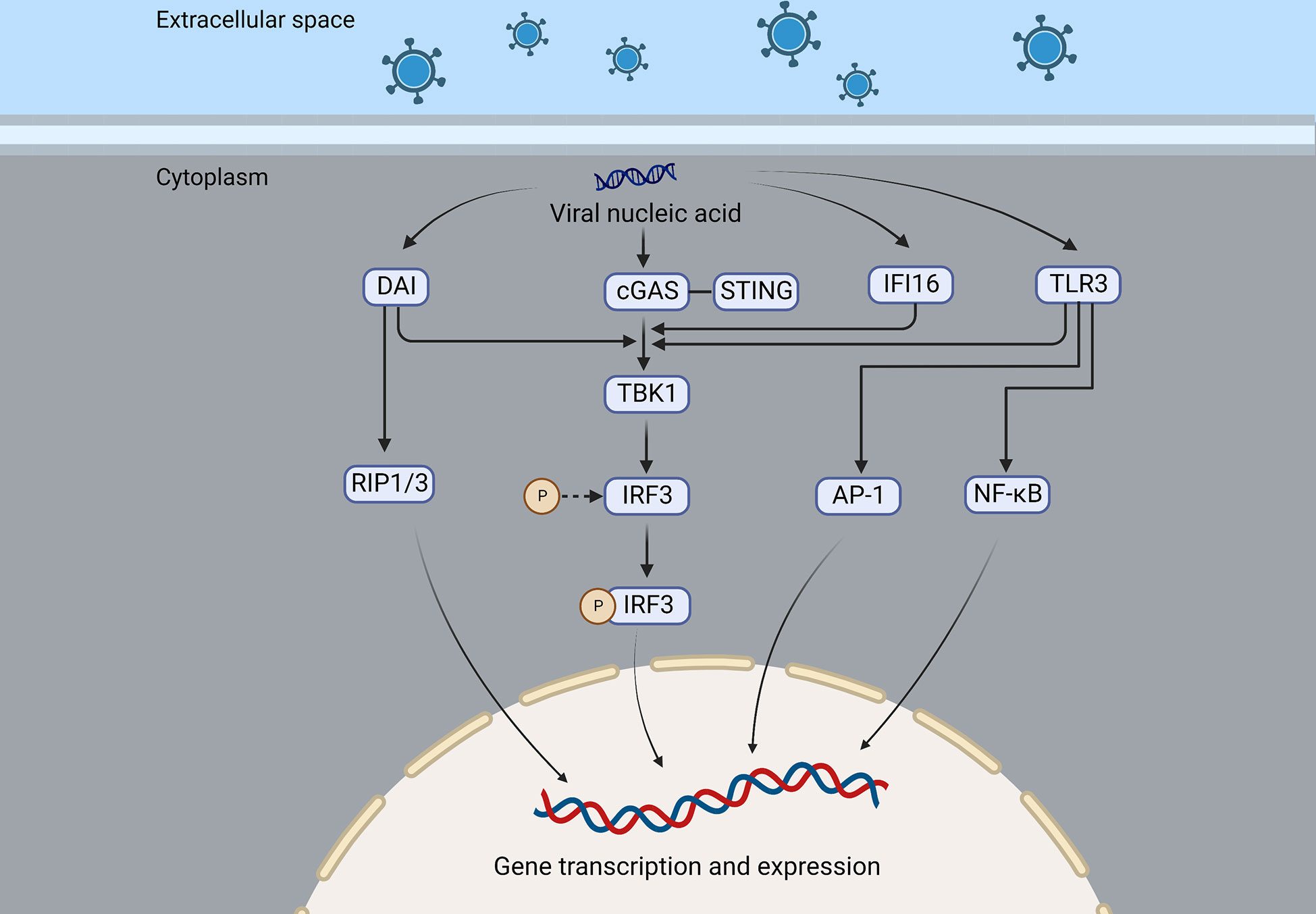
Figure 2 DAI, cGAS, IFI16, and TLR3 are nucleic acid sensing mechanisms of the virus that recruit and activate TBK1, which in turn activates IRF3, resulting in the expression of interferon or cytokines. In addition, DAI can also recruit RIP1/3 to promote gene transcription and expression, and TLR3 can activate NF-κB and AP-1 and induce the production of interferon and inflammatory factors.
Even though the host has many antiviral mechanisms, the virus has developed a powerful immune evasion mechanism (58). The enzymatic activity of cGAS is crucial to antiviral effects by triggering downstream interferon signaling by binding to dsDNA. Interferon mRNA was significantly higher in VP22 knockout HSV-infected cells than in wild-type HSV-infected cells, and ectopic expression of viral proteins VP22 are shown to inhibit cGAS/STING-mediated interferon production (59). VP24 can inhibit cGAS and STING-induced promoter activation and interferon production (60).
Many studies have revealed that the tegument proteins are important in viral gene replication and assembly (61). Among them, UL36 ubiquitin-specific protease (UL36USP) acts as a deubiquitinase that inhibits promoter activation of interferon and NF-κB induced by cGAS and STING, which allows the virus to evade host DNA sensing immune responses (62). Furthermore, UL36USP inhibits the degradation of capsids due to its deubiquitylase activity and prevents the viral genome from entering the cytoplasm, thus preventing DNA sensing-induced antiviral immunity (63). UL24 is a conserved protein among the herpes family but essential for viral replication, it can inhibit interferon and interleukin-6 (IL-6) expression mediated by cGAS-STING, and UL24 is also found to block NF-κB promoter activation. All these lead to viral immune evasion (64). A deamidation of the viral tegument protein UL37 inhibits the synthesis of cGAMP catalyzed by cGAS, interrupts downstream signal transduction, reduces interferon production, and promotes viral survival (65). Even though the cGAS-STING pathway is essential for the host against the virus, HSV-1 has evolved to evade host immune responses. Compared to the UL41-null mutant virus, wild-type HSV-1 infection could inhibit activation of the interferon signaling pathway, and UL41 expression inhibits interferon promoter activation and decreases production (66).
ICP34.5 is the virulence factor of HSV, encoded by a leaky-late gene, which can dephosphorylate eIF2α under the action of protein phosphatase 1-α, thereby allowing the continuous synthesis of viral proteins (67). ICP34.5 inhibits downstream antiviral signaling by preventing STING’s translocation to the Golgi apparatus (68). IFI16 can induce interferon production early in infection and exert antiviral effects (69), whereas later in infection, ICP0 targets IFI16 for degradation through its E3 ubiquitin ligase activity and promote virus replication (70). β-catenin is crucial for regulating the transcription of target genes. However, HSV-1 US3 protein inhibits interferon production by phosphorylating β-catenin in the Wnt signaling pathway and further restricting β-catenin nuclear translocation, thus antagonizing the interferon production and destroying the host antiviral immune response (71).
Infectious, autoimmune, and postinfectious encephalitis is the most common causes of encephalitis, characterized by inflammation of the brain parenchyma with neurological deficits (1), with viral encephalitis accounting for 60% of infectious cases (72). HSVE is one of the most common causes of encephalitis, and although the virus is cleared after regular treatment, patients still experience relapses in neurological symptoms. In some patients, viral DNA was detected in their cerebrospinal fluid, indicating persistent infection or viral reactivation, which signified a true relapse of HSVE.
In some patients, however, the virus was not detected by cerebrospinal fluid PCR after relapse, and the condition improved after Immunotherapy (73), suggesting that the immune mechanism lies at the heart of many of these complications. The authors reviewed a total of 43 patients with herpes simplex encephalitis and anti-N-methyl-D-aspartate receptor (NMDAR) antibody encephalitis, most of whom were children with a biphasic course (74), and anti-NMDAR antibody encephalitis is the most common immune encephalitis after HSVE (75). Additionally, anti-GABAR antibodies, anti-CASPR2 antibodies, and some unknown antibodies will be produced after HSVE (76–79). 27% (14 of 51) of HSVE patients had autoimmune encephalitis (AEs), and all 14 had neuronal antibodies, while 11 of 37 patients without AEs also had neuronal antibodies (80). It was assumed that the viral infection triggered the immune response because none of the patients had these antibodies before developing HSVE. When mice were intranasally injected with HSV-1, Linnoila et al. found that serum NMDAR antibodies were positive, hippocampal NMDAR decreased, and also produced unknown antibodies (81), which had been observed in patients with autoimmune brains after HSVE.
When combined with anti-NMDAR encephalitis (following a non-HSV infection), patients are more likely to develop HSV antibodies than controls (compared with Cytomegalovirus and Epstein-Barr virus), and there are no neuronal or glial markers in the CSF, it is considered that HSV and NMDAR might be connected (82). A molecular mimicry best demonstrates the link between Campylobacter jejuni and Guillain-Barré syndrome (83). Zhao et al. found that, compared with wild-type HSV-1, the virus with protein UL6 gene knockout could not induce autoimmune diseases and that wild-type autoimmune diseases were triggered by autoreactive T cells (84), suggesting that molecular mimicry may contribute to autoimmune disease development following viral infections.
Despite this, molecular modeling alone may not be sufficient to explain immune encephalitis since HSVE is often associated with extensive neuronal damage, leading to the release of antigens from neurons (73, 74, 85), the presence of unknown antibodies could also explain symptoms other than typical NMDAR encephalitis (86, 87). Previously, 33% of patients with anti-NMDAR encephalitis had abnormal brain MRIs, but few had contrast enhancement (88), while most patients with autoimmune encephalitis after herpes simplex encephalitis had contrast enhancement, suggesting disruption of the blood-brain barrier (89) or inflammation (80, 90).
According to Omae et al., the cerebrospinal fluid cytokines or chemokines in patients with NMDAR encephalitis after HSVE increased in the early stage, suggesting immune infiltration into the central nervous system and damage to blood-brain barrier integrity. After treatment, these cytokines or chemokines gradually decreased; then, in the middle stage, they increased again, but NMDAR antibodies were absent; finally, in the late stage, NMDAR antibodies reached their peak, and the cytokines and chemokines gradually decreased (91). In an evaluation of this case, Wesselingh et al. proposed a hypothesis of the pathogenesis of autoimmune brain following herpes simplex encephalitis: HSV infection results in a breach of the blood-brain barrier that allows innate/adaptive immune cells to infiltrate and cause neuroinflammation. Eventually, B cells and T cells are recruited and produce antibodies against neuronal antigens (90, 92, 93) (Figure 3).
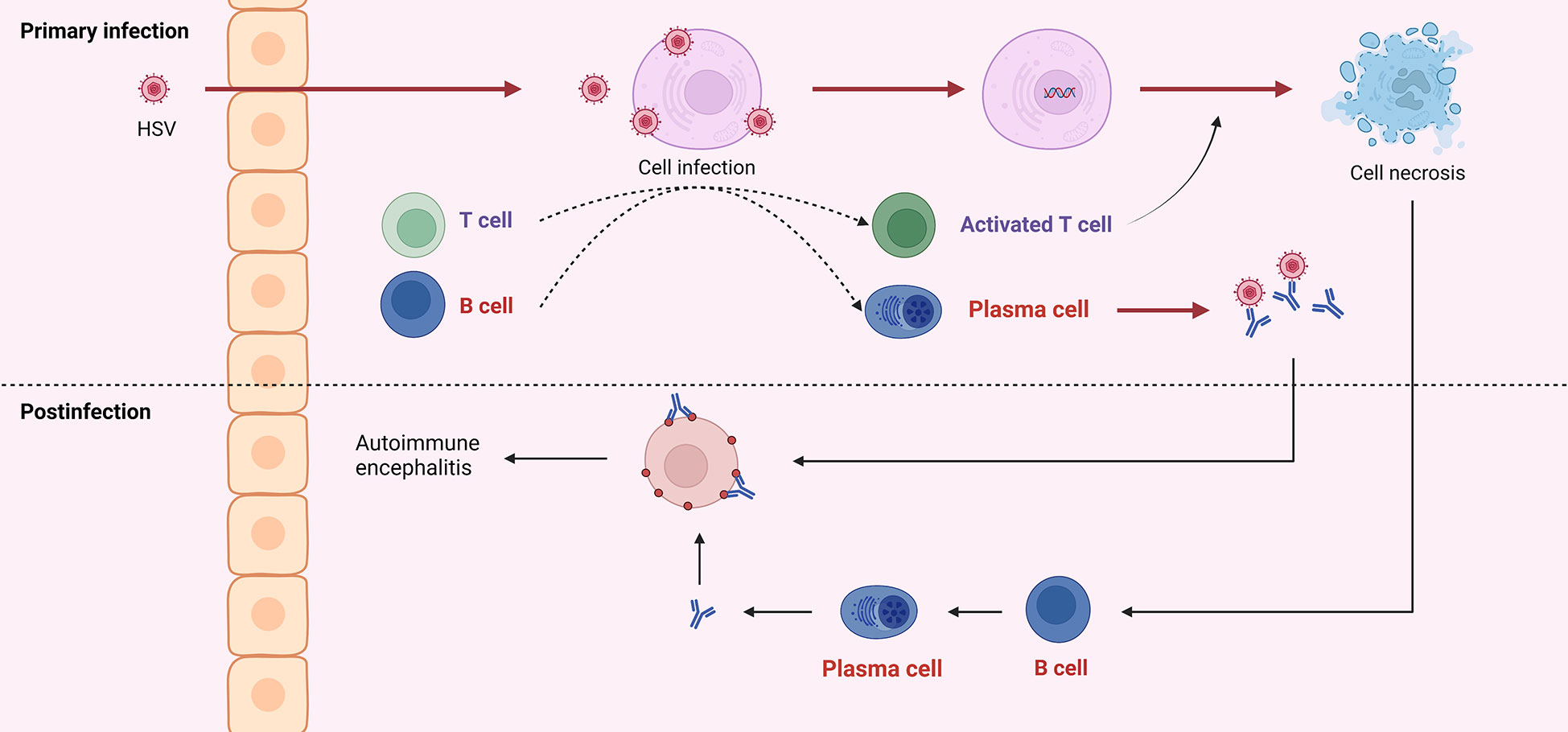
Figure 3 HSV into the central nervous system can cause infection of neurons, which is referred to as primary infection. Both viral particles and infected cells can recruit B and T cells. Antibodies produced by B cells can neutralize virus particles, and T cells can exert cytotoxicity to kill infected cells. Due to the similar structure of viral surface antigens to self-tissue, antibodies derived from the primary infection may attack healthy neurons. And cell disintegration leads to self-antigen exposure, induces B cells to produce antibodies, and further attacks self-cells, which is called post-infectious encephalitis or autoimmune encephalitis.
Patients with herpes simplex encephalitis experience a variety of clinical symptoms. The most common symptoms are headache, fever, and focal neurological symptoms. In severe cases, there may be unconsciousness (94). Early identification and targeted treatment are of great significance to the prognosis of patients.
Aciclovir is a nucleoside analog with potent antiviral properties against herpesviruses. As the first-choice treatment for HSVE, acyclovir has been proven in two previous randomized controlled trials (95, 96). Infectious Diseases Society of America, clinical practice guidelines, recommend treating patients with suspected encephalitis empirically with acyclovir before diagnosis, and for the specific treatment of HSV, acyclovir is also a class III recommendation (97). The British Association of Neurologists and the British Association of Infectious Diseases recommend that if there are no clinical contraindications, cerebrospinal fluid pressure, white blood cell count and classification, protein, and sugar be collected as soon as possible after admission. If there are contraindications, a head CT scan should be performed as soon as possible. When cerebrospinal fluid or imaging suggests viral encephalitis, acyclovir antiviral therapy should be started (10 mg/kg, tid, 14-21 d) (98).
The role of corticosteroids in HSVE is not yet clear, but the treatment is expected to improve cerebral edema, high intracranial pressure, and structural displacements of the brain empirically. In theory, corticosteroids could exacerbate the illness by promoting viral replication. In studies of mice treated with acyclovir combined with corticosteroids, however, the viral load in the brains of mice treated with acyclovir alone did not change significantly, and brain MRI abnormalities in mice treated with corticosteroids decreased significantly (99), demonstrating that corticosteroids can benefit brain injury without affecting viral loads. Acyclovir plus dexamethasone or dexamethasone alone reduced viral load compared to controls (100). A retrospective study describes the benefits of concomitant corticosteroids in patients with HSVE (101). However, British guidelines advise against routinely using corticosteroids for treatment, possibly due to their side effects (98). Corticosteroids have not yet been determined to be the most effective treatment for HSVE. However, if there is obvious edema or mass effect, it is recommended to continue corticosteroids (102). However, animal studies have shown that delayed corticosteroid addition suppresses inflammation and viral genes (103).
In addition to seizures, movement disorders, psychosis, and cognitive changes, NMDAR encephalitis may occur sometime after HSVE (104). The study by Nosadini et al. found that dyskinesia is one of the key symptoms to distinguish HSVE-induced AEs from pure recurrence of HSVE (74). A comprehensive etiology and imaging examination should be performed if symptoms recur and it is impossible to differentiate between virus reactivation and immune induction. New hemorrhage or necrosis on brain MRI often indicates viral replication (74). It is important to consider autoimmune encephalitis if viral testing is negative and to initiate Immunotherapy as soon as possible (105). Immunotherapy is effective in several studies (90, 106–108). AE after HSVE is treated similarly to NMDAR encephalitis, with plasma exchange, corticosteroids, immunoglobulin as first-line treatments, and immunosuppressants, including rituximab, as second-line treatments (109). According to a study, half of the patients with NMDAR encephalitis gradually improved after receiving first-line treatment within 4 weeks. The remaining patients who did not respond well to first-line treatment received a second-line treatment, which was more effective than no treatment (88).
It is a common infectious encephalitis caused by HSV. Although many studies have revealed its etiological mechanism, and many targeted treatment options have been developed, the prognosis is still unsatisfactory, especially for HSVE. The immune system plays an important role in the pathogenesis of herpes simplex virus encephalitis, which is also why corticosteroids play an important role in treating autoimmune encephalitis. Whether it is a molecular simulation or neuron damage, the speculation about the pathogenesis of immune encephalitis is constantly being confirmed. Immunotherapy can have certain curative effects, but the timing of initiation of Immunotherapy is still uncertain, and further research is necessary. In this article, we reviewed the general disease characteristics of herpes simplex virus encephalitis, summarized potential immune mechanisms, and discussed its important complication, autoimmune encephalitis, in the hopes of providing further insight for future research.
LinZ, LijZ, LF, LW, JY and HZ designed and wrote the manuscript. JT, CY and ZX helped with proofreading and revision. All authors contributed to the article and approved the final version.
This work was supported by grants from the Guizhou epilepsy basic and clinical research scientific and technological innovation talent team project (No: CXTD[2022]013), the Collaborative Innovation Center of Chinese Ministry of Education (No: 2020-39), the Guizhou provincial "hundred" level innovative talents funds (No: GCC-2022-038-1), the Guizhou Provincial Science and Technology Foundation (No: ZK2022-656), and the Zunyi City Science and Technology Foundation (No: 2019-71 and 2021-30).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Venkatesan A, Murphy OC. Viral encephalitis. Neurol Clin (2018) 36:705–24. doi: 10.1016/j.ncl.2018.07.001
2. Costa B, Sato DK. Viral encephalitis: a practical review on diagnostic approach and treatment. J Pediatr (Rio J) (2020) 96 Suppl 1:12–9. doi: 10.1016/j.jped.2019.07.006
3. Stahl JP, Mailles A. Herpes simplex virus encephalitis update. Curr Opin Infect Dis (2019) 32:239–43. doi: 10.1097/QCO.0000000000000554
4. Chuquisana O, Strippel C, Gross CC, Melzer N, Kühn J, Kovac S, et al. Antibody response to herpes simplex virus-1 is increased in autoimmune encephalitis. Eur J Immunol (2022) 52:1198–200. doi: 10.1002/eji.202249854
5. Vogrig A, Muñiz-Castrillo S, Desestret V, Joubert B, Honnorat J. Pathophysiology of paraneoplastic and autoimmune encephalitis: genes, infections, and checkpoint inhibitors. Ther Adv Neurol Disord (2020) 13:1756286420932797. doi: 10.1177/1756286420932797
7. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Sci (New York N.Y.) (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
8. Kurtz J. Specific memory within innate immune systems. Trends Immunol (2005) 26:186–92. doi: 10.1016/j.it.2005.02.001
9. Quintin J, Cheng S-C, van der Meer JWM, Netea MG. Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol (2014) 29:1–7. doi: 10.1016/j.coi.2014.02.006
10. Li M, Zhou Y, Feng G, Su SB. The critical role of toll-like receptor signaling pathways in the induction and progression of autoimmune diseases. Curr Mol Med (2009) 9:365–74. doi: 10.2174/156652409787847137
11. Grünewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, et al. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Sci (New York N.Y.) (2003) 302:1396–8.
12. Madavaraju K, Koganti R, Volety I, Yadavalli T, Shukla D. Herpes simplex virus cell entry mechanisms: An update. Front Cell Infect Microbiol (2020) 10:617578. doi: 10.3389/fcimb.2020.617578
13. Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol (2004) 6:401–10. doi: 10.1111/j.1462-5822.2004.00389.x
14. Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, et al. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PloS Pathog (2010) 6:e1000991. doi: 10.1371/journal.ppat.1000991
15. Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol (1997) 136:1007–21. doi: 10.1083/jcb.136.5.1007
16. Richards AL, Sollars PJ, Pitts JD, Stults AM, Heldwein EE, Pickard GE, et al. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PloS Pathog (2017) 13:e1006741. doi: 10.1371/journal.ppat.1006741
17. Zhu S, Viejo-Borbolla A. Pathogenesis and virulence of herpes simplex virus. Virulence (2021) 12:2670–702. doi: 10.1080/21505594.2021.1982373
18. Wysocka J, Herr W. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci (2003) 28:294–304. doi: 10.1016/S0968-0004(03)00088-4
19. Hancock MH, Corcoran JA, Smiley JR. Herpes simplex virus regulatory proteins VP16 and ICP0 counteract an innate intranuclear barrier to viral gene expression. Virology (2006) 352:237–52. doi: 10.1016/j.virol.2006.04.021
20. Jones C. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Microbiol Rev (2003) 16:79–95. doi: 10.1128/CMR.16.1.79-95.2003
21. Pires de Mello CP, Bloom DC, Paixão IC. Herpes simplex virus type-1: replication, latency, reactivation and its antiviral targets. Antivir Ther (2016) 21:277–86. doi: 10.3851/IMP3018
22. Antinone SE, Smith GA. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J Virol (2010) 84:1504–12. doi: 10.1128/JVI.02029-09
23. Warren KG, Brown SM, Wroblewska Z, Gilden D, Koprowski H, Subak-Sharpe J. Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N Engl J Med (1978) 298:1068–9. doi: 10.1056/NEJM197805112981907
24. Nicoll MP, Hann W, Shivkumar M, Harman LER, Connor V, Coleman HM, et al. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PloS Pathog (2016) 12:e1005539. doi: 10.1371/journal.ppat.1005539
25. Marcocci ME, Napoletani G, Protto V, Kolesova O, Piacentini R, Li Puma DD, et al. Herpes simplex virus-1 in the brain: The dark side of a sneaky infection. Trends Microbiol (2020) 28:808–20. doi: 10.1016/j.tim.2020.03.003
26. Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet (2001) 357:1513–8. doi: 10.1016/S0140-6736(00)04638-9
27. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med (2013) 19:1584–96. doi: 10.1038/nm.3407
28. Feige L, Zaeck LM, Sehl-Ewert J, Finke S, Bourhy H. Innate immune signaling and role of glial cells in herpes simplex virus- and rabies virus-induced encephalitis. Viruses 13 (2021). doi: 10.3390/v13122364
29. Reinert LS, Lopušná K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun (2016) 7:13348. doi: 10.1038/ncomms13348
30. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type i interferon pathway. Sci (New York N.Y.) (2013) 339:786–91.
31. Zhang C, Shang G, Gui X, Zhang X, Bai X-C, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
32. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell (2013) 51:226–35. doi: 10.1016/j.molcel.2013.05.022
33. Ishikawa H, Barber GN. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell Mol Life Sci (2011) 68:1157–65. doi: 10.1007/s00018-010-0605-2
34. Ge R, Zhou Y, Peng R, Wang R, Li M, Zhang Y, et al. Conservation of the STING-mediated cytosolic DNA sensing pathway in zebrafish. J Virol (2015) 89:7696–706. doi: 10.1128/JVI.01049-15
35. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448:501–5. doi: 10.1038/nature06013
36. Pham TH, Kwon KM, Kim Y-E, Kim KK, Ahn J-H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J Virol (2013) 87:3076–86. doi: 10.1128/JVI.02860-12
37. Rebsamen M, Heinz LX, Meylan E, Michallet M-C, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep (2009) 10:916–22. doi: 10.1038/embor.2009.109
38. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11 (2010). doi: 10.1038/ni.1932
39. Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A (2012) 109:10558–63. doi: 10.1073/pnas.1203447109
40. Ansari MA, Dutta S, Veettil MV, Dutta D, Iqbal J, Kumar B, et al. Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-β responses. PloS Pathog (2015) 11:e1005019. doi: 10.1371/journal.ppat.1005019
41. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
42. Ma Y, He B. Recognition of herpes simplex viruses: toll-like receptors and beyond. J Mol Biol (2014) 426:1133–47. doi: 10.1016/j.jmb.2013.11.012
43. Liu Y, Goulet M-L, Sze A, Hadj SB, Belgnaoui SM, Lababidi RR, et al. RIG-I-Mediated STING upregulation restricts herpes simplex virus 1 infection. J Virol (2016) 90:9406–19. doi: 10.1128/JVI.00748-16
44. Cheng G, Zhong J, Chung J, Chisari FV, Double-stranded DNA. And double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc Natl Acad Sci U S A (2007) 104:9035–40. doi: 10.1073/pnas.0703285104
45. Crill EK, Furr-Rogers SR, Marriott I. RIG-i is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1. Glia (2015) 63:2168–80. doi: 10.1002/glia.22883
46. Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Sci (New York N.Y.) (2007) 317:1522–7.
47. Mielcarska MB, Bossowska-Nowicka M, Toka FN. Functional failure of TLR3 and its signaling components contribute to herpes simplex encephalitis. J Neuroimmunol (2018) 316:65–73. doi: 10.1016/j.jneuroim.2017.12.011
48. Chen Y, Lin J, Zhao Y, Ma X, Yi H. Toll-like receptor 3 (TLR3) regulation mechanisms and roles in antiviral innate immune responses. J Zhejiang Univ Sci B (2021) 22:609–32. doi: 10.1631/jzus.B2000808
49. Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in toll-like receptor 3-mediated interferon-beta induction. Nat Immunol (2003) 4:161–7. doi: 10.1038/ni886
50. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol (2003) 4:491–6. doi: 10.1038/ni921
51. Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity (2000) 13:539–48. doi: 10.1016/S1074-7613(00)00053-4
52. Sharma S, tenOever BR, Grandvaux N, Zhou G-P, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Sci (New York N.Y.) (2003) 300:1148–51.
53. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol (2011) 11:143–54. doi: 10.1038/nri2937
54. Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A (2004) 101:11416–21. doi: 10.1073/pnas.0403555101
55. He Z, Ye S, Xing Y, Jiu Y, Zhong J. UNC93B1 curbs cytosolic DNA signaling by promoting STING degradation. Eur J Immunol (2021) 51:1672–85. doi: 10.1002/eji.202048901
56. Kim Y-M, Brinkmann MM, Paquet M-E, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452:234–8. doi: 10.1038/nature06726
57. Zhu H, Zhang R, Yi L, Tang Y-D, Zheng C. UNC93B1 attenuates the cGAS-STING signaling pathway by targeting STING for autophagy-lysosome degradation. J Med Virol (2022) 94:4490–501. doi: 10.1002/jmv.27860
58. Zhu H, Zheng C. The race between host antiviral innate immunity and the immune evasion strategies of herpes simplex virus 1. Microbiol Mol Biol Rev (2020) 84. doi: 10.1128/MMBR.00099-20
59. Huang J, You H, Su C, Li Y, Chen S, Zheng C. Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J Virol (2018) 92. doi: 10.1128/JVI.00841-18
60. Zhang D, Su C, Zheng C. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J Virol (2016) 90:5824–9. doi: 10.1128/JVI.00186-16
61. Xu X, Che Y, Li Q. HSV-1 tegument protein and the development of its genome editing technology. Virol J (2016) 13:108. doi: 10.1186/s12985-016-0563-x
62. Ye R, Su C, Xu H, Zheng C. Herpes simplex virus 1 ubiquitin-specific protease UL36 abrogates NF-κB activation in DNA sensing signal pathway. J Virol (2017) 91.
63. Zheng C. Evasion of cytosolic DNA-stimulated innate immune responses by herpes simplex virus 1. J Virol (2018) 92. doi: 10.1128/JVI.00099-17
64. Xu H, Su C, Pearson A, Mody CH, Zheng C. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J Virol (2017) 91.
65. Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe (2018) 24. doi: 10.1016/j.chom.2018.07.004
66. Su C, Zheng C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J Virol (2017) 91. doi: 10.1128/JVI.02414-16
67. Manivanh R, Mehrbach J, Charron AJ, Grassetti A, Cerón S, Taylor SA, et al. Herpes simplex virus 1 ICP34.5 alters mitochondrial dynamics in neurons. J Virol (2020) 94.
68. Pan S, Liu X, Ma Y, Cao Y, He B. Herpes simplex virus 1 γ34.5 protein inhibits STING activation that restricts viral replication. J Virol (2018) 92.
69. Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol (2013) 87:5005–18. doi: 10.1128/JVI.00082-13
70. Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A (2012) 109:E3008–17. doi: 10.1073/pnas.1211302109
71. You H, Lin Y, Lin F, Yang M, Li J, Zhang R, et al. β-catenin is required for the cGAS/STING signaling pathway but antagonized by the herpes simplex virus 1 US3 protein. J Virol (2020) 94. doi: 10.1128/JVI.01847-19
72. Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol (2018) 83:166–77. doi: 10.1002/ana.25131
73. Gelfand JM. Autoimmune encephalitis after herpes simplex encephalitis: insights into pathogenesis. Lancet Neurol (2018) 17:733–5. doi: 10.1016/S1474-4422(18)30279-5
74. Nosadini M, Mohammad SS, Corazza F, Ruga EM, Kothur K, Perilongo G, et al. Herpes simplex virus-induced anti-n-methyl-d-aspartate receptor encephalitis: a systematic literature review with analysis of 43 cases. Dev Med Child Neurol (2017) 59:796–805. doi: 10.1111/dmcn.13448
75. Hara M, Nakajima H. [Autoimmune encephalitis triggered by herpes simplex encephalitis: Main syndrome, diagnosis, and treatment]. Brain Nerve (2022) 74:1163–70.
76. Alexopoulos H, Akrivou S, Mastroyanni S, Antonopoulou M, Dinopoulos A, Giorgi M, et al. Postherpes simplex encephalitis: a case series of viral-triggered autoimmunity, synaptic autoantibodies and response to therapy. Ther Adv Neurol Disord (2018) 11:1756286418768778. doi: 10.1177/1756286418768778
77. Spatola M, Petit-Pedrol M, Simabukuro MM, Armangue T, Castro FJ, Barcelo Artigues MI, et al. Investigations in GABA receptor antibody-associated encephalitis. Neurology (2017) 88:1012–20. doi: 10.1212/WNL.0000000000003713
78. Berek K, Beer R, Grams A, Helbok R, Lindner A, Pfausler B, et al. Caspr2 antibodies in herpes simplex encephalitis: an extension of the spectrum of virus induced autoimmunity? - a case report. BMC Neurol (2022) 22:131. doi: 10.1186/s12883-022-02637-x
79. Armangue T, Moris G, Cantarín-Extremera V, Conde CE, Rostasy K, Erro ME, et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology (2015) 85:1736–43. doi: 10.1212/WNL.0000000000002125
80. Armangue T, Spatola M, Vlagea A, Mattozzi S, Cárceles-Cordon M, Martinez-Heras E, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol (2018) 17:760–72. doi: 10.1016/S1474-4422(18)30244-8
81. Linnoila J, Pulli B, Armangué T, Planagumà J, Narsimhan R, Schob S, et al. Mouse model of anti-NMDA receptor post-herpes simplex encephalitis. Neurol Neuroimmunol Neuroinflamm (2019) 6:e529. doi: 10.1212/NXI.0000000000000529
82. Salovin A, Glanzman J, Roslin K, Armangue T, Lynch DR, Panzer JA. Anti-NMDA receptor encephalitis and nonencephalitic HSV-1 infection. Neurol Neuroimmunol Neuroinflamm (2018) 5:e458. doi: 10.1212/NXI.0000000000000458
83. Moran AP, Prendergast MM. Molecular mimicry in campylobacter jejuni and helicobacter pylori lipopolysaccharides: contribution of gastrointestinal infections to autoimmunity. J Autoimmun (2001) 16:241–56. doi: 10.1006/jaut.2000.0490
84. Zhao ZS, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Sci (New York N.Y.) (1998) 279:1344–7.
85. Höftberger R, Armangue T, Leypoldt F, Graus F, Dalmau J. Clinical neuropathology practice guide 4-2013: post-herpes simplex encephalitis: N-methyl-Daspartate receptor antibodies are part of the problem. Clin Neuropathol (2013) 32:251–4. doi: 10.5414/NP300666
86. De Tiège X, De Laet C, Mazoin N, Christophe C, Mewasingh LD, Wetzburger C, et al. Postinfectious immune-mediated encephalitis after pediatric herpes simplex encephalitis. Brain Dev (2005) 27:304–7. doi: 10.1016/j.braindev.2004.07.007
87. Galli J, Clardy SL, Piquet AL. NMDAR encephalitis following herpes simplex virus encephalitis. Curr Infect Dis Rep (2017) 19:1. doi: 10.1007/s11908-017-0556-y
88. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol (2013) 12:157–65. doi: 10.1016/S1474-4422(12)70310-1
89. Al-Obaidi MMJ, Bahadoran A, Wang SM, Manikam R, Raju CS, Sekaran SD. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol (2018) 62:16–27. doi: 10.4149/av_2018_102
90. Morris NA, Kaplan TB, Linnoila J, Cho T. HSV encephalitis-induced anti-NMDAR encephalitis in a 67-year-old woman: report of a case and review of the literature. J Neurovirol (2016) 22:33–7. doi: 10.1007/s13365-015-0364-9
91. Omae T, Saito Y, Tsuchie H, Ohno K, Maegaki Y, Sakuma H. Cytokine/chemokine elevation during the transition phase from HSV encephalitis to autoimmune anti-NMDA receptor encephalitis. Brain Dev (2018) 40:361–5.
92. Wesselingh R, Butzkueven H, Buzzard K, Tarlinton D, O'Brien TJ, Monif M. Innate immunity in the central nervous system: A missing piece of the autoimmune encephalitis puzzle? Front Immunol (2019) 10:2066.
93. Kothur K, Gill D, Wong M, Mohammad SS, Bandodkar S, Arbunckle S, et al. Cerebrospinal fluid cyto-/chemokine profile during acute herpes simplex virus induced anti-n-methyl-d-aspartate receptor encephalitis and in chronic neurological sequelae. Dev Med Child Neurol (2017) 59:806–14.
95. Whitley RJ, Alford CA, Hirsch MS, Schooley RT, Luby JP, Aoki FY, et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. New Engl J Med (1986) 314:144–9.
96. Sköldenberg B, Forsgren M, Alestig K, Bergström T, Burman L, Dahlqvist E, et al. Acyclovir versus vidarabine in herpes simplex encephalitis. randomised multicentre study in consecutive swedish patients. Lancet (1984) 2:707–11. doi: 10.1016/S0140-6736(84)92623-0
97. Tunkel AR, Glaser CA, Bloch KC, Sejvar JJ, Marra CM, Roos KL, et al. The management of encephalitis: clinical practice guidelines by the infectious diseases society of america. Clin Infect Dis (2008) 47:303–27. doi: 10.1086/589747
98. Solomon T, Michael BD, Smith PE, Sanderson F, Davies NWS, Hart IJ, et al. Management of suspected viral encephalitis in adults–association of british neurologists and british infection association national guidelines. J Infect (2012) 64:347–73. doi: 10.1016/j.jinf.2011.11.014
99. Meyding-Lamadé UK, Oberlinner C, Rau PR, Seyfer S, Heiland S, Sellner J, et al. Experimental herpes simplex virus encephalitis: a combination therapy of acyclovir and glucocorticoids reduces long-term magnetic resonance imaging abnormalities. J Neurovirol (2003) 9:118–25. doi: 10.1080/13550280390173373
100. Thompson KA, Blessing WW, Wesselingh SL. Herpes simplex replication and dissemination is not increased by corticosteroid treatment in a rat model of focal herpes encephalitis. J Neurovirol (2000) 6:25–32. doi: 10.3109/13550280009006379
101. Kamei S, Sekizawa T, Shiota H, Mizutani T, Itoyama Y, Takasu T, et al. Evaluation of combination therapy using aciclovir and corticosteroid in adult patients with herpes simplex virus encephalitis. J Neurol Neurosurg Psychiatry (2005) 76:1544–9. doi: 10.1136/jnnp.2004.049676
102. Bradshaw MJ, Venkatesan A. Herpes simplex virus-1 encephalitis in adults: Pathophysiology, diagnosis, and management. Neurotherapeutics (2016) 13:493–508. doi: 10.1007/s13311-016-0433-7
103. Sergerie Y, Boivin G, Gosselin D, Rivest S. Delayed but not early glucocorticoid treatment protects the host during experimental herpes simplex virus encephalitis in mice. J Infect Dis (2007) 195:817–25. doi: 10.1086/511987
104. Esposito S, Autore G, Argentiero A, Ramundo G, Principi N. Autoimmune encephalitis after herpes simplex encephalitis: A still undefined condition. Autoimmun Rev (2022) 21:103187. doi: 10.1016/j.autrev.2022.103187
105. Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, et al. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry (2021) 92:757–68. doi: 10.1136/jnnp-2020-325300
106. Dorcet G, Benaiteau M, Bost C, Mengelle C, Bonneville F, Martin-Blondel G, et al. Two cases of late-onset anti-NMDAr auto-immune encephalitis after herpes simplex virus 1 encephalitis. Front Neurol (2020) 11:38. doi: 10.3389/fneur.2020.00038
107. Armangue T, Titulaer MJ, Málaga I, Bataller L, Gabilondo I, Graus F, et al. Pediatric anti-N-methyl-D-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr 162 (2013). doi: 10.1016/j.jpeds.2012.10.011
108. Schein F, Gagneux-Brunon A, Antoine J-C, Lavernhe S, Pillet S, Paul S, et al. Anti-N-methyl-D-aspartate receptor encephalitis after herpes simplex virus-associated encephalitis: an emerging disease with diagnosis and therapeutic challenges. Infection (2017) 45:545–9. doi: 10.1007/s15010-016-0959-y
Keywords: herpes simplex virus, autoimmune encephalitis, innate immune, NMDAR encephalitis, immunotherapy
Citation: Zhang L, Zhang L, Li F, Liu W, Tai Z, Yang J, Zhang H, Tuo J, Yu C and Xu Z (2023) When herpes simplex virus encephalitis meets antiviral innate immunity. Front. Immunol. 14:1118236. doi: 10.3389/fimmu.2023.1118236
Received: 07 December 2022; Accepted: 06 January 2023;
Published: 20 January 2023.
Edited by:
Chenhe Su, Wistar Institute, United StatesReviewed by:
Yuelong Yan, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Zhang, Zhang, Li, Liu, Tai, Yang, Zhang, Tuo, Yu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinmei Tuo, dGptdGptMTIyN0AxNjMuY29t; Changyin Yu, eXVjaGFuZ3lpbjY4QDE2My5jb20=; Zucai Xu, ZG9jeHpjQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.